Note: Descriptions are shown in the official language in which they were submitted.
WO g3/10246 PCI`/US92/09~;9
212367 j
--1--
pE8CRIPTIQN
MB~RU~E E~PRE813IQN_Q~HETEROLOGOltJI~ GENE~
S BAC~GROIJND OF ~!HE I~ JTION
Field of the Invention
The invention relates gen~r~lly to the exportation
of heterologous polypeptides to di~crete regions of a
host cell in which it is expressed, to nucleic acid
~eguences encoding exportation polypeptides, to the
preparation of membrane embedded epitopes of immun~genic
antigen~, and to vectors c~nstructed with ~elected
lS exportation sequence~. More particularly, localized
ex~ression of polypeptides may be obtained by providing
exportat~on signals encoded by segment~ of the di~closed
n~cleic acid~ that provide for exportation of expre~ed
heterologous pslypeptides to the inner
membrane/peripla~mic ~pa~e or the outer membrane ~urface
of a ho~t cell.
~escrimtion of Rela~ed Art
Recombinant gene technology has been extensi~ely
i~vestigated in the context of expression o~ foreign
proteins in host cells whi~h harbor recombinant genes,
typically bacterial ho t cell~ 8uch expression is
desirable for producing high value protein~, immunogenic
polypeptides, and in obtaining hybrid proteins t~at are
otherwise difficult to synthesize.
of particular interest is vaccine development. It
i~ potentially feasible to prepare protective vaccines
~rom epitopes of kn~wn antigens of eukaryotic, viral or
prokaryotic pathogens by taking advantage of the
synthetic capacities of transformed host cells. Examples
WO 93/10246 PCI'/US92/09659
--2--
include tumor specific proteins which might be expressed
and utilized to ~timulate an immune response. Oral
vaccines have stimulated re~earch because of the ease of
administration and, more importantly, in some instances
the unsati~actory protection afforded from parenteral
in~-ction. Vaccination against cholera, for example,
g~ve~ ~hort-ter~ protection, thus provoking developmental
work toward an oral vaccine that would presumably
stimulate mucosal inte~tinal immunity more efficiently
~Sanchez et al., 1990).
S~l~onella strains are being studied experimentally
as particularly attractive candidate~ for producing oral
live vaccines. Attenuated strains have been shown to
licit im~une respon~es in several an~al spQcie~
(8t~ugnell et ~1., 1990) and apparently can be highly
l~unogenic in the ho~t. ~umoral antibody re~ponses
includ~ng 108~1 secretory antibody and cellular immune
re~pon~e~ have been ob~erved after oral intake (Dougan et
d ., 1986). Attenuated ~utant~ have been identified via
~¢reening procedure~ ~uch a~ TnphoA mutagenQs~s, which
exclude elimination of mutations in non~ecreted proteins
(Niller st ~1., 1989). However, TnphoA method~ only
indicate asses~ment of integration of the trangposon into
a gene for a secreted or cytoplasmic protein.
Protein expression systems have been developed from
Salmonella strains. A cloning vector useful for
~ntegr~ting DNA into the ~roC gene on Salmonella
cbromo~omes wa~ used to direct expression of heterologous
~ntigens such as tetanus toxin fragment C and Treponema
pall~dum lipoprotein (Strugnell et al., 1990). In some
cases, heterologous polypeptide gene products orally
ad~ini~tered have elicited a serum antibody response, as
3S ~or exa~ple, ,the cholera toxin B subunit protein
expre~sed from a recombinant Yersinia enterocolitica
strain (Sory and Cornelis, 1990). Unfortunately, while
WO 93/10246 2 1 2 3 6 7 ) PCI'/US92/09659
--3--
antibodies were detected in sera of challenged mice, the
response was variable and wa~ directed toward polymer$c
forms of cholera toxin B.
S It i~ recognized that cytopla~mic proteins may not
produce a high immunogenic re~ponse and heterologous
protein~ from recombinant DNA molecules expressed
cytopla~mically o~ten exhibit a diminished antibody
reactivity ~8anchez, ~t al., 1990). Thus ~urface
expre~ed epitopes of bacteria are expected generally to
lioit the greatest humoral re~ponse; however, factors
controlling ~urfacQ expression of heterologous proteins
have not been defined and there is no way to assure that
any given fusion protein will localize to a ho~t cell
lS me~brane sur~ace.
.
VaccineR are the most cost ef~ective medical
intervention known to prevent disease. However,
effec*ive vaccine~ are avail~ble for relatively few
di~ea~es. 8uccessful im~unization again~t infectiou~
organism~ oft~n requires a multicomponent ho~t immune
recponse against a variety of antigenic determin~nts.
Ora}ly admini~tered vaccines, especially live attenuated
~accines, induce specific cell-~ediated effector
re~pon~e~ and elicit ecretory IgA (sIgA) respon~e~.
SIgA i~ ~mportant bec~u~e o~ its e~fecti~ene~s at muco~al
surface~. STgA production and cell e~fector responses
are mediated through the delivery of antigens to gut-
a~sociated lymphoid ti~sue (GALT). Stimulation of GALT
can lead to e~fective cell and humoral defense at all
mNco~al surfaces and provide systemic protection (1,2)~
To deliver antigens to GALT, investigators have
developed avirulent and virulence-attenuated Salmonella
~tain~. Aromatic dependent (aroA (3)), phoP (4), galE
~5), and cya/crp (6) Salmonella mutants have been
reported to interact with GALT in the lamina propria and
W093/10~K PCT/US92/~Sg
st~mul~te an immune respon~e While it is clearly
desirable to use avirulent Salmonella ~trains as carriers
for placmids which expre~Q protective antigen~ of other
pathogens on their surf~ce, it i~ clear that improvements
are needed to develop protective v~ccine~ based on this
~y~te~
~ he u~e of attenuated Salmonella ~train~ to express
hetQrologous antigens and sti~ulate GALT is being
~0 ~xten-ively invostigated In some studies, detectable
leVel8 of 8pecific mucosal and ~erum antibodies to the
h t rologously expressQd antigen have been observed ~7-
10) However, in general results with mo~t antigens have
~sen variable
~It i~ gener~lly believed that the export of
-~h torologous epitopes to the S~lmon~ cell ~urface
nhanco~ their 1D~unogenicity (11) Inve~tig~tors have
u~Qd reco~binant DNA mQthods to expres~ heterologous
epitopes as insert~ in S~l~onell~ flagellin (9) and the
l~mB encoded polypeptide of E col~ (10) In these
studie~, a significant antibody response to the
h~terologous surface-expressed epitope~ was ob~erYed. A
limitation of these systems is the relatively small
number of epitopes which can be inserted into the lamB
and flagellin genes. T~i8 is important as single (or
~ew) epitopes may not result in the broad-baged immune
re~ponse which characterizes today's most successful
vaccines.
There is clearly a need to develop effective systems
to elicit antibody response and in particular to provide
methods of exporting heterologous polypeptides to the
surface of appropriate~host cells. Antigenic peptides
expre~ed on bacterial host cell surfaces may be
signiicant in developing vaccines to such important
antigens as cholera B subunit toxin and HIV antigens.
, ' , ,
WO 93/10246 2 1 2 3 6 7 i PCl/US92/09659
--5--
~RY OF q!~E INVENq!ION
The present invention addressQs one or more of the
foregoing or other problems asso~iated with methods of
controlling surface expression of heterologou~
polypeptides in ~ ho~t cell ~nd provides in particular a
method of directing Qxported polypeptide~ to outer cell
membrane surfaces or to inner membrane/cytoplasmic
regions. ~he invention include~ nucleic acid segments
useful for preparing expression vector~. Such vectors
are suitable $or expressing and directing heterologous
polypeptide~ exported to selected areas of the host cell.
~ransformed cells with surface exprsssed antigene or
epitopic regions ~re expected to be useful as immunogens
lS producing an effective immune respon~e.
The nucleic ~cid segments of the present invention
en¢ode ~ino acid seguQnces associated with particular
targeting o~ fused heterologous polypeptide~ to
p~rticular areas of a tran~formed host cell. It has been
~ound for example that nucleic ~cid ~egments defined by
8EQ ID N0:1 encode a polypeptide product which when fused
to a heterologous polypeptide will direct that
polypeptide to the outer membrane of a bacterial cell.
Z5 By ~eterologous polypeptide i~ mea~t any polxpeptide
other than tho~e normally a~ociated SEQ ID N0:1. It ic
of course understood that such local~zing capabilitie~
are reali~ed under condit~on~ when the exportation
polypeptide i~ incorporated into a ~uit~ble expre~sion
~ector and an appropriate cell host is transfonmed with
that vector~ A preferred embodiment o the DN~ ~egment
i8 defined ~y SEQ ID N0:1. This sequence fused to a phoA
seguence encodes a 46 Kda polypeptide.
Th~ present invention also includes nucleic acid
segment~ encoding amino acid sequences associated with
the transport of heterologous polypep~ides to the
W093/l02~ PCT/USg2/~59
-6-
bacterial inner membrane periplasmic space. Particular
embodiments of these sequences are included in the
nucleic acid sequences defined in SEQ ID NO: 2 . A
preferred inner membrane periplasmic space directing
polypeptide is a 55 Xda polypeptide encoded by the gene
seguence illustrated in F$gure 3 and defined in SEQ ID
N0:2. This preferred embodiment includes gene sequenceg
encoding part of the alkaline pho~phata~e gene, however,
other heterologous genes could be u~ed in place of
alkaline phosphatase.
While particul~r nucleic acid sequence~ have been
defined it is nevertheless contemplated that nucleic acid
s~guQncos will b~ ~ound to vary. It i8 expected that
~n~logou~ seguenoe~ with similar functions may be found
in other gram-negative bacteria such as E. col~.
In certain particular embodiment~, the invention
¢oncerns exp~ession vectors that ~re constructed to
include any of the DNA segments herein disclosed. Such
DNA ~ay be fused dire~tly with a gene of interest and
used in an expre~sion sygtem to produce heterologous
polypeptides ac hybridization probes for, e.g.,
identifying related ~equences, as primers or even as
building blocks ~or the construction of mutant or variant
sequences. A particularly useful application of the DNA
segements of this invention is to achieve directed
~xpression of heterologous polypeptides. Depending on
the DNA segment selected, polypeptides will be expressed
on the inner membrane periplasmic space, the outer
membrane of the host cell, or on the surface of thé outer
membrane of the host cell.
.. ~
In particular embodiments, the pZIP pla~mids of
Figure 2 and Figure 3 have been constructed. Depending
on the plasmid elected, fusion polypeptides are exported
to the inner membrane/periplasmic space or to the ou~er
W093/10246 2 1 2 3 6 7 .i PCT/US92/096sg
membrane of the host cell. In a preferred embodiment,
pZIP-OUT directs the export of fusion polypeptides to the
outer membrane and may also direct a heterologous peptide
to the external surface of a gram-neg~tive ho~t cell.
S pZIP-OUT is a vector which expresses bipartite fusion
which include~ a DNA segment capable of exporting the
fusion product to the external membrane of a gram-
negative cell. The other part of the chimeric gene is a
phoA gene segment lacking signal and expr~ssion segments.
A variety DNA segments m~y be inserted in~o the phoA
~eg~ent at suitable restriction sites to create a
tripartite fusion.
Yet another preferred embodiment is the pZIP-IN
- lS pla~ id ~hown in Figure 3. This pla~id directs the
export o~ polypeptides to the inner ~embrane/periplasmic
~pa¢~. The construction of the plasmid is bipartite.
Part of the aIk~line phosphatase gene lacking signal and
-expression sequen¢eg is fused wi~th a DNA sequence that
¢ontain~ an exportation sequence capable of dir~cting its
fusion polypeptide to an inner membrane/peripl~smic
sp~ce. There are several restriction sites in the phoA
gene segment into which foreign DNA or fr~gment~ of DNA
may be inserted.
Other components of either of these pl~smid~ may
include, in addit$on to the export speci~ying sequences,
resistance genes such as ampicillin or tetracycline
resist~ncé genes. In addition an E. coli phoA gene may
be fused in frame with expression directing DNA
sequences, such as that used to construct the pZIP-IN and
pZIP-OVT plasmids. pZIP-IN additionally encodes a
kana~ycin resistance gene. An advantage of using the
phoA fu~ion is that there ~re variou~ restriction sites
wlthin the phoA gene f~cilita*ing the fusion of
heterologous gene sequences in frame with phoA and the
export specifying sequences.
'
W093/102~ PCT/US92/~659
.~ . .
--8--
Expression vectors may also include a gene encoding
a detectable polypeptide. Typical examples of reporter
genes encoding detectable polypeptides include ~- ~
lactamase and alkaline phosphatase genes. Reporter genes
may be conveniently fused in frame downstream of the
di~clo~ed nucleic acid sequencec with or without other
DNA fragment~/segments. Moreover, restriction sites in
the gene sequence of the reporter gene may be used for
insertion of a desired DNA fragment(s).
Recombinant vectors such as those described are
p~rticularly preferred for tran~forming bacterial host
cells. Several types of bacterial host cell~ may be
employed, most preferred being gram-negative cells such
a~ ~. col~, Salmonella and the like.
Transformed cells may be selected using various
technique~ including ~creening by differential
hybridization, identification of fused reporter gene
produc*~, resistance markers, anti-antigen antibodies,
and ~he like. After identification of an appropriate
clone it may be celected and cultivated under conditions
appropriate to the circumstances, as for example,
conditions favoring expression.
Another aspect of the invention i~ a ~ethod of
preparing heterologous polypeptides. The method
generally inYolves preparing one or more of the
recombinant vectors herein di~closed, transforming a host
c~ll wlth the recombinant vector, then selecting a vector
containing host cell clone and finally isolating from the
cloné the desired polypeptide which will be a
heterologous protein. Examples of useful proteins that
might be used in preparing the recombinant vector include
alkaline phosphatase, cholera toxin B su~unit, fragments
o~ the~e proteins, or any other desired proteins.
W093/10246 2 1 2 3 6 7 ~ PCT/US92/0965g
Depending on the particular recombinant vector
celected for transforming a host cell, recombinant
heterologous polypeptides will be expre~sed in different
compartments of the cell. For tho~e heterologous
S pol~peptides expressed in the inner membrane or
periplasmic space isolation of the heterologous
polypeptide may be affected by cell ly8i~ and other
procedures utili~ed in the isolation of a desired fusion
protein. Heterologous fu~ion proteins exported to the
outer ~embrane of the host cell may be i~olated from the
out~r membrane directly. Typical procedures include
separation of inner and outer cell membrane~ and then
isolation of the fusion polypeptide fxom membranous
material.
~5
In a preferred embodiment, antigenic proteins are
expressed on the surface o~ the host cell. Selected
epitopes of euk~ryotic viral or prokaxyotic pathogens
expressed on the surfa~e of a host cell may be used for
vaccine development. Tu~or specific genes cou'-d be
expressed and utilized to stimulate an immune~response.
Whole cells expressing immunogenic epitopes might be used
for agglu~ination-based screening tests. Surface
expres~ed polypeptides of other organi~ms might be
identi~ied by screen~ng recombinant libraries for
gpecific surface expressed polypeptides. In another
preferred embodiment, cholera toxin B ~ubunit may be
expre~d on the surface of a Salmonell~ harboring the
pZIP-OUT pla~mid vector hereinabove described. When
expre~sed from Salmonella strain TA2362 har~oring plas~id
pRS~18, cholexa toxin B subunits agglutinated in the
presence of specific antibody, indicating exposure of
epi~opic regions on the extexnal membrane surface of
formalin-fixed cells.
Another aspect of the invention involves the
prepaxation of vaccines. Antigens or epitope(s) are
W093~10~K PCT/US92/ ~ 59
,
--10-
~elected and a gene encoding these moieties is inserted
into one or more of the recombinant vectors disclosed.
Appropriate hoct cell~ are transformed and after
screening for transformants one i~ selected which
S ~xpresses the ~ntigen or epitope~ for which a vaccine is
desir~d. Vaccines ~ay then be prepared by a variety of
~ethods. Antigens on the surface o~ appropriate host
cell~ may be ~afely administered orally. For example,
attenuated S~l~onell~ orally administered could stimulate
an i~une regponse on gut ~ucosa. Alternatively, whole
cell8 or cell ~ragmentg containing the ~embrane-bound
antigen ~ay be suitably in~ected into a ~ammal to
generate an ir~une response. In any e~ent, it i8
xpsctQd that th~ immunogenicity of an antigen or epitope
~ay bs ~ignificantly snhanced when expre~ed on the
- ~ur~ace o~ a bacterial cell.
In both im~unodiagno~tic~ and vaccine preparation,
it i~ o~ten po~ible and indeed ~ore practical to prepare
~0 antig ns from s~g~ents of a known lm~n;uMxlunic protsin or
- polypeptide. Certa~n epitopic region~ ~ay be ~sed to
produce responses similar to those produced by the entire
antigeni¢ polypeptide. Often however responses to
epitopic regions are not 80 strong a~ re~pon~es to the
entire polypeptide. HoweYer, ~urface expreg~ion of the~e
epitope~ may generate an enhanced immune response.
In other embodiments, the invention concerns primers
capable of priming amplification of selected portions of
d~clo ed DNA segments~ Primers hybridize to DNA and
serve as initiation sites for synthesis of a portion of
the gene. Nuc}eotide primers are designed to bind at
separate site~ on opposing duplex strains thereby
v~ de~ining the intervening sequence as the portion to be
~pli~ied. Nucleic acid molecules to be employed as
pri~ers whether DNA or RNA will generaily include at
least a lo nucleotide segment of the nucleic acid
wos3/lo~K 2 1 2 3 6 7 t) PCT/US92/~59
sequence of SEQ ID NO: 1 or SEQ ID NO: 2 . The 10 base pair
size is selected as a qeneral lower limit in that ~izes
smaller than 10 bases hybridization stabilization may be
become a problem. However, as the size of the primer
decr-ases too much below 7-8 bases, non-specif~c
hybrld~zation may oc¢ur with other genes hav~ng
complimentary sequences over ~hort stretches.
Primers may be utilized for several purposes. For
1~ -example, primer~ may be used to amplify selected portions
o~ the disclosed DNA segments. Certain primer
co~binations may more effici ntly generate DNA en¢oding
polypeptides that ~ore effectively target to inner or
outer membranes. Additionally, primQrs prepared from the
lS disclosed DNA aay be used to ampli~y regions o~ DNA from
i~ other related organism~ in order to identi~y similar
-~- targeting sequences. Once amplified products are
obtalned probes which referred to nucleic acid molecules
eaployed to detect DNA sequences through hybridization
procedures may be employed to detect~and i~olate selected
DNA ~rag~ents. Like primers, probe~ may be ~NA or RNA
and are generally of similar size usually including at
least a 10 nucleotide ~egment or more, often of 220 or 2
base pairs. Probes may be labeled, for example, by radio
labeling, to a~sist in identification of nucleic acid
seguen¢e~.
As part of the invention, kits useful for the
expre~sion of fusion proteins are al~o envi~ioned
compsising separate containers, each having suitably
aliquoted reagen~s for performing the foregoing methods.
For example, the containers ~ay include one or more
~ectors~ xa~ples being the vectors of claim 19,
particular embodiments of whic~ are ~hown schematically
in Figures 4 and 5. Suitable containers might be ~ials
made o~ plastic or glass,~various tubes such as test
tubes, metal cylinders, ceramic cups or the like.
. ~ ~
W093/10246 PcT/uss2/~sg
-12-
Containers may be prepared with a wide range of suitable
aliquots, depending on applications and on the scale of
the preparation. Generally this will be an amount that
is conveniently handled 80 as to minimize handling and
S subsegu~nt volumetric manipulations. Nost practitioners
will pre~er to select suitable endonuclea~es from common
~upplies usually on hand; however, such restriction
endonucleaQes could also be optionally included in a kit
preparation.
Vectors supplied in kit form are preferably supplied
in lyophilized fo~m, although such DNA fragments may also
be taken up in a suitable solvent such as ethanol,
glycol~ or the like ~nd supplied as suspen~ion~. For
~t ~pplic~tion~, it would be de~ir~ble to remove the
~olv~nt which for ethanol, for example, is a relatively
~imple ~atter of evaporation.
BRIEF DE8CRS N TON OF T~ DRA~ING8
Figure l illustrates the cloning of phoA ~ene fusion
from TnphoA insertion mutants and congtruction of tribrid
gene fw ions. TnphoA is a derivative of TnS which
encodes ~. coli alkaline phosphata~e, minus the 6ignal
sequence and expression signal~, inserted into the left
ISSOL element ~2l). Random trangposition of TnphoA
result~ in an active insertion only when the phoA gene
~eguence i~ fused in frame downstream of the promoter and
~xport signals of a target gene (A). The point at which
the phoA sequence joins the target gene is referred to as
the fusion joint (FJ). The remaining portion of the gene
beging at the distal joint (DJ). Utilizing restriction
enzymes which cut either downstream of the kan~mycin
re~tance gene (e.g., BamHI) or the phoA gene sequence- 35 (e.g., ~indIII), allows cloning of phoA gene fusions ~if
the target gene is not also restricted ("R")). Plasmids
carrying phoA gene fusions can then be used as exposition
WO93/10246 212 3 6 7 a PCT/US92/~ss
vector~ (B). The SspI and PvuII restriction sites in
phoA provide blunt ended sites ~t which in frame
in~ertions (IF) of a gene of interest ( GOI ) can be
in~erted. The GOI must al~o be con~i~tent with the phoA
S fra~e at the in~ertion ~ite. The re~ulting tribrid gene
fusions contain the expression and export signals of the
target gene fu~ed in ~rame with the phoA and GOI
sequences.
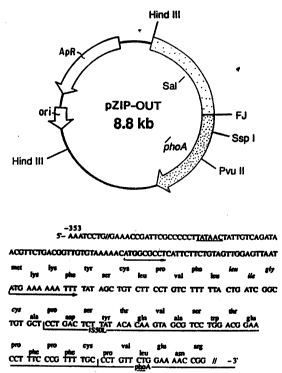
Figure 2B show~ the DNA sequence across the
S~l~onell~: :poA fu~ion ~oint in pZIP-OUT. Dideoxy
sequencin- (Segu~na~e 2.0 USB Bioche~cals) was u~ed to
deter~lne the 353 base pairs (bp) upstre~m of the
S~ln~ phoA ~usion ~oint. A ~lngle open reading
lS fr~e (ORF) wb~ch wa~ in ~rame with that of the
IS50L/p oA ~equence was observed. A stop codon in this
ORF wa~ ob~erved at po~ltion -99. Mult~ple stop codons
in all reading ~rames were present in sequences -l50 to -
200. Two putatlve translation start codons (AUG) were
present at positions -84 and -51.~ A putative Prlbnow box
) was present at posi~ion -120. ~he predic~ed amino
acid s0guence of the coding region is ~hown above the
nucleotide sequence. The IS50L and the beginning of the '
phoA derived ~equences are underlined.
Figure 2 ~chematically shows pla~mid pZIP-OUT
contains a 4.5 Kb ~ndIII chromosomal fragment from
ln~asion-attenuated S. typhimur~um TnphoA insertion
mutant TAP 43 inserted into pBR322 at the H~ndIII site.
It expresses a 46 Kd PHOA fusion prote~n which localizes
to the uter membrane.
i Figure 3 how~ plasmid pZIP-IN which cont~ins a
d I chromosomal fragment from S. typhimur~um TnphoA
~n~ertion ~ut~nt TAG 28, inserted into pBR322 at the
BamHI sité. It expresses a 55 k~ PhoA fusion protein
which localizes to the inner membrane.
,~:
: '
WO93/10246 PCT/USg2/ ~ 59
Figure 4 is an immunoblot analysis of S~lmonella
membrane preparations using mouse anti-alkaline
phosphatase. s. typhimurium TA 2362 harboring pBR322
showed no reaction ~n the total envelope (TE) . TA 2 3 62
S harboring pZIP-OUT showed a 46 Kd PHOA fusion in the TE
and after separation of the inner and outer membrane by
treatm~nt with 0.5% sarkosyl, the majority oS the fusion
protein was a~ociated with the outer membrane (OM). TA
2362 harbosing pZIP-IN showed a 55 Xd PhoA fusion protein
in the TE and after separation of the inner and outer
nembr~ne by treatment with 0.5% sarko~yl, the ma~ority of
the fusion protein W~8 found associatQd with the inner
membrane (IN). ~ll lanes were loaded with membrane
preparations from an equal amount of cell~.
~5
Figure S i8 an i~munoblot analy~i~ of urea extracts
(gURF) using anti-alkaline phosphatase as the primary
antibady. S. ty1~ub~1r~um TA 2362 harboring pB~322 ~howed
no reacting polypeptides to the alkaline phosphatase
antibodie~. TA 2362 harboring pZIP-OUT showed a PhoA
fusion at 46 Kd. TA 2362 harboring pZIP-IN showed no
reacting polypeptides with the ~ame antisera. Lanes were
loaded with an eguivalent amount of extract prepared from~
an eguivalent number of whole cells.
Fi~ure 6 show~ the derivation o~ plasmid pRSPl8^from
pZIP-OUT in which the final 294 base pairs of ctKB have
been inserted in frame (IF) with the phoA gene sequence
at the PvuII site. The ctxB gene seguence is from
pRITl0810 which encodes the entire ctxB gene (22).
Figure 7 ~hows the derivation of plasmid pIMB13 from
pZIP-IN in which the fin~l 294 ~ase pairs of ctxB have
been inserted in frame (IF) with the phoA gene séquence
at the S~pI ~ite. The ctxB gene sequence is from
pRITl0810 which encodes the entire ctxB gene (22).
wos3/lo246 PCT/US92/09659
2123~7.~
Figure 8 is a schematic representation of the CtxB
fusion from pRSP18 and pIMB13 that results in exportation
of the 32 kDa CtxB protein to the outer and inner
membranes, respectively.
Figure 9 is an immunoblot analysis of urea extracts
(SURF) using affinity purified anti-CTB a~ the primary
antibody. S. typh~murium TA 2362 harboring pRITl0810
which encodes cytoplasmically expressed CTB howed no
~0 reaction. TA 2362 harboring pRSP18 showed a CTB tribrid
fusion protein at 32 Kd. TA 2362 harboring p: ~13 showed
no re~ctivity to anti-CTB antibodies. ~anes were loaded
with equal ~mount~ of extract from eguivalent numbers of
whole cells.
Figure 10 is a proposed protocol for insertion of a
~ragment o~ HIV gpl60 gene into pZIP-OUT.
Figure 11 shows the sequence of export specific
signal in pZIP-IN. Promoter and regulatory sequences are
underlined. IS50L and phoA sèquences from pZIP-IN are
shown. The ORF is ghown in capital letters.
PBTAI~D DE8C~IPTION OF ~HB PR~FE~p_EMBODINB~T8
The present invention xelates to nucleic acid
segments encoding particular polypeptide~ capable of
~orming fusion proteins that export to pasticular areas
o~ a host cell. These nucleic acid segments are useful
in constructing vectors that allow expression o~
heterologous proteins in appropriately transformed host
cells. Polypeptides may be localized within the inner
membrane/periplasmic space or on the outer membrane
~ur~ace. Antigens or epitopic regions of antigens
localized on host cell membranes have particular
potential for vaccine development and antibody
production.
WO93/10246 PCT/USg2/~59
-16-
A heterologous gene expression system has been
developed which utilizes a virulence-attenuated
Salmonella as a carrier for a plasmid expression system
~pZIP-OUT) which can direct the products of large
5 segments of heterologous genes to the outer membrane
(Fig. 2). Recombinant DNA techniques are utilized to
fuse the reading frame of the gene to be expressed with
Salmonell~ export specifying sequence~, Figure 1.
Several cloning sites are possible which allow
10 ~aintenance o~ the proper reading frame and prQduce
tribrid fusion polypeptides which contain Salmonella
~xport specifying sequenc~s, the heterologous gene
sequences and phoA gene sequences. Recombinants which
export the tribrid fusion protein are selected through
15 th~ loss o~ phoA act~vity and appearance of the pr~dicted
;~ ~usion polypeptide on the sur~ace o~ the outer membrane.
:: a tribrid ~usion has be~n con~tru¢ted which encodQs
virtually the entire cholera toxin B subunit (¢txB) gene,
Figure 6, and evaluated its subcellular localization in
20 S~l nella. This fusion polypeptide is expres d on the
Salmonella surface as evidenced by: 1) aggl~tination of
-~ tribrid fusion expressing strains by anti-CTB antiserum,
2) localization of the fusion polypeptide in the outer
membrane, and 3) the presence of the fusion polypeptide
25 in cell ~urface preparations.
The DNA of the present invention was i~olated from
Salmonella typh~murium, ~train TAP43, an invasion
attenuated strain. Invasion attenuated refers to species
30 which have lost one or more virulence factor affecting
- the efficiency by which Salmonella invades epithelial
cells. Isolation of an attenuated strain of Salmonella
was considered useful in developing the present invention
x because such strains may be used to deliver heterologous
35 antigens to the gut of an animal. Salmonella given
orally tends to establish an infection in the intestinal
mucosa, leading to an immune response. The presence ~f a
, ~ .
W093/l0246 PCT/US92/09659
21231~7 )
desired antigen is expected to stimulate a response to
that species, as well as to the Salmonella or other host
antigens.
The a~proach to screening for protein export signals
was to use alkaline phosphatase fusions based on the
TnphoA transposon system reviewed by Manoil et al.
~1990). TnpboA i8 a transposon derivative of Tn5 in the
phoA gene which lacks a promoter, tran~lation initiation
site, ~gnal ~equence DNA and the first five amino acids
of its protein. When the transposon, TnphoA, inserts
into a for~ign gen~ in the correct orientation and
reading ~rame, g~-~e fusiong are generated, coding for
hybrid proteins ~hSch have alkaline phosphatase activity
~ tr~n~ported beyond the inner membrane. Detection of
such activity ~s generally accompli~hed with an alkaline
pho~phata~e ind~cator dye, allowing ~i~ualization of
colored colonie~ for ~uccessful gene fusion~ that lead to
export of heterologous gene products.
Part of the present invention contemplate~ ~accine
preparation and use. In general, it is con~emplated that
antigen~ r or epitopes of antigens, will be readily
expre~sed in localized regiong of a host cell u~ing the
~ethod~ disclo~ed. Expres~ion vectorC incorporating the
DNA segment encoding exportation polypeptides directing
products to a host cell outer me~brane surface ar~
expected to be particularly use~ul. Epitoplc regions of
antigens, well ~xposed ~t a ~embrane surface, may elicit
high immunogenic responses, pro~iding a route to vaccines
or antibody production.
General concepts related to ~ethods of vaccine
preparation and use are discussed as applicable to
3S preparations and formulations with antigens, epitopes or
sub~ragments of such antigens obtained from various
W093/lO~K PcT/uss2/~6s9
-18-
~ourceC; for example, cholera B toxin subunit and the
like
vaccine Pre~ration and use
s
Preparation of vaccines which contain peptide
seguenoe~ as active inqredients i8 generally well
understooa in the art, a~ exemplif~ed by U S Patents
4,608,251; 4,60~,903; 4,S99,231; 4,599,230; 4,596,792;
ana 4,578,770, all incorporated herQin by refQrQnce
Typically, such vaccine~ are prepared as in~ectables
eith~r a~ liguid solution~ or suspen~ions; solid forms
suit~ble for solution in, or ~u~pen~ion in, liguid prior
to in~ection ~ay also be prepared The preparation may
lS al-o b ~ul~ified The acti~ immunoqenic inqrediQnt is
oft-n ~ix d with ~xcipien~s whi¢h are phar~aceutically
aoc pt bl- and co~patible witb the activ~ i~gr dient
~uitable~xcipients ar~, for example~, water, saline,
doxtro~e, glycerol, ethanol, or tbe like, and
co bination~ thereof In addition, if aesired, the
vaccine ~ay contain ~inor a~ounts of auxiliar~ sub~tances
~UCh a8 v tting or emul ifying agent4,~pH buffering
agen~8, or ad~uvants which enhance the effectiveness of
the vaccines.
The vaccines are conventionally administered
parenterally, by in~ection, for example, either
subcutaneou~ly or intramuscularly. Additional
formulations which are suitable for other modes of
admlnistr~tion include suppositories and, in some cases,
o~al formulations. For suppositories, traditional
binaers and carriers may include, for example,
polyalkylene glycols or triglycerides; such suppositories
may be~or-ed from mixtures containing the attive
ingredient in the range of 0.5% to 10%, preferably l-Z%.
~,
~; Oral for~ulations include such normally employed
excipients as, for example, pharmaceutical grades of
W093/10246 2 123 67 i PCT/USg2/~59
--19--
mannitol, lactose, ~tarch, magnesium stearate, sodium
saccharine, cellulose, magnesium carbonate, and the like.
These compos~tions take the form of solut~ons,
suspension~, tablet~, pillC, capsule~, ~ustained release
formulations or powder~ and contain 10-95% of active
ingredient, preferably 25-70%.
The protQins may be ~ormulated into the vaccine as
neutral or salt form~. Pharmaceutically acceptable salts
include acid addition salts (formed with the free amino
groups o~ the peptide) and which are formed w~th
inorganic acids such as, for example, hydrochloric or
pho~phoric acids, or such organic acid~ as a¢etic oxalic,
tartaric, ~andelic, and the like. 8~1ts formed with the
lS free boxyl group~ ~ay also be derived from inorganic
b~e~ uch a~, for example, sodium, potassium, ammonium,
c~lciu~, or ~erric hydroxides, and such organi¢ ~ase~ as
l~opropyl~ ine, trimethylamine, 2-ethylamino ethanol,
hi~tidine, procaine, and the like.
The vaccines are administered in a manne~ compatible
with the do~age formulation, and in such amount as will
be therapeutically effective and immunogenic. The
gy~ntity to be administered depends on the sub~ect to be
2~ treated, including, e.g., the capacity of the
individual's immune system to synthesize antibodies, and
the degree of protection de~ired. Precise amount~ of
active ingredient required to be administered depend on
the ~udgment of the practitioner. However, suitable
dosage ranges are of the order of several hundred
- micrograms acti~e ingredient per vaccination. Suitable
r~imes for initial administration and booster shots are
a_~o ~ariable but are typified by an initial
admini~tration followed by subsequent inoculations or
3S other administrations.
wos3/lo~K PcT/uss2~ss
-20-
The manner of application may be varied widely. Any
of the conventional methods for administration of a
vaccine are applicable. These are believed to include
oral application on a solid phy~iologically acceptable
S ba~e or in a phy~iologically acceptable dispersion,
pasenterally, by in~ection or the like. The do~age of
the vaccine will depend on the route of administration
and will vary according to the size of the host.
Various methods of achieving ad~uvant effect for the
vaccine include use of agents ~uch as aluminum hydroxide
or phosphate (alum), commonly used as O.OS to 0.1 percent
~olution in phosphate buffesed saline, admixture with
~ynthetic polymers of sugasC (Carbopol) used as 0.25
p-rcent solution, aggregation of the protein in the
vaccine by heat treatment with temperatures ranging
bstw on 70 to 101C for 30 second to 2 minute period~
r pe¢tively. Aggregation by reactivating with pepsin
treated (Fab) antibodies to album~n, ~ixture with
bacterial cells such as C~ pQrvum or endotoxin~ or
lipopolysaccharide components of gr~m-negative bacteria,
e~Nl~ion in physiologically acceptable oil vehicles ~uch
~ mannide mono-oleate ~Aracel A) or emulsion with a 20
percent ~olution of a perfluorocarbon (Fluosol-DA) used
a~ a block substitute may al~o be employed.
In many instances, it will be desirable to have
multiple admini~trations of the vaccine, usually not
~xceeding six vaccinations, more usually not exceeding
four vaccinations and preferably one or more, usually at
lea~t about three vaccinations. The vaccinations will
normally be admini~tered from two to twelve week
~ntervals, more usually from three to five week
intervals. Periodic boosters at intervals of 1-S years,
u~ually three years, will be desirable to maintain
protective levels of the antibodies. The course of the
immunization may be followed by assays for antibodies for
W093/10246 2 12 3 6 IJ PCT/US92/09659
-21-
the supernatant antigens. ~he assays may be performed by
labeling with conventional labels, such as radionuclides,
enzymes, fluorescers, and the like. These techniques are
well known and may be found in a wide variety of patents,
such as U.S. Patent No~. 3,791,932; 4,~74,384 and
3,949,064, ~s illustrative of these types of assay~.
The invention also contemplates the use of disclosed
nucleic acid segments in the construction of expression
vectors or plasmids and use in host cells. The ~ollowing
is a general discussion relating to such use and the
particular considerations in practicing thi~ aspect of
the invention.
Host Cell Cultures and V~ctors
In general, of course, prokaryotes are preferred for
the initial cloning of DNA sequences and constructing the
vectors useful in the invention. For example, in
addition to the particular strains mentioned in the more
specific disclosure below, one may mention by~way of
example, strains such as E. coli K12 strain 294 (ATCC No.
31446), E. coli B, and E. coli X 1776 (ATCC No. 31537).
These examples aré, of course, in~ended to be
illustrative rather than limiting.
Other prokaryotPs may also be preferred for
expres~ion. The aforementioned strain~, as well ac E.
col~ W3110 (F , lambda-, prototrophic, ATCC No. 273325),
bacilli ~uch as Bacillus subtilus, or other
enterobacteriaceae such as Salmonella -,yphimurium or
Serratia marcesans, and various Pseudomonas species may
be used.
In general, plasmid vectors containing replicon and
control sequences which are derived from species
compat~ble with the host cell are used in connection with
W093/l0246 PCT/US92/ ~ 59
~ ~,
-22-
these hosts. The vector ordinarily carries a replication
site, as well as marking ~equences which are capable of
providing phenotypic selection in transformed cells. For
ex~mple, E. coli is typically transformed using pBR322, a
plasmid derived from an E. coli species (see, e.~.,
~olivar ot al., 1977). The pBR322 plasmid contains genes
for ampicillin and tetracycline re~istance and thus
provides easy means for ident~fying transformed cells.
The pBR plasmid, or other microbial plasmid or phage must
also contain, or be modified to contain, promoters which
can b~ used by the microorganism for expression.
Those promoters most commonly used in recombinant
DNA construction include the B-lactamase (penicillinase)
~nd lactose promoter ~y~tems (Chang et al., 1978; Itakura
et ~1., 1977; GoeddQl ot ~1., 1979) and a tryptophan
(trp) promoter system ~Goeddel et ~1., 1979; EP0 Appl.
~' Publ. No. 0036776). While these are the most ¢ommonly
us~d, other microbial promoters have been dis¢overed and
util~zed, and details concerning their nucleotide
~eguence~ have been published, n~bling a sk~lled worker
to ligate them functionally into plasmid vectors
~Siebwenlist et ~1., 1980). Certain gene~ from
prokaryotes may be expressed efficiently in E. coli from
their own promoter ~eguence~, precluding the need for
addition of another promoter by artificial mean~. ''
In addition to prokaryotes, eukaryotic microbes,
such as yeast cultures may also be used. Saccharomyces
cerevisiae, or common baker's yeast is the most commonly
used among eukaryotic microorganisms, although a number
of other strains are available. For expression in
- Sacch~ro~yc~s, the plas!id YRp7, for example, is commonly
used-(Stinchcomb et al., 1979~; Kingsman et al., 1979;
T~chemper et ~1., 1980). This plasmid already contains
the~trpl gene which provides a selection marker for a
mutant strain of yeast lacking the ability to grow in
W093/1O~K 2 1 2 3 6 7 ~ PCT/US92/09659
-23-
tryptophan, for exa~ple ATCC No. 44076 or PEP4-~ (Jones,
1977). The presence of the trpl lesion as a
oharacteristic of the yeast host cell genome then
provides an effective environment for detecting
S transformation by growth in the absence of tryptophan.
Suitable promoting ~quence~ in yeast vectors
include the promoter~ ~or 3-phosphoglycerate kinase
~Hltzman ot ~1., 1980) or other gly¢olytic enzymes (Hess
et ~1., 1968; Holland et ~1., 1978), such as enolase,
glyoeraldehyde-3-pho~phate dehydrogenase, hexokinase,
pyruvate de¢arboxylase, phosphofructokina~e, glucose-6-
phosphate isomerase, 3-phosphoglycer~te mutase, pyruvate
kina~, trio~ephosph~te isomera~e, phosphoglucose
lS l-oD-ra~e~ and glu¢okina~e. In constsu¢ting suitable
~Ypr ~1on~plasmids, thQ ter~lnation sequences associated
wlth th 8~ ~Qnes are also ligated into the expre~sion
¢tor 3' o~ the sequence desired to be expressed to
provide polyadenylation of the mRNA and termination.
~,,,
Other promoter~, which have the additional advantage
o~ transcription controlled by growth conditions are the
promoter region for alcohol aehydrogenase 2,
i~ocytochrome C, acid phosphatase, degradative enzymes
a~sociated with nitrogen metabolism, and the
a~orementioned glyceraldehyde-3-phosphate dehydrogenase,
and enzymes re~pon~ible ~or maltose and galactose
utilization. Any plasmid vector containing a yeast-
compatible promoter, origin of replication and
term$nation ~equences is suitable.
.
In addition to microorganisms, c~ltures of cells
der$v~d ~rom multicellular organi~ms may also be used as
host~. In principle, any such cell culture is workable,
whether ~rom vertebrate or invertebrate culture.
,. ,
However, interest has been greatest in vertebrate cells,
~-- and propagation of vertebrate ceIls in culture (tissue
,' - .
W093/10246 PCT/US92/~K59
-24-
culture) has become a routine procedure in recent years
~Tissue Culture, 1973). Examples of such useful host
cell lines are VER0 and ~eLa cells, Chinese hamster ovary
(CH0) cell lines, and Wl38, BHK, COS-7 293 and MDCK cell
S lines. Expression vectors fos such cells ordinarily
include (if necessary) an origin of replication, a
promoter located in front of the gene to be expressed,
along with any necessary ribosome binding sites, RNA
splice ~ites, polyadenylation site, and transcriptional
ter~inator sequences.
For use in ~a~malian-cells, the control functions on
the expression v~ctors are often provided by viral
raterial. For example, commonly used promoter~ are
d riv d ~rom polyoma, Adenovirus 2, and most frequently
8~ n Virus 40 (SV40). The early and late promoters of
8V40 virus ~re particularly useful because both are
obtained easily from the virus as a fragment which also
contains the SV40 viral origin of replication (Fier~-et
~l., 1978). Smaller or larger SV40 fragments may al~o be
us~d, provided there is included the approxim~tely 2S0 bp
~ . ,
seguence extending from the HindIII ~ite toward the BglI
~ite located in the viral origin of replication.
Further, it is aiso possible, and often desirable, to
utilize promoter or control sequenoes normally associated
with the desired gene ~eguence, provided &uch control
~eguences are compatible with the ho~t cell 6y~tems.
An origin of replication may be provided either by
con~truction of the vector to include an exogenous
origin, such as may be derived from SV40 or other viral
(e.g., Polyoma, Adeno, VSV, BPV) source, or may be
provided by the host cell chromosomal replication
- mechan~m. If the vector is integrated into the host
cell chromo~ome, the latter is often sufficient.
:
: : ,
W093/10246 2 1 2 3 6 ~ ~ PCT/US92/09659
Also contemplated within the scope of the present
invention is the use of the disclosed DNA as a
hybridization probe. While particular examples are
provided to illustrate such use, the following provides
general background for hybridization applications taking
advantage of the disclosed nucleic acid sequences of the
invention.
~ucleic Acid HYbridization Embodiments
In certain aspects, the DNA sequence information
provided by the invention allows for the preparation of
relatively short DNA (or RNA) sequences having the
~bility to specifically hybridize to S. typh~murium gene
seguences. In these aspects, nucleic acid probes of an
appropriate length ~re prepared based on a consideration
of the sequence, e.g., as shown SEQ ~D N0:1 ~nd SEQ ID
N0:2 or derived from flanking regions of these genes.
The ~bility of cuch nucleic acid probes to specifically
hybridize to the S. typhimurium gene sequences lend them
particular utility in a variety of embodiments. The
probes can be used in a variety of diagnostic assays for
detecting ~he presenoe of pathogenic organisms in a gi~en~
sampl~. However, other uses are envisioned, including
the use of the sequence information for the preparation
of mutant species primexs, or primers for use in
preparing other genetic construct~.
To provide oertain of the advantages in accordance
with the invention, the preferred nucleic acid sequence
e~ployed for hy~ridizations or assays includes ~equences
~hat are compl~mentary to at least a lo to 40, or so,
nucleotide ctretch of the selected ~equence, such as that
~hown in Figure 1 or Figure 2, SEQ ID N0:1 or SEQ ID
N0:2. A ~ize of at least 10 nucleotides in length helps
to ensure that the fragment will be of sufficient length
to form a duplex molecule that is both stable and
W093/~02~ PCT/US92/09659
-26-
selective. Molecules having complementary sequences over
stretches greater than lo bases in length are generally
preferred, though, in order to increase stability and
selectivity of the hybrid, and there~y improve the
quality and degree of specific hybrid molecules obtained.
~hus, one will generally prefer to design nucleic acid
molecules having gene-complementary stretches of 15 to 20
nucleotides, or even longer where desired. Such
fragments may be readily prepared by, for example,
directly synthesizing the fragment by chemical means, by
application of nucleic acid reproduction technology, such
a~ the PCR technology of U.S. Patent 4,603,10~, or by
introducing selected sequences into recombinant vectors
for recombinant production.
~5
Accordingly, the nucleotidæ sequences o~ ~he
invention are important for their ability to selectively
form duplex molecules with compleme~tary stretches of S.
typh~murium gene segments. Depending o~ the application
envisioned, one will desire to employ varying conditions
of hybridization to achieve varying degrees o~
selecti~ity of the probe toward the target ~equence. For
applications requiring a high degree of selectivity, one '
will typically desire to employ relatively stringent
conditions to form the hybrids, for example, one will
select relatively low salt and/or high temperature
conditions, such as provided by 0.02 M-0.15 M NaCl at
temperatures of SOoC to 70C. These conditions are
particularly celective, and tolerate little, if any,
mismatch between the probe and the template or target
strand.
Of course, for some applications, for example, where
one desires to prepare mutants employing a mutant primer
~trand hybridized to an underlying template, less
stringent hybridization conditions are called for in
order to allow formation of the heteroduplex. In these
WOs3/10~K 2 1 2 3 6 ~ ~ PCT/US92/~Sg
-27-
circumstances, one would desire to employ conditions such
as 0.15 M-0.9 M salt, at temperatures ranging from 20OC
to s5Oc. In any case, it is generally appreciated that
conditions can be rendered more stringent by the addition
of inoreasinq amounts of formamide, which serves to
de~tablllze the hybrid duplex in the same manner as
incre~d temperature. Thu~, hybridization conditions
can be re~dily manipulated, and thus will generally be a
method of choice depending on the d~sired result~.
The present invention is envisioned as useful in the
clonlng of nucleic acids encoding cestain exportation
polypeptides. Identification of othes expostation
polypeptldes in addition to the 46 kDa and 55 kDa
lS prot lns chould be possible using methods analogous ~o
tho~e di~clo~ed herein. One method would be to produce a
cDNA ~ibr~ n using mRNA obtained from mutant S.
typbi urluJ strains. Although the production of cDNA
libraries from bacteria is not commonly done because of
the usual absence of poly-A tails on prokaryotic
messages, a cDNA library could be constructe~ from S.
typ imur~um mRNA.
A method of preparing variants of the S. typhimurium
exportation polypept~des is ~ite-directed mutagenesis.
Thi~ technique is useful in the preparation of individual
peptides, or biologically functional equivalent proteins
or peptide~, derived from the 46 kDa or 55 kDa protein
seguence, through specific mutagenesis of the underlying
DNA. The technique further provides a ready ability to
prepare and ~est sequence variant~ ~or example,
inc~rporating one or more of the foregoing
considerations, by introducing one or more nucleotide
~eguence changes into the DNA. Site-specific mutagenesis
'~ 35 allows the production of mutants through the use of
~peci~ic oligonucleotide sequences which encode the DNA
equence of the desired mutation, as well as a sufficient
W093~l0246 PCT/US92/~59
-28-
number of adjacent nucleotides, to provide ~ primer
seguence of sufficient size and sequence complexity to
form a stable duplex on both sides of the deletion
junction being traversed. Typically, a primer of about
17 to 25 nucleotides in length is preferred, with about s
to lO residues on both ~ides of the junction of the
sequence being altered.
In general, the technique of site-specific
~0 mutagQnesis is well known in the art as exemplified by
publications (Adelman ~t ~1., 1983). As will be
appreciated, the technique typically employs a phage
~ector which exi~t~ in both a single stranded and double
stranded ~orm. Typical vectors u~eful in site-directed
lS mutag necis include vector~ such as the M13 ph~ge
(Mb~cing t ~l., 1981). These phage are readily
co~wrcially ~vailable and their use is generally well
known to thoSQ skilled in the art.
In general, slte-directed mutagenesig in accordance
herewith is performed by first obtaining a single-
~tranded vector which includes within its sequence a DNA
~equence whlch encodes an export polypeptide. An
oligonucleotide primer bearing the desired mutated
seguence is prepared, generally synthetically, for
example by the method o~ Crea ct al. (1978). This primer
ic then annealed with the single-~tranded vector, and
sub~ectsd to DNA polymerizing enæymes such as ~. coli
polymerage I Klenow fragment, in order to complete the
~ynthesis of the mutation-bearing strand. Thus, a
heteroduplex is formed wherein one s~rand encodes the
original non-mutated seguence and the second stxand bears
the desired mutation. This heteroduplex vector is then
u~ed to transform appropriate cells, such as E. coli
r; 35 cellg, and clones are selected which include recombinant
vectors bearing the mutated sequence.
~ :
W093/lO~K 2 1 2 3 6 ~ 5 PCT/US92/Og659
-29-
The preparation of sequence variants of the selected
exportation polypeptide gene using site-directed
mutagenesis is provided as a means of producing
potentially useful exportation species and is not meant
S to be limiting as there are other ways in which sequence
variants o~ the exportation polypeptide gene may be
obtain~d. For example, recombinant veotors encoding the
desired gene may be treated with mutagenic agents to
obtain seguence variant~ (see, e.g., a method described
by Eichenlaub, (1979) for the mutagene~is of plasmid DNA
using hydroxylamine).
I~olation of Salmonell~ DNA segment~ was
accosplished by isolation of DNA fragments containing the
phoA g ne phoA fu~ion~. TnphoA i8 a derivative of TnS
which ~ncode~ E. c~lt alkaline phosphatase, minus the
~gn~ qu~nce and exprQ~sion signals, inserted into the
l~t I850L element. Random transposition of TnphoA
r~ults in an active insertion only when the phoA gene
~0gu~nce i~ fused inframe downgtream of the promoter and
export signals of a target gene A, Figure 1.- Plasmids
cont~ining phoA gene fusions can then be used as
exposition vectors, Figure 1, ~B). The SSPl and the
~vuII re~triction ~ites in phoA are blunt ended sites at
which inframe insertions (IF) of a gene of interest (GOI)
can be in~erted. The resulting tribrid gene fusions,
shown as C in Figure 1, contain the expression and export
~ignal~ of the target gene fused inframe with the phoA
and GOI sequences.
Figure 1 is a schematic representation of typical
phoA fusions and illustrating cloning of successful
fu~ions. The point at which the phoA sequence joins the
~` target gene is referred to as the fusion joint (FJ). The
re~aining portion of the gene begins at the distal joint
(DJ). Utilizing restriction enzymes which cut either
downstream of the kanamycin resistance gene (e.g., BamHI)
W093/1O~K PCT/US92/09659
-30-
or the phoA gene sequence (e.g., HindIII) allows cloning
of phoA gene fusions, provided the target gene is not
cleaved ("R"). The fusion joint, including all the phoA
gene fusions and upstream Salmon~lla sequences, were
S cloned into the ~indIII or BamHI site of pBR322, Figures
2 and 3. Plasmids containing phoA gene fusions were then
used as exposition vectors. Cells produced fusion
polypeptide~ that had alkaline phosphatase activity,
indicated by the formation of blue colonies on agar
supplemented with the indicator dye (5-bromo-4-chloro-3-
indolylphosphate).
The following examples are intended to illustrate
the practice of the present invention and are not
intended to be limiting. Although the invention i~
d~m~n~trated with nucleic acid segments isolated from a
strain of S~lmonell~, similar functions may be obtained
~rom nucleic acid ~egments from other Salmonella strains
and even other microorganisms. The nucleic acid
sequences identified and the corresponding encoded
polypeptides are useful in developing methodsl of
producing a wide variety of heterologous proteins as well
as expression vectors for localizing polypeptides in
selected areas of a host cell.
BXAMP~B 1
~ he following illuctrates construction of plasmid
pZIP-IN (Fiqure 3). This plasmid contains a chimeric
gene including a DNA se~ment from a strain of Salmonel ~ a
fused with a segment of the alkaline phosphatase gene
lacking signal and expression sequences. When expressed
in a suitable host cell, the fusion product is localized
to the inner membrane/periplasmic space of the host cell.
W093/102~ 2 1 2 3 ~ 7 i PCT/USg2/09659
Pre~aration of ~-ZIP-IN
pZIP-IN, Figure 3, is a derivative of pBR322
containing a B~mHI fragment encoding alkaline phosphatase
activity and kanamycin resistance inserted at the BamHI
s~te. The BamHI fragment was cloned from a chromosomal
DNA preparation of the TnphoA ~nsertion mutant TAG28,
which was constructed by TnphoA mutagenesis (see above)
of S. typh~mur~um TA2361 (phoN mutant derived from LT2).
Chromosomal DNA was prepared from S0 ml of overnight
growth of TAG28 in L-broth with vigorous shaking at 37C.
The bacterial culture was precipitated and washed once in
pho~phate buffered saline (pH 7.0). The washed b~cterial
pellet was re~u~pend~d in 10 ml of ice cold ET buffer ~10
mM EDTA, 10 mM Tri~-~Cl (pH 8.0)]~ Ly~ozyme was added to
a concentrat~on o~ 0.1 mg/ml and incubated for 15 minutes
at 37C. 1.2 ml of ~arkosyl-pronase ~olution ~10%
sarko~yl, 5 mg/ml pronase in ET buffer) was added and the
solution was incubated for 1 hr at 37C. The solution
was then extracted 3 times with TE ~(10 mN Tr~s HCl, 1 mM
EDTA ~pH 8.0)~ ~aturated phenol followed by 3 extractions
with chloroform:isoamyl alcohol ~4~ he aqueous
phase was trans~erred to a 50 ml beaker on ice and one-
half volume of 7.5 M ammonium acetate was added. Threevolume~ of ice ~old absolute ethanol wa~ gently layered
on top of the ~olution. The chromosomal ~NA was
precipitated onto a glass rod by gently stirring the
~olution to mix the ~nterface. The precipitated DNA was
rinsed once in 70% ice cold ethanol and dissolved
~vernight in 2 ml of TE buffer at 40C. The concentration
of DNA was quantitated by measuring the O.D. at 260 nm.
2 ~g of T~G 28 chromosomal DNA was digested with
B~mHI at 37C for 2 hrs. The solution was extracted once
with TE saturated phenol, followed by 2 extractions with
chloroform:isoamyl alcohol (24:1). The aqueous phase was
wos3/lo~K PCT/US92/09659
-32-
removed and the DNA precipitated by the addition of 1/lo
volume 3 M sodium acetate (pH s.2) and 2 volumes of
ethanol followed by centrifugation in a microcentrifuge.
0.2 ~g of pBR322 DNA was digested with BamHI and prepared
for ligation as above. ~igation of the vector DNA
(p~R322) and TAG 28 chromosomal DNA w~s performed by
overnight incubation at 4C in 20 ~1 of lX commercial
~Promega) liga~e buffer and 2 U of T4 DNA ligase.
pZIP-IN was isolated from the ligation reaction by
tr~nsformation of subcloning efficiency DHS~ competent
cells. S ~1 of the ligation mixture was added to 50 ~1
o~ DHS~ competent cells and incubated on ice for 30
~inutes. Cells were heat shocked for 30 ceconds by
IS ~n¢ubat~ng in a 37C water bath. Cells were cooled on
ice ~or 2 minute~ and O.9S0 ml of L-8roth was added to
the tube.- Cells were incubated for 1 hr at 37C.
Tr~nsformant~ with alkaline phosphatase activity and
Xan~mycin resistance were selected by plating 0.1 ml of
the b~cterial culture on the ~-agar plates containing S0
~g/ml kanamycin and 40 ~g/ml BCIP (5-bromo-4-~hloro-3-
indolyl phosphate), followed by overnight incubation at
37C. The following day, kanamycin resistant colonies
were visible and all were blue, indicating the
transformants had alkaline phosphatase activity. This
was confirmed ~y alkaline phosphatase assays, Western
blotting with monoclonal antibodies to alkaline
phosphatase, and DNA seguencing of the fusion joint.
Figure 3 shows a partial restriction map of pZIP-IN.
E~MPI.E 2
The following example illustrates the construation
- of pZIP-OUT, Figure 2. The plasmid is constructed from a
DNA segment of Salmonella and a PhoA DNA segment lacking
~ignal and expression sequences. When expressed from a
W0931102~ ~ 1 2 3 ~ 7 ~ PCT/US92/096ss
-33-
suitable host cell, the fusion protein is localized to
the outer membrane of the host cell.
Çonstruction of ~ZIP-OUT
Genomic DNA was isolated from strain TAP43. A 25 ml
culture in LB broth was grown overnight at 37C with
shaking. The cells were harvested by centrifugation, and
the pellet washed once in PBS. The washed pellet was
resuspanded in 10 mls of cold TE buffer (10 mM Tris-HCl,
pH 8.0, 1 mN EDTA). One ml of a 1 mg/ml lysozyme
solution was added, and the mixture was incubated in a
37C w~ter bath for fifteen minutes. After this
incub~tion, 1.2 ml of 10% sarkosyl, 5 mg/ml pronase in TE
lS bu~er was added, and incub~tion continued at 37C for 1-
2 hours, until cell lysis occurred. The ly~ate was then
extractQd twice with an equal volume of phenol, once with
phenol/chloroform, and once with chloroform. To the
~in~l extraction, a half-volume of 7.5 M ammonium acetate
wa~ added. The solution w~s mixed gently and placed on
ice. Two volumes of ice-cold ~bsolute ethano~ weré
layered on top of the lysate, and the chromosomal DNA was
collected at the interface by spooling on ~ glass rod.
The ~pooled DNA was rinsed once in 70% ethanol, and then
allowed to di~solve off of the glass rod into TE buffer
overnight at 4~ The buffer, containing the di~solved
DNA, was ~hen ethanol-precipitated. The purified
chromo~ome was collected by centrifugation and
resuspended in a small volume of TE buffer. 1-5 ~g of
the purified DNA was restricted with ~indIII, and then
phenol/chloroform extracted and ethanol precipitated.
The ~ample was collected by centrifugation, the pellet
washed once with 70% ethanol, and dried under vacuum.
Vector pUC18 was also restricted with ~indI,
extracted, and precipitated in the sa~ manner. The
~ndIII fragments of the genomic DNA were then ligated
W093/10246 PCT/US92/Og659
into the HindIII site of pUC18 with T4 DNA ligase. After
ligation, the DNA was transformed into competent DH5~
cells and plated on L-agar supplemented with ampicillin
and BCIP (5-bromo-4-chloro-3-indolyl phosphate), both at
40 ~g/ml. Blue colonies, indicating the presen~e of an
active alkaline phosphatase fusion in the transformant,
were selected and analyzed by restriction mapping.
Transformant 43-17 contained a 4.5 kp ~indIII insert in
the pUC18 vector. 3.1 kp of this insert consisted of
phoA sequences, with the remaining 1.4 kp being derived
from Salmonella chromosomal sequences.
The identity of this clone as a phoA fusion was
confirmed not only by restriction analysis, but also by
80uthern blotting, Figure 4, and seguencing. The
S~lmonella-poA fusion contained within this ~indIII
~ragment was designated as the pZIP-OUT cas~ette. This
cassette was subsequently cloned into the HindI~I sites
of the vectors pBR322 and pAT153. The general structure
of pZIP-OUT is shown in Figure 2.
EXl~NPLE 3
The following çxample illu~trates how DNA may ~e
fused to the gene segments of plasmid pZIP-IN, shown ln
this example with a portion of the cholera su~unit B
gene.
Construction of ~IMB13
39
pIMB13 is a derivative of pZIPoIN in which the final
294 base pairs of ctxB have been inserted in frame with
t~e phoA gene sequence at the SspI site. The inserted
fragment containing the ctxB gene sequence is from
pRIT10810 which encodes the entire ctxB gene. First, the
SspI site in the pBR322 portion of pZIP-IN was eliminated
as follows. 2 ~g of a plas~id preparation of pZIP-IN was
W093/10246 PCT/US92/~59
212367~
-35-
dige~ted with ScaI and EcoRV. Both enzymes cut at a
single site within the pBR322 portion of the vector and
generate compatible blunt ends. The digested DNA was
preoipitated and ligation was performed in 20 ~l of lX
liga~e buffer containing 1 U of T4 DNA ligase overnight
at 4-C. DH5~ frozen competent cells were transformed
with S ~l of the ligation reaction mixture.
.
Transformants were selected on L-agar plates
containing 50 ~g/ml kanamycin. Colonies were then
replicated to L-agar plates containing 40 ~g/ml
~picillin. Loss o~ ampicillin resistance encoded by
pZIP-IN indicated that the sQgment from ScaI (3844) to
~coRV ~185) which contained the SspI site (4168) had been
lS ~ n~t~d. The r~sulting plasmid p~S28-1 contained a
~lngie S~pI site in the phoA seguQnce which generate~ an
in-~r~ blunt end cut.
pINB13 was constr~cted from pAS28-1 as follow~. The
ctxB c~guence encoded by pRIT10810 contains an SspI site
which generates ~n in-fr~me blunt end cut ne~r the 5' end
of the ~tructural gene. pRIT1080 also con~ain~ an SspI
site in the pBR322 portion of the vector. ~igestion of
pRIT101810 with SspI generates 2 fragments, one of which
contains the 3' final 294 ba~e pairs of ctxB. 2 ~g of
p~S28-l and 2 ~g o~ pRIT10810 were dige~ted with SspI.
Following phenol/chlorofo D extraction, the samples were
combined and precipitated with 2 volumes of ethanol.
Ligation of the sample was performed in 20 ~1 of lX
ligase buffer containing 1 U T4 DNA ligase. DH5~ frozen
- competent cells were transformed with 5 ~l of the
ligation mixture. Transformants were selected on L-~gar
plates containing 50 ~g/ml kanamycin and 40 ~gtml BCIP~
Colonies harboring poS28-1 with inserts at the phoA ssp~
~ite appeared white ~ince insertion interrupted the
active phoA gene fusion. White kanamycin resistant
~- colonies were picked for isolation and screened for
W O 93/10246 PC~r/US92/09659
-36-
expression of a ctxB fusion protein by Western blotting
of total envelope fractions with affinity purified anti-
ctxB. A DH5~ strain harboring a derivative of pZIP-IN
encoding a ctxB gene fusion was identified and the
S plasmid was designated pIMB13.
~XAN~s 4
The following example i8 an example of a tripartite
fusion prepared from plasmid pZIP-OUT. This plasmid may
be used to express a fusion polypeptide from suitable
host cellc. The DNA inserted in this example is a
segment from cholera B toxin ~ubunit.
Construction of ~RSP18
The construction of the trihybrid fusion, pRSP18,
wa~ accomplished as follows. Plasmid pRIT10810,
containing the cholera toxin B gene, was first restricted
20 w~th EcoRI and ~stI. The ends generated by these
restrictions were repaired with Klenow, and ~he vector
was ligated back together. This created a .8 kp deletion
in pRIT10~10, eliminating an undesirable SspI site in the'
vector. This deleted pRIT10810 was then re~tricted with
~lndII~ and SspI. pZIP-OUT (in vkctor pUC1~) was doubly
restricted w~th ~indII~ and PvuII. A 2.0 kp fragment
generated ~rom this double restriction, consisting of 1.4
Xp of Salmonella seguence and .6 kbp of phoA, was
i~olated and purified after agarose gel electrophoresis.
This 2.0 kp fragment was then unidirectionally ligated
into the XlndI~I/SspI digested pRIT10810. This generated
an in-frame fusion of the Salmonella-phoA sequences to
the ctxB ~equence (pSP-18). This clone was selected for
on the ba~is of weak tetracycline resistance (1 ~g/ml in
L-agar). To make further manipulations of the plasmid
more e~ficient, a kanamycin gene block (Pharmacia) was
WOg3/10246 PCT/US92/096S9
212367.~)
-37-
cloned into the BamHI site of PSP18, resulting in the
plasmid construction pRSP18.
E~AMP~E 5
Ihis example illustrates the procedure for
extracting and separating bacterial membranes. After
i~olation of the membrane fragments, they were analyzed
for localization of fusion peptide~.
~0
p~D~ration of Bacterial Membranes ~Total En~elo~e) and
~e~aration into Inner and Outer Membrane Fractions
100 ml of overnight bacterial cultures grown in L-
15 Broth with vigorou~ shaking were pelleted and washed lX
~n pho~phate bu~fered ~aline (pH 7.0). Washea pellets
were re~uspended in 3 ml of membrane isolation buffer tlO
rM N~P04, 0.5 mM MgS04 (pH 7.0)]. 8amples were sonicated
~or 20 seconds 3 times with cooling on ice in between.
20 Unbroken cells were removed by centrifugation at 7,000
rpm in a BecXman ultracentrifuge SWS5 rotor for 1 hr.
The supernatants were remo~ed and total envelope pellets
were rinsed lX in sterile deionized water. Pellets were'
re~u~pended ~n 40 ~1 of sterile deionized water. One-
25 ~alf (20 ~1) was saved for We~tern analysis of the total
envelope. A 5% ~olution of sarko~yl in ~terile deionized
water was added to the remaining 20 ~1 to a final
concentration of 0.5%. The samples were incubated for 30
f minuteg at room temperature and centrifuged in a
30 microcetrifuge to pellet the non-soluble fraction
representing the outer membrane. The ~upernatant was
removed for Western analysis of the inner ~embrane
fraction. The outer membrane pellet was rinsed once in
sterile deionized water and saved for Western analysis.
~' 35 Figure 4 shows immunoblot analysis of membrane
preparations using mouse anti-alkaline phosphatase.
W093~l02~ PCT/US92/~659
-38-
EXAMPLE 6
The follcwing example describes the analysis of
alkaline phosphatase activity. For the purpose~ of the
present invention, alkaline phosphatase assays were
performed to test for enzyme activity in membrane
fractions of host cells in which alkaline fusion proteins
were expressed.
Alkaline Phosphatase Assays
Alkaline phosphata~e activity encoded by pZIP-IN and
pZIP-OUT wa~ confirmed by spectrophotometric assay using
the chromogenic alkaline phosphatase substrate para-
nitrophenol phosphate (PNPP). One ml of overnight
bacter~l cultures was pelleted for 15 seconds in a
~icrocentrifuge. The pellet was washed once in 1 N Tris-
HCl ~pH 8.0) and resuspended in 1 ml of 1 M Tris-HCl (pH
8.0). m e O.D. 600 of the bacter~al suspension was
recorded. 50 ~l of chloroform and 50 ~l of 0.1% ~DS were
added to permeabilize the cells. Samples we~ vortexed
briefly. 0.1 ml of a 0.4% solution of PNPP in 1 M Tris-
HCl (pH 8.0) was added and samples were incubated at
37C. After significant yellow color was observed, 10 ~l
o~ 2.5 M XPO4, 0.5 M EDTA was added and ~amples were
- placed on ice to stop the reaction. Cellular debris was
removed by centrifugation in a microcentrifuge. O.D. 420
of the samples were recorded. The units of alkaline
pho~phatase activity were calculated by the following
~ormula:
Units activity =
1,000 X O.D. 420/time of reaction (minutes) X O.D. 600
,
Figure S how~ an immunoblot analysis of urea extracts
using anti-alkaline phosphatase as the primary an~ibody.
No reaction is shown with plasmid pBR322 or with plasmid
WO93~102~ 2 1 2 3 6 7 ~ PCT/US92/09659
-39-
pZIP-IN. A reaction is shown with plasmid pZIP-OUT,
indicating extraction of the alkaline phosphatase fusion
protein.
E~AMP~E 7
The following outlines the general procedure for
extracting proteins from bacterial cells.
Urea Extraction of Bacterial Cells
Ten ml of overnight stationary phase bacterial
cultures grown in L-Broth with vigorous shaking were
cooled on ice for 10 minutes and pelleted at 7,000 rpm in
a Bec~man J2-21 (JA-17 rotor). The bacterial pellet was
washed 3 times in phosphate buffered saline (pH ~.0).
The wa~hed pellet was re~uspended in 0.1 ml of 6 M urea
containing 10 mM ~ris-HCl (pH 7.5) and 5 mM EDTA. The
su~pension was incubated or ~0 minutes on ice. Bacteria
were pelleted in a micxocentrifuge for 1 minute.
Centrifugation of the supernatants wa~ repeated to remove
any traces of debris. Supernatants were frozen and 20 ~1
aliquots were used for SDS-PAGE and Western analysis.
2S EX~PI,B 8
The following example illustrates the expressi~n of
a ctxB polypeptide from an attenuated Salmonella ~train
with localization of the ctxB to the surface of the outer
cell membrane.
Pr~parat~~n of Surface ~xpressed Cholera Toxin Subunit B
The tribrid fusion in pRSP18 contains a 1.4 kb
35 Salmonella DNA seguence which includes expression export
signals, Figure 6. The phoA sequence of the fusion
includes approximately 0. 6 kb from the phoA fusion joint
WO93/102~ PCT/US92/09659
-40-
(FJ) to the inframe insertion (IF) of ctxB. The ctxB
sequence includes the final 294 base pairs of ctxB
beginning at the inframe insertion site IF. Expression
and export result in a 32 kDa tribrid fusion protein
including the final 98 amino acids of ctxB at the C
terminus which localizes to the outer membrane. The
tribrid fusion in a pINB13, Figure 7, contains a l.3 kb
Salmonella DNA sequence which includes the expression and
export signals of the expressed gene. The phoA sequence
~0 of the fusion includes approximately 0.2 kb from the phoA
fusion joint FJ to the inframe insertion IF of ctxB. The
ctx8 ~equence includes the final 294 base pairs of ctxB
beginning at the inframe insertion site IF. Expression
and export result in a 32 kDa tribrid fusion protein
~nclud~ng the final 98 amino acids of ctxB at the C
terminus which localizes to the inner membrane. Figure 8
is a ~chematic representation of the fusion products.
Whole Salmonella TA2362 cells harboring pRSPl8 were
~hown to express cholera B subunit on the outer surface
membrane. Antisera to cholera toxin B subunit were
prepared. Agglutination of TA2632 harboring pRSPl8 was
obtained. No agglutination was observed with strain
TA2362 alone.
An immunoblot ana~ysis of the membrane preparations
was ~un using affinity purified rabbit anti-CTB. S.
typhimurium TA 2362 harboring pRSPl8 showed a 32 kDa CTB
tribrid fusion protein in the total envelope (TE). Upon
separation of the inner and outer membrane by treatment
with 0.5~ sarkosyl, the majority of the fusion protein
was observed associated with the outer membrane ~OM). TA
2362 harboring pINBl3 showed a 32 kDa CTB fusion protein
in the total envelope (TE). Upon separation of the inner
and outer membrane by treatment with 0.5~ sarkosyl, the
majority of the fusion protein was found associated with
the inner membrane (IM). All lanes were loaded with
W093/tO246 2 1 2 3 6 7 ~ PCT/US92/0965g
-41-
membrane preparations prepared fror n equivalent number
of cells.
EX~MPLE 9
s
The following example illustrates the procedures
contemplated as useful for creating an immune response in
a mammal elicited with virulence attenuated Sa~m~nella
strains expressing antigens on the surface of the intact
cell. In this example, CTB is used as an illustra~ion.
Immunoaenic,Responses from Surface-Expr,e~sed CTB
All immune response experimentation will be
conducted using CTB responding C57B/6 mice (15,16). An
virulence attenuated S. typhimurium aroA phoN strain will
be utilized in all experiments. Groups of 10
micelcondition will be challenged with the ~ollowing:
S~l monel l a alone, or Sal mon~l l a with cytoplasmically-
encoded CTB (pRIT~08010), or inner tpINB-~3, Fig. ~) or
outer (pRSP-18, Fig. 6) membrane-expressed t~ibrid fusion
encoding ctrains. I.P. challenge (5X105 cfu) and oral
challenge (5X108 cfu) will be evaluated. These challenge
doses are expected to give op~imal results ~ut may
require adju~ting as neces~ary. Boosting will be 10 days
post-challenge. Mucosal and serum anti-CTB level will
be determined after 1 and 2 challenge by ELISA (15,16)
and by the ability to neutralize cholera toxin activity
on adrenal cells (1). It will also be determined if the
membrane-expressed CTB tribrid polypeptide retains its
potent m~cosal adjuvant activity (17) by comparing
an~ibody titers to Salm~nella and Salmo~ella expressing
CTB. Since CTB mediates Ig class switching, we will also
determine IgA/IgG ratios between the different challenge
protocols by ELISA (17). Alternatively, the adjuvant
activity of membrane expressed CTB will be evaluated
using a purified antigen (i.e., ovalbumin) (18) for
W093/10246 PCT/US92/09659
-42-
concurrent challenge with Salmonell~ or Salmonella
expressing CTB strains. Additional experiments to
further characterize adjuvant activity will be performed
as indicated.
E~AMPLE lo
This example illustrates a contemplated method of
inserting a fragment of HIV gp160 gene into plasmid pZIP-
OUT of Example 2.
Construction of ~ZIP-OUT Encoding a 60 kDa Fraoment of
~IV oD120
A clone containing a 3.~ kb SalI - XhoI fragment
encoding the HIV gp160 gene has been obtained. The
coding regions of gp120 and gp41 are indicated by the
arrow in Figure 10. PvuII dig~stion of this fragmer.t
wil~ yield a 1.8 kb fragment which deletes 0.7 kb of
gp120 coding sequence. The 4.5 Xb pZIP-OUT cassette,
bounded by ~indIII sites, has been cloned intb the
~ndIII site of vector pAT153 (~PvuII site). This
construction has been designated pZIP-0UT-2. pZIP-0UT-2 '
will be digested with PvuII and S~1I, and the E~YII -
XhoI HIV fragment ligated into these ~itas. ~he tribrid
fusion polypeptide predicted from this construction will
yield a 82 kd polypeptide (2000-4000 dal, Salmonella:
20,000 dal, phoA: and 60,000 dal, ~gpl~0/gp41)~
The predicted DNA sequence across the phoA fusion
junction into gp120 is shown in Figure 10. The
p~A::gp120/gp41 reading frame is indicated by the
brackets.~ The amino acid sequence across the fusion
joint is shown,
- 35
WO g3/10246 2 1 2 3 fi 7 ~ PCr/US92/Og659
-43-
EXAMPLE 11
The following outlines general protocols for
sequencing.
s
Pre~aration of Tem~lates
pZIP-IN, pZIP-OUT, and pRSP18 were seguenced by the
Sanger dideoxy protocol for double ~tranded DNA
template~. ~
Purified plasmid preparations for sequencing were
prepared as follows: -
1. Each ~train was grown overnight in 5 ml of L8 broth
(containing the appropriate antibiotic) at 37C with
vigorous aeration.
2. The cultures were harvested by centrifugation. The
cell pellets were resuspended in 100 ~1 of 50 mM
glucose, 10 mM EDTA, and 25 mM Tris-HCl, p~ 8.0, and
incubated at room temperature for 5 minutes.
3. 2D0 ~1 of freshly prepared 0.2N NaOH, 1% D5 were
added to each sample. The samples were mixed by
inversion, and then incu~at d S minutes on ice.
4. 150 ~1 of 3 M potassium acetate (pH 4.8) were added
to each sample. The samples were mixed by inversion
and incubated for 5 minutes on ice.
5. The samples were then centri~uged for 5 minutes, and
the supernatants transferred to fresh tubes. The
~amples were centrifuged a second time for 5 minutes
and the supernatants transferred as before.
WO93~10246 PCT/US92/09659
-44-
6. RNase A was added to a concentration of 20 ~g/ml,
and the samples were incubated at 37C for 20
minutes.
7. Each sample was phenol/chloroform extracted,
chloroform extracted, and then ethanol-precipitated.
8. The DNA precipitates were collected by
centrifugation and each DNA pellet wa~ resu pended
in 16 ~1 deionized water, 4 ~1 4 M NaCl, and 20 ~1
13% polyethylene glycol 8000. The samples were
mixed well and incubated on ice for 2~ minutes.
9. The ~amples were centrifuged 10 minutes and the
supernatant~ discarded. The pellets were washed
twice in 70% ethanol, dried, and resuspended in 20
~1 o~ dH20.
Denaturation. Anneal~nq and_Sequencinq of Tem~lates
For each DNA template prepared as above:~
1. 2 ~1 of 2 m NaOH~ 2 ~M EDTA were added to the entire~
20 ~1 sample and the sample was incubated for 10
minutes at room temperature.
2. The reactions were neutralized by the addition of
4.5 ~1 of 2 M sodium acetate (pH 5.0) and 5.5 ~1 of
distilled H2O. The samples were mixed well, and then
precipitated with 100% ethanol.
3~ The DNA pellets were collected by centrifuging for
15 minutes. ~he pellets were then washed once with
70~ ethanol and dried.
4. All of the following reagents, except primers and
radioactive label, were supplied in the sequenase
W093/102~ PCT/US92/09659
212367~
-45-
sequencing kit, United States Biochemical Co. The
dried pellets were resuspended in 7 ~l dH20, 2 ~l of
5X Sequenase reaction buffer and 1 ~ 20 ng) of
the appropriate primer. For sequencing the
Salmonella ~equences in pZIP-IN and pZIP-OUT,
immediately upstream from the phoA junction, primer
l(AGA ATC ACG CAG AGC G) wa~ used. For extended
sequencing in the Sal monel l a sequences of pZIP-OUT,
primer 2 tTTC AGG AAT GCA TGC) was utilized. To
sequence across the phoA: ctxB junction in pRSP18,
primer 3(AGC GCG ACC AGT GAA A) was used. The
annealing reactions were incubated for 30 minutes at
37C.
5. To each annealing mixture, 1 ~l of .lM
dithiothreitol, 2 ~l of diluted labelling mix, 1 ~l
of tS35~-dATP, and 2 ~l of diluted Sequenase enzyme
were added. The reactions were mixed and incubated
at room temperature for 5 minutes.
6. 3.5 ~l of each labelling reaction were then
transferred to each termination mixture tube,
containing dideoxy ATP, dideoxy GTP, dideo~y CTP,
and dideoxy TTP. The chain termination reactions
were allowed to proceed for 5 minutes at 37C.
7. 4 ~l of stop solution were added to each reaction,
and the reactions were heated to 75C for 2-5
minutes.
8. The reactions were loaded onto a 6% acrylamide-urea
sequencing gel and electrophoresed at 15 mA for 2-6
hours.
5 9. After electrophoresis, the sequencing gel was fixed
in 10% methanol, 10% acetic acid, for 1 hour and
then dried under vacuum for 1 1/2 hours.
W093/l0246 PCT/US92/096S9
-46-
lO. The dried gel was then exposed to autoradiograph
film at room temperature for ~16 hours.
WO93/102~ PCT/US92/09659
212367~
-47-
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT: BOARD OF REGENTS, THE UNIVERSITY ~F
TEXAS SYSTEM
(ii) INVENTORS: NIESEL, David W.
MONCRIEF, J. Scott
PHILLIPS, Linda H.
(iii) TITLE OF INVENTION: MEMBRANE EXPRESSION OF
HETEROLOGOUS GENES
(~v) NUMBER OF SEQUENCES: 2
(v) CORRESPONDENCE ADD~ESS:
(A~ ADDRESSEE: ARNOLD, WHITE & DURKEE
(B3 STREET: P.O. Box 4433
(C~ CITY: Houston
(D) STAT~: Texas 77210
(E) COUNTRY: US
(F) ZIP- 77210
(Yi) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
~C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: WordPerfec~ 5.1
(vii) GURR~NT APPLICATION DATA:
~A) APPLICATION NUMBER: Unknown
(B) F~LING DATE: Unknown
(C) CLASSIFICATION: Unknown
WO93/10246 PCT/US92/09659
~ ,.
-48-
(viii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: 07/792,525 US
(B) FILING DATE: 15 November 1991
(C) CLASSIFICATION: 424
(ix) ATTORNEY/AGENT INFORMATION:
(A) NAME: KTTCHELL, Barbara S.
(B) REGISTRATION NUMBER: 33,928
(C) REFERENCE/DOCKET NUMBER: UTFGlllPCT
(x) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: 512-320-7200
(B) TELEFAX: 713-789-2679
~2~ INFORMATION FOR S~Q ID NO:l:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 213 base pairs
(B) TYPE: nucleic acid
(C~ STRANDEDNE5S: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:l:
GAAACCGATT CGCCCCCTTA TAACTATTGT CAGATAACGT TCTGACGGTT 50
GTGTAAAAAC ATG GCG CCT CAT TCT TCT GT~ GTT GGA GTT AAT 93
Met Ala Pro His Ser Ser Val Val Gly Val Asn
S 10
ATG AAA AAA TTT TAT AGC TGT CTT CCT GTC TTT TTA CTG ATC 135
Met Lys Lys Phe Tyr Ser Cys Leu Pro Val Phe Leu Leu Ile
WO93/102~ 2 1 2 3 6 7 ~ PCT/US92/09659
-49-
GGC TGT GCT CCT GAC TCT TAT ACA CAA GTA GCG TCC TGG ACG l77
Gly Cys Ala Pro Asp Ser Tyr Thr Gln Val Ala Ser Trp Thr
5 GAA CCT TTC CCG TTT TGC CCT GTT CTG GAA AAC CGG 2l3
Glu Pro Phe Pro Phe Cys Pro Val Leu Glu Asn Arg
40 45 50
(2) INFORMATION FOR SEQ ID NO:2:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 387 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D~ ~OPOLOGY: linear
(x~) SEQUENCE DESCRIPTION: SEQ ID NO:2:
GCG~GCATAA TAaGCCCTAC ACAAATTGGG AGATATATCA TGAAAGGCTG 50
GCTTTTTCTT GTTATCGCAA TAGTTGGCGA AGTAATCGCA ACATCCGCAT lO0
TAAAATCTAG CGAGGGCTTT ACTAAGCTTG CCCCTTCCGC CGTTGT~ATA l50
ATCGGTTATG GCATCGCATT TTATTTTCTT TCTCTGGTTC TGAA~TCCAT 200
CCCTGTCGGT GTTGCTTATG CAGTCTGGTC GGGACTCGGC GTCGTCATAA 250
TTACAGCCAT TGCCTGGTTG CTTCATGGGC AAAAGCTTGA TGCGT&GGGC 300
TTTGTAGGTA TGGGGCTCAT AGCTGACTCT TATACACAAG ATGCGC~TGT 350
GACGGAACCT TTCCCGTTTT GCCCTGTTCT GGAAAAC 387
WO93/10246 PCT/US92/09659
--50--
REFERENCB8
The references listed below are incorporated herein
by reference to the extent that they supplement, explain,
S provide a background for or teach methodology, techniques
and/or compositions employed herein.
Sanchez, J., Johansson, S., Lowenadler, B., Svennerholm,
A.M. and Holmgren, J., Res. Microbiol. 141, 971-979
(1990).
Strugnell, R.~., Maskell, D., ~airweather, N., Pickard,
D., Cockayne, A., Penn, C. and dougan, G., Gene 88, 57-63
(lsso) .
~5
Dougan, G., Hormaeche, C.E. and Maskell, D.J., Parasite
Immunol. 9, 151-160 ~1986).
Sory, M.-P. and Cornelis, G.R., Res. Microbiol. 41, 921-
929 (1990).