Note: Descriptions are shown in the official language in which they were submitted.
CA 0221788~ 1997-10-31
W 096/34869 PCT~EP96/01882 ESTERS OF CARBAPENEMS
This invention relates to a class of ~ntib~cttori~l compounds, in particular a
class of c;all,apenems, processes for their preparation, ph~rm~r~e~ltic ~l and veterinary
S compositions compricing such compounds, intermPrli~tf~s thereof, and their use in
~ntibarteri~l therapy.
Carbapenems such as imipenem, the compound of forrnula (A):
HO H H
~--\
~S(CH2)2NHCH=NH
CO2H
(A)
10 have a potent, broad spectrum of antibacterial activity (see US 3 950 357 andUS 4 194 047; Merck and Co). Such carbapenems however tend to be vulnerable to
hydrolysis by the enzyme renal dehydropeptidase-l (DHP-l) and this limits their use
in chemotherapy. In the case of imipenem, this problem may be overcome by the co-
A~lmini~ctration of an inhibitor of DHP-l.
Stability towards DHP-l may also be imparted by ch~miç~l modification of
the carbapenem nucleus, for instance by incorporating a l~B-methyl substitlltent~ as in
~ the compound meropenem, the compound of formula (B):
HO H H CH3 ~ CON(CH3)2
~S --C~NH
CO2H
- (B)
20 (see Shih D.H. et al., Heterocycles, 1984, 21, 29 and Sunagawa M. et al.,
J. Antibiotics, 1990, 43, 519). More recently, this has been extended to a 1~-
~mino~lkyl substituent (see EP 0 433 759, Bristol-Meyers Squibb).
An alternative approach to imparting improved stability to DHP-l utilises 2-
carbon substitllted carbapenems, for in.ct~n~e, 2-aryl, 2-heteroaryl and 2-
25 heteroaromatic carbapenems (US 4 543 257, US 4 260 627, US 4 962 101,
US4978659,EP014493, EP0414489,EP0010316andEP0030032Merck&
Co) and 2-(substituted)methyl carbapenems (Schmidt et al, J.Antibiotics, 41, 1988,
780).
UK Patent 1 593 524, Merck & Co. discloses a number of 5-membered
30 heteroaromatic call,ape-lem derivatives including diazolyl and tetrazolyl compounds.
However, in the case of the pyrazolyl derivatives the heterocyclic compound is
?~ttq~hed to the call,apenem nucleus through the C-4 position.
CA 0221788~ 1997-10-31
WO 96/34869 PCr/EP96/01882
Other structural modifications introduced at position-2 include a snbstitut~qd
vinyl group -C(Ra)=CHRb in which, for in.~t~nce Ra is hydrogen or methyl and Rb is
hydrogen or lower alkyl (EP 0 330 108; Fujisawa) or Ra and Rb are selected from
hydrogen, lower aLkyl, aminocarbonyl, lower alkoxy, cyano, nitro and lower
S aL~ ycall~onyl (EP 0 430 037, Banyu Pharmaceutical Co.). In the absence of a l~-
methyl substituent, such a modification does not however appear to impart DHP- lstability.
Tntern~tion~1 Patent Application No. PCT/GB94/02347 describes compounds
of the general formula (C):
H H
c ~ R
o~N~
CO2 R
(C)
in which R is:
N--N~
~ ~ R~ ;
wl~
R(x is hydrogen, optionally substit11t~od (Cl 6)alkyl or optionally substituted aryl;
Rl~ is hydrogen, optionally substituted (C 1 6)aL~yl or optionally substituted aryl; or
20 R(~ and Rl~ together form an optionally substituted S or 6 membered heterocyclic ring
with or without additional heteroatoms;
Rc is (C 1 6)aLkyl which is unsubstituted or substituted by fluoro, a hydroxy group
which is optionally protected by a readily removable hydroxy protecting group, or by
an amino group which is optionally protected by a readily removable amino
25 protecting group;
Rd is hydrogen or methyl; and
-CO2Re is carboxy or a carboxylate anion or the group Re is a readily removable
carboxy protecting group.
CA 02217885 1997-10-31
W 096/34869 P~ll~lr~ol882
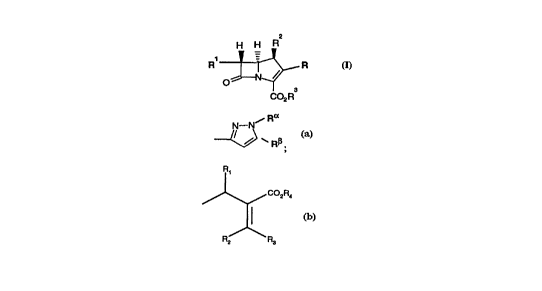
The present invention provides a compound of the general formula (I):
H
R -~ R
0~
CO2R
(I)
S in which R is:
N--N
~ 'R~;
wherein
10 R~ is hydrogen, optionally substitllted (Cl 6)aLkyl or optionally substituted aryl;
Rl~ is hydrogen, optionally substituted (C 1 6)aL~cyl or optionally substituted aryl; or
R(x and Rl3 together form an optionally substituted 5 or 6 membered heterocyclic ring
with or without additional heteroatoms;
R1 is (Cl 6)aLkyl which is unsubstituted or substituted by fluoro, a hydroxy group
15 ~hlch is optci~nally E~otecte~ b~ a rcadily remo~ab~e hyrlroxy proteeting g~tlp, orby
an amino group which is optionally protected by a readily removable amino
protecting group;
R2 is hydrogen or methyl; and
R3 is selected from the group consisting of (a) a group of formula:
R
~C02R~
wherein;
Rl is hydrogen or (Cl 6)aLkyl,
25 R2 is hydrogen, (Cl 6)aLkyl optionally substituted by halogen, (Cl 6)aLkenyl, (C
6)alkoxycarbonyl, aryl, or heteroaryl,
R3 is hydrogen, (Cl 6)aLkyl, or (Cl 6)aLkoxycarbonyl, and
CA 0221788~ 199?-10-31
WO 96/34869 PCTIEP96/01882
R4 is a ph~rm~reutic~lly acceptable ester forming group, and (b) a group of formula
CH(Ra)O.CO.Rb, wherein Ra is hydrogen, (Cl-6)aLkyl, (C3-7)cycloaLcyl, methyl, orphenyl; and Rb is (Cl 6)aL~yl(C3 7)cycloaLcyloxy or (Cl 6)aLcoxy(C1 6)aLcyl.
Compounds of formula (I) have a broad spectrum of anti-bacterial activity and
show good stability towards DHP-l.
Suitable (Cl 6) aL~yl groups for Ra and R13 include straight and branched
chain aL~yl groups having from 1 to 6 carbon atoms, for instance methyl, ethyl, n-
propyl and iso-propyl, preferably ethyl and methyl.
Representative examples of Ra and Rl~ as (Cl 6)aLcyl are when both are
methyl or ethyl. A particularly preferred example is when Ra is ethyl and Rl~ ismethyl .
Suitable optional substituents for the (Cl 6) aL~yl group for Ra and R13
inclucle, for cx~lmrle, halogen, hydroxy, (C 1-6)aL~oxy, carboxy and salt thereof,
(Cl-6)aLkoxycarbonyl, carbamoyl, mono- or di(Cl-6)aLcylcarbamoyl, sll1ph~moyl,
mono- and di(Cl-6)aLLylsulphamoyl, amino, mono- and di(Cl-6)aLcylamino,
(Cl-6)acylamino, ureido, (Cl-6)aL~oxycarbonylamino, aminocarbonyloxy and
mono- and di(Cl-6)aL~ylaminocarbonyloxy, 2,2,2-trichloroethoxycarbonylamino,
aryl, heterocyclyl, oxo, acyl, heteroaryl, (Cl-6)aL~ylthio, arylthio,heterocyclythio,
(Cl-6)aLkane-sulphinyl, arylsulphinyl, (Cl-6)~1k~nPsulphonyl, arylsulphonyl,
(Cl-6)aLco~yi~ o, hydlo~yi,llino, hydrazono, benzohydlo~yhlloyl, and 2-thiophene-
carbohydroxyimoyl. Preferred substituents include carbamoyl, aryl, especially
phenyl, and heteroaryl.
Suitable (Cl 6) aLcyl groups for Rl include straight and branched chain aLcyl
groups having from 1 to 6 carbon atoms. Preferred aL~cyl groups include methyl,
ethyl, iso-propyl, of which ethyl is especially ~l~felled.
Preferably the (Cl 6) aLkyl group of Rl has a hydroxy, fluoro or amino
substituent which is suitably at position- 1 of the aLcyl group. Advantageously R 1 is
(R)- 1 -hydroxyethyl.
Suitably R2 is hydrogen.
When used herein, the term "aryl" includes phenyl and naphthyl.
Suitably an aryl group, including phenyl and naphthyl, may be optionally
substituted by up to five, preferably up to three, substituents.
A representative example of Ra or R~ being an aryl group is phenyl.
Suitable optional substituents for the aryl group include halogen, (Cl 6)aL~yl,
aryl(C1 4)aL~yl, (C1 6)aL~oxy, (C1 6)aLcoxy(C1 6)aL~cyl, halo(C1 6)aL~yl, hydroxy,
amino, mono- and di-N-(Cl 6)aLkylamino, acylamino, carboxy, carboxy salts,
carboxy esters, carbamoyl, mono- and di-N-(Cl 6)aL~ylcarbamoyl,
(C1 6)aL~o~y~ I.onyl, (C1-6) aL~oxycarboxylate, aryloxycarbonyl,
-- 4 --
CA 0221788~ 1997-10-31
W 096/34869 PCTAEP96/01882(Cl 6)alko~ycall,onyl-(Cl 6)alkyl aryl, oxy groups, ureido, guanidino,
sulphonylamino, aminosulphonyl, (Cl 6)alkylthio, (Cl 6)aLkyl sulphinyl
(Cl 6)alkylsulphonyl, heteroeyclyl and heteroeyelyl (Cl 4)alkyl. In addition, two
~dj~< ent ring earbon atoms may be linked by a (C3 s)alkylene ehain, to form a
5 earboeyelie ring.
~, When used herein, the term "heteroatom" ineludes one or more of the
elements oxygen, nitrogen and sulphur.
When used herein, the term "heteroaryl" inl~lucies aromatie single and fused
rings eont~ining up to four heteroatoms in eaeh ring, eaeh of whieh is seleeted from
10 oxygen, nitrogen and sulphur, whieh rings may be unsubstituted or substituted by, for
çy~mple, up to three sllbstiblent.~- Eaeh heteroaryl ring suitably has S or 6 ring atoms.
A fused heteroaryl ring may include carbocyclic rings and need include only one
heteroaryl ring.
When used herein the terms "heterocyclyl" and "heterocyclic" suitably
15 inelude, unless otherwise riefin~d aromatie and non-aromatic, single and fused, rings
suitably containing up to four heteroatoms in each ring, eaeh of whieh is seleeted
from oxygen, nitrogen and sulphur, which rings ,may be unsubstituted or substituted
by, for example, up to three substituents. Each heteroeyclic ring suitably has from 4
to 7, preferably 5 or 6, ling atoms. A fused heteroeyelie ring system may inelude
20 earboeyelie rings and need inelude only one heteroeyelie ring.
Preferably a substituent for a heteroaryl or a heteroeyelyl group is seleeted
from halogen, (C1 6)alkyl, aryl(C1 4)alkyl(C1 6)aLkoxy, (C1 6)aLkoxy(C1 6)aLkyl,halo(Cl 6)alkyl,hydroxy, amino, mono- and di-N-(Cl 6)alkyl-amino,
acylamino,earboxy s~ltc,c~rboxy esters, earbamoyl, mono- and di-N-
25 (Cl 6)alkylearbonyl, (Cl-6) aLko~ycalboxylate, aryloxyearbonyl, (Cl
6)alkoxyearbonyl(Cl:6)aLkyl, aryl, oxy groups, ureido, guanidino, sulphonylamino,
aminosulphonyl, (Cl 6)alkylthio, (Cl 6)aLkylsulphinyl, (Cl 6)alkylsulphonyl,
heterocyelyl and heterocyelyl(C1 4)alkyl.
Suitable hydroxy and amino proteeting groups for use in Rl are those well
30 known in the art and whieh may be removed under eonventional eonditions and
without disrupting the remainder of the moleeule. A eomprehensive discussion of the
ways in which hydroxy and amino groups may be protected and methods for cleavingthe resulting protected clerivatives is given in for example "Protective Groups in
Organic Chemistry" (T.W. Greene, Wiley-Interscience, New York, 2nd edition,
35 1991). Particularly suitable hydroxy protecting groups include, for example,
triorganosilyl groups such as, for inst~n~e7 trialkylsilyl and also organooxycarbonyl
groups such, as for instance, allyloxycarbonyl, trichloroethyloxyearbonyl,
4-methoxybenzyloxyearbonyl and 4-nitrobenzyloxyearbonyl. Partieularly suitable
CA 0221788~ 1997-10-31
W096/34869 PCTAEP96/01882
amino protecting groups include aLkc~yc~l,onyl, 4-meth~ybel.~;ylo~yc~llonyl and
4-nitrobenzylo~y.,~ I,onyl.
Since the carb~penPm compounds of the present invention are intPntled for use
in ph~ relltir~l compositions, it will be further understood that they are each
S provided in substantially pure form, for ex~mplP at least 50% pure, more suitably at
least 75% pure and preferably at least 95% pure (% are on a wt/wt basis). Impurepreparations of the compounds may be used for preparing the more pure forms usedin the ph~rrnaceutic~l compositions.
When some of the compounds of this invention are allowed to crystallise or
10 are recrystallised from organic solvents, solvent of cryst~ tion may be present in
the crystalline product. This invention includes within its scope such solvates.Similarly, some of the compounds of this invention may be crystallised or
recrystallised from solvents cont~ining water. In such cases water of hydration may
be present in the crystalline product. This invention includes within its scope
15 stoichiometric hydrates.
The carbapenem antibiotic compounds according to the invention may be
formulated for a~lmini~tration in any convenient way for use in human or veterinary
mPIlirinP, according to techniques and procedures per se known in the art with
reference to other antibiotics, and the invention therefore includes within its scope a
20 pharm~ceutic~l composition compricing an antibiotic compound according to thepresent invention, together with a ph~rrn~ceutic~lly acceptable carrier or excipient.
The compositions may be fnrmnl~tPd for ~llmini~tration by any suitable route, such as
oral, parenteral or topical application, although the oral route is plefell~d. The
compositions may be in the form of tablets, c~l snlPs, powders, granules, lozenges,
25 creams or liquid preparations, such as oral or sterile parenteral solutions or
suspensions. Tablets and capsules for oral ~-imini.ctration may be in unit dose
present~tion form and may contain conventional excipients such as binding agents,
for example, syrup acacia, gelatin, sorbitol, tr~g;lc~nth, or polyvinylpyrollidone;
fillers, for example lactose, sugar, mai~-starch, c~lcillm phosphate, sorbitol or
30 glycine; tabletting lubric~nt~, for example magnesium stearate, talc, polyethylene
glycol or silica; disintegrants, for example potato starch; or acceptable wetting agents
such as sodium lauryl sulphate. The tablets may be coated according to methods well
known in normal ph~ ceutical practice. Oral liquid preparations may be in the
form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or
35 elixirs, or may be presented as a dry product for reconstitution with water or other
suitable vehicle before use. Such liquid preparations may contain conventional
additives such as suspending agents, for Px~mplP sorbitol, methyl celllllose. glucose
syrup, gelatin, hydlo~LyeLllyl cellulose, carboxymethyl cellulose, alllminium stearate
gel or hydrogenated edible fats; emulsifying agents, for example lPcithin, sorbitan
- 6 --
CA 0221788~ 1997-10-31
W 096/34869 PCTAEP96/01882
monooleate, or acacia; non-aqueous vehicles (which may include edible oils), foreY~mplP almond oil, oily esters, glycerine, propylene glycol, or ethyl alcohol;
preservatives, for example methyl or propyl p-hydroxyben7o~te or sorbic acid; and, if
desired conventional flavouring or colouring agents. Suppositories will contain
convçntion~l suppository base, eg cocoa-butter or other glyceride.
For parenteral ~(imini~tration, fluid unit dosage forms are prepared utili.~ing
the compound and a sterile vehicle, water being preferred. The compound, depending
on the vehicle and concentration used, can be either suspended or dissolved in the
vehicle. In plt;pa illg solutions the compound can be dissolved in water for injection
and filter sterilised before filling into a suitable vial or arnpoule and sealing.
Advantageously, agents such as local ~n~losthetic~ preservative and buffering agents
can be dissolved in the vehicle. To enhance the stability, the composition can be
frozen after filling into the vial and the water removed under vacuum. The dry
lyophilised powder is then sealed in the vial and an accompanying vial of water for
injection may be supplied to reconstitute the liquid prior to use. Parenteral
suspensions are prepared in subst~nti~lly the same manner except that the compound
is suspended in the vehicle instead of being dissolved and sterilisation cannot be
accomplished by filtration. The compound can be sterilised by exposure to ethylene
oxide before suspending in the sterile vehicle. Advantageously, a surfactant or
wetting agent is included in the composition to facilitate uniform distribution of the
compound.
The composition may contain from 0.1% to 99.5% by weight, preferably from
10-60% by weight, of the active material, depending on the method of ~rlmini~tration
Where the compositions comprise dosage units, each unit will preferably contain
from 50-500 mg of the active ingredient. The dosage as employed for adult human
tre~tm.ont will preferably range from 100 mg to 12 g per day for an average adult
patient (body weight 70 kg), for instance 1500 mg per day, depending on the route
and frequency of ~timini~tration. Such dosages correspond to approximately 1.5 to
170 mg/kg per day. Suitably the dosage is from 1 to 6g per day.
The daily dosage is suitably given by ~lminictering a compound of the
invention several times in a 24-hour period. Typically, 250 mg is ~-lmini~t~red 4
times a day although, in practice, the dosage and frequency of ~rlmini~tration which
will be most suitable for an individual patient will vary with the age, weight and
response of the patients, and there will be occasions when the physician will choose a
higher or lower dosage and a different frequency of ~llminictration~ Such dosageregimens are within the scope of this invention.
No toxicological effects are inclic~t~d when a compound of the invention is
mini.ctered in the above mentioned dosage range.
CA 0221788~ 1997-10-31
WO 96/34869 PCT/l~P96101882
The present invention also incln<le.s a method of treating bacteAal infections in
humans and animals which method c-)mpri.cçs ~fimini.cte.rin~ a therapeutically
effective amount of an antibiotic compound of the present invention.
In a further aspect, the present invention also provides for the use of a
compound of formula (I) for the m~nllf~cture of a mediç~ment for treating bacteAal
infection.
The compounds of the present invention are active against a broad range of
Gram-positive and Gram-negative bacteAa, and may be used to treat a wide range of
b~cteri~l infections inr~lu~iin~ those in immunocol.lpl~lllised p~ti~nts.
Amongst many other uses, the compounds of the invention are of value in the
tre~tmçnt of skin, soft tissue, respiratory tract and uAnary tract infections in humans
and may also be used to treat m~stiti.c in cattle.
A particular advantage of the antibacteAally active compounds of this
invention is their stability to ,B-l~rt~m~ce enzymes and they are therefore effective
15 against ~ ct~m~ce producing org~ni.cm.c
The present invention further provides a process for the preparation of a
compound of formula (I) which process compri.ces treating a corresponding
compound of formula (I), wherein R is an alkali metal cation, with a compound offormula (i) or (ii):
R1
X /~/ C02R~
Il
R~R3 (i)
XCH(Ra)O.CO.Rb (ii)
wherein X is a leaving group such as halogen, more particularly bromine or iodine.
The reaction is typically carAed out at between 0 and 60 C, for example at
25 ambient temperature, under an inert, for example argon, atmosphere, in a suitable
organic solvent, for example, N-methylpyrrolidin-2-one. Other suitable solvent
systems include N, N'-dimethylformamide and N, N'-dimethylacetamide.
Compounds of formula (i) can be prepared as descAbed by F. Ameer et al, J.
Che~ Soc., (1983), 2293.
Compounds of formula ICH(Ra)O.CO.Rb can be prepared from the
corresponding chlorAde via the Finkelstein reaction, which is well known to those
skilled in the art.
Compounds of formula ClCH(Ra)O.CO.Rb can be prepared by esteAfying an
acid of formula RbCOOH with chloromethyl chlorosulphate (Binderup et al.,
-- 8 -
CA 0221788~ 1997-10-31
W096/34869 PCTAEP96/01882
Synthet~c Commlm., 14(9), 857-864 (1984)), or by treating a compound of formula
ClCH(Ra)OCOCl with a compound of formula HRb in dichlorometh~n~-/pyridine
(Yoshimura et al., J. Antibiot., 1987, 40(1), 81-90).
Compounds of formula (I) wherein R is an alkali metal cation" such as
5 sodium, are described in International Patent Application No. PCT/GB94/02347, and
hereinbelow in Preparation 1.
A further process for the preparation of a compound of formula (I) comprises
r~ subjecting a compound of formula (II):
R
H H ~
R1_ ~_R
\~ 3
CO2R
(~)
in which R, Rl and R2 are as hereinbefore defined,
R3 is a readily removable carboxy protecting group,
X is oxygen or a group PR4R5R6,
R4, R5 and R6 may be the same or different and is each an optionally substituted(Cl 6)aIkyl or an optionally substituted aryl group, preferably an n-butyl or a phenyl
group;
to c~l,&penem ring forming conditions;
and thereafter, and if n~cecc~ry, carrying out any or all of the following steps:
removing any protecting group(s);
converting a first group R 1 comprising a hydroxyl substituent into a further group R 1
compricing an amino-or fluoro group;
converting the product into a salt; and
esterifying the product as set out above.
Suitable carbapenem ring forming conditions are well known in the art.
When X is oxygen, suitable ring forming conditions include treating the
compound of formula (II) with a trivalent organic phosphorus compound of formula(m)
PR7(oR8)(oR9) (III)
in which:
R7 is (Cl 4)alkyl, (Cl 3)alkoxy or phenyl optionally substitued by (Cl 3)alkyl; and
R8 and R9 which may be the same or different is each (Cl 4)alkyl, allyl, benzyl or
phenyl optionally substitued by (Cl 3)alkyl or (Cl 3)alkoxy; by analogy with theprocess described in EP 0 476 649-A (Hoechst AG). Suitable reagents of formula
(m) include trimethyl phosphite, triethyl phosphite, dimethyl methylphosphonite and
g
CA 0221788~ 1997-10-31
WO 96/34869 PCT/EP96101882
diethyl methylphosphonite. Suitably, the reaction is effected in an organic solvent
such as tetrahydrofuran, ethyl acetate, an aromatic solvent such as benzene, toluene,
xylene or mesitylene or a halogenated hydrocarbon solvent such as dichloromethane,
trichloromethane or 1,1,2-trichloroethane, and at a temperature between 50 and 180~
C, preferably between 70 and 165~C.
When X is a group PR4RSR6, compounds of formula (I) may be obtained by
the well known Wittig cyclisation route to call,apenems (Guthikonda et al, J. Med.
Chem., 1987, 30, 871). For in~t~nce, when R4, R5 and R6 is each phenyl, the process
comprises the ring closing elimin~tion of the elements of triphenylphosphine oxide.
The ring closure may be suitably effected by heating the compound of formula (II, X
= PR4RSR6) at a temperature which is preferably in the range 40 to 145~C, more
preferably 80 to 140~C, in an inert solvent such as benzene, toluene or xylene,
preferably under dry conditions and under an inert atmosphere and optionally in the
presence of a radical scavanger such as hydroquinone. When R4, R5 and R6 is eachn-butyl, cyclisation may be effected at a lower temperature, for instance above 50~C,
by analogy with the process described in WO 92/01695 (Beecham Group, for
analogous penems).
In the substituent Rl, a hydroxyl or an amino group, if present, may
optionally be protected. Suitable hydroxy protecting groups include organosilyl, for
inct:3n~e a triaLkylsilyl group such as trimethylsilyl or t-butyl dimethylsilyl, or
trichloroethyloxycarbonyl, 4-nitrobenzyloxy-carbonyl, 4-methoxybenzyloxy carbonyl
and allyloxycarbonyl. Suitable amino protecting groups include alloxycarbonyl,
4-methoxybenzyloxy carbonyl and 4-nitrobenzyloxycarbonyl.
Suitable values for the protecting group R3 include allyl, 4-methoxybenzyl
and 4-nitrobenzyl. The conditions necessary for removing the protecting group will,
of course, depend upon the precise nature of the protecting group. For instance, when
R3 is 4-methoxybenzyl, aluminium trichloride and anisole in dichloromethane at -30
to -70~C may be used, when R3 is allyl (prop-2-en- l-yl), a combination of
triphenylphosphine, sodium-2-ethylhexanoate in ethyl acetate/MDC and tetrakis-
(triphenylphosphine)palladium (0) may be used and when R3 is p-nitrobenzyl
hydrogenation in the presence of palladium on a carbon catalyst in aqueous solvent
eg, aqueous 1,4,dioxan THF ethanol may be used.
Compounds of forrnula (II) in which X is oxygen may be obtained by a
process which comprises reacting a compound of formula (IV):
R
H H ~
R ~ R
,~NH O
O
- 10-
CA 0221788~ 1997-10-31
W 096134869 PCTAEP96/01882
(IV)
in which R, Rl and R2 are as hereinbefore tiP.finP~l with a compound of
formula (V): --
ClCOCO2R3 (V)
in which R3 is a readily removable carboxy protecting group;
under acylating conditions, by analogy with the process described in TetrahedronLetters, 25, 1984, 2395.
Compounds of formula (~) in which X is a group PR4RSR6 may be obtained
10 from a compound of formula (IV) as hereinbefore defined by the following sequence
of steps:
(a) reacting with a suitably protected glyoxylic acid derivative of formula (VI) or a
functional equivalent thereof such as the hydrate;
(oHC)Co2R3 (VI)
in which R3 is a readily removable carboxy protecting group; under dehydrating
conditions, for in.ct~n-~e azeotropic removal of water;
(b) treating the intermediate formed in step (a) with a halogenating agent, for
in.ct~nr~e thionyl chloride, in the presence of a suitable base such as 2,6-lutidine; and
20 (c) treating the intermediate formed in step (b) with a phosphorus reagent of the
f~nula ~
PR4R5R6 (VII)
in which R4, RS and R6 are as hereinbefore ~1efinP.d, in the presence of a
suitable base such as 2,6-lutidine.
Compounds of formula (IV) may be prepared by treating a compound of
formula (VIII):
,~ CH2R
(VIII)
in which R and R2 are as hereinbefore defined;
with a compound of formula (IX)
-- _ OR
Rl _~
~NH
O
(IX)
35 in which Rl is as hereinbefore defined, and
- 11 -
CA 0221788~ 1997-10-31
WO 96/34869 PCT/EP96/01882
Rll is an acyl group, for in.ct~n~e acetyl;
in the presence of a base, such as, for in.et~n- e, lithium hexamethykli.~ 7.i~ie
(LHMDS);
according to the procedures described in Tet-~h.~ on_Lett., 1987, 28, 507, and Can. J.
S Chem, 1988, ~6, 1537.
Compounds of formula (IV) may also be prepared by treating a compound of
formula (VIIIa):
R 2
J=~ R
oSiR314
(VIIIa)
in which R and R2 are as hereinbefore defined and SiR3 14 is a triaLkylsilyl such as
trimethylsilyl or t-butyldimethylsilyl,
with a compound of formula (IXa):
H H
R _ ,~ OR
13
0~
(~a)
20 in which Rl and R1 1 are as hereinbefore defined and
R13 is either hydrogen or an aminoprotecting group, for in.ct~nçe, a triaLkylsilyl group
such as trimethylsilyl;
in the presence of a Lewis acid, such as, for in.ct~n~e, zinc chloride or trimethylsilyl
trifluoromethane sulphonate, in an inert organic solvent such a halogenated
25 hydrocarbon solvent, for instance dichloromethane at ambient temperature:
Compounds of formula (VIIIa) may be prepared by treating compounds of formula
(VIII) with triaLkylsilyl chloride or triaLkylsilyl triflate, and triethylamine in MDC.
If the aminoprotecting group R13 in (IXa) requires subsequent removal, this
may be achieved by conventional means, such as mild acid treatment eg, methanol
30 and hydrochloric acid or pyridinium p-toluenesulphonate, where Rl3 is
trimethylsilyl.
Compounds of formula (VIII) are well known to those skilled in the art and
may be obtained by standard synthetic procedures as described in the following
Examples.
- 12-
CA 022l788~ l997-lO-3l
W 096/34869 PCTnEP96/01882
Compounds of formula (IX) are well known to those skilled in the art and
may be obtained by standard synthetic procedures such as described in, for example,
Het., 1982, 17, 201 (IX, R1 is l-hy~ yelhyl) and EP 0 234 484 (~, Rl is 1-
fluoroethyl).
Compounds of formula (I) in which Rl is an amino-substituted alkyl or
- cycloaLkyl may be conveniently prepared from a corresponding compound of formula
(I) in which Rl includes a hydroxy group by a Mitsunobu-type azide displacement of
, the hydroxy group thereof, followed by catalytic reduction, according to the
procedure described in J Chem Soc, Perkin I, 1982, 3011.
Compounds of formula (I) may also be prepared by a process which
comprises reacting a cornpound of formula (X)
H H R
0
CO2R
(X)
in which Rl and R2 are as hereinbefore ~lç~lnPti, R3 is a readily removable carboxy
protecting group and Xl is a leaving group,
with a compound of forrnula (XI):
M-R (XI)
in which M is a metallo group and R is as hereinbefore defined;
25 in a cross-coupling reaction in the presence of a cross-coupling reaction catalyst
selected according to the identity of M and thereafter and if nçce.~C.S~ry removing any
protecting group and/or converting the product into a salt and/or esterifying the
product as set out hereinabove.
~uitable values for the protecting group R3 include 4-methoxybenzyl
30 4-nitrobenzyl.
Examples of suiLable leaving groups X1 include for instance
trifluoromçth~nçsulphonyloxy, methanesulphonyloxy, 4-toluene sulphonyloxy,
fluorosulphonyloxy, chloro, bromo, iodo and diphenoxyphosphoryloxy.
Suitable metals for use in the metallo group M are well known in the art and
35 include tin, alllminium, zinc, boron, mercury and zirconium.
- 13 -
CA 022l788~ l997-lO-3l
W 096/34869 PCTAEP96/01882 Preferred ç~mplçs of the metaIlo group M include for inct~nre
R14R15R16Sn, B(OR)2 and ZnCl in which R14, R15 and R16 may the same or
different and are each (Cl 6~ alkyl. Preferably, the metallo group M is an
organl-st~nn~nP R14RlSR16Sn, and R14=R15=R16= methyl or n-butyl.
S Suitable cross-coupling catalysts are well known in the art and includep~ inm compounds, in particular p~ rlinm (0) and p~ lm (II) compounds,
such as those described in "p~ lm E~ç~gçnt~ in Organic Synthesis", RF Heck,
~rademiC Press Ltd, 1985. Examples thereof include
tris(dibenzyli~1PnPacetone)-lir~ lillm (O), tetrakis(triphenylphosphine)ps~ rlillm (O),
trans dimethyl bis(triphenylphosphine)p~ nm (II), and p~ lm (II) acetate,
benzyl bis(triphenylphosphine)p~ illm (II) chloride,
bis(triphenylphosphine)p~ lillm (II) dichloride. Such p~ linm reagents are
preferably used in combination with a halide source such as zinc chloride or lithum
chloride and optionally in the presence of a phosphine ligand of palladium, for
instance a compound such as a triarylphosphine, for example,
tris(4-methoxyphenyl)phosphine or tris(2,4,6-trimethoxyphenyl) phosphine; a tri-heteroarylphosphinP, for example, trifurylphosphine, or a triarylarsine, for example
triphenylarsine.
When M is an organost~nn~nP R14 R15 R16 Sn-, a preferred catalyst system
is tris(dibenzyli-lPnPacetonç)dip~ (lillm (0), in the presence of zinc chloride and a
phosphine compound. When M is ZnCl a preferred catalyst is
tris(dibenzylidenP~c etone ~lip~ lillm (O), in the presence of a phosphine compound.
Suitably the reaction is effected in an inert aprotic polar coordin~ting solventsuch as tetrahydrofuran, diethylether, dioxane, 2-dimethoxyethane, acetonitrile,dimethyl formamide, dimethyl sulphoxide and the like, and under a dry, inert
atmosphere such as argon. Suitably, the reaction is effected initially at a low
temperature, for in.ct~nre about -78~C, with the final phase of the reaction then being
effected at ambient temperature.
Analogous procedures in which M is organoct~nn~np are described in
EP 0 444 889 (Merck & Co.) and EP 0 430 037 (Banyu Pharm~ce~ltic~l Co.).
Compounds of formula (X) are well known in the art and may be obtained
according to the procedures described in EP 0 444 889 (Merck & Co.), EP 0 430 037
(Banyu Ph~rm~ceutical Co.) and by Rano et al, Tet. Letters, 1990, 31, 2853.
Compounds of formula (XI) are well known in the art and may be obtained
according to the procedure described in Heterocycles, 1992, 33(2), 813.
The following Examples illustrate the invention but are not intended to limit the
scope in any way.
CA 02217885 1997-10-31
W 096/34869 PCTAEP96101882General Instruclions - Solutions were dried using anhydrous m~gn~Si~.... s-llrh
and solvents were removed by evaporation under reduced pressure using a rotary
evaporator. Colllmn chromatography on silica gel used Merck silica gel 60, particle
size <0.063mm.
- 15-
CA 0221788~ 1997-10-31
W 096/34869 PCT/EP96101882
F.Y~lnr'~ 1
2-Ethoxy~ onyl-E-but-2-enyl (5R,65)-2-(1-Ethyl-5-methyl-1,2-pyrazol-5-yl)-6-
S t(lR)-l-l~y~ xyell~yl]carbapen-2-em-3 c~rl,ox,~late
Sodium (5R,65)-2-(1-ethyl-5-methyl-1,2-pyrazol-5-yl)-6-[(lR)-l-
hydroxyethyl]carbapen-2-em-3-carboxylate (200mg) in N-methylpyrrolidinone (2ml)
at room temperature under argon was treated with a solution of ethyl E-2-
10 bromomethylbut-2-enoate [F. Ameer et al, J. Chern Soc., (1983), 2293], (253mg) in
N-methylpyrrolidinone (lml).
After 2h the solution was diluted with ethyl acetate, washed with water (3x),
dried over anhydrous m~gnecillm sulphate and concentrated. The product was
purified by flash silica gel chromatography eluting with 50% ethyl acetate/hexane,
15 then ethyl acetate to give the title com~ound as a pale yellow foam, (153mg, 58%);
(Found: M+, 431.2065, C22H29N3O6 requires M431.2056); nll"" (CH2C12) 3605(w),
1774, 1712 cm~l; dH(CDCl3) 1.24-1.41 (9H, m), 1.82 (lH, d, J4.9Hz), 1.98 (3H, d,n.3Hz), 2.28 (3H, s), 3 16 (lh, dd, J2.8,6.6Hz), 3.27 (lH, dd, J9.0,18.5Hz), 3.57
(lH, dd, J9.9,18.5Hz), 4.07 (2H, q, J7.3Hz), 4.17-4.29 (3H, m), 5.03 and 5.11 (2H
20 ABq, J11.9Hz), 6.94 (lH, s) and 7.21 (lh, q, J7.3Hz).
FY~mplc 2
l-Methylcydohexyloxy~rl,onyloxy,..~ll,yl (5R,6S)-6-t(R)-l-l-y.ll~xyetl,yl~-2-(1-ethyl-5-methyl-pyrazol-3-yl)carl,a~en-2-em-3-carboxylate
(a) Io(lon~thyl l-methylcydohexyl carbonate
1-Methylcyclohexanol (4.57 g) in dichlorometh~n~ (40 ml) was cooled in an ice-bath
and treated with pyridine (3.24 ml). Chloromethyl chloroformate (3.65 ml) in
dichlort-meth~n~ (10 ml) was added dropwise through a pressure-equalising funnel.
The mixture was stirred for 2 h and then washed with water (2 x), dried (MgSO4) and
then evaporated to leave a reddish-coloured oil, which was dissolved in
dichlorom~fh~n.o and filtered through silica gel (230 - 400 mesh ASTM) to give (after
evaporation) chloromethyl 1-methylcyclohexyl carbonate as a colourless oil (8.97 g), o
maX(CH2C12) 2940, 2865, 1763, l~l~ll, 1346, 1292, and 1237 cm~l; o(CDC13) 1.25 -1.70 (llH, m), 2.12 -2.18 (2H, m), 5.69 (2H,s) ppm.
Chloromethyl 1-methylcyclohexyl carbonate (2.07 g) in acetone (7.5 ml) was treated
with 2,6-lutidine (0.116 ml). Sodium iodide (2.25 g) was then added and the mixture
was stirred for 1.5 h. More sodium iodide (1.5 g) was then added and stirring was
- 16 -
CA 0221788~ 1997-10-31
W 096/34869 PCT~EP96101882
contin~le~i After a total of 2.5 h the acetone was removed and CH2C12 / water were
added and the layers separated. The dichlor~ m~oth~ne Iayer was washed with 5%
aqueous Na2S2O3, dried (MgSO4~ and evaporated to leave an oil. This was taken upin hexane (50 ml) and loaded onto a silica gel column, and the column was eluted with
5 5% ethyl acetate in hexane to give iodomethyl l-methylcyclohexyl carbonate
co..l~ g some of the starting chloromethyl compound, ~(CDC13) 1.26 - 1.35 (2H,
m), 1.40 - 1.65 (9H, m, including s at ~ 1.52), 2.10 -2.20 (2H, m), 5.69 (s) and 5.91 (s)
(together 2H, ratio 1: 5) ppm.
(b) 1-MethyIcydohexyloxycarbonyloxyJnethyl (5R,6S)-6-t(R)-1-l~y~ yethyl]-2-
(1-ethyl-5-methyl-pyrazol-~yl)carbapen-2-em-~rl~yla~
Sodium (SR,65)-6-[(R)-1-hydroxyethyl]-2-(1-ethyl-5-methylpyrazol-3-yl)carbapen-2-
em-3-carboxylate (200 mg) in N-methylpyrrolidinone (2 ml) cont~ining 2,6-lutidine
(0.07 ml) was treated with iodomethyl l-methylcyclohexyl carbonate (363 mg) in
15 tetrahydrofuran (0.5 ml). The mixture was stirred under argon for 20 min. Ethyl
acetate (20 ml) was added and washed with water (3 x 25 ml), then with 5%
Na2S2O3 (20 ml), then with 0.05M HCI, followed by saturated brine. After drying
(MgSO4) the ethyl acetate was evaporated to leave a gum which was purified by
chromatography on silica gel (230 - 400 mesh ASTM) (2.5 x 12 cm column), loading20 in CH2C12 / hexane, and eluting with ethyl acetate / hexane mixtures, followed by ethyl
acetate. Fractions containing the product were combined and evaporated and then
rechromatographed on silica gel (230 - 400 mesh ASTM) (2 x), eluting with acetone /
hexane (1: 1). Fractions containing the product were combined and evaporated.
Hexane was added and evaporated to give l-methylcyclohexylo~y~ onyloxymethyl
(SR,65)-6-[(R)-1-hydroxyethyl]-2-(1-ethyl-5-methylpy~razol-3-yl)carbapen-2-em-3-carboxylate (187 mg) as a solid foam, ~maX(cH2cl2) 3678, 3601, 2939, 1773, 1598,1546, 1450, 1380, 1317, and 1241 cm~l; ~max(EtOH/nm 325 (~/dm3mol~lcm~l
10,924), 213 (~/dm3mol~lcm~l 9,391); ~(CDC13) 1.2 - 1.5 (14H, m, including d, J
6.4 at ~ 1.35, t, J7.3 Hz at ~ 1.39 and s at ~ 1.51), 1.93 (lH,d, J4.7 Hz), 2.05 - 2.2
(2H, m), 2.28 (3H, s), 3.18 (lH, dd, J2.7 & 6.5 Hz), 3.29 (lH, dd, J9.1 & 18.7 Hz),
3.64 (lH, dd, J9.9 & 18.8 Hz), 4.08 (2H, q, J7.3 Hz), 4.15 - 4.30 (2H, m), 5.90
(2H, s), 7.05 (lH, s) ppm.
FY~mple 3
~Methoxyprop-2-ylcarbonyloxymethyl (SR,6S)-2-(1-ethyl-~-methylpyrazol-~
yl)-6-[(lR)-1-hydroxyethyl]carbapen-2-em-3-carboxylate
CA 0221788~ 1997-10-31
WO 96/34869 PCT/EP96/01882
The product from Preparation 1 (200 mg, 0.61 mmol) was dissolved in N-
methylpyrrolidin-2-one (2 ml). A solution of 2-metho~y~rop-2-ylcarbonyloxymethyliodide (0.34 g, 1.3 mmol) in N-methylpyrrolidin-2-one (0.5 ml) was added to thissolution at room temperature under argon and after 0.5 h the reaction mixture was
5 diluted with ethyl acetate (15 ml) and the solution was washed with water (3 x 15 ml),
5% sodium thiosulfate solution (15 ml) and then s~t-lr~t~d brine (15 ml). The organic
extract was dried (Na2SO4) and concentrated to an oil which was purified by
chromatography on silica gel eluting with an acetone/toluene gradient mixture to yield
a pale yellow solid which was recrystallised from acetone/diethyl ether to afford the
10 title com~ound as a colourless oil (0.23 g, 87%); (Found: M+ 435.2007.
C21H2gN3O7 requiresM435.2006); vmaX (CHCl3) 3612, 3016, 2981, 2937, 1771,
1734(sh), 1596 cm~l; ~H (CDC13) 1.38-1.45 (12H, m), 1.81 (lH, d, J4.9 Hz), 2.29
(3H,s),3.18(1H,dd,J2.8,6.6Hz),3.30(1H,dd,J9.1, 18.8Hz),3.64(1H,dd,J9.9,
18.8 Hz), 4.08 (2H, q, J7.3 Hz), 4.16-4.29 (2H, m), 5.96 (lH, d, J5.5 Hz), 6.03 (lH,
J5.5 Hz) and 7.02 (lH, s) ppm.
Preparation 1
Sodium (5R,6S)-6-[(R)-l-hyuro~y~tl~yl]-2-(1-ethyl-5-methylpyrazol-3-
yl)carbapen-2-em-3-carboxylate
(a) Ethyl l-ethyl-S-methyl~yl ~zole-3-carboxylate
N-Ethylhydrazine oxalate (12 g) in glacial acetic acid (100 ml) was cooled in an ice-
bath and treated with ethyl 2,4-dioxovalerate (11.24 ml). After addition was complete
the mixture was stirred at room temperature; after ca. 45 min the mixture was
warmed to dissolve insoluble ethylhydrazine oxalate. The mixture was stirred for a
further 2 h and then poured into water( ca. 300 ml) / ethyl acetate (ca. 700 ml) and
solid K2CO3 was carefully added, with stirring, until the pH was neutral. After
separation the aqueous layer was re-extracted with ethyl acetate. The combined ethyl
acetate extracts were dried (MgS04), and the solvents removed to leave an oil.
Chromatography on silica gel, loading in CH2Cl2/hexane and eluting with a gradient
elution of ethyl acetate/hexane mixtures (from 2:8 to 1:1) gave ethyl 1-ethyl-5-methylpyrazole-3-carboxylate as an oil (13.2 g); vmaX (CH2Cl2) 1717, 1446, 1389,and 1219 cm~1; ~(CDCl3) 1.38 (3H, t, J7.2 Hz), 1.42 (3H, t, J7.3 Hz), 2.30 (3H, s),
4.17 (2H, q, J7.3 Hz), 4.38 (2H, q, J7.1 Hz), 6.55 (lH, s); (Found n2/z 182.1055.
CgH14N2O2 requires n2/z 182.1055).
- 18-
CA 022l788~ l997-lO-3l
W096/34869 PCT~P96/01882
(b) l-Ethyl-5-~ ll-yl~ ole-3-carboxylicacid
Ethyl l-ethyl-5-methylpyrazole-3-carboxylate (10.93 g) in ethanol (70 ml) was
treated with KOH (3.69 g), followed by water (30 ml), and the mixture was stirred
z S and heated under reflux for 6 h. The ethanol was removed using a rotary evaporator
and ethyl acetate/water were added. The pH of the mixture was adjusted to 3.0 and
the layers were separated. The aqueous layer was re-extracted with ethyl acetate.
The combined ethyl acetate layers were extracted with excess aqueous NaHCO3. TheNaHCO3 extract was poured into excess acid, and the pH was then adjusted to 3, and
NaCl was added to the solution. The mixture was then repeatedly extracted with
ethyl acetate, and the combined extracts were dried (MgSO4) and evaporated. The
residue was triturated with diethyl ether to give the acid as a solid (5.65 g); ~max
(CH2C12) 2754, 2598, 1698, 1498, 1464, 1387, and 1233 cm~l; o(CDC13) 1.40
(3H, t, J7.3 Hz), 2.32 (3H,s), 4.19 (2H, q, J7.3 Hz), 6.61 (lH,s) ppm; (Found m/z
154.0740. C7HloN202requires m/z 154.0742).
(c)N-Methoxy-N-methyl- l-ethyl-5-methylpyrazole-3-carboy~mi~le
l-Ethyl-S-methylpyrazole-3-carboxylic acid (5.25 g) in dry dichloromethane (100 ml)
cont~ining N,N-dimethylformamide (0.26 ml) was cooled in an ice-bath and treatedwith a solution of oxalyl chloride (3.27 ml) in dichloromethane (25 ml), added
dropwise. The mixture was stirred in the cold for 25 min, and then allowed to warm
to room temperature, when evolution of a gas was observed. After 10 min the solvent
was removed by evaporation in vacuo and toluene was added and removed (x 2) to
ensure any residual HCl and oxalyl chloride had been removed. The resultant acidchloride was redissolved in dry dichloromethane and then treated with N,O-
dimethylhydroxylamine hydrochloride (3.61 g). The mixture was cooled in an ice-
bath and treated with pyridine (6.0 ml). the mixture was then allowed to stir at room
temperature for l.S h and then diluted with ether (100 ml) and washed with brine.
The organic layer was then dried (Mg~04) and evaporated to leave an oil. This was
the chromatographed on silica gel, loading in dichloromethane, and eluting with ethyl
acetate / hexane mixtures to give, after evaporation of requisite fractions, thehydroxamate (5.2 g) as a solid;
l)maX(CH2C12) 2982, 2937, 1641, 1489, 1445, 1379,and 975 cm~l; ~(CDC13) 1.43
e (3H, t, J 7.3 Hz), 2.29 (3H, s), 3.42 (3H, s), 3.76 (3H, s, ), 4.13 (2H, q, J 7.3 Hz),
6.49(1H,s); (Foundm/zl97.1164. CgHlsN302requiresm/zl97.1164).
- 19-
CA 0221788~ 1997-10-31
W096/34869 PCT~EP96/01882
(d) 3-Acetyl-l-ethyl-5-m~Ll.~ ~ole
N-Methoxy-N-methyl-l-ethyl-5-methylpyrazole-3-carbox~mi~le (3.12 g) in dry
tetrahydrofuran (60 ml) was cooled in an ice-bath and treated with a 3.0M solution of
S methylm~gn~.cillm bromide in ether (11.08 ml). After stirring for 1.5 h the mixture
was poured into a mixture of methanol (100 ml) and SM aqueous HCI (10 ml) in an
ice-bath. The mixture was then evaporated to lower volume and treated with a
mixture of dichlorom~th~ne, water and saturated brine. After separation the aqueous
layer was re-extracted with dichloromethane. The combined dichloromPth~ne
10 extracts were dried (MgSO4) and evaporated to leave an oil (2.26 g), which solidified
on standing; ~max (CH2C12) 1680, 1446, 1425, 1380, 1324, 1208, and 945 cm~1;
(CDC13) 1.44 (3H, t, J7.3 Hz), 2.30 (3H, s), 2.53 (3H, s), 4.13 (2H, q, J7.3 Hz,),
6.51 (lH,s); (Found~ z 152.0949. CgH12N2O requires m~z 152.090).
(e) (3S,4R)-4-[(1-ethyl-5-mell~ y-~zol-3-yl)carbonylmethyl]-3-t(R)-l-tert-
butyldimethylsilyloxyethyl]~7~t-Ain-2-one
3-Acetyl-1-ethyl-5-methylpyrazole (3.51 g) in dry tetrahydrofuran (THF) (150
ml) under an argon atmosphere was cooled in an acetone / solid carbon dioxide bath
and then treated with a lM solution of lithium bis(trimethylsilyl)amide (50 ml). The
mixture was stirred f~r 45 minutes and then (3R,4R)-4-acetoxy-3-[(lR)- 1-tert-
butyldimethylsilyloxyethyl]azetidinone (6.6 g) was added as a solid under a blanket
of argon. The mixture was stirred in the cold for 3.5h. Saturated aqueous ammonium
chloride was then added, followed by ethyl acetate, and the mixture was allowed to
warm to room temperature. ~ little water was added and the layers were separatedand the aqueous layer was re-extracted with ethyl acetate. The combined ethyl
acetate extracts were washed with saturated brine, dried and evaporated.
Chromatography on silica gel, eluting with ethyl acetate/hexane mixtures gave the
title compound (3.65 g), l)max (CH2C12) 3411, 1761, 1678, 1376, 1151, and 838 cm~
1; ~(CDC13) 0.064 (6H, s), 0.86 (9H, s), 1.20 (3H,d, J 6.3 Hz), 1.44 (3H, t, J 7.3
Hz), 2.31 (3H, s), 2.89 (lH, dd, J 1.8 & 4.9 Hz), 3.15 (lH, dd, J 10.0 & 17.1 Hz),
3.50 (lH, dd, J 3.5 & 17.0 Hz), 4.06 - 4.25 (4H, m), 6.11 (lH, s), 6.53 (lH, s).(Found nz/z 379.2296. ClgH33N303Si requires ~~/z 379.2291).
- 20 -
CA 0221788~ 1997-10-31
W 096/34869 PCTAEP96/01882
(f) Allyl (2R and 2S)-2-{(3S,4R) 1 t(l-ethyl-5-mell.yl~yl ~ ol-3
yl)carbonylmethyl]-3-t(R)-l-tert-butyldh--ell.yl~ilyloxy~:ll.yl]-2--)~o~7etiAinyl}-2-
hydroxy~~etq~e.
(35,4R)-4-[(1-Ethyl-5-methylpyrazol-3-yl)carbonylmethyl]-3-[(R)-l-tert-
butyldimethylsilyloxyethyl]~7eti-1in-2-one (3.6 g) and allyl glyoxylate hydrate (1.66
g) in toluene (100 ml) were heated under reflux in a Dean and Stark apparatus under
a~l aullosphere of argOn for 3.5h. T.i.c. or ~ihe reacuon mixture showed the reaGtion
had almost prceeded to completion, so more allyl glyoxylate hydrate (190 mg) wasadded and the mixture was heated under reflux for a further 45 min. The mixture was
cooled, the toluene was removed to give crude allyl (2R and 2S)-2-{(35,4R)-4-[(1-
ethyl-5-methylpyrazol-3-yl)carbonylmethyl]-3-[(R)- l-tert-
butyldimethylsilyloxyethyl]-2-oxoazetidinyl}-2-hydroxyacetate, which was used inthe next stage; ~max (CH2C12) 3681, 3518, 1758, 1676, 1448, 1376, 1326, 1209,
1148, 1092, 954, and 836 cm~l; ~(CDC13) inter alia 0.035(S), 0.061(S) (together
6H,), 0.858 (s), 0.865 (s) (together 9H), 1.21 (d, J 6.2 Hz), 1.24 (d, J 6.2 Hz),
(together 3H), 1.44 (3H, t, J 7.2 Hz), 2.31 (3H, s), 2.95 - 3.00 (lH, m), 3.25 - 3.64
(2H, m), 6.53 (s), 6.56 (s) ppm.
(g) Allyl 2-{(3S,4R)-4-[~1-ethyl-5-mell-yl~ ~ol-3-yl)carbonylmethyl]-3-[(R)-l-
tert-butyldimethylsilyl~xyelllyl]-2-~o~7et~ nyl}-2-(tri-n
l~ulyll~hosphoranylidene)~ret~te
Allyl (2R and 2S)-2-{(3S,4R)-4-[(1-ethyl-5-methylpyrazol-3-
yl)carbonylmethyl]-3-[(R)- l-tert-butyldimethylsilyloxyethyl] -2-oxoazetidinyl } -2-
hydroxyacetate (crude from the above preparation) in dry THF (125 ml) under argon
was cooled to -20~C and treated with 2,6-lutidine (1.98 ml), followed by thionylchloride (1.24 ml). The mixture was stirred at -20~C for 30 min~tes~ and then
allowed to warm to room temperature and filtered, washing the residue with THF (20
ml). The filtrate was evaporated in vacuo, toluene (70ml) was added and removed in
vacuo and the residual oil was dried in vacuo. The oil was then taken up in 1,4-dioxan (40 ml) under an argon atmosphere, and treated with tri-n-butylphosphine
(3.11 ml). The mixture was stirred for 1 h. 2,6-Lutidine (1.59 ml) was then added
and the mixture was stirred for a further 30 min~lt.os The mixture was diluted with
ethyl acetate, washed with water, then with brine, and dried (MgSO4). After
removal of the ethyl acetate the crude product was chromatographed on silica gel,
- 21 -
- = =
CA 0221788~ 1997-10-31
W O~/3~ PCTAEP96/01882
eluting with ethyl acetate/hexane mixtures to give the phosph-)r~nP, which was used
in the next stage.
S (h) Allyl 2-{(3S,4R)~-[(l-ethyl-~-mt:ll.yl~yl~ol-3-yl)carbonylmethyl]-3-[(R)-I-
I~y~lr~>xy~thyl]-2-o~o~7~h~linyl}-2-(tri-n-b~ly~ horanylidene)~
The phosphorane prepared above was taken up in 1,4-dioxan (60 ml) and
treated with SM HCl (20 ml). After 1 h the mixture was carefully treated with ca. 40
ml saturated aqueous NaHCO3, followed by solid NaHCO3 until the pH was slightly
~lk~1ine Saturated brine was added and the mixture was extracted twice with ethyl
acetate. The combined extracts were dried (MgSO4) and evaporated. The residue
was chromatographed on silica gel, eluting with ethyl acetate/hexane mixtures to give
the hydroxy compound, (2.60 g), vmaX (CH2C12) 3454, 1741, 1667, 1606, 1448,
1403, 1379, 1155, 1087, 953, and 811 cm~1.
(i) A!lyl (SR~6S)-6-[(R)-l-hydl ~y~ yl]-2-(l-efflyl-S-meLhyll~yl ~ol-3-
yl)carbapen-2-em-3-carboxylate
Allyl 2- { (35,4R)-4-[(1 -ethyl-5-methylpyrazol-3-yl)carbonylmethyl]-3-[(R)- 1 -hyd~ y~thyl~-2-oxo~7eti~1inyl}-2-(tri-n-butylphosphoranylidene)acetate (2.6 g), in
toluene (120 ml) containing hydroquinone (20 mg) was heated under reflux in an
argon atmosphere for 4 h, allowed to stand for 64 h, and then heated under reflux for
a further 2 h. The mixture was cooled and then loaded onto a column (4.5 x 12 cm)
of silica gel (particle size 0.040 -0.063 mm), eluting with ethyl acetate/hexanemixtures; 1:1; 6:4; 7:3; 8:2; 9:1 (250 ml of each), followed by ethyl acetate. This gave
the carbapenem (436 mg); 1~maX(CH2Cl2) 3604, 2976, 1774, 1716, 1600, 1546,
1311, 1189 cm~l; ~max(EtOH)/nm 321.5 (~/dm3mol~lcm~l 14,856), â(CDCl3)
1.36 (d, J 6.3 Hz), 1.39 (t, J 7.3 Hz) (together 5H), 1.80 (lH, d, J 5.0 Hz), 2.28
(3H,s), 3.19 (lH, dd J 2.7 & 6.7 Hz), 3.28 (lH, dd, J 9.0 & 18.6 Hz), 3.60 (lH, dd, J
9.9 & 18.5 Hz), 4.08 (2H, q, J7.3 Hz), 4.16 - 4.30 (2H, m), 4.68 - 4.90 (2H, m), 5.27
- 22 -
CA 0221788~ 1997-10-31
W 096/34869 PCT~EP96101882(lH, m, approx d, J ca. 12 Hz), 5.46 (m, approx d, J ca. 17 Hz), 5.93 - 6.08 (lH, m),
7.00 (lH, s) ppm; [Found )n/z 345.1693. ClgH23N3O4 requires m/z 345.1689].
~j) Sodium (~R,6S)-6-[(R)-1-lly~ll o~y~ yl]-2-(1-ethyl-5-methyl-pyrazol-3-
r 5 yl)carbapen-2-em-3-carboxylate
Allyl (5R,65)-6-[(R)-l-hyd~ y~ yl]-2-(1-ethyl-5-methylpyrazol-3-
yl)carbapen-2-em-3-carboxylate (267 mg) in dichlorometh~n~ (3ml) and ethyl acetate
(3 ml) under argon was treated with sodium 2-ethylhexanoate (183 mg), followed by
10 triphenylphosphine (24 mg), followed by tetrakis(triphenylphosphine)palladium(0)
(35 mg) and the mixture was stirred for 45 min. Diethyl ether (lOOml) was then
added, and after stirring for 90 minutes, the mixture was centrifuged. The residual
solid was dried under a stream of argon, and then in a desiccator. The solid was then
taken up in water containing sodium chloride and chromatographed on DIAION
15 HP20S~ resin, eluting with water, followed by water/THF mixtures; 1%, 2%, and3%,THF. Fractions were monitored by HPLC, and those containing the product were
combined, reduced in volume and freeze-dried to give sodium (SR,65)-6-[(R)-l-
hydLo~yethyl]-2-(1-ethyl-5-methylpyrazol-3-yl)carbapen-2-em-3-carboxylate as a
solid (168 mg); ~max(KBr) 1761, 1608, 1577, 1381, 1225 cm~l; ~max(H2O)/nm 298
20 (~/dm3mol~lcm~l 8,531); ~(D20) 1.26 (d, J ca. 6 Hz), 1.27 (d, J ca. 7 Hz) (together
SH), 2.23 (3H, s), 3.17 (2H, approx d, J ca. 9 Hz), 3.44 (lH, dd, J 2.9 & 6.0 Hz),
4.04 (2H, q, J7.3 Hz), 4.15 - 4.25 (2H, m), 6.41 (lH, s) ppm.