Note: Descriptions are shown in the official language in which they were submitted.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
GENETIC VACCINE VECTOR ENGINEERING
CROSS-REFERENCE TO RELATED APPLICATIONS
This application claims benefit of US Provisional Application No.
60/074,294, filed February 11, 1998, which application is incorporated herein
by reference
for all purposes.
BACKGROUND OF THE INVENTION
Field of the Invention
This invention pertains to the field of genetic vaccines. Specifically, the
invention provides multicomponent genetic vaccines that contain components
that are
optimized for a particular vaccination goal.
Background
Genetic immunization represents a novel mechanism of inducing protective
humoral and cellular immunity. Vectors for genetic vaccinations generally
consist of DNA
that includes a promoter/enhancer sequence, the gene of interest and a
polyadenylation/
transcriptional terminator sequence. After intramuscular or intradermal
injection, the gene
of interest is expressed, followed by recognition of the resulting protein by
the cells of the
immune system. Genetic immunizations provide means to induce protective
immunity even
in situations when the pathogens are poorly characterized or cannot be
isolated or cultured in
laboratory environment.
Elicitation of a desired in vivo response by a genetic vaccine generally
requires multiple cellular processes in a complex sequence. Several potential
pathways exist
along which a genetic vaccine can exert its effect on the mammalian immune
system. In one
pathway, the genetic vaccine vector enters cells that are the predominant cell
type in the
tissue that receives vaccine (e.g., muscle or epithelial cells). These cells
express and release
the antigen encoded by the vector. The vaccine vector can be engineered to
have the antigen
released as an intact protein from living transfected cells (i.e., via a
secretion process) or
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
2
directed to a membrane-bound form on the surface of these cells. Antigen can
also be
released from an intracellular compartment of such cells if those cells die.
Extracellular
antigen derived from any of these situations interacts with antigen presenting
cells (APC)
either by binding to the cell surface (specifically via IgM or via other non-
immunoglobulin
receptors) and subsequent endocytosis of outer membrane, or by fluid phase
micropinocytosis wherein the APC internalizes extraceliular fluid and its
contents into an
endocytic compartment. Interaction with APC may occur before or after partial
proteolytic
cleavage in the extracellular environment. In any case, the antigen derived
from vaccine
vector internalization and antigen expression within the predominant cell type
in the tissue
ends up within APC. The APC then process the antigen internally to prime MHC
Class I
and or Class II, essential steps in activation of CD4+ T-helper cells (TH1
and/or TH2) and
development of potent specific immune responses.
In a parallel pathway, the genetic vaccine plasmid enters APC (or the
predominant cell type in the tissue) and, instead of antigen derived from
plasmid expression
being directed to extracellular export, antigen is proteolytically cleaved in
the cell cytoplasm
(in a pmteasome dependent or independent process). Often, intracellular
processing in such
cells occurs via proteasomal degradation into peptides that are recognized by
the TAP-1 and
TAP-2 proteins and transported into the lumen of the rough endoplasmic
reticulum (RER).
The peptide fragments transported into the RER complex with MHC Class I. Such
antigen
fragments are then expressed on the cell surface in association with Class I.
CD8+ cytotoxic
T lymphocytes (CTL) bearing specific T cell receptor then recognize the
complex and can,
in the presence of appropriate additional signals, differentiate into
functional CTLs.
In addition, poorly characterized pathways, which are generally not dominant,
exist in APC for trafficking of cytoplasmically generated peptides into
endosomal
compartments where they can end up complexed with MHC Class II, and thereby
act to
present antigen peptides to CD4+ TH 1 and TH2 cells. Because activation,
proliferation,
differentiation and immunoglobulin isotype switching by B lymphocytes requires
help of
CD4+ T cells, antigen presentation in the context of MHC Class II molecules is
crucial for
induction of antigen-specific antibodies. By virtue of this pathway, a genetic
vaccine vector
can lead to CD4+ T cell stimulation in addition to the dominant CD8+ CTL
activation
CA 02320626 2000-08-10
WO 99/41369 PGTNS99/03022
process described above. This alternative pathway is, however, of little
consequence in
muscle cells where levels of MHC Class II expression are very low or zero.
Genetic vaccination can also elicit cytokine release from cells that bind to
or
take up DNA. So-called immunostimulatory or adjuvant properties of DNA are
derived
from its interaction with cells that internalize DNA. Cytokines can be
released from cells
that bind and/or internalize DNA in the absence of gene transcription.
Separately, interaction
of antigen with APC followed by presentation and specific recognition also
stimulates
release of cytokines that have positive feedback effects on these cells and
other immune
cells. Chief among these effects are the direction of CD4+ TH cells to
differentiate/
proliferate preferentially to TH 1 or TH2 phenotypes. Furthermore, cytokines
released at the
site of DNA vaccination, regardless of the mechanism of their release,
contribute to
recruitment of other immune cells from the immediate local area and more
distant sites such
as draining lymph nodes. In recognition of the importance of cytokines in
elicitation of a
potent immune response, some investigators have included the genes for one or
more
cytokines in the DNA vaccine plasmid along with the target antigen for
immunization. In
this case cytokines are derived not only from processes intrinsic to the
interaction of DNA
with cells, or specific cell responses to the antigen, but via synthesis
directed by the vaccine
plasmid.
Immune cells are recruited to the site of immunization from distant sites or
the bloodstream. Specific and non-specific immune responses are then greatly
amplified.
Immune cells, including APC, bearing antigen fi~agments complexed to MHC
molecules or
even expressing antigen from uptake of plasmid, also move from the
immunization site to
other sites (blood, hence to all tissues; lymph nodes; spleen) where
additional immune
recruitment and qualitative and quantitative development of the immune
response ensue.
While these pathways often compete, previously available genetic vaccines
have incorporated all components for influencing each of the pathways into a
single
polynucleotide molecule. Because separate cell types are involved in the
complex
interactions required for a potent immune response to a genetic vaccine
vector, mutually
incompatible consequences can arise from administration of a genetic vaccine
that is
incorporated in a single vector molecule. Current genetic vaccine vectors
employ simple
methods for expression of the desired antigen with few if any design elements
that control
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
4
the precise intracellular fate of the antigen or the immunological
consequences of antigen
expression. Thus, although genetic vaccines show great promise for vaccine
research and
development, the need for major improvements and several severe limitations of
these
technologies are apparent.
Largely due to the lack of suitable laboratory models, none of the existing
genetic vaccine vectors have been optimized for human tissues. The existing
genetic vaccine
vectors typically provide low and short-lasting expression of the antigen of
interest, and even
large quantities of DNA do not always result in sufficiently high expression
levels to induce
protective immune responses. Because the mechanisms of the vector entry into
the cells and
transfer into the nucleus are poorly understood, virtually no attempts have
been made to
improve these key properties. Similarly, little is known about the mechanisms
that regulate
the maintenance of vector functions, including gene expression. Furthermore,
although there
is increasing amount of data indicating that specific sequences alter the
immunostimulatory
properties of the DNA, rational engineering is a very laborious and time-
consuming
approach when using this information to generate vector backbones with
improved
immunomodulatory properties.
Moreover, presently available genetic vaccine vectors do not provide
suffcient stability, inducibility or levels of expression in vivo to satisfy
the desire for
vaccines which can deliver booster immunization without additional vaccine
administration.
Booster immunizations are typically required 3-4 weeks after the primary
injection with
existing genetic vaccines.
Therefore a need exists for improved genetic vaccine vectors and
formulations, and methods for development of such vectors. The present
invention fulfills
these and other needs.
~~I~ARY OF THE INVENTION
The present invention provides multicomponent genetic vaccines that include
at least one, and preferably two or more genetic vaccine components that
confer upon the
vaccine the ability to direct an immune response so as to achieve an optimal
response to
vaccination. For example, the genetic vaccines can include a component that
provides
optimal antigen release; a component that provides optimal production of
cytotoxic T
lymphocytes; a component that directs release of an immunomodulator; a
component that
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
directs release of a chemokine; and/or a component that facilitates binding
to, or entry into, a
desired target cell type. For example, a component can confer improved
improves binding
to, and uptake of, the genetic vaccine to target cells such as antigen-
expressing cells or
antigen-presenting cells.
5 Additional components include those that direct antigen peptides derived
from uptake of an antigen into a cell to presentation on either Class I or
Class II molecules.
For example, one can include a component that directs antigen peptides to
presentation on
Class I molecules and comprises a polynucleotide that encodes a protein such
as tapasin,
TAP-1 and TAP-2, and/or a component that directs antigen peptides to
presentation on Class
II molecules and comprises a polynucleotide that encodes a protein such as an
endosomal or
lysosomal protease.
In some embodiments, one or more of the genetic vaccine components is
obtained by a method that involves: ( 1 ) recombining at least first and
second forms of a
nucleic acid which can confer a desired property upon a genetic vaccine,
wherein the first
and second forms differ from each other in two or more nucleotides, to produce
a library of
recombinant nucleic acids; and (2) screening the library to identify at least
one optimized
recombinant component that exhibits an enhanced capacity to confer the desired
property
upon the genetic vaccine. If fi~rther optimization of the component is
desired, the following
additional steps can be conducted: (3) recombining at least one optimized
recombinant
component with a further form of the nucleic acid, which is the same or
different from the
first and second forms, to produce a further library of recombinant nucleic
acids; (4)
screening the further library to identify at least one further optimized
recombinant
component that exhibits an enhanced capacity to confer the desired property
upon the
genetic vaccine; and (5) repeating (3) and (4), as necessary, until the
further optimized
recombinant component exhibits a further enhanced capacity to confer the
desired property
upon the genetic vaccine.
In some embodiments of the invention, the first form of the nucleic acid is a
first member of a gene family and the second form of the nucleic acid
comprises a second
member of the gene family. Additional forms of the module nucleic acid can
also be
members of the gene family. As an example, the first member of the gene family
can be
obtained from a first species of organism and the second member of the gene
family
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
6
obtained from a second species of organism. If desired, the optimized
recombinant genetic
vaccine component obtained by the methods of the invention can be backcrossed
by, for
example, recombining the optimized recombinant genetic vaccine component with
a molar
excess of one or both of the first and second forms of the substrate nucleic
acids to produce a
S further library of recombinant genetic vaccine components; and screening the
further library
to identify at least one optimized recombinant genetic vaccine component that
further
enhances the capability of a genetic vaccine vector that includes the
component to modulate
the immune response.
Additional embodiments of the invention provide methods of obtaining a
genetic vaccine component that confers upon a genetic vaccine vector an
enhanced ability to
replicate in a host cell. These methods involve creating a library of
recombinant nucleic
acids by subjecting to recombination at least two forms of a polynucleotide
that can confer
episomal replication upon a vector that contains the polynucleotide;
introducing into a
population of host cells a library of vectors, each of which contains a member
of the library
of recombinant nucleic acids and a polynucleotide that encodes a cell surface
antigen;
propagating the population of host cells for multiple generations; and
identifying cells which
display the cell surface antigen on a surface of the cell, wherein cells which
display the cell
surface antigen are likely to harbor a vector that contains a recombinant
vector module
which enhances the ability of the vector to replicate episomally.
Genetic vaccine components that confer upon a vector an enhanced ability to
replicate in a host cell can also be obtained by creating a library of
recombinant nucleic acids
by subjecting to recombination at least two fornls of a polynucleotide derived
from a human
papillomavirus that can confer episomal replication upon a vector that
contains the
polynucleotide; introducing a library of vectors, each of which contains a
member of the
library of recombinant nucleic acids, into a population of host cells;
propagating the host
cells for a plurality of generations; and identifying cells that contain the
vector.
In additional embodiments, the invention provides methods obtaining a
genetic vaccine component that confers upon a vector an enhanced ability to
replicate in a
human host cell by creating a library of recombinant nucleic acids by
subjecting to
recombination at least two forms of a polynucleotide that can confer episomal
replication
upon a vector that contains the polynucleotide; introducing a library of
genetic vaccine
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
7
vectors, each of which comprises a member of the library of recombinant
nucleic acids, into
a test system that mimics a human immune response; and determining whether the
genetic
vaccine vector replicates or induces an immune response in the test system. A
suitable test
system can involve human skin cells present as a xenotransplant on skin of an
immunocompromised non-human host animal, for example, or a non-human mammal
that
comprises a functional human immune system. Replication in these systems can
be detected
by determining whether the animal exhibits an immune response against the
antigen.
The invention also provides methods of obtaining a genetic vaccine
component that confers upon a genetic vaccine an enhanced ability to enter an
antigen-
presenting cell. These methods involve creating a library of recombinant
nucleic acids by
subjecting to recombination at least two forms of a polynucleotide that can
confer episomal
replication upon a vector that contains the polynucleotide; introducing a
library of genetic
vaccine vectors; each of which comprises a member of the library of
recombinant nucleic
acids, into a population of antigen-presenting or antigen-processing cells;
and determining
the percentage of cells in the population which contain the nucleic acid
vector. Antigen-
presenting or antigen-processing cells of interest include, for example, B
cells,
monocytes/macrophages, dendritic cells, Langerhans cells, keratinocytes, and
muscle cells.
In additional embodiments, the invention provides methods of obtaining a
recombinant genetic vaccine component that confers upon a genetic vaccine an
enhanced
ability to induce a desired immune response in a mammal. These methods
involve: (1)
recombining at least first and second forms of a nucleic acid which comprise a
genetic
vaccine vector, wherein the first and second forms differ from each other in
two or more
nucleotides, to produce a library of recombinant genetic vaccine vectors; (2)
transfecting the
library of recombinant vaccine vectors into a population of mammalian cells
selected from
the group consisting of peripheral blood T cells, T cell clones, freshly
isolated
monocytes/macrophages and dendritic cells; (3) staining the cells for the
presence of one or
more cytokines and identifying cells which exhibit a cytokine staining pattern
indicative of
the desired immune response; and (4) obtaining recombinant vaccine vector
nucleic acid
sequences from the cells which exhibit the desired cytokine staining pattern.
Also provided by the invention are methods of improving the ability of a
genetic vaccine vector to modulate an immune response by: ( 1 ) recombining at
least first
CA 02320626 2000-08-10
WO 99/41369 PCT/US99I03022
8
and second forms of a nucleic acid which comprise a genetic vaccine vector,
wherein the
first and second forms differ from each other in two or more nucleotides, to
produce a library
of recombinant genetic vaccine vectors; (2) transfecting the library of
recombinant genetic
vaccine vectors into a population of antigen presenting cells; and (3)
isolating from the cells
optimized recombinant genetic vaccine vectors which exhibit enhanced ability
to modulate a
desired immune response.
Another embodiment of the invention provides methods of obtaining a
recombinant genetic vaccine vector that has an enhanced ability to induce a
desired immune
response in a mammal upon administration to the skin of the mammal. These
methods
involve: ( 1 ) recombining at least first and second forms of a nucleic acid
which comprise a
genetic vaccine vector, wherein the first and second forms differ fibm each
other in two or
more nucleotides, to produce a library of recombinant genetic vaccine vectors;
(2) topically
applying the library of recombinant genetic vaccine vectors to skin of a
mammal; (3)
identifying vectors that induce an immune response; and (4) recovering genetic
vaccine
vectors from the skin cells which contain vectors that induce an immune
response.
The invention also provides methods of inducing an immune response in a
mammal by topically applying to skin of the mammal a genetic vaccine vector,
wherein the
genetic vaccine vector is optimized for topical application through use of DNA
shuffling. In
some embodiments, the genetic vaccine is administered as a formulation
selected from the
group consisting of a transdermal patch, a cream, naked DNA, a mixture of DNA
and a
transfection-enhancing agent. Suitable transfection-enhancing agents include
one or more
agents selected from the group consisting of a lipid, a liposome, a protease,
and a lipase.
Alternatively, or in addition, the genetic vaccine can be administered after
pretreatment of
the skin by abrasion or hair removal.
In another embodiment, the invention provides methods of obtaining an
optimized genetic vaccine component that confers upon a genetic vaccine
containing the
component an enhanced ability to induce or inhibit apoptosis of a cell into
which the vaccine
is introduced. These methods involve: (1) recombining at least first and
second forms of a
nucleic acid which comprise a nucleic acid that encodes an apoptosis-
modulating
polypeptide, wherein the first and second forms differ from each other in two
or more
nucleotides, to produce a library of recombinant nucleic acids; (2)
transfecting the library of
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
9
recombinant nucleic acids into a population of mammalian cells; (3) staining
the cells for
the presence of a cell membrane change which is indicative of apoptosis
initiation; and (4)
obtaining recombinant apoptosis-modulating genetic vaccine components from the
cells
which exhibit the desired apoptotic membrane changes.
Other embodiments of the invention provide methods of obtaining a genetic
vaccine component that confers upon a genetic vaccine reduced susceptibility
to a CTL
immune response in a host mammal. These methods can involve: (1) recombining
at least
first and second forms of a nucleic acid which comprises a gene that encodes
an inhibitor of
a CTL immune response, wherein the first and second forms differ from each
other in two or
more nucleotides, to produce a library of recombinant CTL inhibitor nucleic
acids; (2)
introducing genetic vaccine vectors which comprise the library of recombinant
CTL
inhibitor nucleic acids into a plurality of human cells; (3) selecting cells
which exhibit
reduced MHC class I molecule expression; and (4) obtaining optimized
recombinant CTL
inhibitor nucleic acids from the selected cells.
The invention also provides methods of obtaining a genetic vaccine
component that confers upon a genetic vaccine reduced susceptibility to a CTL
immune
response in a host mammal. These methods involve: (1) recombining at least
first and
second forms of a nucleic acid which comprises a gene that encodes an
inhibitor of a CTL
immune response, wherein the first and second forms differ from each other in
two or more
nucleotides, to produce a library of recombinant CTL inhibitor nucleic acids;
(2) introducing
viral vectors which comprise the library of recombinant CTL inhibitor nucleic
acids into
mammalian cells; (3) identifying mammalian cells which express a marker gene
included in
the viral vectors a predetermined time after introduction, wherein the
identified cells are
resistant to a CTL response; and {4) recovering as the genetic vaccine
component the
recombinant CTL inhibitor nucleic acids from the identified cells.
gltlFF DESCRIPTION OF THE DRAWINGS
Figure 1 shows a schematic representation of a multimodule genetic vaccine
vector. A typical genetic vaccine vector will include one or more of the
components
indicated, each of which can be native or optimized using the DNA shuffling
methods
described herein. The components can be present on the same vaccine vector, or
can be
included in a genetic vaccine as separate molecules.
CA 02320626 2000-08-10
WO 99/41369 PGTNS99/03022
Figure 2 shows a scheme for in vitro shuffling, "recursive sequence
recombination," of genes.
Figure 3 shows a diagram of the application of DNA shuffling to evolution of
genetic vaccines. Different forms of nucleic acids having known functional
properties (e.g.,
5 regulatory, coding, and the like), are shuffled and screened to identify
variants that exhibit
improved properties for use as genetic vaccines.
Figure 4 is a diagram of flow cytometry-based screening methods (FACS) for
selection of optimized promoter sequences evolved using recursive shuffling. A
cytomegalovirus (CMV) promoter is used for illustrative purposes.
10 Figure 5 shows an apparatus that is suitable for microinjection of genetic
vaccines and other reagents into tissue such as skin and muscle. The apparatus
is
particularly useful for screening large numbers of agents in vivo, being based
on a 96-well
format. The tips of the apparatus are movable to allow adjustment so that the
tips fit into a
microtiter plate. After obtaining a reagent of interest is obtained from a
plate, the tips are
adjusted to a distance of about 2-3 mm apart, enabling transfer of 96
different samples to an
area of about 1.6 cm by 2.4 cm to about 2.4 cm by 3.6 cm. If desired, the
volume of each
sample transferred can be electronically controlled; typically the volumes
transferred range
from about 2 pl to about 5 ~.1. Each reagent can be mixed with a marker agent
or dye to
facilitate recognition of the injection site in the tissue. For example, gold
particles of
different sizes and shaped can be mixed with the reagent of interest, and
microscopy and
immunohistochemistry used to identify each injection site and to study the
reaction induced
by each reagent. When muscle tissue is injected, the injection site is first
revealed by
surgery.
Figure 6 shows an example of family shuffling. Four different strains of a
virus are used in this illustration, but the technique is applicable to any
nucleic acid for
which different strains, species, or gene families have homologous nucleic
acids that have
one or more nucleotide changes compared to other homologous nucleic acids. The
different
variant nucleic acids are shuffled as described herein, and screened or
selected to identify
those variants that exhibit the desired property. The shuffling and screening
can be repeated
one or more times to obtain further improvement.
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/030Z2
11
Figure 7 shows an example of a vector that is useful for screening to identify
improved promoters from a library of shuffled promoter nucleic acids. Shuffled
putative
promoters are inserted into the vector upstream of a reporter gene for which
expression is
readily detected. For many applications, it is desirable that the product of
the reporter gene
be a cell surface protein so that cells which express high levels of the
reporter gene can be
sorted using flow cytometry-based cell sorting using the reporter gene
product. Examples of
suitable reporter genes include, for example, B7-2 and mAb179 epitopes. A
polyadenylation
region is typically placed downstream of the reporter gene (SV40 polyA is
illustrated). The
vector can also include a second reporter gene an internal control (GFP; green
fluorescent
protein); this gene is linked to a promoter (SRaP). The vector also typically
includes a
selectable marker (kanamycin/neomycin resistance is shown), and origins of
replication that
are functional in mammalian (SV40 ori) and/or bacterial (pUC ori) cells.
Figure 8 shows a diagram of a scheme for cycling evolution of inducible
promoters using DNA shuffling and flow cytometry-based selection. A library of
shuffled
promoter nucleic acids present in appropriate vectors is transfected into the
cells, and those
cells which exhibit the least expression of marker antigen when grown in
uninduced
conditions are selected. The vectors are recovered, introduced into cells, and
grown in
inducing conditions. Those cells that express the highest level of marker
antigen are
selected.
Figure 9 provides a schematic diagram of a method for evolving a genetic
vaccine vector for improved oral delivery.
Figure 10 is an alignment of the nucleotide sequences of the immediate/early
gene of two human cytomegalovirus (CMV) strains and two monkey strains.
Figure 11 is an alignment of Intron A nucleotide sequences from CMV strains
Towne and AD169.
Figure 12 shows a schematic presentation of the promoter/enhancer/intron
sequences derived from human (AD 169 and Towne) and monkey (rhesus and vervet
monkey) cytomegaloviruses by PCR amplification. These amplified fragments are
suitable
for use in family shuffling.
Figure 13 shows the enrichment of a library by subjecting shuffled CMV
promoter sequences to fluorescence-activated cell sorting.
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
12
Figure 14 shows the functional diversity and enrichment of high activity
CMV promoters obtained by DNA shuffling followed by fluorescence-activated
cell sorting.
Figure 15 shows the level of transgene expression obtained upon
intramuscular injection of a vector that contained a luciferase gene under the
contml of a
shuffled versus a control CMV promoter.
Figure 16 shows a schematic representation of the use of DNA shuffling to
generate promoter sequences in which unnecessary CpG sequences are deleted.
DF'~AILED DESCRPTION
Definitions
The term "screening" describes, in general, a process that identifies optimal
antigens. Several properties of the antigen can be used in selection and
screening including
antigen expression, folding, stability, immunogenicity and presence of
epitopes from several
related antigens. Selection is a form of screening in which identification and
physical
separation are achieved simultaneously by expression of a selection marker,
which, in some
genetic circumstances, allows cells expressing the marker to survive while
other cells die (or
vice versa). Screening markers include, for example, luciferase, beta-
galactosidase and
green fluorescent protein. Selection markers include drug and toxin resistance
genes, and
the like. Because of limitations in studying primary immune responses in
vitro, in vivo
studies are particularly useful screening methods. In these studies, the
antigens are first
introduced to test animals, and the immune responses are subsequently studied
by analyzing
protective immune responses or by studying the quality or strength of the
induced immune
response using lymphoid cells derived from the immunized animal. Although
spontaneous
selection can and does occur in the course of natural evolution, in the
present methods
selection is performed by man.
A "exogenous DNA segment", "heterologous sequence" or a "heterologous
nucleic acid", as used herein, is one that originates from a source foreign to
the particular
host cell, or, if from the same source, is modified from its original form.
Thus, a
heterologous gene in a host cell includes a gene that is endogenous to the
particular host cell,
but has been modified. Modification of a heterologous sequence in the
applications
described herein typically occurs through the use of DNA shuffling. Thus, the
terms refer to
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
13
a DNA segment which is foreign or heterologous to the cell, or homologous to
the cell but in
a position within the host cell nucleic acid in which the element is not
ordinarily found.
Exogenous DNA segments are expressed to yield exogenous polypeptides.
The term "gene" is used broadly to refer to any segment of DNA associated
with a biological function. 'Thus, genes include coding sequences and/or the
regulatory
sequences required for their expression. Genes also include nonexpressed DNA
segments
that, for example, form recognition sequences for other proteins. Genes can be
obtained from
a variety of sources, including cloning from a source of interest or
synthesizing from known
or predicted sequence information, and may include sequences designed to have
desired
parameters.
The term "isolated", when applied to a nucleic acid or protein, denotes that
the nucleic acid or protein is essentially free of other cellular components
with which it is
associated in the natural state. It is preferably in a homogeneous state
although it can be in
either a dry or aqueous solution. Purity and homogeneity are typically
determined using
analytical chemistry techniques such as polyacrylamide gel electrophoresis or
high
performance liquid chromatography. A pmtein which is the predominant species
present in a
preparation is substantially purified. In particular, an isolated gene is
separated from open
reading frames which flank the gene and encode a protein other than the gene
of interest.
The term "purified" denotes that a nucleic acid or protein gives rise to
essentially one band
in an electrophoretic gel. Particularly, it means that the nucleic acid or
protein is at least
about 50% pure, more preferably at least about 85% pure, and most preferably
at least about
99% pure.
The term "naturally-occurring" is used to describe an object that can be found
in nature as distinct from being artificially produced by man. For example, a
polypeptide or
polynucleotide sequence that is present in an organism (including viruses,
bacteria, protozoa,
insects, plants or mammalian tissue) that can be isolated from a source in
nature and which
has not been intentionally modified by man in the laboratory is naturally-
occurring.
The term "nucleic acid" refers to deoxyribonucleotides or ribonucleotides and
polymers thereof in either single- or double-stranded form. Unless
specifically limited, the
term encompasses nucleic acids containing known analogues of natural
nucleotides which
have similar binding properties as the reference nucleic acid and are
metabolized in a manner
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
14
similar to naturally occurring nucleotides. Unless otherwise indicated, a
particular nucleic
acid sequence also implicitly encompasses conservatively modified variants
thereof (e.g.
degenerate codon substitutions) and complementary sequences and as well as the
sequence
explicitly indicated. Specifically, degenerate codon substitutions may be
achieved by
generating sequences in which the third position of one or more selected (or
all) codons is
substituted with mixed-base and/or deoxyinosine residues (Batzer et al. (1991)
Nucleic Acid
Res. 19: 5081; Ohtsuka et al. (1985) J. Biol. Chem. 260: 2605-2608; Cassol et
al. (1992) ;
Rossolini et al. (1994) Mol. Cell. Probes 8: 91-98). The term nucleic acid is
used
interchangeably with gene, cDNA, and mRNA encoded by a gene.
"Nucleic acid derived from a gene" refers to a nucleic acid for whose
synthesis the gene, or a subsequence thereof, has ultimately served as a
template. Thus, an
mRNA, a cDNA reverse transcribed from an mRNA, an RNA transcribed from that
cDNA, a
DNA amplified from the cDNA, an RNA transcribed from the amplified DNA, etc.,
are all
derived from the gene and detection of such derived products is indicative of
the presence
and/or abundance of the original gene and/or gene transcript in a sample.
A nucleic acid is "operably linked" when it is placed into a fiuictional
relationship with another nucleic acid sequence. For instance, a promoter or
enhancer is
operably linked to a coding sequence if it increases the transcription of the
coding sequence.
Operably linked means that the DNA sequences being linked are typically
contiguous and,
where necessary to join two protein coding regions, contiguous and in reading
frame.
However, since enhancers generally function when separated from the promoter
by several
kilobases and intronic sequences may be of variable lengths, some
polynucleotide elements
may be operably linked but not contiguous.
A specific binding affinity between two molecules, for example, a ligand and
a receptor, means a preferential binding of one molecule for another in a
mixture of
molecules. The binding of the molecules can be considered specific if the
binding affinity is
about 1 x 104 M -1 to about 1 x 106 M -~ or greater.
The term "recombinant" when used with reference to a cell indicates that the
cell replicates a heterologous nucleic acid, or expresses a peptide or protein
encoded by a
heterologous nucleic acid. Recombinant cells can contain genes that are not
found within
the native (non-recombinant) form of the cell. Recombinant cells can also
contain genes
CA 02320626 2000-08-10
WO 99/41369 PC"fNS99/03022
found in the native form of the cell wherein the genes are modified and re-
introduced into
the cell by artificial means. The term also encompasses cells that contain a
nucleic acid
endogenous to the cell that has been modified without removing the nucleic
acid from the
cell; such modifications include those obtained by gene replacement, site-
specific mutation,
S and related techniques.
A "recombinant expression cassette" or simply an "expression cassette" is a
nucleic acid construct, generated recombinantly or synthetically, with nucleic
acid elements
that are capable of effecting expression of a structural gene in hosts
compatible with such
sequences. Expression cassettes include at least promoters and optionally,
transcription
10 termination signals. Typically, the recombinant expression cassette
includes a nucleic acid
to be transcribed (e.g., a nucleic acid encoding a desired polypeptide), and a
promoter.
Additional factors necessary or helpful in effecting expression may also be
used as described
herein. For example, an expression cassette can also include nucleotide
sequences that
encode a signal sequence that directs secretion of an expressed protein from
the host cell.
15 Transcription termination signals, enhancers, and other nucleic acid
sequences that influence
gene expression, can also be included in an expression cassette.
A "multivalent antigenic polypeptide" or a "recombinant multivalent
antigenic polypeptide" is a non-naturally occurring polypeptide that includes
amino acid
sequences from more than one source polypeptide, which source polypeptide is
typically a
naturally occurring polypeptide. At least some of the regions of different
amino acid
sequences constitute epitopes that are recognized by antibodies found in a
mammal that has
been injected with the source polypeptide. The source polypeptides from which
the different
epitopes are derived are usually homologous (i.e., have the same or a similar
structure and/or
function), and are often from different isolates, serotypes, strains, species,
of organism or
from different disease states, for example.
The terms "identical" or percent "identity," in the context of two or more
nucleic acid or polypeptide sequences, refer to two or more sequences or
subsequences that
are the same or have a specified percentage of amino acid residues or
nucleotides that are the
same, when compared and aligned for maximum correspondence, as measured using
one of
the following sequence comparison algorithms or by visual inspection.
CA 02320626 2000-08-10
WO 99/41369 PC'f/US99/03022
16
The phrase "substantially identical," in the context of two nucleic acids or
polypeptides, refers to two or more sequences or subsequences that have at
least 60%,
preferably 80%, most preferably 90-95% nucleotide or amino acid residue
identity, when
compared and aligned for maximum correspondence, as measured using one of the
following
sequence comparison algorithms or by visual inspection. Preferably, the
substantial identity
exists over a region of the sequences that is at least about 50 residues in
length, more
preferably over a region of at least about 100 residues, and most preferably
the sequences are
substantially identical over at least about 150 residues. In some embodiments,
the sequences
are substantially identical over the entire length of the coding regions.
For sequence comparison, typically one sequence acts as a reference sequence
to which test sequences are compared. When using a sequence comparison
algorithm, test
and reference sequences are input into a computer, subsequence coordinates are
designated,
if necessary, and sequence algorithm program parameters are designated. The
sequence
comparison algorithm then calculates the percent sequence identity for the
test sequences)
1 S relative to the reference sequence, based on the designated program
parameters.
Optimal alignment of sequences for comparison can be conducted, e.g., by
the local homology algorithm of Smith & Waterman, Adv. Appl. Math. 2:482
(1981), by the
homology alignment algorithm of Needleman & Wunsch, J. Mol. Biol. 48:443
(1970), by the
search for similarity method of Pearson & Lipman, Proc. Nat'1. Acad. Sci. USA
85:2444
(1988), by computerized implementations of these algorithms (GAP, BESTFIT,
FASTA,
and TFASTA in the Wisconsin Genetics Software Package, Genetics Computer
Group, 575
Science Dr., Madison, WI), or by visual inspection (see generally Ausubel et
al., infra).
One example of an algorithm that is suitable for determining percent
sequence identity and sequence similarity is the BLAST algorithm, which is
described in
Altschul et al., J. Mol. Biol. 215:403-410 (1990). Software for performing
BLAST analyses
is publicly available through the National Center for Biotechnology
Information
(http://www.ncbi.nlm.nih.gov/). This algorithm involves first identifying high
scoring
sequence pairs (HSPs) by identifying short words of length W in the query
sequence, which
either match or satisfy some positive-valued threshold score T when aligned
with a word of
the same length in a database sequence. T is referred to as the neighborhood
word score
threshold (Altschul et al., supra). These initial neighborhood word hits act
as seeds for
CA 02320626 2000-08-10
WO 99/41369 PGT/US99103022
17
initiating searches to find longer HSPs containing them. The word hits are
then extended in
both directions along each sequence for as far as the cumulative alignment
score can be
increased. Cumulative scores are calculated using, fvr nucleotide sequences,
the parameters
M (reward score for a pair of matching residues; always > 0) and N (penalty
score for
mismatching residues; always < 0). For amino acid sequences, a scoring matrix
is used to
calculate the cumulative score. Extension of the word hits in each direction
are halted when:
the cumulative alignment score falls off by the quantity X from its maximum
achieved
value; the cumulative score goes to zero or below, due to the accumulation of
one or more
negative-scoring residue alignments; or the end of either sequence is reached.
The BLAST
algorithm parameters W, T, and X determine the sensitivity and speed of the
alignment. The
BLASTN program (for nucleotide sequences) uses as defaults a wordlength (W) of
11, an
expectation (E) of 10, a cutoff of 100, M=5, N=-4, and a comparison of both
strands. For
amino acid sequences, the BLASTP program uses as defaults a wordlength (W) of
3, an
expectation (E) of 10, and the BLOSUM62 scoring matrix (see Henikoff &
Henikoff (1989)
Proc. Natl. Acad. Sci. USA 89:10915).
In addition to calculating percent sequence identity, the BLAST algorithm
also performs a statistical analysis of the similarity between two sequences
(see, e.g., Karlin
& Altschul (1993) Proc. Nat'1. Acad. Sci. USA 90:5873-5787). One measure of
similarity
provided by the BLAST algorithm is the smallest sum probability (P(N)), which
provides an
indication of the probability by which a match between two nucleotide or amino
acid
sequences would occur by chance. For example, a nucleic acid is considered
similar to a
reference sequence if the smallest sum probability in a comparison of the test
nucleic acid to
the reference nucleic acid is less than about 0.1, more preferably less than
about 0.01, and
most preferably less than about 0.001.
Another indication that two nucleic acid sequences are substantially identical
is that the two molecules hybridize to each other under stringent conditions.
The phrase
"hybridizing specifically to", refers to the binding, duplexing, or
hybridizing of a molecule
only to a particular nucleotide sequence under stringent conditions when that
sequence is
present in a complex mixture {e.g., total cellular) DNA or RNA. "Bind(s)
substantially"
refers to complementary hybridization between a probe nucleic acid and a
target nucleic acid
and embraces minor mismatches that can be accommodated by reducing the
stringency of
CA 02320626 2000-08-10
WO 99/41369 PCT/US99103022
18
the hybridization media to achieve the desired detection of the target
polynucleotide
sequence.
"Stringent hybridization conditions" and "stringent hybridization wash
conditions" in the context of nucleic acid hybridization experiments such as
Southern and
northern hybridizations are sequence dependent, and are different under
different
environmental parameters. Longer sequences hybridize specifically at higher
temperatures.
An extensive guide to the hybridization of nucleic acids is found in Tijssen
(1993)
Laboratory Techniques in Biochemistry and Molecular Biology--Hybridization
with Nucleic
Acid Probes part I chapter 2 "Overview of principles of hybridization and the
strategy of
nucleic acid probe assays", Elsevier, New York. Generally, highly stringent
hybridization
and wash conditions are selected to be about 5° C lower than the
thermal melting point (Tm)
for the specific sequence at a defined ionic strength and pH. Typically, under
"stringent
conditions" a probe will hybridize to its target subsequence, but to no other
sequences.
The Tm is the temperature (under defined ionic strength and pH) at which
50% of the target sequence hybridizes to a perfectly matched probe. Very
stringent
conditions are selected to be equal to the Tm for a particular probe. An
example of stringent
hybridization conditions for hybridization of complementary nucleic acids
which have more
than 100 complementary residues on a filter in a Southern or northern blot is
50%
formamide with 1 mg of heparin at 42°C, with the hybridization being
carried out overnight.
An example of highly stringent wash conditions is O.15M NaCI at 72°C
for about 15
minutes. An example of stringent wash conditions is a 0.2x SSC wash at
65°C for 15
minutes (see, Sambrook, infra., for a description of SSC buffer). Often, a
high stringency
wash is preceded by a low stringency wash to remove background probe signal.
An example
medium stringency wash for a duplex of, e.g., more than 100 nucleotides, is lx
SSC at 45°C
for 15 minutes. An example low stringency wash for a duplex of, e.g., more
than 100
nucleotides, is 4-6x SSC at 40°C for 15 minutes. For short probes
(e.g., about 10 to 50
nucleotides), stringent conditions typically involve salt concentrations of
less than about 1.0
M Na+ ion, typically about 0.01 to 1.0 M Na+ ion concentration (or other
salts) at pH 7.0 to
8.3, and the temperature is typically at least about 30°C. Stringent
conditions can also be
achieved with the addition of destabilizing agents such as formamide. In
general, a signal to
noise ratio of 2x (or higher) than that observed for an unrelated probe in the
particular
CA 02320626 2000-08-10
WO 99141369 PCT/US99/03022
19
hybridization assay indicates detection of a specific hybridization. Nucleic
acids which do
not hybridize to each other under stringent conditions are still substantially
identical if the
polypeptides which they encode are substantially identical. This occurs, e.g.,
when a copy of
a nucleic acid is created using the maximum codon degeneracy permitted by the
genetic
code.
A further indication that two nucleic acid sequences or polypeptides are
substantially identical is that the polypeptide encoded by the first nucleic
acid is
immunologically cross reactive with, or specifically binds to, the polypeptide
encoded by the
second nucleic acid. Thus, a polypeptide is typically substantially identical
to a second
polypeptide, for example, where the two peptides differ only by conservative
substitutions.
The phrase "specifically (or selectively) binds to an antibody" or
"specifically
(or selectively) immunoreactive with", when referring to a protein or peptide,
refers to a
binding reaction which is determinative of the presence of the protein, or an
epitope from the
protein, in the presence of a heterogeneous population of proteins and other
biologics. Thus,
under designated immunoassay conditions, the specified antibodies bind to a
particular
protein and do not bind in a significant amount to other proteins present in
the sample. The
antibodies raised against a multivalent antigenic polypeptide will generally
bind to the
proteins from which one or more of the epitopes were obtained. Specific
binding to an
antibody under such conditions may require an antibody that is selected for
its specificity for
a particular protein. A variety of immunoassay formats may be used to select
antibodies
specifically immunoreactive with a particular protein. For example, solid-
phase ELISA
immunoassays, Western blots, or immunohistochemistry are routinely used to
select
monoclonal antibodies specifically immunoreactive with a protein. See Harlow
and Lane
(1988) Antibodies, A Laboratory Manual, Cold Spring Harbor Publications, New
York
"Harlow and Lane"), for a description of immunoassay formats and conditions
that can be
used to determine specific immunoreactivity. Typically a specific or selective
reaction will
be at least twice background signal or noise and more typically more than 10
to 100 times
background.
"Conservatively modified variations" of a particular polynucleotide sequence
refers to those polynucleotides that encode identical or essentially identical
amino acid
sequences, or where the polynucleotide does not encode an amino acid sequence,
to
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
essentially identical sequences. Because of the degeneracy of the genetic
code, a large
number of functionally identical nucleic acids encode any given polypeptide.
For instance,
the codons CGU, CGC, CGA, CGG, AGA, and AGG all encode the amino acid
arginine.
Thus, at every position where an arginine is specified by a codon, the codon
can be altered to
5 any of the corresponding codons described without altering the encoded
polypeptide. Such
nucleic acid variations are "silent variations," which are one species of
"conservatively
modified variations." Every polynucleotide sequence described herein which
encodes a
polypeptide also describes every possible silent variation, except where
otherwise noted.
One of skill will recognize that each codon in a nucleic acid (except AUG,
which is
10 ordinarily the only codon for methionine) can be modified to yield a
fimctionally identical
molecule by standard techniques. Accordingly, each "silent variation" of a
nucleic acid
which encodes a polypeptide is implicit in each described sequence.
Furthermore, one of skill will recognize that individual substitutions,
deletions or additions which alter, add or delete a single amino acid or a
small percentage of
15 amino acids (typically less than 5%, more typically less than 1%) in an
encoded sequence are
"conservatively modified variations" where the alterations result in the
substitution of an
amino acid with a chemically similar amino acid. Conservative substitution
tables providing
functionally similar amino acids are well known in the art. The following five
groups each
contain amino acids that are conservative substitutions for one another:
20 Aliuhatic: Glycine (G), Alanine (A), Valine (V), Leucine (L), Isoleucine
(i);
o ti : Phenylalanine (F), Tyrosine (Y), Tryptophan (W);
cmlWr_con lnin~: Methionine (M), Cysteine (C);
basic: Arginine (R), Lysine (K), Histidine (H);
A ' 'c: Aspartic acid (D), Glutamic acid (E), Asparagine (N), Glutamine (Q).
See also, Creighton (1984) Proteins, W.H. Freeman and Company, for additional
groupings
of amino acids. In addition, individual substitutions, deletions or additions
which alter, add
or delete a single amino acid or a small percentage of amino acids in an
encoded sequence
are also "conservatively modified variations".
A "subsequence" refers to a sequence of nucleic acids or amino acids that
comprise a part of a longer sequence of nucleic acids or amino acids (e.g.,
polypeptide)
respectively.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99103022
21
~eQ~ ip~,on of the Preferred Embodiments
I, ever 1
The present invention provides multicomponent genetic vaccines that include
one or more component modules, each of which provides the genetic vaccine with
the
acquisition of or an improvement in a property or characteristic useful in
genetic vaccination.
The invention provides significant advantages over previously used genetic
vaccines.
Through use of a multicomponent vaccine, one can obtain an immune response
that is
particularly effective for a particular application. A multicomponent genetic
vaccine can, for
example, contain a component that is optimized for optimal antigen expression,
as well as a
component that confers improved activation of cytotoxic T lymphocytes (CTLs)
by
enhancing the presentation of the antigen on dendritic cell MHC Class I
molecules.
Additional examples are described herein.
In additional embodiments, the present invention provides methods of
obtaining components for use in genetic vaccines, including the multicomponent
vaccines,
that are more effective in conferring a desired immune response property upon
a genetic
vaccine. The methods involve creating a library of recombinant nucleic acids
and screening
the library to identify those library members that exhibits an enhanced
capacity to confer a
desired property upon a genetic vaccine. Those recombinant nucleic acids that
exhibit
improved properties can be used as components in a genetic vaccine, either
directly as a
polynucleotide or as a protein that is obtained by expression of the component
nucleic acid.
The properties or characteristics that can be sought to be acquired or
improved vary widely, and, of course depend on the choice of substrate. For
genetic
vaccines, improvement goals include higher titer, more stable expression,
improved stability,
higher specificity targeting, higher or lower frequency of integration,
reduced
immunogenicity of the vector or an expression product thereof, increased
immunogenicity of
the antigen, higher expression of gene products, and the like. Other
properties for which
optimization is desired include the tailoring of an immune response to be most
effective for a
particular application. Examples of genetic vaccine components are shown in
Figure 1. Two
or more components can be included in a single vector molecule, or each
component can be
present in a genetic vaccine formulation as a separate molecule.
CA 02320626 2000-08-10
WO 99141369 PCT/US99/03022
22
In the methods of the invention, at least two variant forms of a nucleic acid
are recombined to produce a library of recombinant nucleic acids, which is
then screened to
identify at least one recombinant component that is optimized for the
particular vaccine
property. Sequence recombination can be achieved in many different formats and
permutations of formats, as described in further detail below. These formats
share some
common principles. A family of nucleic acid molecules that have some sequence
identity to
each other, but differ in the presence of mutations, is typically used as a
substrate for
recombination. In any given cycle, recombination can occur in vivo or in
vitro, intracellularly
or extracellularly. Furthermore, diversity resulting from recombination can be
augmented in
i 0 any cycle by applying prior methods of mutagenesis (e.g., error-prone PCR
or cassette
mutagenesis) to either the substrates or products of recombination. In some
instances, a new
or improved property or characteristic can be achieved after only a single
cycle of in vivo or
in vitro recombination, as when using different, variant forms of the
sequence, as homologs
from different individuals or strains of an organism, or related sequences
from the same
organism, as allelic variations. However, recursive sequence recombination,
which entails
successive cycles of recombination, can generate further improvement.
In a presently preferred embodiment, DNA shuffling is used to obtain the
library of recombinant nucleic acids. DNA shuffling, which is diagrammed in
Figure 2, can
result in optimization of a desired property even in the absence of a detailed
understanding
of the mechanism by which the particular property is mediated. The substrates
for this
modification, or evolution, vary in different applications, as does the
property sought to be
acquired or improved. Examples of candidate substrates for acquisition of a
property or
improvement in a property include viral and nonviral vectors used in genetic
vaccination, as
well as nucleic acids that are involved in mediating a particular aspect of an
immune
response. The methods require at least two variant forms of a starting
substrate. The variant
forms of candidate components can have substantial sequence or secondary
structural
similarity with each other, but they should also differ in at least two
positions. The initial
diversity between forms can be the result of natural variation, e.g., the
different variant foams
(homologs) are obtained from different individuals or strains of an organism
(including
geographic variants; termed "family shuffling") or constitute related
sequences from the
same organism (e.g., allelic variations). Alternatively, the initial diversity
can be induced,
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
23
e.g., the second variant form can be generated by error-prone transcription,
such as an error-
prone PCR or use of a polymerase which lacks proof reading activity (see, Liao
(1990) Gene
88:107-111), of the first variant form, or, by replication of the first form
in a mutator strain
(mutator host cells are discussed in further detail below).
A recombination cycle is usually followed by at least one cycle of screening
or selection for molecules having a desired property or characteristic. If a
recombination
cycle is performed in vitro, the products of recombination, i.e., recombinant
segments, are
sometimes introduced into cells before the screening step. Recombinant
segments can also
be linked to an appropriate vector or other regulatory sequences before
screening.
Alternatively, products of recombination generated in vitro are sometimes
packaged as
viruses before screening. If recombination is performed in vivo, recombination
products can
sometimes be screened in the cells in which recombination occurred. In other
applications,
recombinant segments are extracted from the cells, and optionally packaged as
viruses,
before screening.
The nature of screening or selection depends on what property or
characteristic is to be acquired or the property or characteristic for which
improvement is
sought, and many examples are discussed below. It is not usually necessary to
understand
the molecular basis by which particular products of recombination (recombinant
segments)
have acquired new or improved properties or characteristics relative to the
starting
substrates. For example, a genetic vaccine vector can have many component
sequences each
having a different intended role (e.g., coding sequence, regulatory sequences,
targeting
sequences, stability-conferring sequences, immunomodulatory sequences,
sequences
affecting antigen presentation, and sequences affecting integration). Each of
these
component sequences can be varied and recombined simultaneously.
Screening/selection
can then be performed, for example, for recombinant segments that have
increased episomal
maintenance in a target cell without the need to attribute such improvement to
any of the
individual component sequences of the vector.
Depending on the particular screening protocol used for a desired property,
initial rounds) of screening can sometimes be performed in bacterial cells due
to high
transfection efficiencies and ease of culture. Later rounds, and other types
of screening
which are not amenable to screening in bacterial cells, are generally
performed in
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
24
mammalian cells to optimize recombinant segments for use in an environment
close to that
of their intended use. Final rounds of screening can be performed in the
precise cell type of
intended use (e.g., a human antigen-presenting cell). In some instances, this
cell can be
obtained from a patient to be treated with a view, for example, to minimizing
problems of
immunogenicity in this patient. In some methods, use of a genetic vaccine
vector in
treatment can itself be used as a round of screening. That is, genetic vaccine
vectors that are
successively taken up and/or expressed by the intended target cells in one
patient are
recovered from those target cells and used to treat another patient. The
genetic vaccine
vectors that are recovered from the intended target cells in one patient are
enriched for
vectors that have evolved, i.e., have been modified by recursive
recombination, toward
improved or new properties or characteristics for specific uptake,
immunogenicity, stability,
and the like.
The screening or selection step identifies a subpopulation of recombinant
segments that have evolved toward acquisition of a new or improved desired
property or
properties useful in genetic vaccination. Depending on the screen, the
recombinant segments
can be screened as components of cells, components of viruses or other
vectors, or in free
form. More than one round of screening or selection can be performed after
each round of
recombination.
If further improvement in a property is desired, at least one and usually a
collection of recombinant segments surviving a first round of
screening/selection are subject
to a further round of recombination. These recombinant segments can be
recombined with
each other or with exogenous segments representing the original substrates or
further
variants thereof. Again, recombination can proceed in vitro or in vivo. If the
previous
screening step identifies desired recombinant segments as components of cells,
the
components can be subjected to further recombination in vivo, or can be
subjected to further
recombination in vitro, or can be isolated before performing a round of in
vitro
recombination. Conversely, if the previous screening step identifies desired
recombinant
segments in naked form or as components of viruses or other vectors, these
segments can be
introduced into cells to perform a round of in vivo recombination. The second
round of
recombination, irrespective how performed, generates further recombinant
segments which
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
encompass additional diversity compared to recombinant segments resulting from
previous
rounds.
The second round of recombination can be followed by a further round of
screening/selection according to the principles discussed above for the first
round. The
stringency of screening/selection can be increased between rounds. Also, the
nature of the
screen and the property being screened for can vary between rounds if
improvement in more
than one property is desired or if acquiring more than one new property is
desired.
Additional rounds of recombination and screening can then be performed until
the
recombinant segments have sufficiently evolved to acquire the desired new or
improved
10 property or function.
II. Formats for Recombination
A number of different formats are available by which one can create a library
of recombinant nucleic acids for screening. In some embodiments, the methods
of the
invention entail performing recombination ("shuffling") and screening or
selection to
15 "evolve" individual genes, whole plasmids or viruses, multigene clusters,
or even whole
genomes (Stemmer (1995) BiolTechnology 13:549-$53). Reiterative cycles of
recombination and screening/selection can be performed to further evolve the
nucleic acids
of interest. Such techniques do not require the extensive analysis and
computation required
by conventional methods for polypeptide engineering. Shuffling allows the
recombination
20 of large numbers of mutations in a minimum number of selection cycles, in
contrast to
traditional, pairwise recombination events (e.g., as occur during sexual
replication). Thus,
the sequence recombination techniques described herein provide particular
advantages in
that they provide recombination between any or all of the mutations, thereby
providing a
very fast way of exploring the manner in which different combinations of
mutations can
25 affect a desired result. In some instances, however, structural and/or
functional information
is available which, although not required for sequence recombination, provides
opportunities
for modification of the technique.
The DNA shuffling methods can involve one or more of at least four different
approaches to improve immunogenic activity as well as to broaden specificity.
First, DNA-
shuffling can be performed on a single gene. Secondly, several highly
homologous genes can
be identified by sequence comparison with known homologous genes. These genes
can be
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
26
synthesized and shuffled as a family of homologs, to select recombinants with
the desired
activity. The shuffled genes can be introduced into appropriate host cells,
which can include
E. coli, yeast, plants, fimgi, animal cells, and the like, and those having
the desired properties
can be identified by the methods described herein. Third, whole genome
shuffling can be
performed to shuffle genes that can confer a desired property upon a genetic
vaccine (along
with other genomic nucleic acids). For whole genome shuffling approaches, it
is not even
necessary to identify which genes are being shuffled. Instead, e.g., bacterial
cell or viral
genomes are combined and shuffled to acquire recombinant nucleic acids that,
either itself or
through encoding a polypeptide, have enhanced ability to induce an immune
response, as
measured in any of the assays described herein. Fourth, polypeptide-encoding
genes can be
codon modified to access mutational diversity not present in any naturally
occurring gene.
Exemplary formats and examples for sequence recombination, sometimes
referred to as DNA shuffling, evolution, or molecular breeding, have been
described by the
present inventors and co-workers in co-pending applications U.S. Patent
Application Serial
No. 08/198,431, filed February 17, 1994, Serial No. PCT/US95/02126, filed,
February 17,
1995, Serial No. 08/425,684, filed April 18, 1995, Serial No. 08/537,874,
filed October 30,
1995, Serial No. 08/564,955, filed November 30, 1995, Serial No. 08/621,859,
filed March
25, 1996, Serial No. 08/621,430, filed March 25, 1996, Serial No.
PCT/US96/05480, filed
April 18, 1996, Serial No. 08/650,400, filed May 20, 1996, Serial No.
08/675,502, filed July
3, 1996, Serial No. 08/721, 824, filed September 27, 1996, Serial No.
PCT/LTS97/17300,
filed September 26, 1997, and Serial No. PCT/US97/24239, filed December 17,
1997;
Stemmer, Science 270:1510 (1995); Stemmer et al., Gene 164:49-53 (1995);
Stemmer,
BiolTechnology 13:549-553 (1995); Stemmer, Proc. Natl. Acad. Sci. U.S.A.
91:10747-10751
(1994); Stemmer, Nature 370:389-391 (1994); Crameri et al., Nature Medicine
2(1):1-3
(1996); Crameri et al., Nature Biotechnology 14:315-319 (1996), each of which
is
incorporated by reference in its entirety for all purposes.
Other methods for obtaining libraries of recombinant polynucleotides and/or
for obtaining diversity in nucleic acids used as the substrates for shuffling
include, for
example, homologous recombination (PCT/US98/05223; Publ. No. W098/42727);
oligonucleotide-directed mutagenesis (for review see, Smith, Ann. Rev. Genet.
19: 423-462
(1985); Botstein and Shortle, Science 229: 1193-1201 (1985); Carter, Biochem.
J. 237: 1-7
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
27
( 1986); Kunkel, "The efficiency of oligonucleotide directed mutagenesis" in
Nucleic acids &
Molecular Biology, Eckstein and Lilley, eds., Springer Verlag, Berlin (1987)).
Included
among these methods are oligonucleotide-directed mutagenesis (Zoller and
Smith, Nucl.
Acids Res. 10: 6487-6500 (1982), Methods in Enzymol. 100: 468-500 (1983), and
Methods in
Enzymol. 154: 329-350 (1987)) phosphothioate-modified DNA mutagenesis (Taylor
et al.,
Nucl. Acids Res. 13: 8749-8764 (1985); Taylor et al., Nucl. Acids Res. 13:
8765-8787
(1985); Nakamaye and Eckstein, Nucl. Acids Res. 14: 9679-9698 (1986); Sayers
et al., Nucl.
Acids Res. 16: 791-802 (1988); Sayers et al., Nucl. Acids Res. 16: 803-814
(1988)),
mutagenesis using uracil-containing templates (Kunkel, Proc. Nat'1. Acad. Sci.
USA 82: 488-
492 (1985) and Kunkel et al., Methods in Enzymol. 154: 367-382)); mutagenesis
using
gapped duplex DNA (Kramer et al., Nucl. Acids Res. 12: 9441-9456 (1984);
Kramer and
Fritz, Methods in Enzymol. 154: 350-367 (1987); Kramer et al., Nucl. Acids
Res. 16: 7207
(1988)); and Fritz et al., Nucl. Acids Res. 16: 6987-6999 (1988)). Additional
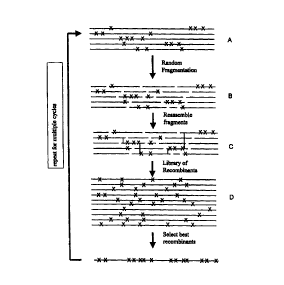
suitable
methods include point mismatch repair {Kramer et al., Cell 38: 879-887
(1984)),
mutagenesis using repair-deficient host strains (Carter et al., Nucl. Acids
Res. 13: 4431-4443
(1985); Carter, Methods in Enzymol. 154: 382-403 (1987)), deletion mutagenesis
(Eghtedarzadeh and Henikoff, Nucl. Acids Res. 14: 5115 (1986)), restriction-
selection and
restriction-purification (Wells et al., Phil. Traps. R. Soc. Lond. A 317: 415-
423 (1986)),
mutagenesis by total gene synthesis (Nambiar et al., Science 223: 1299-1301
(1984);
Sakamar and Khorana, Nucl. Acids Res. 14: 6361-6372 (1988); Wells et al., Gene
34: 315-
323 (1985); and Grundstrbm et al., Nucl. Acids Res. 13: 3305-3316 (1985). Kits
for
mutagenesis are commercially available (e.g., Bio-Rad, Amersham International,
Anglian
Biotechnology).
The breeding procedure starts with at least two substrates that generally show
substantial sequence identity to each other (i.e., at least about 30%, 50%,
70%, 80% or 90%
sequence identity), but differ from each other at certain positions. The
difference can be any
type of mutation, for example, substitutions, insertions and deletions. Often,
different
segments differ from each other in about 5-20 positions. For recombination to
generate
increased diversity relative to the starting materials, the starting materials
must differ from
each other in at least two nucleotide positions. That is, if there are only
two substrates, there
should be at least two divergent positions. If there are three substrates, for
example, one
CA 02320626 2000-08-10
WO 99/41369 PC'T/US99103022
2$
substrate can differ from the second at a single position, and the second can
differ from the
third at a different single position. The starting DNA segments can be natural
variants of
each other, for example, allelic or species variants. The segments can also be
from
nonallelic genes showing some degree of structural and usually functional
relatedness (e.g.,
different genes within a superfamily, such as the family of Yersinia V-
antigens, for
example). The starting DNA segments can also be induced variants of each
other. For
example, one DNA segment can be produced by error-prone PCR replication of the
other,
the nucleic acid can be treated with a chemical or other mutagen, or by
substitution of a
mutagenic cassette. Induced mutants can also be prepared by propagating one
(or both) of
the segments in a mutageruc strain, or by inducing an error-prone repair
system in the cells.
In these situations, strictly speaking, the second DNA segment is not a single
segment but a
large family of related segments. The different segments forming the starting
materials are
often the same length or substantially the same length. However, this need not
be the case;
for example; one segment can be a subsequence of another. The segments can be
present as
part of larger molecules, such as vectors, or can be in isolated form.
The starting DNA segments are recombined by any of the sequence
recombination formats provided herein to generate a diverse library of
recombinant DNA
segments. Such a library can vary widely in size from having fewer than 10 to
more than
105, 109, 1012 or more members. In some embodiments, the starting segments and
the
recombinant libraries generated will include full-length coding sequences and
any essential
regulatory sequences, such as a promoter and polyadenylation sequence,
required for
expression. In other embodiments, the recombinant DNA segments in the library
can be
inserted into a common vector providing sequences necessary for expression
before
performing screening/selection.
A further technique for recombining mutations in a nucleic acid sequence
utilizes "reassembly PCR". This method can be used to assemble multiple
segments that
have been separately evolved into a full length nucleic acid template such as
a gene. This
technique is performed when a pool of advantageous mutants is known from
previous work
or has been identified by screening mutants that may have been created by any
mutagenesis
technique known in the art, such as PCR mutagenesis, cassette mutagenesis,
doped oligo
mutagenesis, chemical mutagenesis, or propagation of the DNA template in vivo
in mutator
CA 02320626 2000-08-10
WO 99/41369 PCTIUS99/03022
29
strains. Boundaries defining segments of a nucleic acid sequence of interest
preferably lie in
intergenic regions, introns, or areas of a gene not likely to have mutations
of interest.
Preferably, oligonucleotide primers (oligos) are synthesized for PCR
amplification of
segments of the nucleic acid sequence of interest, such that the sequences of
the
oligonucleotides overlap the junctions of two segments. The overlap region is
typically
about 10 to 100 nucleotides in length. Each of the segments is amplified with
a set of such
primers. The PCR products are then "reassembled" according to assembly
protocols such as
those discussed herein to assemble randomly fragmented genes. In brief, in an
assembly
protocol the PCR products are first purified away from the primers, by, for
example, gel
electrophoresis or size exclusion chromatography. Purified products are mixed
together and
subjected to about 1-10 cycles of denaturing, reannealing, and extension in
the presence of
polymerase and deoxynucleoside triphosphates (dNTP's) and appropriate buffer
salts in the
absence of additional primers ("self priming"). Subsequent PCR with primers
flanking the
gene are used to amplify the yield of the fully reassembled and shuffled
genes.
In a further embodiment, PCR primers for amplification of segments of the
nucleic acid sequence of interest are used to introduce variation into the
gene of interest as
follows. Mutations at sites of interest in a nucleic acid sequence are
identified by screening
or selection, by sequencing homologues of the nucleic acid sequence, and so
on.
Oligonucleotide PCR primers are then synthesized which encode wild type or
mutant
information at sites of interest. These primers are then used in PCR
mutagenesis to generate
libraries of full length genes encoding penmutations of wild type and mutant
information at
the designated positions. This technique is typically advantageous in cases
where the
screening or selection process is expensive, cumbersome, or impractical
relative to the cost
of sequencing the genes of mutants of interest and synthesizing mutagenic
oligonucleotides.
III. Vectors Used in Genetic Vaccination
The invention provides multicomponent genetic vaccines, and methods of
obtaining genetic vaccine components that improve the capability of the
genetic vaccine for
use in nucleic acid-mediated immunomodulation. A general approach for
evolution of
genetic vaccines and components by DNA shuffling is shown schematically in
Figure 3.
Broadly speaking, a genetic vaccine vector is an exogenous polynucleotide
which produces a
medically useful phenotypic effect upon the mammalian cells) and organisms
into which it
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
is transferred. A vector may or may not have an origin of replication. For
example, it is
useful to include an origin of replication in a vector to allow for
propagation of the vector in
order to obtain sufficient quantities of the vector prior to administration to
a patient. If the
vector is designed to integrate into host chromosomal DNA or bind to host mRNA
or DNA,
5 or if replication in the host is otherwise undesirable, the origin of
replication can be removed
before administration, or an origin can be used that functions in the cells
used for vector
production but not in the target cells. However, in certain situations,
including some of those
discussed herein, it is desirable that the genetic vaccine vector be capable
of replicating in
appropriate host cells.
10 Vectors used in genetic vaccination can be viral or nonviral. Viral vectors
are
usually introduced into a patient as components of a virus. Illustrative viral
vectors into
which one can incorporate nucleic acids that are modified by the DNA shuffling
methods of
the invention include, for example, adenovirus-based vectors (Cantwell (1996)
Blood
88:4676-4683; Ohashi {1997} Proc. Nat'l. Acad. Sci USA 94:1287-1292), Epstein-
Ban
15 virus-based vectors {Mazda (1997) J. Immunol. Methods 204:143-151),
adenovirus-
associated virus vectors, Sindbis virus vectors (Stmng (1997) Gene Ther. 4:
624-627),
herpes simplex virus vectors (Kennedy (1997) Brain 120: 1245-1259) and
retroviral vectors
(Schubert (1997) Curr. Eye Res. 16:656-662).
Nonviral vectors, typically dsDNA, can be transferred as naked DNA or
20 associated with a transfer-enhancing vehicle, such as a receptor-
recognition protein,
liposome, lipoamine, or cationic lipid. This DNA can be transferred into a
cell using a
variety of techniques well known in the art. For example, naked DNA can be
delivered by
the use of liposomes which fuse with the cellular membrane or are endocytosed,
i.e., by
employing ligands attached to the liposome, or attached directly to the DNA,
that bind to
25 surface membrane protein receptors of the cell resulting in endocytosis.
Alternatively, the
cells may be permeabilized to enhance transport of the DNA into the cell,
without injuring
the host cells. One can use a DNA binding protein, e.g., HBGF-1, known to
transport DNA
into a cell. Furthermore, DNA can be delivered by bombardment of the skin by
gold or other
particles coated with DNA which are delivered by mechanical means, e.g.,
pressure. These
30 procedures for delivering naked DNA to cells are useful in vivo. For
example, by using
liposomes, particularly where the liposome surface carries ligands specific
for target cells, or
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
31
are otherwise preferentially directed to a specific organ, one may provide for
the introduction
of the DNA into the target cells/organs in vivo.
A. Yiral Vectors
Various viral vectors, such as retroviruses, adenoviruses, adenoassociated
viruses and herpes viruses, are commonly used in genetic vaccination. They are
often made
up of two components, a modified viral genome and a coat structure surrounding
it (see
generally Smith (1995) Annu. Rev. Microbiol. 49, 807-838), although sometimes
viral
vectors are introduced in naked form or coated with proteins other than viral
proteins. Most
current viral vectors have coat structures similar to a wildtype virus. This
structure packages
and protects the viral nucleic acid and provides the means to bind and enter
target cells. In
contrast, the viral nucleic acid in a vector designed for genetic vaccination
can be changed in
many ways. The goals of these changes can be, for example, to enhance or
reduce replication
of the virus in target cells while maintaining its ability to grow in vector
form in available
packaging or helper cells, to incorporate new sequences that encode and enable
appropriate
expression of a gene of interest (e.g., an antigen-encoding gene), and to
alter the
immunogenicity of the viral vector itself. Viral vector nucleic acids
generally comprise two
components: essential cis-acting viral sequences for replication and packaging
in a helper
line and a transcription unit for the exogenous gene. Other viral functions
can be expressed
in trans in a specific packaging or helper cell line.
(1) ~d_enoviruses
Adenoviruses comprise a large class of nonenveloped viruses that contain
linear double-stranded DNA. The normal life cycle of the virus does not
require dividing
cells and involves productive infection in permissive cells during which large
amounts of
virus accumulate. The productive infection cycle takes about 32-36 hours in
cell culture and
comprises two phases, the early phase, prior to viral DNA synthesis, and the
late phase,
during which structural proteins and viral DNA are synthesized and assembled
into virions.
In general, adenovirus infections are associated with mild disease in humans.
Adenovirus vectors are somewhat larger and more complex than retrovirus or
AAV vectors, partly because only a small fraction of the viral genome is
removed from most
current vectors. If additional genes are removed, they are provided in trams
to produce the
vector, which so far has proved difficult. Instead, two general types of
adenovirus-based
CA 02320626 2000-08-10
WO 99/41369 PC'f/US99/03022
32
vectors have been studied, E3-deletion and E1-deletion vectors. Some viruses
in laboratory
stocks of wild-type lack the E3 region and can grow in the absence of helper.
This ability
does not mean that the E3 gene products are not necessary in the wild, only
that replication
in cultured cells does not require them. Deletion of the E3 region allows
insertion of
exogenous DNA sequences to yield vectors capable of productive infection and
the transient
synthesis of relatively large amounts of encoded protein.
Deletion of the E1 region disables the adenovirus, but such vectors can still
be grown because there exists an established human cell line (called "293")
that contains the
E1 region of Ad5 and that constitutively expresses the E1 proteins. Most
recent gene-therapy
applications involving adenovirus have utilized E1 replacement vectors grown
in 293 cells.
The main advantages of adenovirus vectors are that they are capable of
efficient episomal gene transfer in a wide range of cells and tissues and that
they are easy to
grow in large amounts. Adenovirus-based vectors can also be used to deliver
antigens after
topical application onto the skin, and induction of antigen-specific immune
responses can be
observed following delivery to the skin (Tang et al. (1997) Nature 388: 729-
730). The main
disadvantage is that the host response to the virus appears to limit the
duration of expression
and the ability to repeat dosing, at least with high doses of first-generation
vectors.
In one embodiment, the recombination methods of the invention are used to
construct a novel adenovirus-phagemid capable of packaging DNA inserts over 10
kilobases
in size. Incorporation of a phage fl origin in a plasmid using the methods of
the invention
also generates a novel in vivo shuffling format capable of evolving whole
genomes of
viruses, such as the 36 kb family of human adenoviruses. The widely used human
adenovirus type S (Ad5) has a genome size of 36 kb. It is difficult to shuffle
this large
genome in vitro without creating an excessive number of changes which may
cause a high
percentage of nonviable recombinant variants. To minimize this problem and
achieve whole
genome shuffling of AdS, an adenovirus-phagemid was constructed. The Ad-
phagemid has
been demonstrated to accept inserts as large as 15 and 24 kilobases and to
effectively
generate ssDNA of that size. In a further embodiment, larger DNA inserts, as
large as 50 to
100 kb are inserted into the Ad-phagemid of the invention; with generation of
full length
ssDNA corresponding to those large inserts. Generation of such large ssDNA
fragments
provides a means to evolve, i.e. modify by the recursive recombination methods
of the
CA 02320626 2000-08-10
WO 99!41369 PCT/US99/03022
33
invention, entire viral genomes. Thus, this invention provides for the first
time a unique
phagemid system capable of cloning large DNA inserts (>10 I~B) and generating
ssDNA in
vitro and in vivo corresponding to those large inserts.
The genomes of related serotypes of human adenovirus are shuffled in vivo
using this unique phagemid system, as described in International Application
No.
PCT/US97/17302 (Pub!. No. W098/13485). The genomic DNA is first cloned into a
phagemid vector, and the resulting plasmid, designated an "Admid," can be used
to produce
single-stranded {ss) Admid phage by using a helper M13 phage. To achieve in
vivo
recombination, ssAdmid phages containing the genome of homologous human
adenoviruses
are used to perform high multiplicity of infection (MOI) on F+ mutS E. coli
cells. The
ssDNA is a better substrate for recombination enzymes such as RecA. The high
MOI
ensures that the probability of having multiple cross-ovens between copies of
the infecting
ssAdmid DNA is high. The shuffled adenovirus genome is generated by
purification of the
double stranded Admid DNA from the infected cells and is introduction into a
permissive
human cell line to produce the adenovirus library. This genomic shuffling
strategy is useful
for creation of recombinant adenovirus variants with changes in multiple
genes. This allows
screening or selection of recombinant variant phenotypes resulting from
combinations of
variations in multiple genes.
(2) Adeno-Associated Virus IAAVI
AAV is a small, simple, nonautonomous virus containing linear single-
stranded DNA. See, Muzycka, Current Topics Microbiol. Immunol. 158, 97-129
(1992).
The virus requires co-infection with adenovirus or certain other viruses in
order to replicate.
AAV is widespread in the human population, as evidenced by antibodies to the
virus, but it
is not associated with any known disease. AAV genome organization is
straightforward,
comprising only two genes: rep and cap. The termini of the genome comprises
terminal
repeats (ITR) sequences of about 145 nucleotides.
AAV-based vectors typically contain only the ITR sequences flanking the
transcription unit of interest. The length of the vector DNA cannot greatly
exceed the viral
genome length of 4680 nucleotides. Currently, growth of AAV vectors is
cumbersome and
involves introducing into the host cell not only the vector itself but also a
plasmid encoding
rep and cap to provide helper functions. The helper plasmid lacks ITRs and
consequently
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
34
cannot replicate and package. In addition, helper virus such as adenovirus is
often required.
The potential advantage of AAV vectors is that they appear capable of long-
term expression
in nondividing cells, possibly, though not necessarily, because the viral DNA
integrates.
The vectors are structurally simple, and they may therefore provoke less of a
host-cell
response than adenovirus.
{3) Pa~illoma Virus
Papillomaviruses are small, nonenveloped, icosahedral DNA viruses that
replicate in the nucleus of squamous epithelial cells. Papillomaviruses
consist of a single
molecule of double-stranded circular DNA about 8,000 by in size within a
spherical protein
coat of 72 capsomeres. Such papillomaviruses are classified by the species
they infect (e.g.,
bovine, human, rabbit) and by type within species. Over 50 distinct human
papillomaviruses
("HPV") have been described. See, e.g., Fields Virology (3rd ed., eds. Fields
et al.,
Lippincott-Raven, Philadelphia, 1996). Papillomaviral vectors are described in
detail in
copending, commonly owned US Patent Application No. 08/958822, filed October
28, 1997,
which is incorporated herein by reference in its entirety for all purposes.
Papillomaviruses display a marked degree of cellular tropism for epithelial
cells. Specific viral types have a preference for either cutaneous or mucosal
epithelial cells.
All papillomaviruses have the capacity to induce cellular proliferation. The
most common
clinical manifestation of proliferation is the production of benign warts.
However, many
papillomaviruses have capacity to be oncogenic in some individuals and some
papillomaviruses are highly oncogenic. Based on the pathology of the
associated lesions,
most human papillomaviruses (FIPVs) can be classified in one of four major
groups, benign,
low-risk, intermediate-risk and high-risk (Fields Virology, (Fields et al.,
eds., Lippincott-
Raven, Philadelphia, 3d ed. 1996); DNA Tumor Viruses: Papilloma in
(Encyclopedia of
Cancer, Academic Press) Vol. 1, p 520-531). For example, viruses HPV-1, HPV-2,
HPV-3,
HPV-4, and HPV-27 are associated with benign cutaneous lesions. Viruses HPV-6
and
HPV-11 are associated with vulval, penile, and laryngeal warts and are
considered low-risk
viruses as they are rarely associated with invasive carcinomas. Viruses HPV-
16, HPV-18,
HPV-31, and HPV-45 are considered high risk virus as they are associated with
a high
frequency with adeno- and squamous carcinoma of the cervix. Viruses HPV-5 and
HPV-8
are associated with benign cutaneous lesion in a multifactorial disease
Epidermodysplasia
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
Verruciformis (EV). Such lesions, however, can progress into squamous cell
carcinomas.
These viruses do not fall under one of the four major risk groups. Newly
discovered HPVs
can classified for risk based on the frequency of cancerous lesions relative
to that of HPVs
that have already been classified for risk.
HPV vectors can be subjected to iterative cycles of recombination and
screening (i.e., shuffling) with a view to obtaining vectors with improved
properties.
Improved properties include increased tissue specificity, altered tissue
specificity, increased
expression level, prolonged expression, increased episomal copy number,
increased or
decreased capacity for chromosomal integration, increased uptake capacity, and
other
10 properties as discussed herein. The starting materials for shuffling are
typically vectors of
the kind described above constructed from different strains of human
papillomaviruses, or
segments or variants of such generated by e.g., error-prone PCR or cassette
mutagenesis.
The human papillomaviruses, or at least the E1 and E2 coding regions thereof
are preferably
human cutaneous papillomaviruses.
15 (4) Retroviruses
Retroviruses comprise a large class of enveloped viruses that contain single-
stranded RNA as the viral genome. During the normal viral life cycle, viral
RNA is reverse-
transcribed to yield double-stranded DNA that integrates into the host genome
and is
expressed over extended periods. As a result, infected cells shed virus
continuously without
20 apparent harm to the host cell. The viral genome is small (approximately 10
kb), and its
prototypical organization is extremely simple, comprising three genes encoding
gag, the
group specific antigens or core proteins; pol, the reverse transcriptase; and
env, the viral
envelope protein. The termini of the RNA genome are called long terminal
repeats (LTRs)
and include promoter and enhancer activities and sequences involved in
integration. The
25 genome also includes a sequence required for packaging viral RNA and splice
acceptor and
donor sites for generation of the separate envelope mRNA. Most retroviruses
can integrate
only into replicating cells, although human immunodeficiency virus (HIV)
appears to be an
exception.
Retrovirus vectors are relatively simple, containing the 5' and 3' LTRs, a
30 packaging sequence, and a transcription unit composed of the gene or genes
of interest,
which is typically an expression cassette. To grow such a vector, one must
provide the
CA 02320626 2000-08-10
WO 99/41369 PGTNS99/03022
36
missing viral functions in trans using a so-called packaging cell line. Such a
cell is
engineered to contain integrated copies of gag, pol, and env but to lack a
packaging signal so
that no helper virus sequences become encapsidated. Additional features added
to or
removed from the vector and packaging cell line reflect attempts to render the
vectors more
efficacious or reduce the possibility of contamination by helper virus.
For some genetic vaccine applications, retroviral vectors have the advantage
of being able integrate in the chromosome and therefore potentially capable of
long-term
expression. They can be grown in relatively large amounts, but care is needed
to ensure the
absence of helper virus.
B. Non-Yiral Genetic Vaccine Vectors
Nonviral nucleic acid vectors used in genetic vaccination include plasmids,
RNAs, polyamide nucleic acids, and yeast artificial chromosomes (YACs), and
the like.
Such vectors typically include an expression cassette for expressing a
polypeptide against
which an immune response is induced. The promoter in such an expression
cassette can be
constitutive, cell type-specific, stage-specific, and/or modulatable (e.g., by
tetracycline
ingestion; tetracycline-responsive promoter). Transcription can be increased
by inserting an
enhancer sequence into the vector. Enhancers are cis-acting sequences,
typically between 10
to 300 base pairs in length, that increase transcription by a promoter.
Enhancers can
effectively increase transcription when either 5' or 3' to the transcription
unit. They are also
effective if located within an intron or within the coding sequence itself.
Typically, viral
enhancers are used, including SV40 enhancers, cytomegalovirus enhancers,
polyoma
enhancers, and adenovirus enhancers. Enhancer sequences from mammalian systems
are
also commonly used, such as the mouse immunoglobulin heavy chain enhancer.
Nonviral vectors encoding products useful in gene therapy can be introduced
into an animal by means such as lipofection, biolistics, virosomes, liposomes,
immunoliposomes, polycation:nucleic acid conjugates, naked DNA injection,
artificial
virions, agent-enhanced uptake of DNA, ex vivo transduction. Lipofection is
described in
e.g., US Patent Nos. 5,049,386, 4,946,787; and 4,897,355) and Iipofection
reagents are sold
commercially (e.g., TransfectamTM and LipofectinTM). Cationic and neutral
lipids that are
suitable for efficient receptor-recognition lipofection of polynucleotides
include those of
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
37
Felgner, WO 91/17424, WO 91/16024. Naked DNA genetic vaccines are described
in, for
example, US Patent No. 5,589,486.
IV. Multicom~onent Genetic Vaccines
The invention provides multicomponent genetic vaccines that are designed to
obtain an optimal immune response upon administration to a mammal. In these
vaccines,
two or more separate genetic vaccine components are used for immunization,
preferably in
the same formulation. Each component can be optimized for particular functions
that will
occur in some cells and not in others, thus providing a means for eliciting
differentiated
responses in different cell types. When mutually incompatible consequences are
derived
from use of one plasmid, those activities are separated into different vectors
that will have
different fates and effects in vivo. Genetic vaccines are ideal for the
formulation of several
biologically active entities into one preparation. The vectors are preferably
all of the same
chemical type so there is no incompatibility of this nature, and can all be
manufactured by
the same chemical and/or biological processes. The vaccine preparation can
consist of a
defined molar ratio of the separate vector components that can be formulated
exactly and
repeatedly.
Several genetic vaccine vector components that can be used as components of
a multicomponent genetic vaccine are described below. The methods of the
invention greatly
simplify the development of such vector components, because the mechanism by
which a
particular feature is controlled and the properties of a molecule that, when
modified, will
enhance that feature, need not be known. Even in the absence of such
knowledge, by
carrying out the recombination and screening methods of the invention, one can
obtain
vector components that are improved for each of the properties listed.
A. Vector "AR'; designed to provide optimal antigen release
Genetic vaccine vector component "AR" is designed to provide optimal
release of antigen in a form that will be recognized by antigen presenting
cells (APC) and
taken up by those cells for efficient intracellular processing and
presentation to T helper (TH)
cells. Cells transfected with AR plasmid can be considered as an antigen
factory for APC.
AR plasmids typically have one or more of the following properties, each of
which can be
optimized using the DNA shuffling methods of the invention:
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
38
(a) optimal plasmid binding to and uptake by the chosen antigen expressing
cells (e.g., myocytes for intramuscular immunization or epithelial cells for
mucosal
immunization). This is a critical property which differentiates AR from other
vector
components in the multicomponent DNA vaccine. Optimal vector binding to the
target cell
includes not only the concept of very avid binding and subsequent
internalization into target
cells, but relative inability to bind to and enter other cells. Optimization
of this ratio of
desired binding to undesired binding will significantly increase the number of
target cells
transfected. This property can be optimized using DNA shuffling according to
the present
invention as described herein. For example, variant vector component sequences
obtained
by DNA shuffling, combinatorial assembly of vector components, insertion of
random
oligonucleotide sequences, and the like, can first be selected for those that
bind to target
cells, after which this population of cells is depleted for those that bind to
other cells. Vector
components for targeting genetic vaccine vectors to particular cell types, and
methods of
obtaining improved targeting, are described in copending, commonly assigned US
Patent
Application No. , filed February 10, 1999 as TTC Attorney Docket No. 18097-
030200US, which is entitled "Targeting of Vaccine Vectors."
(b) optimal trafficking of the vector DNA to the nucleus. Again, the present
invention provides methods by which one can obtain genetic vaccine components
that are
optimal for such properties.
(c) optimal transcription of the antigen gene(s). This can involve, for
example, the use of optimized promoters, enhancers, introns, and the like. In
a preferred
embodiment, cell-specific promoters are used that only allow transcription of
the genes when
the vector is within the nucleus of the target cell type. In this case,
specificity is derived not
only from selective vector entry into target cells.
(d) optimal trafficking of mRNA to the cytoplasm and optimal longevity of
the mRNA in the cytoplasm. To achieve this property, the methods of the
invention are used
to obtain optimal 3' and 5' non-translated regions of the mRNA.
(e) optimal translation of the mRNA. Again, the DNA shuffling methods are
used to obtain optimized recombinant sequences which exhibit optimal ribosome
binding
and assembly of translational machinery, plus optimal codon preference.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
39
(f) optimal antigen structure for efficient uptake by APC. Extracellular
antigen is taken up by APC by at least five non-exclusive mechanisms. One
mechanism is
sampling of the external fluid phase by micropinocytosis and internalization
of a vesicle.
The first mechanism has, as far as is presently known, no structural
requirements for an
antigen in the fluid phase and is therefore not relevant to considerations of
designing antigen
structure. A second mechanism involves binding of antigen to receptors on the
APC surface;
such binding occurs according to rules that are only now being studied (these
receptors are
not immunoglobulin family members and appear to represent several families of
proteins
and glycoproteins capable of binding different classes of extracellular
proteins/glycoproteins). This type of binding is followed by receptor-mediated
internalization, also in a vesicle. Because this mechanism is poorly
understood at present,
elements of antigen design cannot be incorporated in a rational design
process. However,
application of gene shuffling, an empirical process of selection of variant
DNA molecules
most successful at entry into APC, can select for variants that are improved
throughout this
mechanism.
The other three mechanisms all relate to specific antibody recognition of the
extracellular antigen. The first mechanism involves immunoglobulin-mediated
recognition
of the specific asitigen via IgG that is bound to Fc receptors on the cell
surface. APC such
as monocytes, macrophages and dendritic cells can be decorated with surface
membrane IgG
of diverse specificities. In a primary response, this mechanism will not be
operative. In
previously immunized animals, IgG on the surface of APC can specifically bind
extracellular
antigen and mediate uptake of the bound antigen into an intracellular
endosomal
compartment. Another mechanism involves binding to clonally-derived surface
membrane
immunoglobulin which is present on each B cells (IgM in the case of primary B
cells and
IgG when the animal has been previously exposed to the antigen). B cells are
efficient
APC. Extracellular antigen can bind specifically to surface Ig and be
internalized and
processed in a membrane compartment for presentation on the B cell surface.
Finally,
extracellular antigen can be recognized by specific soluble immunoglobulin
(IgM in the case
of a primary immunization and IgG in the previously immunized animals).
Complexing
with Ig will elicit binding to the surface of APC (via Fc receptor recognition
in the case of
IgG) and internalization.
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/030Z2
In each of these latter three mechanisms, the extent to which the conformation
of the antigen is the same as the recognition specificity of the pre-existing
antibody is critical
to the efficiency of the process of antigen presentation. Antibodies can
recognize linear
protein epitopes as well as conformational epitopes determined by the three
dimensional
5 structure of the protein antigen. Protective antibodies that will recognize
an extracellular
virus or bacterial pathogen and by binding to its surface prevent infection or
mediate its
immune destruction (complement mediated lysis, immune complex formation and
phagocytosis) are almost exclusively generated against conformational
determinants on the
proteins with native structure displayed on the surface of the pathogen.
Hence, it is
10 imperative for generation of host protective humoral immunity, to have
those naive B cells
which bear antibody specific for conformational epitopes present on the
pathogen be
stimulated by direct contact with T helper cells after intracellular
processing of the antigen
and presentation of degradation peptides in the context of MHC Class II. This
T help will
allow selective proliferation of the relevant B cells with consequent mutation
of antibody
15 and antigen driven selection for antibodies with increased specificity, as
well as antibody
class switching.
To summarize, optimal uptake of antigen by APC to elicit humoral immunity,
as well as specific CD4+ cytotoxic T cells, requires that the antigen be in
native protein
conformation (as presented subsequently to the immune system upon natural
infection) and
20 recognized by naive B cells bearing the appropriate membrane antibody.
Native protein
conformation includes appropriate protein folding, glycosylation and any other
post-
translational modifications necessary for optimal reactivity with the
receptors
(immunoglobulin and possibly non-immunoglobulin) on APC. In addition to the
three
dimensional structure of the expressed antigen required for recognition by
specific antibody
25 and elicitation of the required immune responses, the structure (and
sequence) can be
optimized for increased protein stability outside the expressing cell, until
the time when it is
recognized by immune cells, including APCs. The recombination and screening
methods of
the invention can be used to optimize the antigen structure (and sequence) for
subsequent
processing after uptake by APC so that intracellular processing results in
derivation of the
30 required peptide fragments for presentation on Class I or Class II on APC
and desired
immune responses.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
41
(g) optimal partitioning of the nascent antigen into the desired subcellular
compartment or compartments. This can be directed by signal and trafficking
signals
embodied in the antigen sequence. It may be desirable for all of the antigen
to be secreted
from these cells; alternatively, all or part of the antigen could be directed
to be expressed on
the cell surface of these factory cells. Signals to direct vesicles containing
the antigen to
other subcellular compartments for post-translational modifications, including
glycosylation,
can be embodied in the antigen sequence.
(h) optimal display of the antigen on the cell surface or optimal release of
the
antigen from the cells. A variation on items (f) and (g) is to design the
expression of the
antigen within the cytoplasm of the factory cell followed by lysis of that
cell to release
soluble antigen. Cell death can be engineered by expression on the same
genetic vaccine
vector of an intracellular pmtein that will elicit apoptosis. In this case,
the timing of cell
death is balanced with the need for the cell to produce antigen, as well as
the potential
deleterious effect of killing some cells in a designed process.
In combination, items (a) -(h) lead to a variety of scenarios for~the
optimizing
the longevity and extent of antigen expression. It is not always desirable
that the antigen be
expressed for the longest time at the highest level. In certain clinical
applications, it will be
important to have antigen expression that is short time-low expression, short
time-high
expression, long time-low expression, long time-high expression or somewhere
in between.
Plasmid AR can be designed to express one or more variants of a single
antigen gene or several quite different targets for immunization. Methods for
obtaining
optimized antigens for use in genetic vaccines are described in copending,
commonly
assigned US Patent Application No. , filed February 10, 1999 as TTC
Attorney Docket No. 18097-028710US, which is entitled "Antigen Library
Immunization".
Multiple antigens can be expressed from a monocistronic or multicistronic form
of the
vector.
B, rector components "CTL-DC'; "CTL-LC" and "CTL-MM'; designed for
optimal production of CTLs
Genetic vector components "CTL-DC", "CTL-LC" and "CTL-MM" are
designed to direct optimal production of cytotoxic CD8+ lymphocytes (CTL) by
dendritic
cells (CTL-DC), Langerhan's cells (CTL-LC), and monocytes and macrophages (CTL-
MM).
CA 02320626 2000-08-10
WO 99/41369 PC'T/US99/03022
42
These vector components direct presentation of optimal antigen fragments in
association
with MHC Class I, thereby ensuring maximal cytotoxic T cell immune responses.
Cells
transfected with CTL vector components can be considered as the direct
activators of this
arm of specific immunity that is usually critically important for protection
against viral
diseases.
CTL vector components are typically designed to have one or more of the
following properties, each of which can be optimized using the DNA shuffling
methods of
the invention:
(a) optimal vector binding to, and uptake by, the chosen antigen presenting
cells (e.g., dendritic cells, monocytes/macrophages, Langerhan's cells). This
is a critical
property to differentiate CTL series vectors from other vectors in the
multicomponent DNA
vaccine. CTL series vectors preferably do not bind to or enter cells that are
chosen to be the
extracellular antigen expression host via AR vectors. This separation of
functions is critical,
as the intracellular fate and trafficking of antigen destined for stimulation
of immune cells
after release from an antigen expressing cell is quite different than the fate
of antigen
destined to be presented on the cell surface in association with MHC Class I.
In the former
case, antigen is directed via a signal secretion sequence to be delivered
intact to the lumen of
the rough endoplasmic reticulum (RER) and then secreted. In the latter case,
antigen is
directed to remain in the cytoplasm and there be degraded into peptide
fragments by the
proteasomal system followed by delivery to the lumen of the RER for
association with MHC
Class I. These complexes of peptide and MHC Class I are then delivered to the
cell surface
for specific interaction with CD8+ cytotoxic T cells. Vector components, and
methods for
obtaining optimized vector components, that are optimized for targeting to
desired cell types
are described in copending, commonly assigned US Patent Application No.
, filed February 10, 1999 as TTC Attorney Docket No. 18097-
030200US, which is entitled "Targeting of Genetic Vaccine Vectors."
(b) optimal transcription of the antigen gene(s). This can be accomplished by
optimizing promoters, enhancers, introns, and the like, as discussed herein.
Cell specific
promoters are valuable in such vectors as an additional level of selectivity.
(c) optimal longevity of the mRNA. Optimal 3' and 5' non-translated regions
of the mRNA can be obtained using the methods of the invention.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
43
(d) optimal translation of the mRNA. Again, the DNA shuffling and
selection methods of the invention can be used to obtain polynucleotide
sequences for
optimal ribosome binding and assembly of translational machinery, as well as
optimal codon
preference.
(e) optimal protein conformation. In this case, the optimal protein
conformation yields appropriate cytoplasmic proteolysis and production of the
correct
peptides for presentation on MHC Class I and elicitation of the desired
specific CTL
responses, rather than a conformation that will interact with specific
antibody or other
receptors on the surface of APC.
(f) optimal proteolysis to generate the correct peptides. The order of
specific
proteolytic cleavages will depend on the nature of protein folding and the
nature of proteases
either in the cytoplasm or in the proteasome.
(g) optimal transport of the antigen peptides across the endoplasmic reticulum
membrane to be delivered into the RER lumen. This may be mediated by
recognition of the
peptides by TAP proteins or by other membrane transporters
(h) optimal association of the peptides with the Class I-(32 microglobulin
complex and trafficking to the cell surface via the secretory pathway.
(i) optimal display of the MHC-peptide complex with associated accessory
molecules for recognition by specific CTL.
Vector CTL can be designed to express one or more variants of a single
antigen gene or several different targets for immunization. Multiple optimized
antigens can
be expressed from a monocistronic or multicistronic form of the vector.
C. Vectors "M'; designed for optimal release ojimmune modulators
Vectors "M" are designed to direct optimal release of immune modulators,
such as cytokines and other growth factors, from target cells. Target cells
can be either the
predominant cell type in the immunized tissue or immune cells such dendritic
cells (M-DC),
Langerhan's cells (M-LC), monocytes & macrophages (M-MM)". These vectors
direct
simultaneous expression of optimal levels of several immune cell "modulators"
(cytokines,
growth factors, and the like) such that the immune response is of the desired
type, or
combination of types, and of the desired level. Cells transfected with M
vectors can be
considered as the directors of the nature of the vaccine immune response (CTL
vs T~-,1 vs
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
44
TH2 vs NK cell, etc.) and its magnitude. The properties of these vectors
reflect the nature of
the cell in which the vectors are designed to operate. For example, the
vectors are designed
to bind to and enter the desired cell type, and/or can have cell-specific
regulated promoters
that drive transcription in the desired cell type. The vectors can also be
engineered to direct
maximal synthesis and release of the cell modulator proteins from the target
cells in the
desired ratio.
"M" genetic vaccine vectors are typically designed to have one or more of the
following properties, each of which can be optimized using the DNA shuffling
methods of
the invention:
(a) optimal vector binding to and uptake by the chosen modulator expressing
cell. Suitable expressing cells include, for example, muscle cells, epithelial
cells or other
dominant (by number) cell types in the target tissue, antigen presenting cells
(e.g. dendritic
cells, monocytes/macrophages, Langerhans cells). This is a critical property
which
differentiates M series vectors from those designed to bind to and enter other
cells.
(b) optimal transcription of the immune modulator gene(s). Again,
promoters, enhancers, introns, and the like can be optimized according to the
methods of the
invention. Cell specific promoters are very valuable here as an additional
level of
selectivity.
(c} optimal longevity of the mRNA. Optimal 3' and 5' non-translated regions
of the mRNA can be obtained using the methods of the invention.
(d) optimal translation of the mRNA. Again, the DNA shuffling and
selection methods of the invention can be used to obtain polynucleotide
sequences for
optimal ribosome binding and assembly of translational machinery, as well as
optimal codon
preference.
(e) optimal trafficking of the modulator into the lumen of the RER (via a
signal secretion sequence). An alternative strategy for modulation of the
immune response
uses membrane anchored modulators rather than secretion of soluble modulator.
Anchored
modulator can be retained on the surface of the synthesizing cell by, for
example, a
hydrophobic tail and phosphoinositol glycan linkage.
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
(f j optimal protein conformation for each modulator. In this case, the
optimal
protein conformation is that which allows extracellular modulator and/or cell
membrane
anchored modulator to interact with the relevant receptor.
(g) the ratio of modulators and their type can be determined empirically. One
5 will test sets of modulators that are known to work in concert to direct the
immune response
in the direction of a TH1 response (e.g., production of IL-2 and/or IFNy) or
TH2 response
(e.g., IL-4, IL-5, IL-13), for example.
Vector M can be designed to express one or more modulators. Optimized
immunomodulators, and methods for obtaining optimized immodulators, are
described in
10 copending, commonly assigned US Patent Application No. , filed February 10,
1999 as TTC Attorney Docket No. 18907-0303US, which is entitled "Optimization
of
Immunomodulatory Molecules." These optimized immunomodulatory sequences are
particularly suitable for use as components of the multicomponent genetic
vaccines of the
invention. Multiple modulators can be expressed from a monocistronic or
multicistronic
15 form of the vector.
D. Vectors "CK'; designed to direct release of cheniokines
Genetic vaccine vectors designated "CK" are designed to direct optimal
release of chemokines from target cells. Target cells can be either the
predominant cell type
in the immunized tissue, or can be immune cells such as dendritic cells (CK-
DC),
20 Langerhan's cells (CK-LC), or monocytes and macrophages (CK-MM). These
vectors
typically direct simultaneous expression of optimal levels of several
chemokines such that
the recruitment of immune cells to the site of immunization is optimal. Cells
transfected with
CK vectors can be considered as the traffic police, regulating the immune
cells critical for
the vaccine immune response. The properties of these vectors reflect the
nature of the cell in
25 which the vectors are designed to operate. For example, the vectors are
designed to bind to
and enter the desired cell type, and/or can have cell-specific regulated
promoters that drive
transcription in the desired cell type. The vectors are also engineered to
direct maximal
synthesis and release of the chemokines from the target cells in the desired
ratio. Genetic
vaccine components, and methods for obtaining components, that provide optimal
release of
30 chemokines are described in commonly assigned, copending US Patent
Application No.
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
46
filed February 10, 1999 as TTC Attorney Docket No. 18097-0303US,
entitled "Optimization of Immunomodulatory Molecules."
CK vectors are typically designed to have one or more of the following
properties, each of which can be optimized using the DNA shuffling methods of
the
invention:
(a) optimal vector binding to and uptake by the chosen chemokine expressing
cell. Suitable cells include, for example, muscle cells, epithelial cells, or
cell types that are
dominant (by number) in the particular tissue of interest. Also suitable are
antigen
presenting cells (e.g. dendritic cells, monocytes and macrophages, Langerhans
cells). This is
a critical property which differentiates CK series vectors fi-om those
designed to bind to and
enter other cells.
(b) optimal transcription of the chemokine gene(s). Again, promoters,
enhancers, introns, and the like can be optimized according to the methods of
the invention.
Cell specific promoters are very valuable here as an additional level of
selectivity.
(c) optimal longevity of the mRNA. Optimal 3' and 5' non-translated regions
of the mRNA can be obtained using the methods of the invention.
(d) optimal translation of the mRNA. Again, the DNA shuffling and
selection methods of the invention can be used to obtain polynucleotide
sequences for
optimal ribosome binding and assembly of translational machinery, as well as
optimal codon
preference.
(e) optimal trafficking of the chemokine into the lumen of the RER (via a
signal secretion sequence). An alternative strategy for modulation of the
immune response
via recruitment of cells will use membrane anchored chemokine rather than
secretion of
soluble chemokine. Anchored chemokine will be retained on the surface of the
synthesizing
cell by a hydrophobic tail and phosphoinositol glycan linkage.
(f) optimal protein conformation for each chemokine. In this case, the
optimal protein conformation is that which allows extracellular chemokine/cell
membrane
anchored chemokine to interact with the relevant receptor.
(g) the ratio of diverse chemokines can be determined empirically. One can
test sets of chemokines that are known to work in concert to direct
recruitment of CTL, TH
cells, B cells, monocytes/macrophages, eosinophils, and/or neutrophils as
appropriate.
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
47
Vector CK can be designed to express one or more chemokines. Multiple
chemokines can be expressed from a monocistronic or multicistronic form of the
vector.
E. Other vectors
Genetic vaccines which contain one or more additional component vector
moieties are also provided by the invention. For example, the genetic vaccine
can include a
vector that is designed to specifically enter dendritic cells and Langerhans
cells, and will
migrate to the draining lymph nodes. This vector is designed to provide for
expression of
the target antigen(s), as well as a cocktail of cytokines and chemokines
relevant to elicitation
of the desired immune response in the node. Depending on the clinical goals
and nature of
the antigen, the vector can be optimized for relatively long lived expression
of the target
antigen so that stimulation of the immune system is prolonged at the node.
Another example
is a vector that specifically modulates MHC expression in B cells. Such
vectors are
designed to specifically bind to and enter B cells, cells either resident in
the injection site or
attracted into the site. Within the B cell, this vector directs the
association of antigen
peptides derived from specific uptake of antigen into the endocytic
compartment of the cell
to either association with Class I or Class II, hence directing the
elicitation of specific
immunity via CD4+ T helper cells or CD8+ cytotoxic lymphocytes. Numerous means
exist
for this intracellular direction of the fate of processed peptide that are
discussed herein.
Examples of molecules that direct Class I presentation include tapasin, TAP-1
and TAP-2
(Koopman et al. (1997) Curr. Opin. Immunol. 9: 80-88), and those affecting
Class II
presentation include, for example, endosomal/lysosomal proteases (Peters
(1997) Curr.
Opin. Immunol. 9: 89-96). Genetic vaccine components, and methods for
obtaining
components, that provide optimized Class I presentation are described in
commonly
assigned, copending US Patent Application No. , filed February 10, 1999 as
TTC Attorney Docket No. 18097-0303US, entitled "Optimization of
Immunomodulatory
Molecules."
An optimal DNA vaccine could, for example, combine an AR vector (antigen
release), a CTL-DC vector (CTL activation via dendritic cell presentation of
antigen peptide
on MHC Class I), an M-MM vector for release of IL-12 and IFNg from resident
tissue
macrophages, and a CK vector for recruitment of TH cells into the immunization
site.
CA 02320626 2000-08-10
WO 99/41369 1'CT/US99/03022
48
DNA vaccination can be used for diverse goals that can include the following,
among others:
~ stimulation of a CTL response and/or humoral response ready to react
rapidly and aggressively against an invading bacterial or viral pathogen at
some time in the distant future
~ a continuous but non-aggressive response to prevent inappropriate
responses to allergens
~ a continuous non-aggressive and tolerization of immunity to an
autoantigen in autoimmune disease
~ elicitation of an aggressive CTL response as rapidly as possible against
tumor cell antigens
~ redirection of the immune response away from a strong but inappropriate
immune response to an on-going chronic infection in the direction of
desired responses to clear the pathogen and/or prevent pathology.
These goals cannot always be met by the format of a single vector DNA
vaccine, particularly wherein competing goals are embodied within one DNA
sequence. A
multicomponent format allows the generation of a portfolio of DNA vaccine
vectors, some
of which will be reconstructed on each occasion (e.g., those vectors
containing antigen)
while others will be used as well characterized and understood reagents for
numerous
different clinical applications (e.g., the same chemokine-expressing vector
can be used in
different situations).
IV. r ni s V V r ul s
Recombinant nucleic acid libraries that are obtained by the methods described
herein are screened to identify those DNA segments that have a property which
is desirable
for genetic vaccination. The particular screening assay employed will vary, as
described
below, depending on the particular property for which improvement is sought.
Typically,
the shuffled nucleic acid library is introduced into cells prior'to screening.
If the DNA
shuffling format employed is an in vivo format, the library of recombinant DNA
segments
generated already exists in a cell. If the sequence recombination is performed
in vitro, the
recombinant library is preferably introduced into the desired cell type before
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
49
screening/selection. The members of the recombinant library can be linked to
an episome or
virus before introduction or can be introduced directly.
A wide variety of cell types can be used as a recipient of evolved genes.
Cells of particular interest include many bacterial cell types that are used
to deliver vaccines
or vaccine antigens (Courvalin et al.(1995) C. R. Acad. Sci. 11118: 1207-12),
both gram-
negative and gram-positive, such as salmonella (Attridge et al. (1997) Vaccine
15: 155-62),
clostridium (Fox et al. (1996) Gene Ther. 3: 173-8), lactobacillus, shigella
(Sizemore et al.
(1995) Science 270: 299-302), E. coli, streptococcus (Oggioni and Pozzi (1996)
Gene 169:
85-90), as well as mammalian cells, including human cells. In some embodiments
of the
invention, the library is amplified in a first host, and is then recovered
from that host and
introduced to a second host more amenable to expression, selection, or
screening, or any
other desirable parameter. The manner in which the library is introduced into
the cell type
depends on the DNA-uptake characteristics of the cell type, e.g., having viral
receptors,
being capable of conjugation, or being naturally competent. If the cell type
is unsusceptible
to natural and chemical-induced competence, but susceptible to
electroporation, one would
usually employ electroporation. If the cell type is unsusceptible to
electroporation as well,
one can employ biolistics. The biolistic PDS-1000 Gene Gun {Biorad, Hercules,
CA) uses
helium pressure to accelerate DNA-coated gold or tungsten microcarriers toward
target cells
The process is applicable to a wide range of tissues, including plants,
bacteria, fungi, algae,
intact animal tissues, tissue culture cells, and animal embryos. One can
employ electronic
pulse delivery, which is essentially a mild electroporation format for live
tissues in animals
and patients (Zhao, Advanced Drug Delivery Reviews 17:257-262 (1995)). Novel
methods
for making cells competent are described in International Patent Application
PCT/US97/04494 (Publ. No. W097/35957). After introduction of the library of
recombinant
DNA genes, the cells are optionally propagated to allow expression of genes to
occur.
In many assays, a means for identifying cells that contain a particular vector
is necessary. Genetic vaccine vectors of all kinds can include a selectable
marker gene.
Under selective conditions, only those cells that express the selectable
marker will survive.
Examples of suitable markers include, the dihydrofolate reductase gene (DHFR),
the
thymidine kinase gene (TK), or prokaryotic genes conferring drug resistance,
gpt (xanthine-
guanine phosphoribosyltransferase, which can be selected for with mycophenolic
acid; neo
CA 02320626 2000-08-10
WO 99/41369 PCT/US99103022
(neomycin phosphotransferase), which can be selected for with 6418,
hygromycin, or
puromycin; and DHFR (dihydrofolate reductase), which can be selected for with
methotrexate (Mulligan & Berg (1981) Proc. Nat'1. Acad. Sci. USA 78: 2072;
Southern &
Berg (1982) J. Mol. Appl. Genet. 1: 327).
As an alternative to, or in addition to, a selectable marker, a genetic
vaccine
vector can include a screenable marker which, when expressed, confers upon a
cell
containing the vector a readily identifiable phenotype. For example, gene that
encodes a cell
surface antigen that is not normally present on the host cell is suitable. The
detection means
can be, for example, an antibody or other ligand which specifically binds to
the cell surface
10 antigen. Examples of suitable cell surface antigens include any CD (cluster
of
differentiation) antigen (CD1 to CD163) from a species other than that of the
host cell which
is not recognized by host-specific antibodies. Other examples include green
fluorescent
protein (GFP, see, e.g., Chalfie et al. (1994) Science 263:802-805; Crameri et
al. (1996)
Nature Biotechnol. 14: 315-319; Chalfie et al. (1995) Photochem. Photobiol.
62:651-656;
15 Olson et al. (1995) J. Cell. Biol. 130:639-650) and related antigens,
several of which are
commercially available.
A. Screening for Vector Longevity or Translocation to Desired Tissue
For certain applications, it is desirable to identify those vectors with the
greatest longevity as DNA, or to identify vectors which end up in tissues
distant from the
20 injection site. This can be accomplished by administering to an animal a
population of
recombinant genetic vaccine vectors by the chosen route of administration and,
at various
times thereafter excise the target tissue and recover vector from the tissue
by standard,
molecular biology procedures. The recovered vector molecules can be amplified
in, for
example, E. coli and/ or by PCR in vitro. The PCR amplification can involve
further gene
25 shuffling, after which the derived selected population used for
readministration to animals
and further improvement of the vector. After several rounds of this procedure,
the selected
vectors can be tested for their capacity to express the antigen in the correct
conformation
under the same conditions as the vector was selected in vivo.
Because antigen expression is not part of the selection or screening process
30 described above, not all vectors obtained axe capable of expressing the
desired antigen. To
overcome this drawback, the invention provides methods for identifying those
vectors in a
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/0302Z
51
genetic vaccine population that exhibit not only the desired tissue
localization and longevity
of DNA integrity in vivo, but retention of maximal antigen expression (or
expression of other
genes such as cytokines, chemokines, cell surface accessory molecules, MHC,
and the like).
The methods involve in vitro identification of cells which express the desired
molecule using
cells purified from the tissue of choice, under conditions that allow recovery
of very small
numbers of cells and quantitative selection of those with different levels of
antigen
expression as desired.
Two embodiments of the invention are described, each of which uses a library
of genetic vaccine vectors as the starting point. The goal of each method is
to identify those
vectors that exhibit the desired biological properties in vivo. The
recombinant library
represents a population of vectors that differ in known ways (e.g., a
combinatorial vector
library of different functional modules), or has randomly generated diversity
generated either
by insertion of random nucleotide stretches, or has been shuffled in vitro to
introduce low
level mutations acmss all or part of the vector.
(1) Selec 'on for expression of cell surface-localized antieen
In a first embodiment, the invention method involves selection for expression
of cell surface-localized antigen. The antigen gene is engineered in the
vaccine vector library
such that it has a region of amino acids which is targeted to the cell
membrane. For
example, the region can encode a hydrophobic stretch of C-terminal amino acids
which
signals the attachment of a phosphoinositol-glycan (PIG) terminus on the
expressed protein
and directs the protein to be expressed on the surface of the transfected
cell. With an antigen
that is naturally a soluble protein, this method will likely not affect the
three dimensional
folding of the protein in this engineered fusion with a new C-terminus. With
an antigen that
is naturally a transmembrane protein (e.g., a surface membrane protein on
pathogenic
viruses, bacteria, protozoa or tumor cells) there are at least two
possibilities. First, the
extracellular domain can be engineered to be in fusion with the C-terminal
sequence for
signaling PIG-linkage. Second, the protein can be expressed in toto relying on
the signalling
of the host cell to direct it efficiently to the cell surface. In a minority
of cases, the antigen
for expression will have an endogenous PIG terminal linkage (e.g., some
antigens of
pathogenic protozoa):
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03012
52
The vector library is delivered in vivo and, after a suitable interval of time
tissue and/or cells from diverse target sites in the animal are collected.
Cells can be purified
from the tissue using standard cell biological procedures, including the use
of cell specific
surface reactive monoclonal antibodies as affinity reagents. It is relatively
facile to purify
isolated epithelial cells from mucosal sites where epithelium may have been
inoculated or
myoblasts from muscle. In some embodiments, minimal physical purification is
performed
prior to analysis. It is sometimes desirable to identify and separate specific
cell populations
from various tissues, such as spleen, liver, bone marrow, lymph node, and
blood. Blood
cells can be fractionated readily by FACS to separate B cells, CD4+ or CD8+ T
cells,
dendritic cells, Langerhans cells, monocytes, and the like, using diverse
fluorescent
monoclonal antibody reagents.
Those cells expressing the antigen can be identified with a fluorescent
monoclonal antibody specific for the C-terminal sequence on PIG-linked forms
of the
surface antigen. FACS analysis allows quantitative assessment of the level of
expression of
the correct form of the antigen on the cell population. Cells expressing the
maximal level of
antigen are sorted and standard molecular biology methods used to recover the
plasmid DNA
vaccine vector that conferred this reactivity. An alternative procedure that
allows purification
of all those cells expressing the antigen (and that may be useful prior to
loading onto a cell
sorter since antigen expressing cells may be a very small minority
population), is to rosette
or pan-purify the cells expressing surface antigen. Rosettes can be formed
between antigen
expressing cells and erythrocytes bearing covalently coupled antibody to the
relevant
antigen. These are readily purified by unit gravity sedimentation. Panning of
the cell
population over petri dishes bearing immobilized monoclonal antibody specific
for the
relevant antigen can also be used to remove unwanted cells.
Cells expressing the required conformational structure of the target antigen
can be identified using specific conformationally-dependent monoclonal
antibodies that are
known to react specifically with the same structure as expressed on the target
pathogen.
Because one monoclonal antibody cannot define all aspects of correct folding
of the target
antigen, one can minimize the possibility of an antigen which reacts with high
affinity to the
diagnostic antibody but does not yield the correct conformation as defined by
that in which
the antigen is found on the surface of the target pathogen or as secreted from
the target
CA 02320626 2000-08-10
WO 99/41369 PCTIUS99/03022
53
pathogen. One way to minimize this possibility is to use several monoclonal
antibodies,
each known to react with different conformational epitopes in the correctly
folded protein, in
the selection process. This can be achieved by secondary FACS sorting for
example.
The enriched plasmid population that successfully expressed sufficient of the
antigen in the correct body site for the desired time is then used as the
starting population for
another round of selection, incorporating gene shuffling to expand the
diversity. In this
manner, one recovers the desired biological activity encoded by plasmid from
tissues in
DNA vaccine-immunized animals.
This method can also provide the best in vivo selected vectors that express
immune accessory molecules that one may wish to incorporate into DNA vaccine
constructs.
For example, if it is desired to express the accessory protein B7.1 or B7.2 in
antigen-
presenting-cells (APC) (to promote successful presentation of antigen to T
cells) one can sort
APC isolated from different tissues (at or different to the inoculation site)
using
commercially available monoclonal antibodies that recognize functional B7
proteins.
(2) ~e~ecti~~ ~'or e~ression of secreted antigen/cytokine/chemokine
The invention also provides methods to identify plasmids in a genetic vaccine
vector population that are optimal in secretion of soluble proteins that can
affect the
qualitative and quantitative nature of an elicited immune response. For
example, the methods
are useful for selecting vectors that are optimal for secretion of particular
cytokines, growth
factors and chemokines. The goal of the selection is to determine which
particular
combinations of cytokines, chemokines and growth factors, in combination with
different
promoters, enhancers, polyA tracts, introns, and the like, elicits the
required immune
response in vivo.
Combinations of the genes for the soluble proteins of interest can be present
in the vectors; transcription can be either from a single promoter, or the
genes can be placed
in multicistronic arrangements. Typically, the genes encoding the polypeptides
are present in
the vaccine vector library in combination with optimal signal secretion
sequences, such that
the expressed proteins are secreted from the cells.
The first step in these methods is to generate vectors that are capable of
secreting high (or in some case low) levels of different combinations of
soluble factors in
vitro and that will express those factors for a short or long time as desired.
This method
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
54
allows one to select for and retain an inventory of plasmids which can be
characterized by
known patterns of soluble protein expression in known tissues for a known
time. These
vectors can then be tested individually for in vivo efficacy, after being
placed in combination
with the genetic vaccine antigen in an appropriate expression construct.
The vector library is delivered to a test animal and, after a chosen interval
of
time, tissue and/or cells from diverse sites on the animal are collected.
Cells are purified
from the tissue using standard cell biological procedures, which often include
the use of cell
specific surface reactive monoclonal antibodies as affinity reagents. As is
the case for cell
surface antigens described above, physical purification of separate cell
populations can be
performed prior to identification of cells which express the desired protein.
For these studies,
the target cells for expression of cytokines will most usually be APC or B
cells or T cells
rather than muscle cells or epithelial cells. In such cases FACS sorting by
established
methods will be preferred to separate the different cell types. The different
cell types
described above may also be separated into relatively pure fractions using
affinity panning,
rosetting or magnetic bead separation with panels of existing monoclonal
antibodies known
to define the surface membrane phenotype of marine immune cells.
Purified cells are plated onto agar plates under conditions that maintain cell
viability. Cells expressing the required conformational structure of the
target antigen are
identified using conformationally-dependent monoclonal antibodies that are
known to react
specifically with the same structure as expressed on the target pathogen.
Release of the
relevant soluble protein from the cells is detected by incubation with
monoclonal antibody,
followed by a secondary reagent that gives a macroscopic signal (gold
deposition, color
development, fluorescence, luminescence). Cells expressing the maximal level
of antigen
can be identified by visual inspection, the cell or cell colony picked and
standard molecular
biology methods used to recover the plasmid DNA vaccine vector that conferred
this
reactivity. Alternatively, flow cytometry can be used to identify and select
cells harboring
plasmids that induce high levels of gene expression. The enriched plasmid
population that
successfully expressed sufficient of the soluble factor in the correct body
site for the desired
time is then used as the starting population for another round of selection,
incorporating gene
shuffling to expand the diversity, if further improvement is desired. In this
manner, one
CA 02320626 2000-08-10
WO 99/41369 PGTNS99/03022
recovers the desired biological activity encoded by plasmid from tissues in
DNA vaccine-
immunized animals.
Several monoclonal antibodies, each known to react with different
conformational epitopes in the correctly folded cytokine, chemokine or growth
factor, can be
used to confirm that the initial results from screening with one monoclonal
antibody reagent
still hold when several conformational epitopes are probed. In some cases the
primary probe
for functional cytokine released from the cell/cell colony in agar could be a
soluble domain
of the cognate receptor.
B. Flow Cytometry
10 Flow cytometry provides a means to efficiently analyze the fimctional
properties of millions of individual cells. The cells are passed through an
illumination zone,
where they are hit by a laser beam; the scattered light and fluorescence is
analyzed by
computer-linked detectors. Flow cytometry provides several advantages over
other methods
of analyzing cell populations. Thousands of cells can be analyzed per second,
with a high
15 degree of accuracy and sensitivity. Gating of cell populations allows
multiparameter analysis
of each sample. Cell size, viability, and morphology can be analyzed without
the need for
staining. When dyes and labeled antibodies are used, one can analyze DNA
content, cell
surface and intracytoplasmic proteins, and identify cell type, activation
state, cell cycle stage,
and detect apoptosis. Up to four colors (thus, four separate antigens stained
with different
20 fluorescent labels) and light scatter characteristics can be analyzed
simultaneously (four
colors requires two-laser instrument; one-laser instrument can analyze three
colors). The
expression levels of several genes can be analyzed simultaneously, and
importantly, flow
cytometry-based cell sorting ("FACS sorting") allows selection of cells with
desired
phenotypes. Most of the vector module libraries, including the promoter,
enhancer, intron,
25 episomal origin of replication, expression level aspect of antigen,
bacterial origin and
bacterial marker, can be assayed by flow cytometry to select individual human
tissue culture
cells that contain the recombined nucleic acid sequences that have the
greatest improvement
in the desired property. Typically the selection is for high level expression
of a surface
antigen or surrogate marker protein, as diagrammed in Figure 4. The pool of
the best
30 individual sequences is recovered from the cells selected by flow cytometry-
based sorting.
CA 02320626 2000-08-10
WO 99141369 PCTNS99/03022
56
An advantage of this approach is that very large numbers (>10~) can be
evaluated in a single
vial experiment.
C. In Vitro Screening Methods
Genetic vaccine vectors and vector modules can be screened for improved
vaccination properties using various in vitro testing methods that are known
to those of skill
in the art. For example, the optimized genetic vaccines can be tested for
their effect on
induction of proliferation of the particular lymphocyte type of interest,
e.g., B cells, T cells,
T cell lines, and T cell clones. This type of screening for improved adjuvant
activity and
immunostimulatory properties can be performed using, for example, human or
mouse cells.
A library of genetic vaccine vectors (obtained either from shuffling of random
DNA or of vectors harboring genes encoding cytokines, costimulatory molecules
etc.) can be
screened for cytokine production (e.g., IL-2, IL-4, IL-5, IL-6, IL-10, IL-12,
IL-13, IL-15,
IFN-r, TNF-a) by B cells, T cells, monocytes/macmphages, total human PBMC, or
(diluted)
whole blood. Cytokines can be measured by ELISA or and cytoplasmic cytokine
staining
and flow cytometry (single-cell analysis). Based on the cytokine production
profile, one can
screen for alterations in the capacity of the vectors to direct TH1/TH2
differentiation (as
evidenced, for example, by changes in ratios of IL-4/1FN-y, IL-4/IL-2, IL-
5/IFN-y, IL-5/IL-
2, IL-13/IFN-y, IL-13/IL-2).
Induction of APC activation can be detected based on changes in surface
expression levels of activation antigens, such as B7-1 (CD80}, B7-2 (CD86),
MHC class I
and II, CD14, CD23, and Fc receptors, and the like.
In some embodiments, genetic vaccine vectors are analyzed for their capacity
to induce T cell activation. More specifically, spleen cells from injected
mice can be
isolated and the capacity of cytotoxic T lymphocytes to lyse infected,
autologous target cells
is studied. The spleen cells are reactivated with the specific antigen in
vitro. In addition, T
helper cell differentiation is analyzed by measuring proliferation or
production of TH1 (IL-2
and TFN-'y) and TH2 (IL-4 and IL-5) cytokines by ELISA and directly in CD4~ T
cells by
cytoplasmic cytokine staining and flow cytometry.
Genetic vaccines and vaccine components can also be tested for ability to
induce humoral immune responses, as evidenced, for example, by induction of B
cell
production of antibodies specific for an antigen of interest. These assays can
be conducted
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
57
using, for example, peripheral B lymphocytes from immunized individuals. Such
assay
methods are known to those of skill in the art. Other assays involve detection
of antigen
expression by the target cells. For example, FACS selection provides the most
efficient
method of identifying cells which produce a desired antigen on the cell
surface. Another
advantage of FACS selection is that one can sort for different levels of
expression;
sometimes lower expression may be desired. Another method involves panning
using
monoclonal antibodies on a plate. This method allows large numbers of cells to
be handled
in a short time, but the method only selects for highest expression levels.
Capture by
magnetic beads coated with monoclonal antibodies provides another method of
identifying
cells which express a particular antigen.
Genetic vaccines and vaccine components that are directed against cancer
cells can be screened for their ability to inhibit proliferation of tumor cell
lines in vitro.
Such assays are known in the art.
An indication of the efficacy of a genetic vaccine against, for example,
cancer
or an autoimmune disorder, is the degree of skin inflammation when the vector
is injected
into the skin of a patient or test animal. Stmng inflammation is correlated
with strong
activation of antigen-specific T cells. Improved activation of tumor-specific
T cells may
lead to enhanced killing of the tumors. In case of autoantigens, one can add
immunomodulators that skew the responses towards TH2. Skin biopsies can be
taken,
enabling detailed studies of the type of immune response that occurs at the
sites of each
injection (in mice large numbers of injections/vectors can be analyzed)
Other suitable screening methods can involve detection of changes in
expression of cytokines, chemokines, accessory molecules, and the like, by
cells upon
challenge by a library of genetic vaccine vectors.
D. Screening for Optimal Induction of Protective Immunity
To select genetic vaccine vectors that provide efficient protective immunity,
one can screen the vector libraries in a test mammal using lethal infection
models, such as
Pseudomonas aeruginosa, Salmonella typhimurium, Escherichia coli, Klebsiella
pneumoniae, Toxoplasma gondii, Plasmodium yoelii, Herpes simplex, influenza
virus (e.g.,
Influenza A virus), and Vesicular Stomatitis Virus. Pools of genetic vaccine
vectors or
individual vectors are introduced into the animals intradermally,
intramuscularly,
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
58
intravenously, intratracheally, anally, vaginally, orally, or
intraperitoneally and vectors that
can prevent the disease are chosen for further rounds of shuffling and
selection.
As an example, optimal vectors can be screened in mice infected with
Leishmania major parasites. When injected into footpads of BALB/c mice, these
parasites
cause a progressive infection later resulting in a disseminated disease with
fatal outcome,
which can be prevented by anti-IL-4 mAbs or recombinant IL-12 (Chatelain et
al. (1992) J.
Immunol. 148: 1182-1187). Pools of plasmids can be injected intravenously,
intraperitoneally or into footpads of these mice, and pools that can prevent
the disease are
chosen for further analysis and screened for vectors that can cure existing
infections. The
size of the footpad swelling can be followed visually providing simple yet
precise
monitoring of the disease progression. Mice can be infected intratracheally
with Klebsiella
pneumoniae resulting in lethal pneumonia, which can be prevented by
recombinant IL-12
(Crreenberger et al. (1996) J. Immunol. 157: 3006-3012). The advantage of this
model is that
the infection occurs through the lung, which is a common route of human
pathogen invasion.
The vectors can be given to the lung together with the pathogen or they can be
administered
after symptoms are evident in order to screen for vectors that can cure
established infections.
In another example, the genetic vaccines are a mouse vaccination model for
Influenza A virus. Influenza was one of the first models in which the efficacy
of genetic
vaccines was demonstrated (Uhner et al. (1993) Science 259: 1745-1749).
Several Influenza
strains are lethal in mice providing an easy means to screen for efficacy of
genetic vaccines.
For example, Influenza virus strain A/PR/8/34, which is available through the
American
Type Culture Collection (ATCC VR-95), causes lethal infection, but 100%
survival can be
obtained when the mice are immunized with and influenza hemagglutinin (HA)
genetic
vaccine (Deck et al. (1997) Vaccine 15: 71-78). This model provides a way to
screen for
vectors that provide protection at very low quantities of DNA and/or high
virus
concentrations, and it also allows one to analyze the levels of antigen
specific Abs and CTLs
induced in vivo.
The genetic vaccine vectors can also be analyzed for their capacity to provide
protection against infections by Mycobacterium tuberculosis. This is an
example of a
situation where genetic vaccines have provided partial protection, and where
major
improvements are required.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
59
Once a number of candidate vectors has been identified, these vectors can be
subjected to more detailed analysis in additional models. Testing in other
infectious disease
models (such as HSV, Mycoplasma pulmonis, RSV and/or rotavirus) will allow
identification of vectors that are optimal in each infectious disease.
In each case, the optimal plasmids from the first round of screening can be
used as the starting material for the next round of shuffling, assembly and
selection. Vectors
that are successful in animal models are sequenced and the corresponding human
genes are
cloned into genetic vaccine vectors. These vectors are then characterized in
vitro for their
capacity to induce differentiation of TH1/TH2 cells, activation of TH cells,
cytotoxic T
lymphocytes and monocytes/macrophages, or other desired trait. Eventually, the
most
potent vectors, based on in vivo data in mice and comparative in vitro studies
in mice and
man, are chosen for human trials, and their capacity to counteract various
human infectious
diseases is investigated.
In addition to determining whether a vector pool provides protective
immunity, one can measure immune parameters that correlate to protective
immunity, such
as induction of specific antibodies (particularly IgG) and induction of
specific CTL
responses. Spleen cells can be isolated from vaccinated mice and measured for
the presence
of antigen-specific T cells and induction of TH1 cytokine synthesis profiles.
ELISA and
cytoplasmic cytokine staining, combined with flow cytometry, can provide such
information
on a single-cell level.
E. Screening of Genetic Vaccine Vectors that Activate Human Antigen-
specific Lymphocyte Responses
To screen for vectors with optimal immunostimulatory properties for the
human immune system, peripheral blood mononuclear cells (PBMCs) or purified
professional antigen-presenting cells (APCs) can be isolated from previously
vaccinated or
infected individuals or from patients with acute infection with the pathogen
of interest.
Because these individuals have increased frequencies of pathogen-specific T
cells in
circulation, antigens expressed in PBMCs or purified APCs of these individuals
will induce
proliferation and cytokine production by antigen-specific CD4+ and CD8+ T
cells. Thus,
genetic vaccine vectors encoding the antigen for which the individuals have
specific T cells
can be transfected into PBMC of the individuals, after which induction of T
cell proliferation
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
and cytokine synthesis can be measured. Alternatively, one can screen for
spontaneous entry
of the genetic vaccine vector into APCs, thus providing a means by which to
screen
simultaneously for improved transfection efficiency, improved expression of
antigen and
improved induction of activation of specific T cells. Vectors with the most
potent
5 immunostimulatory properties can be screened based on their capacity to
induce B cell
proliferation and immunoglobulin synthesis. One huffy coat derived from a
blood donor
contains PBMC lymphocytes from 0.5 liters of blood, and up to 104 PBMC can be
obtained,
enabling very large screening experiments using T cells from one donor.
When healthy vaccinated individuals (lab volunteers) are studied, one can
10 make EBV-transformed B cell lines from these individuals. These cell lines
can be used as
antigen presenting cells in subsequent experiments using blood from the same
donor; this
reduces interassay and donor-to-donor variation). In addition, one can make
antigen-specific
T cell clones, after which genetic vaccines are transfected into EBV
transformed B cells.
The efficiency with which the transformed B cells induce proliferation of the
specific T cell
15 clones is then studied. When working with specific T cell clones, the
proliferation and
cytokine synthesis responses are significantly higher than when using total
PBMCs, because
the frequency of antigen-specific T cells among PBMC is very low.
CTL epitopes can be presented by most cells types since the class I major
histocompatibility complex (MHC) surface glycoproteins are widely expressed.
Therefore,
20 transfection of cells in culture by libraries of shuffled DNA sequences in
appropriate
expression vectors can lead to class I epitope presentation. If specific CTLs
directed to a
given epitope have been isolated from an individual, then the co-culture of
the transfected
presenting cells and the CTLs can lead to release by the CTLs of cytokines,
such as IL-2,
IFN-y, or TNFa, if the epitope is presented. Higher amounts of released TNFa
will
25 correspond to more efficient processing and presentation of the class I
epitope from the
shuffled, evolved sequence.
A second method for identifying optimized CTL epitopes does not require the
isolation of CTLs reacting with the epitope. In this approach, cells
expressing class I MHC
surface glycoproteins are transfected with the library of evolved sequences as
above. After
30 suitable incubation to allow for processing and presentation, a detergent
soluble extract is
prepared from each cell culture and after a partial purification of the MHC-
epitope complex
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
61
(perhaps optional) the products are submitted to mass spectrometry (Henderson
et al. (1993)
Proc. Nat'1. Acad. Sci. USA 90: 10275-10279). Since the sequence is known of
the epitope
whose presentation to be increased, one can calibrate the mass spectrogram to
identify this
peptide. In addition, a cellular protein can be used for internal calibration
to obtain a
quantitative result; the cellular protein used for internal calibration could
be the MHC
molecule itself. Thus one can measure the amount of peptide epitope bound as a
proportion
of the MHC molecules.
F. SCID-human Skin Model for Vaccination Studies
Successful genetic vaccinations require transfection of the target cells after
injection of the vector, expression of the desired antigen, processing the
antigen in antigen
presenting cells, presentation of the antigenic peptides in the context of MHC
molecules,
recognition of the peptide/MHC complex by T cell receptors, interactions of T
cells with B
cells and professional APCs and induction of specific T cell and B cell
responses. All these
events could be differentially regulated in mouse and man. A limitation of
mouse models in
vaccine studies is the fact that the MHC molecules of mice and man are
substantially
different. Therefore, proteins and peptides that effectively induce protective
immune
responses in mice do not necessarily function in humans.
To overcome these limitations mouse models can be used to study human
tissues in mice in vivo. Live pieces of human skin are xenotransplanted onto
the back of
immunodeficient mice, such as SCID mice, allowing screening of the vector
libraries for
optimal properties in human cells in vivo. Recursive selection of episomal
vectors provides
strong selection pressure for vectors that remain episomal, yet provide high
level of gene
expression. These mice provide an excellent model for studies on transfection
efficiency,
transfer sequences and gene expression levels. In addition, antigen presenting
cells (APCs)
derived from these mice can also be used to assess the level of antigens
delivered to
professional APCs, and to study the capacity of these cells to present
antigens and induce
activation of antigen-specific CD4+ and CD8+ T cells in vitro. Significantly,
although SCID
mice have severely deficient T and B cell components, antigen presenting cells
(dendritic
cells and monocytes) are relatively normal in these mice.
In one embodiment of this model system, immunocompetent mice are
rendered immunodeficient in order to enable transplantation of human tissue.
For example,
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
62
blocking of CD28 and CD40 pathways promotes long-term survival of allogeneic
skin grafts
in mice (Larsen et al. (1996) Nature 381: 434). Because the in vivo
immunosuppression is
transient, this model also enables vaccine studies in human skin
xenotransplanted into mice
with genetically normal immune systems. Several methods of blocking CD28-B7
interactions and CD40-CD40 ligand interactions are known to those of skill in
the art,
including, for example, administration of neutralizing anti-B7-1 and B7-2
antibodies, soluble
CTLA-4, a soluble form of the extracellular portion of CTLA-4, a fusion
protein that
includes CTLA-4 and an Fc portion of an IgG molecule, and neutralizing anti-
CD40 or anti-
CD40 ligand antibodies. Additional methods by which one can improve transient
immunosuppression include administration of one or more of the following
reagents:
cyclosporin A, anti-IL-2 receptor a-chain Ab, soluble IL-2 receptor, IL-10,
and
combinations thereof.
A model in which SCID-mice transplanted with human skin are injected with
HLA-matched PBMC can be used to analyze vectors that provide long lasting
expression in
vivo. In this model, the vectors are injected, or topically applied, into the
human skin.
Thereafter, HLA-matched PBMC are injected into these mice. If the PBMC
contains
lymphocytes specific for the vector, the transfected cells will be recognized,
and eventually
destroyed, by these vector-specific lymphocytes. Therefore, this model
provides
possibilities to screen for vectors that efficiently escape destruction by the
immune cells. It
has been shown that human PBLs injected into mice with human skin transplants
reject the
organ, indicating that the CTLs reach the skin in this model. Obtaining HLA-
matching skin
and blood is possible (e.g. blood sample and skin graft from a patient
undergoing skin
removal due to malignancy, or blood and foreskin from the same infant).
An additional model that is suitable for screening as described herein is the
modified SCII7hu mouse model, in which pieces of human fetal thymus, liver and
bone
marrow are transplanted into SCID mice providing functional human immune
system in
mice (Roncarolo et al. (1996) Semin. Immunol. 8: 207). Functional human B and
T cells,
and APCs can be observed in these mice. When additionally human skin is
transplanted, it
is likely to allow studies on the efficacy of genetic vaccine vectors
following injection into
the skin. Cotransplantation of skin is likely to improve the model because it
will provide an
additional source of professional APCs.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
63
G. Mouse model for studying the eff:ciency of genetic vaccines in transfecting
human muscle cells and inducing human immune responses in vivo
A lack of suitable in vivo models has hampered studies of the efficiency of
genetic vaccines in inducing antigen expression in human muscle cells and in
inducing
specific human immune responses. The vast majority of studies on the capacity
of genetic
vaccines to transfect muscle cells and to induce specific immune responses in
vivo have
employed a mouse model. Because of the complexity of events occurnng after
genetic
vaccination, however, it is sometimes difficult to predict whether results
obtained in the
mouse model reliably predict the outcome of similar vaccinations in humans.
The events
required in successful genetic vaccination include transfection of the cells
after delivery of
the plasmid, expression of the desired antigen, processing the antigen in
antigen presenting
cells, presentation of the antigenic peptides in the context of MHC molecules,
recognition of
the peptide/MIiC complex by T cell receptors, interactions of T cells with B
cells and
professional antigen presenting cells and finally induction of specific T cell
and B cell
responses. All these events are likely to be somewhat differentially regulated
in mouse and
man.
The invention provides an in vivo model for human muscle cell transfection.
This model system is especially valuable because there is no in vitro culture
system available
for normal muscle cells. Muscle tissue, obtained for example from cadavers, is
transplanted
subcutaneously into immunodeficient mice. Immunodeficient mice can be
transplanted with
tissues from other species without rejection. Mice suitable for
xenotransplantations include,
but are not limited to, SCID mice, nude mice and mice rendered deficient in
their genes
encoding RAGl or RAG2 genes. SCID mice and RAG deficient mice lack functional
T and
B cells, and therefore are severely immunocompromised and are unable to reject
transplanted organs. Previous studies indicate that these mice can be
transplanted with
human tissues, such as skin, spleen, liver, thymus or bone, without rejection
(Roncarolo et
al. (1996) Semin. Immunol. 8: 207). After transplantation of human fetal
lymphoid tissues
into SCID mice, functional human immune system can be demonstrated in these
mice, a
model generally referred to as SCID-hu mice. When human muscle tissue is
transplanted
into SCID-hu mice, one can not only study transfection efficiency and
expression of the
desired antigen, but one can also study induction of specific human immune
responses
induced by genetic vaccines in vivo. In this case, muscle and lymphoid organs
from the
CA 02320626 2000-08-10
WO 99/41369 PCf/US99/03022
64
same donor are used. Fetal muscle also has an advantage in that it contains
few mature
lymphocytes of donor origin decreasing likelihood of graft versus host
reaction.
Once the human muscle tissue is established in the mouse, genetic vaccine
vectors are introduced into the human muscle tissue to study the expression of
the antigen of
interest. When studying transfection efficiency only, RAG deficient mice are
preferred,
because these mice never have mature B or T cells in the circulation, whereas
"leakiness" of
SCID phenotype has been demonstrated which may cause variation in the
transplantation
e~ciency.
The survival of human muscle tissue in mice is likely to be limited even in
immunocompromised mice. However, because expression studies can be performed
within
one or two days, this model provides an efficient means to study gene
expression in human
muscle cells in vivo. A modified SCID-hu mouse model with human muscle
transplanted
into these mice can be used to study human immune responses in mice in vivo.
H. Screening for Improved Delivery of Vaccines
For certain applications, it is desirable to identify genetic vaccine vectors
that
are capable of being administered in a particular manner, for example, orally
or through the
skin. The following screening methods provide suitable assays; additional
assays are also
described herein in conjunction with particular genetic vaccine properties for
which the
assays are especially suitable.
Screening for oral delivery can be performed either in vitro or in vivo. An
example of an in vitro method is based on Caco-2 (human colon adenocarcinoma)
cells
which are grown in tissue culture. When grown on semipermeable filters, these
cells
spontaneously differentiate into cells that resemble human small intestine
epithelium, both
structurally and functionally. Genetic vaccine libraries and/or vectors can be
placed on one
side of the Caco-2 cell layer, and vectors that are able to move through the
cell layer are
detected on the opposite side of the layer.
Libraries can also be screened for amenability to oral delivery in vivo. For
example, a library of vectors can be administered orally, after which target
tissues are
assayed for presence of vectors. Intestinal epithelium, liver, and the
bloodstream are
examples of tissues that can be tested for presence of library members.
Vectors that are
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
successful in reaching the target tissue can be recovered and, if further
improvement is
desired, used in succeeding rounds of shuffling and selection.
For screening a library of genetic vaccine vectors for ability to transfect
cells
upon injection into skin or muscle, the invention provides an apparatus which
permits large
5 numbers of vectors to be screened efficiently. This apparatus (Figure 5) is
based on 96-well
format and is designed to transfer small volumes (2-5 ~.1) from a microtiter
plate to skin or
muscle of laboratory animals, such as mice and rats. Moreover, human muscle or
skin
transplanted into immunodeficient mice can be injected.
The apparatus is designed in such a way that the tips move to fit a microtiter
10 plate. After the reagent of interest has been obtained from the plate, the
distance of the tips
from each other is decreased to 2-3 mm, enabling transfer of 96 reagents to an
area of 1.6 cm
x 2.4 cm to 2.4 cm x 3.6 cm. The volume of each sample transferred is
electronically
controlled. Each reagent is mixed with a marker agent or dye to enable
recognition of
injection site in the tissue. For example, gold particles of different sizes
and shapes are
15 mixed with the reagent of interest, and microscopy and immunohistochemistry
can be used
to identify each injection site and to study the reaction induced by each
reagent. When
muscle tissue is injected the injection site is first revealed by surgery.
This apparatus can be used to study the effects of large numbers of agents in
vivo. For example, this apparatus can be used to screen efficiency of large
numbers of
20 different DNA vaccine vectors to transfect human skin or muscle cells
transplanted into
immunodeficient mice.
V. Outim ration of Genetic Vaccine Components
Many factors can influence the efficacy of a genetic vaccine in modulating an
immune response. The ability of the vector to enter a cell, for example, has a
significant
25 effect on the ability of the vector to modulate an immune response. The
strength of an
immune response is also mediated by the immunogenicity of an antigen expressed
by a
genetic vaccine vector and the level at which the antigen is expressed. The
presence or
absence of costimulatory molecules produced by the genetic vaccine vector can
affect not
only the strength, but also the type of immune response that arises due to
introduction of the
30 vector into a mammal. An increase in the persistence of a vector in an
organism can lengthen
the time of immunomodulation, and also makes feasible self boosting vectors
which do not
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
66
require multiple administrations to achieve long-lasting protection. The
present invention
provides methods for optimizing many of these properties, thus resulting in
genetic vaccine
vectors that exhibit improved ability to elicit the desired effect on a
mammalian immune
system.
Genetic vaccines can contain a variety of functional components, whose
preferred sequences are best determined by DNA shuffling, the empirical
sequence evolution
described in detail herein. The methods of the invention involve, in general,
constructing a
separate library for each of the major vector components by DNA shuffling of
multiple
homologous starting sequences, or other methods of generating a population of
t0 recombinants, resulting in a complex mixture of chimeric sequences. The
best sequences are
selected from these libraries using the high-throughput assays described
below. After one or
more cycles of selection from each of the single module libraries, the pools
of the best
sequences of different modules can be combined by shuffling as long as the
screens are
compatible. The screens for promoter, enhancer, intron, transfer sequences,
mammalian ori,
15 bacterial on and bacterial marker, and the like, can eventually be
combined, resulting in co-
optimization of the context of each sequence. An important aspect in these
experiments is
the selection from large libraries using recursive cycles of shuffling to
maximally access all
the fortuitous but complex mechanisms that cannot be approached rationally,
such as DNA
transfer into the cell.
20 A library of different vectors can be generated by assembling vector
modules
that provide promoters, cytokines, cytokine antagonists, chemokines,
immunostimulatory
sequences, and costimulatory molecules using assembly PCR and combinatorial
molecular
biology. Assembly PCR is a method for assembly of long DNA sequences, such as
genes
and fragments of plasmids. In contrast to PCR, there is no distinction between
primers and
25 template, because the fragments to be assembled prime each other. The
library of vector
modules obtained as described herein can be fused with promoters, which can
themselves be
optimized by the DNA shuffling methods of the invention. The resulting genes
can be
assembled combinatorially into DNA vaccine vectors, where each gene is
expressed under a
different promoter (e.g., a promoter derived from a library of shuffled CMV
promoters), and
30 the vector library is screened as described herein to identify vectors
which exhibit the desired
effect on the immune system.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
67
The methods of the invention are useful for obtaining genetic vaccines that
are optimized for one or more of many properties that influence the efficacy
or desirability
of the vaccine. These properties include, but are not limited to, the
following.
A. Episomal vector maintenance
One property that one can optimize using the sequence recombination
methods of the invention is the ability of a genetic vaccine vector to
replicate episomally in a
mammalian cell. Episomal replication of a vaccine vector is advantageous in
many
situations. For example, episomally replicating vectors are maintained in a
cell for a longer
period of time than non-replicating vectors, thus resulting in an increased
length of immune
response modulation or increased delivery of a therapeutically useful protein.
Episomal
replication also permits the development of self boosting vaccines which,
unlike traditional
vaccines, do not require multiple vaccine administrations. For example, a self
boosting
vaccine vector can include an antigen-encoding gene which is under the control
of an
inducible control element which allows induction of antigen expression, and
the
1 S corresponding immune response, in response to a specific stimulus.
However, screening for
naturally occurring vector modules which result in enhanced episomal
maintenance using
traditional approaches or attempts to rationally design mutants with improved
properties
would require many person-years of research. The invention provides methods
for
generating and screening orders of magnitude more diversity in a short time
period.
The ability of a genetic vaccine vector to replicate episomally can be
optimized by using DNA shuffling to recombine at least two forms of a nucleic
acid which is
capable of conferring upon a genetic vector the ability to replicate
autonomously in
mammalian cells. The two or more forms of the episomal replication vector
module differ
from each other in two or more nucleotides. A library of recombinant episomal
replication
vector modules is produced, and the library is screened to identify one or
more optimized
replication vector modules which, when placed in a genetic vaccine vector,
confer upon the
vector an enhanced ability to replicate autonomously compared to a vector
which contains a
non-optimized episomal replication vector module.
In one embodiment, the DNA shuffling process is repeated at least once using
as a substrate an optimized episomal replication vector module obtained from a
previous
round of DNA shuffling. The optimized vector module obtained in the earlier
round is
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
68
recombined with a further form of the vector module, which can be the same as
one of the
forms used in the earlier round, or can be a different form of a nucleic acid
that functions as
an episomal replication element. Again, a library of recombinant episomal
replication vector
modules is produced, and the screening process is repeated to identify those
episomal
replication modules which exhibit enhanced ability to confer episomal
maintenance upon a
vector containing the module.
Nucleic acids which are useful as substrates for the use of DNA shuffling to
optimize episomal replication ability include any nucleic acid that is
involved in conferring
upon a vector the ability to replicate autonomously in eukaryotic cells. For
example,
papillomavirus sequences E1 and E2, simian virus 40 (SV40) origin of
replication, and the
like.
Exemplary episomal replication vector modules that can be optimized using
the methods of the invention are genes from human papillomaviruses (HPV) which
are
involved in episomal replication. HPV are non-tumorigenic viruses which
replicate
episomally in skin and are stably expressed in vivo for years. Bernard and Apt
(1994) Arch.
Dermatol. 130: 210. Despite these in .vivo properties, it has not been
possible to maintain
HPV episomally in tissue culture due to underreplication. The invention
provides methods
by which HPV genes involved in episomal maintenance can be optimized for use
in genetic
vaccine vectors. HPV genes involved in episomal replication include, for
example, the E1
and E2 genes. Thus, according to one embodiment of the invention, either or
both of the
HPV E1 and E2 genes are subjected to DNA shuffling to obtain a recombinant
episomal
replication module which, when placed in a nucleic acid vaccine vector,
results in increased
maintenance of the vector in mammalian cells. In a preferred embodiment, the
HPV E1 and
E2 genes from different, but closely related, benign HPVs are used in a
"family shuffling"
procedure, as shown in Figure 6. For example, family shuffling of HPV E1 and
E2 genes
from closely related strains of HPV (such as, for example, HPV 2, 27, and 57)
can be used to
obtain a library of recombinant E1 and E2 genes which are then subjected to an
appropriate
screening method to identify those that exhibit improved episomal maintenance
properties.
To identify recombinant episomal replication vector modules that exhibit
improved ability to mediate episomal maintenance, members of the library of
recombinant
vector modules are inserted into vectors which are introduced into mammalian
cells. The
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/030Z2
69
cells are propagated for at least several generations, after which cells that
have maintained
the vector are identified. Identification can be accomplished, for example,
employing a
vector that includes a selectable marker. Cells containing the library members
are
propagated in the absence of selection for the selectable marker for at least
several
generations, after which selective pressure is added. Cells which survive
selection are
enriched for cells that harbor vectors which contain a recombinant vector
module which
enhances the ability of the vector to replicate episomally. DNA is recovered
from the
selected cells and introduced into bacterial host cells, allowing recovery of
episomal, non-
integrated vectors.
In another embodiment of the invention, the screening step is accomplished
by introducing members of the library of recombinant episomal replication
vector modules
into a vector that includes a polynucleotide that encodes an antigen which,
when expressed,
is present on the surface of a cell. The library of vectors is introduced into
mammalian cells
which are propagated for at least several generations, after which cells which
display the cell
surface antigen on the surface of the cell are identified. Such cells most
likely harbor a
genetic vaccine vector which enhances the ability of the vector to replicate
autonomously.
Upon identifying cells which contain an episomally maintained vector, the
optimized
recombinant episomal replication vector module is obtained and used to
construct genetic
vaccine vectors. Cell surface antigens which are suitable for use in the
screening methods are
described above, and others are known to those of skill in the art.
Preferably, an antigen is
used for which a convenient means of detection is available.
Cells which are suitable for use in the screening methods include both
cultured mammalian cells and cells which are present in an animal. To screen
for
recombinant vector modules that are intended for use in humans, the preferred
cells for
screening purposes are human cells. Generally, initial screening is
accomplished in cell
culture, where processing of large libraries of shuffled material is feasible.
In a preferred
embodiment, cells which display a vector-encoded cell surface antigen on the
cell surface are
identified by flow cytometry based cell sorting methods, such as fluorescence
activated cell
sorting. This approach allows very large numbers (>10~) cells to be evaluated
in a single vial
experiment.
CA 02320626 2000-08-10
WO 99/41369 PC'fNS99/03022
Constructs which replicate autonomously in cell culture and give rise to
strong marker gene expression can be further tested for durability in vivo in
an animal model.
For example, mouse models for studies of human tissues in mice in vivo are
described in
copending US Patent Application No. 08/958,822, which was filed on October 28,
1997.
5 Live pieces of human skin are xenotransplanted onto the back of SCID mice,
allowing
screening of the vector libraries for optimal properties in human cells in
vivo. Recursive
selection of episomal vectors will provide strong selection pressure for
vectors that remain
episomal, yet provide high level of gene expression.
In another embodiment, the screening step involves introducing a genetic
10 vaccine vector which includes the recombinant episomal replication vector
module, as well
as polynucleotide that encodes an antigen or pharmaceutically useful protein,
into a mammal
that has a functional human immune system. The animal is then tested for the
existence of an
immune response against the antigen. In a preferred embodiment, the mammals
used for
such assays are non-human mammals that have a functional human immune system.
For
1 S example, a functional human immune system can be created in an
immunodeficient mouse
by introducing one or more of a human fetal tissue selected from the group
consisting of
liver, thymus, and bone marrow (Roncarolo et al. (1996) Semin. Immunol. 8:
207).
Stable episomal vectors which are obtained using the methods of the
invention are useful not only as genetic vaccines, but also are useful tools
in other library
20 screening applications. In contrast to randomly integrating and transient
vectors, episomally
maintained vectors result in high signal-to-noise ratios upon FACS selection,
and they also
significantly improve the possibility to recover the plasmids from a small
number of selected
cells.
B. Evolution of Optimized Promoters for Expression of an Antigcn
25 In another embodiment, the invention provides methods of optimizing vector
modules such as promoters and other gene expression control signals. Usually,
a coding
sequence for an antigen that is delivered by a genetic vaccine is operably
linked to an
additional sequence, such as a regulatory sequence, to ensure its expression.
These
regulatory sequences can include one or more of the following: an enhancer, a
promoter, a
30 signal peptide sequence, an intron and/or a polyadenylation sequence. A
desirable goal is to
increase the level of expression of functional expression product relative to
that achieved
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
71
with conventions! vectors. The efficacy of a genetic vaccine vector often
depends on the
level of expression of an antigen by the vaccine vector. An optimized promoter
and/or other
control sequence is likely to result in improved efficacy of genetic
vaccinations, reduce the
amount of DNA required for protective immunity and thereby the cost of
vaccination.
Moreover, it is sometimes desirable to have control over the type of cell in
which a gene is
expressed, and/or the taming of antigen expression. The methods of the
invention provide
for optimization of these and other factors which are influenced by promoters
and other
control sequences.
Improved expression of selection markers can be achieved by performing
DNA shuffling, for example. Expression can effectively be improved by a
variety of means,
including increasing the rate of production of an expression product,
decreasing the rate of
degradation of the expression product or improving the capacity of the
expression product to
perform its intended function. The methods involve subjecting to DNA shuffling
polynucleotides which are involved in control of gene expression. At least
first and second
forms of a nucleic acid that comprises a control sequence, which forms differ
from each
other in two or more nucleotides, are recombined as described above. The
resulting library of
recombinant transfer modules are screened to identify at least one optimized
recombinant
control sequence that exhibits enhanced strength, inducibility, or
specificity.
The substrates for recombination can be the full-length vectors, or fragments
thereof, which include a coding sequence and/or regulatory sequences to which
the coding
sequence is operably linked. The substrates can include variants of any of the
regulatory
and/or coding sequences) present in the vector. If recombination is effected
at the level of
fragments, the recombinant segments should be reinserted into vectors before
screening. If
recombination proceeds in vitro, vectors containing the recombinant segments
are usually
introduced into cells before screening. An example of a vector suitable for
use in screening
of shuffled promoters and other regulatory regions is shown in Figure 7.
Cells containing the recombinant segments can be screened by detecting
expression of the gene encoded by the selection marker. For purposes of
selection and/or
screening, a gene product expressed from a vector is sometimes an easily
detected marker
rather than a product having an actual therapeutic purpose, e.g., a green
fluorescent protein
(see, Crameri {1996) Nature Biotechnol. 14: 315-319) or a cell surface
protein. For example,
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
72
if this marker is green fluorescent protein, cells with the highest expression
levels can be
identified by flow cytometry-based cell sorting. If the marker is a cell
surface protein, the
cells are stained with a reagent having affinity for the protein, such as
antibody, and again
analyzed by flow cytometry-based cell sorting. However, some genes having a
therapeutic
purpose, e.g., drug resistance genes, themselves provide a selectable marker,
and no
additional or substitute marker is required. Alternatively, the gene product
can be a fusion
protein comprising any combination of detection and selection markers.
Internal reference
marker genes can be included on the vector to detect and compensate for
variations in copy
number or insertion site.
Recombinant segments from the cells showing highest expression of the
marker gene can be used as some or all of the substrates in a further round of
recombination
and screening, if additional improvement is desired.
1. Constitutive promoters
The invention provides methods of evolving nucleotide sequences that are
capable of directing constitutive expression of a gene of interest which is
operably linked to
the control sequence. Typically, the control sequences, which can include
promoters,
enhancers, and the like, are evolved so that a gene of interest is expressed
at a higher level
than is a gene operably linked to a non-evolved control sequence. To screen
for control
sequences which are of increased strength, a recombinant library of control
sequences can be
introduced into a population of cells and the level of expression of a
detectable marker
operably linked to the control sequences determined. Preferably, the optimized
promoter is
capable of expressing an operably linked gene at a level that is at least
about 30% greater
than that of a control promoter construct, more preferably the optimized
promoter is at least
about 50% stronger than a control, and most preferably at least about 75% or
more stronger
than a control promoter.
Examples of promoters which can be used as substrates in the methods
include any constitutive promoter that functions in the intended host cell.
The major
immediate-early (IE) region transcriptional regulatory elements, including
promoter and
enhancer sequences (the promoter/enhancer region), of cytomegalovirus (CMV) is
widely
used for regulating transcription in vectors used for gene therapy because it
is highly active
in a broad range of cell types. Optimized CMV transcriptional regulatory
elements which
CA 02320626 2000-08-10
WO 99/4369 PCT/US99/03022
73
direct increased levels of antigen expression is generated by the recursive
recombination
methods of the invention, resulting in improved efficacy of gene therapy. As
the CMV
promoter and enhancer is active in human and animal cells, the improved CMV
promoter/enhancer elements are used to express foreign genes both in animal
models and in
clinical applications. Other constitutive promoters that are amenable to use
in the claimed
methods include, for example, promoters from SV40 and SRa, and other promoters
known
to those of skill in the art.
In a preferred embodiment, a library of chimeric transcriptional regulatory
elements is created by DNA shuffling of wild-type sequences from two or more
of the five
related strains of CMV. The promoter, enhancer and first intron sequences of
the IE region
are obtained by PCR from the CMV strains: human VR-538 strain AD169 (Rowe
(1956)
Proc. Soc. Exp. Biol. Med. 92:418; human V-977 strain Towne (Plotkin (1975)
Infect.
Immunol. 12:521-527); rhesus VR-677 strain 68-1 (Asher (1969) Bacteriol. Proc.
269:91);
vervet VR-706 strain CSG (Black (1963) Proc. Soc. Exp. Biol. Med. 112:601);
and, squirrel
monkey VR-1398 strain SqSHV (Rangan (1980) Lab. Animal Sci. 30:532). The
promoter/
enhancer sequences of the human CMV strains are 95% homologous, and share 70%
homology with the sequences of the monkey isolates, allowing the use of family
shuffling to
generate a library great diversity. Following shuffling, the library is cloned
into a plasmid
backbone and used to direct transcription of a marker gene in mammalian cells.
An internal
marker under the control of a native promoter is typically included in the
plasmid vector,
which will allow analysis and sorting of cells harboring equal numbers of
vectors.
Expression markers, such as green fluorescent protein (GFP) and CD86 (also
known as
B7.2, see Freeman (1993) J. Exp. Med. 178:2185, Chen (1994) J. Immunol.
152:4929) can
also be used. In addition, transfection of SV40 T antigen-transformed cells
can be used to
amplify a vector which contains an SV40 origin of replication. The transfected
cells are
screened by FACS sorting to identify those which express high levels of the
marker gene,
normalized against the internal marker to account for differences in vector
copy numbers per
cell. If desired, vectors carrying optimal, recursively recombined promoter
sequences are
recovered and subjected to further cycles of shuffling and selection.
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
74
2. Cell-specific promoters
One of the safety concerns associated with genetic vaccines has been the
possibility of autoimmune disorders following introduction of foreign antigens
into host
cells. This risk can be reduced if the pathogen antigen is specifically
expressed in
professional APCs that express the proper costimulatory molecules. Although it
is
somewhat debatable which cells are the most important cells expressing the
pathogen
antigen following genetic vaccinations, it is likely that professional APCs
are involved. It
has been shown that blood monocytes express antigen following intnnnuscular
injection of
genetic vaccine vectors, and dendritic cells derived from lymph nodes of
vaccinated animals
efficiently induced antigen-specific T cell activation (C. Bona, The First
Gordon Conference
on Genetic Vaccines, Plymouth, NH, July 21, 1997). These data, together with
previous
studies indicating that small number of dendritic cells expressing antigen or
antigenic
peptides is sufficient to induce activation of antigen-specific T cells
(Thomas and Lipsky,
Stem Cells 14: 196, 1996), support the conclusion that genetic vaccines
specifically
expressed in professional APC, such as dendritic cells and macrophages, are
likely to
provide efficient induction of protective immunity with minimized chance of
adverse effects.
The present invention provides methods of obtaining promoters and
enhancers that induce high expression levels specifically in professional
APCs. Previously
existing APC-specific vectors did not provide sufficient expression levels
following genetic
vaccinations. The methods involve performing DNA shui~ling as described above
using as
substrates different forms of a nucleic acid that comprises an APC-specific
promoter or other
control signal: Suitable promoters include, for example, the MHC Class II, and
the CD1 lb,
CD1 lc, and CD40 promoters. Natural diversity of the promoters can be
exploited as a
highly appropriate source of substrates for the DNA shufrling. For example,
genomic DNA
from monkeys, pigs, dogs, cows, cats, rabbits, rats and mice, can be obtained,
and the proper
sequences obtained by using multiple PCR primers specific for the most
conserved regions
based on known sequence information. The selection of the optimal promoters
can be done
in monocytic or B cell lines, such as U937, HL60 or Jijoye, using FACS-
sorting. In
addition, SV40+ cell lines, such as COS-1 and COS-7, can be used to improve
the recovery
of the plasmids. Further analysis can be undertaken in human dendritic cells
obtained by
culturing peripheral blood monocytes in the presence of IL-4 and GM-CSF as
described
(Chapuis et al. (1997) Eur. J. Immunol. 27: 431).
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
3. Inducible promoters
A particularly desirable property of a genetic vaccines would be an ability to
induce the promoter controlling transgene expression simply by taking an
innocuous oral
drug, resulting in a boost of the immune response. Essential requirements for
inducible
5 promoters are low base-line expression and strong inducibility. Several
promoters with
exquisite in vitro regulation exist, but the expression level and inducibility
of each is too low
to be useable in vivo. The invention overcome these problems by DNA shuffling
using as
substrates two or more variants of a nucleic acid that functions as an
inducible control
sequence. Suitable substrates include, for example, tetracycline and hormone
inducible
10 expression systems, and the like. Hormones that have been used to regulate
gene expression
include, for example, estrogen, tomoxifen, toremifen and ecdysone (Ramkumar
and Adler
(1995) Endocrinology 136: 536-542). Libraries of recombinant inducible
promoters are
screened as described above in the presence and absence of the inducer.
The most commonly used inducible gene expression protocol is the
15 tetracycline responsive system, which provides possibilities to both induce
and turn off gene
expression (Gossen and Bujard (1992) Proc. Nat'1. Acad. Sci. U~'A 89: 5547;
Gossen et al.
(1995) Science 268: 1766). A repressor gene is located on the plasmid and
binds to an
operator in the promoter. Tetracycline or doxycycline modulates the binding
ability of the
repressor. Interestingly, four amino acid changes convert the repressor into
an activator. In
20 addition to the tetracycline responsive system, other candidates for
inducible promoter
evolution include the ecdysone responsive element (No et al., Proc. Nat'1.
Acad. Sci. USA
93: 3346, 1997).
Inducible promoters such as those obtained using the methods of the
invention are useful in autoboost vaccines. Particularly when combined with a
stably
25 maintained episomal vector obtained as described above, the inducible
promoters provide a
means by which a vaccine dose can be administered subsequent to the initial
administration
simply by ingestion of a reagent that causes induction of the inducible
promoter. Figure 8
demonstrates a flow cytometry-based screening protocol that is suitable for
optimization of
inducible promoters.
30 The functionality of autoboosting vaccines can be tested in a mouse model
such as that described above. Genetic vaccine vectors are injected into the
skin of normal
mice and into human skin in SCID-human skin mice. A gene encoding hepatitis B
surface
CA 02320626 2000-08-10
WO 99/41369 PCT/US99I03022
76
antigen (HBsAg) or other surface antigen is incorporated into these vectors
enabling direct
measurements of the levels of antigen produced, because HBsAg levels can be
measured in
cell culture supernates and in the circulation of the mice. The drug inducing
the expression
of the antigen is given after 1, 2, 4 and 6 weeks, and the expression levels
of HBsAg are
studied. Moreover, the levels of anti-HBsAg antibodies are measured. The mice
are also
injected with a vector containing a pathogen antigen discovered by ELI, and
specific
immune responses are followed.
Combining the SCID-human skin model with traditional SCID-hu mouse
model (Roncarolo et al., Semin. Immunol. 8: 207, 1996) allows the assessment
of
functionality of autoboosting generic vaccines in human immune system in vivo,
and also
allows measurements of human Ab responses in vivo. This model can also be used
to assess
production of HBsAg after oral boosting of novel genetic vaccine vectors
harboring the gene
encoding HBsAg.
G Evolution of Genetic Vaccine Vectors for Increased Vaccination E, fflcacy
and Ease of Vaccination
This section discusses the application of the invention to some specific goals
in genetic vaccination. Many of these goals relate to improvements in vectors
used in
vaccine delivery. Unless otherwise indicated the methods are applicable to
both viral and
nonviral vectors.
1. Topical annlication of genetic vaccine vectors
The invention provides methods of improving the ability of genetic vaccine
vectors to induce a desired response after topical application of the vector.
Adenoviral
vectors topically applied to bare skin have been shown to be capable of acting
as vaccine
antigen delivery vehicles (Tang et al. (1997) Nature 388: 729-730). An
adenoviral vector
that encoded carcinoembryonic antigen (CA) was shown to induce antibodies
specific for
CA after application to the skin. However, the efficiency of topical
application is generally
quite low, and pmtecdve immune responses have not been demonstrated after
topical
application.
The invention provides methods of obtaining vectors that exhibit improved
efficiency when topically administered. Several factors can influence topical
application
efficiency, each of which can be optimized using the methods of the invention.
For
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
77
example, the invention provides methods of improving vector affinity for skin
cells,
improved skin cell transfection efficiency, improved persistence of the vector
in skin cells
(both through improved replication or through avoidance of destruction by
immune cells),
and improved antigen expression in skin cells, and improved induction of an
immune
response.
These methods involve performing DNA shuffling using as substrates
plasmid, naked DNA vectors, or viral vector nucleic acids, including, for
example,
adenoviral vectors. Libraries of shuffled nucleic acids are screened to
identify those nucleic
acids that confer upon a vector an enhanced ability to induce an immune
response upon
topical administration. Screening can be conducted by, for example, topically
applying a
library of shuffled vectors to skin, either mouse skin, monkey skin, or human
skin that has
been transplanted to immunodeficient mice, or to normal human skin in vivo.
Vectors that
persist and/or provide efl~cient and long-lasting expression of marker gene
are recovered
from the skin samples. In a preferred embodiment, the desired cells are first
selected by cell
I S sorting, magnetic beads, or panning. For example, recovery can be effected
through
expression of a marker gene (e.g., GFP) and detecting cells that are
transfected using
fluorescence microscopy or flow cytometry. Cells that express the marker gene
can be
isolated using flow cytometry based cell sorting. Screening can also involve
selection of
vectors that induce the highest specific antibody or CTL responses upon
administration to a
test mammal, or the identification of vectors that provide an enhanced
protective immune
response to challenge with a corresponding pathogen. Shuffled polynucleotides
are then
recovered, e.g., by polymerase chain reaction, or the entire vectors can be
purified from
these selected cells. If desired, further optimization of topical application
efficiency can be
obtained by subjecting the recovered shuffled polynucleotides to new rounds of
shuffling
and selection.
Genetic vaccine vectors that are optimized for topical application can be
applied topically to the skin, or by intramuscular, intravenous, intradenmal,
oral, anal, or
vaginal delivery. The vector can be delivered in any of the suitable forms
that are known to
those of skill in the art, such as a patch, a cream, as naked DNA, or as a
mixture of DNA and
one or more transfection-enhancing agents such as liposomes and/or lipids.. In
preferred
embodiments, the genetic vaccine vector is applied after the skin or other
target is rendered
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
78
more susceptible to uptake of the vector by, for example, mechanical abrasion,
removal of
hair (e.g., by treatment with a commercially available product such as NairTM,
NeetTM, and
the like). In one embodiment, the skin is pretreated with professes or lipases
to make it more
susceptible to DNA delivery. In addition, the DNA can be mixed with the
professes or
lipases to enhance gene transfer. Alternatively, a droplet containing the
vector and other
vaccine components, if any, can simply be administered to the skin.
2. Enhanced ability to escape host immune system
Immunogenicity is a particular concern with viral vectors, since a host
immune response can prevent a virus from reaching its intended target
particularly in
repeated administrations. The efficacy of some viral vectors which are used
for genetic
vaccination and gene delivery is limited by host immune responses directed
against the viral
vector. For example, most individuals have pre-existing antibodies against
adenovirus.
Adenoviral vectors can sometimes induce strong immune responses which can
destroy cells
harboring adenoviral vectors or clear adenoviral vectors from the host even
before target
cells are entered. Cellular immune responses can also be induced against
nonviral vectors
administered in naked form or shielded with a coat such as liposomes.
The invention provides methods to create genetic vaccine vectors that can
escape immune responses that would otherwise be detrimental to obtaining the
desired
effect. These methods are useful for prolonging expression and secretion of
pathogen
antigen or pharmaceutically useful protein by genetic vaccine vectors. Several
strategies are
provided by which one can improve a genetic vaccine vector's ability to avoid
the humoral
(Ab) and cellular (CTL) immune systems. These strategies can be used in
combination to
obtain optimal avoidance such as may be required for highly immunogenic
vectors such as
adenovirus.
In one embodiment, the invention provides methods of obtaining viral vectors
that are capable of escaping a host CTL immune response. This method can be
used in
conjunction with methods for obtaining genetic vaccine vectors that can escape
the humoral
response; the combination of approaches is often desirable, as different viral
serotypes often
have CTL epitopes in common, suggesting that virus variants which are not
recognized by
antibodies still are likely to be recognized by CTLs. This embodiment of the
invention
involves incorporating into genetic vaccines one or more components that
inhibit peptide
CA 02320626 2000-08-10
WO 99/41369 PCTNS99I03022
79
transport and/or MHC class I expression. An essential element in the
activation of cytotoxic
T lymphocyte (CTL) responses is an interaction between T cell receptors on
CTLs and
antigenic peptide-MHC class I molecule complexes on antigen presenting cells.
Expression
of MHC class I molecules on thymocytes and antigen presenting cells is a
requirement for
maturation and activation of antigen-specific CD8+ T lymphocytes. Thus, genes
that encode
inhibitors of MHC class I-mediated antigen presentation can be shuffled as
described herein
and placed into viral vectors to obtain vectors that, when present in target
cells, do not
induce destruction of the target cells by the cells of the immune system. This
can result in
prolonged survival of cells harboring genetic vaccine vectors, including those
that express a
pathogen antigen, as well as vectors that express a pharmaceutically useful
protein. In the
case of genetic vaccines, reduced expression of MHC class I molecules will
allow secretion
of the pathogen antigen, which then will be presented by professional antigen
presenting
cells elsewhere. In the case of vectors encoding pharmaceutical proteins,
reduced expression
of MHC class I molecules prevents recognition by the immune system prolonging
the
survival of the cells expressing the gene.
Among the proteins involved in MHC class I molecule expression and
antigen presentation are those encoded by TAP genes {transporters associated
with antigen
processing), which are described above. In one embodiment of the invention,
genes that
encode inhibitors of TAP activity are shul~led to obtain genes that encode
optimized TAP
inhibitors. The substrates for these methods can include, for example, one or
more of the
viral genes that are known to regulate levels of MHC class I molecule
expression. TAP 1 and
TAP2 gene expression is 5-10-fold and 100-fold reduced, respectively, in cells
transformed
by adenovirus 12, which results in reduced class I expression and thus leads
to reduced
virus-specific cytotoxic T lymphocyte responses. Similarly, TAP gene
expression is
downregulated in 49% of HPV-16+ cervical carcinomas (Seliger et al. (1997)
Immunol.
Today 18: 292). Thus, adenovirus and HPV viral nucleic acids provide examples
of suitable
substrates for carrying out the methods of the invention. Additional examples
of suitable
DNA shuffling substrates for this embodiment of the invention include the
human
cytomegalovirus (CMV) encoded genes US2, US3 and US11, which can downregulate
MHC
class I expression {Wiertz et al. (1996) Nature 384: 432 and Cell (1996) 84:
769; Ahn et al.
(1996) Proc. Nat'l. Acad. Sci. USA 93: 10990). Another human CMV gene that
encodes an
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
inhibitor of TAP-dependent peptide translocation is US6 (Lehner et al. (1997)
Proc. Nat'l.
Acad. Sci. USA 94: 6904-9). Cells transfected with US6 had reduced expression
of MHC
class I molecules on their surface and reduced capacity to activate cytotoxic
T lymphocytes.
Thus, in one embodiment, the invention involves DNA shuffling of this cluster
of genes
5 (approximately 7kb), or fragments thereof, in order to identify the
sequences that are most
potent in inhibiting the expression of MHC class I molecules. Such optimized
TAP inhibitor
polynucleotide sequences are useful not only for use in constructing vectors
that can escape
CTL immune responses, but also for generation of animal models for use with
human
viruses that normally are eliminated in laboratory animals due to their
immunogenicity. The
10 desired expression levels and functional properties of TAP inhibitors may
vary depending on
whether genetic vaccine vector, gene therapy vector or animal model is
evolved.
Alternative embodiments of the invention involve DNA shuffling of other
genes that are involved in downregulating expression of MHC class I molecules
and/or
antigen presentation. Examples of other possible target genes include genes
encoding
15 adenoviral E3 protein, herpes simplex ICP47 protein, and tapasin
antagonists (Seliger et al.
(1997) Immunol. Today 18:292-299; Galoncha et al. (1997) J. Exp. Med. 185:
1565-1572; Li
et al. (1997) Proc. Nat'1. Acad. Sci. USA 94: 8708-8713; Ortmanxi et al.
(1997) Science 277:
1306-1309.
Because reduced expression of MHC class I molecules on cell surfaces may
20 act as a stimulus for NK cells, it may be useful to include in genetic
vaccine vectors a gene
that encodes an MHC like molecule that inhibits NK cell function but is unable
to present
antigens to T lymphocytes. An example of such molecule is MHC class I
homologue
encoded by cytomegalovirus (Farrell et al. (1997) Nature 386: 510-S 14).
The invention also provides methods of obtaining viral vectors that exhibit an
25 enhanced capability of avoiding attack by CD4+ T lymphocytes. Such vectors
are
particularly useful in situations where the target cells are capable of
expressing MHC class II
molecules, such as in the case of vaccinations and gene therapy targeted to
the cells of the
immune system. Substrates for DNA shuffling include genes that encode
inhibitors of MHC
class II molecules such as, for example, IL-10 and antagonists of IFN-y (such
as soluble
30 IFN-y receptor).
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
81
Vectors that have the greatest capability of escaping the host immune system,
will typically include DNA sequences that result in inhibition of MHC class I
expression and
MHC class II expression, and additional sequences that encode homologs of MHC
class I
molecules. The properties of all these can be further improved by DNA
shuffling according
to the methods of the invention.
Once a library of shuffled DNA molecules is obtained, any of several
methods are available for screening the library to identify those
polynucleotides that, when
present in a viral vector (or in an animal model) exhibit the desired effect
on the host
immune response. For example, to obtain shuffled polynucleotides that inhibit
MHC class I
expression and/or antigen presentation, a library of shuffled genes can be
incorporated into
genetic vaccine or gene therapy vectors and transfected into human cell lines,
such as, for
example, HeLa, U937 or Jijoye, in a single tube transfection. Primary human
monocytes, or
dendritic cells generated by culturing human cord blood cells or monocytes in
the presence
of IL-4 and GM-CSF, are also suitable. Initial screening can be done using
FACS-sorting.
Cells expressing the lowest levels of MHC class I molecules are selected, the
polynucleotides that encode the MHC inhibitors, or whole plasmids containing
the
sequences, are recovered. If desired, the selected sequences can be subjected
to new rounds
of shuffling and selection. Cells expressing the lowest levels of MHC class I
molecules are
expected to have the lowest capacity to induce CTL responses.
Another screening method involves incorporating libraries of shuffled
polynucleotides that encode inhibitors of MHC class I expression are
incorporated into
human papillomavirus (HPV) vectors. This library is injected into the skin of
mice.
Normally, marine cells expressing HPV are destroyed by the host immune system.
However, cells expressing potent inhibitors of peptide transportation andlor
MHC class
expression will be able to escape the immune response. The cells that express
a marker gene
present on the vector, such as GFP, for extended periods of time are selected,
the sequences
or whole plasmids are recovered, and, if further optimization is desired, the
selected
sequences are subjected to new rounds of shuffling and selection. Long-lasting
maintenance
of HPV in mice will allow drug screening and vaccine studies, which to date
have not been
possible due to high immunogenicity of HPV in mice.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
82
In another embodiment, the libraries of shuffled polynucleotides encoding
inhibitors of MHC class I expression are incorporated into human adenovirus
vectors. This
library is transfected into human cell lines, such as HeLa cells, and cells
expressing the
lowest levels of MHC class I molecules are selected as described above. The
sequences that
provide the lowest levels of MHC class I expression are further tested by
analyzing the
capacity of antigen-presenting cells transfected with adenovirus harboring
evolved inhibitors
of MHC class I expression to activate specific T cell lines or clones. These
inhibitors will
block efficient presentation of immunogenic peptides, and hence, will strongly
downregulate
activation of antigen-specific CTLs allowing long-lasting transgene expression
in vivo.
Methods to screen for improved inhibitors of MHC class II expression
include detection of MHC class II molecules on the surface of the target cells
by fluorescent
labeled specific monoclonal antibodies, fluorescence microscopy, and flow
cytometry. In
addition, the inhibitors can be analyzed in functional assays by studying the
capacity of the
inhibitors to block activation of MHC class II restricted antigen-specific
CD4+ T
lymphocytes. For example, one can determine the capacity of the inhibitor to
inhibit
induction of CD4+ T cell proliferation induced by autologous antigen
presenting cells, such
as monocytes, dendritic cells, B cells or EBV-transformed B cell lines, that
harbor genes
encoding the MHC class II inhibitor or have been treated with supernatant
containing the
inhibitor.
3. Fnh~nced Antiviral Activity
The invention also provides methods of obtaining a recombinant viral vector
which has an enhanced ability to induce an antiviral response in a cell. DNA
shuffling is
used to produce a library of recombinant viral vectors. The library is
transfected into a
population of mammalian cells, which are then tested for ability to induce an
antiviral
response. One suitable test involves staining the cells for the presence of Mx
protein, which
is produced by cells that are exhibiting an antiviral response (see, e.g.,
Hallimen et al. (1997)
Pediatric Research 41: 647-650; Melen et al. (1994) J. Biol. Chem. 269: 2009-
2015).
Recombinant viral vectors can be isolated from cells which stain positive for
Mx protein.
These recombinant viral vectors from positive staining cells are enriched for
those that
exhibit enhanced ability to induce an antiviral response. Viral vectors for
which this method
is useful include, for example, influenza virus.
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
83
4. Evolution of vectors having increased copv number in production cells
The invention provides methods for obtaining vector components that, when
present in a genetic vaccine vector (such as a plasmid) the ability to
replicate to a high copy
number in a cell used to produce the vector. Plasmids can incorporate various
heterologous
DNA sequences, however the size or the nature of the cloned sequences in a
given plasmid
vector may render that vector less able to grow to high copy number in the
bacteria in which
it is propagated. It is therefore desirable to have a method to increase the
plasmid copy
number after all elements have been cloned into the vector. This is especially
important
when the plasmid is to be manufactured on a large scale as will be the case
for genetic
vaccines.
The methods of the invention involve incorporating into the plasmid one or
more polynucleotide sequences that bind proteins which would otherwise be
toxic to the
bacterium. One suitable toxic moiety and binding site combination is the
transcription factor
GATA-l and its recognition site. It has been shown that expression of a DNA-
binding
fragment of GATA-1 is toxic to bacteria; this toxicity apparently results from
inhibition of
bacterial DNA replication. Trudel et al. ((1996) Biotechniques 20: 684-693)
have described a
plasmid (pGATA) that expresses the Z2B2 region of GATA-1 as a GST fusion
protein. The
expression of the fusion protein in this plasmid is under the control of the
IPTG-inducible
lac promoter. The GST-GATA-1 fragment also binds strongly to a sequence from
the
mouse ~i-globin gene promoter as well as to the C-oligonucleotide from the (3-
globin gene 3'
enhancer; either or both of these are suitable for use as binding sites in the
methods of the
invention.
The plasmids preferably also include a selectable marker such as, for
example, kanamycin resistance (aminoglycoside 3'-phosphotransferase (EC
2.7.1.95)) and
the like. The plasmid backbone polynucleotide sequence is subjected to DNA
shuffling as
described herein to generate a library of plasmids which have different
backbone sequences
and possibly different supercoil densities. In order to introduce sufficient
sequence diversity
to search for improved function, it is preferable to perform family DNA
shuffling. This can
be accomplished in the context of the present invention by including in the
shuffling reaction
only a single form of the selectable marker. In this way, significant
diversity can be achieved
in the shuffled library to recover a plasmid which is improved in its growth
properties while
fully retaining the appropriate selection function of the plasmid.
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
84
The selection for high copy number plasmids is performed by introducing the
library of shuffled recombinant plasmids into the desired host cell. The host
cells also
express the toxic moiety, preferably under the control of a promoter which is
inducible. For
example, the pGATA plasmid is suitable for use in E. coli host cells. The
shuffled plasmids
are introduced into the cells under non-inducing conditions. Transformed cells
are then
placed under conditions which induce expression of the toxic moiety. For
example, E. coli
cells that contain pGATA can be placed on media containing increasing
concentrations of
IPTG. Those target plasmids which grow to high copy number in the bacteria
will express
correspondingly higher numbers of the binding sequences for GATA-1. The target
plasmids
will bind the GST-GATA-1 fusion protein and thus neutralize the toxic effects
on the
bacteria.
Plasmids with the highest copy number are detected as those which confer the
best growth to bacteria on the inducer-containing gmwth media. Such plasmids
can be
recovered and transformed into bacteria which lack the gene that encodes the
toxic moiety;
these plasmids should retain their high copy number characteristics. Further
rounds of
shuffling can be used to isolate high copy number plasmids by the above
selection
procedure. Alternatively, manual screening can be done in the bacterial host
of choice,
lacking the toxic moiety-encoding plasmid, to avoid any effects due to the
presence of this
extraneous plasmid. .
VI. Genetic Vaccine Pharmaceutical Composi 'ons and lVlethods of
Administration
The vector components and multicomponent genetic vaccines of the invention
are useful for treating and/or preventing various diseases and other
conditions. For example,
genetic vaccines that employ the reagents obtained according to the methods of
the invention
are useful in both prophylaxis and therapy of infectious diseases, including
those caused by
any bacterial, fungal, viral, or other pathogens of mammals. The reagents
obtained using the
invention can also be used for treatment of autoimmune diseases including, for
example,
rheumatoid arthritis, SLE, diabetes mellitus, myasthenia gravis, reactive
arthritis, ankylosing
spondylitis, and multiple sclerosis. These and other inflammatory conditions,
including IBD,
psoriasis, pancreatitis, and various immunodeficiencies, can be treated using
genetic
vaccines that include vectors and other components obtained using the methods
of the
invention. Genetic vaccine vectors and other reagents obtained using the
methods of the
CA 02320626 2000-08-10
WO 99!41369 PCTNS99/03022
invention can be used to treat allergies and asthma. Moreover, the use of
genetic vaccines
have great promise for the treatment of cancer and prevention of metastasis.
By inducing an
immune response against cancerous cells, the body's immune system can be
enlisted to
reduce or eliminate cancer.
5 In presently preferred embodiments, the optimized genetic vaccine
components are used in conjunction with other optimized genetic vaccine
reagents. For
example, an antigen that is useful for a particular condition can be optimized
by methods
analogous to the recombination and screening methods described herein (see,
copending,
commonly assigned US Patent Application Ser. No. - entitled "Antigen Library
10 Immunization", which was filed on February 10, 1999 as TTC Attorney Docket
No. 18097-
028710US). The polynucleotide that encodes the recombinant antigenic
polypeptide can be
placed under the control of a promoter, e.g., a high activity or tissue-
specific promoter. The
promoter used to express the antigenic polypeptide can itself be optimized
using
recombination and selection methods analogous to those described herein. The
vector can
15 contain immunostimulatory sequences such as are described in copending,
commonly
assigned US Patent Application Serial No. , entitled "Optimization of
Immunomodulatory Molecules," filed as TTC Attorney Docket No. 18097-030300US
on
February 10, 1999. It is sometimes advantageous to employ a genetic vaccine
that is targeted
for a particular target cell type (e.g., an antigen presenting cell or an
antigen processing cell);
20 suitable targeting methods are described in copending, commonly assigned US
patent
application Serial No. , entitled "Targeting of Genetic Vaccine Vectors,"
filed on
February 10, 1999 as TTC Attorney Docket No. 18097-030200US.
Genetic vaccines, including the multicomponent genetic vaccines described
herein, can be delivered to a mammal (including humans) to induce a
therapeutic or
25 prophylactic immune response. Vaccine delivery vehicles can be delivered in
vivo by
administration to an individual patient, typically by systemic administration
(e.g.,
intravenous, intraperitoneal, intramuscular, subdermal, intracranial, anal,
vaginal, oral,
buccal route or they can be inhaled) or they can be administered by topical
application.
Alternatively, vectors can be delivered to cells ex vivo, such as cells
explanted from an
30 individual patient (e.g., lymphocytes, bone marrow aspirates, tissue
biopsy) or universal
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
86
donor hematopoietic stem cells, followed by reimplantation of the cells into a
patient,
usually after selection for cells which have incorporated the vector.
A large number of delivery methods are well known to those of skill in the
art. Such methods include, for example liposome-based gene delivery (Debs and
Zhu (1993)
WO 93/24640; Mannino and Gould-Fogerite (1988) BioTechniques 6(7): 682-691;
Rose
U.S. Pat No. 5,279,833; Brigham (1991) WO 91/06309; and Felgner et al. {1987)
Proc. Natl.
Acad. Sci. USA 84: 7413-7414), as well as use of viral vectors (e.g.,
adenoviral (see, e.g.,
Berns et al. (1995) Ann. NYAcad Sci. 772: 95-104; Ali et al. (1994) Gene Ther.
1: 367-384;
and Haddada et al. (1995) Curr. Top. Microbiol. Immunol. 199 ( Pt 3): 297-306
for review),
papillomaviral, retroviral (see, e.g., Buchscher et al. (1992) J. Virol. 66(5)
2731-2739;
Johann et al. (1992) J. Virol. 66 (5):1635-1640 (1992); Sommerfelt et al.,
(1990) Virol.
176:58-59; Wilson et al. (1989) J. Yirol. 63:2374-2378; Miller et al., J.
Virol. 65:2220-2224
(1991); Wong-Staal et al., PCT/LTS94105700, and Rosenburg and Fauci (1993) in
Fundamental Immunology, Third Edition Paul (ed) Raven Press, Ltd., New York
and the
references therein, and Yu et al., Gene Therapy (1994) supra.), and adeno-
associated viral
vectors (see, West et al. (1987) Virology 160:38-47; Carter et al. (1989) U.S.
Patent No.
4,797,368; Carter et al. WO 93/24641 (1993); Kotin (1994) Human Gene Therapy
5:793-
801; Muzyczka (1994) J. Clin. Invst. 94:1351 and Samulski (supra) for an
overview of AAV
vectors; see also, Lebkowski, U.S. Pat. No. 5,173,414; Tratschin et al. (1985)
Mol. Cell.
Biol. 5(11):3251-3260; Tratschin, et al. {1984) Mol. Cell. Biol., 4:2072-2081;
Hermonat and
Muzyczka (1984) Proc. Natl. Acad. Sci. USA, 81:6466-6470; McLaughlin et al.
{1988) and
Samulski et al. (1989) J. Virol., 63:03822-3828), and the like.
"Naked" DNA and/or RNA that comprises a genetic vaccine can be
introduced directly into a tissue, such as muscle. See, e.g., USPN 5,580,859.
Other methods
such as "biolistic" or particle-mediated transformation (see, e.g., Sanford et
al., USPN
4,945,050; USPN 5,036,006) are also suitable for introduction of genetic
vaccines into cells
of a mammal according to the invention. These methods are useful not only for
in vivo
introduction of DNA into a mammal, but also for ex vivo modification of cells
for
reintroduction into a mammal. As for other methods of delivering genetic
vaccines, if
necessary, vaccine administration is repeated in order to maintain the desired
level of
immunomodulation.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99103022
87
Genetic vaccine vectors (e.g., adenoviruses, liposomes, papillomaviruses,
retroviruses, etc. ) can be administered directly to the mammal for
transduction of cells in
vivo. The genetic vaccines obtained using the methods of the invention can be
formulated as
pharmaceutical compositions for administration in any suitable manner,
including parenteral
(e.g., subcutaneous, intramuscular, intradermal, or intravenous), topical,
oral, rectal,
intrathecal, buccal (e.g., sublingual), or local administration, such as by
aerosol or
transdermally, for prophylactic and/or therapeutic treatment. Pretreatment of
skin, for
example, by use of hair-removing agents, may be useful in transdermal
delivery. Suitable
methods of administering such packaged nucleic acids are available and well
known to those
of skill in the art, and, although more than one route can be used to
administer a particular
composition, a particular route can often provide a more immediate and more
effective
reaction than another route.
Pharmaceutically acceptable carriers are determined in part by the particular
composition being administered, as well as by the particular method used to
administer the
composition. Accordingly, there is a wide variety of suitable formulations of
pharmaceutical
compositions of the present invention. A variety of aqueous carriers can be
used, e.g.,
buffered saline and the like. These solutions are sterile and generally free
of undesirable
matter. These compositions may be sterilized by conventional, well known
sterilization
techniques. The compositions may contain pharmaceutically acceptable auxiliary
substances
as required to approximate physiological conditions such as pH adjusting and
bui~ering
agents, toxicity adjusting agents and the like, for example, sodium acetate,
sodium chloride,
potassium chloride, calcium chloride, sodium lactate and the like. The
concentration of
genetic vaccine vector in these formulations can vary widely, and will be
selected primarily
based on fluid volumes, viscosities, body weight and the like in accordance
with the
particular mode of administration selected and the patient's needs.
Formulations suitable for oral administration can consist of (a) liquid
solutions, such as an effective amount of the packaged nucleic acid suspended
in diluents,
such as water, saline or PEG 400; (b) capsules, sachets or tablets, each
containing a
predetermined amount of the active ingredient, as liquids, solids, granules or
gelatin; (c)
suspensions in an appropriate liquid; and (d) suitable emulsions. Tablet forms
can include
one or more of lactose, sucrose, mannitol, sorbitol, calcium phosphates, corn
starch, potato
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
88
starch, tragacanth, microcrystalline cellulose, acacia, gelatin, colloidal
silicon dioxide,
croscarmellose sodium, talc, magnesium stearate, stearic acid, and other
excipients,
colorants, fillers, binders, diluents, buffering agents, moistening agents,
preservatives,
flavoring agents, dyes, disintegrating agents, and pharmaceutically compatible
carriers.
Lozenge forms can comprise the active ingredient in a flavor, usually sucrose
and acacia or
tragacanth, as well as pastilles comprising the active ingredient in an inert
base, such as
gelatin and glycerin or sucrose and acacia emulsions, gels, and the like
containing, in
addition to the active ingredient, carriers known in the art. It is recognized
that the genetic
vaccines, when administered orally, must be protected from digestion. This is
typically
accomplished either by complexing the vaccine vector with a composition to
render it
resistant to acidic and enzymatic hydrolysis or by packaging the vector in an
appropriately
resistant carrier such as a liposome. Means of protecting vectors from
digestion are well
known in the art. The pharmaceutical compositions can be encapsulated, e.g.,
in liposomes,
or in a formulation that provides for slow release of the active ingredient.
The packaged nucleic acids, alone or in combination with other suitable
components, can be made into aerosol formulations (e.g., they can be
"nebulized") to be
administered via inhalation. Aerosol formulations can be placed into
pressurized acceptable
propellants, such as dichlorodifluoromethane, propane, nitrogen, and the like.
Suitable formulations for rectal administration include, for example,
suppositories, which consist of the packaged nucleic acid with a suppository
base. Suitable
suppository bases include natural or synthetic triglycerides or paraffin
hydrocarbons. In
addition, it is also possible to use gelatin rectal capsules which consist of
a combination of
the packaged nucleic acid with a base, including, for example, liquid
triglycerides,
polyethylene glycols, and paraffin hydrocarbons.
Formulations suitable for parenteral administration, such as, for example, by
intraarticular (in the joints), intravenous, intramuscular, intradermal,
intraperitoneal, and
subcutaneous routes, include aqueous and non-aqueous, isotonic sterile
injection solutions,
which can contain antioxidants, buffers, bacteriostats, and solutes that
render the formulation
isotonic with the blood of the intended recipient, and aqueous and non-aqueous
sterile
suspensions that can include suspending agents, solubilizers, thickening
agents, stabilizers,
and preservatives. In the practice of this invention, compositions can be
administered, for
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
89
example, by intravenous infusion, orally, topically, intraperitoneally,
intravesically or
intrathecally. Parenteral administration and intravenous administration are
the preferred
methods of administration. The formulations of packaged nucleic acid can be
presented in
unit-dose or mufti-dose sealed containers, such as ampoules and vials.
Injection solutions and suspensions can be prepared from sterile powders,
granules, and tablets of the kind previously described. Cells transduced by
the packaged
nucleic acid can also be administered intravenously or parenterally.
The dose administered to a patient, in the context of the present iiwention
should be sufllcient to effect a beneficial therapeutic response in the
patient over time. The
dose will be determined by the efficacy of the particular vector employed and
the condition
of the patient, as well as the body weight or vascular surface area of the
patient to be treated.
The size of the dose also will be determined by the existence, nature, and
extent of any
adverse side-effects that accompany the administration of a particular vector,
or transduced
cell type in a particular patient.
In determining the effective amount of the vector to be administered in the
treatment or prophylaxis of an infection or other condition, the physician
evaluates vector
toxicities, progression of the disease, and the production of anti-vector
antibodies, if any. In
general, the dose equivalent of a naked nucleic acid from a vector is from
about 1 ~.g to 1 mg
for a typical 70 kilogram patient, and doses of vectors used to deliver the
nucleic acid are
calculated to yield an equivalent amount of therapeutic nucleic acid.
Administration can be
accomplished via single or divided doses.
In therapeutic applications, compositions are administered to a patient
suffering from a disease (e.g., an infectious disease or autoimmune disorder)
in an amount
sufficient to cure or at least partially arrest the disease and its
complications. An amount
adequate to accomplish this is defined as a "therapeutically effective dose."
Amounts
effective for this use will depend upon the severity of the disease and the
general state of the
patient's health. Single or multiple administrations of the compositions may
be administered
depending on the dosage and frequency as required and tolerated by the
patient. In any
event, the composition should provide a sufficient quantity of the proteins of
this invention
to effectively treat the patient.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
In prophylactic applications, compositions are administered to a human or
other mammal to induce an immune response that can help protect against the
establishment
of an infectious disease or other condition.
The toxicity and therapeutic efficacy of the genetic vaccine vectors provided
5 by the invention are determined using standard pharmaceutical procedures in
cell cultures or
experimental animals. One can determine the LDso (the dose lethal to 50% of
the
population) and the EDso (the dose therapeutically effective in 50% of the
population) using
procedures presented herein and those otherwise known to those of skill in the
art.
A typical pharmaceutical composition for intravenous administration would
10 be about 0.1 to 10 mg per patient per day. Dosages from 0.1 up to about 100
mg per patient
per day may be used, particularly when the drug is administered to a secluded
site and not
into the blood stream, such as into a body cavity or into a lumen of an organ.
Substantially
higher dosages are possible in topical administration. Actual methods for
preparing
parenterally administrable compositions will be known or apparent to those
skilled in the art
15 and are described in more detail in such publications as Remington's
Pharmaceutical
Science, 15th ed., Mack Publishing Company, Easton, Pennsylvania (1980).
The genetic vaccines obtained using the methods of the invention can be
packaged in packs, dispenser devices, and kits for administering genetic
vaccines to a
mammal. For example, packs or dispenser devices that contain one or more unit
dosage
20 forms are provided. Typically, instructions for administration of the
compounds will be
provided with the packaging, along with a suitable indication on the label
that the compound
is suitable for treatment of an indicated condition. For example, the label
may state that the
active compound within the packaging is useful for treating a particular
infectious disease,
autoimmune disorder; tumor, or for preventing or treating other diseases or
conditions that
25 are mediated by, or potentially susceptible to, a mammalian immune
response.
1T°~ of Genetic Vaccines
Genetic vaccines which include optimized vector modules and other reagents
provided by the invention are useful for treating many diseases and other
conditions that are
either mediated by a mammalian immune system or are susceptible to treatment
by an
30 appropriate immune response. Representative examples of these diseases are
listed below;
antigens appropriate for each are described in copending, commonly assigned US
patent
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
91
application Ser. No. , filed February 10, 1999 as TTC Attorney
Docket No. 18097-028710US.
A. Infectious Diseases
Genetic vaccine vectors obtained according to the methods of the invention
are useful in both prophylaxis and therapy of infectious diseases, including
those caused by
any bacterial, fungal, viral, or other pathogens of mammals. In some
embodiments,
protection is conferred by use of a genetic vaccine vector that will express
an antigen (either
or both of a humoral antigen or a T cell antigen) of the pathogen of interest.
In preferred
embodiments, the antigen is evolved using the methods of the invention in
order to obtain
optimized antigens as described herein. The vector induces an immune response
against the
antigen. One or several antigens or antigen fragments can be included in one
genetic vaccine
delivery vehicle. Examples of pathogens and corresponding polypeptides from
which an
antigen can be obtained include, but are not limited to, HIV (gp120, gp160),
hepatitis B, C,
D, E (surface antigen), rabies (glycoprotein), Schistosoma mansoni (calpain;
Jankovic (1996)
J. Immunol. 157: 806-14). Other pathogen infections that are treatable using
genetic vaccine
vectors include, for example, herpes zoster, herpes simplex -1 and -2,
tuberculosis (including
chronic, drug-resistant), lyme disease (Borrelia burgorferii~, syphilis,
parvovirus, rabies,
human papillomavirus, and the like.
B. Inflammatory and Autoimmune Diseases
Autoimmune diseases are characterized by immune response that attacks
tissues or cells of ones own body, or pathogen-specific immune responses that
also are
harmful for ones own tissues or cells, or non-specific immune activation which
is harmful
for ones own tissues or cells. Examples of autoimmune diseases include, but
are not limited
to, rheumatoid arthritis, SLE, diabetes mellitus, myasthenia gravis, reactive
arthritis,
ankylosing spondylitis, and multiple sclerosis. These and other inflammatory
conditions,
including IBD, psoriasis, pancreatitis, and various immunodeficiencies, can be
treated using
genetic vaccines that include vectors and other components obtained using the
methods of
the invention.
These conditions are often characterized by an accumulation of inflammatory
cells, such as lymphocytes, macrophages, and neutrophils, at the sites of
inflammation.
CA 02320626 2000-08-10
WO 99/41369 PG"T/US99I03022
92
Altered cytokine production levels are often observed, with increased levels
of cytokine
production. Several autoimmune diseases, including diabetes and rheumatoid
arthritis, are
linked to certain MHC haplotypes. Other autoimmune-type disorders, such as
reactive
arthritis, have been shown to be triggered by bacteria such as Yersinia and
Shigella, and
evidence suggests that several other autoimmune diseases, such as diabetes,
multiple
sclerosis, rheumatoid arthritis, may also be initiated by viral or bacterial
infections in
genetically susceptible individuals. '
Current strategies of treatment generally include anti-inflammatory drugs,
such as NSAID or cyclosporin, and antiproliferative drugs, such as
methotrexate. These
therapies are non-specific, so a need exists for therapies having greater
specificity, and for
means to direct the immune responses towards the direction that inhibits the
autoimmune
process.
The present invention provides several strategies by which these needs can be
fulfilled. First, the invention provides methods of obtaining vaccines which
exhibit
improved delivery of tolerogenic antigens, antigens which have improved
antigenicity,
genetic vaccine-mediated tolerance, and modulation of the immune response by
inclusion of
appropriate accessory molecules. In a preferred embodiment, the vaccines
prepared
according to the invention exhibit improved induction of tolerance by oral
delivery. Oral
tolerance is characterized by induction of immunological tolerance after oral
administration
of large quantities of antigen (Chen et al. (1995) Science 265: 1237-1240; Haq
et al. (1995)
Science 268: 714-716). In animat models, this approach has proven to be a very
promising
approach to treat autoimmune diseases, and clinical trials are in progress to
address the
efficacy of this approach in the treatment of human autoimmune diseases, such
as
rheumatoid arthritis and multiple sclerosis (Chen et al. (1994) Science
265:1237-40;
Whitacre et al. (1996) Clin. Immunol. Immunopathol. 80: S31-9; Hohol et al.
(1996) Ann. N.
Y. Acad. Sci. 778:243-50). It has also been suggested that induction of oral
tolerance against
viruses used in gene therapy might reduce the immunogenicity of gene therapy
vectors.
However, the amounts of antigen required for induction of oral tolerance are
very high and
improved methods for oral delivery of antigenic proteins would significantly
improve the
efficacy of induction of oral tolerance.
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
93
Expression library immunization (Barry et al. (1995) Nature 377: 632) is a
particularly useful method of screening for optimal antigens for use in
genetic vaccines. For
example, to identify autoantigens present in Yersinia, Shigella, and the like,
one can screen
for induction of T cell responses in HLA-B27 positive individuals. Complexes
that include
epitopes of bacterial antigens and MHC molecules associated with autoimmune
diseases,
e.g., HLA-B27 in association with Yersinia antigens can be used in
the.prevention of
reactive arthritis and ankylosing spondylitis in HL,A-B27 positive
individuals.
Treatment of autoimmune and inflammatory conditions can involve not only
administration of tolerogenic antigens, but also the use of a combination of
cytokines,
costimulatory molecules, and the like. Such cocktails are formulated for
induction of a
favorable immune response, typically induction of autoantigen-specific
tolerance. Cocktails
can also include, for example, CD1, which is crucially involved in recognition
of self
antigens by a subset of T cells (Porcelli (1995) Adv. Immunol. 59: 1). Generic
vaccine
vectors and cocktails that skew immune responses towards the TH2 are often
used in treating
autoimmune and inflammatory conditions, both with antigen-specific and antigen
non-
specific vectors.
Screening of genetic vaccines and accessory molecules can be done in animal
models which are known to those of skill in the art. Examples of suitable
models for various
conditions include collagen induced arthritis, the NFS/sld mouse model of
human Sjogren's
syndrome; a 120 kD organ-specific autoantigen recently identified as an analog
of human
cytoskeletal pmtein a-fodrin (Haneji et al. (1997) Science 276: 604), the New
Zealand
Black/White F1 hybrid mouse model of human SLE, NOD mice, a mouse model of
human
diabetes mellitus, fas/fas ligand mutant mice, which spontaneously develop
autoimmune and
lymphoproliferative disorders (Watanabe-Fukunaga et al. (1992) Nature 356:
314), and
experimental autoimmune encephalomyelitis (EAE), in which myelin basic protein
induces a
disease that resembles human multiple sclerosis.
Autoantigens that are useful in genetic vaccines for treating multiple
sclerosis
include, but are not limited to, myelin basic protein (Stinissen et al. (1996)
J. Neurosci. Res.
45: 500-511) or a fusion protein of myelin basic protein and proteolipid
protein in multiple
sclerosis (Elliott et al. (1996) J. Clin. Invest. 98: 1602-1612), proteolipid
protein (PLP)
(Rosener et al. (1997) J. Neuroimmunol. 75: 28-34), 2',3'-cyclic nucleotide 3'-
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
94
phosphodiesterase (CNPase) (Rosener et al. (1997) J. Neuroimmunol. 75: 28-34),
the
Epstein Barr virus nuclear antigen-1 (EBNA-1) in multiple sclerosis (Vaughan
et al. (1996)
J. Neuroimmunol. 69: 95-102), HSP70 in multiple sclerosis (Salvetti et al.
(1996) J.
Neuroimmunol. 65: 143-53; Feldmann et al. (1996) Cell 85: 307).
C. Allergy and Asthma
Genetic vaccine vectors and other reagents obtained using the methods of the
invention can be used to treat allergies and asthma. Allergic immune responses
are results of
complex interactions between B cells, T cells, professional antigen-presenting
cells (APC),
eosinophils and mast cells. These cells take part in allergic immune responses
both as
modulators of the immune responses and are also involved in producing factors
directly
involved in initiation and maintenance of allergic responses.
Synthesis of polyclonal and allergen-specific IgE requires multiple
interactions between B cells, T cells and professional antigen-presenting
cells (APC).
Activation of naive, unprimed B cells is initiated when specific B cells
recognize the
allergen by cell surface immunoglobulin (sIg). However, costimulatory
molecules expressed
by activated T cells in both soluble and membrane-bound forms are necessary
for
differentiation of B cells into IgE-secreting plasma cells. Activation of T
helper cells
requires recognition of an antigenic peptide in the context of MHC class II
molecules on the
plasma membrane of APC, such as monocytes, dendritic cells, Langerhans cells
or primed B
cells. Professional APC can efficiently capture the antigen and the peptide-
MHC class II
complexes are formed in a post-Golgi, proteolytic intracellular compartment
and
subsequently exported to the plasma membrane, where they are recognized by T
cell
receptor (TCR) (Monaco (1995) J. Leuk. Biol. 57: 543-547). In addition,
activated B cells
express CD80 (B7-1) and CD86 (B7-2, 870), which are the counter receptors for
CD28 and
which provide a costimulatory signal for T cell activation resulting in T cell
proliferation and
cytokine synthesis (Bluestone (1995) Immunity 2: 555-559). Since allergen-
specific T cells
from atopic individuals generally belong to the TH2 cell subset, activation of
these cells also
leads to production of IL-4 and IL-13, which, together with membrane-bound
costimulatory
molecules expressed by activated T helper cells, direct B cell differentiation
into IgE-
secreting plasma cells (de Vries and Punnonen, In CytoJcine Regulation of
Humoral
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
Immunity: Basic and Clinical Aspects, Ed. CM Snapper, John Wiley 8c Sons Ltd,
West
Sussex, UK, p. 195-215, 1996).
Mast cells and eosinophils are key cells in inducing allergic symptoms in
target organs. Recognition of specific antigen by IgE bound to high-affinity
IgE receptors on
5 mast cells, basophils or eosinophils results in crosslinking of the
receptors leading to
degranulation of the cells and rapid release of mediator molecules, such as
histamine,
prostaglandins and leukotrienes, causing allergic symptoms.
Immunotherapy of allergic diseases currently includes hyposensibilization
treatments using increasing doses of allergen injected to the patient. These
treatments result
10 skewing of immune responses towards THl phenotype and increase the ratio of
IgG/IgE
antibodies specific for allergens. Because these patients have circulating IgE
antibodies
specific for the allergens, these treatments include significant risk of
anaphylactic reactions.
In these reactions, flee circulating allergen is recognized by IgE molecules
bound to high-
aff nity IgE receptors on mast cells and eosinophils. Recognition of the
allergen results in
15 crosslinking of the receptors leading to release of mediators, such as
histamine,
prostaglandins, and leukotrienes, which cause the allergic symptoms, and
occasionally
anaphylactic reactions. Other problems associated with hyposensibilization
include low
efficacy and difficulties in producing allergen extracts reproducibly.
Genetic vaccines provide a means of circumventing the problems that have
20 limited the usefulness of previously known hyposensibilization treatments.
For example, by
expressing antigens on the surface of cells, such as muscle cells, the risk of
anaphylactic
reactions is significantly reduced. This can be achieved by using genetic
vaccine vectors
that encode transmembrane forms of allergens. The allergens can also be
modified in such a
way that they are efficiently expressed in transmembrane forms, further
reducing the risk of
25 anaphylactic reactions. Another advantage provided by the use of genetic
vaccines for
hyposensibilization is that the genetic vaccines can include cytokines and
accessory
molecules which further direct the immune responses towards the TH1 phenotype,
thus
reducing the amount of IgE antibodies produced and increasing the efficacy of
the
treatments. Vectors can also be evolved to induce primarily IgG and IgM
responses, with
30 little or no IgE response. Furthermore, DNA shuffling can be used to
generate allergens that
are not recognized by the specific IgE antibodies preexisting in vivo, yet are
capable of
CA 02320626 2000-08-10
WO 99/41369 PC'T/US99/03022
96
inducing efficient activation of allergen-specific T cells. For example, using
phage display
selection, one can express shuffled allergens on phage, and only those that
are not
recognized by specific IgE antibodies are selected. These are fiuther screened
for their
capacity to induce activation of specific T cells. An efficient T cell
response is an indication
that the T cell epitopes are functionally intact, although the B cell epitopes
were altered, as
indicated by lack of binding of specific antibodies.
In these methods, polynucleotides encoding known allergens, or homologs or
fragments thereof (e.g., immunogenic peptides) are inserted into DNA vaccine
vectors and
used to immunize allergic and asthmatic individuals. DNA shuffling can be used
to obtain
antigens that activate T cells but cannot induce anaphylactic reactions.
Examples of allergies
that can be treated include, but are not limited to, allergies against house
dust mite, grass
pollen, birch pollen, ragweed pollen, hazel pollen, cockroach, rice, olive
tree pollen, fungi,
mustard, bee venom.
The invention also provides a solution to another shortcoming of genetic
vaccination as a treatment for allergy and asthma. While genetic vaccination
primarily
induces CD8+ T cell responses, induction of allergen-specific IgE responses is
dependent on
CD4+ T cells and their help to B cells. TH2-type cells are particularly
efficient in inducing
IgE synthesis because they secrete high levels of IL-4, IL-5 and IL-13, which
direct Ig
isotype switching to IgE synthesis. IL-5 also induces eosinophilia. The
methods of the
invention can be used to develop genetic vaccines that efficiently induce CD4+
T cell
responses, and direct differentiation of these cells towards the TH1
phenotype.
The invention also provides methods by which the level of antigen release by
a genetic vaccine vector is regulated. Regulation of the antigen dose is
crucial at the onset of
hyposensibilization for safety reasons. Low antigen levels are preferably used
at first, with
the antigen level increasing once evidence has been obtained that the antigen
does not induce
adverse effects in the individual. The DNA shui~ling methods of the invention
allow
generation of genetic vaccine vectors that induce expression of different
(high and low)
levels of antigen. For example, two or more different evolved promoters can be
used for
antigen expression. Alternatively, the antigen gene itself can be evolved for
different levels
of expression by, for example, altering codon usage. Vectors that induce
different levels of
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
97
antigen expression can be screened by use of specific monoclonal antibodies,
and cell
sorting (e.g, FACS).
D. Cancer
Immunotherapy has great promise for the treatment of cancer and prevention
of metastasis. By inducing an immune response against cancerous cells, the
body's immune
system can be enlisted to reduce or eliminate cancer. Genetic vaccines
prepared using the
methods of the invention, as well as accessory molecules described herein,
provide cancer
immunotherapies of increased effectiveness compared to those that are
presently available.
One approach to cancer immunotherapy is vaccination using genetic vaccines
that encode antigens that are specific for tumor cells. The methods of the
invention can be
used for enhancement of immune responses against known tumor-specific
antigens, and also
to search for novel protective antigenic sequences. Genetic vaccines that
exhibit optimized
antigen expression, processing, and presentation can be obtained as described
herein. The
methods of the invention are also suitable for obtaining optimized cytokines,
costimulatory
molecules, and other accessory molecules that are effective in induction of an
antitumor
immune response, as well as for obtaining genetic vaccines and cocktails that
include these
and other components present in optimal combinations. The approach used for
each
particular cancer can vary. For treatment of hormone-sensitive cancers (for
example, breast
cancer and prostate cancer), methods of the invention can be used to obtain
optimized
hormone antagonists. For highly immunogenic tumors, including melanoma, one
can screen
for genetic vaccine vectors that optimally boost the immune response against
the tumor.
Breast cancer, in contrast, is of relatively low immunogenicity and exhibits
slow
progression, so individual treatments can be designed for each patient.
Prevention of
metastasis is also a goal in design of genetic vaccines.
EXAMPLES
The following examples are offered to illustrate, but not to limit the present
invention.
CA 02320626 2000-08-10
WO 99141369 PCTNS99103022
98
Example 1
Animal Model for Screening Genetic Vaccine Vectors
This Example provides a mouse model system that is useful for screening and
testing genetic vaccine vectors in human skin in vivo. Pieces of human skin
are
xenotransplanted onto the back of SCID mice. Pieces of human skin can be
obtained from
infants undergoing circumcision, from skin removal operations due to, for
example,
cosmetic reasons, or from patients undergoing amputation due to, for example,
accidents.
These pieces are then transplanted onto the backs of C.B-17 scidlscid (SCID)
mice as
described by others (Deng et al. (1997) Nature Biotechnology 15: 1388-1391;
Khavari et al.
(1997) Adv. Clin. Res. 15:27-35; Choate and Khavari (1997) Human Gene Therapy
8:895-
901 ).
The vector libraries are selected, for example, after topical application to
the
skin. However, in an analogous manner, depending on the optimal route of
immunization,
the evolved vectors can also be selected after i.m., i.v., i.d., oral, anal or
vaginal delivery.
The DNA delivered onto the skin can be in the form of a patch, in a form of a
cream, in a
form of naked DNA or mixture of DNA and transfection-enhancing agent (such as
proteases,
lipases or lipids/liposomes), and it can be applied after mechanical abrasion,
after removal of
the hair, or simply by adding a droplet of DNA or DNA-lipid/liposome mixture
onto the
skin. Similar delivery methods apply to small animals, such as mice or rat,
large animals,
such as cat, dog, cow, horse or monkey, as well as humans.
Suitable proteases and lipases that enhance the delivery include, but are not
limited to, individuals or mixtures of the following: a protease (such as
Alcalase or Savinase)
with or without an alpha-amylase, a lipase (such as Lipolase) (Sarlo et al.
(1997) J. Allergy
Clin. Immunol. 100:480-7).
'The recovery of the optimal vectors can be done from the transfected cells
by,
for example, PCR, or by recovering entire vectors. One can either select
vectors based
purely on their capacity to enter the cells or by selecting only cells that
express the antigen
encoded by the vector in normal mice, monkeys or SCID mice transplanted with
human
skin. One can use, for example, GFP as a marker gene, and after delivery
detect cells that
are transfected by fluorescence microscopy or flow cytometry. The positive
cells can be
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
99
isolated for example by flow cytometry based cell sorting. This format allows
selection of
vectors that optimally express antigens in and transfect human cells in vivo.
Additionally, one can screen in mice by selecting vectors that are able to
induce effective immune responses after delivery onto the skin. One can select
vectors that
induce highest specific antibody or CTL responses, or one can select based on
induction of
protective immune response following challenge by the corresponding pathogen.
Example 2
Episomally-Replicating Nucleic Acid Vaccine Vector
This Example describes a procedure for obtaining stable, episomally
maintained genetic vaccine vectors by applying DNA shui~ling to human
papillomavirus
(HPV) genes. HPV can be maintained in human skin for extended periods (Bernard
and Apt
(1994) Arch. Dermatol. 130: 210). Despite these in vivo properties, it has not
been possible
to maintain HPV episomally in tissue culture due to underreplication. The
primary goal of
the procedure described in this Example is to improve the stability and copy
number of
vector constructs. Screening for natural HPV variants using traditional
approaches or
attempts to rationally design mutants with improved properties would require
many person-
years of research.
To obtain improved mutants in an efficient manner, family shuffling is
performed using the HPV E1 and E2 genes from different, but closely related,
benign HPVs.
Family shuffling allows one to generate and screen orders of magnitude more
diversity than
traditional mutagenesis approaches in much shorter time periods than are
required for the
traditional approaches. Libraries of HPV E1 and E2 genes are generated by
using family
shuffling of three closely related cutaneous HPV strains (HPV 2, 27, and 57).
Alternatively,
large libraries of vector sequences are generated by incorporation of random
DNA
sequences, for example derived from human or mouse genomic DNA, into genetic
vaccine
vectors. Green fluorescent protein (GFP) is used as a marker gene to detect
the most stable
vectors with superior expression levels. The best chimeric constructs from a
library of
millions of vectors are selected by flow cytometry-based cell sorting.
Episomal vectors are
then recovered providing an additional selection pressure towards
nonintegrating vectors.
CA 02320626 2000-08-10
WO 99/41369 PGT/US99/03022
100
Initial screening is performed in cell culture, where processing of large
libraries of shuffled material is feasible. Stable episomal vectors are also
likely to prove to
be very useful tools in other library screening applications. In contrast to
randomly
integrating and transient vectors, episomally maintained vectors result in
high signal-to-noise
ratios upon FACS selection, and they also significantly improve the
possibility to recover the
plasmids from a small number of selected cells. Alternatively or additionally,
the vectors are
screened and analyzed for durability in vivo in SCID mice transplanted with
live human skin
(see, Example 1 ).
To directly screen for optimal properties in human cells in vivo, the vector
libraries are screened in an animal model, in which SCID mice are transplanted
with human
skin. In this model, live human skin is xenotransplanted onto the back of SCID
mice
without any signs of rejection, providing a possibility to optimize and evolve
genetic vaccine
vector directly in human tissue in vivo. Recursive selection of episomal
vectors will provide
strong selection pressure for vectors that remain episomal, yet provide a high
level of gene
expression. Moreover, despite their immunodeficient phenotype, SCID mice have
normal
levels of monocytes and macrophages. Therefore, antigen presenting cells (APC)
derived
from these mice can be used to assess the level of antigens delivered to
professional antigen
presenting cells, and to study the capacity of these cells to present antigens
and induce
activation of antigen-specific CD4+ and CD8+ T cells in vitro.
Ezamele 3
Evolution Of The Major Immediate Earl~r~'romoter/Enhancer
R~ion Of Cytomegalovirus
The major immediate-early (IE) region promoter/enhancer of
cytomegalovirus (CMV) is widely used for regulating transcription of genes,
because it is
highly active in a broad range of cell types. An optimized CMV promoter
(generated by
DNA shuffling) which directs increased levels of gene expression, can improve
the efficacy
of genetic vaccines. The fact that the CMV promoter is active in human and
animal cells
means that it can be used to express foreign genes both in animal models and
in clinical
applications.
A library of chimeric promoter/enhancer sequences was created by DNA
shuffling of wild-type sequences from four related strains of CMV. The
promoter, enhancer
CA 02320626 2000-08-10
WO 99/41369 PCTNS99/03022
101
and first intron sequences of the IE region are obtained by PCR from the AD169
and Towne
human CMV strains, and from rhesus and vervet monkey CMVs (Figure 12). The
promoter/enhancer sequences of the human CMV strains are 95% identical, and
share
approximately 70% identity with the sequences of the monkey isolates, allowing
the use of
S family shuffling to generate a library with greater diversity than would be
achieved using the
conventional shuffling procedure. Alignments and the sequence similarities of
the
promoter/enhancer regions of these sequences are shown in Figure 10.
Alignments and
sequence similarities of the intron A sequences in the PCR products from the
human CMV
strains, Towne and AD169 are shown in Figure 11, and schematic diagrams of the
PCR
products obtained upon amplification of these promoters are shown in Figure
12.
The following primers can be used to amplify promoter sequences from
human and monkey CMVs:
Primers for promoters ~n human CMI~stra~xs Towne and AD169:
5'-primer: 5'-ATA TGA GGC TAT ATC GCC GAT A-3'
3' primer: 5'-AAG GAC GGT GAC TGC AGA AAA-3'
Primers for Rhesus Monkey CMV promoter:
5'-primer: 5"-AAT GGC GAC TTG GCA TTG AGC CAA TT-3"
3' primer: 5'-TAT CCG CGT TCC AAT GCA CCC TT-3'
Prinurs for Vervet Monkey CMV promoter:
5'-primer: 5'-ACT TGG CAC GGT GCC AAG TTT-3'
3' primer: 5'-TAT CCG CAT TCC AAT GCA CCG T-3'
Following shuffling, the library was cloned into a plasmid backbone and used
to direct transcription of a marker gene in mammalian cells. An internal
marker under the
control of a native promoter was included in the piasmid vector, enabling
analysis and
selection of cells expressing equal numbers of vectors. An example of a
suitable vector for
use in screening shuffled promoter sequences is shown in Figure 7.
The transfected cells were screened by flow cytometric cell sorting to
identify
those which express highest levels of the marker gene, normalized against the
internal
marker to account for differences in vector copy numbers per cell. Vectors
carrying optimal
promoter sequences are then recovered and subjected to further cycles of
shuffling and
CA 02320626 2000-08-10
WO 99/41369 PCT/US99/03022
102
selection. Results shown in Figure 13 demonstrate that recombination followed
by
fluorescence-activated cell sorting resulted in the promoter library being
enriched for
promoters having strong activity. Figure 14 shows the distribution of antigen
expression, as
measured by flow cytometry, of individually analyzed shuffled clones. Again,
the FACS-
sorted library enriched the population for high-activity promoters.
A_ vector that contains a shuffled CMV promoter (S171 operable linked to a
luciferase
gncodin~ gene was infected intramuscularly into a mouse f??l, and the amount
of
luciferase eauression was determined at various time Doints after infection.
Results are
shown in Figure 15
Eaamge 4
Shuffling Of Oligo-Directed Cps Knock-Outs
A common problem associated with genetic vaccine vectors is that the
expression induced by the vectors is often short-lasting due to downregulation
of promoter
activity. One reason for downregulation of promoter activities is methylation
(Robertson
and Ambinder ( 1997) 71:6445-54). CpG sequences are particularly prone to
methylation
and this example describes the use of DNA shuffling method to generate
promoter sequences
where all unnecessary CpG sequences have been deleted. The approach is
illustrated in
Figure 16.
It is understood that the examples and embodiments described herein are for
illustrative purposes only and that various modifications or changes in light
thereof will be
suggested to persons skilled in the art and are to be included within the
spirit and purview of
this application and scope of the appended claims. All publications, patents,
and patent
applications cited herein are hereby incorporated by reference for all
purposes.