Note: Descriptions are shown in the official language in which they were submitted.
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
HETERO-ASSOCIATING COILED-COIL PEPTIDES
The present invention relates to methods for the identification of novel
hetero-
associating coiled-coil peptides and uses of these peptides for hetero-
dimerization of
fusion proteins. It furthermore relates to vectors, host cells useful for the
production of
these novel hetero-association peptides and (poly)peptides/proteins comprising
these
peptides.
Increasingly, there is a need for proteins which combine two or more
functions, such as
binding to two different specificities, or binding and enzymatic activity, in
a single
structure. Typically, proteins which combine two or more functions are
prepared either
as fusion proteins or through chemical conjugation of the component functional
domains. Both of these approaches suffer from disadvantages. Genetic "single
chain"
fusions suffer the disadvantages that (i) only a few (two to three) proteins
can be fused
(1 ), (ii) mutual interference between the component domains may hinder
folding, and (iii)
the size of the fusion protein may make it difficult to prepare. The
alternative, chemical
cross-linking in vitro following purification of independently expressed
proteins, is difficult
to control and invariably leads to undefined products and to a severe loss in
yield of
functional material.
A third approach takes advantage of using genetic fusions of functional to
association
domains which lead to a self-association on co-expression in appropriate host
cells. To
assemble at least two different fragments fused to association domains, the
domains
must have a tendency to form hetero-multimers. In one approach, a natural
protein or
protein domain was dissected and fused to protein partners to achieve hetero-
association of the fusion proteins via the reconstitution of the native-like
structure of the
dissected protein or protein domain (WO 96/13583).
In principle, hetero-association can be achieved with complementary helices
such as the
hetero-dimerizing Jun and Fos zippers of the AP-1 transcription factor (2) or
other helical
coiled-coil structures which are involved in the oligomerization of a wide
variety of
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
2
proteins. Because of their small size and structural regularity, they have
also been used
as artificial domains to mediate oligomerization of various proteins (3, 4).
For example,
the association of two separately expressed scFv antibody fragments by C-
terminally
fused amphipathic helices in vivo provides homo-dimers of antibody fragments
in E. coil
(W093/15210; 5, 6). Coiled coils consist of two or more amphipathic helices
wrapping
around each other with a slight supercoil. They contain a characteristic
heptad repeat
(abcdefg)~ with a distinct pattern of hydrophobic and hydrophilic residues
(Fig. 1 A, (7,
8)). The positions a and d, which form the hydrophobic interface between the
helices,
are usually aliphatic and a have profound effect on the oligomerization state
(9, 10). The
positions b, c, e, g, and f are solvent-exposed and usually polar. The
positions a and g,
which flank the hydrophobic core, can make interhelical interactions between
g; and e'.+5
residues, and thereby mediate heterospecific pairing (11-14).
As most naturally-occurring coiled coils are homodimeric, synthetic sequences
have
been designed to promote specific hetero oligomerization (11, 13-15). However,
it was
observed that designed coiled coils which behave well as synthetic peptides
failed in
fusion proteins expressed in E. coli, as they were proteolytically degraded.
Thus, the clear disadvantage of association domains based on hetero-
associating
helices is their pseudo-symmetry and their similar periodicity of hydrophobic
and
hydrophilic residues. This structural similarity resulted in a strong tendency
to form
homo-dimers and thus to lower significantly the yield of hetero-dimers (2,
16).
Furthermore, the formation of Jun/Fos hetero-dimers is kinetically disfavoured
and
requires a temperature-dependent unfolding of the kinetically favoured homo-
dimers,
especially Jun/Jun homo-dimers (WO 93/15210; 2, 16). Because of the need for
additional purification steps to separate the unwanted homo-dimers from hetero-
dimers
and the resulting decrease in yield, hetero-association domains based on
amphipathic
helices have so far not resulted in practical advantages compared to
conventional
chemical coupling.
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
3
Further, it is not currently possible to predict sequences of coiled coil-
forming peptides
that will simultaneously have high stability and heterospecificity as well as
advantageous
in-vivo properties, such as resistance to proteases. This is crucial to
practical
applications of optimal interacting heterodimers for in vivo studies of
protein
oligomerization, e.g. the design of bispecific miniantibodies (17).
Thus, the technical problem underlying the present invention is to provide
association
domains based on helical coiled-coil structures which lead to hetero-
association. The
solution to the above technical problem is achieved by the embodiments
characterized
in the claims. Accordingly, the present invention provides a method which
allows to
identify hetero-associating (poly)peptides. The technical approach, i.e. the
design of an
appropriate coiled-coil library and screening by using a library-vs-library
approach is
neither provided nor suggested by the prior art.
Thus, the present invention relates to a method for the identification of
hetero-
associating (poly)peptides comprising the steps of:
(a) providing a library A of (poly)peptides/proteins comprising (poly)peptides
Am
having the general formula:
VAQLXEXVKTLXAXZYELXSXVQRLXEXVAQL
wherein X represents a mixture of E, K, Q, and R, and wherein Z represents a
mixture of N and V,
(b) providing a library B of (poly)peptides/proteins comprising (poly)peptides
B~
having the general formula:
VDELXAXVDQLXDXZYALXTXVAQLXKXVEKL
wherein X represents a mixture of E, K, Q, and R, and wherein Z represents a
mixture of N and V;
(c) combining in a common medium the (poly)peptides/proteins of said libraries
A
and B; and
(d) screening or selecting for a screenable or selectable property caused by
the
hetero-association of a (poly)peptide Am with a (poly)peptide B".
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
4
The term "(poly)peptide" relates to molecules consisting of one or more chains
of
multiple, i. e. two or more, amino acids linked via peptide bonds.
The term "protein" refers to (poly)peptides where at least part of the
(poly)peptide has or
is able to acquire a defined three-dimensional arrangement by forming
secondary,
tertiary, or quaternary structures within and/or between its (poly)peptide
chain(s). This
definition comprises proteins such as naturally occurring or at least
partially artificial
proteins, as well as fragments or domains of whole proteins, as tong as these
fragments
or domains have a defined three-dimensional arrangement as described above.
In this context, the commonly known one-letter code for amino acid residues is
used.
The term " screenable or selectable property" refers to a property which is
generated in
the event of a successful interaction taking place during screening or
selection.
Examples for screenable selectable properties include, but are not limited to,
binding to
a target or presentation of a target for ligand-binding, enzymatic activity,
transactivation
of transcription of a reporter gene such as beta-galactosidase, alkaline
phosphatase or
nutritional markers such as his3 and leu, or resistance genes giving
resistance to an
antibiotic such as ampicillin, chloramphenicol, kanamycin, zeocin, neomycin,
tetracycline
or streptomycin. In another embodiment, the selectable or screenable property
can be
restoration of phage infectivity to a filamentous phage rendered non-
infectious by
deletion of the N-terminal domains) of the genelll protein (U.S. Patent No.
5,514,548).
In a preferred embodiment, the invention relates to a method wherein said
libraries A
and B are provided by providing libraries of nucleic acid sequences encoding
said
(poly)peptides/proteins, followed by causing or allowing the expression of
said libraries
of (poly)peptides/proteins.
Methods for providing libraries of nucleic acid sequences, and for causing or
allowing
their expression, either in vivo after transformation, transfection or
transduction of
appropriate host cells, using appropriate vectors, containing all elements
necessary for
transcription and translation such as promotor/operator elements etc., or in
vitro using in
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
vitro expression systems are well known to anyone of ordinary skill in the art
(see e.g.
Sambrook et al., 1989, Molecular Cloning: a Laboratory Manual, 2nd ed.).
Further preferred is a method wherein said common medium are host cells, each
cell
harbouring nucleic acid sequences encoding a (poly)peptide/protein of each of
said
libraries A and B.
Most preferred is a method wherein said (poly)peptides/proteins of said
libraries A and B
further comprise either a N- or a C-terminal fragment of the murine DHFR
enzyme, and
wherein said screenable or selectable property is insensitivity of the host
cell to
trimethoprim by reconstitution of the DHFR enzyme on hetero-association of
(poly)peptides Am and B".
The DHFR assay has been published (WO 98/34120; 19) and is further exemplified
in
the examples.
In another embodiment, the present invention relates to a hetero-associating
(poly)peptide Am taken from the list of:
WINZIPA1: VAOLEEKVKTLRAQNYELKSRVORLREQVAQL
WINZIPA2: VAQLRERVKTLRAQNYELESEVQRLREOVAOL
WINZIPA3: VAQLQEKVKTLRARNYELKSEVQRLEEKVAQL
WINZIPA4: VAQLEEQVKTLQARNYELKSKVORLKEKVAQL
WINZIPAS: VAQLEERVKTLRAQNYELKSKVORLEEOVAOL
WINZIPA6: VAOLEEQVKTLEAENYELKSKVORLRERVAQL
WINZIPA7: VAOLOEOVKTLEAQNYELESEVQRLKEQVAQL
WINZIPAB: VAOLEERVKTLKAENYELESEVORLKERVAQL
WINZIPA9: VAOLEEKVKTLKAKNYELKSKVORLKEKVAQL
WINZIPA10: VAQLOEEVKTLOAENYELRSEVORLEEEVAQL
WINZIPA11: VAOLRERVKTLRARNYELQSKVQRLKERVAQL
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
6
Furthermore, the present invention relates to a hetero-associating
(poly)peptide B~ taken
from the list of:
WINZIPB1: VDELQAEVDQLQDENYALKTKVAQLRKKVEKL
WINZIPB2: VDELKAEVDQLQDQNYALRTKVAQLRKEVEKL
WINZIPB3: VDELEAEVDQLKDQNYALKTKVAQLQKQVEKL
WINZIPB4: VDELRAKVDQLQDENYALETEVAQLQKRVEKL
WINZIPBS: VDELEAEVDQLEDQNYALQTRVAQLEKRVEKL
WINZIPB6:VDELKAKVDQLKDKNYALRTKVAQLRKKVEKL
WINZIPB7: VDELRAQVDQLQDKNYALRTRVAQLKKRVEKL
WINZIPBB: VDELQAEVDQLQDQNYALRTQVAQLKKKVEKL
WINZIPB9: VDELRAQVDQLEDQNYALETQVAQLEKEVEKL
WINZIPB10: VDELQAKVDQLKDENYALQTKVAQLQKRVEKL
WINZIPB11: VDELRAEVDQLEDENYALRTRVAQLRKQVEKL
Particularly preferred is the use of a hetero-associating (poly)peptide
according to the
present invention for the identification of optimized hetero-associating
(poly)peptides in a
method according to the present invention, wherein one of the hetero-
associating
peptide WinZipAm as listed hereinabove is used instead of library A of
(poly)peptides/proteins comprising (poly)peptides Am in step (a) above, or
wherein a
hetero-associating peptide WinZip B~ as listed hereinabove is used instead of
library B
of (poly)peptides/proteins comprising (poly)peptides Bn in step (b) avove.
In a still further preferred embodiment, the present invention relates to an
optimized
hetero-associating (poly)peptide obtainable by the method of the present
invention.
In a most preferred embodiment, the present invention relates to a pair of
hetero-
associating (poly)peptides taken from the list of:
WinZipA1 and WinZipB1
WinZipA2 and WinZipB1
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
7
WinZipA1 and WinZip B2
WinZipA3 and WinZip B3
WinZipA4 and WinZip B4
WinZipA5 and WinZip B5
WinZipA6 and WinZip B6
WinZipA7 and WinZip B7
WinZipA8 and WinZip B8
WinZipA9 and WinZip B9
WinZipAlO and WinZip B10
WinZipAl1 and WinZip B11
In a yet further preferred embodiment, the invention relates to a
(poly)peptide/protein
comprising one of the hetero-associating (poly)peptides, or an optimized
hetero-
associating (poly)peptide of the present invention, and a further
(poly)peptide/protein.
In that context, "(poly)peptide/protein comprising one of the hetero-
associating
(poly)peptides, or an optimized hetero-associating (poly)peptide of the
present invention,
and a further (poly)peptide/protein" refers to all constructs which comprise
one of the
hetero-association peptides according to the present invention and additional
moieties.
This comprises (poly)peptides/proteins which are expressed from a contiguous
nucleic
acid coding sequence. Additionally, this comprises constructs where the
individual
components are expressed from different nucleic acid coding sequences, or
where the
components are produced by peptide synthesis, and where the separate
components
are linked by the formation of disulfide bonds or by chemical conjugation.
Still further preferred is a (poly)peptide/protein wherein said further
(poly)peptide/protein
is an enzyme, a toxin, a cytokine, a metal binding domain, a transcription
factor, a
member of the immunoglobulin superfamily, a bioactive peptide of 5 to 15 amino
acid
residues, a peptide hormone, a growth factor, a lectin, a lipoprotein, a
peptide which is
able to bind to an independent binding entity, or a functional fragment of any
said further
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
8
(poly)peptide/protein.
In a most preferred embodiment, the present invention relates to a hetero-
associated
(poly)peptide/protein comprising at least two (poly)peptide/proteins of the
present
invention, associated by hetero-association of a hetero-associating
(poly)peptide Am and
a hetero-associating (poly)peptide B~.
The term "hetero-associated (poly)peptide/protein" refers to all bispecific
and/or bivalent
complexes formed by taking advantage of the hetero-associating peptides
according to
the present invention. These include, for example, constructs where said
"further
(poly)peptides/proteins" are two antibody fragments directed against different
specificities. Such bispecific constructs can be used to increase selectivity
of antibody-
based approaches in therapy of diseases where the target cells exhibit a
pattern of two
cell-surface markers distinct from that of non-target cells which may present
one of the
two markers. Furthermore, one of the antibody specificities may be directed to
a target
cells, whereas the second may be used to target a drug carrier moiety
selectively to the
target. Additionally, antibody fragments as targeting vehicles may be combined
with
(poly)peptides/proteins which serve as effector domains, such as enzymes or
signalling
molecules.
Particularly preferred is a DNA sequence encoding a hetero-associating
(poly)peptide
taken from the list of WINZIPA1 to WINZIPA11 and WinZipB1 to WinZipBll, or
encoding an optimized hetero-associating (poly)peptide or a
(poly)peptide/protein of the
present invention.
Further preferred is a DNA sequence encoding a hetero-associating
(poly)peptide
wherein said DNA sequence hybridizes under stringent conditions to a DNA
sequence
encoding a hetero-associating (poly)peptide taken from the list of WinZipA1 to
WinZipAl1 and WinZipB1 to WinZipBll.
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
9
As used herein, the term "hybridizes under stringent conditions" is intended
to describe
conditions for hybridization and washing under which nucleotide sequences at
least 60%
homologous to each other typically remain hybridized to each other.
Preferably, the
conditions are such that at least sequences at least 65%, more preferably at
least 70%,
and even more preferably at least 75% homologous to each other typically
remain
hybridized to each other. Such stringent conditions are known to those skilled
in the art
and can be found in Current Protocols in Molecular Biology, John Wiley & Sons,
New
York. (1989), 6.3.1-6.3.6. A preferred, non-limiting example of stringent
hybridization
conditions is hybridization in 6 x sodium chloride/sodium citrate (SSC) at
about 45°C,
followed by one or more washes in 0.2 x SSC, 0.1 % SDS at 50°-
65°C.
In still another embodiment the invention relates to a vector comprising a DNA
sequence
according to the invention.
In a further embodiment, the invention relates to a vector comprising DNA
sequences
encoding at least two (poly)peptide/, comprising at least a hetero-associating
(poly)peptide Am and a hetero-associating (poly)peptide Bn.
Vectors which can be used in accordance with the present invention are well-
known to
the practitioner in the art.
In another embodiment, the invention relates to a host cell containing at
least one vector
of the present invention.
In a further preferred embodiment the host cell is a mammalian, preferably
human cell, a
yeast cell, an insect cell, a plant cell, or a bacterial, preferably E.coli
cell.
In a highly preferred embodiment, the invention relates to a method for the
production of
a hetero-associating (poly)peptide, an optimized hetero-associating (poly), a
(poly)peptide/protein, or a hetero-associated (poly)peptide/protein of the
present
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
invention, which comprises culturing the host cell of the present invention in
a suitable
medium, and recovering said (poly)peptide or said (poly)peptide/protein
produced by
said host cell.
In a most preferred embodiment, the invention relates to a pharmaceutical
composition
comprising the hetero-associated (poly)peptide/protein or the present
invention.
Still further preferred is a diagnostic composition comprising the hetero-
associated
(poly)peptide/protein of the present invention.
Further preferred is a kit containing at least one of
a hetero-associating (poly)peptide, an optimized hetero-associating
(poly)peptide, or a
(poly)peptide/protein of claims, or a hetero-associated (poly)peptide/protein
of the
present invention; or
a vector of claims according to the invention.
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
11
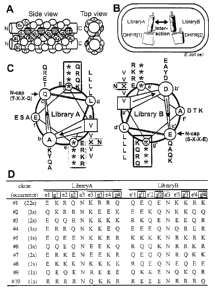
FIGURE CAPTIONS
Figure 1: (A) Schematic representation of a parallel dimeric coiled coil.
Hatched bars
indicate the possible interhelical interactions between a and g positions. (B)
Schematic
representation of the protein complementation assay as described in the
Examples.
Introduction of a mutation at the DHFR interface (1114A) was used to increase
selection
stringency (19). (C) Overview of the library design depicted as a helical
wheel plot from
the N- to the C-terminus (inside to outside. The randomized positions are
indexed (*:
equimolar mixture of Q, E, K, R, x: equimolar mixture of N, V). The selected
residues
from the predominant pair, WinZip-A1 B1 (clone #1, Fig. 1 D), are next to the
randomized
positions in the respective box. (D) Partial sequences (randomized positions)
of clones
from the highest stringency selection which survived at least until passage 10
(clone #1:
W inZip-A1 B1, clones #2 to #10: W inZip-A3B3 to W inZip-A11 B11 ). Clone #1
(named
WinZip-A1 B1 ) was identified 18/22 times in passage 12 and 4/11 times in
passage 10
(the full amino acid sequences are shown in Fig. 11 ).
Figure 2: (A) DNA constructs code for fusions between library proteins (shown
as a-
helical leucine zippers) and either fragment of murine DHFR (mDHFR). Fusions
were
created using either the wild-type or the mutant mDHFR fragment 2 (11e1
l4Ala), yielding
LibA-DHFR[1] and LibB-DHFR[ 2] or LibB-DHFR[2:1114A], respectively. (B)
Principle of
the mDHFR-fragment complementation assay: E, coli cells are cotransformed with
both
fusion libraries in minimal medium, in the presence of IPTG (for induction of
expression)
and trimethoprim (for inhibition of the bacterial DHFR). If the library
proteins
heterodimerize, mDHFR can fold from the individual fragments resulting in
active
enzyme and bacterial growth. Both mDHFR fragments must be present, and
dimerization of the fused proteins is essential, in order for cell propagation
to be
possible. No growth is observed if any of these conditions is not fulfilled
(19) . The
surviving colonies are the result of "single-step selection" and can be
directly analyzed
by DNA sequencing. (C) "Competition selection" is undertaken by pooling
colonies from
(B) in selective, liquid culture (passage 0 or PO), propagating the cells and
diluting into
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
12
fresh selective medium for further passages. An aliquot can be plated and the
resulting
colonies analyzed by DNA sequencing.
Figure 3: Competition selection and chain shuffling. (A) Approximately 1.42 x
104 clones
resulting from single-step, 1114A-mutant selection were pooled (=PO) and
competition
selection was undertaken as described in Figure 2C, and in the Examples. At
each
passage, some cells were plated and colony sizes were quantitated. (B)
Quantitation of
the colony sizes from (A). For comparative purposes, quantitation of colony
sizes of cells
transformed with DNA of WinZip-A1 B1 (but not passaged in liquid culture) is
shown. (C)
Quantitation of the colony sizes from passages of the chain shuffling
experiment:
WinZip-B1-DHFR[2:1114A] + LibA-DHFR[1]. In (B) and (C) the numbers of colonies
were
normalized such that passages could be directly compared.
Figure 4: (A) Schematic representation of a leucine zipper pair visualized
from the N-
terminus illustrating e/g-interactions and the hydrophobic core formed by the
a- and d-
positions. (B) Distribution of residues at the semi-randomized positions
throughout
selection. The number of zipper pairs sequenced is given in parentheses, save
"Before
selection" where the theoretical distribution is reported. Each pair carries
one core a-pair
and 6 e/g-pairs. Neutral e/g-pairs have one or both residues as Gln. In
"Competition
(1114A)" only clones from P6 to P12 (not from earlier passages) were
considered for
analysis. Thus, 37 individual clones were sequenced, and most of the resulting
sequences were identical in two or more clones. The distributions were
calculated
according to the frequency of sequence occurence (n=37). (C) Leucine zipper
sequences obtained after competition selection and chain shuffling. The heptad
positions (a to g) are followed by the heptad number (1 to 5). Invariant
residues from
GCN4 are underlined. Clear boxes indicate the semi-randomized e- and g-
positions and
core a-position (a3). Circled residues were designed to contribute to helix
capping.
Shaded residues were designed for the introduction of restriction sites. Other
residues
are from c-Jun (LibA) or c-Fos (LibB). Arrows indicate putative e/g-
interactions.
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
13
Figure 5: Efficiency of competition in a model selection. The selection was
set up by
mixing known numbers of cells expressing either GCN4-DHFR[1]/GCN4-
DHFR[2:1114A]
fusions or one of 7 LibA-DHFR[1]/LibB-DHFR[2:1114A] pairs previously selected
by
single-step selection. The starting ratio was 2.9 x 104: 1 (GCN4 to Lib).
Competition
selection was undertaken as described in Figure 2C, and in the Examples. The
appearance of the library pairs in the pool was monitored by restriction
analysis. A Pwll
fragment (1138 bp) is unique to the LibB sequence of the LibB-DHFR[2] plasmid,
while
another (762 bp) is from pRep4 (repressor plasmid) and remains approximately
constant. The bands were quantitated using the NIH Image gel analysis function
to
calculate the ratio of LibB/pRep4 (indicated below each lane).
Figure 6: (A) Deviation of observed e/g-interactions in the selected
heterodimer and the
two putative homodimers from the statistically expected distribution in the
absence of
selection. Interactions are grouped in potentially attractive (E-K, K-E, E-R,
R-E; left black
bars), neutral (Q Q, Q-E, E-Q, Q-K, K-Q, Q-R, R-Q; grey bars) and repulsive (E-
E, K-K,
K R, R-K, R-R; right black bars). (i) Low stringency selection: clones were
subdivided
into those with an N-N pair in the core a-position (n=8) and those with an N-V
or V-V pair
(n=6), (ii) medium stringer ;y selection: only clones with an Asn-pair in the
core a-
position were considered (n=23), and (iii) highest stringency selection:
clones, which
survived the competition selection at least up to passage 10 (Fig. 1 D) were
considered.
These were analyzed counting each sequence once (not weighted, n=10) or
according
to their frequence of appearance (weighted, n=37). (B) Number of selected
pairs having
a certain difference of attractive (grey bars) or repulsive interactions
(black bars),
respectively, between the heterodimer and its constituting homodimers. The
definition of
(i), (ii) and (iii) is as in (A).
Figure 7: Positional distribution of amino acids at each e- and g-position in
sequences
obtained from the highest stringency selection. The statistically expected
random
occurrence of each amino acid at each position was subtracted from the
relative
occurrence observed in the selection (Q left (black), E middle (grey), K/R
right (black)).
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
14
Figure 8: (A) Determination of the molecular weight of WinZip-A1 B1 by
sedimentation
equilibrium. The upper panel shows the residuals between measured data
obtained at
NM peptide concentration at 25°C and data fitted as monomer (top),
dimer (middle),
or trimer (bottom). The lower panel shows the residuals for a dimeric fit to
the data set
obtained at 150 ,uM peptide concentration, 10°C. (B) Native gel
electrophoresis. 1:
WinZip-A1 (p1 11.2); 2: WinZip-B1 (p1 4.9); 3: WinZip-A1 B1. (C) Urea
titration of the
heterodimer WinZip-A1 B1 (~), and the homodimers WinZip-A1 (~) and WinZip-B1
(~).
Figure 9: CD-measurements of the synthesized peptides of WinZip-A1 B1. (A)
Temperature dependence of []222 for WinZip-A1 B1 (~), WinZip-A1 (~), WinZip-B1
(~),
and the calculated average of both homodimers (- - -). (B) Dependence of Tm
and oTm
(-o -) on pH, and (C) on salt. It must be noted that thermal denaturation of
WinZip-A1
was not completely reversible at 1 M salt concentration, and only 71 % of the
starting
signal was regained.
Figure 10: Sequencing profile of pools from passages of the chain shuffling
WinZip-B1-
DHFR[2:1114A] + LibA-DHFR[1]. Representative semi-randomized positions (see
Fig. 4)
were taken from a single competition experiment, such that the selection rates
can be
directly compared. The ratio of the individual triplet codons (central three
nucleotides of
each frame) was visually estimated (GAG = Gln; GAG = Glu; AAG = Lys; CGT =
Arg; the
equimolar random mix of the 4 codons results in the predominance of C at the
first
position, A at the second and G at the third). Mixed positions are marked by
(NNN),
positions where a single codon is dominant (> 50%) are marked in lower case
and those
where the codon is clear (> 90%) are marked in upper case. For passages 0, 2
and 8,
two independent sequencing reactions were performed, which yielded identical
results.
Figure 11: (A) Sequences of (poly)peptides WinZipA1 to WinZipAl1
(B) Sequences of (poly)peptides WinZipB1 to WinZipBl1
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
The examples illustrate the invention:
EXAMPLES
All reagents used were of the highest available purity. Sequencing was carried
out either
by cycle sequencing with fluorescence labeling (MWG-Biotech, Ebersberg,
Germany)
using a LiCor detection system or by automated sequencing with an ABI
sequencer.
Restriction endonucleases and DNA modifying enzymes were from Pharmacia and
New
England Biolabs. E. coli strain XL1-Blue (Stratagene) was used for subcloning
and
propagation of the libraries. E. coli strain BL21 harboring the lack' plasmid
pRep4
(Qiagen) was cotransformed with the appropriate DNA constructs for the
survival
assays.
Abbreviations: CD, circular dichroism; mDHFR, murine dihydrofolate reductase;
WinZip: dominant zipper pairs obtained from competition selection; WinZip-A1
B1:
original pair selected, comprising peptide A1 from IibraryA and peptide B1
from IibraryB;
WinZip-A1 B2 and WinZip-A2B1: optimized pairs comprising the original partner
Al or B1
and the new partner B2 or A2, respectively.
Example 1: Selection of hetero-association peptides (see WO 98/34120, Example
7)
1.1 Introduction
Here we present a strategy for library-vs-library screening in intact cells
based on the
folding of murine enzyme dihydrofolate reductase (mDHFR) from complementary
fragments (18, 19). DHFR was genetically dissected into two rationally
designed
fragments, each of which can be fused to a library of proteins or peptides
(Fig. 2A).
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
16
Members of one library which heterodimerize with a member of the other library
drive
the reassembly of the mDHFR fragments, resulting in reconstitution of
enzymatic activity
(Fig. 2B). Activity is detected in vivo using an E. coli-based selection
assay, where the
bacterial DHFR is specifically inhibited with trimethoprim, preventing
biosynthesis of
purines, thymidylate, methionine and pantothenate, and therefore cell
division. The
reconstituted mDHFR, which is insensitive to the low trimethoprim
concentration present
in selection, restores the biosynthetic reactions required for bacterial
propagation. As a
result, the interaction between library partners is directly linked to cell
survival and
detected by colony formation. We have previously demonstrated the utility of
this
strategy with GCN4 leucine zipper-forming peptides, as well as with larger
heterodimerizing partner proteins (19) with Kps ranging between 3 and 160 nM
(20, 21 ),
although the affinity limits have not been determined.
1.2 Constructs for DHFR fragment complementation
The DNA constructs encoding the N-terminal (1-107) and C-terminal (108-186)
mDHFR
fragments have been previously described (19). The vectors are variants of
plasmids Z-
F[1,2], encoding the N-terminal DHFR fragment, and Z-F[3] or Z-F[3:1114A],
respectively, encoding the C terminal DHFR fragment with or without the 1114A
mutation
(19), Briefly, each fragment was amplified by PCR with appropriate unique
flanking
restriction sites and subcloned into a bacterial expression vector (pQE-32
from Qiagen).
Each plasmid encodes an N-terminal hexahistidine tag, followed by a designed
flexible
linker and the appropriate DHFR fragment. Unique restriction sites between the
hexahistidine tag and the flexible linker allow subcloning of the desired
library.
1.3 Library design
Our goal was to select for metabolically stable dimeric coiled-coils with high
heterospecificity. Thus, two libraries were designed to meet the requirements
of genetic
diversity to prevent recombination, high helix stability and a high
probability of
complementarity, all within a reasonable library size (Fig. 1 C).
As templates for the outer, solvent exposed residues (positions b, c, f) we
chose the
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
17
leucine-zipper regions of the proto-oncogenes c-Jun and c-Fos for IibraryA and
B,
respectively, thus minimizing potential recombination despite the repetitive
pattern of the
heptads. Indeed, no recombination was found in any of the 80 clones sequenced.
We
chose a helix length of 4.5 heptads as good compromise between stability and
size. For
the hydrophobic core residues (positions a, d) we chose the residues of the
parallel,
homodimeric leucine zipper GCN4 (Val at a, Leu at d). A single a-position in
the middle
of each helix is often occupied by a polar residue, most often an Asn, which
forms a
hydrogen bond inside the hydrophobic core (22, 23). Replacement of this Asn
pair by a
non-polar one increases the stability significantly, but leads to helices
packing in
different registers and orientations, as well as forming higher order
oligomers (24-26).
Since we could not ascertain a priori whether higher specificity or stability
would be more
advantageous, we included both by allowing Asn and Val at the core a-position
with
equal probability. A difficult problem in library design is to encode only the
desired amino
acids with a predetermined ratio. We solved this problem by using defined
trinucleotide
mixtures in the oligonucleotides, where each trinucleotide codes for one
specific amino
acid (27).
The solvent-accessible residues at the e- and g-positions can form
interhelical salt
bridges or hydrogen bonds which can contribute to stability and heteromeric
specificity
(13, 14, 28, 29). Based on these results and on commonly occurring amino acids
at
these positions (30, 31 ), we simultaneously randomized all eight e- and g-
positions with
equimolar mixtures of Gln, Glu, Lys, Arg, also using trinucleotide codons in
DNA
synthesis. Including the Asn-Val combination at the core a-position, each
library had a
theoretical diversity of 1.3x105.
To increase the stability of the helices, we introduced helix capping residues
in both
libraries to saturate the missing hydrogen bonds at the helix ends with their
side chains.
Based on studies of helix-capping propensities in proteins (32, 33) and
peptides (34,
35), we chose T-X-X-Q (Ncap-N1-N2-N3) for IibraryA, and S-X-X-E for IibraryB.
The C-
cap has only a minor effect on helix stability (35). As Gly has a high
preference for the
C-cap position (32, 33), we added a C terminal Gly. This may contribute to
helix
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
18
termination or extend the linker which connects the coiled coil to the DHFR
fragments
(Fig. 1 B).
1.4 Library synthesis
Trinucleotide codons (27) were used to code for randomized positions, all
other
positions were made with mononucleotides.
LibraryA: TACTGTGGCGCAACTGNNNGAANNNGTGAAAACCCTTNNNGCTNNNXXX-
TATGAACTTNNNTCTNNNGTGAGCGCTTGNNNGAGNNNGTTGCCCAGCTTGCTA
(encoding VAQLXEXVKTLXAXZYELXSXVQRLXEXVAQL, wherein X represents a
mixture of E, K, Q, and R, and wherein Z represents a mixture of N and V);
IibraryB: CTCCGTTGACGAACTGNNNGCTNNNGTTGACCAGCTGNNNGACNNNXXX-
TACGCTCTGNNNACCNNNGTTCGCAGCTGNNNAAANNNGTGGAAAAGCTGTGATA
(encoding VDELXAXVDQLXDXZYALXTXVAQLXKXVEKL, wherein X represents a
mixture of E, K, Q, and R, and wherein Z represents a mixture of N and V)
(NNN = equimolar mixture of the trinucleotides AAG, CAG, GAG, CGT; XXX =
equimolar
mixture of the trinucleotides AAT, GTT).
Generation of the second strand and introduction of Sall and Nhel restriction
sites were
achieved by PCR using the primers prA-fwd: GGAGTACTGGCATGCAGTCGACTACT-
GTGGCGCAACTG and prA-rev: GGACTAGTACCTTCGCTAGCAAGCTGGGCAAC or
prB-fwd: GGAGTACTGGCATGCAGTCGACCTCCGTTGACGAACTG and prB-rev:
GGACTAGTGCTAGCTTCTGACAGCTTTTCCAC, respectively. This resulted in a 142
by double-stranded oligonucleotide for either library.
1.5 Expression plasmids
LibraryA and B were both digested with Sall and Nhel, gel purified and ligated
to the
appropriate vector (Fig 2) yielding the plasmids LibA-DHFR[1], LibB-DHFR[2],
LibB-
DHFR[2:1114A] (Fig. 2A). After subcloning, the resulting linker between either
library and
DHFR fragment was: A(SGTS)2 STSSGI for LibA and SEA(SGTS)2STS for LibB. To
achieve maximal library representation, the ligation mixes were individually
electroporated into XL1-Blue cells and selected with ampicillin on rich medium
(LB). A 2-
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
19
to 7-fold over-representation of each library was obtained. The resulting
colonies were
pooled and the plasmid DNA purified such that supercoiled plasmid DNA was
obtained
for cotransformation. The supercoiled DNA was cotransformed in BL21 cells
yielding
about 4x106 double-transformants. We used BL21 cells with a transformation
efficiency
of no less than 5 x 10' transformants per mg of DNA using 200 pg of DNA, or 2
x 10'
transformants per mg using 500 ng of DNA. In cotransformations, the occurrence
of
double transformation was calculated as the number of colonies growing under
selective
pressure with trimethoprim (described below) divided by the number growing in
the
absence, when cotransformed with equal amounts of each DNA of a given, pre-
selected
pair. In order to verify that the library populations encode the designed
amino acids with
the expected frequency, single clones from each library were randomly picked
and
sequenced before selection. No statistically significant biases were detected.
Seventy to
80% of each library had no mutations or frame-shifts, and thus the library-vs-
library
combination yielded approximately 50% correct sequence combinations. Thus, the
experimental library-vs-library size of correct pairs is estimated as 2x106.
1.6 Selection procedure.
Three selection strategies were tested here, each having a different level of
stringency.
In the lowest stringency selection, we screened two expressed libraries
against each
other in a single-step selection (Fig. 2B), where cells cotransformed with
complementary
libraries were directly plated on selective medium plates (M9 medium with 1
Ng/ml
trimethoprim), and resulting colonies were analyzed, thereby identifying all
interacting
polypeptide partners. In the second strategy, we increased the selection
stringency by
using a mutant DHFR fragment (IIe114A1a) containing the destabilizing 1114A
mutation
in DHFR[2] which occurs at the interface between both DHFR fragments. This
mutation
prevents stable reassembly of DHFR from its fragments (19) and should thus
require
more efficiently heterodimerizing, as opposed to homodimerizing, interacting
partners to
drive enzyme reconstitution. Finally, we introduced competitive metabolic
selection,
where clones obtained with the second strategy were pooled and passaged
through
several rounds of competition selection, in order to enrich for the optimally
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
heterodimerizing partners. Thereby, the most stable heterodimers should have
higher
mDHFR activity and thus a growth advantage.
Selective pressure for DHFR was maintained throughout all steps by inhibiting
the
bacterial DHFR with trimethoprim (1 mg/ml) in minimal medium. Ampicillin and
kanamycin (100 mg/ml and 50 mg/ml, respectively) were also included in all
steps to
retain the library plasmids and the lack repressor-encoding plasmid (pRep4),
respectively. Expression of the proteins was induced with 1 mM IPTG. When
selecting
on solid medium, growth was allowed for 45 hrs at 37°C.
When it was necessary to control precisely the starting number of cells in a
competition,
the number of viable cells in the starter cultures was quantitated as follows.
The
appropriate clones were propagated in liquid media under selective conditions
and dilute
aliquots were frozen at -80°C with 15% glycerol. One aliquot for each
clone was thawed
and plated under selective conditions, and the colonies counted after 45 hrs.
The
volume of cells to use for PO was then calculated, such that each clone should
be over-
represented by a factor of at least 2000. Colony sizes (in Fig. 3) were
evaluated using
the NIH Image Particle Analysis Facility. When selecting in liquid medium, the
starting
O.D. (600 nm) was either 0.0005 or 0.0001. Cells were propagated either in
Erlenmeyer
flasks or in a 10 liter New Brunswick fermentor, depending on the volume
required to
ensure adequate representation of all clones present, at 37°C with
shaking, or stirring at
250 RPM. After 10 to 24 hrs, O.D. (600 nm) reached 0.2 to 1.0 and cells were
harvested.
1.7 Single-step selection
As a first step in selection of heterodimerizing leucine zippers, a single-
step selection
was undertaken, using the wild-type mDHFR fragments, by cotransforming the
libraries
LibA-DHFR[ 1] and LibB-DHFR[2] and plating on selective media (Fig. 2B). This
strategy
applies only a low stringency of selection to the potential pairs, thus many
library
combinations were expected to be selected. Approximately 1.7% of the resulting
ampicillin-resistant cells were doubly transformed, harboring (at least) one
plasmid from
each library when using 5 ng of each DNA, or 8% were doubly transformed when
using
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
21
20 ng of each DNA, as seen from control transformations (calculated as
described
above). Of the doubly transformed cells which harbor no mutations or frame-
shifts,
approximately 35% formed colonies under selective conditions (Table 1 ). This
result
immediately demonstrates that even with relatively low stringency of
selection, only a
fraction of the possible combinations of the two libraries allows zipper
heterodimerization
leading to efficient mDHFR reassembly. Fourteen colonies resulting from two
independent cotransformations were picked and the sequences encoding the
zippers
were determined. Even under these low stringency conditions there exist
important
sequence biases in these sequences relative to the unselected ones (Fig. 4B).
A
reduction in same-charged e/g-pairs from 31.3% (unselected) to 19% (selected)
and an
increase in opposite-charged pairs from 25% (unselected) to 31 % (selected)
were seen.
As well, a strong enrichment of N-N pairing at the core a-position (25%
unselected vs
57% selected) was observed. The characteristics that have been enriched are
consistent with the selection of stable leucine zipper heterodimers.
1.8 Increased stringency: use of the mDHFR IIe114A1a mutation.
We repeated the single-step selection, using the IIe114A1a mutant of mDHFR
(18, 19), in
order to increase the stringency of selection. We reasoned that only library
partners that
form the most stable heterodimers can compensate for the reduced ability of
the
mDHFR(IIe114A1a) fragments to fold into active enzyme, resulting in higher
enzyme
activity and growth rates. When bacteria were cotransformed with LibA-DHFR[1]
and
LibB-DHFR[ 2:1114A], we observed a 50-fold decrease in the number of colonies
upon
selective plating compared to the wild-type DHFR fragments (Table 1 ). Twenty-
five
colonies were picked from 3 independent cotransformations and the DNA
sequences
were analyzed. The increase in selectivity was concomitant with an extremely
strong
selection for N-N pairing at the core a-position (92%; Fig. 4B), illustrating
that the
specificity of in-register parallel alignment provided by N-N pairing is more
highly favored
under these in-vivo selection conditions than the higher stability afforded by
V-V pairing.
Reassembly of mDHFR from its fragments requires that in the final structure,
the two
fragment N-termini be brought close enough together to allow native-like
refolding of
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
22
DHFR (see Fig. 1 ) (19, 36). The peptide linkers that connect the library
sequences to the
DHFR fragments must be sufficiently flexible to allow DHFR to fold from its
fragments,
but not so long that any C-terminal to N-terminal orientation of the final
folded leucine
zipper would be allowed. As a result of this structural requirement, parallel
in-register
heterodimerization of the library peptides is the only configuration possible.
Other biases
in these sequences were also more pronounced than with the wt DHFR fragments
(Fig.
4B). In particular, an additional increase in opposite-charged e/g-pairs from
31 % to 37%
was seen. In one case, a point-mutation resulted in a single clone (1/25) with
a V-T pair
at the core a-position.
1.9 Competition selection: Efficiency of selection
To further increase the selection pressure, we applied the principle of
competition
selection. We reasoned that, among selected zipper pairs, those which result
in more
stable heterodimerization will allow the most efficient enzyme reconstitution,
leading to
higher DHFR activity. If DHFR activity is limiting for growth, the higher
activity should
result in more rapid bacterial propagation, hence these cells would become
enriched in
a pool. Thereby, after sequential rounds of growth-competition, subtle
differences in
growth rate can be amplified, increasing the stringency of selection relative
to the single-
step selection.
To determine the rate at which competition can enrich for particular partner
pairs, we
first set up a model competition with a limited number of clones as described
in Figure
1C. The initial cell mixture (PO) contained known amounts of viable cells
expressing
either GCN4- DHFR[1]/GCN4-DHFR[2:1114A] or one of seven LibA-DHFR[1]/LibB-
DHFR[2:1114A] pairs previously obtained in a single-step selection of those
libraries,
mixed at a ratio of 2.9 x 10 4 : 1 (GCN4 : library clones). Productive
association of the
homodimeric GCN4 pair should occur only 50% of the time versus up to 100% for
heterodimerizing library clones, thus is disadvantaged. Within 3 passages, the
library
pairs were already visibly enriched (Fig. 5), and after 5 passages the
measured ratio
between a restriction fragment indicative of the library and a constant
fragment from the
repressor plasmid had reached its maximium, showing that enrichment was
maximal.
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
23
Colonies resulting from passage 9 (P9) were sequenced. No GCN4 leucine zippers
were
present among 24 sequences analyzed. Therefore, enrichment of the library
pairs over
GCN4 by a factor of at least 24 x 2.9 x 104 = 7 x 105 was achieved. Four out
of the 7
library clones initially present survived until P9, with varying distributions
(data no
shown). The experiment was also repeated at a lower starting ratio of GCN4 and
the
same library clones were enriched, consistent with their enrichement being
truly the
result of selection (and not of unrepresentative sampling). This indicated
that selection
among the pre-selected clones was not as rapid as that seen between pre-
selected and
GCN4 zippers, but that the smaller differences between the pre-selected ones
can still
be amplified in selection. These results demonstrate that there is a direct
link between
reconstitution of mDHFR and growth rate.
1.10 Competition selection for optimal pairs
Our ultimate goal was to select for the "best" among the zipper pairs obtained
by single-
step selection. We obtained a large initial number of clones by cotransforming
bacteria
with 0.5 mg of DNA each from LibA-DHFR[1] and LibB-DHFR[ 2:1114A].
Approximately
50% of cells were at least doubly transformed (52% + 10%, average of 2
independent
control experiments, calculated as described in the Experimental Protocol). We
obtained
approximately 1.42 x 104 clones on selective medium, which arise from a 1.4 x
102-fold
selection factor (see Table 1 ), and were thus selected from (1.42 x 104) x
(1.4 x 102) _
2.0 x 106 library-vs-library cotransformants. These were pooled and passaged.
There
was a clear increase in colony sizes with subsequent passages, indicating that
faster-
growing clones were taking over (Fig. 3A, B). At P12, the colonies are
homogeneously
large, showing similar growth rates among the clones. Twenty-two individual
colonies
from P12 were picked and sequenced, as well as 11 from P10 and 2 from each
previous
second passage. A single pair (WinZip-A1 B1, composed of WinZip-A1-DHFR[1] and
WinZip-B1-DHFR[2:1114A]) was identified 18/22 times (82%) in P12, 4/11 (33%)
in P10,
but not in previous passages (Fig. 4C). While other sequences were found in
early and
late passages, none was as enriched as WinZip-A1 B1. In order to verify that
the growth
rate recorded after competition (P12) was independent of bacteria-specific
factors
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
24
resulting from passaging, we cotransformed DNA from a pure clone of WinZip-A1
B1 into
fresh bacteria. The colony size distribution is similar for P12 and for the
transformants
(Fig. 3B), illustrating that the growth rate is a direct product of mDHFR
reconstitution
directed by the WinZip-A1 B1 pair. The sequence bias observed at the core-a
position
was yet stronger here: only N-N pairing was recorded at the core a-position.
When the
biases at the e/g-positions were calculated according to the occurrence of
each
sequence (n=37), there was no significant change in opposite charged pairing
(37%),
while a small increase in same-charged pairing was observed (from 23% to 26%)
as a
result of the two same-charged pairs which occur in the predominant WinZip-A1
B1 (Fig.
4B, C). However, when each unique sequence was considered only once (n=10) a
further increase of opposite-charged e/g-pairing was observed.
Example 2: Analysis of clones resulting from library screening
2.1 Introduction
Clones resulting from the three selection strategies with increasing
stringencies were
analyzed and compared: (i) lowest stringency: 14 clones analyzed, (ii) medium
stringency: 25 clones analyzed (see Table 2), (iii) highest stringency: 41
clones analyzed
from various passages. The last passage (P12) yielded a population dominated
by a
single pair of coiled-coil sequences, WinZip-A1 B1, as described above (Fig.
1C, clone
#1 in Fig. 1 D), which was biophysically analyzed (see below). The sequences
of clones
surviving at least up to passage 10 are reported in Figure 1 D. By comparing
the
selected clones from the three strategies, we analyzed the preferences for the
core a-
position, the distribution of e/g-pair combinations, and the presence of any
bias for
certain amino acids within the individual helices.
2.2 Selection in the core a-position
Sequencing of 16 clones prior to selection showed an equal distribution of Asn
and Val
at the core a position. After selection a strong bias was found toward Asn-
pairs, which
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
became stronger with increasing selection stringency. No bias was seen between
Val-
Val or Val-Asn combinations. An Asn-pair was found in 57% (n=14) of the lowest
stringency, in 92% (n=25) of the medium and in 100% (n=41 ) of the highest
stringency
selection. Thus, the specificity gained by this polar interaction clearly
outweighs the
more energetically stable Val-pairs. This seems to be a very important
feature, since a
strong selection occurred even at the lowest stringency.
2.3 Selected e/g-pairs in hetero- and putative homodimers
All selected clones must form a heterodimer to reconstitute DHFR activity,
however, the
heterospecificity, and thus the ratio of heterodimers (IibraryA/IibraryB) to
homodimers
(IibraryA/IibraryA and IibraryB/IibraryB) can vary, and a mixture of both may
still generate
sufficient amounts of active enzyme to allow cell propagation under low
stringency
conditions. We analyzed the average occurrence of all e/g-pair combinations in
the
heterodimers and the putative homodimers arising from the various selections
in relation
to the random statistical distribution (Fig. 6A). The selected heterodimers
show on
average more attractive and less repulsive interactions than expected in a
random
population, indicating selection for stability. This trend, although
increasing with higher
stringency, is already observed in the lowest stringency selection, indicating
that a
certain threshold of stability is needed to induce enzyme dimerization.
Selection for
heterospecificity is achieved by a higher stability of the heterodimer
relatively to the
homodimers and is only observed in the medium and highest stringency
selections
using the destabilizing 1114A DHFR[2]-mutant (compare Fig. 6A, (i) vs (ii) and
(iii)).
Interestingly, this effect is more pronounced for IibraryA homodimers than for
IibraryB
homodimers, and the biophysical characterization of WinZip-A1 B1 indicated
that the
IibraryA homodimer is more stable (see below), and thus might have a stronger
influence on titrating out fragments than the IibraryB homodimer.
To determine the degree of heterospecificity achieved in the various
selections, the
relative stability of heterodimer versus homodimer was estimated for each
single clone.
We calculated the difference of attractive or repulsive interactions,
respectively, between
the heterodimer and the average of the two corresponding homodimers and
displayed a
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
26
histogram of pairs as a function of this difference (Fig. 6B). The results
clearly show that
heterospecificity is achieved not only by a decrease of repulsive interactions
but also by
an increase of attractive interactions in the heterodimer relative to the
homodimers.
However, the lowest and medium stringency selection yielded still a certain
fraction of
pairs with no difference or even a reversed ratio, whereas the highest
stringency
selection exclusively yielded pairs with distinct heterospecificity. In
addition, in no case
were more than three repulsions found in the heterodimers, although in a
random
combination 8% of all pairs should have 4 to 6 repulsions.
2.4 Positional distribution of the selected amino acids
Intrahelical electrostatic interactions can influence stability and may even
promote
selection of apparently repulsive e/g-pairs. Interactions with the helix
macrodipole, for
example, can modulate stability in coiled coils (37). Indeed, we observed a
bias for
negatively charged and neutral amino acids in the N-terminal part and
positively charged
amino acids in the C-terminal part (Fig. 7). This positional preference may at
least
partially compensate the loss of stability resulting from a repulsive e/g-
interaction. In
addition, interactions with adjacent residues on the outside of the helix (b-
and c
positions) may influence the contributions of charges at the e- and g
positions (38). This
may explain why at position e1 in IibraryB a negatively charged amino acid is
not
favored, contrary to the expected counterbalancing of the helix dipole, since
this position
is adjacent to two aspartates in positions b1 and b2. On a more general note,
the
predominantly selected sequence with the residues from c-Jun at the outer
positions
(IibraryA) bears remarkable similarity to the e- and g sequences in the
naturally-
occurring c-Jun.
2.5 Library complexity
Although the predominantly selected sequence pair WinZip-A1 B1 showed all the
desired
properties in vivo as well as in vitro (see below), we were not able to cover
all theoretical
library vs-library combinations in our selection. Nonetheless, we covered all
possible
electrostatic interaction combinations (+/+, -/-, +/-, +/n, -/n, n/n;
n=neutral) in all six
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
27
interacting e/g-positions, when grouping the core a-position into favored (Asn-
Asn) or
disfavored (Asn-Val, Val-Val) combinations. This reduces the theoretical
library size from
1.7x10'° to 9x104 possibilities, which was well covered by the
experimental library of
2x106. It is therefore likely that WinZip-A1 B1 contains the most important
features for
stability and heterospecificity. Furthermore, the random probability of
finding pairs with
no repulsive interactions was 1:40, and with solely attractive interactions
was 1:1.6x104.
Thus, our selection covered a representative sequence space and the same-
charged
interactions in WinZip-A1 B1 are not a result of incomplete library sampling
but must
have more subtle reasons, including in-vivo factors, which we cannot fully
address.
Furthermore, in the medium as well as in the highest stringency selection 13
out of 38
pairs sequenced had no repulsive e/g pairs, but none competed successfully
against
WinZip-A1 B1 in the selection.
Example 3: Biophysical characterization of WinZip-A1, WinZip-B1 and WinZip-
A1B1
3.1 Secondary structure and oligomerization state of the predominant pair
WinZip-A1 B1
We investigated the stability and specificity of the predominantly selected
peptides
WinZip-A1 and WinZip-B1 alone and in an equimolar mixture (WinZip-A1B1). All
experiments were performed with N-acetylated and C-amidated synthetic
peptides.
3.1.1 Peptide synthesis and purification
The peptides WinZip-A1: Ac-STTVAQLEEKVKTLRAQNYELKSRVQRLREQVAQLAS-
NH2 and WinZip-B1: Ac-STSVDELQAEVDQLQDENYALKTKVAQLRKKVEKLSE-NH2
were synthesized (Applied Biosystem 431 A) and purified by reversed-phase
HPLC.
Electrospray mass spectrometry confirmed purity and identity of the peptides
with a
mass deviation of less than 1 Da. Peptide concentrations were determined by
tyrosine
absorbance in 6 M GdnHCI (39).
3.1.2 Circular dichroism measurements
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
28
All peptides formed stable a-helical coiled coils as demonstrated by CD-
spectra.
CD spectra were recorded at 5°C at a total peptide concentration of 150
,uM (1 mm
cuvette, Aviv 62DS spectrometer). The standard buffer was 10 mM K2HP0~/KH2P04,
pH
7.0, 100 mM KF; salt concentration and pH were varied as indicated in the
respective
experiments. Thermal denaturations were measured at 222 nm in steps of
2.5°C (2 min
equilibration, 30 s averaging). Thermal transitions were >91% reversible
except where
indicated. Apparent Tm were determined by least-squares curve fitting of the
denaturation curves (40), assuming a two-state model. oTm was calculated as
Tm(WinZip-A1 B1 )-'h[Tm(WinZip-A1 )+Tm(WinZip-B1 )]. Urea denaturation
equilibria were
determined at 20°C by automated titration of native peptide with
denatured peptide in 6
M urea (30 ,uM WinZip-A1 or WinZip A1 B1, respectively, or 60 ,uM WinZip-B1)
measuring the CD signal at 222 nm (300 s equilibration, 30 s averaging). Kp
values were
calculated by linear extrapolation to 0 M denaturant assuming a two-state
model
(Kp=[unfolded monomer]2/[folded dimerJ).
The helical content was in the range of 90% (WinZip B1) to 100% (WinZip-A1 and
WinZip-A1B1). Peptide WinZip-A1 as well as the mixture WinZip-A1B1 (Fig. 4A)
were
dimeric at 10°C and 25°C over a concentration range from 10 NM
to 150 ,uM as
determined by equilibrium sedimentation. WinZip-B1 was partially unfolded as
seen both
by CD (Fig. 8C at 0 M urea) and equilibrium sedimentation.
3.1.3 Equilibrium sedimentation.
Equilibrium sedimentation experiments were performed using a Beckman XL-A
Ultracentrifuge. Absorbance was monitored at 220 and 275 nm at peptide
concentrations of 10, 50 and 150 ,uM in 10 mM K2HP0~/KH2P04, pH 7.0, 100 mM
KCI.
Partial specific volumes and solvent densities were determined as described
(41 ). The
data sets were fitted to single molecular masses of monomer, dimer and trimer.
Equilibrium sedimentation indicated a mixture of monomers and dimers, with
decreasing
amount of dimer at increasing temperature.
3.1.4 Structural stability and heterospecificity
Thermal denaturation studies at neutral pH (Fig. 9A) revealed apparent Tm
values of
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
29
28°C (WinZip-B1), 49°C (WinZip-A1), and 55°C for the
equimolar mixture of both
(WinZip-A1 B1 ). The large difference between the denaturation curve of the
heterodimer
and the average of the curves from WinZip-A1 and WinZip-B1 indicates that
heterodimers form preferentially at equilibrium. This high heterospecificity
is best
reflected in a large and positive oTm value, and indeed, we observed a oTm of
16.5°C.
To probe the mechanism of specificity, the effects of pH (Fig. 9B) and ionic
strength (Fig.
9C) were investigated. All peptides were more stable at high pH, most likely
because all
have at least one e/g-pair with two positive charges which are neutralized at
high pH.
The increased stability of WinZip-B1 at low pH could be due to the shielding
of
electrostatic repulsions resulting from its high concentration of acidic
residues. However,
the oTm is positive over the whole pH range indicating heterospecificity. The
maximum
degree of heterospecificity was observed at neutral to slightly basic pH,
consistent with
the intracellular pH of E, coli (42). High salt concentrations increased the
absolute Tm
values (Fig. 9C), presumably due to increased hydrophobic interaction or
reduced
electrostatic repulsion. However, the oTm is reduced compared to low salt
concentrations (0-100 mM), most likely due to the decreased influence of ionic
interactions at higher ionic strength.
Interestingly, the overall stability did not correlate directly with the
number of potentially
repulsive e/g-interactions. The homodimer WinZip-B1 has two same-charged ion
pairs,
but is significantly less stable than the homodimer WinZip-A1 with four same-
charged
pairs (Fig. 9). Since the overall helical propensity is comparable for both
peptides
according to (43), the difference is probably due to intrahelical
interactions. LibraryB
might be destabilized by its high local concentration of acidic residues at
the N-terminus.
This may also explain why IibraryA homodimers have generally more repulsive
and less
attractive e/g-interactions than IibraryB homodimers, since the elg-positions
play a
bigger role in the destabilization of the intrinsically more stable IibraryA
in order to
reduce homodimerization.
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
3.1.5 Native gel electrophoresis
Heterospecificity also was observed by native gel electrophoresis (Fig. 8B).
Gels (7.5% polyacrylamide (19:1 ), 375 mM f3-alanine acetate buffer, pH 4.5)
were run
with 500 mM f3-alanine acetate buffer, pH 4.5. Samples (~l0,ug peptides per
lane) were
two-fold diluted with 600 mM f3-alanine acetate, pH 4.5, 0.2% methylgreen, 10%
glycerol. Gels were prerun at 100 V for at least 45 min, run for 2-3 h at
5°C, and fixed
with 2% glutaraldehyde or 20% (w/v) TCA, respectively, before staining with
Coomassie
blue.
To obtain a significant migration, an acidic buffer (pH 4.5) had to be used,
and thus
conditions where the heterospecificity is the lowest (oTm of only 7°C,
compared to
16.5°C at neutral pH, Fig. 9C). Nevertheless, even under these
stringent conditions
heterodimers were obtained almost exclusively from the equimolar mixture (Fig.
8B),
suggesting very high heterospecificity at neutral pH, and thus indicating how
strongly
heterospecificity was selected for.
3.1.6 Kp determination
Dissociation constants of the peptides were derived from equilibrium urea
denaturations
(Fig. 8C). The heterodimer WinZip A1B1 was the most stable species with a Kp
of
approximately 24 nM, while the homodimer WinZip-A1 had a Ko of approximately
63 nM.
The accuracy of the Kp determination of WinZip-B1 is lower since it is already
partially
unfolded without denaturant (see above). The Kp was estimated to be in the 10-
5 M
range. Calculations were confirmed by determining the Kp values from thermal
denaturation curves by a van't Hoff analysis, assuming as a first
approximation a
constant off (40). We found reasonable agreement to the data obtained by urea
denaturation with a maximal deviation of Kp by a factor of 2.6.
3.1.7 Comparison to other coiled coils
Designed coiled coils are usually only judged for being stable in vitro and,
in certain
cases, for heterospecificity, whereas naturally-occurring coiled coils must
also function
reliably in a cellular environment. Similar demands are imposed on our
selection and on
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
31
further in-vivo applications, and therefore WinZip-A1B1 is best compared with
other
naturally-occurring coiled coils. The homodimeric coiled coil of the yeast
transcription
factor GCN4 has an equal or slightly higher Tm, depending on the length and
concentration of the peptides chosen (44, 45). The N terminal homodimeric
coiled coil of
the APC protein has a Tm lower by at least 9°C than WinZip-A1 B1 (46).
The
heterodimeric coiled coil from c-Jun/c-Fos shows comparable Tm and oTm values
(2).
However, those data were derived from disulfide-bridged peptides. The coiled
coil from
c-Myc/Max also heterodimerizes to a fairly high extent, but peptides of
comparable
length have a Tm of only 31 °C and a Kp of 60,uM (25°C) (45),
whereas our WinZip-A1 B1
has a Tm of 55°C and a Kp of approximately 24 nM (20°C). Thus,
WinZip-A1 B1
compares successfully with naturally-occurring coiled coils and will therefore
be very
useful for a variety of in-vivo applications.
Example 4: Chain shuffling of the WinZip-A1 B1 sequences
3.1 Introduction
In the above experiment, WinZip-A1 B1 was selected from a sample representing
2.0 x
106 library-vs-library cotransformants. As the theoretical library-vs-library
diversity is
(1.31 x 105)2 - 1.72 x 10'°, approximately 0.01 % of the library-vs-
library space was
sampled. However, we obtained a very high coverage of either single library
(theoretical
complexity of 1.31 x 105), where the probability of all members being present
at least
once is P=0.973. Thus, each polypeptide sampled only a small portion of the
opposite
library (2.0 x 106/ 1.31 x 105 - 15.4 polypeptides of the other library with
P=0.999,
assuming equal transformation rates for both libraries) and it is likely that
better
combinations for the WinZip-A1 B1 peptides may be found. Using WinZip-A1 B1 as
a
partially optimized starting point, we combined each of the two WinZip-A1 B1
polypeptides with the opposite library (W inZip-A1-DHFR[1 ] + LibB-
DHFR[2:1114A] and
WinZip-B1-DHFR[2:1114A] + LibA-DHFR[1]).
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
32
3.2 Chain shufifling
DNA from the WinZip-A1 B1 clone was isolated and retransformed into bacteria
in order
to obtain clones carrying either plasmid WinZip-A1-DHFR[1] or WinZip-B1-
DHFR[2:1114A]. A pure clone (for each) was electroporated with the appropriate
library.
Library representation was calculated by comparison with control
transformations of the
same cells with DNA from the other WinZip-A1 B1 polypeptide (calculated as the
number
of colonies growing in the presence of trimethoprim divided by the number
growing in
the absence).
Single-step selection yielded pre-selected pools for either competition. In
both cases,
the library (1.3 x 105) was over-represented by a factor of 24 and 14,
respectively, and
the probability that all members were present at least once as partners of the
"constant"
peptide is P > 0.999 and 0.882, respectively. With passages of selection
competition, a
clear increase in colony sizes was again observed, indicating that faster-
growing clones
were taking over (Fig. 3C).
3.2 Analysis
At PO and each second passage, DNA from the entire pool of cells was sequenced
in
order to follow the rate of evolution of each library against a constant
partner. Figure 10
illustrates the results from representative semi-randomized positions. It is
clear that the
rate of selection is not constant at all positions: some positions showed a
dominant
residue ( > 50%) already at P4 and clear selection ( > 90%) at P6 (see
position e2) while
others remained mixed (<50%) until P6 and became clear only at P10 (see
position g3).
This was observed in both selections. The sequences from individual colonies
were
analyzed. In both selections, a predominant clone was identified (Table 1 and
Fig. 4C),
which is similar, but not identical, to the originally selected WinZip-A1 B1
pair. The
selection of the predominant clone WinZipA2B1 (selection of LibA-DHFR[1]
against
WinZip-B1- DHFR[2:1114A]) was achieved before P10, as P10 (4 clones analyzed)
and
P12 (4 clones analyzed) revealed only this clone. The selection of the
predominant
clone WinZipAlB2 (selection of LibB-DHFR[2:1114A] against WinZip-A1-DHFR[1])
was
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
33
clear but not complete after 12 passages, as it was identified 4/6 times in
P12 and 3/5
times in P10.
Analysis of the biophysical properties of the peptide pairs selected in the
chain shuffling
experiments, performed as described in Examples 2 and 3, indicated that the
observed
e/g-interaction pattern is similar to that of W inZip-A1 B1.
Conclusion
We applied a fast and simple strategy, a library-vs-library selection with the
fragment
complementation assay, to select for a metabolically stable, dimeric and
highly
heterospecific coiled coil with high affinity. Comparison of the outcome of
various
selections performed with different stringencies revealed insight into which
properties
are selected already at lower stringency, and are thus the most crucial for
successful
heterodimerization, and those which only become apparent at higher selection
stringency, and thus represent a more subtle optimization. The most striking
selection
occurred at the core a-position for Asn-pairs, revealing that structural
uniqueness is
essential for efficient and selective heterodimerization. Furthermore,
comparison of
selected e/g-pairs from hetero- and putative homodimers indicated selection
for stability
even at the lowest stringency, whereas selection for heterospecificity was
more
pronounced at higher stringency. Heterospecificity was achieved not only by
decreasing
the numbers of repulsive e/g-interactions but also by increasing the number of
attractive
interactions in the heterodimer relative to the homodimers.
The selection for heterospecificity (and thus against homodimers) may be a
unique
feature of this selection system. Not only is active enzyme exclusively formed
by parallel
heterodimers, but homodimers and higher oligomers are likely to have a
negative effect
by unproductively wasting fragments and perhaps even harmfully accumulating
non-
functional enzyme. Dimer stability, in turn, is dependent not only on e/g-pair
interactions,
but also on helical propensity, intrahelical interactions and helix dipole
stabilization.
Indeed, our analysis revealed that the most successful variants do not simply
consist of
complementary charges in the e/g-positions, but show a more complicated
pattern,
presumably fulfilling a variety of naturally conflicting demands on the
sequence, whose
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
34
optimum would have been extremely challenging to predict.
The biophysical characterization of the predominantly selected pair WinZip-A1
B1
revealed the formation of a stable dimeric coiled coil with very high
heterospecificity,
confirming the results from the sequence analysis and the validity of the
selection
strategy. The results obtained with WinZip-A1 B1 and the peptide pairs
identified in the
chain shuffling experiment, are supporting the view that idealized sequences,
based on
the single principle of merely relieving repulsive e/g-interactions in the
homodimers with
complementary charges in the heterodimer, may not be optimal for biological
applications
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
TABLE 1
0
~~ O ~ '~ ~
w 0 0 0 > o0 y
i o ~ .~ ~~
o . y ~
--. ~ M b ,
U N ~, N ~ '
U ~ O ~C V
) ,~ N M o 1 o .b Y w
o O
~ O
N o b O .~ U . O
~ U
~ w ..C'~ p
N O O .~
O 0 i~, ~...~ ~ b 3. .
~ " ~ y ~ >G
00 ~ ~ U O Y
~ O 0
N ~ 'b y
N ~ N y ~ 0
_. p ~ ~ ' G
'-' ~ ~ ~ y ~
~ p~ O. ~ fl v~ ~ ~=._O
CL ~N N .~
. N N o ~ 'd
~ ~ ~ ~ ~ ,~ N
3 3 :n a, w
~ w o
, n ~ ~_ O O o U
N ~ y ~ .O GADU
. >_ O O O U Gj '~ O O O ~S
O ~ ~G ~ ~ '-' ~ G. ~ O
cUC c_C G1 M M cd ~+ b v' N 4~
-" ,--,~d ~ .~ p U t~.
p r0-i .Ur. O N .: cn N
O w ~ c3 ~
U U O O ~ y ~ U
'O U U ~ ' v~
Y ap U ~' v~
0 O i-~-~00 U ~,Oct3
U ~ Y ~ 3
., ~~ 0 ~ ~ .x w
G . z ~ o ~
.~ " ~
_ _
o ~ x
~ o n L ~f/7
~ O x ' 3 0, c U
' n. ~ ~ ~ o _; >
w o ~ ' ~' ~ ~ b
Q b
~ ~ ~_ ~_ b0 O 'v-'N U '~",O
"'~ ~"~~ '~ ~ ''~~ U O
U ' y N O O
~ ~ + .G O .~ V U
O O O ~ f.. U c~ Q
U b0 ;r Q ~ O U b U ~
y CL L~,V > ,
cn ~ ' N N ,~ ~ U ~
, x on ~ b
rx O ~ 3 3 ~' ~ ~' '?
H w :~ ~
y G C a~ i ~ ~.
~ -' o N ~ by A"
3 ~ G. ~ ~ ' ~ ittO
~ U ~ y ~ ~ U
v v ~ ~ ~' tl7~ H o ~ ~,
.~
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
36
REFERENCES
1. Rock et al. (1992), Prot. Eng. 5, 583-591.
2. O'Shea, E. K., Rutkowski, R. & Kim, P. S. (1992) Cell 68, 699 708.
3. Pack, P. & Pluckthun, A. (1992) Biochemistry 31, 1579-84.
4. Weissenhorn, W., Calder, L. J., Dessen, A., Laue, T., Skehel, J. J. &
Wiley, D. C.
(1997) Proc. Natl. Acad. Sci. USA 94, 6065-6069.
5. Pack et al. (1993) Bio/Technology 11, 1271-1277
6. Pack, P., Muller, K., Zahn, R. & Pluckthun, A. (1995) J. Mol. Biol. 246, 28-
34.
7. Hurst, H. C. (1995) Protein Profile 2, 101-68.
8. Hodges, R. S. (1996) Biochem. Cell Biol. 74, 133-54.
9. Harbury, P. B., Kim, P. S. & Alber, T. (1994) Nature 371, 80-3.
lO.Harbury, P. B., Zhang, T., Kim, P. S. & Alber, T. (1993) Science 262, 1401-
7.
ll.Jelesarov, I. & Bosshard, H. R. (1996) J. Mol. Biol. 263, 344-58.
l2.Zhou, N. E., Kay, C. M. & Hodges, R. S. (1994) Protein Eng. 7, 1365-72.
13.0'Shea, E. K., Lumb, K. J. & Kim, P. S. (1993) Curr. Biol. 3, 658 667.
14. Graddis, T. J., Myszka, D. G. & Chaiken, I. M. (1993) Biochemistry 32,
12664-71.
15. Nautiyal, S., Woolfson, D. N., King, D. S. & Alber, T. (1995) Biochemistry
34, 11645-
51.
16. Pack (1994) Ph.D. Thesis, Ludwig-Maximilians-Universitat Munich, Germany.
17. Muller, K. M., Arndt, K. M., Strittmatter, W. & Pliackthun, A. (1998) FEBS
Lett. 422,
259-264.
18. Pelletier, J.N., Remy, I. and Michnick, S. W. (1998) J. Biomol. Tech.,
http://www.abrf.org/JBT/Articles/JBT0012/jbt0012.html.
19. Pelletier, J. N., Campbell-Valois, F. X. & Michnick, S. W. (1998) Proc.
Natl. Acad.
Sci. USA 95, 12141-12146.
20.Sydor, J.R., Engelhard, M., Wittinghofer, A., Goody, R.S. & Herrmann, C.
(1998)
Biochemistry 37, 14292-14299.
2l.Chen, J., Zheng, X.F., Brown, E.J. & Schreiber, S.L. (1995) Proc. Natl.
Acad. Sci. U
S A 92, 4947-4951.
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
37
22.Junius, F. K., Mackay, J. P., Bubb, W. A., Jensen, S. A., Weiss, A. S. &
King, G. F.
(1995) Biochemistry 34, 6164-74.
23.0'Shea, E. K., Klemm, J. D., Kim, P. S. & Alber, T. (1991 ) Science 254,
539-44.
24. Potekhin, S. A., Medvedkin, V. N., Kashparov, I. A. & Venyaminov, S.
(1994) Protein
Eng. 7, 1097-101.
25. Lumb, K. J. & Kim, P. S. (1995) Biochemistry 34, 8642-8.
26.Ogihara, N. L., Weiss, M. S., DeGrado, W. F. & Eisenberg, D. (1997) Protein
Sci. 6,
80-8.
27.Virnekas, B., Ge, L., Pliackthun, A., Schneider, K. C., Wellnhofer, G. &
Moroney, S.
E. (1994) Nucleic Acids Res. 22, 5600-7.
28. Krylov, D., Mikhailenko, I. & Vinson, C. (1994) EMBO J. 13, 2849 61.
29. John, M., Briand, J. P., Granger-Schnarr, M. & Schnarr, M. (1994) J. Biol.
Chem.
269, 16247-53.
30.Cohen, C. & Parry, D. A. (1990) Proteins 7, 1-15.
31. Woolfson, D. N. & Alber, T. (1995) Protein Sci. 4, 1596-1607.
32.Dasgupta, S. & Bell, J. A. (1993) Int. J. Pept. Protein Res. 41, 499-511.
33. Richardson, J. S. & Richardson, D. C. (1988) Science 240, 1648 52.
34. Doig, A. J. & Baldwin, R. L. (1995) Protein Sci. 4, 1325-36.
35. Chakrabartty, A., Doig, A. J. & Baldwin, R. L. (1993) Proc. Natl. Acad.
Sci. USA 90,
11332-6.
36.Kohn, W. D., Kay, C. M. & Hodges, R. S. (1997) J. Pept. Sci. 3, 209-23.
37. Yu, Y., Monera, O. D., Hodges, R. S. & Privalov, P. L. (1996) Biophys.
Chem. 59,
299-314.
38. Edelhoch, H. (1967) Biochemistry 6, 1948-54.
39. Becktel, W. J. & Schellman, J. A. (1987) Biopolymers 26, 1859-77.
40. Laue, T. M., Shah, B. D., Ridgeway, T. M. & Pelletier, S. M. (1992) in
Analytical
Ultracentrifugation in Biochemistry and Polymer Science, eds. Harding, S. E.,
Rowe,
A. J. & Horton, J. C. (Royal Society of Chemistry, Cambridge), pp. 90-125.
4l.Oliver, D. B. (1996) in Escherichia coli and Salmonella, ed. Neidhardt, F.
C. (ASM
Press, Washington, D. C., U.S.A.), Vol. 1, pp. 88-103.
CA 02377513 2001-12-21
WO 01/00814 PCT/EP00/05922
38
42.0'Neil, K. T. & DeGrado, W. F. (1990) Science 250, 646-51.
43.Thompson, K. S., Vinson, C. R. & Freire, E. (1993) Biochemistry 32, 5491-6.
44.Lumb, K. J., Carr, C. M. & Kim, P. S. (1994) Biochemistry33, 7361-7.
45.Joslyn, G., Richardson, D. S., White, R. & Alber, T. (1993) Proc. Natl.
Acad. Sci.
USA 90, 11109-13.
46. Muhle-Goll, C., Nilges, M. & Pastore, A. (1995) Biochemistry 34, 13554-64.