Note: Descriptions are shown in the official language in which they were submitted.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-1-
METHODS, DEVICES, ARRAYS AND KITS
FOR DETECTING AND ANALYZING BIOMOLECULES
Reference to Related Applications
This application is a Continuation in Part (CIP) of U.S. Patent Application
No. 09/7S3,S74
(filed January 4, 2001), which is a CIP of U.S. Patent Application No.
09/718,990 (Filed November
20, 2000), which is a CIP of International Patent Application No. US00/20354,
filed July 26, 2000
and published in the English language, and claims the benefit of U.S.
Provisional Patent Application
No. 60/145,613 (filed July 26, 1999). The current application further claims
the benefit of U.S.
Provisional Patent Application Nos. 60/286,258 (filed April 25, 2001),
60/304,031 (filed July 9,
2001), and 60/296,475 (filed June 8, 2001). Each of these related applications
is incorporated herein
in their entirety.
Statement of Government Rights
At least one of the inventors is an employee of an agency of the Government of
the United
States, and the Government may have certain rights in this invention.
Field of the Disclosure
The present disclosure is directed to methods, devices, arrays, and kits for
identifying and
analyzing large numbers of biomolecules in a sample, such as a biological
sample. The disclosure
further relates to using these methods, devices, arrays, and kits to help
determine the function and
role of biomolecules in disease, and to correlating the presence, absence, or
quantity of a
combination of biomolecules with particular diseases, prognoses, or responses
to therapies.
2S Background of the Disclosure
Now that the 50,000 or so genes that make up the human genome have been
sequenced,
tools are needed to determine when and in what type of tissue those genes are
active so as to ascertain
their function and role, particularly in disease. This effort, often referred
to as "functional genomics"
and "proteomics," is especially important in efforts to discover new drugs
since new pharmaceutical
agents axe being designed to target specific enzymes, receptors, and other
proteins. Eventually,
proteomic information will be used in clinical diagnostics to help guide
treatment selection in the
emerging era of "personalized medicine:'
Some believe that the 100,000 human genes may turn out to produce up to a
million
different protein variants due to post-translational and other modifications.
Within the next decade
3S the pharmaceutical industry is expected to identify up to 10,000 proteins
against which human
therapeutics can be directed. Additional therapeutics, gene modifiers,
expression modifiers, and
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
_2_
valuable biomolecules also are expected to be developed or identified through
the extension of
proteomics to the analysis of non-human animals and plants.
Although there may be up to a million different protein variants in humans,
only about
10,000-15,000 proteins are expressed in any particular cell type. Thus, for
example, liver cells have
essentially the same genome as skin cells taken from the same individual, but
the two cell
populations express substantially different sets of proteins. It is often
desirable, therefore, to profile
and compare the patterns of proteins (i.e., the "proteome" of a cell) in
different cell populations (e.g.,
diseased and normal tissue; fetal and mature tissue; human and non-human
tissue, etc.) to identify
targets for drugs.
One common approach to establishing or confirming the association of gene
activity with
disease is through expression analysis. DNA microarrays are used to survey
differential expression
patterns of thousands of genes from extracts taken from samples of tissues
representing various
diseases. If particular genes are expressed in diseased tissue but not in
normal tissue they may be
relevant as diagnostic markers and targets of pharmaceutical intervention. One
disadvantage with
this approach is that the sample being tested is disassociated from the tissue
from which it was
isolated, thereby losing the ability to observe gene expression patterns in
the context of the tissue in
which the genes are active. Since the morphological relationship is not
preserved in microarray
analysis, it is hard to know what component of the sample is responsible for
the changes observed in
gene expression. Also, microarray analysis is usually performed on a
homogenized sample of tissue,
making it virtually impossible to ascribe expression to a specific cell type,
let alone a specific cell.
In situ detection and visualization of proteins traditionally has been
accomplished through
immuno-histochemistry (IHC). This technique involves the mounting a thin
tissue section on the
glass slide and visualizing a protein of interest with a detectable antibody
that has specific binding
affinity for the target protein. Because of certain technical limitations of
IHC, only one or two
proteins from a single tissue section can be achieved. Also, proteins are
still embedded in the tissue
and are not presented to the antibodies in the most appropriate way (proteins
are not highly
denatured) lowering the success rate of the antibody reactivity.
The most widely used method for identifying and measuring proteins and nucleic
acids that
have been removed from tissue samples is gel electrophoresis. Electrophoresis
generally refers to
techniques for separating or resolving molecules in a mixture under the
influence of an applied
electric field. Separation is based on difference in (usually) the size and/or
charge of the molecules.
Molecules separated by electrophoresis are often visualized by staining with a
non-specific dye, such
as Coomassie blue (for proteins) or ethidium bromide (for nucleic acids). Such
dye staining does not
specifically identify individual molecules. Furthermore, ubiquitous dye
staining is generally not very
sensitive.
More sensitive detection methods exist, such as antibody-based detection for
proteins. In
particular, immunoblotting, also known as "Western blotting," is often used to
detect gel-separated
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-3-
proteins. This technique uses detectable antibodies specific to the proteins
of interest in lieu of a
ubiquitous stain. A key limitation of the technique is its low throughput; at
most only a handful of
proteins can be identified from a single lane of an immunoblot on a single
blot, due to overlapping
banding patterns and cross reactivity of antibodies with different proteins in
the sample. Thus,
immunoblotting is typically performed using only one antibody per membrane to
ensure specificity.
Though it is possible to strip and re-probe an immunoblot, stripping will also
remove
protein of the sample that had been bound to the membrane, thus encumbering
quantitative analysis
of the sample. Moreover, the proportion of each individual protein removed
from the membrane by
such treatment will vary depending upon the nature of the protein, which
further clouds efforts to
quantitate the relative amounts of protein initially present in the sample.
There remains a clear need
to develop blotting techniques that permit larger numbers of antigens to be
detected simultaneously
from a single test sample.
It would be desirable to have high-throughput approaches for detecting,
identifying and
comparing large numbers of biomarkers that is relatively inexpensive, can be
used by ordinary
laboratory personnel, and readily permits the capture, organization, and
analysis of the data generated
thereby.
Summary of the Disclosure
The present disclosure is directed to devices and methods for detecting
biomolecules in a
substantially two-dimensional sample (e.g., tissue section, tissue array,
electrophoretic gel, and so
forth) by creating substantial copies of the biomolecules eluted from the
sample. The biomolecules
then can be visualized on the copies using detectors, for example antibodies
or DNA probes, having
specific affinity for the biomolecules of interest.
The present disclosure is further directed to methods and devices for
identifying the pattern
of biomolecules (e.g., proteins and nucleic acids) expressed in tissue
samples, and for correlating the
expression pattern with, for instance, various diseases, prognoses, or
responses to therapies.
Provided herein are methods of detecting biomolecules in a sample, which
methods involve
providing a stack of at least two layered membranes; applying the sample to
the stack under
conditions that permit movement of the biomolecules through multiple layered
membranes of the
stack, and allow capture of at least a portion of the biomolecules on the
membranes, and detecting the
biomolecules on one or more of the multiple membranes. In specific examples of
such methods, the
biomolecules are captured directly by the membranes. Certain membranes for use
in such methods
have a high affinity but low capacity for biomolecules, for instances
proteins, nucleic acids, lipids,
carbohydrates, or combinations thereof.
Another embodiment of the disclosure is a method of making multiple
substantial copies
(which need not be identical) of a biological sample. These methods involve
providing a stack of
layered membranes, wherein the membranes permit biomolecules applied to the
stack to move
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-4-
through a plurality of the membranes, while capturing (for instance, directly)
at least a portion of the
biomolecules on multiple membranes and applying the biological sample to the
stack, under
conditions that allow the multiple membranes to capture at least a portion of
the biomolecules from
the sample. This forms the multiple substantial copies of the biological
sample.
Samples for use in examples of provided methods are (or can be made)
substantially two-
dimensional; representative non-limiting types of samples include tissue
sections, tissue microarrays,
tissue macroarrays, laser capture microdissected tissue samples, and
electrophoretic gels (e.g., 1-D or
2-D electrophoretic gels).
Yet a further embodiment is a method of creating a set of microarray copies,
which method
involves providing a stack of layered membranes, and applying a plurality of
molecules (e.g., DNA
probes, antibodies, or a combination thereof), to the stack of layered
membranes. In examples of
such methods, the stack of layered membranes includes a plurality of
substrates through which the
molecules move, and in which a portion of the molecules are directly captured
by one or more of the .
substrates.
Another specific embodiment is a method of analyzing biomolecules in a tissue
sample,
which method involves providing at least one membrane (in some embodiments, a
plurality of
membranes), positioning the at least one (or more) membrane in contact with
the tissue sample, and
applying heat and/or pressure to the tissue sample, whereupon biomolecules are
transferred from the
tissue sample onto the at least one membrane (referred to generally as contact
transfer). One or more
of the biomolecules can then be analyzed on the at least one membrane.
Another example of a provided method is a method of replicating biomolecular
content of a
tissue array (such as a micro- or macroarray), which method involves providing
the tissue array and
transferring biomolecules from the tissue array onto a plurality of membranes
so as to produce at
least one replicate of the biomolecular content of the tissue array.
The disclosure also provides a method of analyzing cellular material embedded
on an LCM
transfer film, which method involves providing one or more membranes,
positioning the one or more
membranes adjacent to the LCM transfer film, transferring biomolecules from
the cellular material to
the one or more membranes, and detecting the biomolecules on the membranes.
Further encompassed methods include methods for analyzing the proteome of a
biological
sample. Examples of such methods involve separating at least one protein from
another protein
present in the biological sample, transferring a portion of the separated
protein to a plurality of
membranes in a stacked configuration, incubating each of the membranes in the
presence of one or
more different species of predetermined ligand molecules (or detector
molecules) under conditions
sufficient to permit binding between the separated protein and a
ligand/detector capable of binding to
such protein; and analyzing the proteome by determining the occurrence of
binding between the
protein and any of the species of predetermined ligand molecules.
A further embodiment is a method for identifying biomolecules that have been
separated on
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
_S_
a solid support (e.g., a 1-D or 2-D gel). Such methods involve providing a
solid support containing
the separated biomolecules, wherein the support has an upper side and a lower
side, applying a first
stack of membranes to the upper side and a second stack of membranes to the
lower side, permitting
the biomolecules to be transferred from the support to the first and second
membrane stacks, and
separating the membranes. The transferred biomolecules can then be detected,
identified, or
otherwise analyzed on at least one of the membranes.
The disclosure also provides kits. Examples of kits include a membrane array
for detecting
biomolecules in a sample, and one or more containers of detector molecules for
detecting molecules
captured on membranes of the array. Arrays included in such kits contain a
plurality of membranes,
each of which has substantially a same affinity for biomolecules that may be
analyzed using the kit.
Another kit embodiment is a kit for comparing the molecular profiles of tissue
samples.
Such kits contain at least one tissue microarray, and at least one replicate
of the tissue microarray.
Replicates contained in such kits may be made, for instance, using methods
described herein.
Also provided are kits for replicating a pattern of biomolecules from a tissue
sample, which
kits include a plurality of membranes, each having a coating on its upper
and/or lower surfaces to
increase specific binding of a target biomolecule, a quantity of transfer
buffer, and a fluid impervious
enclosure (for instance, a heat-sealable bag).
Another example of a described kit is a kit for analyzing a proteome, which
kit contains a
plurality of membranes, each having a affinity for at least one protein, and a
plurality of reagent
species (such as detector molecules, particularly labeled detectors), each
species is adapted to detect
one or more specific proteins bound to the membranes.
Further embodiments are membranes unit for use in blotting, which unit
includes a stack of
at least two porous membranes (examples of which have a thickness no greater
than about 30
microns), and a frame, mounted to the membranes, which has a thickness no
greater than about 300
microns.
Also provided are porous membranes having a high affinity but low capacity for
biomolecules. Examples of such membranes include a core substrate and a
coating, and generally are
no more than about 30 microns thick. Specific examples of such membranes
contain polycarbonate
in the core substrate and nitrocellulose in the coating.
The foregoing and other advantages and features will become hereinafter
apparent, and may
be more clearly understood by reference to the following detailed description,
the appended claims,
and the several views illustrated in the drawings.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-6-
Brief Description of the Drawings
FIG. 1 is a perspective view of a membrane array, shown transferring molecules
from a
tissue section using wicking transfer.
FIG. 2A is an oblique view of an apparatus shown transferring molecules from a
tissue
S section onto a membrane stack. FIG. 2B is a front view of an assembled
contact transfer stack,
prepared for transfer in the apparatus illustrated in FIG. 2A.
FIG. 3 is a photograph of a typical tissue microarray on a slide.
FIG. 4A is a schematic illustration showing the components of a kit according
to one
embodiment. FIG. 4B is a perspective view of a membrane stack.
FIG. 5 is a schematic illustration showing a method according to one provided
gel-transfer
embodiment.
FIG. 6 is a sectional view of a stack of membranes shown operatively engaged
with an
apparatus to transfer proteins from a gel onto the membranes.
FIG. 7A is a perspective view of a typical prior art LCM instrument. FIG 7B is
an enlarged
perspective view of an LCM cap shown engaged with a glass slide via a
transport arm. FIGS. 7C and
7D are side elevation views showing the transfer of cellular material from a
tissue section on a glass
slide to an LCM cap.
FIG. 8 is a longitudinal section view of one embodiment, in which LCM samples
have been
prepared for transfer through a membrane stack.
FIG. 9 is perspective view of one LCM transfer embodiment, shown in use and
operation.
FIG. 10A is a side elevation view of a modified LCM cap according to provided
embodiments. FIG. l OB is a section view taken along line B-B of FIG. 4A.
FIG. 11 is a perspective view of a transfer array shown in use with a
microtiter plate.
FIG. 12 is a longitudinal sectional view of an individual membrane according
to one
provided embodiment.
FIG. 13 is a schematic drawing, illustrating direct capture.
FIG. 14 is a schematic drawing, illustrating indirect capture.
FIG. 15 is a schematic illustration showing a method according to another gel-
transfer
embodiment.
FIG. 16 is a perspective view of a representative framed membrane stack.
FIG. 17 is a front elevation view of a single framed membrane.
FIG. 18 is a sectional view of the single membrane taken along line 115-115 of
FIG. 17.
FIG. 19 is a schematic illustration showing a hypothetical example
illustrating the method of
creating the antibody cocktails. The Gel (A) shows proteins as detected by
Coomassie Blue staining
prior to transfer. Membrane -Layer #1 (B), Membrane -Layer #2 (C), and
Membrane -Layer #3 (C)
show proteins detected on membranes with antibodies.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
_7_
FIG. 20 is a schematic illustration showing the components of a kit according
to one
embodiment.
FIG. 21 is an oblique view of a pressure heater apparatus.
FIG. 22 is a longitudinal section view of a stack of membranes shown with
apparatus to
transfer proteins from' a gel onto the membranes.
FIG. 23 is a schematic illustration of one embodiment in use and operation,
showing the
transfer of proteins from a gel to the membrane stack so as to create multiple
replicates of the protein
content of the gel.
FIG. 24 is a sectional view of a stack of membranes shown operatively engaged
with an
apparatus to transfer proteins from a gel onto the membranes.
FIG. 25 is a schematic illustration showing a comparison between a template
image with a
sample membrane.
FIG. 26 is a schematic illustration comparing the binding capacity of
membranes
constructed of nitrocellulose and polycarbonate, both coated and uncoated.
FIG. 26A shows scanned
images of the membranes incubated in protein comparing the intensity of
signal. FIG. 26B is a chart
plotting the amount of protein bound to different membrane materials.
FIG 27 shows images of tissue sections that show that portions of total
biomolecules can be
successfully transferred through a stack of polycarbonate (PC) layers onto the
trap. FIG. 27A shows
transfer through polycarbonate membranes. FIG. 27B shows transfer through
polycarbonate coated
with nitrocellulose. FIG. 27C shows transfer through polycarbonate coated with
poly-L-lysine
membranes.
FIG. 28 is a series of images showing immunodetection of different proteins
from two
regions (healthy and cancerous) of a breast tissue using the membrane array.
FIG. 29 is a series of photographs of four membrane replicates of a tissue
microarray. The
top row shows total protein staining of the replicates with a ubiquitous
stain; the bottom row shows
immunodetection of two specific proteins, keratin and prostate specific
antigen (PSA).
FIG. 30 is a series of photographs showing a tissue microarray before transfer
(stained with
hematoxylin and eosin (H&E)) and four replicates thereof immunodetected with
antibodies to four
different proteins (keratin, PSA, p53, and p300) as indicated.
FIG. 31 is a series of photographs showing total proteins captured on the
membranes (first
column) and immunodetection of cytokeratin (second column).
FIG. 32 is a photograph of images of the membranes with biotinylated protein
bound to
them. Proteins were separated by 1-D PAGE, transferred through the membrane
stack and visualized
with streptavidin-allcaline phosphatase complex (strep-AP) and enhanced
chemiluminescence
(ECL)reagent.
FIG. 33 is a photograph of images of the membranes with Rsk and p300 proteins
bound to
them. Protein separation and blotting was performed as stated in FIG.15.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
_g_
FIG. 34 is a photograph of images of the membranes with GAPDH, Alpha-tubulin
and Beta-
actin bound to them. Proteins were separated by 2-D PAGE, transferred through
the membrane stack
and visualized with primary-secondary antibody-alkaline phosphatase
complex and ECL reagent.
FIG. 35 is a photograph of images of the membranes with protein or DNA
attached to them
and a diagram that explains the relationship between different protein-DNA
complexes and their
position in the gel.
FIG. 36 is line graph showing the relationship between protein loading on the
gel, protein
size, and uniformity of transfer to the membranes.
FIG. 37 is a photograph showing differential expression of gel-separated
proteins from three
cell samples (Jurkat, HN12, and SW480) blotted onto a seven-layer stack of
membranes.
FIG. 38 is a photograph showing differential expression of gel-separated
proteins from four
cell samples blotted onto a ten-layer stack of membranes. The upper row
(marked "Total Staining")
shows the membranes stained ubiquitously with a dye. The bottom row (marked
"ECL") shows the
membranes probed with the indicated antibodies.
FIG. 39 is a photograph showing distribution of total protein transferred by a
method
provided herein.
Brief Description of the Sequence Listing
SEQ ID NO: 1 shows the nucleic acid sequence of a 43-residue synthetic
hybridization
oligonucleotide.
Detailed Description of the Disclosure
1. Explanation of Certain Ternas
"Addressable" refers to that which is capable of being reliably and
consistently located and
identified, as in an addressable location on an array or a gel.
"Affinity" means the chemical attraction or force between molecules.
"Antibody cocktails" means mixtures of between two to about 100 different
detector
antibodies.
"Array" means two or more.
"Biological sample" means any solid or fluid sample obtained from, excreted by
or secreted
by a living organism (including microorganisms, plants, animals, and humans).
"Biomolecules" are molecules typically produced by living organisms including
peptides,
proteins, glycoproteins, nucleic acids, fatty acids, and carbohydrates.
"Capacity" means the ability to receive, hold, or absorb biomolecules from the
sample.
"Captor" means a molecule, such as an antibody or DNA probe, that is anchored
to a
membrane and has an affinity (such as a specific affinity) for one of the
biomolecules.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-9-
biomolecule is not directly conjugated to the membrane.
"cDNA" refers to a DNA molecule lacking internal, non-coding segments
(introns) and
regulatory sequences which determine transcription. cDNA may be synthesized in
the laboratory by
reverse transcription from messenger RNA extracted from cells.
"Counter-ligand staining" is intended to refer to any detection technique that
detects the
presence of ligand that is not bound to a protein of the biological sample,
and thus reveals (as, for
example, by an absence of staining, etc.) the presence of ligand that is bound
to a protein of the
biological sample
"Detector" means a molecule, such as an antibody or DNA probe, that is free in
solution
(i. e. not anchored to a membrane) and has an affinity for one of the sample
components.
"Direct capture" means the conjugation or binding of a biomolecule directly
onto the
surface of the membrane without the aid of a captor antibody or the like.
"DNA" is a long chain polymer that contains the genetic material of most
living organisms
(the genes of some viruses are made of ribonucleic acid (RNA)). The repeating
units in DNA
polymers are four different nucleotides, each of which includes one of the
four bases (adenine,
guanine, cytosine and thymine) bound to a deoxyribose sugar to which a
phosphate group is attached.
Triplets of nucleotides (referred to as codons) code for each amino acid in a
polypeptide, or for a
stop signal. The term "codon" is also used for the corresponding (and
complementary) sequences of
three nucleotides in the mRNA into which the DNA sequence is transcribed.
"EST" (Expressed Sequence Tag) is a partial DNA or cDNA sequence, typically of
between
500 and 2000 sequential nucleotides, obtained from a genomic or cDNA library,
prepared from a
selected cell, cell type, tissue or tissue type, organ or organism, which
corresponds to an mRNA of a
gene found in that library. An EST is generally a DNA molecule sequenced from
and shorter than
the cDNA from which it is obtained.
"Fluorophore" refers to a chemical compound, which when excited by exposure to
a
particular wavelength of light, emits light (i.e., fluoresces), for example at
a different wavelength.
Fluorophores can be described in terms of their emission profile, or "color."
Green fluorophores, for
example Cy3, FITC, and Oregon Green, are characterized by their emission at
wavelengths generally
in the range of 515-540 ~,. Red fluorophores, for example Texas Red, Cy5 and
tetramethylrhodamine, are characterized by their emission at wavelengths
generally in the range of
590-690 ~,.
Examples of fluorophores that may be used are provided in U.S. Patent No.
5,866,366 to
Nazarenko et al., and include for instance: 4-acetamido-4'-
isothiocyanatostilbene-2,2'disulfonic acid,
acridine and derivatives such as acridine and acridine isothiocyanate, 5-(2'-
aminoethyl)aminonaphthalene-1-sulfonic acid (EDANS), 4-amino-N-[3-
vinylsulfonyl)phenyl]naphthalimide-3,5 disulfonate (Lucifer Yellow VS), N-(4-
anilino-1-
naphthyl)maleimide, anthranilamide, Brilliant Yellow, coumarin and derivatives
such as coumarin, 7-
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-10-
amino-4-methylcoumarin (AMC, Coumarin 120), 7-amino-4-trifluoromethylcouluarin
(Coumaran
151); cyanosine; 4',6-diaminidino-2-phenylindole (DAPI); 5', 5"-
dibromopyrogallol-sulfonephthalein
(Bromopyrogallol Red); 7-diethylamino-3-(4'-isothiocyanatophenyl)-4-
methylcoumarin;
diethylenetriamine pentaacetate; 4,4'-diisothiocyanatodihydro-stilbene-2,2'-
disulfonic acid; 4,4'-
diisothiocyanatostilbene-2,2'-disulfonic acid; 5-[dimethylamino]naphthalene-1-
sulfonyl chloride
(DNS, dansyl chloride); 4-(4'-dimethylaminophenylazo)benzoic acid (DABCYL); 4-
dimethylaminophenylazophenyl-4'-isothiocyanate (DABITC); eosin and derivatives
such as eosin
and eosin isothiocyanate; erythrosin and derivatives such as erythrosin B and
erythrosin
isothiocyanate; ethidium; fluorescein and derivatives such as 5-
carboxyfluorescein (FAM), 5-(4,6-
dichlorotriazin-2-yl)aminofluorescein (DTAF), 2'T-dimethoxy-4'S'-dichloro-6-
carboxyfluorescein
(JOE), fluorescein, fluorescein isothiocyanate (FITC), and QFITC (XRITC);
fluorescamine; IR144;
IR1446; Malachite Green isothiocyanate; 4-methylumbelliferone; ortho
cresolphthalein;
nitrotyrosine; pararosaniline; Phenol Red; B-phycoerythrin; o-
phthaldialdehyde; pyrene and
derivatives such as pyrene, pyrene butyrate and succinimidyl 1-pyrene
butyrate; Reactive Red 4
(Cibacron ® Brilliant Red 3B-A); rhodamine and derivatives such as 6-
carboxy-X-rhodamine
(ROX), 6-carboxyrhodamine (R6G), lissamine rhodamine B sulfonyl chloride,
rhodamine (Rhod),
rhodamine B, rhodamine 123, rhodamine X isothiocyanate, sulforhodamine B,
sulforhodamine 101
and sulfonyl chloride derivative of sulforhodamine 101 (Texas Red); N,N,N',N'-
tetramethyl-6-
carboxyrhodamine (TAMRA); tetramethyl rhodamine; tetramethyl rhodamine
isothiocyanate
(TRITC); riboflavin; rosolic acid and terbium chelate derivatives.
Other suitable fluorophores include GFP (green fluorescent protein),
LissamineTM,
diethylaminocoumarin, fluorescein chlorotriazinyl, naphthofluorescein, 4,7-
dichlororhodamine and
xanthene and derivatives thereof. Other fluorophores known to those skilled in
the art may also be
used.
"High throughput genomics" refers to application of genomic or genetic data or
analysis
techniques that use microarrays or other genomic technologies to rapidly
identify large numbers of
genes or proteins, or distinguish their structure, expression, or function
from normal or abnormal
cells or tissues.
"Hybridization" refers to an interaction between nucleic acid molecules that
are
complementary to each other. Hybridization is based on hydrogen bonding, which
includes Watson-
Crick, Hoogsteen or reversed Hoogsteen hydrogen bonding between complementary
nucleotide
units. For example, adenine and thymine are complementary nucleobases that
pair through formation
of hydrogen bonds. "Complementary" refers to sequence complementarity between
two nucleotide
units. For example, if a nucleotide unit at a certain position of an
oligonucleotide is capable of
hydrogen bonding with a nucleotide unit at the same position of a DNA or RNA
molecule, then the
oligonucleotides are complementary to each other at that position. The
oligonucleotide and the DNA
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-11-
or RNA are complementary to each other when a sufficient number of
corresponding positions in
each molecule are occupied by nucleotide units which can hydrogen bond with
each other.
"Specifically hybridizable" and "complementary" are terms that 'indicate a
sufficient degree
of complementarity such that stable and specific binding occurs between the
oligonucleotide and the
DNA or RNA target. An oligonucleotide need not be 100% complementary to its
target DNA
sequence to be specifically hybridizable.
Hybridization conditions resulting in particular degrees of stringency will
vary depending
upon the nature of the hybridization method of choice and the composition and
length of the
hybridizing DNA used. Generally, the temperature of hybridization and the
ionic strength (especially
the Na+ concentration) of the hybridization buffer will determine the
stringency of hybridization.
Calculations regarding hybridization conditions required for attaining
particular degrees of
stringency are discussed by Sambrook et al. Molecular Cloning: A Laboratory
Manual, Cold Spring
Harbor Laboratory Press (1989), chapters 9 and 1 l, herein incorporated by
reference
"Indirect capture" means the conjugation or binding of a biomolecule onto a
captor antibody
or the like which in turn is bound to the surface of the membrane. Thus, with
indirect capture the
biomolecule is not directly conjugated to the membrane.
"Identical" means having substantially the same affinity for biomolecules.
"Label" refers to detectable markers or reporter molecules, which can be
attached for
instance to a specific biomolecule (e.g., a protein or nucleic acid). Typical
labels include
fluorophores, radioactive isotopes, ligands, chemiluminescent agents, metal
sols and colloids, and
enzymes. Methods for labeling and guidance in the choice of labels useful for
various purposes are
discussed, e.g , in Sambrook et al., in Molecular Cloning: A Laboratory
Manual, Cold Spring Harbor
Laboratory Press (1989) and Ausubel et al., in Current Protocols in Molecular
Biology, Greene
Publishing Associates and Wiley-Intersciences (1987).
"Nucleic acid" refers to a deoxyribonucleotide or ribonucleotide polymer in
either single or
double stranded form, and unless otherwise limited, and encompasses known
analogues of natural
nucleotides that hybridize to nucleic acids in a manner similar to naturally
occurring nucleotides.
"Membrane" means a thin sheet of natural or synthetic material that is porous
or otherwise
at least partially permeable to biomolecules.
"Microarray" is an array that is miniaturized so as to require microscopic
examination for
visual evaluation.
"Polypeptide" means any chain of amino acids, regardless of length or post-
translational
modification (e.g., glycosylation or phosphorylation).
"Proteomics" means the identification or analysis of a proteome. A proteome is
the group
of proteins expressed and/or present in a biological sample.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-12-
"Sample" means a material that contains biomolecules including tissue, gels,
bodily fluids,
and individual cells in suspensions or in pellet, as~well as materials in
containers of biomolecules
such as microtiter plates.
"Stack" refers to adjacent substrates, whether oriented horizontally,
vertically, at an angle,
or in some other direction. The substrates (e.g., membranes) may be spaced or
touching, for example
contiguous.
"Subject" refers to living, multicellular vertebrate organisms, a category
that includes both
human and veterinary subjects for example, mammals, birds, and more
particularly primates.
Il. General Description ofSeveral Embodiments
Particular embodiments are especially useful in connection with archival
tissue samples that
have been fixed and embedded, for instance in paraffin. Whole tissue sections,
tissue macroarrays,
and arrays of minute tissue sections, e.g., in the format of a tissue
microarray, all may be analyzed
according to the disclosed methods, as can other samples from which
biomolecules are to be detected
(e.g., gels produced from 1- or 2-D separation of proteins or nucleic acids).
The biomolecules on the
copies can be visualized using detector molecules ("probes"), for example
antibodies, lectins, or
DNA hybridization probes, having specific affinity for the biomolecule(s) of
interest.
Specific embodiments provided herein include direct layered expression
scanning
techniques, which utilize a stack of "blank" membranes that are not specific
for any particular target
molecule. Instead, all (or a subset, e.g., proteins or nucleic acid)
biomolecules in a sample
ubiquitously bind to such membranes so as to give the user the flexibility of
detecting a wide variety
of biomolecules in an open format.
Thin membranes in a stacked or layered configuration are applied to the
sample, such as a
tissue section, or protein or nucleic acid gel, and reagents and reaction
conditions are provided so that
at least a portion of the biomolecules are eluted from the sample and
transferred onto a plurality of
the stacked membranes. This produces multiple substantial replicas of the
biomolecular content of
the sample. The resultant loaded (treated) membranes (or layers) are then
separated. Each
membrane may be incubated with one or more different detectors (for example
antibodies) specific
for a particular biomolecule (such as a protein) of interest. The detectors
employed are labeled or
otherwise detectable using any of a variety of techniques, for instance
chemiluminescence.
In an example in which proteins are detected, each membrane has essentially
the same
pattern of proteins bound to it, but different combinations of proteins are
made visible (detectable) on
each membrane due to the particular detectors (e.g., antibodies) selected to
be applied. For example,
one membrane layer may display proteins involved in programmed cell death
(apoptosis) while an
adjacent layer may display enzymes involved in cell division such as tyrosine
kinases.
In addition to proteins, nucleic acids may be targeted by using labeled DNA
probes as
detectors in lieu of antibodies. Moreover, different types of target
biomolecules may be detected in
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-13-
different layers. For example, both protein and nucleic acid targets can be
detected in parallel by
applying protein-specific detectors (e.g., antibodies) and nucleic acid
detectors (e.g., hybridization
probes) to different layers of the array.
According to certain methods of the present disclosure, a sample from which
biological
molecules are to be transferred (e.g., a tissue section or gel) is positioned
in contact with a face of a
stack of membranes and both the sample and stack (an assembled "contact
transfer stack") are placed
inside a fluid impervious enclosure such as a plastic bag or the like. In
certain embodiments, the
sample is supported by a substantially fluid impervious support, such as a
glass slide; in these
embodiments, the stack of membranes is placed on the other side of the sample.
In other
embodiments, the sample from which biomolecules are to be transferred is not
supported by an
impervious support, and the sample is placed between members of the membrane
stack, such that one
or more membranes is placed adjacent to each of two faces of the sample.
Also within the enclosure is a liquid transfer reagent. Heat and/or pressure
are applied to the
contents of the enclosure (from one or both sides) so as to permit proteins
and other molecules to be
transferred from the sample to the membrane stack. This produces multiple
copies or replicas of the
biomolecular content of the tissue sample. The processed membranes (or layers)
then may be
separated and incubated with one or more different probes (e.g., nucleic acid
hybridization probes or
antibodies) specific for particular targets of interest. The probes employed
are labeled or otherwise
detectable using any of a variety of techniques such as chemiluminescence.
While each membrane has essentially the same pattern of biomolecules
(including proteins
and/or nucleic acids) bound to it, different combinations of such biomolecules
are made visible on
each membrane due to the particular probes or antibodies selected to be
applied. For example, one
membrane layer may be used to detect proteins involved in programmed cell
death (apoptosis), while
an adjacent layer may be used in detecting enzymes involved in cell division,
such as tyrosine
kinases. In addition to proteins, nucleic acids may be targeted by using
labeled DNA probes in lieu
of antibodies. Moreover, both protein and nucleic acid targets can be detected
in parallel by applying
both antibodies and probes to different layers of the array of membranes to
which the biomolecules
have been transferred.
In one embodiment, the disclosed methods may be used for a side-by-side
comparison of the
protein expression patterns in different archival tissue samples, for instance
from patients with
different diseases, disease outcomes, or responses to therapies. Thus, for
example, where patient
response to a particular drug can be correlated to a specific protein
expression pattern from the
diseased organ this provides a useful tool for predicting whether future
patients likely will benefit or
be harmed by that drug.
Advantageously, provided methods may be used to screen archival tissue, which
is usually
formalin fixed and paraffin embedded. Provided methods may also be used for
examination of
proteins that cannot be detected with antibodies in situ but can be detected
after the protein has been
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-14-
transferred onto a membrane. Furthermore, provided methods enable the
quantitative analysis of
targets in tissue, for example, the quantification of cell surface receptor
density on malignant cells.
Beneficially, the methods, device, arrays, and kits provided herein can be
used with laser
capture microdissected samples, permitting molecular analysis of tissue
without protein or nucleic
acid purification as a prerequisite. These embodiments retain the two-
dimensional relationship of
distinct cell populations within the same tissue section so as to preserve the
spatial relationships
between the dissected cells and permit different cell types to be processed
and analyzed in parallel.
Thus, methods are provided for detecting biomolecules in a sample collected by
LCM, by
eluting the biomolecules away from the microdissected sample and binding them
to one or more
membranes in a layered or stacked configuration, then visualizing the
biomolecules on the
membranes.
In examples of such methods, cellular samples embedded in/on an LCM transfer
film (or the
like) are positioned adjacent to a stack of one or more membranes, and
reagents and reaction
conditions are provided so that the biomolecules are eluted from the cellular
sample and transferred
1 S onto the membrane(s). Biomolecules on the membrane then can be detected
and visualized using
detector molecules (e.g., antibodies or DNA probes) having specific affinity
for the biomolecule(s) of
interest.
Also provided are methods for identifying and analyzing biomolecules that have
been
resolved via electrophoretic, chromatographic, or fractionating means.
Examples of such methods
are sensitive enough to detect proteins in low abundance, yet able to detect
large numbers of proteins
in a high-throughput manner preferably without requiring expensive and
sophisticated laboratory
equipment.
Thus, according to one aspect of a method of the present disclosure,
biomolecules (e.g.,
proteins or nucleic acids) that have been electrophoretically separated on a
gel are transferred from
the gel onto a stack of membranes. In certain examples, these membranes are
constructed and/or
chemically treated to have a high affinity but low capacity for the
biomolecules. This allows the
creation multiple replicates of the molecular content of the gel. After
transfer, the membranes are
separated and each is incubated with a one or a unique mixture (also referred
to as a "cocktail") of
detectors (e.g., antibodies specific for a particular subset of proteins,
nucleic acid probes, etc). Thus,
while each membrane has essentially the same pattern of biomolecules bound to
it, different
combinations are made visible on each membrane due to the particular detector
(or set of detectors)
selected to corresponds to the particular layer. In specific examples, the
detector cocktail is an
antibody cocktail that has been carefully formulated so that no two antibodies
in a cocktail bind
overlapping or adjacent protein spots. Thus, protein spots that are too close
together to be
discriminated on a single membrane are detected on separate membranes
according to the inventive
method herein.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-15-
According to certain disclosed methods, proteins that have been separated
(either by ifz situ
synthesis, electrophoretically, chromatagraphically, etc.) on a gel, tissue or
other support are
transferred from the gel/support onto the membrane stack to allow the creation
of multiple replicates
or imprints of the protein content of the gel/support. With regard to gels,
the amount of protein
loaded into the wells is greater than the amount conventionally loaded so as
to permit a more even
and uniform distribution of the proteins throughout the stack.
Since antibodies can be used to detect many post-translational protein
modification (e.g.
phosphorylation), certain examples of disclosed methods can be employed to
identify or analyze
protein function as well as structure. In addition to 2-D gels, described
methods can be used for one-
dimensional gels such as the identification of transcription factors separated
by a gel-shift assay.
In detail, one specific embodiment is a method of analyzing the proteome of a
biological
sample. Such a method involves separating the protein from another protein
present in the sample;
transferring a portion of the separated protein to a plurality of membranes
(for instance, 2, 10, 20 or
more) in a stacked configuration; incubating each of the membranes in the
presence of one or more
species of predetermined ligand molecules (e.g., 2, 10, 20 or more) under
conditions sufficient to
permit binding between the separated protein and a ligand capable of binding
to such protein; and
analyzing the proteome by determining the occurrence of binding between the
protein and any of the
species of predetermined ligand molecules.
Another embodiment is a method for analyzing the extent of similarity between
the
proteomes of two or more samples. Such a method involves, for each such
sample, separating a
protein of such sample from another protein present in the sample;
transferring a portion of the
separated protein to a plurality of membranes (e.g., 2, 10, 20 or more) in a
stacked configuration;
incubating two or more of the membranes in the presence of one or more species
of predetermined
ligand molecules (e.g , 2, 10, 20 or more) under conditions sufficient to
permit binding between the
separated protein and a ligand capable of binding to such protein; and
analyzing the extent of
similarity between the proteomes by comparing the separated proteins of each
such sample with the
separated proteins of another such sample for the occurrence of binding
between the separated
protein and any of the species of predetermined ligand molecules.
Another embodiment is a method for uniquely visualizing a desired
predetermined protein if
present in a biological sample. This method involves separating the proteins
present in the sample
from one another; transferring a portion of the separated proteins of the
sample to a plurality of
membranes (for instance, 2, 10, 20 or more) in a stacked configuration;
incubating two or more of the
membranes in the presence of one or more species of predetermined
detector/ligand molecules (e.g.,
2, 10, 20 or more) under conditions su~cient to permit binding between desired
predetermined
protein and a ligand capable of binding to such protein; and visualizing any
binding between the
protein and any of the species of predetermined ligand molecules.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-16-
Also provided are embodiments of all such methods wherein the separation of
the protein
from another protein present in the sample is accomplished by electrophoresis
(for instance, 2-
dimensional (2-D) gel electrophoresis).
Further embodiments include all such methods wherein the sample is obtained
from
mammalian cells or tissue, and particularly from human cells or tissue, and
the embodiments wherein
the mammalian cells or tissue are human cells or tissue and the separated
protein is a product of a
human gene.
It is contemplated that the detector/ligand species can be any of a variety of
molecule types.
Thus, also provided are embodiments of all such methods wherein at least one
of the species of
detector/ligand is an antibody, an antibody fragment, a single chain antibody,
a receptor protein, a
solubilized receptor derivative, a receptor ligands, a metal ion, a virus, a
viral protein, an enzyme
substrate, a toxin, a toxin candidate, a pharmacological agent, a
pharmacological agent candidate, a
hybridization probe, a oligonucleotide, and others as discussed herein.
Other embodiments include all such methods wherein the binding of at least one
of the
species of detector/ligand is dependent upon the structure of the separated
biomolecule (e.g., protein
or nucleic acid). It still further provides the embodiments of all such
methods wherein the binding of
at least one of the species of detector/ligand is dependent or upon the
function of the separated
biomolecule (e.g., protein or nucleic acid).
The disclosure also provides all such methods wherein at least one of the
membranes is
incubated with more than one species of ligand or detector molecule. Also
provided are
embodiments of all such methods wherein at least two membranes are employed,
at least 10
membranes are employed, or at least 20 membranes are employed.
Further provided are the embodiments of all such methods wherein at least at
least two
ligand species or detector molecules are employed, wherein at least 10 are
employed, or at least 20 or
more are employed.
Additional embodiments are membranes that have a high affinity but a low
capacity for
proteins and/or other biomolecules so as to allow the creation of multiple
replicates or imprints of the
proteins eluted from a gel. Examples of these membranes are substantially
thinner than those
conventionally used for blotting. The membranes are optionally provided with
(or within) a frame,
so that they may be easily handled and manipulated when separated from that
stack. The frame
optionally defines a channel to permit release of air and fluid trapped
between adjacent membranes.
Removable tabs or the like also may be provided on each frame to permit the
stack to be held
together, for instance when it is applied to the gel.
Loaded membranes may be scanned or otherwise digitally imaged using one of
several
commercially available scientific imaging instruments. Imaging instrumentation
and software, such
as those described herein, may be employed to permit viewing, analysis, and/or
interpretation of the
expression patterns from the sample (e.g., a tissue sample or other two-
dimensional source, such as a
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-17-
gel). Software may be provided with template images corresponding to each of
the membrane
images. This allows the identity of the biomolecule in each defined locus
(e.g., a spot on a 2-D gel, a
band on a 1-D gel, or a localized molecular deposit in a tissue sample) to be
confnmed based on its
vertical and horizontal position. The software also can allow the density of
each locus to be
calculated so as to provide a quantitative read-out. The software may also
have links to a database of
images generated from other gels to allow comparisons to be made between
different diseased and
normal samples. In addition to computerized analysis of membranes, the source
sample (e.g., actual
tissue sections or other substantially two-dimensional source) or a
substantially similar sample (e.g.,
an adjacent tissue slice) may be analyzed with conventional techniques (e.g.,
histochemical
techniques) to confirm or compare the digital analysis.
Also provided herein are kits that include a plurality of membranes (e.g., 3
or more, for
instance 5, 10, 15, 25, 50, or 100 or more membranes) in a stack or other
configuration that permits
them to be stacked. Optionally, the provided kits may further include one or
more different
detectors, such as cocktails of antibodies or hybridization probes, to be
applied to the treated
membranes for biomolecule detection/analysis. The kits may also provide one or
more additional
components, such as a volume of a transfer reagent, a fluid impervious
enclosure (for instance, a
sealable bag), one or more pieces of filter paper, and/or a tissue array
contained on a slide or other
comparison sample or control sample. Optionally such kits may also include
instructions for how to
use the kit to detect, analyze, and/or identify one or more biomolecules.
Detection chemistries may
be included, which are tailored to coincide with the detector molecules
provided with the kit or
anticipated for use with the other kit components. The aforementioned software
may also be
included in the kit, or may be accessible via modem or the Internet.
In certain embodiments, the methods and kits according to the present
disclosure allow up to
several thousand discrete biomolecule (e.g., protein) loci to be identified,
annotated, and, at the user's
option, compared to the pattern of loci generated from other samples stored in
a database.
One specific example of a provided kit for analyzing a proteome includes a
plurality of
membranes, each having a specific affinity for at least one protein, and a
plurality of detector/ligand
species (e.g., species such as an antibody, an antibody fragment, a single
chain antibody, a receptor
protein, a solubilized receptor derivative, a receptor ligands, a metal ion, a
virus, a viral protein, an
enzyme substrate, a pharmacological agent, and a pharmacological agent
candidate), each adapted to
detect one or more specific proteins bound to the membranes.
Also provided in another embodiment is a kit for uniquely visualizing a
desired
predetermined protein if present in a biological sample. Such a kit includes a
plurality of
membranes, each having a specific affinity for at least one protein, and a
plurality of detector/ligand
species (e.g., species such as an antibody, an antibody fragment, a single
chain antibody, a receptor
protein, a solubilized receptor derivative, a receptor ligands, a metal ion, a
virus, a viral protein, an
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-18-
enzyme substrate, a pharmacological agent, and a pharmacological agent
candidate), each adapted to
detect the desired predetermined protein if bound to the membranes.
In particular embodiments, the membranes provided in kits described herein
include a
porous substrate having a thickness of less than about 30 microns. Particular
examples of such a kit
include membranes that are polycarbonate membranes, especially polycarbonate
membranes coated
with a material for increasing the affinity of the membrane to biomolecules,
for instance
nitrocellulose, poly-L-lysine, or mixtures thereof.
111. Transfer Modes
Provided herein are multiple methods for transferring biomolecules from a
sample that is
generally substantially two-dimensional into one or more thin membranes,
usually arranged in a
stack. Several different specific transfer modes are provided. Some of these
modes overlap, in that
wicking or contact transfer can be used to transfer biomolecules from both
tissue- and gel-based
samples, and so forth. Even though perhaps not explicitly enumerated, all
variations and
combinations of the described methods are encompassed herein.
Wicking Transfer
In particular embodiments, a transfer liquid (such as a buffer) is passed
through the
membranes to encourage movement of the biomolecules from the sample to the
membranes and
through them. A distal or downstream wick may also be provided to help move
liquid (such as the
buffer) through the membranes in a desired direction of movement.
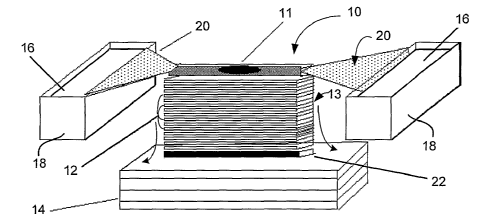
There is illustrated in FIG. 1 a perspective view of a representative
disclosed membrane
array transfer apparatus designated generally by reference numeral 10.
Apparatus 10 includes a
plurality of membranes 12 shown in a layered or stacked configuration such as
array 13. While only
about a dozen membranes are shown in array 13 of FIG. 1, it will be
appreciated that many more
membranes (e.g., 10, 50, 100 or more) may be employed depending on the number
of targets sought
to be identified, the quantity of biomolecules present in the sample, and the
thickness of the material
employed to construct membranes 12. Optionally, membranes 12 may be packaged
in a suitable
sealed enclosure or frame (not shown), for instance to maintain their
integrity and/or prevent
contamination. .
Membrane array 13 is placed atop a stack of one or more sheets of blotting
paper 14 that
acts as a lower wick pulling buffer out of buffer chambers 18 though upper
wicks 20 and membrane
array 12 in the direction of the arrows shown in FIG. 1. A biomolecule trap 22
may be positioned
intermediate membrane array 12 and blotting paper 14 to help the user
ascertain whether and/or to
what extent transfer has occurred.
In use and operation, apparatus 10 may be employed to create "carbon copies"
or substantial
replicas of the biomolecular contents of the sample applied to the stack.
Membranes 12 are arrayed
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-19-
in a layered or stacked configuration as shown in FIG. 1 as reference numeral
13. In a particular
embodiment, a substantially two-dimensional sample 11 (such as a conventional
frozen tissue section
as illustrated) is placed on a support substrate (e.g., a layer of
polycarbonate) and then sandwiched
between two slices of 2% agarose (not shown). The entire preparation is
positioned adjacent to the
membrane array 13. Buffer 16 is applied using buffer chambers 18 and upper
wicks 20 to elute and
transfer proteins from the frozen section. About 50-100 milliliters of buffer
per square centimeter are
used in each transfer with average length of the transfer being about 1-2
hours.
After transfer the membranes are separated and incubated with the detector
antibody.
Antibodies are selected based on the types of targets sought. Membranes are
washed in a buffer and
the protein / detector complex can be visualized using a number of techniques
such as ECL, direct
fluorescence, or colorimetric reactions. Commercially available flatbed
scanners and digital imaging
software can be employed to display the images according to the preference of
the user.
The specific example illustrated in FIG. 1 shows a device and a method for
detecting
biomolecules in a tissue section 11 or other two-dimensional sample (e.g., an
electrophoretic gel) by
creating "carbon copies" (substantial copies that are not necessarily
identical copies, they may have
slight differences but can be identical or nearly identical) of the
biomolecules eluted from the sample,
and visualizing the biomolecules on the copies using antibodies or other
molecules having specific
affinity for the biomolecules of interest. Thin membranes 12 in a stacked or
layered configuration
are brought into contact with the sample and reagents, and reaction conditions
are provided so that
the biomolecules are eluted from the sample onto the membranes, whereupon the
biomolecules can
be visualized using a variety of techniques, as set forth herein.
Certain embodiments of the disclosure include a method of detecting an analyte
in a
biological sample using stacked contiguous layered membranes that permit
biomolecules to move
through a plurality of the membranes, while directly capturing the
biomolecules on one or more of
the membranes. Biomolecules from the sample are moved through the membranes
under conditions
that allow one or more of the membranes to directly capture the biomolecules,
and biomolecules of
interest are concurrently or subsequently detected on the membranes, for
example by exposing the
biomolecules of interest to a detector, such as a specific capture molecule
(for example an antibody
or a nucleic acid probe).
Alternatively, the biomolecule itself may be a detector (such as a nucleic
acid probe) to
which a sample is exposed. In this case, the biological sample is one or more
purified nucleic acid
probes placed in assigned locations on a surface of the stack, which are
allowed to migrate through
membranes (for example in a direction of movement transverse to the layers) to
produce multiple
substantial "copies" of the original probes in corresponding locations on the
multiple membranes.
The layers then can be separated and exposed to a target biological specimen,
which may have
nucleic acid molecules that hybridize to the probes.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
_20-
In some examples, the biological sample is a tissue specimen that is placed on
the stack of
layered membranes, and biomolecules from the tissue specimen are directly
captured by the
membranes as the biomolecules move through the membranes. The membranes may,
for example,
be separated prior to detecting the biomolecules of interest, and the
separated membranes are
exposed to the detectors. Alternatively, the biological molecules of interest
may be contained in a
biological specimen to which the membranes are exposed. For example, the
biomolecules directly
captured by the membranes may themselves be nucleic acid probes or antibodies,
and the membranes
may be exposed to a biological specimen in which a nucleic acid or peptide
(such as a protein) is to
be detected.
Biomolecules detected on the membrane copies may be correlated with a
biological
characteristic of the sample. For example, a tissue specimen may be placed in
a position on top of
the stack, and a biomolecule of interest (such as a particular protein) may be
detected in one of the
membrane copies at a position that corresponds to the position in which the
tissue specimen (or one
of its substructures such as an organelle) was placed. The presence of that
biomolecule in the tissue
specimen can then be correlated with a biological characteristic of the
sample. For example, a highly
malignant tissue specimen may be found to contain a protein that may then be
associated with the
highly malignant phenotype of the specimen.
In particular examples, the method can be used to create a set of microarray
substantial
"copies" by applying a plurality of detectors, such as DNA probes, antibodies,
or a combination
thereof, to the stack of layered membranes. The stack of layered membranes
provide a plurality of
substrates through which the probes or antibodies (generally, detector
molecules) move, and in which
a portion of the probes or antibodies are directly captured by one or more of
the substrates. The
substrates can be subsequently separated to provide corresponding substrates
having a plurality of
DNA probes, antibodies or a combination thereof, in corresponding positions of
each of said
substrates. The multiple membranes maintain a substantially coherent
relationship between the
probes and/or antibodies as they move through the substrate. This coherent
relationship may or may
not be a direct spatial correspondence, but the relative relationship between
the biomolecules may be
maintained in such a way that the identity of the biomolecules on the
membranes can be known from
the relationship in which the biomolecules were placed on the stack of layered
membranes.
Contract Transfer
There is illustrated in FIG. 2A an alternative embodiment of an apparatus 10
for transferring
biomolecules from a substantially two-dimensional sample 11 onto a membrane
stack 13, which
stack in some embodiments is provided in the form of a kit. Apparatus 10
generally includes a
membrane stack 13 upon which a sample 11 (illustrated as a tissue section) may
be placed, a pair of
filter pads 24 and 26, and a fluid impervious enclosure 28, such as a plastic
bag or the like.
Optionally, the sample 11 (e.g., a tissue section) may be presented on a
support 30 (as illustrated in
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-21-
FIG. 2B). In particular embodiments, the support 30 is a microscope slide or
other fluid impervious
support such as a piece of tape.
More specifically, in a first embodiment, membrane stack 13 comprises one or
more
membranes 12, for instance up to five membranes, generally constructed as
described herein. The
membranes 12 in stack 13 should have a high affinity for proteins and other
biomolecules but have a
low capacity for retaining such molecules. This feature permits the molecules
to pass through the
membrane stack with only a limited number being trapped on each of the
successive layers, thereby
allowing multiple "carbon copies" (substantial copies that are not necessarily
identical copies, they
may have slight differences but can be identical or nearly identical) to be
generated. In other words,
the low capacity allows the creation of multiple replicates as only a limited
quantity of the
biomolecules are trapped on each layer.
First and second filter pads 24, 26 are preferably constructed of a blotting
paper such as
GB004 Blotter Paper available from Schleicher and Schuell. Filter pads 24, 26
are saturated with a
transfer buffer such as Tris or phosphate base buffers.
Enclosure 28 may comprise any collapsible, fluid impervious material adapted
to envelop
the sample 11, membrane stack 13, and filter pads 24, 26, which may be kit
components. Enclosure
28 is preferably a plastic bag, such as a heat sealable pouch. By way of
example, such a bag may be
made of a resin, such as a polyester or other resin. In certain embodiments,
enclosure 28 is a heat
sealable pouch such as those available from Kapak Corp. (Minneapolis, MN).
In use and operation, the sample 11 (e.g., a tissue section sample or tissue
microarray 31,
shown in FIG. 3) is positioned in contact with a face of a membrane stack 13
and both the sample
and stack are placed between two filter pads 24, 26, which have been saturated
with transfer buffer,
to for an assembled contact transfer stack. The assembled contact transfer
stack is placed inside fluid
impervious enclosure 28, such as a plastic bag. The membranes are pre-wetted
in the aforementioned
transfer solution.
Fluid impervious enclosure 28 is placed between a pair of substantially flat
surfaces 32, at
least one of which also serves as a source of heat. By way of example, the
pair of substantially flat
surfaces 32 can be surfaces of a pair of heating elements such as those
provided in gel dryers
manufactured by Bio-Rad Laboratories (Hercules, CA). In other embodiments, the
pair of flat
surfaces 32 may be provided by MJ Research devices, such as the PTC-200
Peltier thermal cycler,
which provide a separate heated lid and a thumbwheel to adjust height and
pressure of the lid and
thereby provide pressure.
In embodiments where heat is applied only from one side of the assembled
sample and
stack, the heat is preferentially applied from the side of the sample rather
than the membrane stack
side, such that a heat gradient is created with the heat applied on the sample
side.
To effect transfer, the bag and its contents are heated to a temperature of 60
to 95 °C, in
some embodiments 60 to 80 °C, or more particularly in some embodiments
70 °C. The bag and its
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-22-
contents are heated for at least about an hour, and in some embodiments about
two hours or more.
Sufficient pressure is applied throughout the heating process to ensure that
there is adequate contact
between the sample and the membrane stack to facilitate transfer of
biomolecules to the membrane
stack. By way of example, such pressure can be applied using a weight 34 of
0.5 to 2 pounds, which
may optionally be included as a kit component. Springs, clamps, or clips
capable of applying
pressure may be employed instead of a weight.
The combination of heat and pressure being applied causes biological
components,
including proteins and/or nucleic acids and/or carbohydrates and/or lipids, to
be transferred from the
sample 11 to membrane stack 13. This produces multiple copies or replicas of
the biomolecular
content of the tissue sample, due at least in part to the binding
characteristics of the membranes.
To ensure that the binding capacity of the membranes is sufficiently low to
prevent trapping
of too much of the sample, in some embodiments the thickness of membrane
substrate should be less
than 30 microns, in some embodiments from 4 to 20 microns, and particular
embodiments from 8 to
10 microns. The pore size of the substrate should be from 0.1 to 5.0 microns,
in particular
embodiments 0.4 microns. Another advantage of using such a thin membrane is
that is lessens the
phenomenon of lateral diffusion. The thicker the stack of membranes, the wider
the diffusion of
biomolecules moving through the stack.
The substrate includes a coating on its upper and/or lower surfaces to
increase specific
binding of the proteins or other targeted biomolecules. The coating in certain
embodiments is
nitrocellulose, but other materials such as poly-L-lysine may also be
employed.
Tissue section sample 11 may be derived from fresh/frozen tissue or tissue
that has been
fixed in formalin (or another fixative) and paraffin embedded tissue. The
section is created by
conventional methods, for instance using a microtome. The thickness of a
tissue section can vary
from 3 to 30 microns depending on the desired number of membrane replicates to
be created. As a
rule of thumb, the thickness of the section should be one micron for each
replicate sought. Thus, for
example, a 10 micron section would be used to create ten membrane copies.
As used herein "tissue" means any material containing cells, proteins, or
nucleic acids
including plant, animal, and human material. In lieu of tissue section sample
11, a tissue microarray
31 (FIG. 3) may be employed. Tissue microarrays are described in Kononen et
al., Nature Medicine,
4:844-847, 1998) and are provided by several commercial entities, such as the
Vast ArrayTM tissue
arrays available from Research Genetics (Huntsville, AL). Tissue macroarrays
are similarly
constructed, except that they contain tissue sections that are generally
larger than microarray
samples; the tissue samples used in tissue macroarrays may optionally be
dissected by hand.
Alternately, in some embodiments the biomolecules on a gel (e.g., an
electrophoretic gel) or other
substantially two-dimensional sample are transferred to a membrane stack using
similar methods, in
place of tissue section 14.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
- 23 -
Gel-Based Transfer
The most widely used method for identifying and measuring biological molecules
is gel
electrophoresis, a collection of techniques for separating or resolving
molecules in a mixture under
the influence of an applied electric field based on (usually) the difference
in their size and/or charge.
Electrophoretic separation is most commonly performed using porous polymer
gels. During one-
dimensional electrophoresis, a mixture of proteins is applied to a gel and
exposed to the flow of an
electric current. Since smaller proteins migrate faster through the gel than
larger ones, separation
based on their size is achieved. By way of example, this one-dimensional
approach can only
generate about 100 distinct protein bands, which is inadequate for many
applications since the
estimated number of proteins expressed in a typical mammalian cell is between
about 10,000-15,000
proteins.
In order to improve the resolving power of electrophoresis gels, a two-
dimensional gel
technique was introduced in the 1970s, wherein electrophoretic separation of
the proteins based on
their size is preceded by charge-based separation. Isoelectric focusing (IEF)
electrophoresis, which
separates proteins according to their charge (pH), is run in one direction and
mass separation is
carried out in a perpendicular direction. Such two-dimensional (2-D) gel
electrophoresis (often
abbreviated as "2-D PAGE," for two dimensional polyacrylamide gel
electrophoresis) has become
the backbone of proteomics. The technique is routinely employed for
characterizing the proteome of
different classes of tissues, cells, cell lysates, body fluids or exudates.
The end result of 2-D PAGE is
the production and separation of various protein "spots" in a two dimensional
Cartesian plane where
the coordinates of each spot are represented by charge and molecular weight.
However, the major
challenge of 2-D electrophoresis is the identification of the proteins after
they have been separated on
the gel.
Proteins that have been separated on gels are usually identified, detected,
and analyzed by
one of several different techniques. If the protein spot represents an unknown
protein, the most
common approach is to physically remove or excise the spot from the gel,
digest it with an enzyme,
and characterize the protein by mass spectroscopy. A computer generates a plot
of protein fragments
according to their mass, and this plot serves as a fingerprint that may be
used to facilitate the
identification of the original protein. As in the analysis of actual
fingerprints, the ability of mass
spectroscopy to identify a detected protein relies on the prior recovery and
analysis of a reference
protein whose fragments match those of the detected protein. The
identification of a truly new
protein by mass spectroscopy remains a significant challenge.
Although mass spectroscopy provides the most incontrovertible data, the method
is time
consuming, expensive and cannot be accomplished in the absence of expensive
core facilities and
highly trained personnel. Furthermore, the technique is used only to analyze
the proteins that can be
stained with a ubiquitous stain such as Coomassie blue. Unfortunately,
ubiquitous stains are not
sensitive and permit only a small fraction of the proteins in the sample to be
visualized. In other
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-24-
words, mass spectroscopy of ubiquitously stained gels does not yield a broad
"dynamic range" as it
fails to identify certain low abundance - but potentially important -
proteins. Among the low
abundance proteins that may be left behind by these techniques are tyrosine
kinases, cytokines, and
transcription factors, which play a key role in many diseases.
An alternative approach to identifying gel separated protein's is immuno-blot
analysis, which
uses a detectable antibody specific to a protein of interest in lieu of a
ubiquitous stain. The proteins
are transferred onto a membrane, typically constructed of either
nitrocellulose or of polyvinylidene
difluoride (PVDF) and antibodies are applied to the membranes. Immuno-blotting
is rapid and can
be accomplished in less than a day. Also, it is estimated to be about 1000-
fold more sensitive than
Coomassie blue staining, allowing even low abundance proteins to be
identified. It is significantly
more specific as well. However, a key limitation of immuno-blotting is that at
most only a handful of
proteins can be identified on a single blot due to overlapping spots and cross-
reactivity with different
proteins in the sample. Since the 2-D gel process requires approximately 24
hours to complete, it
would be prohibitively time consuming to create enough immuno-blots to
identify the large quantity
of proteins needed for most proteomics applications.
Thus, there is a clear need to develop techniques that permit large numbers of
proteins
across a wide dynamic range to be identified in parallel. Information
potentially relevant to attempts
to address this need can be found in the following references: Sanchez et al.,
Electrophoresis,
18:638-641, 1997; Neumann & Mullner, Eleetrophoresis, 19:752-757, 1998; Manabe
et al., Annal.
Bioehern., 143:39-45, 1984; Legocki & Verma, Annal. Biochem., 111:385-345,
1981; and PCT
International Publication No. W000 045168, all herein incorporated by
reference.
However, each of the techniques described in these references suffers from one
or more of
the following disadvantages: (i) not sensitive enough to detect low abundance
proteins, (ii) cannot
identify large numbers of proteins in a high-throughput manner, and (iii)
requires specialized or
sophisticated hardware that leads to loss of protein and a decrease in the
resolution the protein spots
during the transfer.
According to methods provided herein, biomolecules that have been
electrophoretically
separated on a gel, or via chromatography, etc. are transferred from the gel
onto a stack of
membranes. Examples of such membranes are membranes that are constructed and
chemically
treated to have a high affinity but low capacity for proteins. Suitable
membranes and methods for
their construction and preparation are described herein. The use of such
membranes allows the
creation of multiple replicates of the protein content of the gel.
The membranes are then incubated with a unique ligand species (a detector
molecule) or
mixture or cocktail of such, to assist in and permit detection and/or analysis
of biomolecules on the
membranes. The membranes are generally separated one from another prior to
such incubation.
Detector molecules/ligands can be any of a number of molecules that have
binding specificity for a
target molecule of interest, and include antibodies (such as monoclonal
antibodies), antibody
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
_25-
fragments (e.g., FAB, F(AB)Z, single chain antibodies, receptor proteins,
solubilized receptor
derivatives, receptor ligands, metal ions (particularly paramagnetic or
radioactive ions), viruses, viral
proteins (e.g., human rhinovirus or proteins thereof that bind to ICAM-1, or
HIV or proteins thereof
that bind to CD44), enzyme substrates, toxins, toxin candidates,
pharmacological agents,
pharmacological agent candidates, other small molecules that bind to specific
proteins, as well as
molecules that bind or hybridize to nucleic acids (e.g., nucleic acid probes
or specific binding
proteins or fragments thereof) etc. While each membrane has essentially the
same pattern of
biomolecules bound to it, different combinations of such biomolecules can be
detected on each
membrane due to the particular ligand or cocktail of ligands selected to
corresponds to the particular
layer.
The nature of the species of ligand(s) in the cocktail provided to the
membrane determines
the nature of information that can be obtained from that membrane. For
example, by incubating a
membrane with an antibody or antibody fragment, one is able to identify the
presence or absence of
protein molecules of the sample that bind to such molecules. In this way, for
example, a membrane
could be incubated with an antibody that specifically binds a protein kinase,
in order to determine
whether a particular protein is a protein kinase, or possesses an epitope that
mimics that of a protein
kinase. Similarly, by employing as the ligand, a cellular receptor protein,
solubilized receptor
derivative, or receptor ligand, the membrane would enable one to identify
whether a particular
protein was a receptor or receptor ligand. Since viruses and other pathogens
are capable of binding
to cellular receptor proteins, a cocktail containing a virus or viral protein
could be employed in the
same manner as a receptor ligand to identify whether a particular protein was
a cellular receptor or
receptor ligand. In an alternative embodiment, the cocktail could comprise one
or more
pharmacological agents to identify proteins that interact with such agents.
Likewise,
pharmacological agent candidates could be incubated with the membranes,
thereby revealing the
ability of such candidate molecules to bind to specific proteins. For example,
an acetylcholinesterase
inhibitor or a monoamine oxidase inhibitor (MAOI) could be incubated with a
membrane to identify
proteins that bind the inhibitor and which thus might be additional
therapeutic targets of the inhibitor.
Likewise, a compound suspected of possessing therapeutic potential could be
incubated with a
membrane to reveal whether it binds to proteins expressed, for example, in the
liver or kidney,
thereby revealing its potential to treat diseases affecting these organs.
Examples of the methods and
kits permit the further analysis of such binding to determine, for example,
whether such proteins are
expressed in other organs and tissues (e.g., the brain).
In one embodiment, a membrane will be incubated in the presence of a single
ligand, or a
cocktail of different ligands of the same class of ligands (e.g., antibodies,
receptors, hybridizing
probes, etc.). Alternatively, a membrane may be incubated with different
classes of ligands. For
example, a membrane that is incubated with antibodies that bind protein
kinases and with a
therapeutic candidate, can be employed to reveal therapeutic candidates that
bind to protein kinases.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-26-
Where mixtures or cocktails of ligands are employed, the cocktails are
preferably formulated so that
no two ligands bind overlapping or adjacent protein spots. Thus, for example
protein spots that are
too close together to be discriminated on a single membrane may be detected on
separate membranes.
In an alternative embodiment, the ligand is permitted to bind to proteins of
the sample prior
to the transfer to a membrane. Thus, in some examples the ligand is provided
to a living or deceased
subject, to a tissue or cell, to a tissue or cell preparation, or to a tissue
or cell extract, prior to the
fractionation or separation of protein. The proteins are then transferred to
membranes and the
proteins and ligand are visualized. In such an embodiment, one can detect
whether binding between
a ligand and a protein of the sample and occurs in situ, and/or under
physiological conditions.
Optionally, the membranes can be incubated in the presence of additional
ligand (which may be the
same or different from the initially employed ligand) in order to detect
competition between or
among ligands for binding sites, to evaluate the avidity of binding, to
examine binding complexes of
three or more molecules, etc.
Particular embodiments provide a method and a kit 36 for identifying (i.e.
detecting,
annotating, and/or characterizing) groups of proteins (not shown) that have
been separated by gel
electrophoresis. As illustrated in FIG. 4A, in one example kit 36 generally
comprises the following
components: (i) a stack of membranes 13 upon which the proteins are
transferred, (ii) primary
antibody cocktails 38, for instance one for each of the membranes 13, and
(iii) other reagents 40
including (as in illustrated in kit 36) protein transfer buffer 42 and
antibody detection chemistries 44.
The kit 36 may also include software 46 that allows the user to analyze and
manipulate the images
produced so as to yield a "proteomic image" of the biological sample being
tested and compare it to
proteomic images from other samples in a database. Alternatively the software
may be acquired or
accessed independent of the kit.
In a specific embodiment, and with reference to FIG. 4A, membrane stack 12
comprises a
plurality of membranes 13 adapted to be removably stacked atop one another, as
shown.
According to the method of a particular embodiment (as illustrated in FIG. 5),
proteins 48
that have been electrophoretically separated on gel 50 are transferred from
the gel through membrane
stack 13. This allows the creation of multiple replicate blots 52 of the
protein content of the gel. The
membranes are then separated and each is incubated with one of the unique
cocktails 38 (a-c) of
ligands, e.g., antibodies. The antibodies employed are labeled or otherwise
detectable using any of a
several techniques such as enhanced chemiluminescence (ECL). This produces
unique spot patterns
54 (a-c) on each of the membranes. The membranes with unique spot patterns 54
are then scanned
or digitally imaged using an imaging instrument (not shown) so that the
density of the spot may be
calculated, compared to other samples, and displayed on a computer using
software 46, as described
herein.
One advantage of specific embodiments provided herein is that they provide a
third
dimension of protein separation for a biological sample, one additional
dimension from the size and
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
_27_
charge separations obtainable from 2-D gels. The layered membranes provide a
cost-effective tool
for selecting groups of compatible antibodies that can be used to detect
subsets of proteins on the
same membrane. Once selected these ligand combinations can be packaged in a
kit and used
repeatedly for the controlled analysis of proteomes displayed on stacked
membranes. Since 15-20
replicates or copies can be generated from a single gel and ten or more
ligands can be applied to each
membrane several thousand different proteins can be identified from a single
gel according herein
described methods.
Since ligands can be used to detect many post-translational modification of
proteins (e.g.
phosphorylation) the present disclosure can be employed to identify protein
function as well as
structure.
Although these embodiments have been described with respect to 2-D gels, it is
also
contemplated that the methods and devices described can be employed with one
dimensional gels
(e.g., as for the identification of transcription factors separated by a gel-
shift assay), or proteins may
be separated from other proteins of a sample, by other means, as by
chromatography. It is also
contemplated that these methods can be used to generate duplicate copies of
non-protein
biomolecules, such as nucleic acids, lipids, sugars (such as polysaccharides)
and combinations or
complexes of two or more types of biomolecules.
In certain embodiments, buffer reagent for eluting proteins from a gel to a
membrane stack
comprises a mixture of glycine, methanol, and SDS as described herein. For 1-D
gel analysis,
protein staining can be carried out using FastBlue Stain (Chemicon).
Bi-Direetional Transfer
In alternative embodiments of the provided methods, the sample from which
biomolecules
are to be transferred is not supported by an impervious support and the sample
is placed between
members of the membrane stack. Thus, in such embodiments one or more membranes
is placed
adjacent to each of two faces of the substantially two-dimensional sample, and
transfer of the
biomolecules from the sample to the membranes occurs in two directions (bi-
directional transfer).
By way of example, this technique is illustrated schematically in FIG. 6. Here
first and
second membrane stacks 13a and 13b sandwich gel slab 54, which contains sample
11. A pair of
filter pads 24 and 26, preferably constructed of a blotting paper such as
GB004 Blotter Paper
available from Schleicher and Schuell are provided adjacent to the membrane
stacks as shown. Filter
pads 24 and 26 are saturated with a transfer buffer such as TRIS or phosphate
base buffers.
A collapsible, fluid impervious enclosure 28 is provided to envelop the pads,
membrane
stacks, and gel as shown in FIG. 7. Enclosure 28 (which in some instances is a
plastic bag) is
preferably a heat sealable pouch/bag such as those available from Kapak Corp.
(Minneapolis, MN).
Preferably, most of the air is removed from enclosure 28 by gentle squeezing
and/or vacuum suction
and it is sealed by a heat sealer such as the Impulse Sealer (American
International Electric).
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
_28_
Enclosure 28 is then placed between a pair of heating elements 56a and 56b
such as those provided
in Gel Dryers manufactured by Bio-Rad Laboratories (Hercules, CA). The
enclosure 28 and its
contents are optionally heated to a temperature of between about 50 to
90° C, preferably to about
80° C for about 2-4 hours. In some embodiments, pressure is applied
throughout the heating process
using a weight 34.
The heat and pressure applied to contents of the enclosure permit proteins and
other
molecules to be transferred from the gel or other two-dimensional sample to
the membrane stack.
This produces multiple copies or replicas of the biomolecular content of the
sample.
Transfer from Laser Capture Microdissection Samples
Under the microscope, tissues are heterogeneous, complicated structures with
hundreds of
different cell types locked in morphologic units exhibiting strong adhesive
interactions with adjacent
cells, connective stroma, blood vessels, glandular and muscle components,
adipose cells, and
inflammatory or immune cells. In normal or developing organs, specific cells
express different genes
and undergo complex molecular changes both in response to internal control
signals, signals from
adjacent cells, and humoral stimuli. In diseased tissues the cells of
interest, such as pre-cancerous
cells or invading groups of cancer cells, are typically surrounded by these
heterogeneous tissue
elements. Cell types undergoing similar molecular changes, such as those
thought to be most
definitive of the disease progression, may constitute less than 5% of the
volume of the tissue biopsy
sample. Therefore, a need arose to "microdissect" diseased cells from
surrounding normal cells to
permit molecular analysis of disease lesions in actual tissue.
To address this need researchers at the U.S. National Institutes of Health
developed a
technique known as "Laser Capture Microdissection" ("LCM") for procuring pure
cells from specific
microscopic regions of tissue sections. See Emmert-Buck, et al., Seience
274:998-1001,1996;
Bonner, et al., Science 278:1481-1483,1997, incorporated herein in their
entirety. LCM allows small
groups of cells to be isolated from tissue sections thereby allowing an
investigator to collect only
cells of interest so as to achieve high purity of the sample. Once collected,
cells are homogenized
and genomic DNA, total cellular RNA or total proteins can be isolated. Details
of LCM are
described, for example, in PCT International Patent Applications publications
WO 09917094A2 and
WO 098352A1, which are incorporated herein and are illustrated in FIG. 7.
In short, a laser beam focally activates a special transfer film which bonds
specifically to
cells identified and targeted by microscopy within the tissue section. The
transfer film with the
bonded cells is then lifted off the thin tissue section, leaving all unwanted
cells behind (which would
contaminate the molecular purity of subsequent analysis). This allows multiple
homogeneous
samples within the tissue section or cytological preparation to be targeted
and pooled for extraction
of molecules and analysis.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-29-
In order to simplify the process of handling the transfer filin, the film may
be permanently
bonded to the underside of a transparent vial cap, such as those available
from Arcturus Engineering
Inc. (Mountain View, California). After the targeted cells are transferred to
the cap surface the cap is
placed directly onto a centrifuge tube to extract biomolecules from the cap
and purify biomolecules
for subsequent analysis, for instance using electrophoresis gels, DNA
microarrays and the like.
Unfortunately, many molecular biology assays such as Western blotting are
difficult to
perform on LCM-collected samples since the amount of material collected per
unit of time is very
small. While analysis of nucleic acids from LCM collected material is aided by
the amplification
techniques such as the polymerize chain reaction (PCR), protein amplification
is not possible.
Proteomics studies on LCM collected samples are thus particularly difficult.
Another current limitation of LCM is that different cell subtypes (e.g.
epithelium and
connective tissue) must. be transferred to different caps. Since the
biomolecules (proteins and nucleic
acid) are removed from the cap for further analysis, different cell types
cannot be mixed on the same
cap since it could not be determined from which cell type a particular
biomolecule originated. Thus
users of LCM typically must process a different cap for each cell type in a
tissue section, a procedure
that is time consuming and creates variability in experimental design.
Embodiments provided herein include methods and apparatuses for detecting and
analyzing
biomolecules in a sample collected by LCM by eluting biomolecules away from
the sample and
binding them to one or more membranes in a layered or stacked configuration,
then visualizing the
biomolecules on the membranes.
In general, cellular samples embedded in/on an LCM transfer film or the like
are positioned
adjacent to a stack of one or more membranes, and reagents and reaction
conditions are provided so
that biomolecules are eluted from the cellular sample and transferred onto the
membrane(s).
Biomolecules on the membrane are then detected and visualized using one or
more detector
molecules, for instance antibodies or DNA probes having specific affinity for
the biomolecules of
interest.
There is illustrated in FIG. 8 a longitudinal section view of one embodiment,
preferably in
the form of a kit, designated generally by reference numeral 58. Kit 58
generally comprises a
membrane stack 13, LCM cap holder assembly 60, a pair of filter pads 24 and
26, and a fluid
impervious enclosure 28 such as a plastic bag or the like.
In some embodiments, membrane stack 13 comprises up to 20 membranes, generally
constructed as described herein. Representative membranes 12 in stack 13 have
a high affinity for
proteins and other biomolecules, but have a low capacity for retaining such
molecules. In another
embodiment, a single membrane is used in lieu of a plurality of membranes. If
only one membrane
is used it need not have the low capacity requirements of certain other
embodiments, and it can be
constructed of any of a variety of materials conventionally employed as
blotting membranes, such as
nitrocellulose or PVDF.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-30-
LCM cap holder assembly 60 is preferably constructed of a heat conductive
material such as
metal and has generally rectangular dimensions. A plurality of apertures 62
are defined by cap
holder assembly 60 with each aperture adapted to receive a standard LCM cap 64
such as those
available from Arcturus Engineering, Inc. (Mountain View, California) or a
modified LCM cap 66
(FIG. 10). Mounted to caps 64 (or 66) is a standard LCM transfer film 68
having adhered thereto the
selected cellular material 70 that serves as the transfer sample 11 from the
tissue sample following an
LCM procedure. By way of example, LCM is performed on tissue sections (such as
frozen or
fixed/para~n embedded sections) using the equipment such as that illustrated
in FIG. 7 according to
known methods, such as those recommended by Arcturus Engineering, Inc.
First and second filter pads 24, 26 are preferably constructed of a blotting
paper such as
GB004 Blotter Paper available from Schleicher and Schuell. Filter pads 24, 26
are saturated with a
transfer buffer such as Tris or phosphate base buffers.
Enclosure 28 may comprise any collapsible, fluid impervious material adapted
to envelop
the other kit components. Plastic bag 28 is preferably a heat sealable pouch
such as those available
from Kapak Corp. (Minneapolis, MN).
After microdissection, caps 26 can be stored frozen until transfer of the
molecules is desired.
Cellular material 70 embedded within transfer film 68 is hydrated through
gradient of ethanol and
optionally mildly digested with proteases. Caps 64 (or 66) are then inserted
within apertures 62
defined in cap holder assembly 60 and the cap holder is placed adjacent to
membrane stack 13 so that
the transfer film 68 is in direct contact with a membrane. First filter pad 24
is placed above cap
holder assembly 62 and second filter pad 26 is placed below membrane stack 13.
(Both pads are
soaked in a transfer buffer.) Pads 24 and 26, sandwiching the other components
of the assembled
stack of kit 58, are placed within enclosure 28. Most of the air is removed
from enclosure 28 by
gentle squeezing and/or vacuum suction and it is sealed by a heat sealer such
as the Impulse Sealer
(American International Electric).
With reference to FIG. 9 plastic bag 28 is placed between a pair of heating
elements 56 such
as those provided in Gel Dryers manufactured by Bio-Rad Laboratories
(Hercules, CA). The bag and
its contents are heated to a temperature of between about 60 to 80° C,
preferably to about 70° C for
about two hours. Pressure is applied throughout the heating process using a
weight 34, which may
optionally be added as a kit component.
In other embodiments, multiple caps are created from a single cell type and
the
biomolecules (proteins and/or nucleic acids) are transferred to the single
membrane or membrane
stack in the manner described herein. One membrane (or more) can then be cut
into pieces
corresponding to the number of caps so that the biomolecular content from each
cap may be
separately incubated with a different detector molecule or detection system.
It may be desirable to prevent rotation of the LCM caps during the transfer
process so that
positions of the cellular samples remain fixed relative to the membranes. This
would be useful when
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-31 -
particular regions of the film 68 are allocated to particular cell types (e.g.
epithelium vs. connective
tissue or diseased vs. normal cells). By preventing rotation of the LCM caps
the user can correlate
the position of the biomolecules on the membranes with the region of film 68
and cell type from
which the biomolecules originated. Lines or other indicia (not shown) may be
provided on the
membranes and caps 64 to aid the user in this process. In order to prevent
rotation, the standard
LCM cap may be modified as shown in FIG. 10. Modified cap 66 has a shank
portion 72 that defines
a flat surface 74 (shown in FIG. l OB) that is adapted to engage an similarly
shaped aperture in the
LCM cap holder assembly (not shown). The size of cap 66 and corresponding
transfer film may be
enlarged so that cells of interest from an entire tissue section may be
microdissected or otherwise
transferred onto a single cap, thereby saving time and reducing experimental
variability as compared
to using different caps for each cell type as is the practice currently in
use.
"Microarray" Transfer
Another use of the membrane arrays provided herein is to make multiple copies
of a cDNA
or other microarray in a manner that is less expensive and labor-intensive
than robotic systems. In
particular embodiments, the plurality of DNA probes, antibodies, or
combination thereof, is applied
to the stack of membranes from a plate having a plurality of wells (e.g., a
microtiter or like plate),
each containing a different DNA probe or antibody. The DNA probes or
antibodies are transferred
from the wells to the stack so as to create a set of substantially replicate
microarrays.
With reference to FIG. 11, DNA sequences representing different genes are
placed into
individual microtiter wells 80 of a microtiter plate 82 (e.g., a 96-well
plate). The microtiter plate 82
is placed adjacent to a stack 13 of membranes 12, to allow the contents of the
microtiter wells 80 to
be transferred from the respective wells to the stack of membranes 13. In the
illustrated embodiment,
contents of the wells are transferred from the wells 80 to a top surface of
the stack of membranes 13,
so that the contents are applied in a pattern that corresponds to a pattern of
the wells.
The DNA is transferred through the membranes in a direction of movement from
the wells
toward a wick member 84, and the spatial orientation of the samples is
maintained. Because of the
high affinity, low capacity characteristics of membranes 12, as the nucleic
acids traverse the capture
membrane stack 13, a small percentage of DNA hybridizes to each membrane,
creating a series of
replicate copies, each one containing a grid of DNA spots that match the
orientation of the DNA
samples in the microtiter plate. Thus, a set of cDNA arrays may be created in
a very rapid and
inexpensive fashion. Antibody and tissue lysate arrays can also be created by
this method.
IV. Types ofSamples
Any two-dimensional sample material that contains releasable biomolecules can
be used as a
source of biomolecules in the provided transfer processes. By "two-
dimensional" it is meant that the
material is, or can be formulated so that it is, substantially flat and
relatively thin. Representative
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-32-
examples of substantially two-dimensional samples include tissue samples such
as thin section slices
(e.g., archival or frozen tissue samples), tissue arrays, cDNA or other
nucleic acid microarrays,
protein microarrays, 1-D protein gels, 1-D nucleic acid gels, 2-D protein
gels, and so forth.
It is further contemplated that the described transfer methods, arrays, and
devices can be
used in forensic procedures to detect and study biological material such as
bodily fluids; to detect
biological (e.g., microbial) contamination of food or other substances; and so
forth. In order to
provide the sample in a substantially flat and thin format, substances may be
suspended in a liquid or
gas, then run through and optionally affixed to a filter such as a sheet of
filter paper, with the filter
then used as the transfer sample. By way of example, a soil sample or fluid
sample could be so
prepared for transfer. Some substances may be compressed into a substantially
flat form, for instance
by rollers or another spreading mechanism; by way of example, a food sample
(e.g., ground meat)
could be so prepared. Generally these samples can be referred to as
structurally transformed
samples, because their format is altered to render them substantially two
dimensional prior to transfer
onto a membrane stack.
Embodiments provided herein may be used to identify biomolecules (e.g.,
proteins or
nucleic acids) in any biological sample including bodily fluids (e.g. blood,
plasma, serum, urine, bile,
cerebrospinal fluid, aqueous or vitreous humor, or any bodily secretion), a
transudate, an exudate
(e.g. fluid obtained from an abscess or any other site of infection or
inflammation), fluid obtained
from a joint, and so forth. Additionally, a biological sample can be obtained
from any organ or tissue
(including or autopsy specimen) or may comprise cells.
V. Membranes
Also provided herein are membranes, which can be used in the described methods
of
biomolecule separation.
In particular embodiments, the membranes comprise a material that non-
specifically
increases the affinity of the membranes to the biological molecules (or a
class of biomolecules such
as proteins or nucleic acids) that are moved through the membranes. For
example, the membranes
may be dipped in, coated with, or impregnated with nitrocellulose, poly-L-
lysine, or mixtures
thereof. In certain examples the membranes are not treated with a material
that blocks the non-
specific binding of the biomolecules to the membranes, at least during
transfer of the biomolecules
through the membranes. However, in other embodiments, some such blocking
agents can be added
to the membranes, as long as the amount of blocking agent minimizes the amount
of biomolecules
bound, without blocking it altogether. In certain examples, blocking agent may
be added to the
membranes after transfer of the biomolecules through the membranes, but before
or during exposure
to the detectors.
In particular examples, the membranes are sufficiently thin to allow the
biomolecules to
move through the plurality of membranes (for example 10, 50, 100 or more) in
the stack. Such
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
- 33 -
membranes, for example, have a thickness of less than 30 microns. The
membranes may be made of
a material that does not substantially impede movement of the biomolecules
through the membranes,
such as polycarbonate, cellulose acetate, or mixtures thereof.
The material of the membranes may maintain a relative relationship of
biomolecules as they
move through the membranes, so that the same biomolecule (or group of
biomolecules) move
through the plurality of membranes at corresponding positions. In such
examples, this coherence of
relative relationships allows the different membranes to be substantial
"copies" of one another, much
like a "carbon copy" would be. However, like a "carbon copy" there may be some
differences
between the different "copies" present in the different membranes.
In some embodiments, a membrane stack will include a number of individual
membranes,
for instance at least 2, at least 5, at least 10, at least 20, at least 50, or
even more in some instances.
Membranes in the stack are generally constructed as described herein. Examples
of the membranes
are constructed of a porous substrate coated with a material that increases
the affinity of the
membrane to the biomolecules being transferred. The substrate may be
constructed of polycarbonate
or a similar polymeric material that maintains sufficient structural integrity
despite being made
porous and very thin. Representative membranes for use in the methods,
devices, and apparatuses
have a high affinity for proteins and/or other biomolecules, but have a low
capacity for retaining such
molecules. This binding profile permits biomolecules to pass through the
membrane stack with only
a limited number being trapped on each successive layer, thereby allowing
multiple "carbon copies"
of the biomolecules in the sample to be generated. In other words, the low
capacity allows the
creation of multiple replicates as only a limited quantity of the biomolecules
is trapped on each layer.
With reference to FIG. 12, individual membranes 12 are constructed of a porous
substrate
90 coated with a material that increases the affinity of the membrane to the
biomolecules being
transferred. Substrate 90 is, for example, constructed of polycarbonate or a
similar polymeric
material that maintains sufficient structural integrity despite being made
porous and very thin.
However, in lieu of polycarbonate the substrate 90 may for example be
constructed of cellulose
derivatives such as cellulose acetate, as well as polyolefms, (e.g.,
polyethylene, polypropylene, etc.).
The illustrated membrane 12 includes a coating 92 on its upper and lower
surfaces to
increase non-specific binding of the proteins or other targeted biomolecules.
Although the binding to
the coating is "non-specific" in the sense that it does not recognize
particular proteins or other
biomolecules, _such as particular nucleic acids, it may be specific in that it
recognizes and specifically
binds classes of biomolecules, such as proteins or nucleic acids, or
combinations thereof. Coating 92
in specific disclosed embodiments is nitrocellulose, but other materials such
as poly-L-lysine may
also be employed.
Before being applied to substrate 90, the nitrocellulose is dissolved in
methanol or other
appropriate solvent in concentration from 0.1%-1.0%. The membranes are
immersed in this solution
as described more fully in the Examples, below. In lieu of coating 92,
nitrocellulose or other
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-34-
materials with an affinity for biomolecules can be mixed with the
polycarbonate before the substrate
is formed thereby providing an uncoated substrate having all of the desired
characteristics of the
membrane. Alternative coating methods known in the art may be used in lieu of
dip coating
including lamination. Alternatively, only one surface may be coated, such as
the surface that faces
the sample, instead of both surfaces.
It is a particular feature of certain embodiments that membranes 12 have a
high affinity for
proteins and other biomolecules, but have a low capacity for retaining such
molecules. This feature
permits the molecules to pass through the membrane stack with only a limited
number being trapped
on each of the successive layers thereby allowing multiple "carbon copies" to
be generated. In other
words, the low capacity allows the creation of multiple substantial replicates
as only a limited
quantity of the biomolecules are trapped on each layer. If a membrane were
used that had a high
binding capacity for biomolecules-such as with nitrocellulose membranes
conventionally used with
gel blotting-multiple replicas could not as easily be made. More specifically,
the affinity and
capacity of membrane 12 should be such that when at least five and preferably
more than ten
membranes are stacked and applied to a sample according to a disclosed method,
most of the
biomolecules of interest can be detected on any and all of the membranes,
including those positioned
furthest from the sample.
With reference to FIG. 13, the aforementioned technique may be described as
"direct
capture" since the target biomolecules 100 are captured directly on the
surface of membranes (or
within the membrane), instead of being captured indirectly by a binding agent
(such as an antibody
or nucleic acid probe) applied to the membrane. During this disclosed process
different components
of the sample bind to the membrane with the same affinity, but directly
proportional to their
concentration in the sample (a component with a higher concentration will
leave more molecules on
each membrane, and a component with a lower concentration will leave less
molecules on each
membrane). A detector molecule 104, such as a labeled antibody that
specifically binds to the
biomolecule 100 at illustrated epitopes 102, may be utilized to detect
biomolecule bound to the
membrane. In examples in which the amount of a component bound to the membrane
is proportional
to the amount of the component in the sample, an amount of the detector
molecule can be correlated
to an amount (or relative amount) of the biomolecule detected.
In order to achieve high affinity and high capacity for a particular group of
biomolecules
from a sample, coating of the membranes with a captor molecule 106 is
performed in the method
described by Englert et al. (Cancer Researeh 60:1526-1530, 2000). This may be
referred to as
"indirect capture" and is illustrated in FIG. 14. Captor 106 can be cDNA,
antibody, or any other
protein specific for the target of interest. Single-stranded cDNA molecules
generated by number of
means (Polymerase Chain Reaction, nick translation, reverse transcription,
oligonucleotide synthesis)
or proteins (like immunoglobulin) can be directly attached to the membrane.
Alternatively, the
linker-arms that would allow spatial control of the captor binding could be
directly attached to the
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-35-
membrane followed by captor attachment to them. This will expose the majority
of the active target
recognition sites increasing that way capacity of the indirect capture.
Streptavidin coated membranes
may be employed to bind end-biotinylated nucleic acids and randomly
biotinylated proteins, or
protein A and protein G to bind immunoglobulins.
In another embodiment (illustrated in FIG. 15), each of the membranes 108
comprise a
ligand coating (e.g., a unique ligand coating, in that it is different from
the others in the stack) that
selectively binds to proteins in the biological sample based on a particular
characteristic of the
protein chemistry (e.g. hydrophobicity, carbohydrate content, etc.) As a
result, the membranes 108
function to fractionate the proteins rather than replicate them as with
membranes 12 in other
described embodiments. The coating could be made in many different ways so
that each membrane
binds a selective subset of the total protein content in the sample. For
example, carbon chains of
increasing length, starting with a small carbon molecule can be used in the
coating. As the number of
carbons increases the ability to bind to proteins increases. Thus, for
example, the first layer may
have a six carbon-chain coating and will only bind to the most hydrophobic
proteins in the sample,
the remaining proteins will pass through to the next layer; the second layer
has an eight-carbon chain
and will pull out slightly less hydrophobic proteins while the remaining
proteins pass through; the
third layer has a ten carbon-chain, etc.
Thus, with another embodiment, each of the membranes will bind to a different
group of
proteins essentially permitting "3-D gel electrophoresis" by allowing proteins
to be separated into
three dimensions: in the X and 1' dimensions by charge and mass, and then in
the Z dimension by an
additional chemical characteristic. The proteins on the membranes according to
the second
embodiment can be visualized by the immuno-staining and imaging methods set
forth below. They
may also be advantageously analyzed by mass spectrometry either without
additional cleavage or
after such cleavage (see, e.g., W000 045168), or by other means. Examples of
the methods and kits
facilitate such analysis because the stratification by the different membranes
helps to expose
moderate and low abundance protein spots that would otherwise be undetectable
on standard 2-D
gels. The more spots that are available for analysis, the more data can be
generated by mass
spectroscopy or by such other approaches.
Other Membrane Characteristics
It is a particular feature of some embodiments that membranes used for the
transfer have a
high affinity for proteins and/or other biomolecules, but have a low capacity
for retaining such
molecules. This feature permits the molecules to pass through the membrane
stack with only a
limited number being trapped on each of the successive layers, thereby
allowing multiple replicate
"carbon copies" to be generated. In other words, the low capacity of the
membrane material allows
creation of multiple replicates, since only a limited quantity of the
biomolecules (e.g., proteins) are
trapped on each layer.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-36-
More specifically, in specific embodiments the affinity and capacity of
membrane should be
such that when at least five and preferably more than ten membranes are
stacked and applied to a
sample according to one of the provided methods, most of the biomolecules of
interest can be
detected on any and all of the membranes, including those positioned furthest
from the sample. If a
membrane were used that had a high binding capacity- such as the transfer
membranes used with
conventional gel blotting, multiple replicas could not be made in this manner
unless the binding
capacity of the membrane was overwhelmed by the amount of biomolecule applied
to the membrane.
To maintain the binding capacity of membrane sufficiently low to avoid
trapping of too
much of the sample, the thickness of the substrate is, for example, less than
about 30 microns, and in
particular embodiments is between about 4-20 microns, for example between
about 8 to 10 microns.
The pore size of the substrate is, for example, between about 0.1 to 5.0
microns, such as about 0.4-
0.6 microns, and more specifically 0.4 microns. Another advantage of using a
thin membrane is that
is lessens the phenomenon of lateral diffusion. The thicker the overall stack,
the wider the lateral
diffusion of biomolecules moving through the stack.
It will be appreciated that because the size of the membranes in the
stack/array can be
varied, the user has the option of analyzing a large number of different
samples in parallel, thereby
permitting direct comparison between different patient samples (e.g.,
different patient samples, or
patient samples and a reference standard, or samples of different tissues or
species, etc.). For
example, different samples from the same patient at different stages of
disease can be compared in a
side-by-side arrangement, as can samples from different patients with the same
disease. By way of
alternative example, the area of protein separation resulting from most 2-D
gels is generally between
about 10x10 cm to 20x20 cm; membranes used for transfers of 2-D gels may vary
accordingly.
Membrane Construction
The membrane substrate includes a coating on its upper and lower surfaces to
increase
specific binding of the proteins or other targeted proteins. The coating is
preferably nitrocellulose
but other materials such as poly-L-lysine may also be employed. Before being
applied to substrate,
the nitrocellulose is dissolved in methanol or other appropriate solvent in
concentration from 0.1%-
1.0%. The membranes are immersed in this solution as described more fully in
the Examples, below.
In lieu of coating, nitrocellulose, or other materials with an affinity for
proteins, can be mixed with
the polycarbonate before the substrate is formed thereby providing an uncoated
substrate having all
of the desired characteristics of the membrane. Alternative coating methods
known in the art may be
used in lieu of dip coating, including lamination. In all instances it should
be understood that
preferably only one surface - the surface that faces the sample -is coated or
treated, instead of both.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-37-
Framed membranes
In another embodiment, with reference to FIGS. 16-18, framed membrane stack
110
comprises a plurality of individual membrane units 112 releasably secured to
one another. Each
membrane unit 112 comprises a membrane 12 having a frame 114 mounted about the
periphery
thereof. Membrane unit 112 can vary in size but should be large enough so that
membrane 12 can
overlay a typical electrophoresis gel.
The number of membrane units 112 included in stack 110 can vary depending on
the
number of proteins to be detected from the gel. For most applications, from 3
to 25 or more
membranes will be sufficient, preferably from 5 to 15 and most preferably
about 10 to 12. The entire
thickness, Ts, of stack 110 (FIG. 16) is in some embodiments no more than
about 0.25 cm.
In some embodiments, in order to give each membrane sufficient rigidity to
enable it to be
separated the other membranes in stack 110 and individually processed, a frame
114 is mounted onto
the periphery of membrane 12 thereby forming membrane unit 112. Frames 114
preferably comprise
a generally "Z" shaped configuration covering three sides of the membranes
while defining an open
space or gap 120 that functions as a channel to permit the manual removal of
air pockets or fluids in
the manner described.
The composition and dimensions of frame 114 should be such that the frame
provides
sufficient rigidity for the user to grip the frame with one hand and
manipulate the membranes as
needed. At the same time, the frames must be sufficiently thin so that when
stacked they do not
interfere with protein transfer from the gel onto the membrane stack 110. Each
membrane 12 in
stack 110 should be capable of making direct contact with.adjacent membranes
during the transfer
process described herein.
The width W (FIG. 18) of frame 114 is preferably between about 0.3 to 0.7 cm
and the
thickness of the frame, Tf, is between about 0.005 to 0.03 cm., most
preferably about 0.01 cm thick.
Thus, frame 114 is about ten times thicker than membrane 12. In certain
embodiments, the materials
that comprise frames 114 are able to maintain their structure and function at
temperatures of up to
80° C but are able to melt when applied to a typical heat-sealing
apparatus. One skilled in the
relevant art will readily appreciate that a variety of compositions and
configurations of frames 114
could meet these requirements. Examples of materials that may be employed to
make frames 114 are
transparency film available from Canon or any thin plastic sheet made of
polycarbonate, polyester,
polyvinylchoride or polyvinilechloride.
As best viewed in FIG. 17, a pair of outwardly depending tabs 116 is defined
by frame 114.
Each tab is adapted to be sealed to the corresponding tab on an adjacent
membrane so as to hold
stack 110 together during the gel transfer process. After the proteins are
transferred onto the
membranes tabs 116 are cut with a scissors so that the membranes may be
separated and incubated in
separate detection solutions.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-38-
At least one side of frame 114 defines a surface 118 upon which indicia may be
imprinted.
The indicia may include the name of the product or manufacturer or the
membrane number.
Machine-readable indicia such as a bar code or the like (not shown) may also
be provided.
Frames 114 may be mounted to the perimeter of membranes 12 by various means
readily
familiar to those skilled in the art including use of adhesives such as rubber
cement or 3M adhesive
or conventional heat-sealing or laminating techniques.
V1. Analysis of Membrane Replicates
After transfer, the processed membranes (or layers) can be separated and each
incubated
with one or more different detector molecules (such as nucleic acid
hybridization probes, lectins, or
antibodies) specific for particular targets of interest. In certain
embodiments, the detectors/probes
employed are labeled or otherwise detectable using any of a variety of
techniques such as
chemiluminescence. Thus, while each membrane has essentially the same pattern
of biomolecules
bound to it, different combinations of biomolecules can be made observable on
each membrane by
selecting particular probes to be applied and detected.
By way of example, one membrane layer may display proteins involved in
programmed cell
death (apoptosis) while an adjacent layer may display enzymes involved in cell
division such as
tyrosine kinases.
In addition to proteins, nucleic acids may be targeted and detected by using
labeled DNA
hybridization probes rather than antibodies. Moreover, both protein and
nucleic acid targets can be
detected in parallel by applying both antibodies and nucleic acid probes to
different layers of the
stack. Similarly, carbohydrates can be detected using carbohydrate-binding
molecules such as
lectins.
Digital images of membranes may be created using a variety of instruments
including the
Image Station~ CCD instrument available from Kodak Scientific Imaging (New
Haven, CT).
Alternatively images may be captured on film (such as X-ray film) and
digitalized by flat bed
scanners. Software is preferably provided to align the images and perform
densitometry functions.
The user can select the region of interest for analysis and the signal
intensities are recorded and
normalized. The numerical intensity values are then compared.
For analysis of transferred proteins, after the transfer by any of the herein-
described protein-
transfer techniques, the membranes are separated from stack and each is
incubated in a separate
solution of primary antibody specific for a desired protein. Only the band
containing this protein
binds the antibody, forming a layer of antibody molecules. After incubation
for about 1-8 hours, the
membranes are usually washed in buffer to remove unbound antibody.
For detection of the proteins on the membranes (in the form of bands, spots,
or "in situ"
from tissue transfers), the loaded membranes are incubated in a secondary
antibody that binds to the
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-39-
primary antibody. The secondary antibody may be covalently linked to an enzyme
such as ' .
horseradish peroxidase (HRf) or allealine phosphatase (AP) that catalyzes
substrate and protein /
antibody complex can be visualized using a number of techniques such as ECL,
direct fluorescence,
or colorimetric reactions. ECL is preferred. Commercially available flatbed
scanners may be
employed in conjunction with film. Alternatively, specialized imaging
instrumentation for ECL,
such as the Kodak IMAGE STATION available from NEN may be utilized and digital
imaging
software can be employed to display the images according to the preference of
the user.
In lieu of antibodies, other ligands may be employed as detectors. Ligands can
be antibody
fragments, receptors, receptor ligands, enzymes, viruses or viral particles,
enzyme substrates or other
small molecules that bind to specific proteins. Moreover, in addition to
identifying proteins of
interest structurally, kits can also be employed to identify the functional
state of proteins. One way
to do so is to use phospho-specific antibodies to determine the
phosphorylative state of proteins) of
interest. Another approach to identifying protein function is to first
renature the proteins on the
membranes by any of a number of techniques known in the art such as incubating
the membrane in
Triton-X~ (octylphenol polymerized with ethylene oxide). Once renatured,
proteins will regain their
enzymatic activity and one of several substrate degradation assays known in
the art can be used.
With this approach the activity of kinases, phosphates and metalloproteinases
can be determined.
Panels for scientific research may be grouped by the proteins involved in a
particular
cellular phenomenon such as apoptosis, cell cycle, signal transduction, etc.
Panels for clinical
diagnostics may be grouped by proteins associated with a particular disease
such as Alzheimer's
disease, prostate cancer, etc.
In many embodiments, the detectors/ligands employed are labeled or otherwise
made
detectable using any of several techniques, such as enhanced chemiluminescence
(ECL),
fluorescence, counter-ligand staining, radioactivity, paramagnetism, enzymatic
activity, differential
staining, protein assays involving nucleic acid amplification, etc. The
membrane blots are preferably
scanned, and more preferably, digitally imaged, to permit their storage,
transmission, and reference.
Such scanning and/or digitalization may be accomplished using any of several
commercially
available scientific imaging instruments (see, e.g., Patton et al.,
Electrophoresis 14:650-658, 1993;
Tietz et al., Electrophoresis 12:46-54, 1991; Spragg et al., Anal Biochern.
129:255-268, 1983;
Garrison et al., JBiol. Chem. 257:13144-13149, 1982; all herein incorporated
by reference).
Example Detection Chemistries with Detector Cocktails
In certain embodiments, after proteins have been transferred through the
membrane stack,
individual membranes layers are separated and each is incubated in a separate
antibody (or other
detector molecule) cocktail. A key advantage of creating multiple replicate
blots is that many more
detector molecules (e.g., antibodies) can be usefully employed than if all of
the detectors had to be
crowded onto a single blot.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-40-
An exemplary process for designing the ligand cocktails - and for determining
which
proteins will be identified on each membrane layer - is provided below. First
the panel of proteins of
interest is selected. These can be randomly selected proteins and/or proteins
that are not directly
related to one another or may be groups of known proteins previously
implicated to play a role in one
or more particular cellular phenomena (e.g. apoptosis, cell cycle progression)
or a particular disease
(e.g. prostate cancer specific antigen, PSA). These should be proteins that
have been characterized
by sequence or coordinates on 2-D gels or for which ligands have been or could
be generated. Data
bases of annotated 2-D gels include the Quest Protein Database Center
(http:/lsiva.cshl.org), the
Swiss 2-D PAGE database (http://expasy.cbr.nrc.ca/ch2d), Appel et al.
Eleetrophoresis. 14(11):1232-
1238, 1993; the Danish Centre for Human Genome Research (http://biobase.dk/cgi-
bin/celis), Celis
et al., FEBS Lett. 398(2-3):129-134, 1996, etc. Antibodies may be obtained
from a variety of sources
such as BD Transduction Laboratories (Lexington, KY) or Santa Cruz
Biotechnology (Santa Cruz,
CA, USA).
Although, as discussed above, any of a broad class of ligands may be employed,
for
simplicity the embodiment is illustrated with reference to the use of antibody
ligands.
Immunological identification of the proteins on the membranes thus preferably
involves the selection
of antibodies having a high affinity and specificity for their targets.
However, antibodies, both
monoclonal or polyclonal, frequently recognize more then one protein in
Western blotting detection.
This cross-reactivity phenomenon becomes increasingly apparent as the
concentration of antibody
increases relative to that of the sample proteins. Hence, the first step in
the antibody selection
process preferably involves choosing antibodies (and their working
concentrations) that consistently
visualize preferably 1 but no more then 5 proteins on the same membrane. When
the detector
antibody binds to more than one spot, the undesired proteins ("false spots")
can be eliminated based
on their X-Y positions on the membranes. Since the molecular weight and charge
(pI) of a given
protein is generally constant, it should appear at about the same coordinates
on the gel each time it is
run.
If two or more proteins in a sample are of similar size and charge - and
therefore migrate to
the same general vicinity on the gel - they would likely create overlapping
spots if detected on the
same membrane. In a preferred embodiment, examples of the method avoid this
problem by
designing the antibody cocktail to detect adjacent or overlapping proteins on
different membranes.
The cocktail design process can be readily understood with reference to the
following
hypothetical example (illustrated in FIG. 19). For simplicity in this example,
thirteen proteins
annotated as A-M in FIG. 19A are sought to be identified using only a three-
layer membrane stack.
The ligands employed in the example are antibodies, and three cocktails, one
for each stack, each
with four to six different antibodies, are employed.
For the first membrane cocktail (corresponding to layer one) antibodies are
screened for
protein spot A and the most specific antibody is selected. Antibodies for
spots B-E are picked the
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-41-
same way. Because spots F and G overlap with spot E these are put aside for
other layers. The
second and third cocktails (corresponding to membrane layers two and three)
are created using the
same considerations: (1) if the spot position generated by any two antibodies
cannot be easily
distinguished, the antibodies will not be used in the same cocktail; (2) if
any antibody results in a
background spot near the spot generated by another antibody, the two
antibodies will not be included
in the same cocktail unless the background spot is remote from other spots on
that layer (e.g. spots B
and D on layer 2 created due to cross-reactivity from antibodies directed to
other spots), in which
case such cross-reactivity is simply ignored when the membrane spots are
compared to the template.
Applying these considerations to the hypothetical example results in three
cocktails corresponding to
the layers illustrated in FIG. 19B-D.
Once assembled, the antibody cocktails will be additionally tested for their
specificity by
two different control tests. In a first test, membranes made from the transfer
of a single gel (or from
several gels that contain the same sample and were prepared in the same
manner) will be probed with
cocktails that differ in only one antibody component (each cocktail will lack
one of the antibodies).
As a result of this procedure, immunoblotted membranes should differ from each
other in only one
spot. In a second test, antibody cocktail will be incubated for 0.5-12 hours
at 4-25 °C with a mixture
of epitopes (peptides or proteins) that are used for immunization. During this
incubation, free
antibodies bind to the appropriate epitopes and become immobilized and
functionally inactive. Since
the cocktail becomes depleted of free antibodies subsequent incubation of the
membrane with this
free antibody depleted mixture should yield no specific signal.
Each cocktail will also include one or more antibodies against "housekeeping"
proteins (i.e.,
abundant structural proteins found in all eukaryotic cells such as actin,
tubulin, etc.). Thus, for
example, the antibodies employed with respect to membrane Layer #1 of FIG. 19
will contain an
antibody to actin, which will result in the production of a spot. These
antibodies serve as internal
landmarks to normalize samples for loading differences and to compensate for
any distortion caused
by gel running process. Once the cocktails are designed, they can be reused in
any kit that seeks to
identify the same panel of proteins that were identified in creating the
cocktails, regardless of the
origin of the sample.
It will be appreciated that the present disclosure allows not only the
simultaneous
characterization of a large number of different proteins but also permits the
characterization of a
large number of characteristics of a single protein based on number of
different characteristics. For
example, the protein p70 S6 kinase, required for cell growth and cell cycle
progression, is activated
by phosphate group attachments (phosphorylation) to threonine on position 229
and/or 389 of the
protein. Identification of this kinase would provide not only a determination
of its presence or
absence but also a demonstration of its activity. By way of example, with a
kit containing at least a
four-membrane stack, four copies can be made of a 2-D gel. The first membrane
would be incubated
in antibody specific for the whole protein to determine if this enzyme is
present in the sample or not.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-42-
The second membrane can be used in kinase assay to determine if the enzyme is
active or not. The
third membrane can be probed with phospho-p70 S6 kinase (Thr229) antibody to
determine if
activity of the enzyme is due to activation of this site. The fourth membrane
can be probed with
phospho-p70 S6 Kinase (Thr389) antibody to determine if the activity of the
enzyme is due to
activation of that site. And since all of these tests are done on the single
sample (rather than different
batches of the same sample) the information obtained is more reliable.
Antibody cocktails (such as those illustrated in FIG. 4 and 5, reference
number 38) are
preferably stored in vials, preferably made of plastic or glass, and are
optionally combined in a kit to
create a "panel" of protein targets of interests. Panels for scientific
research may be grouped by the
proteins involved in a particular cellular phenomenon such as apoptosis, cell
cycle, signal
transduction, etc. Panels for clinical diagnostics may be grouped by proteins
associated with a
particular disease such as Alzheimer's, prostate cancer, etc.
V11. Ifits
Other embodiments of the disclosure include kits that contain a membrane array
for
detecting biomolecules (such as proteins or nucleic acids) in a sample. The
array includes a plurality
of membranes, each of which has a non-specific or substantially same affinity
for the biomolecules.
Certain provided kits also include one or more containers of detector
molecules, such as antibodies or
probes (or mixtures of antibodies, mixtures of probes, or mixtures of the
antibodies and probes), for
detecting biomolecules captured on at least one of the membranes. In
particular examples of the kit,
the membranes are polymer substrates containing or coated with a material
(such as nitrocellulose)
for increasing an affinity of the substrate to the biomolecules.
Kits may additionally contain reagents for effecting the detection of
detector/ligand-
biomolecule binding, buffer, and/or instructions or labels that indicate the
particular detector or
detector cocktail to be applied to a particular membrane. Software such as
that discussed herein may
also be included in the kit or may be accessible via modem, the Internet, by
mail, or by other means.
Primary antibodies to particular groups of proteins, such as biochemical
pathways may be
optionally included with a kit. Alternatively the user can supply primary
antibodies.
The methods and kits allows up to several thousand discrete protein spots to
be identified,
annotated, and, at the user's option, compared to the pattern of protein spots
generated from other
biological samples stored in a database.
Certain kit embodiments have been discussed above, including first kit 36 and
second kit 58.
Also provided is another specific embodiment, directed to a method and a kit
122 for identifying (i.e.
detecting, annotating, and/or characterizing) groups of proteins that have
been separated by gel
electrophoresis. As illustrated in FIG. 20, representative kit 122 comprises
the following
components: (i) a membrane stack 13 or framed membrane stack 110 (as
illustrated) upon which the
proteins are transferred, (ii) protein transfer reagents) 124 and (iii)
protein detector molecules, such
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
- 43 -
as stain 126 and protein-specific detector molecule 128. The kit may also
include software 46 (not
shown in FIG. 20) that allows the user to analyze and manipulate the images
produced so as to yield
a "proteomic image" of the biological sample being tested and compare it to
proteomic images from
other samples in a database. Alternatively the software may be acquired or
accessed independent of
the kit.
In some embodiments, transfer reagent is also provided with a kit. Examples of
transfer
reagents include Tris, Phosphate, Tris/Glycine or Phosphate/Glycine buffers
with an alkaline pH
(e.g., 8.0-9.5), with or without methanol (usually 20% or less) and/or SDS (in
some embodiments
0.05% or less, and in particular embodiments 0.025% or less). Specific
examples of transfer reagent
suitable for use in examples of such kits are in the Examples.
In addition to identifying proteins of interest structurally, kits are
provided that can be
employed to identify the functional state of proteins. One way to do so is to
use phospho - specific
antibodies to determine the phosphorylation state of proteins) of interest.
Another approach to
identifying protein function is to first renature the proteins on the
membranes by any of a number of
techniques known in the art (such as incubating the membrane in Triton-X-100 ~
(octylphenol
ethylene oxide condensate). Once renatured, some proteins will regain their
functional activity and
one of several substrate degradation or modification assays known in art can
be used. With this
approach the activity of kinases, phosphates and metalloproteinases, etc., can
be determined.
VIII. Devices and Apparatuses
In certain provided embodiments, particularly those which employ contact
transfer, the
transfer can be effected by placing the assembled membrane stack into a gel
drier-type apparatus,
which applies heat and/or pressure to the stack. The combination of heat and
pressure being applied
causes biological components, including proteins and/or nucleic acids and/or
carbohydrates and/or
lipids, to be transferred from the sample 11 to membrane stack 13. This
produces multiple copies or
replicas of the biomolecular content of the tissue sample, due at least in
part to the binding
characteristics of the membranes.
In lieu of gel dryers, a specialized instrument 130 (FIG. 21) may be employed
to provide
heat and/or pressure to the sample and membrane stack. The instrument
comprises a body 134 and a
lid 136, each having a face 132a, 132b which serves as one of the
substantially flat surfaces 132.
The surfaces may be provided by the upper face 132b of the body 134 and the
lower face 132a of the
lid 136 directly, or may be provided by a substantially flat panel or other
flat object disposed on a
face 132a, 132b of the body 134 or lid 136.
One or both of the substantially flat surfaces may protrude in order to ensure
adequate
contact to provide pressure between them. In the illustrated embodiment, for
instance, the upper
substantially flat face 132a is a surface of a member that protrudes from the
lower face 132a of the
lid 136. In some embodiments, one or the other or both of the substantially
flat surfaces 132a, 132b
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-44-
may be compressible (for instance, somewhat spongy), to further ensure that
pressure applied to the
sample and membrane stack is relatively complete and even across the surface
of the stack.
The lid 136, in some provided embodiments including that illustrated in FIG.
21 may be of
sufficient weight to provide sufficient pressure to a sample and membrane
stack placed under the lid
136 so that it facilitates biomolecule transfer as described herein. Such
weight is not required, but in
those embodiments wherein the lid 136 does not provide sufficient weight,
another mechanism fox
applying pressure is included. Such means includes for instance a separate
weight (not shown), such
as a weight 34 placed on the upper surface of the lid 136, or clips, springs,
clamps or the like that
urge the lid 136 toward the base 134 with sufficient force to provide the
amount of pressure needed
to facilitate transfer.
In some embodiments, the lid 136 may be hingedly attached to the body 134,
such that
when the lid 136 is lifted it does not come fully away from the body 34 but
remains connected in at
least one place. In the illustrated example (FIG. 21), two hinges 138 are
provided to maintain the
connection between the body 134 and the lid 136. In particular embodiments,
the hinge or hinges
138 are "loose" or "floating," in that they permit some play between the lid
136 and the body 134.
This play permits the device to accommodate assembled contact transfer stacks
of different thickness,
while still adequately applying sufficient and even pressure to the stack.
Though some embodiments
are large enough to accommodate multiple stacks in side-by-side arrays, it is
not recommended that
stacks of substantially different thickness be transferred in the same device
at the same time, as the
applied pressure may not be adequate on thinner stacks when a substantially
thicker stack is present
between the faces 132a, 132b.
Some embodiments of the device 130 are capable of supplying heat as well as
pressure to
the contact transfer stack. These embodiments may contain, for instance, a
heater element (not
shown) in the body 134 or the lid 136, or both, that provides heat to one or
both of the substantially
flat faces 132a, 132b. Examples of such heated devices 130 will be equipped
with an internal or
external power source, for instance a battery (not shown) or connection to a
source of alternating
current (not shown). Methods of and mechanisms for providing heat to a surface
are well known, as
are thermostats for controlling the level of heat provided. Specific examples
of heated devices 130
will include a mechanism for controlling whether or not heat is generated
(e.g., an "ON/OFF" switch
140 as shown in FIG. 21), a mechanism for regulating the level of heat
produced (e.g., a thermostat,
with or without a user control), and/or an indicator that indicates when the
device is heating or
heated. In the illustrated embodiment, an indicator light 142 is provided,
which is capable of
indicating when the device reaches a factory-set temperature (e.g., 80
°C), and is thus ready for use.
Specific examples of the heated device 130 that include a heater element in
both the lid 136
and the body 134 may include a mechanism or control (such as dial 144) for
selecting whether one,
the other, or both heater elements are engaged when the device is turned on.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
- 45 -
IX Applications
The heat and pressure applied to contents of the enclosure permit proteins and
other
molecules to be transferred from the embedded cellular material to the
membrane stack. This
produces multiple copies or replicas of the biomolecular content of the
cellular sample. The
processed membranes (or layers) are then separated and each is incubated with
one or more different
probes or antibodies specific for particular targets of interest. The probes
employed are labeled or
otherwise detectable using any of a variety of techniques such as
chemiluminescence. Thus, while
each membrane has essentially the same pattern of proteins bound to it,
different combinations of
proteins are made visible on each membrane due to the particular probes or
antibodies selected to be
applied. For example, one membrane layer may display proteins involved in
programmed cell death
(apoptosis) while an adjacent layer may display enzymes involved in cell
division such as tyrosine
kinases. In addition to proteins, nucleic acids may be targeted by using
labeled DNA probes in lieu
of antibodies. Moreover, both protein and nucleic acid targets can be detected
in parallel by applying
both antibodies and probes to different layers of the stack. Commercially
available flatbed scanners
and digital imaging software can be employed to display the images according
to the preference of
the user.
With reference to FIG. 20, kit 122 may be used to identify proteins that have
been separated
on electrophoresis gels, both two-dimensional gels and one-dimensional gels.
Proteins are isolated
from a biological sample and separated on the gel according to techniques well
known in the art,
such as those described herein and in Manabe, Electrophoresis. 21(6):1116-
1122, 2000; Oh et al.,
Electrophoresis. 20:766-774, 1999; Dunn, JChromatogr. 418:145-185, 1987,
In some.embodiments, after gel 50 is run, it is removed from the
electrophoresis apparatus
and sandwiched and placed in a transfer apparatus such as the type typically
used in creating Western
blots. Such devices are available, for example, from Biorad Laboratories,
Inc., Novex, Inc. and
Amersham Pharmacia. Membrane stack 13 is positioned between the electrodes
adjacent to gel 50 as
illustrated in FIG. 22. While only about a half dozen membranes are shown in
FIG. 22 it will be
appreciated that up to one hundred may be employed depending on the number of
targets sought to
be identified in a panel, the quantity of proteins present in the sample, and
the thickness of the
material employed to construct membranes 12. Optionally, membranes 12 may be
packaged in a
suitable sealed enclosure or frame (not shown) to maintain their integrity and
prevent contamination.
Sponge pads 130, preferably constructed of foam, rubber or filter paper and
layers of filter paper 14
are also sandwiched between the electrodes as shown in FIG. 22.
Transfer buffer (25 mM Tris pH 8.3, 192 mM glycine, 0.025% SDS and 20%
methanol) is
applied to elute and transfer proteins from the gel 50 to the membranes 12.
Any of a variety of
conventional methods for effecting such transfer may be employed, including
wet tank transfer, and
semi-dry transfer. In a wet tank transfer, the membranes are immersed into a
tank containing buffer;
in a semi-dry transfer, the membranes are blotted with moist pads. In both
cases, the membranes are
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-46-
subjected to a voltage potential (e.g., 125-150 mAmps for 1-10 hours). In such
transfer, it is
important that a tight contact be created between the membranes and the gel.
Wet tank transfer is
preferred. For a membrane of 10 x 10 cm2, a tank containing 400-500 ml of
buffer may be
employed. Preferred transfer conditions are 60-110 mAmps for 1-2 hours.
After transfer the membranes are separated and incubated with detector
antibody(s).
Antibodies are selected based on the types of target molecules sought.
Membranes are washed in a
buffer, and the protein / detector complex can be visualized using a number of
techniques such as
ECL, direct fluorescence, or colorimetric or calorimetric reactions.
Commercially available flatbed
scanners may be employed in conjunction with film, to detect or record
signals. Alternatively,
specialized imaging instrumentation (e.g., for ECL), such as the Kodak IMAGE
STATION (NEN)
may be utilized. Digital imaging software can be employed to display the
images according to the
preference of the user, as discussed herein.
In addition to use with 2-D gels, provided methods may be employed to identify
proteins
that have been separated by a 1-D gel such as conventional gels for separating
proteins by size, and
gel shift assays. Gel shift assays (also known as "mobility shift assays") are
the most commonly
used tool for studying protein - DNA interactions. The assay is based on
labeling of the DNA
fragment that contains presumptive protein binding site and incubation of that
labeled fragment with
protein that binds to that site. If they interact, complex will be formed. If
source of protein is a cell
extract (rather than a solution of in vitro synthesized proteins), the complex
may contain number of
proteins, of unknown identity, that interact with each other. After binding, a
mixture of DNA and
proteins is loaded onto a non-denaturing polyacrylamide gel and the proteins
are separated based on
their size. DNA-protein complexes are visualized by exposure to X-ray filin,
or by other means. The
higher the bands are in the gel, the larger the size of the DNA-protein
complex. In most cases, this
type of analysis does not reveal identity of the proteins) in the complex.
As illustrated in FIG. 23, membrane stack 110 may be used to identify
biomolecules that
have been separated on electrophoresis gels, including proteins that have been
separated on one-
dimensional (1-D) gels 132 or two dimensional (2-D) gels (such as 50, not
shown in this figure) as
well as nucleic acids that have been separated on agarose gels. The following
description relates to
use of embodiments in conjunction with protein detection of 1-D gels.
Proteins are isolated from a biological sample and applied and separated onto
a gel 132,
typically a sodium dodecyl sulfate - polyacrylamide gel, which is cast, for
example, as a square slab
gel with a thickness between 0.5 to 2.0 mm. Pre-cast gels useful with the
present disclosure can be
obtained from a variety of suppliers including InVitrogen (Carlsbad, CA).
Unlike with conventional blotting, wherein less than 30 micrograms of protein
is loaded into
each well of the gel, according to specific methods herein between about 50 to
100 micrograms of
protein is loaded into each well. The amount loaded will depend upon the
number of "copies" of
membranes to be created and size of the protein one wishes to detect (see
Example 11).
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-47-
In some embodiments, the components of transfer buffer 124 are provided in
separate
containers, which are combined and applied to elute and transfer proteins from
the gel 132 to the
membranes 12. About 500 milliliters may be used in each transfer, with average
length of the
transfer being about 1-2 hours. With reference to FIG. 23, 24, and 6,
separated proteins on gel 132
are transferred to framed membrane stack 110 (though a stack of unframed
membranes could be
used) by one of several alternative techniques.
A first technique, illustrated in FIG. 24, involves electric transfer using a
standard gel
electro-blotting apparatus such as the MiniCell unit (Bio-Rad Laboratories,
CA). Gel 132 is removed
from the electrophoresis apparatus and placed adjacent to membrane stack 110.
The gel 132 and
membrane stack 110 are then placed between the anode 134 and cathode 136 of
electro-blotting
apparatus 138 with sponge pads 130 positioned as shown. The electro-blotting
apparatus 138 is
activated with a voltage of about 59-63 volts for about 60-70 minutes.
A second transfer technique (referred to as bi-directional contact transfer)
is illustrated in
FIG. 6. Here first and second membrane stacks 13a and 13b sandwich gel slab
54. A pair of filter
pads 24 and 26, for instance constructed of a blotting paper such as GB004
Blotter Paper available
from Schleicher and Schuell, are provided adjacent to the membrane stacks as
shown. Filter pads 24
and 26 are saturated with a transfer buffer such as TRIS or phosphate base
buffers.
A collapsible, fluid impervious enclosure 28 is provided to envelop the pads,
membrane
stacks, and gel as shown in FIG. 6. Plastic bag enclosure 28 is preferably a
heat sealable pouch such
as those available from I~apak Corp. (Minneapolis, MN). In many embodiments,
it is best to remove
most of the air from bag 28, for instance by gentle squeezing and/or vacuum
suction. The bag is then
sealed by a heat sealer such as the Impulse Sealer (American International
Electric). Enclosure 28 is
then placed between a pair of heating elements 56a and 56b such as those
provided in Gel Dryers
manufactured by Bio-Rad Laboratories (Hercules, CA). The bag and its contents
are preferably
heated to a temperature of between about 50 to 90° C, preferably to
about 80° C for about 2-4 hours.
Pressure is preferably applied throughout the heating process using a weight
34. Alternatively, a
specific device for applying heat andlor pressure (such as that illustrated in
FIG. 21) can be
employed.
The heat and pressure applied to contents of the enclosure permit proteins and
other
molecules to be transferred from the gel to the membrane stack. This produces
multiple copies or
replicas of the biomolecular content of the gel.
In addition to their use in identifying the proteins of the proteome, the
methods and kits
provided herein can be used to measure the concentration of a protein (either
in absolute terms, or
relative to the concentration of another protein). Likewise, they can be used
to measure the
distribution of variants of a protein, and to identify proteins that are
analogous in structure or
function to identified (e.g., human) proteins, or to identify plant clones or
transgenic animals that
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-48-
express a particular protein or protein variant (which may be linked to, or
associated with, a trait or
phenotype).
X. Image Analysis Software
Software 46 is made available to users of any of the provided kits by
providing it on a
diskette to be included within the kit (e.g., kit 36, 58, or 122) or by making
it accessible for
downloading over the Internet or a private Intranet network, or by other
means. The function of
software 46 is to translate the visible spots generated by detector molecules
(such as antibody
cocktails 38) into useful information about the proteome of the sample being
tested. This
information primarily includes the quantity of the proteins in the test sample
relative to a control and,
in some cases, information about certain functional aspects of these proteins.
Suitable software can
be obtained from, or adapted from, any of a variety of sources (e.g.,
http://www.2dgels.com/home.html and http://expasy.proteome.org.au).
After it is determined which molecules (e.g., proteins) will be identified on
each layer for a
given panel/kit, a template image such as that shown for a 2-D gel (reference
numeral 140) is created
corresponding to each layer (FIG. 25) and stored in software 46. In this
example, the 2-D gel X-Y
coordinates of each protein can be ascertained from any of a number of
references and databases.
Thus, referring to FIG. 25, template image 140 is the image of what a membrane
would look like if
all of the targeted proteins assigned to the layer are present in the sample
being tested along with the
landmark "housekeeping" proteins 142a, 142b, 142c. Each antibody cocktail
generates a unique dot
pattern on the corresponding membrane to which it is applied as a result of
the selection process
outlined above. A template membrane 140 will be used by image processing
software to analyze
experimental membranes generated by users. Important feature of the template
is existence of the
internal landmarks 142. These spots will originate from the set of antibodies
targeted against
housekeeping proteins present in every sample regardless of origin. Since
their relationship always
stay the same these landmarks will serve to normalize samples for loading
differences and to
compensate for any distortion caused by gel running process.
Image analysis will start with digitalized images) of the experimental
membranes. As the
first step, the user matches templates with the membranes. Software then
compares an image of the
template and an image of the membrane and performs alignment of spots/bands.
The user has
options of visual alignment control and the ability to hand correct minor
discrepancies. The second
step of analysis will include densitometric readings of the spots on
experimental membranes. This
data is stored in the database. The third step includes numerical data
manipulation. Intensity values
of each experimental spot on the membrane are divided with values of the
landmark spots. This step
generates normalized intensity values for each spot on the membrane. All the
spots of interest can
thus be compared with each other.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-49-
Software 46 preferably allows the user to select the kind of comparative
analysis to be
performed (i.e. comparing the spots or bands present in one sample with those
in another sample or
comparing those present on one membrane with those of another membrane within
the same
membrane stack). Results of the analysis are displayed in, for instance,
tabular format and user is
given the option to go back and compare magnified sections of the images of
interest.
In one embodiment, software is provided with template images corresponding to
each of the
membrane images. Such software allows the identity of the protein in each spot
to be confirmed
based upon the vertical and horizontal position of the protein's spot on the
gel. Examples of such
software also allow the density of each spot to be calculated so as to provide
a quantitative or semi-
quantitative read-out as described herein. Such software may also have links
to a database of images
generated from other gels to allow comparisons to be made between different
diseased and normal
samples, or links to images or data (structure, sequence, function, etc.).
In some embodiments, software is also provided to overlay images of the bands
or spots or
cells onto a master image of a ubiquitously stained sample or gel. A key
feature of examples of such
software is the ability to quantify the biomolecules by determining the
density of the bands or spots
and comparing them to a control. This process is known as "normalization." For
analysis of 1-D
gels a variety of commercially available programs may be employed such as the
1-D Image Analysis
Software available from Eastman Kodak Co.
Having now generally described the invention, the same will be more readily
understood
through reference to the following examples, which are provided by way of
illustration, and are not
intended to be limiting of the present invention, unless specified.
EXAMPLES
Example 1
Construction of Polycarbonate Membranes for Protein Binding
Native, non-coated polycarbonate membrane (Millipore, MA) has low affinity and
low
binding capacity for proteins. To improve its protein binding characteristics,
polycarbonate
membranes were coated with either poly-L lysine (referred to as PC+Lysin in
FIG. 26) or
nitrocellulose (referred to as PC+NC in FIG. 26). Membranes (177 square
centimeters) were
immersed for one minute in 5 ml of either aqueous solution of 0.1% poly-L-
lysine or 0.1-1.0%
nitrocellulose solution in 100% methanol. After coating, membranes were
suspended in vertical
position and air-dried at room temperature for 5-10 minutes. Poly-L-lysine
treated membranes were
before use additionally baked for two hours at 50° C. Small squares
(0.25 square centimeters) of
both treated and non-treated membranes were incubated in TBST solution (50 mM
TRIS pH 8.0, 150
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-50-
mM NaCI and 0.05% Tween-20) with 1.0-100.0 ng/~1 of goat immunoglobulin
labeled with Cy3
fluorescent dye (Amersham Pharmacia Biotech, USA) for 0.5-2 hours at room
temperature.
Membranes were washed in TBST and examined on STORM scanner (Molecular
Dynamics, USA). The results are shown in FIG. 26A. The intensity of the signal
was quantified by
ImageQuant (Molecular Dynamics, USA) and data points from five different
experiments were
plotted using Microsoft Excel. The results shown in FIG. 26B demonstrate that
polycarbonate
membranes have a low protein binding potential that can be considerably
enhanced by coating the
membrane with poly-L-lysine (PC+Lysin) or nitrocellulose (PCNC).
Example 2
Testing the Porosity of Prepared Polycarbonate Membrane Layers
To demonstrate porosity of manufactured layers, native, poly-L-lysine or
nitrocellulose
coated membranes were blocked in 5% bovine serum albumen solution in 50 mM
TRIS pH 8.0 to
prevent any protein binding. Fifty-one square centimeter pieces were cut out
and stacked together to
make a pile. A non-blocked pure nitrocellulose layer was used at the bottom to
capture proteins
(NC-trap). Three adjacent 20 micrometer thick frozen sections of normal breast
tissue were collected
on a polycarbonate membrane with 5.0 um pore size and embedded in a 2% agarose
gel and
transferred side by side through each stack. Between 50 and 100 milliliters of
TBST buffer was used
per square centimeter of the membrane stack with average length of the
transfer being 1 hour. After
transfer, proteins remaining in the tissue sections and total proteins on the
NC-trap were visualized
by Ponceau S staining (SIGMA, MO).
As shown in FIG. 27, the outline of the total proteins transferred through the
stack and
trapped on the nitrocellulose layer very closely resembled the outline of the
applied tissue section.
This suggests that not only were membranes porous enough to allow for the
proteins to be
transferred, but also that at least up to 50 polycarbonate membranes can be
used in this kind of assay
without apparent distortion of the image due to lateral diffusion.
Example 3
Demonstration of Low Capacity Protein Binding to Nitrocellulose
Coated Polycarbonate Layers
Examples l and 2 demonstrate that proteins in solution can bind to a single
nitrocellulose
coated polycarbonate layer and that complete saturation of the layer with
proteins does not affect its
porosity. To ascertain how much of the total protein would be trapped on each
individual layer
during the tissue section transfer, 20 micron thick frozen sections of normal
and tumor breast tissue
were placed adjacent to each other on a polycarbonate membrane with 5.0 um
pore size, embedded in
2% agarose gel and transferred through 14 layers of nitrocellulose coated
polycarbonate to the NC-
trap on the bottom, in 100 ml/cm2 of buffer containing 25 mM TRIS pH 8.3, 192
mM glycine, 0.05%
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-51-
SDS and 20% methanol. After transfer, proteins left over in the tissue
sections were visualized by
Ponceau S staining (SIGMA, U.S.A.) and total eluted proteins captured on the
NC-trap were
visualized by BLOT FastStain (Chemicon, USA). The image formed on the trap
demonstrated
successful transfer of the protein through the membranes.
To determine whether sufficient total protein trapped on each membrane during
the transfer
to perform immunological detection 14 arbitrarily selected antibodies were
used. Antibodies were:
Anti-GAPDH, 1:100 (Chemicon, MAB374); Anti-Rsk, 1:1,000 (Transduction
Laboratories,
R23820); Anti-StatSa, 1:500 (Santa Cruz Biotechnology, sc-1081); Anti-
IFNalpha, 1:500 (Biosource,
AHC4814); Anti-RARalpha, 1:1,000 (Biomol, sa-178); Anti-phospho-EGFR, 1:1,000
(Upstate, 05-
483); Anti-non-phospho EGFR, 1:1,000 (Upstate, OS-484); Anti-phospho-NRl,
1:500 (Upstate, 06-
651); Anti-Statl, 1:2,000 (Transduction Laboratories, G16920); Anti-Rb,
1:1,000 (Santa Cruz
Biotechnology, sc-50); Anti-Jakl, 1:500 (Santa Cruz Biotechnology, sc-295);
Anti-tubulin-alpha,
1:500 (Santa Cruz Biotechnology, sc-5546); Anti-beta-actin, 1:2,000 (SIGMA,
A5441).
Polycarbonate layers were first blocked in lx casein solution (Vector Labs,
U.S.A.) for one
hour at room temperature and incubated overnight at 4° C in primary
antibodies as listed in FIG. 28
followed by TBST washes and incubation in alkaline phosphatase (AP) conjugated
secondary
antibodies (1:2,000 dilution) (Rockland, U.S.A.). Membranes were then
incubated for five minutes
in enhanced chemiluminescence (ECL) substrate (Vector Labs, USA) followed by
visualization of
the protein by exposing the membranes to X-ray film (Kodak, USA).
The results showed that methods provided herein allow detection of a number of
different
proteins. To ascertain how the membranes performed with respect to the amount
of total protein
captured, the membranes were each incubated with the same antibody, allowing
determination of the
protein content on each of them. Anti-GAPDH antibody was used for three hours
at room
temperature, washed in TBST, incubated with anti-mouse secondary antibody
conjugated to
horseradish peroxidase (HRP) and visualized in enhanced chemiluminescence
substrate specific only
for HRP (Pierce, USA). After ECL reaction membranes were exposed to film as
stated before. The
results confnmed that all of the membranes did capture a similar portion of
the total protein and
differences seen in the first part of the experiment are not the result of
differences in membrane
"loading." For documentation purposes, the X-ray film was scanned on the flat
bed scanner (Lacie,
USA) and images were processed using ADOBE PhotoShop 4Ø
Exahaple 4
Transferring Proteins from Tissue Microarrays
A five microns (5 ~M) thick paraffin section of a tissue microarray
originating from the
National Institutes of Health (NIH) Tissue Array Research Program (TARP) was
collected on tape
and transferred through four membranes in the manner described above. The
membranes were as
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-52-
provided above. The transfer solution contained 50 mM TRIS and 380 mM glycine.
This yields a
buffer with approximately pH 8.6, but this can be adjusted to anywhere in a
range of pH 8.0 to 9.5.
Plastic pouch enclosing membrane stack and tissue array was placed in a Gel
Drier
(BioRad) and lid of the drier was used as a pressure and heating (80
°C) source. Heat and pressure
were applied for two (2) hours.
After transfer, membranes were gently washed in TBST buffer (50 mM TRIS pH
8.0, 150
mM NaCI and 0.05% Tween-20) and stained with FAST Blue Stain (Chemicon)
according to
manufacturer instructions. Scanning on an Astra 2200 scanner (UMAX)
digitalized images of the
individual layers. After staining, membranes were rinsed in TBST buffer,
blocked for 15 minutes in
lx casein solution (Vector Laboratories, Inc.) and incubated overnight at
4° C in primary antibody
(anti-cytokeratin (1:5,000, Sigma) or anti-PSA (1:500, Script)). The membranes
where then washed
in TBST, incubated in the complex of secondary antibody and alkaline
phosphatase, and washed
again. Localization of the target protein (cytokeratin or PSA) was visualized
by enhanced
chemiluminescence (ECL) (DuoLux, Vector Laboratories, Inc.) and Biomax MR film
(Kodak). The
images were digitalized by scanning on an Astra 2200 scanner (UMAX).
The results, shown in FIG. 29, demonstrate that membrane replicas can be made
from tissue
arrays by using the described techniques without loosing spatial resolution.
It also demonstrate that
immunodetection of a single protein is possible on these membranes.
Example S
Differential Protein Expression in Different Tumors
Membrane copies of the TARP array were prepared and assayed as stated in the
previous
example. For detection, the following primary antibodies were used: anti-
cytokeratin (1:5,000,
Sigma), anti-PSA (1:500, Script), anti-p53 (1:1,000, Transduction
Laboratories) and anti-p300
(1:500, Transduction Laboratories).
The results, shown in FIG. 30, demonstrate that different tumor types express
different
amounts of the same protein (for instance, PSA is primarily expressed in the
prostate cancer samples)
and that the same tumor type can express different amount of the same protein
(for instance, p53 and
p300 are expressed in only a subset of colon carcinoma samples).
Example 6
Immunodetection on Membranes Using Antibodies Ineffective in ITiC
Membrane copies obtained from the transfer of normal human tonsilar tissue,
normal human
kidney tissue, and TARP tissue array were produced as described in the
previous examples. In some
cases, the membranes were subjected to antigen retrieval, by immersing them in
a solution of 0.1 M
sodium citrate containing 10 mM EDTA pH 8, for 5 minutes, at 95 °C.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-53-
Following blocking in a lx casein solution (Vector Laboratories) for 30
minutes, the
membranes were incubated with monoclonal antibodies diluted at 1:20-1:50 for
16 hours at 4 °C.
Primary antibodies were used essentially as directed in the manufactures'
instructions; each of the
antibodies selected are noted by the manufacture to be ineffective when used
to detect proteins in
formalin preserved tissue samples, even when the samples are subjected to
antigen retrieval. The
following antibodies were used: anti-CD3 (CALTAG); anti-EGFR; anti-
Progesterone Receptor;
(Dako); and anti-erbB2 (Zymed). Following TBST washes, proteins were
visualized as described in
Example 4.
In each case, the antibody yielded clear signals on the transferred membranes
but would not
yield signals when used for IHC on adjacent sections, directly on a
corresponding microarray. Thus,
transfer of biomolecules to membranes using the described contact transfer
method is effective for
immunodetection visualization using antibodies that are ineffective in IHC.
Example 7
Transferring Proteins from Cells Collected by LCM to Membranes
Five microns thick frozen section of squamous carcinoma of the head and neck
was
collected on plain glass slide. The slide was fixed in 100% ethanol for three
minutes, immersed in
0.5% ethanol solution of Azure Blue (SIGMA) for one minute followed by five
minutes incubation
in xylene. LCM was performed as recommended by the manufacturer (Arcturus).
Each LCM cap
received approximately 50 laser hits, corresponding to 250-300 cells during a
15-20 minute time
period. Immediately after this, caps were stored at -80° C until
transfer.
Just prior to transfer, caps were hydrated through an ethanol gradient and
transfer was
assembled as shown in Fig. 8. Five different membrane layers were used.
Transfer buffer contained
mM TRIS, 192 mM glycine and 0.025% SDS. The assembled package was placed in a
gel drier
25 (BioRad) and lid of the drier was used as a pressure and temperature
(80° C) source. The transfer
process took about two hours.
After transfer, the stack was disassembled, membranes were washed in TBST
buffer (50
mM TRIS pH 8.0, 150 mM NaCl and 0.05% Tween-20) and then stained with FAST
Blue Stain
(Chemicon) according to manufacturer instructions. Scanning on Astra 2200
scanner (IJMAX)
produced digitalised images of the layers. After staining, membranes were
rinsed in TBST buffer,
blocked for 15 minutes in lx casein solution (Vector Laboratories, Inc.) and
incubated overnight at
+4° C in anti-cytokeratin antibody (1:5,000, Sigma), washed in TBST,
incubated in the complex of
secondary antibody and alkaline phosphatase, washed again and location of the
protein was
visualized by ECL (DuoLux, Vector Laboratories, Inc.) and Biomax MR film
(Kodak). The resultant
image was digitalised by scanning on an Astra 2200 scanner (UMAX). FIG. 31
shows "copies" that
were made on five membranes, and that antibodies were effectively used to
detect proteins on each
layer.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-54-
Exafnple 8
Transfer and Capture of Proteins From a 1-D Gel
This example demonstrates that polycarbonate coated nitrocellulose (PCNC)
membranes,
with their high binding affinity but low capacity for the proteins eluted from
the gel, can be used to
make multiple copies of a gel.
1.0 pg/lane of biotinylated protein marker (Vector Laboratories, Inc) was
separated by 15%
PAGE and electro-transferred in 25 mM Tris, 192 mM glycine, 0.025% SDS and 20%
methanol (60-
110 V for 1-2 hours) through a stack of PCNC membranes; the number of
membranes per stack was
5-20, depending on the experiment. At the end of the stack, one pure
nitrocellulose membrane was
used to capture proteins that were not bound to PCNC layers ("NC trap").
Transfer was performed
from 0.5-3 hours at 60-110 V in a Ready Gel Cell apparatus (BioRad).
After transfer, membranes were rinsed in 50 mM Tris pH 8.0 and 150 mM NaCI
(TBS
buffer), blocked for 10-60 minutes in lx casein solution (Vector Laboratories,
Inc.), and incubated
for 30 minutes in VECTASTAIN ABC-AmP reagent (Vector Laboratories, Inc.).
Membranes were
washed again in TBST, rinsed in 0.1 M TRIS pH 9.5, incubated in DuoLux reagent
(Vector
Laboratories, Inc.) for 3-5 minutes, and exposed to Biomax MR filin (Kodak).
An example of one
representative experiment is shown in FIG. 32.
Results demonstrated that:
1. PCNC stack of membranes did not interfere with post-transfer Western
blotting
procedure - proteins were transferred from the gel through the stack and to
the NC trap;
A wide range of protein sizes were transferred through the stack with very
similar
transfer efficiency - 7 kDa-200 kDa proteins were detected on the NC trap; and
3. PCNC layers captured proteins regardless of their size.
In order to determine compatibility of PCNC membranes with immunodetection,
Jurkat cell
were lysed in buffer (50 mM TRIS pH 8.0 and 1% SDS) and 20 pg/lane of total
protein was
separated by 15% PAGE. The resultant gel was electro-transferred through a
stack of PCNC
membranes in 25 mM TRIS, 192 mM glycine, 0.025% SDS and 20% methanol. Transfer
was
carried out at 60-110 V for one to two hours.
All of the membranes were incubated in primary anti-Rsk (1:100, Transduction
Laboratories) and anti-p300 (1:500, Transduction Laboratories) antibody,
washed in TBST buffer,
incubated with the complex of secondary antibody and alkaline phosphatase, and
washed again. The
location of the protein on the blots was visualized using ECL (DuoLux, Vector
Laboratories, Inc.)
and Biomax MR film (Kodak). The results, shown in FIG. 33, demonstrated that
PCNC membranes
are suitable for this type of protein detection. Each membrane captured
sufficient protein to be
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-55-
detected by immunological methods, but each single membrane did not capture
too much protein,
enabling a number of copies of the same gel to be generated.
Example 9
Transfer and Capture of Proteins From a 2-D Gel
2-D protein gels have greater separation capabilities than 1-D gels. Two-
dimensional
separation allows identification of hundreds or even thousands of proteins on
the same gel. Proteins
separated by 2-D gels are identified by protein sequencing or immunological
features. Sequencing
requires expensive equipment, highly trained operators, and its use is limited
to a small number of
privileged groups. Immunodetection is easier to do but it is a low throughput
technique, since
traditional blotting procedures generate only one membrane per gel.
As described above, one can make 10 or more 1-D gel copies using PCNC
membranes. In
order to fmd out if 2-D gels can be "copied" the same way, the proteins
present in 500 pg of Jurkat
cell protein lysate were separated by 2-D PAGE. A commercial immobilized pH
gradient (IPG) from
3.0 to 10.0 (Pharmacia Biotech, Uppsala, Sweden) was used for first-dimension
separation. Eight to
12 hours of in-gel sample rehydration was used for protein loading. Proteins
were separated for a
total of 15,000-30,000 Vhrs. After equilibration, the IPG strips were
transferred onto vertical
gradient gel (4-20%, Novex) for second dimension separation.
After electrophoresis, the 2-D gel was transferred in 25 mM Tris, 192 mM
glycine, 0.025%
SDS and 20% methanol (60-110 V for 1-2 hours) through a stack of five PCNC
membranes. The
membranes were then rinsed in TBST buffer, then blocked for 10-60 minutes in
lx casein solution
(Vector Laboratories, Inc.) prior to probing with specific antibodies.
Individual membranes were
probed by incubating them overnight at 4 °C in anti-GAPDH (1:5,000,
Chemicon), anti-beta-actin
(1:5,000, Sigma) and/or anti alpha-tubulin (1:1,000, Calbiochem) antibody. The
membranes were
then washed in TBST, incubated in the complex of secondary antibody and
alkaline phosphatase, and
washed again. The location of the protein was visualized by ECL (DuoLux,
Vector Laboratories,
Inc.) and Biomax MR film (Kodak).
Antibodies were first applied separately to three different membranes (from
three different
gels) to fmd the precise spatial location of specific proteins in the 2-D gel.
These three proteins
(GAPDH, actin, and tubulin) differ in their size and charge, and were
spatially separated from each
other on the gel.
In oxder to increase the throughput of immuno-detection, all three antibodies
were mixed
together and applied as a detector cocktail to all five membranes from the
same gel. The results of
this experiment are shown in FIG. 34. Generating multiple replicas of the same
gel and using an
antibody cocktail approach increased throughput of the immunological protein
identification on 2-D
gels.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-56-
Example 10
Use of Layered Membranes for Protein-DNA Complexes Identification
The following example demonstrates the ability of the layered membranes of the
present
invention to speed up and simplify the identification of the proteins of a
protein-DNA complex. It
shows that copies of the gel were made and each of the membranes was
successfully immuno-probed
with a different antibody of interest.
250 ng of recombinant his6-c-rel and 120 ng of purified recombinant his6-CREB
were
incubated alone or in combination with 0.2 ng of'ZP-5' labeled duplex
oligonucIeotide (SEQ ID NO:
1), in 10 p1 of buffer containing 10 mM HEPES, 50 mM NaCl, 20% glycerol, and 4
mM (3ME. The
hybridization reaction was allowed to proceed at room temperature for 30
minutes. Samples were
separated by electrophoresis on 4% polyacrylamide gel at 180 Volts for one
hour, then transferred in
25 mM TRIS, 192 mM glycine, 0.025% SDS and 20% methanol (60-110 V for 1-2
hours) through a
stack of four PCNC membranes (as described herein) and one NA45 DEAE membrane
(Schleicher &
Schuell). This last layer of charged cellulose was used to trap DNA released
from the gel that
transferred through the entire thickness of the stack. After transfer,
registration (orientation) marks
were made using a 19G needle. The DEAE membrane was dried, exposed overnight
to a
phosphoimager screen, and visualized on a Phosphorimager: SI (Molecular
Dynamics).
First and second PCNC membranes were rinsed in TBST buffer, blocked for 10-60
minutes
in lx casein solution (Vector Laboratories, Inc.) and incubated overnight at 4
°C in anti-rat antibody
(1:200, NCI Laboratory of Pathology, Transcription Regulation Unit Chief, Dr.
Kevin Gardner) and
anti-His (1:10,000, Stratagene). The membranes were washed in TBST, incubated
in the complex of
secondary antibody and allcaline phosphatase, then washed again. The location
of the specific
proteins was visualized by ECL (DuoLux, Vector Laboratories, Inc.) and Biomax
MR film (Kodak).
Images of all of the membranes were aligned in Adobe Photoshop (FIG. 35).
The results demonstrated that the layered membrane array provides fast and
reliable
identification of proteins from a protein complex.
Example Il
Uniformity of Protein Capture on Multiple Membranes
During electrotransfer, proteins are pushed (or pulled) out of the gel onto
the membrane
substrate. The speed of their migration is influenced by the magnitude of the
electric current and size
of the protein. A higher voltage will push proteins out of the gel faster then
a lower voltage. Even
with fixed current flow, smaller proteins generally move faster then larger
ones. The length of the
transfer is another variable that can influence quality of membrane copies. If
transfer is too short, not
enough of the protein will leave the gel and be accessible for binding onto
the membranes.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-57-
An analysis of the results obtained with the methods and materials described
herein
indicates that, regardless of the amount of protein that is present in the
gel, more uniform membrane
copies can be generated if transfer is performed for shorter time with higher
voltage. All of the
transfers in this Example are performed for 60-70 minutes at 59-63 volts.
Keeping the transfer
conditions constant, the influence of protein load amount on the ability to
create membrane copies
was examined.
Total protein extracted from the Jurkat cell line (cells of lymphatic origin),
the HN12 cell
line (epithelial cells of keratinocyte origin) and the SW480 cell line (cells
of adeno-epithelial origin)
were used for this Example. All cell lines were cultured in a 37° C
humidified incubator in DMEM
media with 10% added serum. At about 80-90% confluence, cells were harvested
by scraping them
from the dish; the cells were then resuspended in phosphate buffered saline
(PBS) with 1% added
SDS. The concentration of the total protein was determined by BCA Protein
Assay Reagent (Pierce).
Approximately 30-100 micrograms of total protein was separated by 4-20%
polyacrylamide gel
electrophoresis (PAGE) (BioRad). A suitable protein gel running buffer was
used in the
electrophoresis to separate the proteins (for instance, 25 mM TRIS pH 8.3, 192
mM glycine, 0.1%
SDS). In addition, protein size markers (Bio-Rad Kaleidoscopic Standard,
catalog number 161-
0324) were loaded on the gel.
After electrophoretic separation, proteins were transferred through a 10-
layered array by
electroblotting (Bio-Rad catalog number 170-3930). A fiber pad, or more than
one fiber pad was
used at the anode and the cathode during electroblotting. Thus, starting from
the cathode side of the
electroblotting cassette, the fiber pad (on the bottom of the sandwich),
filter paper, gel, and
membrane stack are layered in order, with one membrane (the first membrane,
denoted membrane
"1") in contact with the gel. When assembled, the electroblotting cassette
tightly squeezes the
"transfer sandwich" (unlike a single membrane transfer, which can be gently
squeezed). Fiber pads
may be added on the outside of the sandwich until the cassette seems
"overfilled." When the
sandwich has the proper thickness, it may be necessary to force the cassette
closed.
The electroblotting procedure will vary depending on the system used (for Bio-
Rad devices,
transfer is accomplished at 59-63 volts for 60-70 minutes; for Novex devices,
transfer is
accomplished at 25 volts for 120 minutes). To facilitate later labeling of
individual membranes,
holes can be punched (for instance, using a 23g-25g needle) distinguishable
locations, such as in the
center of each protein standard band, and in the center of each well.
After transfer, the membrane stack was removed from the gel by gently peeling
up one
corner, and the frames were opened or removed. The membrane stack was then
washed in Tris or
phosphate buffered solution, and the membranes separated while they are still
in the solution. Before
immunodetection, the membranes are immersed in Blocking Reagent (20/20
GeneSystems) for 15
minutes.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-58-
Membranes were separately stained with Sypro Ruby (Molecular Probes) as
recommended
by the manufacturer and visualized on an Image Station 440CF (Kodak).
Fluorescence intensities
were taken from three different regions of every sample on every membrane
using KODAK 1D
Image Analysis Software (Kodak). The first region included proteins from 20-40
kDa in size. The
second region included proteins from 40-100 kDa in size. The third region
included proteins 100-
150 kDa in size. The relationship between different groups was analyzed using
Microsoft Excel~.
A data plot (Fig. 36) demonstrates that the smallest variability in total
protein loading per
membrane was seen for proteins 40-100 kDa in size. The data also suggest that
the amount of
protein loaded was an important variable in this system. For proteins that are
40-100 kDa in size, it
was determined that loading of 70-100 pg per lane kept variability between the
membranes in the less
than 10% range.
Example 12
Detecting Presence and Functional State of Multiple
Proteins Separated on a Single Gel
To determine the feasibility of detecting the presence and functional state of
multiple
proteins from the same gel, the presence and functional state of EGFR and c-
myc protein was
checked in parallel. Samples used were from the Jurkat cell line, HN12 cell
line, and SW480 cell
line; cells were cultured and harvested as stated in Example 11. Thirty
micrograms of total protein
was loaded per lane of 4-20% polyacrylamide gel (BioRad) and separated for two
hours at 50 V.
After electrophoresis, the gel was equilibrated for 10 minutes in lx transfer
buffer from 20/20 Gene
Systems, Inc. and electrotransfer was assembled with a seven-layered membrane
stack (20/20 Gene
Systems, Inc). A MiniCell blotter (BioRad) was used for the electrotransfer.
Transfer was
performed for 60-70 minutes at 59-63 V. After transfer, membranes were
separated in 50 mM Tris
pH 8.0, 150 mM NaCI and 0.05% Tween-20 (TBST), blocked in lx casein solution
(Vector) for 15
minutes at room temperature and incubated with antibodies diluted in TBST as
indicated in
TABLE 1 for 12 hours at 4° C.
TABLE 1
ayer Protein ManufacturerPart NumberAb Dilution
L
-
Number
1 Total EGFR Neomarker MS-610 1:500
2 Total EGFR Santa CruzSC-03 1:200
3 Phospho-EGFRUpstate OS-484 1:1,000
4 Phospho-EGFRUpstate OS-483 1:1,000
5 Total c-mycSanta CruzSC-764 1:200
6 Total c-mycNeomarkersMS-127 1:500
7 Phospho-mycCell Signaling9401L 1:1,000
1-7 Alpha-tubulinCalbiochemCP06 1:500
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-59-
After incubation with primary antibody, each membrane was washed separately
three times
for five minutes each in TBST and incubated in 1:4,000 dilution of horseradish
peroxidase (HRP)
conjugated secondary antibody (Amersham) in lx casein solution for 30 minutes
at room
temperature. Membranes were then washed for five minutes in TBST and twice for
five minutes
each in 50 mM TRIS pH 8.0, 150 mM NaCI (TBS), incubated for five minutes in
ECL PLUS
substrate (Amersham) and exposed to Biomax MR film (Kodak) from 1 - 45
minutes.
The image of the filin was digitized on an Astra 2200 scanner (Umax) and
manipulated in
ADOBE Photoshop 5Ø Following incubation in the first set of antibodies, all
of the membranes
were incubated in anti-alpha-tubulin antibody for two hours at room
temperature and signal
visualized as stated above, with the exception that the secondary antibody was
conjugated with
alkaline phosphatase (AP) (Vector) and the ECL reagent used was DuoLux
(Vector).
The result of this Example, shown in FIG. 37, demonstrated that multiple
membrane copies
made from the same gel could be used to determine the presence and functional
state of multiple
proteins from the same sample: In this Example, both total and activated forms
of EGFR and c-myc
protein were detected in extracts prepared from the SW480 cell line. Results
also demonstrated that
different samples could be compared to each other to reveal the presence of
total protein (for
instance, EGFR was expressed in HN12 and SW480 cells, but not in Jurkat cells)
and that the
presence of total protein does not necessarily mean functional activity (c-myc
was present in both
Jurkat and SW480 cell lines but only Jurkat cells had an active, functional
form).
Example 13
Detecting Proteins Involved in Epidermal Growth
Factor Receptor (EGFR) Signaling Pathway
Advantages of certain of the encompassed embodiments include that they permit
analysis
and comparison, in parallel, of a number of different proteins from multiple
samples. The value of
this parallel approach is even greater where the proteins of interest belong
to a single biological
system (e.g., all are component s of a receptor signaling pathway). Since
analysis for all of the
proteins is done on a single sample, comparative studies are easier to
perform, and it is expected that
the results are more consistent and reliable.
In this Example, the functional state of nine proteins that are involved in
signaling through
the EGFR pathway were analyzed and compared. Four different keratinocyte cell
lines were cultured
and harvested as described above (see Example 11 ). One hundred micrograms of
total protein from
each cell line was separated on a 4-20% acrylamide gradient gel (BioRad) and
transferred through a
ten-layered array as described above (see Example 11).
Membranes were stained with the ubiquitous dye Sypro Ruby (Molecular Probes)
and
images captured and stored on Image Station 440CF (Kodak). Following
visualization of the total
protein, membranes were blocked in lx casein solution (Vector) for 15 minutes
at room temperature,
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-60-
then incubated with antibodies diluted in TBST as indicated in TABLE 2 for 12
hours at 4° C. The
control membrane layer was incubated in no primary antibody.
TABLE 2
Protein ManufacturerPart NumberAb DilutionDetected
Phospho-RafBiosource 44-504 1:1,500 Yes
Phosho-AktCell Signaling92765 1:1,000 Yes
Phosho-ErkCell Signaling9106S 1:1,000 Yes
Phosho-MycCell Signaling9401L 1:1,000 Yes
Phosho-EGFRUpstate OS-493 1:1,000 Yes
Total EGFRNeomarkers MS610 1:500 Yes
Phospho-Stat3Biosource 44-384 1:1,000 Yes
Phospho-PKCCell Signaling2261 1:1,000 No
Phospho-SrcBiosource 44-660 1:1,000 Yes
I I
Following incubation with primary antibodies, membranes were processed as
described above (see
EXAMPLE 12).
FIG. 38 shows that eight of the nine proteins tested could be detected on the
stacked
membranes using this method. The phosphorylated form of PKC was not detected
in these samples.
Follow up experiments also failed to detect this form of protein when the same
amount of sample
was blotted on single nitrocellulose membrane with positive control cellular
extract being positive
(PDGF treated 3T3 cells, 10 pg/lane, provided by Cell Signaling). This
suggests that the failure to
detect the phosphorylated form of PKC was not due to a deficiency in the
transfer system but to the
very small (if any) amount of this protein present in the tested cell lines.
The results also clearly
illustrate differential expression between different cell lines for all of the
proteins tested.
Example 14
Contact Transfer of Proteins From a 1-D Gel
Diffusion based transfer of proteins from an acrylamide gel onto single
membrane substrate
was previously discussed by Bowen et al. (Nucleic Acid Res., 8:1-20, 1980).
The apparent advantage
of this system is that it does not require special blotting equipment. This
Example was carried out in
order to determine if it is possible to use contact transfer (without applying
an electric current) with
the provided membrane arrays.
A 10% gel (BioRad) with 25 and 50 micrograms of total protein was sandwiched
between
two five-membrane membrane stacks as shown in FIG. 6, with five membranes on
each side of the
gel. Three layers of Whatman~ filter paper soaked in lx transfer buffer from
20/20 Gene Systems,
Inc. were added on each side of the sandwich and the whole, assembled stack
was sealed in a plastic
bag. Three parallel sample stacks were assembled and placed in a gel drier
(BioRad) with the lid
closed at 80° C. Individual sample stacks were removed after 30, 60 or
120 minutes of transfer.
After transfer, the membranes were washed in TBST and stained with FastBlue
Stain (Calbiochem)
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
-61-
as recommended by the manufacturer. Stained membranes were scanned using an
Astra 2200
scanner (LTmax) to detect total transferred protein, and the images were
manipulated in ADOBE
Photoshop 5Ø
The results of this procedure are shown in FIG. 39. Proteins were effectively
transferred
from the gel into the membranes on both sides of the gel (bi-directional
transfer). The amount of the
protein transferred was dependent on the length of the transfer (more protein
was transferred after
two hours compared to half an hour) and the size of the protein (transfer of
the large proteins was less
efficient). Thus, contact transfer is an effective alternative to
electrotransfer of proteins and other
biomolecules onto/into membrane stacks.
Although certain embodiments have been described herein, it will be apparent
to those
skilled in the art to which the invention pertains that variations and
modifications of the described
embodiments may be made without departing from the spirit and scope of the
disclosure.
Accordingly, it is intended that the invention be limited only to the extent
required by the appended
claims and the applicable rules of law. The references cited above are
incorporated herein in their
entirety.
CA 02428441 2003-05-09
WO 02/48674 PCT/USO1/44009
SEQUENCE LISTING
<l10> 20/20 GENE. SYSTEMS, TNC.
THE GOVERNMENT OF THE UNITED STATES OF AMERICA, AS REPRESENTED BY THE
SECRETARY, DEPARTMENT OF HEALTH & HUMAN SERVICES, THE NATIONAL INSTITUTES OF
HEALTH
KNEZEVIC, Vladimir
EMMERT-BUCK, Michael R.
BAIBAKOVA, Galina
HARTMANN, Dan-Paul
HEWITT, Stephen
MITCHELL, Capre
GARDNER, Kevin
<120> METHODS, DEVICES, ARRAYS AND KITS FOR DETECTING AND ANALYZING
BIOMOLECULES
<130> 6457-61282
<150> US 09/753,574
<151> 2000-01-04
<150> 09/718,990
<151> 2000-11-20
<150> PCT/US00/20354
<151> 2000-07-26
<150> 60/145,613
<151> 1999-07-26
<l50> 60/286,258
<151> 2001-04-25
<150> 60/304,031
<151> 2001-07-09
<150> 60/296,475
<151> 2001-06-08
<160> 1
<170> PatentIn version 3.1
<210> 1
<211> 43
<212> DNA
<213> Artificial sequence
<220>
<223> Synthetic oligonucleotide
<400> 1
tcgacctctt ctgatgactc tttggaattt ctttaaaccc cca 43
1