Note: Descriptions are shown in the official language in which they were submitted.
CA 02515865 2005-08-12
1
TITLE: COMPOSITIONS CONTAINING WATER CONTROL
TREATMENTS AND FORMATION DAMAGE CONTROL
ADDITIVES, AND METHODS FOR THEIR USE
SPECIFICATION
This application claims the benefit of U.S. Provisional Patent Application
Serial No 60/601,466 filed August 13, 2004, the contents of which are
incorporated
herein by reference.
s FIELD OF THE INVENTION
The invention relates to treatments of subterranean formations to control
water
production and inhibit formation damage occurrence, and, more specifically,
provides
methods and compositions for controlling water production and formation damage
occurrence simultaneously with a minimum number of steps.
io BACKGROUND OF THE INVENTION
When a wellbore is initially drilled in a hydrocarbon-producing field, the
hydrocarbon (i.e., oil or gas) extracted is typically dry, being substantially
free of
aqueous impurities. However, as the hydrocarbon reserves dwindle, or the
formation
becomes less producing, a progressively greater quantity of aqueous impurities
begin
~s to become mixed with the hydrocarbons. Certain locations are more prone to
incorporating undesirable amounts of water with the produced hydrocarbon
product,
further raising the post-production costs involved in removing the water and
purifying
the hydrocarbon product and significantly reducing both the economic return
and life
of the well.
ao While there are numerous causes for this production including channeling
through the cement casing, coning from bottom water drive reservoirs, high
permeable and water-saturated streaks in the producing interval allowing water
migration from water floods or edge water drive reservoirs, produced water is
generally considered to be an inevitable consequence of water injection when
water
CA 02515865 2005-08-12
2
flooding is used to develop a hydrocarbon reservoir or when the field drive
mechanism involves strong aquifer support.
During this same period of decreasing productivity, changes in physical
conditions of the formation can generate both direct and indirect costs to the
operator,
s and the water can simultaneously induce and sustain production
changing/reducing
formation damage within the hydrocarbon bearing formation.
For example, locations rich in minerals cause the formation of solid deposits
in the production tubing and pipes, slowing the production of hydrocarbon
product.
Removal of these solid deposits ("scale") slows production and increases the
unit cost
io of the produced hydrocarbon product. Such occurrences of formation damage
further
compound the production problems. This is especially true in the instance of
unwanted water production occurring at the same time, or just prior to, one or
more
additional formation damage occurrences.
Various chemical treatments have been developed for the treatment or
is prevention of water production and scale formation. A representative
summary of
these treatments follows.
U.S. Patent No. 6,228,812 B1 (issued May 8, 2001) describes compositions
that modify the permeability of subterranean formations, thereby reducing the
production of water. The compositions contain copolymers such as a copolymer
Zo having a hydrophilic monomer, an anchoring monomer, and a filler monomer.
The
anchoring monomer is based on N-vinylformamide or N,N-diallylacetamide.
U.S. Patent No. 4,708,974 (issued November 24, 1987) offers injection of gel-
forming phenolic compositions into a formation to reduce the amount of water
produced with the desired hydrocarbon products. The gelled compositions have
zs selective oil to water permeability, where the compositions are more
permeable to oil
than to water.
U.S. Patent No. 6,720,291 (issued April 13, 2004) describes a composition
useful for prevention of downhole salt erosion in high-iron environments. The
composition contains a first ingredient of sodium ferrocyanide and/or
potassium
CA 02515865 2005-08-12
3
ferrocyanide, and a second ingredient of phosphonic acid, and/or alkali and
alkaline
earth metal salts of a phosphonic acid.
U.S. Patent No. 6,613,899 (issued September 2, 2003) offers the use of
carboxyl-containing fructans such as carboxymethylinulin for the prevention of
scale
s deposits during oil production.
U.S. Patent No. 6,527,983 (issued March 4, 2003) presents a series of
phosphonate compounds useful for the inhibition of scale formation in waters
having
a high barium content and low pH.
Clariant Oil Services (Aberdeen, UK) announced a new product named
io RELTREAT on October 31, 2003. According to Clariant, "RELTREAT is a polymer
based system which selectively shuts-off or slows the production of unwanted
water"
during oil and gas production. North Sea uses were reported to increase oil
production from 413 to 1147 bbl/day. The water to oil ratio was reduced from
over
20:1 to 8:1. Representatives of Clariant Oil Services presented information on
the use
is of RELTREAT on the BP Miller field at the PEA Water and Gas Shut Off Forum
on
November 6, 2003 in Sandnes, Norway.
While these various references describe either the reduction of water content
or the inhibition of scale formation, none of them describe the simultaneous
achievement of both desired results.
zo Other types of formation damage or damage to the various parts of the
producing systems can also occur, such as hydrate formation, paraffin or wax
deposition, and fine migration. For example, multiphase production and
transport of
hydrocarbon resources induces the coexistence of gas, water and eventually a
liquid
hydrocarbon phase (condensate or crude) at low temperature and under
relatively high
zs pressure. Such operation conditions can lead to hydrate crystals formation
which can
plug the equipment. Similarly, asphaltene coagulation and deposition can occur
as a
result of changes in parameters such as reservoir pressure, reservoir fluid
temperature,
and oil composition brought about by normal recovery operations. Likewise,
oils
with high wax content may cause pipeline plugging and deterioration of
equipment,
3o resulting in production shutdowns and economic losses.
CA 02515865 2005-08-12
4
While there are a number of approaches to these production problems in the
patent and journal literature, nearly all of them deal with the problem on an
individual
basis as they form, which can be costly when numerous problems arise during
production. A further problem with such an approach is the cost off tracking
and
s monitoring such production problems. For example, the technique of "downhole
squeezing" is commonly used to address oil field scale formation, wherein
control
agent chemicals in a solution are injected into the near-wellbore area, using
a pre-
flush, squeeze, and overflush treatment before the well can be returned to
normal
function. However, the overflush process often flushes a significant portion
of the
~o control agent chemical from the rock surface, and the remaining control
agent is
gradually removed from the rock surface as oil production continues. Thus,
further
descaling treatments will be required.
There exists a need for compositions and methods which can minimize the
steps of treating these conditions, while simultaneously achieving comparable
or
~s improved treatment results. The simultaneous inhibition of both of these
challenges
would greatly improve the efficiency and cost-effectiveness of a hydrocarbon
producing well.
SUMMARY OF THE INVENTION
The present invention is directed generally to aqueous compositions
zo comprising a relative permeability modifier (RPM) macromolecule and one or
more
formation damage control additives, for use in treating hydrocarbon-producing
wells,
formations, and equipment. Such compositions, comprising the RPM macromolecule
and the one or more formation damage control additive, can result in the
formation of
a composition wherein the components exhibit a "synergistic" effect, whereby
the
zs ability of the formation damage control additive to prevent formation
damage and to
prevent the formation of particulates and deposits on pipes and tubing is
enhanced
relative to the use of the same additive separately.
The present invention is also directed to methods for controlling formation
damage in hydrocarbon-producing formations, wells, and equipment, such as
3o production tubing, wherein the method comprises introducing an aqueous
treating
CA 02515865 2005-08-12
solution comprising a RPM macromolecule and at least one formation damage
control
additive into the producing formation, well, or equipment, for a time and in
an amount
sufficient to control, reduce, or inhibit one or more types of formation
damage.
According to such methods, the aqueous treating solution can be pre-
manufactured, or
prepared directly on-site, immediately prior to the introducing step.
DESCRIPTION OF THE DRAWINGS
The following figures form part of the present specification and are included
to further demonstrate certain aspects of the present invention. The invention
may be
better understood by reference to one or more of these figures in combination
with the
~o detailed description of specific embodiments presented herein.
Figure 1 shows a graph of pressure (psi; y-axis) versus time (seconds; x-axis)
for a 2% KCl brine control solution (mixture A).
Figure 2 shows a graph of pressure (psi; y-axis) versus time (seconds; x-axis)
for a sample containing scale control agent but no RPM (mixture B).
is Figure 3 shows a graph of pressure (psi; y-axis) versus time (seconds; x-
axis)
for a sample containing 90% scale control agent and 10% RPM (mixture C).
Figure 4 shows a graph of pressure (psi; y-axis) versus time (seconds; x-axis)
for a sample containing 75% scale control agent and 25 % RPM (mixture D).
Figure 5 shows a graph of pressure (psi; y-axis) versus time (seconds; x-axis)
zo for a sample containing 50% scale control agent and 50 % RPM (mixture E).
Figure 6 shows a graph of pressure (psi; y-axis) versus time (seconds; x-axis)
for a sample containing 10% scale control agent and 90 % RPM (mixture F).
Figure 7 shows a graph of pressure (psi; y-axis) versus time (seconds; x-axis)
for a sample containing RPM but no scale control agent (mixture G).
as DETAILED DESCRIPTION OF THE INVENTION
The present invention is directed generally to methods and compositions for
controlling formation damage in a hydrocarbon-producing formation, as well as
the
CA 02515865 2005-08-12
6
tubing and equipment associated with producing a hydrocarbon from such
formation.
The method generally comprises introducing an aqueous treating composition
into the
formation, tubing, or equipment, wherein the aqueous treating composition
comprising a relative permeability modifier (RPM) macromolecule and at least
one
s formation damage control additive. Typically, the total amount of RPM
macromolecule and formation damage control additives) in the aqueous treating
composition is between from about 0.01 to about 70 volume percent. In a
preferred
mode, the weight ratio of RPM macromolecule to formation damage control
additive
in the composition is between from about 200:1 to about 1:200.
io Suitable RPM macromolecules include, for example, anionic RPM
macromolecules, anionic microgels which are crosslinked, and RPM
macromolecules
which are optionally deformable, all of which have a K-value from about 200 to
about
1,000 and used at a concentration such that the viscosity of the treating
fluid can
range from about 1 to about 20 cP.
is In one aspect of the present invention, a method for controlling formation
damage in a hydrocarbon-producing formation that is susceptible to such
formation
damage is described, wherein the method comprises introducing an aqueous
treating
composition comprising an RPM macromolecule and at least one formation damage
control additive into the hydrocarbon-producing formation. The RPM
macromolecule
zo and the formation damage control additive can have a synergistic
relationship, in
accordance with this aspect of the invention.
In a further aspect of the present invention, a method for treating formation
damage in a hydrocarbon-producing well susceptible to such damage is
described,
wherein the method comprises injecting into the well an aqueous treating
composition
zs comprising a RPM macromolecule and at least one formation damage control
additive. In accordance with this aspect of the invention, the RPM
macromolecule
and at least one of the formation damage control additives can have a
synergistic
relationship.
In yet another aspect of the present invention, a composition comprising a
3o RPM and at least one formation damage control additive is described. The
RPM
macromolecule may be ionic or non-ionic. For instance, in accordance with this
CA 02515865 2005-08-12
7
aspect of the invention, such RPM macromolecules include anionic RPMs,
crosslinked RPM microgels, anionic RPM microgels, and anionic RPM microgels
which are crosslinked. The formation damage control additives include scale
control
agents, salt control agents, fines control agents, organic deposit control
agents, such
s as paraffin control agents and asphaltene control agents, hydrate control
agents,
corrosion control agents and/or mixtures thereof.
In accordance with a further aspect of the present invention, a composition
comprising a RPM macromolecule and at least one scale control agent is
described.
In accordance with this particular aspect, the RPM macromolecule can be an
anionic
io RPM macromolecule, such as an anionic crosslinked microgel, combined with
the
scale control agent(s).
Compositions can further comprise water or an aqueous solvent mixture. The
water can generally be any aqueous solution such as distilled water, fresh
water, salt
water ("brine"), acid, etc. The composition can further comprise one or more
~s additional solvents.
The compositions may be used in concentrate or working forms. The
concentrate compositions can be prepared with or without water. The working
compositions can be prepared by adding an appropriate amount of water or
aqueous
solution to a concentrate composition. Alternatively, the working composition
can be
ao prepared by sequentially or simultaneously adding at least one formation
control
additive and at least one RPM to water without f rst preparing a concentrate
composition.
The concentrate compositions can generally contain the formation control
additive and RPM at any concentration higher than the desired final working
2s composition. The amount of concentrate added to water to prepare a desired
concentration in the working composition can be easily calculated using the
ratios of
the two concentrations. For example, a 250 ppm concentrate would require a ten-
fold
dilution to achieve a 25 ppm working composition. The concentrate compositions
can
generally have any concentration greater than the working composition. For
example,
so the concentration can be about Sx, about 10x, about 15x, about 20x, about
25x, about
CA 02515865 2005-08-12
g
50x, about 100x, or ranges between any two of these values. Higher
concentrations
are possible, and would require higher dilution values.
I. Relative permeability modifiers
The compositions of the present invention are aqueous treatment compositions
s containing one or more RPM macromolecules. The RPM macromolecule may be any
relative permeability modifier. For instance, a suitable RPM for use in the
invention
may be a polymer which can either impede the production of water and/or
redirect
water through permeable formation materials. Further, a RPM can be a
macromolecule effective to selectively reduce the production of water from
~o subterranean formations by modifying the permeability of the formation.
Suitable as
RPM macromolecule are deformable, polymeric compositions that comprise at
least
one hydrophilic monomer which aids in the RPM adhering to the formation and
adds
to the water/brine solubility; and at least one anchoring monomeric unit to
cause the
RPM to adhere to the formation. Further general characteristics of the RPM
i s macromolecules as used herein include those RPM macromolecules having K-
values
(Fikentscher's K-value, is a measure of a polymer's average molecular weight)
from
about 200 to about 1,000 (which can be controlled by the concentration of the
starting
monomers and/or the amount of crosslinking), and those RPM macromolecules that
are crosslinked, or both. Typically, the concentration of the RPM
macromolecule in
zo the aqueous treatment composition is from about 100 ppm to about 5,000 ppm.
Such RPM macromolecules also include soft microgels, which as used herein
refers to those RPM macromolecules which may be crosslinked during their
manufacture and have a weight average molecular weight of from about 104 to
10g
g/mol. Typically, the microgel is crosslinked in an amount from about 0.25 to
about
as 2.5 weight percent, based on the weight of the microgel. Further, such
microgels can
optionally and equally be described as having a size of from about 0.001
microns in
diameter to about 100 microns in diameter.
In another aspect of the present invention, the RPM macromolecules as used
herein are anionic, crosslinked, water-soluble macromolecules which are
readily
so pumpable and injectable, homogenous, and have a K-value, measured according
to
standard test methods (e.g., ISO 1628-2 (DIN 53726)), in the range from about
200 to
CA 02515865 2005-08-12
9
about 1,000. Typically, the RPM macromolecules will have a K-value in the
range
from about 200 to about 500, and more typically a K-value in the range from
about
250 to about 300.
Suitable RPMs include copolymers, homopolymers, or terpolymers comprised
s of hydrophilic monomers, at least one anchoring monomeric unit, an optional
secondary anchoring unit, and one or more filler/spacer monomer units.
Optionally,
the RPMs can provide grafting sites for the inclusion of organosilicon
compounds.
Suitable RPM include those described in U.S. Patent Nos. 5,735,349; 6,169,058;
6,465,397; and 6,228,812, herein incorporated by reference. Optionally, and in
~o accordance with the present invention, the RPM can include one or more
organosilicon compounds. Preferred RPM macromolecules suitable for use within
the present invention include AquaConTM, AQUATROLTM I, and AQUATROLTM V,
all available from BJ Services Company (Houston, TX).
The RPM macromolecules suitable for use in the present invention have
~s weight average molecular weights ranging from about 10,000 g/mol to about
50,000,000 g/mol, preferably from about 50,000 g/mol to about 5,000,000 g/mol,
and
more preferably from about 100,000 g/mol to about 2,000,000 g/mol. The RPM
macromolecules for use herein are used at concentrations in the treating fluid
such
that the treating fluid has a viscosity from about 1 cP (0.001 Pa-s) to about
20 cP
zo (0.020 Pa-s), and more preferably from about 1 cP (0.001 Pa-s) to about 5
cP (0.005
Pa-s), as measured by standard techniques.
The RPM macromolecule compositions of the present invention include
copolymers, homopolymers or terpolymers comprising a hydrophilic monomeric
unit;
at least one first anchoring monomeric unit; and may also include at least one
zs optionally selected second anchoring monomeric unit. A filler monomeric
unit may
also be employed. These copolymer compositions may be advantageously used in
aqueous-based water control treatment fluids to selectively control water
production
from hydrocarbon production wells.
As used herein, the term "monomeric anchoring unit" refers to components of
3o a polymer that will preferentially bind, by either physical or chemical
processes, to
subterranean formation material and which therefore tend to retain the polymer
to the
CA 02515865 2005-08-12
formation material. Anchoring groups are typically selected to prevent a
polymer
from washing out of the formation due to fluid flow. Primary anchoring sites
for the
monomeric anchoring units are typically clay and feldspar surfaces existing in
formation pores, channels and pore throats. With benefit of this disclosure,
those of
s skill in the art will understand that particularly useful anchoring
monomeric units are
those having carboxylate, quaternary nitrogen or functional groups capable of
hydrolyzing to form amine-based anchoring groups on the polymer. Examples
include
amide-containing monomeric units.
Advantageously, the disclosed co-polymers having the first anchoring
io monomeric units described herein may be utilized in well treatment methods
to
selectively reduce the permeability of a subterranean formation to water by a
factor of
about 10 or more, while at the same time leaving the permeability of the
formation to
oil virtually unchanged. Furthermore, the disclosed compositions, when
introduced
into a formation, tend to exhibit a high resistance to removal from water
bearing areas
~ s of the formation over time.
Hydrophilic monomers may include both ionic and nonionic monomers. The
term "nonionic monomer" refers to monomers that do not ionize appreciably in
aqueous solution at near-neutral pH. Examples of suitable nonionic hydrophilic
monomers include, but are not limited to, vinyl acrylamide comonomers
including,
zo but not limited to, acrylamide, N-vinyl acetamide, diallylacetamide, N-
vinyl-N-
methyl acetamide, N,N-dimethyl acetamide, N-vinyl-2-pyrrolidone, N-vinyl
formamide (VF), and N-ethenyl-N-alkyl acetamide, as well as mixtures of two or
more of such comonomers. Ionic monomers may be either anionic or cationic.
Examples of anionic monomers include, but are not limited to, alkaline salts
of acrylic
zs acid, ammonium or alkali salts of acrylamidomethylpropane sulfonic acid
("AMPS"),
malefic acid, itaconic acid, styrene sulfonic acid, and vinyl sulfonic acid
(or its
ammonium or alkali metal salts). Examples of suitable cationic monomers
include,
but are not limited to, dimethyldiallyl ammonium chloride and quaternary
ammonium
salt derivatives from acrylamide or acrylic acid such as
acrylamidoethyltrimethyl
so ammonium chloride.
CA 02515865 2005-08-12
11
In one embodiment, one or more hydrophilic monomeric units are typically
employed and are based on AMPS (such as at least one of ammonium or alkali
metal
salt of AMPS, including sodium and/or potassium salts of AMPS), acrylic acid,
an
acrylic salt (such as sodium acrylate, N-vinyl pyrrolidone, ammonium or alkali
metal
s salts of styrene sulfonic acid, etc.), or a mixture thereof. It may be
desirable to
employ ammonium or alkali metal salts of AMPS for added stability, with or
without
one or more other hydrophilic monomers, in those cases where aqueous treatment
and/or formation fluids contain high concentrations of divalent ions, such as
Ca+z,
Mg+z, and the like.
io Optional second anchoring monomeric units may include any monomeric unit
that will adsorb onto formation material. In one embodiment, examples of
optional
second anchoring monomeric units include at least one of
dimethyldiallylammonium
chloride, ammonium or alkali metal salts of acrylic acid, (such as sodium
salts), vinyl
phosphonate or vinyl phosphinate, or vinyl phosphate or a mixture thereof.
is Optional filler monomeric units may include any monomeric unit suitable for
copolymerization with the other monomers in the composition. Desirable
characteristics of filler monomer units are the ability to retain water
solubility and/or
relative low cost compared to other monomer units present in a copolymer.
Filler
monomer units may be based on, for example, monomers such as acrylamide,
zo methylacrylamide, etc. In one embodiment, optional filler monomeric units
include
monomers such as acrylamide, methylacrylamide, and the like.
With benefit of the present disclosure, the disclosed compositions may be
prepared using any method suitable for preparing co-polymers known to those of
skill
in the art. In one embodiment, monomers corresponding to the desired monomeric
as units in the copolymer are selected and polymerized in an aqueous monomer
solution.
In one exemplary embodiment, a first N-vinylformamide monomer is
combined with a hydrophilic monomer (such as ammonium or alkali metal salts of
AMPS) and a filler monomer (such as acrylamides), in an aqueous base fluid,
typically water. Other additives may include disodium ethylenediamine
tetraacetate
30 (NazEDTA), pH adjusting chemicals (such as potassium or sodium hydroxide,
CA 02515865 2005-08-12
12
carbonate, or bicarbonate), and a catalyst to initiate polymerization.
Monomers with
other anchoring groups may also be present.
In addition to one or more salts selected from alkali metal salts, alkali
earth
salts, formate salts, bicarbonate salts, chloride salts, bromide salts,
sulfate salts and
s acetate salts, the composition for use in the invention may further include
an aqueous
acid, such as hydrochloric, hydrobromic, acetic, formic, citric, glycolic,
gluconic, and
hydrofluoric, and/or mixtures thereof.
Any relative proportion of the disclosed monomers that is suitable for
polymerization and use in a water control treatment fluid may be combined in
an
io aqueous solution for polymerization. However, in one embodiment, a first
anchoring
monomer is combined to be present in an amount of from about 2% to about 30%
by
weight of the total polymer composition, alternatively from about 5% to about
15%
by weight of the total polymer composition. In another embodiment a first
anchoring
monomer is combined to be present in an amount from about 2% to about 30%,
is alternatively from about 5% to about 15%, by weight of the total polymer
composition; ammonium or alkali metal salts of AMPS is combined so that AMPS-
based monomer is present in an amount from about 0% to about 50%,
alternatively
from about 20% to about 30%, by weight of the total polymer composition; and
acrylamide is combined to be present in an amount from about 20% to about 98%,
Zo alternatively from about 40% to about 65% by weight of the total polymer
composition. In one embodiment, N-vinylformamide is utilized as the first
anchoring
monomer.
Where necessary or desirable, the pH of a monomer solution may be adjusted
or neutralized prior to polymerization by, for example, addition of a base
such as
zs sodium or potassium hydroxide, carbonate or bicarbonate. For example, the
pH of an
aqueous solution containing ammonium or alkali metal salts of AMPS may be
adjusted to, for example, about 10 prior to the addition of N-vinylformamide
and/or a
second anchoring monomer or a filler monomer such as acrylamide. In one
embodiment, a copolymer may be prepared by mixing the appropriate monomers
into
3o a tank of fresh water, followed by addition of a NaZEDTA, pH adjuster and
catalyst
CA 02515865 2005-08-12
13
system to initiate polymerization. In one embodiment, ultimate pH range may be
from
about 4.5 to about 10.0 and alternatively from about 7.5 to about 9.5.
As indicated previously, and in accordance with the present invention, the
RPM macromolecules of the present invention used as water redirecting agents
can be
s crosslinked either internally, externally, or both. Such crosslinking is
preferably
performed using one or more chemical cross-linking techniques (vs. UV
irradiation,
biological crosslinking, etc.), and can occur during the synthesis of the RPM
macromolecules, at the wellsite just prior to injection into an injector well
(in the case
of external crosslinking), or both. Crosslinkers suitable for use with the RPM
~o macromolecules/microgels of the present invention include aldehydes,
amides,
acrylamides, isocyanates, metal salts, di- or poly-allyl based monomers,
carbodiimide
cross-linkers, and polyepoxide compounds. Most preferably, the RPM
macromolecules of the present invention are crosslinked using aldehyde-based
crosslinking techniques, acrylamide-based crosslinking techniques, or using
~s polyepoxide compounds.
Examples of useful multifunctional crosslinking monomers include
multifunctional acrylamides, and (meth)acrylates containing unsaturation at
preferably 2, and optionally 3 or more sites on each copolymerizable comonomer
molecule. In one embodiment, the multifunctional crosslinking monomers are
zo selected from the group consisting of monomeric polyesters of acrylic or
methacrylic
acids and polyhydric alcohols; and monomeric polyalkenyl polyethers of
polyhydric
alcohols containing from 2 to about 6 polymerizable alkenyl ether groups per
polyether molecule. Another exemplary crosslinking monomer is a monomeric
polyester of an acrylic or methacrylic acid and a polyhydric alcohol
containing from 2
Zs to about 6 polymerizable a,(3-unsaturated acrylic groups per polyester
molecule.
Other copolymerizable crosslinking monomers include divinyl ether, ethylene
glycol
dimethacrylate, (m)ethylene-bisacrylamide, allylpentaerythritol, and the like.
The
preferred crosslinking comonomers are somewhat water soluble and monomer
soluble. Preferably, the acrylamide crosslinking agent used with the RPM
3o macromolecules suitable for use in the methods of the present disclosure is
methylene
bis-acrylamide, or combinations of crosslinkers including methylene bis-
acrylamide.
CA 02515865 2005-08-12
14
Aldehyde-based cross-linking techniques includes those techniques using a
reagent containing two reactive aldehyde groups to form covalent cross-links
between
neighboring amino groups of monomer residues in the RPM macromolecules
described herein [Khor, E., Biomaterials, Vol. 18: pp. 95-105 (1997)].
Aldehydes
s suitable for use with the present invention include but are not limited to
glutaraldehyde, formaldehyde, propionaldehyde, butyraldehyde and dialdehydes
such
as glyoxal. Preferably, the aldehydes are glutaraldehyde or formaldehyde.
Polyepoxy-based cross-linking techniques and agents include the use of
compounds, such as short, branched polymers, terminating in reactive epoxy
io functionalities. Polyepoxy compounds suitable for use as cross-linking
agents in the
present invention include but are not limited to the epoxides of glycerol
ethers,
glycols, and glycerol polyglycidyl ethers.
Isocyanates are also suitable for use as cross-linking agents in the present
invention. Generally, the isocyanates (R-NCO) react with primary amines to
form a
i s urea bond (R-H-CO NH-R); difunctional isocyanates therefore have the
ability
to cross-link RPMs via lysine-like side chains. Isocyanates suitable for use
as cross-
linking agents in the present invention are preferably diisocyanates,
including
biphenyl diisocyanate, dimethoxy-4,4'-biphenyl diisocyanate, dimethyl-4,4'-
biphenyl
diisocyanate, 1,3-bis(isocyanatomethyl)benzene, phenyl diisocyanate, toluene
2o diisocyanate, tolylene diisocyanate, diisocyanato hexane, diisocyanato
octane,
diisocyanato butane, isophorone diisocyanante, xylene diisocyanate,
hexamethylene
diisocyanante, octamethylene diisocyanante, phenylene diisocyanate, and
poly(hexamethylene diisocyanate). Preferably, the isocyanate used as a cross-
linking
agent of the RPM macromolecules of the present invention is hexamethylene
zs diisocyanate.
Carbodiimide cross-linking agents and techniques can also be used within the
scope of the present invention. These agents react with the carboxyl groups of
monomers within the RPM macromolecules/microgels to form isoacylurea
derivatives/iso-peptide bonds [Khor, E., ibid.]. Carbodiimides suitable for
use as
3o cross-linking agents with the RPM macromolecules of the present invention
include
but are not limited to N,N'-dicyclohexylcarbodiimide (DCC); N,N'-
CA 02515865 2005-08-12
diisopropylcarbodiimide (DIC); N,N'-di-tert-butylcarbodiimide; 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide (EDC; EDAC); water-soluble EDC (WSC); 1-
tert-butyl-3-ethylcarbodiimide; 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide;
bis(trimethylsilyl)carbodiimide; 1,3-bis(2,2-dimethyl-1,3-dioxolan-4-
s ylmethyl)carbodiimide (BDDC, as described in U.S. Patent No. 5,602,264); N-
cyclohexyl-N'-(2-morpholinoethyl) carbodiimide; N,N'-diethylcarbodiimide
(DEC);
1-cyclohexyl-3-(2-morpholinoethyl)carbodiimide methyl-p-toluenesulfonate
[e.g.,
Sheehan, J.C., et al., J. Org. Chem., Vol. 21: pp. 439-441 (1956)]; oligomeric
alkyl
cyclohexylcarbodiimides, such as those described by Zhang, et al. [J. Org.
Chem.,
io Vol. 69: pp. 8340-8344 (2004)]; polymer bound DCC; and polymer bound EDC,
such
as cross-linked N-ethyl-N'-(3-dimethylaminopropyl)carbodiimide on JANDAJELTM.
Additionally, N-hydroxysuccinimide (NHS), 1-hydroxy-7-azabenzotriazole (HOAt),
or similar reagents can be utilized in conjunction with the carbodiimide to
minimize
internal rearrangement of the activated isoacylurea derivative and provide
more
is efficient cross-linking.
Other chemical cross-linking agents suitable for use in the present invention
to
provide cross-linked RPM macromolecules for use in redirecting formation water
to
improve hydrocarbon recovery from subterranean formations include but are not
limited to homobifunctional cross-linkers such as BMME, BSOCOES, DSP (a thio-
Zo cleavable cross-linker), DSS, EGS, water-soluble EGS, and SATA, as well as
heterobifunctional cross-linking agents including GMB, MBS, PMPI, SMCC, SPDP,
and MPH (maleimidopropionic acid hydrazide), MCH, EMCH (maleimidocaprionic
acid hydrazide), KMUH (N-(k-Maleimidoundecanoic acid)hydrazide), and MPBH (4
(4-N-MaleimidoPhenyl)butyric acid hydrazide), all available from Interchim
(Cedex,
Zs France).
Specific examples of other crosslinking monomers suitable for use herein
include but are not limited to trimethylol propane triacrylate (TMPTA),
trimethylol
propane trimethacrylate (TMPTMA); diethylene glycol diacrylate (DEGDA),
diethylene glycol dimethacrylate (DEGDMA), trimethylene glycol diacrylate,
3o butylene glycol diacrylate, methylene-bis-acrylamide, pentamethylene glycol
diacrylate, octylene glycol diacrylate, glyceryl diacrylate, glyceryl
triacrylate,
CA 02515865 2005-08-12
16
neopentyl glycol diacrylate, the tetraacrylate ester of pentaerythritol, as
well as
combinations thereof.
It is understood that certain monounsaturated monomers may act in varying
degrees to crosslink or branch the water soluble copolymer of the invention.
For
s example, acrylate monomers with abstractable hydrogens, which can function
as
radical reactive sites, can in some embodiments of this invention, form a more
branched or crosslinked polymer, thus affecting the preferred levels of the
polyethylenic unsaturated crosslinking comonomers. An example of a
monounsaturated monomer with an abstractable hydrogen is 2-ethylhexyl
acrylate.
io Optional heat-reactive, latent carboxy- or hydroxy-reactive internal
crosslinking systems can be provided by the incorporation of carboxylic-group
containing comonomers, and N-alkylol amides, for example, N-methylol
acrylamide,
N-propylol acrylamide, N-methylol methacrylamide, N-methylol maleimide, N-
methylol maleamic acid esters, N-methylol-p-vinyl benzamide, and the like.
Is Known methods for optional post-polymerization crosslinking of carboxylic
acid containing copolymers include, for example, U.S. Pat. No. 4,666,983
(crosslinking agent without any carrier solvent), using e.g. polyhydric
alcohols,
polyglycidyl ethers, polyfunctional amines and polyfunctional isocyanates.
U.S. Pat.
No. 4,507,438 and 4,541,871 utilize a difunctional compound in water with
inert
Zo solvent or mixture of solvents. The difunctional compounds include glycidyl
ethers,
haloepoxies, aldehydes and isocyanates with ethylene glycol diglycidyl ether
crosslinker. The solvents include polyhydric alcohols with ethylene glycol,
propylene
glycol and glycerine enumerated as preferred polyhydric alcohols. U.S. Pat.
No.
5,140,076 teaches a water-solvent-crosslinker mixture. Crosslinkers such as
as polyhydric alcohol, diglycidyl ether, polyaziridene, urea, amine and ionic
crosslinkers
are suggested.
II. Formation damage control additives
In accordance with the compositions of the present invention, as well as the
associated methods employing such compositions, at least one formation damage
3o control additive is included in the composition. As used herein, the term
"formation
damage control additive" refers broadly to those compounds or compositions
which
can reduce the presence of damage, inhibit the formation of damage, or
generally
CA 02515865 2005-08-12
17
improve the well production profile following treatment of the well with such
a
compound or compositions. These formation damage control additives include,
but
are not limited to, scale control agents, salt control agents, fines control
agents,
paraffin or wax control agents, asphaltene control agents, hydrate control
agents,
s corrosion control agents and mixtures thereof. Optionally, and in accordance
with the
present invention, one or more of these control additives can have an
observable
synergistic effect when combined with one or more RPM macromolecules.
A. Scale control agents
Scale formation, as used herein, can generally be thought of as a deposit of
io predominantly inorganic compounds. In this regard, a common process leading
to
scale formation in hydrocarbon production operations is the precipitation of
sparingly
soluble salts from oilfield brines. Some oilfield brines contain sufficient
sulfate ion in
the presence of barium, calcium, and/or strontium ions that the potential for
forming
barium sulfate (BaS04) and/or strontium sulfate (SrS04) scale exists. Often,
the
~s formation of scale results in reduced production and increased maintenance
costs
associated with the hydrocarbon production. Further, in some locations,
naturally
occurring radioactive materials have been found to incorporate themselves into
the
scale, resulting in health, safety, and liability concerns and increased scale
disposal
costs, in addition to the removal and/or inhibition of scale formation.
Accordingly,
Zo scale control agents, as used herein, refer to those classes of compounds,
specific
compounds or mixtures, which act to reduce the occurrence, formation, or
recurrence
of scale, such as inorganic sulfate buildup and the like.
Scale control agents suitable for use herein can generally be any of the known
scale control agents in the art. Typically, such scale control agents are
ionic
as compounds and polymers. Examples of scale control agents suitable for use
with the
compositions of the present invention include, but are not limited to,
polyelectrolytes,
phosphonates, such as DETPMP, polyphosphinocarboxylic acids (PPCA),
phosphinates, phosphates, and polymers such as polyacrylate (PAA), polyvinyl
sulphonate (PVS), sulfonated polyacrylates, phosphomethylated polyamines
(PMPA),
3o and the like. Typically, one class of ionic polymer suitable for use within
the
compositions of the present invention is the group of phosphinocarboxylic acid
CA 02515865 2005-08-12
18
polymers. Suitable ionic polymers also include polyphosphonates,
polyphosphinates,
and polyphosphates. Specific examples of scale control agents suitable for use
herein
include diethylenetriamine penta(methylphosphonic acid), (DETPMP);
hexamethylenediamine tetramethylene phosphonic acid (HMDP); phosphino
s polycarboxylic acid (PPCA); nitrilotris(methylene) triphosphonic acid (NTP);
Bellasol 30TM (an aqueous solution of a phosphinocarboxylic acid commercially
available from OSP Microcheck, Inc.; Calgary, Alberta, Canada); the ACUMERTM
polymer products, such as ACUMERTM 2100, a carboxylate / sulfonate copolymer
commercially available from Rohm and Haas Company (Philadelphia, PA); and PSI-
io 720 (BJ Services Company; Houston, TX), a phosphinocarboxylic acid polymer.
Mixtures of phosphonates, phosphinates, phosphates, polyphosphonates,
polyphosphinates, polyphosphates, phosphinocarboxylic acid and its polymers,
and
polyalkylenephosphonic acid, such as BJ Services SCB-100 are also applicable.
In
addition, compounds such as glycolic acid, citric acid, gluconic acid,
is ethylenediaminetetraacetic acid, and other aminomethylcarboxylate compounds
and
derivatives used as metal chelating agents. All of the above-mentioned scale
control
agents can be used in either their acid or salt forms. Salts are generally
ammonium or
alkali metal salts.
The concentration of scale control agents) in the compositions of the present
ao invention can generally be any concentration suitable for inhibiting the
formation of
scale in a subterranean formation, in a wellbore, in pipe or tubing, or within
hydrocarbon-producing equipment. Typical ranges of concentration of one or
more
scale control agents for use in the compositions described herein are from
about 1
ppm to about 1000 ppm, more typically from about 5 ppm to about 100 ppm, for
is example from about 5 pm to about 25 ppm. Specific examples of
concentrations of
scale control agents in the compositions of the present invention include
about 1 ppm,
about 5 ppm, about 10 ppm, about 15 ppm, about 20 ppm, about 25 ppm, about 30
ppm, about 35 ppm, about 40 ppm, about 45 ppm, about 50 ppm, as well as ranges
between any two of these values. In one aspect, the concentration of scale
control
3o agent in the composition is about 25 ppm.
B. Salt control agents
CA 02515865 2005-08-12
19
Salt precipitation control agents, as used herein, refers to
nitrilotriacetonitrile,
nitrilotriacetarnide or its derivatives, including salts of
nitrilotriacetamides such as
those described in US Patent Application 2003/0173087. Generally
concentrations of
25 to 1000 parts per million are used, or an amount necessary to inhibit salt
(e.g.,
s halite) crystallization.
C. Fines control agents
The formation fines, broadly defined as particles having a diameter of less
than 44 microns, are ubiquitous in reservoir sandstones. These fines are
mineralogically diverse and range in composition from clay minerals to non-
clay
io siliceous minerals (i.e. quartz, feldspars, zeolites, silt, etc.). The
significance of fines
in hydrocarbon production and their potential permeability damage have been
reported extensively. The two mechanisms by which clays cause permeability
damage
are swelling and migration. In swelling, fines such as clays (e.g. smectite,
mixed-layer
clays) imbibe water into their crystalline structures and subsequently
increase in
is volume, plugging the pores in which they reside. In migration, clay and/or
silt
minerals (e.g. kaolinite, illite, chlorite) can be dispersed by contact with a
foreign
fluid or can be entrained by produced fluids and transported until a
restriction, usually
a pore throat, is encountered, where entrained particles bridge and restrict
fluid flow.
High fluid velocities, low salinities and high pH values are known to cause
fines
zo entrainment. Other non-clay fines known to cause permeability damage to
sandstones
are migrating quartz, feldspars, and zeolites particles.
Fines control agents, as used herein, refers to those agents which act to
control, inhibit, suspend, or otherwise stabilize potentially mobile fines
material.
Such control agents include, but are not limited to, organosilane additives
which react
zs in situ to stabilize potentially mobile siliceous fines; organic polymer
clay-control
agents, and mixtures thereof, and mixtures of organophosphonate or
organophosphinate compounds, and polyalkylenephosphonic acid, such as BJ
Services SCB-100, or sodium acid pyrophosphate (SAPP).
D. Asphaltene control agents
CA 02515865 2005-08-12
Asphaltenes are commonly defined as that portion of crude oil which is
insoluble in heptane, are soluble in toluene, and typically exist in the form
of colloidal
dispersions stabilized by other components in the crude oil. Asphaltenes are
often
brown to black amorphous solids with complex structures, involving carbon,
s hydrogen, nitrogen, and sulfur. Asphaltenes are typically the most polar
fraction of
crude oil, and will often precipitate out upon pressure, temperature, and
compositional
changes in the oil resulting from blending or other mechanical or
physicochemical
processing. Asphaltene precipitation can occur in pipelines, separators, and
other
equipment, as well as downhole and in the subterranean hydrocarbon-bearing
~o formation itself. Once deposited, these asphaltenes generally present
numerous
problems for crude oil producers, such as plugging downhole tubulars and/or
wellbores, choking off pipes, and interfering with the functioning of
separator
equipment, all of which compound the production costs and require the need for
remediation.
is Asphaltene deposition/precipitation control agents suitable for use with
the
compositions and methods described herein include 2-hexadecyl naphthalene,
sorbitan mono-oleate, decaglycerol tetraoleate, esters formed from the
reaction of
polyhydric alcohols with carboxylic acids, ethers formed from the reaction of
glycidyl
ethers or epoxides with polyhydric alcohols, esters formed from the reaction
of
Zo glycidyl ethers or epoxides with carboxylic acids, combinations thereof,
and the like.
In addition, organic solvents based on terpenes such as d-limonene, a-pinene,
and the
like, are applicable.
Application of one or more asphaltene control agents according to the methods
described herein may be made in combination with at least one RPM
macromolecule
zs and, optionally, one or more other formation damage inhibiting agents, by
continuous
or batch injection into the hydrocarbon-bearing formation itself, the well or
wellhead
system, or the hydrocarbon pipeline. It will be appreciated by those of skill
in the art
that there are a number of complex and interrelated factors which can
determine the
range of dosage of asphaltene control agent in a particular hydrocarbon
stream,
3o including but not limited to the chemical composition of the hydrocarbon or
crude oil,
and the temperature and pressure of the stream and the nature of any
mechanical or
CA 02515865 2005-08-12
21
physiochemical process the stream will be subjected to. The latter factor
includes, but
is not limited to, depressurization, cooling or heating, mixing with other
produced
fluids, shearing, the use of other additives, and the like. While it is nearly
impossible
to generalize about dosage levels due to these numerous and complex factors,
it will
s be appreciated that in one non-limiting aspect, the proportion of asphaltene
deposition
control agent used in the compositions of the present invention can be at
least about
ppm, typically in a range from about 15 ppm to about S00 ppm, and more
typically
in a range from about 100 ppm to about 300 ppm. Specific amounts of asphaltene
control agents include about 20 ppm, about 25 ppm, about 50 ppm, about 75 ppm,
~o about 100 ppm, about 150 ppm, about 200 ppm, about 250 ppm, about 300 ppm,
about 350 ppm, about 400 ppm, about 450 ppm, about 500 ppm, as well as ranges
and
values between any two of these values, e.g., about 230 ppm, or a range from
about
25 ppm to about 300 ppm.
E. Hydrate control agents
is Natural gas hydrates comprise "cages" of water molecules enclosing "guest"
molecules of natural gas, which occurs with sufficient combinations of
temperature
and pressure. Typical hydrate guest molecules include methane, ethane,
propane, light
hydrocarbons, methane-to-heptanes, nitrogen, hydrogen sulfide (H2S), and
carbon
dioxide (COz). Natural gas hydrates can form during the production, gathering,
and
zo transportation of hydrocarbons in the presence of water at high pressures
and low
temperatures. Depending on the pressure and gas composition, gas hydrates can
build
up at any place where water coexists with natural gas at temperatures as high
as $0 °F
(about 30° C). Once formed, hydrates can deposit in the tubing, flow
lines, and/or
process equipment, thus restricting flow. In many cases, these restrictions
eventually
zs form plugs. Gas transmission lines and new gas wells are especially
vulnerable to
being at least partially blocked by hydrates. Hydrate plugs represent safety
hazards as
they contain significant volumes of compressed natural gas and have been known
to
destabilize, release large volumes of gas, and create safety concerns. As
such, many in
the industry feel it prudent to prevent hydrate plugs whenever possible,
rather than
3o trying to remediate them once they form.
CA 02515865 2005-08-12
22
Formation of gas hydrates can be eliminated or hindered by several methods.
The thermodynamic prevention methods control or eliminate elements necessary
for
hydrate formation: the presence of hydrate forming guest molecules, the
presence of
water, high pressure and low temperature. The elimination of any one of these
four
s factors from a system would preclude the formation of hydrates.
Unfortunately,
elimination of these hydrate elements is often impractical or even impossible.
Transmission lines heating and insulating is a common mechanical solution to
hydrate
problems often encountered in long subsea pipelines. Hydrates will never form
if the
gas/water system is kept above the hydrate formation temperature. Gas
dehydration is
~o another method of removing a hydrate component. However, in a practical
field
operation, water can be economically removed to a certain vapor pressure only
and
residual water vapors axe always present in a dry gas. Hydrate plugs in "dry"
gas lines
have been reported in the past.
Hydrate control agents, as used herein, refer generally to those chemicals
is added to the gas/water streams to prevent or inhibit hydrate formation.
Typically the
hydrate control agents in accordance with the present invention include
methanol
(MeOH) or ethylene glycol (MEG), both of which have been used to prevent
natural
gas hydrates in systems which operate inside the hydrate stability region.
These
chemicals function as thermodynamic hydrate control agents, shifting the
conditions
zo at which hydrates are stable and thus allowing the system to operate
hydrate-free.
Also suitable for use as hydrate control agents, in accordance with the
present
invention, are low-dosage hydrate control agents.
Low-dosage hydrate control agents (LDHAs) have been applied in numerous
applications worldwide and continue to gain acceptance as alternatives to
traditional
as methods of controlling hydrates. Whether used as retrofits to systems
designed for use
of MeOH or MEG, or used in systems specifically designed for LDHAs, LDHAs
offer the distinct advantage of low dosage rates, often 1 - 10% that of MeOH
or MEG.
There are two commercially available types of LDHA chemicals available to
control
hydrates in hydrocarbon production, gathering, and transportation systems:
anti-
3o agglomerants (AAs) and kinetic hydrate control agents (KHAs). Both AAs and
KHAs can be used as hydrate control agents in accordance with the present
invention.
CA 02515865 2005-08-12
23
Anti-agglomerates (AAs) function by allowing hydrates to form but keeping
the particles small and well-dispersed in the hydrocarbon liquid. Fluid
viscosities
remain low, allowing the hydrate particles to be transported along with the
produced
fluids without depositing. Because of this dispersion mechanism, true AAs
function
s well, regardless of cooling and independent of how long the system remains
in the
hydrate region. These products do require the presence of sufficient liquid
hydrocarbon to allow transportation of the hydrate particles, and as such are
best
suited to oil or gas condensate systems.
Kinetic control agents are a relatively new class of control agents developed
to
io overcome gas hydrate problems, and are suitable for inclusion as formation
damage
control additives in accordance with the compositions and methods of the
present
invention. These additives have been designed to delay nuclei formation and to
slow
down their growth to form larger crystals. Kinetic control agents are
generally water
soluble polymers and are effective at low concentrations (<1% by weigh with
respect
is to water). A kinetic control agent suitable for use herein is HYTREATTM, a
polymeric composition available from Clariant. Other useful compounds include
polyether ammonium salts such as those described in US Patent 6,025,302.
F. Wax/paraffin control agents
The phenomenon of wax deposits formation is common in petroleum industry
2o and it occurs consequent to modifications in the thermodynamic variables
that change
the solubility of wax fractions present in petroleum. The paraffin deposition
phenomenon involves saturated hydrocarbons of linear chain and high molecular
weight during production, flow and treatment of petroleum. The deposition in
subsea
lines, surface equipment, production column, or even in reservoir rock can
cause
is significant loss of petroleum production.
Precipitation and deposition of wax are associated with phase equilibrium of
hydrocarbons and with fluid dynamic conditions of flow, respectively. The
paraffining phenomenon is a function of the petroleum intrinsic
characteristics and the
temperature, velocity and pressure variations during production. The
appearance of a
3o solid phase in petroleum and the subsequent wax deposition are related to
changes in
CA 02515865 2005-08-12
24
the phase equilibrium, caused by petroleum cooling and/or separation of
lighter
fractions, originally dissolved in petroleum.
There are several preventive and/or corrective methods to control wax
deposition: use of chemical control agents, injection of hot organic solvents
and
s mechanical removal. In many cases, chemical treatments of paraffin or wax
deposits
have been stopgap or band-aid approaches that worked sporadically, if at all.
The
problem has been made particularly difficult because paraffin deposits vary
significantly from reservoir to reservoir. Chemicals that are effective in one
producing
field are not always applicable in other reservoirs and even in various wells
within the
~o same reservoir (i.e., case specific). In association with the present
invention, it has
been found that chemical treatments of paraffin or wax deposits using
compositions
of the present invention can elicit improved results.
Chemicals suitable for use as paraffin/wax deposition remediation agents
include solvents, dispersants, and control agents, such as crystal modifiers
and crystal
rs disrupters, and are used to prevent paraffin crystals from forming massive
crystal
lattice structures which may plug tubing and process equipment. Suitable
solvents
include such hydrocarbons, as xylene, toluene and terpenes like d-limonene and
a
pinene. Terpenes can be dispersed or emulsified in aqueous solution in
concentrations
ranging from 2 to 70 vol. percent, or water, acid and brine solutions can be
dispersed
zo or emulsified in terpenes.
G. RPM Retention
The compositions described herein can also optionally include one or more
additives for enhancing the anchoring capabilities of the RPM macromolecule to
the
formation substrate. Such additives include organosilane compounds, such as
zs aminopropyltriethoxysilanes, or phosphonates, phosphinates, phosphates,
polyphosphonates, polyphosphinates, polyphosphates, phosphinocarboxylic acids
and
their polymers, or mixtures thereof.
Organosilane compounds suitable for use in the compositions of the present
invention include, but are not limited to, organosilane alkoxides such as
3o methyltriethoxysilane, dimethyldiethoxysilane, methyltrimethoxysilane,
CA 02515865 2005-08-12
divinyldimethoxysilane, divinyldi-2-methoxyethoxy silane, di(3-
glycidoxypropyl)
dimethoxysilane, vinyltriethoxysilane, vinyltris-2-methoxy-ethoxysilane, 3-
glycidoxypropyltrimethoxysilane, 3-methacryloxy-propyltrimethoxy-silane, 2-
(3,4-
epoxycyclohexyl) ethyltrimethoxysilane, N-2-aminoethyl-3-
s propylmethyldimethoxysilane, N-2-aminoethyl-3-propyltrimethoxysilane, N-2-
aminoethyl-3-aminopropyltrimethoxysilane, 3-aminopropyltriethoxysilane,
tetraethoxysilane and the like; and organosilane halides such as
methyldiethylchlorosilane, dimethyldichlorosilane, methyltrichlorosilane,
dimethyldibromosilane, diethyl-diiodosilane, dipropyldichlorosilane,
~o dipropyldibromosilane, butyltrichlorosilane, phenyltribromosilane,
diphenyldichlorosilane, tolyltribromosilane, methylphenyl-dichlorosilane,
propyldimethoxychlorosilane and the like; amino silanes including 3-
aminopropyltriethoxy silane and N-2-aminoethyl-3-aminopropyltrimethoxy silane;
Vinyl silanes including vinyl tris-(2-methoxyethoxy) silane, aminopropyl
triethoxy
~ s silane, aminoethyl triethoxy silane, aminopropyl trimethoxy silane,
aminoethyl
trimethoxy silane, ethylene trimethoxy silane, ethylene triethoxy silane,
ethyne
trimethoxy silane, ethyne triethoxy silane, and the like; and combinations of
such
organo silanes.
The weight ratio of RPM macromolecule to organosilicon compound in the
ao aqueous composition, when such organosilicon compound is present, is
generally
from about 1:1 to about 200:1. The weight percentage of the RPM and
organosilicon
compound composite in the aqueous composition is generally from about 0.01 to
about 25 weight percent. For instance, where the RPM macromolecule is PVA, the
concentration ratio in parts per million of PVA RPM macromolecule to silicon
in the
as organosilicon compound in the aqueous composition is generally from about
1:1 to
about 200:1, preferably from about 30:1 to about 400:1. The weight percentage
of the
PVA RPM and silicon in the organosilicon compound composite in the aqueous
composition is generally from about 1.0% to 24.0%, preferably from 3.0% to
10.25%,
weight percentage. The concentration ratio in parts per million of
polyacrylamide
3o RPM macromolecule to silicon in the organosilicon compound in the aqueous
composition is generally from about 10:1 to about 1:10, preferably from about
0.05:1
to about 1:05. The weight percentage of the polyacrylamide RPM and silicon in
the
CA 02515865 2005-08-12
26
organosilicon compound composite in the aqueous composition is generally from
about 0.03% to 0.3%, preferably from 0.09% to 0.18%, weight percent.
The compositions as described herein, in addition to the RPM macromolecule
and one or more formation damage control additives, can further comprise one
or
s more additional components. For example, the compositions can further
comprise
one or more salts, including inorganic salts, such as alkali metal, alkali
earth, and
transition metal salts, and organic salts. Such salts include, but are not
limited to,
ammonium salts (i.e., tetramethylammonium chloride; ammonium chloride),
calcium
salts (i.e., CaClz, CaBrz), magnesium salts, sodium salts, potassium salts
(i.e., KCI,
~o KBr), acetate salts (i.e., sodium acetate), formate salts, sulfate salts,
carbonate salts,
bicarbonate salts, and halide salts. Numerous examples of such suitable salts
are
know in the art. Further additives suitable for use in the compositions of the
presently
disclosed invention include additives for preventing, eliminating, or reducing
emulsification of the treating fluids.
~s The pH of the compositions as described herein can generally be any pH
compatible with the components of the composition, as well as with the piping,
machinery, and equipment used during the drilling and production process
and/or that
the compositions will come into contact with. For example, the pH can be in
the
range from about 1 to about 9.5. Specific examples of suitable pH values
include a
zo pH of about 1, a pH of about 2, a pH of about 3, a pH of about 4, a pH of
about 5, a
pH of about 6, a pH of about 7, and ranges between any two of these pH values.
A
typical pH range in accordance with the presently described compositions is
from
about 4.5 to about 7. Such compositions are typically stable at room
temperature (i.e.,
about 25 °C), and consequently do not form significant amounts of gel
or precipitate
zs following 3 or more days of storage at room temperature.
II. Methods of Use
The above-described compositions can be used to treat oil and gas producing
wells, as well as subterranean, hydrocarbon-producing formations. The
compositions
described above, comprising at least one relative permeability modifier
3o macromolecule and one or more formation damage control additive, are
generally
referred to herein as the "working compositions". Such compositions can be
prepared
CA 02515865 2005-08-12
27
in advance and stored until use, or they can be prepared "on demand" at the
work site.
In accordance with this latter aspect, for example, the RPM macromolecule,
formation damage control additive (i.e., scale control agent, asphaltene
control agent,
or both), and any additional components can be separate, and then are mixed to
the
s desired concentration and pH at the wellsite as needed. This allows for the
overall
composition of the "working composition" to be adjusted as necessary,
depending
upon the specific problems or characteristics of the individual wellsites.
The working compositions can be introduced into a hydrocarbon producing
well or hydrocarbon producing subterranean formation using any means, such as
by
~o pumping through a wellbore, or injecting through an injector or producer
well into a
subterranean formation. The pumping or injecting can be through coiled tubing,
conventional pipes, or other delivery systems. Typically, the working
compositions
are pumped at a sufficient volume and pressure such that they contact the
downhole,
subterranean formation for a time sufficient to act. For example, the
compositions
~s can be allowed to contact the formation for about 12 hours, by being "shut
in". Times
of contact suitable for use with the compositions described herein will vary
depending
upon the individual composition, the needs of the well, and the desired result
of the
contact, but contact times can include ranges from about 2 hours to about 12
hours,
from about 4 hours to about 10 hours, and from about 6 hours to about 8 hours.
ao Specific examples of contact times suitable in association with the working
compositions include about 2 hours, about 4 hours, about 6 hours, about 8
hours,
about 10 hours, about 12 hours, and ranges between any two of these values.
These
treatments can also be pumped with carbon dioxide or nitrogen gases, either
during
the pumping of the treatment or after the treatment, as a flush. Pumping with
the
zs gases allows better coverage of the treatment interval, particularly as a
means to
increase contact with the formation, reduce the treatment volume and hence,
the
treatment cost.
Treatment of a hydrocarbon-producing well with the above-described
compositions preferably results in at least the reduction of water production.
Such
3o treatments also preferably result in a reduction or inhibition of one or
more types of
CA 02515865 2005-08-12
28
formation damage, such as asphaltene deposition/precipitation, scale
formation, salt
precipitation, hydrate formation, and the like.
The reduction of water can be determined by any number of means, but
typically it is determined by comparing water production before and after the
s treatment. The reduction of water is typically in the range from more than
0% to
100%.
The inhibition, prevention, or reduction in formation damage in association
with the compositions of the present invention comprising both a RPM
macromolecule and one or more formation damage control agents can be
determined
Io by comparing formation damage before and after treatment, or by comparing
formation damage in two similar systems, one treated with the compositions of
the
present invention and one not treated with such a composition. For example,
the
installation of test nipples or other suitable monitoring devices within a
flow line
would enable visual inspection of the system to determine if scale formation
or
Is asphaltene deposition was reduced or inhibited. Similarly, periodic water
analyses of
water from the production system can indicate mineral content in the water,
such as
for example the calcium or barium content of the water in the production
system. A
decrease in the amount of specific, target minerals (e.g., barium or calcium)
could
indicate that the composition of the present invention was inhibiting scale
formation.
ao Formation damaging problems, such as scale formation, hydrate formation,
salt
precipitation, asphaltene deposition, and wax deposition can also be
monitored, in
accordance with the present disclosure, by monitoring parameters such as
pressure, a
method described specifically in the Examples below.
The compositions of the present invention can be used to treat, in addition to
zs subterranean formation damage and wellbore damage, pipes and tubing
associated
with the production of oil and gas from wells. The presently described
compositions
can be passed through the pipe or tubing to prevent, reduce, or inhibit the
formation of
damage, e.g., scale formation, asphaltene precipitation, or salt
precipitation, on or in
the pipe or tubing. Typically, the passage of the "working composition"
through the
3o pipe or tubing can be achieved using pumping means known in the art. Other
means
CA 02515865 2005-08-12
29
of passing such compositions through the tubing or pipes include gravity flow
means,
vacuum, or combinations of such methods.
Use of the above-described compositions provides several advantages over the
typical, step-wise application of well treatments, such as first injecting a
water control
s composition into the formation, and after completion of such treatment
injecting a
scale control agent composition into the formation. A first advantage is that
the
simultaneous application of two or more materials (e.g., an RPM macromolecule
and
one or more formation damage control additives) is convenient, requires only
one
application, and is potentially more cost effective, allowing the well to
return to
~o production more rapidly. Another advantage, as described above and as
specifically
shown in the Examples below, the simultaneous application of an RPM
macromolecule and at least one other material (e.g., a scale control agent)
results in a
synergistic effect, wherein the water control effect and/or the formation
damage
control effect (i.e., scale inhibition) is better than that achieved by
applying either of
is the compositions separately. As a result, water control and improved
formation
damage control can be accomplished simultaneously, using one composition.
The following examples are included to demonstrate preferred embodiments
of the invention. It should be appreciated by those of skill in the art that
the
techniques disclosed in the examples which follow represent techniques
discovered
zo by the inventors to function well in the practice of the invention, and
thus can be
considered to constitute preferred modes for its practice. However, those of
skill in
the art should, in light of the present disclosure, appreciate that many
changes can be
made in the specific embodiments which are disclosed and still obtain a like
or similar
result without departing from the scope of the invention.
EXAMPLES
Example 1 ~ Preparation of Compositions Containi~ Scale Control Agent and RPM
PSI-720 (BJ Services Company; Houston, TX), a phosphinocarboxylic acid
polymer (prepared as a 50 wt. % solution in water) was selected as an
exemplary
3o scale control agent. AquaConTM (BJ Services Company; Houston, TX), a
polyacrylamide-based terpolymer containing sulfonated functional groups, was
CA 02515865 2005-08-12
selected as an exemplary relative permeability modifier. A series of mixtures,
A-G,
were prepared at room temperature, as shown in Table 1, below.
Table 1. RPM and Scale Control Agent Mixtures
Mixture Amt. PSI-720 (wt. Amt. AquaConTM (wt.
%) %)
A (control) ' 0 0
B 100 0
C 90 10
D 75 25
E 50 50
F 10 90
G 0 100
n i
'2 wt. % KCl in water.
s Example 2: Tube Blocking Test Results
A tube blocking test was used to assay the effect caused by combining a scale
control agent and a relative permeability modifier, if any. The test system
comprised
peristaltic pumps, a mixing "tee", a 1/8-inch (0.3175 cm) coil of stainless
steel tubing,
and a 0.02 inch (0.05 cm) test coil of PEEK tubing.
io Each of the mixtures described in Example 1 was mixed with test brine (2
wt.
potassium chloride in water) in an amount sufficient to give a final
concentration of
about 25 ppm PSI-720. This value was selected as it is slightly under the
minimum
scale control agent concentration.
The two brine solutions were used to form a calcium carbonate scale
is precipitate. The first was a "cationic brine", containing about 24,000 ppm
sodium
chloride, about 1,050 ppm sodium sulfate, about 6,610 ppm sodium bicarbonate,
and
about 4,610 ppm sodium acetate. The second brine prepared was an "anionic
brine",
containing about 24,000 ppm sodium chloride, about 1,340 ppm magnesium
chloride,
and about 1,470 ppm calcium chloride.
CA 02515865 2005-08-12
31
The mixture and the brines were pumped into the testing system and into the
mixing "tee", then into the stainless steel tubing, and finally into the test
coil.
Premature precipitation of the calcium carbonate scale was prevented by
combining
the cationic brine and the anionic brine immediately before the test coil. The
pressure
s on the system was measured using a pressure transducer. The transducer
recorded the
pressure at ten second intervals. A computer plotted the pressure against the
elapsed
time, as shown graphically in Figures 1-7. Figures 1-7 and their corresponding
compositions are shown in Table 2.
io Table 2.
Mixture Figure Amt. PSI-720 Amt. AquaConTM
(ppm) (ppm)
A (Control)' 1 0 0
B 2 25 0
C 3 25 524
D 4 25 712
E 5 25 1600
F 6 25 4000
G 7 0 4000
'2 wt. % KC1 in water.
The blank control composition (containing brine but no scale control agent or
relative permeability modifier; Figure 1) exhibited a significant increase in
pressure
(in psi) after 600-800 seconds (about 10-14 minutes). The composition
containing
is scale control agent (B; Figure 2) effectively prevented any pressure
increase in the
test coil for a period of at least one hour (the tests were terminated after
one hour,
measured in seconds, s). A mixture was deemed to be "acceptable" if no
significant
increase in pressure (e.g., less than about 0.3 psi) was observed after one
hour.
CA 02515865 2005-08-12
32
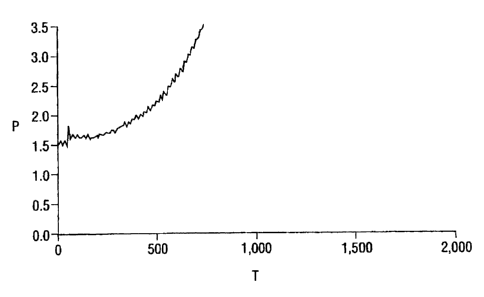
Figures 3, 4, 5 and 6 all show that the addition of a relative permeability
modifier to the compositions did not adversely effect the ability of the scale
control
agent to prevent a significant increase in pressure (the concentration of
scale control
agent was held at about 25 ppm for these tests). A significant increase in
pressure
s would suggest that the scale control agent was not working, or not working
effectively. Figures 4, 5, and 6 (mixtures D, E, and F, respectively)
exhibited
remarkable inhibition of pressure increase. Figure 7 (mixture G, containing
only
relative permeability modifier) showed an increase in pressure greater than
the
mixture containing scale control agent only (mixture B). However, both
mixtures G
io and B exhibited a pressure increase less than that observed with the
control (mixture
A, containing only brine) shown in Figure 1.
These results show that the addition of a relative permeability modifier to a
brine solution containing a fixed concentration of scale control agent
improves the
inhibition of scale (as evidenced by the lack of pressure increase). These
results
is suggest a "synergistic" effect of the two components.
Example 3: Stability of Compositions at Room Temperature
The mixtures prepared as described in Example 1 were kept at room
temperature for three days. At the end of this period, no precipitation,
gelling, or
other signs of incompatibility were observed.
zo All of the compositions, methods and/or processes disclosed and Claimed
herein can be made and executed without undue experimentation in light of the
present disclosure. While the compositions and methods of this invention have
been
described in terms of preferred embodiments, it will be apparent to those of
skill in
the art that variations may be applied to the compositions, methods and/or
processes
zs and in the steps or in the sequence of steps of the methods described
herein without
departing from the concept and scope of the invention. More specifically, it
will be
apparent that certain agents which are chemically related may be substituted
for the
agents described herein while the same or similar results would be achieved.
All such
similar substitutes and modifications apparent to those skilled in the art are
deemed to
3o be within the scope and concept of the invention.