Note: Descriptions are shown in the official language in which they were submitted.
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
ONCE-A-DAY, ORAL, CONTROLLED-RELEASE,
OXYCODONE DOSAGE FORMS
I. CROSS REFERENCE TO RELATED APPLICATIONS
[0001 ] This application claims the benefit under 35 USC ~ 119(e) of U.S.
Provisional Application No. 60/515,880 filed October 29, 2003, the contents of
which
in its entirety is hereby incorporated by reference.
(0002] This application is a Continuation-In-Part of U.S. Application No.
10/423,454 filed April 25, 2003, which claims the benefit under 35 USC ~119(e)
of
U.S. Provisional Application No. 60/376,470 filed April 29, 2002, and which
was
published as U.S. Patent Publication No. 2004-0010000 Al on January 15, 2004
and as
WO 03/092648 on November 13, 2003, the contents of all of which in their
entireties
are hereby incorporated by reference.
[0003] This application is also a Continuation-In-Part of U.S. Application No.
10/447,910 filed May 28, 2003, which claims the benefit under 35 USC ~119(e)
of U.S.
Provisional Application No. 60/384,442 filed May 31, 2002, and which was
published
as U.S. Patent Publication No. 2003-0224051 Al on December 4, 2003, and as WO
03/101384 on December 11, 2003, the contents of all of which in their
entireties are
hereby incorporated by reference.
II. FIELD OF THE INVENTION
[0004] This invention relates to ih vitro and ira vivo profiles, i.e., in
vitro
dissolution/release profiles and in vivo single dose and in vivo steady state
plasma
profiles, for the opioid analgesic, oxycodone, when administered orally using
a
controlled-release dosage form. In particular, the invention relates to ih
vitf°o and ifa
vivo oxycodone profiles designed to produce effective pain management and a
reduced
probability of "lilting" when oxycodone is orally administered to a patient on
a once-a-
day basis.
III. BACKGROUND OF THE INVENTION
A. OXYCODONE
[0005] Oxycodone, a Schedule II drug, is an opioid for the management of
moderate to severe chronic pain, such as, pain due to surgery, cancer, trauma,
biliary
colic, renal colic, myocardial infarction and burns. Oxycodone has been
marlceted as
1
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
an analgesic for more than 70 years. It is currently available in immediate
release (IR)
forms, as well as in a controlled release (CR) formulation indicated for
b.i.d. dosing.
[0006] The pharmacological and medical properties of analgesic opioids
including
oxycodone are known in Pharmaceutical Sciences, Remington, 17th Ed., pp. 1099-
1107
(1985), and The Phannacolo~ical Basis of Therapeutics, Goodman and Rall, 8th
Ed.,
pp. 485-518 (1990). Generally, the analgesic action of parenterally
administered
oxycodone is apparent within 15 minutes, while the onset of action of orally
administered oxycodone is somewhat slower with analgesia occurring within
about 30
minutes. In human plasma, the half life of orally administered immediate
release
oxycodone is about 3.2 hours. Physicians' Desk Reference, Thompson Healthcare,
5611'
Ed., pp. 2912-2918 (2002).
[0007] In the past, oxycodone has been administered in conventional forms,
such as
nonrate-controlling, dose-dumping immediate release tablets, or by dose-
dumping
capsules, and usually at multiple, repetitive dosing intervals throughout the
day.
Oxycodone is also administered on a twice-a-day basis with a controlled
release matrix
system, OXYCONTIN~ (Purdue Pharma LP, Stamford, CT). The OXYCONTIN~
mode of therapy, however, continues to lead to an initial high dose of
oxycodone in the
blood after administration, followed by decreased levels of oxycodone in the
blood.
Moreover, this peals and trough pattern occurs twice during a 24-hour period
due to the
twice-a-day dosing regimen. The concentration differences in dosing patterns
are
related to the presence and absence of administered drug, which is a major
disadvantage associated with these prior dosage forms. Conventional dosage
forms and
their mode of operation, including dose peaks and valleys, are discussed in
Pharmaceutical Sciences, Remington, 18th Ed., pp. 1676-1686 (1990), Mack
Publishing Co.
B. TOLERANCE TO OXYCODONE
[0008] Previous studies in rats and mice with opioids have shown the
development
of tolerance to analgesia (antinociception) after bolus dosing, intermittent
dosing, and
constant rate infusions (Ekblom et al 1993, Gardmark et al 1993, Ouellet &
Pollaclc
1995, 1997, Duttaroy & Yoburn 1995).
[0009] In connection with the OXYCONTIN~ product, employees of Purdue
Pharma and its associated companies have published scientific articles in
which
biphasic profiles are described as being better than flat profiles with regard
to the
2
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
development of oxycodone tolerance. Thus, in the Journal of Pain and Symptom
Management, Purdue employees wrote (Benziger et al. 1997 at page 81):
[00010] "Although the benefits of controlled-release dosage forms that
permit less frequent dosing are well established, it has been
suggested that the maintenance of nearly constant plasma
concentrations of opioids may lead to tolerance development.
The CR oxycodone tablets under study [OXYCONTIN~] were
developed to reduce the number of Cm;"/CmaX fluctuations during
the 12-hr dosing interval while matching the degree of
fluctuation (Cm;"/CmaX) in plasma oxycodone concentrations
observed during steady-state dosing with comparable doses of IR
oxycodone. By retaining the degree of fluctuation in plasma
concentrations the possibility of diminished pharmacodynamic
effects over time may be diminished as compared to CR
formulations that maintain comparable constant blood levels."
(citations omitted.)
[00011 ] Similarly, Dr. Robert Kaiko, an inventor of OXYCONTIN~, wrote in Acta
Annesthesiol Scand (Kaiko 1997 at page 172):
[00012] "Another rationale basis for the biphasic opioid absorption
profile is to produce a peak-to-trough fluctuation comparable to
the conventional immediate-release opioid. Because it has been
suggested that very steady plasma opioid concentrations may
lead to tolerance development, it is anticipated that alteration of
the rate of fluctuation without alteration of the degree of
fluctuation would minimize tolerance development." (citations
omitted.)
[00013] Further teachings against flat plasma profiles and, in particular,
against flat
plasma profiles for once-a-day dosage forms, can be found in the patent
literature.
Thus, U.S. Patent No. 5,478,577, assigned to Euroceltique, S.A., a company
related to
Purdue Pharma, states (column 5, lines 34-42):
[00014] "It has now been surprisingly discovered that quiclcer and greater
analgesic efficacy is achieved by 24 hour oral opioid
formulations which do not exhibit a substantially flat serum
3
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
concentration curve, but which instead provide a more rapid
initial opioid release so that the minimum effective analgesic
concentration can be more quickly approached in many patients
who have measurable if not significant pain at the time of
dosing."
[00015] See also Euroceltique's U.S. Patent No. 5,672,360, colmnn 5, lines 40-
47.
[00016] In view of these explicit warnings against flat profiles by the
leading
manufacturer of controlled-release oxycodone products, persons of ordinary
skill in the
art have been led away from the use of oxycodone dosage forms having
substantially
zero order isa vitf°o release profiles. In particular, such persons
would expect that flat
profiles would generate higher levels of tolerance than biphasic profiles.
[00017] As discussed fully below (see Example 8), it has been found that
notwithstanding Purdue Pharma's teachings, oxycodone tolerance levels
associated with
biphasic profiles and flat profiles (substantially zero order release
profiles) are, in fact,
not statistically different. In addition, as illustrated in Figure 5 (see
discussion below),
substantially zero order oxycodone release profiles produce low single dose
CmaX values
and thus are expected to have lower levels of "liking" than profiles that are
not
substantially zero order, such as biphasic profiles. As has been well-
documented in the
literature, including the popular press, Purdue Pharma's biphasic OXYCONTIN~
product has serious abuse problems, substantially beyond any issue of
"liking."
[00018] hnportantly, as illustrated by the efficacy data of Example 7 below,
substantially zero order oxycodone release profiles achieve effective pain
management.
Accordingly, in accordance with the invention, it has been shown that
oxycodone
dosage forms which have substantially zero order in vitro release profiles can
be used
to achieve effective pain management without exaggerated tolerance problems
and with
a reduced probability of "lilting" -- a combination of benefits not previously
known or
expected from the existing state of the art.
IV. SUMMARY OF THE INVENTION
[00019] In accordance with a first aspect, the invention provides a controlled-
release
oxycodone formulation for once-a-day oral administration to human patients
comprising a dose D of:
(i) oxycodone,
4
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
said formulation providing (a) a mean, single dose, maximum plasma
concentration
CmaX and (b) a mean, single dose, area under a plasma concentration-time curve
for
0-48 hours AUCo_48 which satisfy the relationships:
3.5 x 10-4 liter 1 < CmaX/D ~ 6.8 x 10-4 liter 1, and
7.6 x 10-3 hour/liter <_ AUCa_48/D <_ 16.7 x 10-3 hour/liter,
wherein said formulation provides pain relief for about 24 hours or more after
administration to the patient.
[00020] In accordance with a second aspect, the invention provides a
controlled-
release oxycodone formulation for once-a-day oral administration to human
patients
comprising a dose D of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
wherein:
(a) the formulation provides a mean, single dose, plasma concentration
profile that increases substantially monotonically over 24 hours or more;
(b) the formulation provides a mean, single dose, area under a plasma
concentration-time curve for 0-48 hours AUCo_48 which satisfies the
relationship:
7.6 x 10-3 hour/liter <_ AUCo_481D <_ 16.7 x 10-3 hour/liter; and
(c) the formulation provides pain relief for about 24 hours or more after
administration to the patient.
In accordance with a third aspect, the invention provides a controlled-release
oxycodone formulation for once-a-day oral administration to human patients
comprising a dose D of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
5
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
said formulation providing (a) a mean, single dose, 12 hour plasma
concentration Clz
and (b) a mean, single dose, area under a plasma concentration-time curve for
0-48
hours AUCo_48 which satisfy the relationships:
2.7 x 10-4 liter 1 < Ciz/D <_ 5.7 x 10-4 liter 1, and
7.6 x 10-3 hour/liter <_ AUCo_48/D <_ 16.7 x 10-3 hour/liter,
wherein said formulation provides pain relief for about 24 hours or more after
administration to the patient.
[00021 ] In accordance with a fourth aspect, the invention provides a
controlled-
release oxycodone formulation for once-a-day oral administration to human
patients
comprising a dose D of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
said formulation providing mean, steady state, areas under a plasma
concentration-time
curve for 0-6 hours AUCo_6, 6-12 hours AUC6_lz, 12-18 hours AUCIZ_ls, 18-24
hours
AUC18_z4, and 0-24 hours AUCo_z4 which satisfy the relationships:
AUCo_6/AUCo_z4 > 0.18,
AUC6_iz/AUCo_z4 > 0.18,
AUCIZ_18/AUCo_z4 > 0.18, and
AUCIS_z4/AUCo_z4 > 0.18,
wherein said formulation provides pain relief for about 24 hours or more after
administration to the patient.
[00022] In accordance with a fifth aspect, the invention provides a controlled-
release
oxycodone formulation for once-a-day oral administration to human patients
comprising a dose of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
6
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
said formulation having an ira vitro release profile in which:
(a) 0-20% of the dose is released in 0-2 hours;
(b) 30-65% of the dose is released in 0-12 hours; and
(c) 80-100% of the dose is released in 0-24 hours;
wherein the release profile is determined using a USP Type VII bath indexer in
a
constant temperature water bath at 37°C and wherein said formulation
provides pain
relief for about 24 hours or more after administration to the patient.
[00023] In accordance with a sixth aspect, the invention provides a controlled-
release oxycodone formulation for once-a-day oral administration to human
patients
comprising a dose of:
(i) oxycodone, .
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
wherein:
(a) the dose comprises a first component for immediate release and a second
component for sustained release; and
(b) the weight ratio W of the first component to the sum of the first and
second components is less than about 0.25.
[00024] In accordance with a seventh aspect, the invention provides a method
of
treating pain in humans comprising orally administering to a human patient on
a once-
a-day basis a controlled-release dosage form comprising a dose D of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
said dosage form providing (a) a mean, single dose, maximum plasma
concentration
Cmax ~d (b) a mean, single dose, area under a plasma concentration-time curve
for
0-48 hours AUCo_48 which satisfy the relationships:
7
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
3.5 x 10~ liter 1 < Cn.,aX/D <_ 6.8 x 10-4 liter 1, and
7.6 x 10-3 hour/liter _< AUCo_48/D <_ 16.7 x 10-3 hour/liter,
wherein the dosage form provides pain relief for about 24 hours or more after
administration to the patient.
[00025] In accordance with an eight aspect, the invention provides a method of
treating pain in humans comprising orally administering to a human patient on
a once-
a-day basis a controlled-release dosage form comprising a dose D of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
wherein:
(a) the dosage form provides a mean, single dose, plasma concentration
profile that increases substantially monotonically over 24 hours or more;
(b) the dosage form provides a mean, single dose, area under a plasma
concentration-time curve for 0-48 hours AUCo_48 which satisfies the
relationship:
7.6 x 10-3 hour/liter <_ AUCo_48/D <_ 16.7 x 10-3 hour/liter; and
(c) the dosage form provides pain relief for about 24 hours or more after
administration to the patient.
In accordance with a ninth aspect, the invention provides a method of treating
pain in humans comprising orally administering to a human patient on a once-a-
day
basis a controlled-release dosage form comprising a dose D of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
said dosage form providing (a) a mean, single dose, 12 hour plasma
concentration Clz
and (b) a mean, single dose, area under a plasma concentration-time curve for
0-48
hours AUCo_4$ which satisfy the relationships:
2.7 x 10-4 liter 1 < ClzlD <_ 5.7 x 10-4 liter 1, and
7.6 x 10-3 hour/liter <_ AUCn_4$/D <_ 16.7 x 10-3 hour/liter,
8
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
wherein said dosage form provides pain relief for about 24 hours or more after
administration to the patient.
[00026] In accordance with a tenth aspect, the invention provides a method of
treating pain in humans comprising orally administering to a human patient on
a once-
a-day basis a controlled-release dosage form comprising a dose D of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
said dosage form providing mean, steady state, areas under a plasma
concentration-time
curve for 0-6 hours AUCo_6, 6-12 hours AUC6_12, 12-18 hours AUC12_ls, 18-24
hours
AUCIg_24, and 0-24 hours AUCo_z4 which satisfy the relationships:
AUCo_6/AUCo_z4 > 0.18,
AUC6_12/AUCo_2a > 0.18,
AUCIZ_i8/AUCo_Zø > 0.18, and
AUC1$_24/AUCo_24 > 0.18,
wherein said dosage form provides pain relief for about 24 hours or more after
administration to the patient.
[00027] In accordance with an eleventh aspect, the invention provides a method
of
treating pain in humans comprising orally administering to a human patient on
a once-
a-day basis a controlled-release dosage form comprising a dose D of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
said dosage form providing pain relief for about 24 hours or more after
administration
to the patient and having an ira vitro release profile in which:
(a) 0-20% of the dose is released in 0-2 hours;
(b) 30-65% of the dose is released in 0-12 hours; and
(c) 80-100% of the dose is released in 0-24 hours;
9
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
where the release profile is determined using a USP Type VII bath indexer in a
constant
temperature water bath at 37°C.
[00028] In accordance with a twelfth aspect, the invention provides a method
of
treating pain in humans comprising orally administering to a human patient on
a once-
a-day basis a controlled-release dosage form comprising a dose D of:
(i) oxycodone,
(ii) one or more pharmaceutically-acceptable acid addition salts of
oxycodone, or
(iii) a combination of oxycodone and one or more pharmaceutically-
acceptable acid addition salts of oxycodone,
wherein:
(a) the dose comprises a first component for immediate release and a second
component for sustained release;
(b) the weight ratio W of the first component to the sum of the first and
second components is less than about 0.25; and
(c) the dosage form provides pain relief for about 24 hours or more after
administration to the patient.
The various AUC and C values referred to above can be determined using
plasma samples from individuals to whom one or more opioid antagonists (e.g.,
naltrexone) have been administered or by using samples from individuals to
whom an
antagonist has not been administered. For higher dosage levels, antagonists
are
normally used, especially in studies involving healthy volunteers. For
example, various
of the numerical values set forth above are based on the pharmacolcinetic data
of
Example 6, which used healthy volunteers and a dosage form which contained 80
mg
of oxycodone HCI. As described in Example 6, naltrexone was administered in
this
study. As known in the art, naltrexone has a tendency to increase plasma
oxycodone
concentrations. Accordingly, somewhat lower AUC and C values would be expected
if
naltrexone had not been used, but the changes would not be expected to move
the mean
values substantially outside of the ranges specified.
[00029] Additional features and advantages of the invention are set forth in
the
detailed description which follows, and in part will be readily apparent to
those skilled
in the art from that description or recognized by practicing the invention as
described
herein. It is to be understood that both the foregoing general description and
the
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
following detailed description are merely exemplary of the invention, and are
intended
to provide an overview or framework for understanding the nature and character
of the
invention as it is claimed. Also, the above listed aspects of the invention,
as well as the
preferred and other embodiments of the invention discussed below, can be used
separately or in any and all combinations.
[00030] The accompanying drawings are included to provide a further
understanding
of the invention, and are incorporated in and constitute a part of this
specification. The
drawings illustrate various embodiments of the invention, and together with
the
description serve to explain the principles and operation of the invention.
The drawings
and, in particular, Figures 1-4, are not intended to indicate scale or
relative proportions
of the elements shown therein. In the drawings and the specification, like
parts in
related figures are identified by like numbers.
V. BRIEF DESCRIPTION OF THE DRAWINGS
[00031 ] Figure 1 illustrates one type of dosage form that can be used in the
practice
of the invention. The dosage form is shown in Figure 1 prior to administration
to a
subj ect.
[00032] Figure 2 illustrates a first embodiment of the,dosage form of Figure 1
in
opened section. As shown, the dosage form comprises an internally-housed,
pharmaceutically-acceptable therapeutic oxycodone composition.
[00033] Figure 3 illustrates a second embodiment of the dosage form of Figure
1 in
opened section. As shown, the dosage form comprises an internally-housed,
pharmaceutically-acceptable therapeutic oxycodone composition and a separate
and
contacting displacement composition comprising means for pushing the
pharmaceutical
oxycodone composition from the dosage form.
[00034] Figure 4 illustrates a dosage form which further includes an immediate-
release overcoat of a pharmaceutically-acceptable therapeutic oxycodone
composition.
[00035] Figure 5 is a plot of simulated single dose plasma concentrations for
a
substantially zero order (SZO) release rate (curve 100), a fast-followed-by-
slow release
rate (curve 102), and a slow-followed-by-fast release rate (curve 104).
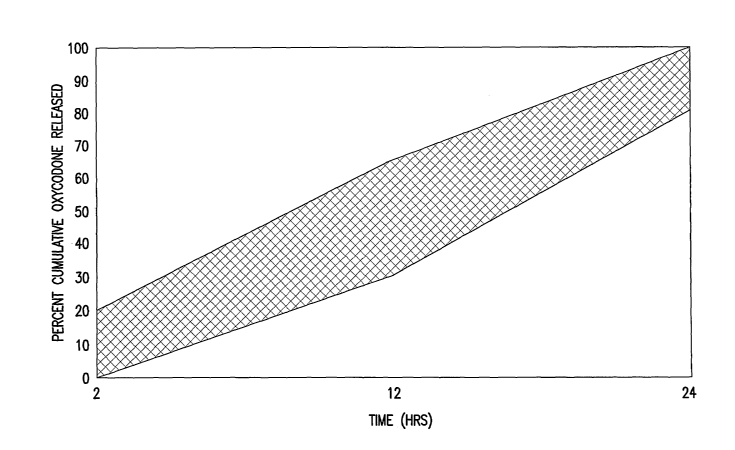
[00036] Figure 6 is a plot of a preferred cumulative release range for the
dosage
forms of the invention. The vertical axis plots percent cumulative release of
oxycodone
and/or one or more of its pharmaceutically-acceptable acid addition salts
(e.g., % of
11
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
label claim for a dosage form that has received regulatory approval) and the
horizontal
axis plots time.
[00037] Figures 7A and 7B are plots of in vitro release profiles for the 17 mg
oxycodone HCl dosage form identified as the "fast system" in Example 1. Figure
7A
(curve 106) plots the percent released per hour (e.g., % of label claim
released per
hour), while Figure 7B (curve 108) plots the cumulative release in percent
(e.g.,
cumulative % of label claim).
[00038] Figures 8A and 8B are plots of ira vitro release profiles for the 17
mg
oxycodone HCl dosage form identified as the "slow system" in Example 1. Figure
8A
(curve 110) plots the percent released per hour (e.g., % of label claim
released per
hour), while Figure 8B (curve 112) plots the cumulative release in percent
(e.g.,
cumulative % of label claim).
[00039] Figures 9A and 9B are plots of in vitro release profiles for the 20 mg
oxycodone HCl dosage form of Example 2. Figure 9A (curve 114) plots the
percent
released per hour (e.g., % of label claim released per hour), while Figure 9B
(curve
116) plots the cumulative release in percent (e.g., cumulative % of label
claim).
[00040] Figures 10A and lOB are plots of in vitro release profiles for the 80
mg
oxycodone HCl dosage form of Example 3. Figure 10A (curve 118) plots the
percent
released per hour (e.g., % of label claim released per hour), while Figure lOB
(curve
120) plots the cumulative release in percent (e.g., cumulative % of label
claim).
[00041 ] Figure 11 is a plot of pupil diameter in millimeters (mm) versus time
in
hours for healthy male subjects who received placebo (curve 122), morphine
(curve
124), or the dosage form of Example 2 (curve 126).
[00042] Figure 12 is a plot of plasma concentrations in nanograms/milliliter
(ng/mL)
of oxycodone (curve 128), noroxycodone (curve 130), and oxymorphone (curve
132)
versus time in hours for healthy male subjects who received the dosage form of
Example 2.
[00043] Figure 13 is a plot of simulated pharmacolcinetics, specifically,
single dose
plasma concentrations, for immediate release (IR) dosing (q6h) (curve 134), as
well as
experimental data for the dosage form of Example 2 and a best-fit curve to
that data
(curve 136).
[00044] Figure 14 is a plot of simulated pharmacokinetics, specifically,
steady-state
plasma concentrations, for immediate release (IR) dosing (q6h) (curve 140),
12
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
OXYCONTIN biphasic dosing (curve 138), and substantially zero order/once-a-day
(SZO-24) dosing using a dosage form having the overcoat/sustained release drug
distribution of Example 2, i.e., 5% of the drug in the overcoat (curve 142).
The y-axis
in this figure shows oxycodone concentration.
[00045] Figures 15A and 15B are plots of mean in vivo plasma oxycodone
concentration profiles for immediate release (IR) dosing (q6h) (curve 144;
n=16),
dosing with the 17 mg oxycodone HCl dosage form identified as the "fast
system" in
Example 1 (curve 146; n=17), and dosing with the 17 mg oxycodone HCl dosage
form
identified as the "slow system" in Example 1 (curve 148; n=17). Figure 15A
shows the
single dose profiles and Figure 15B shows the steady-state profiles. The error
bars
associated with the data points show the standard deviation (SD) in one
direction.
[00046] Figures 16A, 16B, 16C, and 16D are plots of mean in vivo plasma
oxycodone concentration profiles for substantially zero order (SZO) dosing
with the 80
mg oxycodone HCl dosage form of Example 3 (curve 150; n=37), and biphasic
dosing
with 40 mg OXYCONTIN tablets (curve 152; n=38). Figure 16A shows single dose
and steady-state profiles, Figures 16B and 16C show single dose profiles, and
Figure
16D shows steady-state profiles. The error bars associated with the data
points show
the standard deviation (SD) in one direction.
[00047] Figure 17A and 17B are plots of the data of Tables 12A and 12B, with
Figure 17A plotting all of the data of these tables and Figure 17B plotting
Day +3 data
for tail flick testing doses of 0, 0.25, 0.5, 0.75, and 1.0 mg/kg.
VI. DEFINITIONS
[00048] As used in this specification and in the claims, the following terms
and
phrases shall have the following meanings.
[00049] By "dosage form" is meant a pharmaceutical composition or device
comprising an active pharmaceutical agent, such as oxycodone and/or one or
more of
its pharmaceutically-acceptable acid addition salts, the composition or device
also
containing inactive ingredients, i.e., pharmaceutically acceptable excipients
such as
suspending agents, surfactants, disintegrants, binders, diluents, lubricants,
stabilizers,
antioxidants, osmotic agents, colorants, plasticizers, coatings and the like,
that are used
to manufacture and deliver active pharmaceutical agents.
[00050] By "active agent", "drug", or "compound" is meant an agent, drug, or
compound having the characteristics of oxycodone and/or one or more of its
13
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
pharmaceutically-acceptable acid addition salts. If desired, other analgesics
or, more
generally, other medicaments, can be included in the dosage forms of the
invention.
[00051 ] By "pharmaceutically-acceptable acid addition salts" are meant those
salts
in which the anion does not contribute significantly to the toxicity or
pharmacological
activity of the salt, and, as such, they are the pharmacological equivalents
of the bases
of the oxycodone compound. Examples of pharmaceutically acceptable acids that
are
useful for the purposes of salt formation include but are not limited to
hydrochloric,
hydrobromic, hydroiodic, citric, acetic, benzoic, mandelic, phosphoric,
nitric, mucic,
isethionic, palinitic, and others.
[00052] By "sustained release" is meant predetermined substantially continuous
release of active agent to an environment over a prolonged period.
[00053] The expressions "exit," "exit orifice," "delivery orifice" or "drug
delivery
orifice," and other similar expressions, as may be used herein include one or
more
members selected from the group consisting of a passageway; an aperture; an
orifice;
and a bore. The expressions also include orifices that are formed or formable
from a
substance or polymer that erodes, dissolves or is leached from the dosage form
to
thereby form an exit orifice.
[00054] A drug "release rate" refers to the quantity of drug released from a
dosage
form per unit time, e.g., milligrams of drug released per hour (mg/hr). Drug
release
rates for drug dosage forms are typically measured as an i~ vitro rate of
release, i.e., a
quantity of drug released from the dosage form per unit time measured under
appropriate conditions and in a suitable fluid. The release rate tests
utilized in the
examples described herein were performed on dosage forms placed in metal coil
sample holders attached to a USP Type VII bath indexer in a constant
temperature
water bath at 37°C. Aliquots of the release rate solutions were inj
ected into a
chromatographic system to quantify the amounts of drug released during the
testing
intervals.
[00055] By "release rate assay" is meant a standardized assay for the
determination
of the release rate of a compound from a dosage form tested using a USP Type 7
interval release apparatus. It is understood that reagents of equivalent grade
may be
substituted in the assay in accordance with generally accepted procedures.
14
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[00056] For clarity and convenience herein, the convention is utilized of
designating
the time of drug administration as zero hours (t = 0 hours) and times
following
administration in appropriate time units, e.g., t = 30 minutes or t = 2 hours,
etc.
As used herein, unless otherwise specified, a drug release rate obtained at a
specified
time "following administration" refers to the ih vitro drug release rate
obtained at the
specified time following implementation of an appropriate dissolution test.
The time at
which a specified percentage of the drug within a dosage form has been
released may
be referenced as the "TX" value, where "x" is the percent of drug that has
been released.
For example, a commonly used reference measurement for evaluating drug release
from dosage forms is the time at which 70% of drug within the dosage form has
been
released. This measurement is referred to as the "T~o" for the dosage form.
[00048] An "immediate-release dosage form" refers to a dosage form that
releases
drug substantially completely within a short time period following
administration, i.e.,
generally within a few minutes to about 1 hour.
[00049] By "sustained release dosage form" is meant a dosage form that
releases
drug substantially continuously for many hours (the "sustained release time
period").
Sustained release dosage forms in accord with the present invention preferably
exhibit
Tao values of at least about 10 to 20 hours and preferably 15 to 18 hours. The
dosage
forms preferably continuously release drug for sustained periods of at least
about 10
hours, more preferably 12 hours or more and, even more preferably, 16-20 hours
or
more.
[00050] Dosage forms in accord with the present invention preferably exhibit
uniform release rates of oxycodone for a prolonged period of time within the
sustained
release time period.
[00051 ] By "uniform release rate" is meant an average hourly release rate
from the
core that varies positively or negatively by no more than about 30% and
preferably no
more than about 25% and most preferably no more than 10% from either the
preceding
or the subsequent average hourly release rate as determined in a USP Type 7
Interval
Release Apparatus where the cumulative release is between about 25% to about
75%.
[00052] By "prolonged period of time" is meant a continuous period of time of
at
least about 4 hours, preferably 6-8 hours or more and, more preferably, 10
hours or
more. For example, the exemplary osmotic dosage forms described herein
generally
begin releasing oxycodone at a uniform release rate within about 2 to about 6
hours
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
following administration and the uniform rate of release, as defined above,
continues
for a prolonged period of time from about 25% to until at least about 75% and
preferably at least about 85% of the drug is released from the dosage form.
Release of
oxycodone continues thereafter for several more hours although the rate of
release is
generally slowed somewhat from the muform release rate.
[00053] By the phrase "a dosage form having a substantially zero order in
vitro
release profile" and similar phrases is meant a dosage form which overall has
substantially zero order ira vitro release kinetics, i.e., the overall ih
vitro release rate is
substantially constant over a 24 hour period. For example, for a dosage form
which has
both a controlled-release component and an initial loading dose (initial
loading
component), a substantially zero order in. vitro release profile means that
the in vitro
release rate resulting from the combined release of drug from the two
components is
substantially constant over a 24 hour period. At steady state, a dosage form
that has a
substantially zero order in vitro release profile produces an iya vivo plasma
profile that is
substantially flat as opposed to being biphasic as with the OXYCONTIN product
(see
below).
[00054] By "C" is meant the concentration of drug in the blood plasma of a
subject,
generally expressed as mass per unit volume, typically nanograms per
milliliter. For
convenience, this concentration may be referred to herein as "plasma drug
concentration" or "plasma concentration" which is intended to be inclusive of
drug
concentration measured in any appropriate body fluid or tissue. The plasma
drug
concentration at any time following drug administration is referenced as
Ctime~ as in C9h
or C24h, etc.
[00055] By "steady state" is meant the condition in which the profile of drug
present
in the blood plasma of a subj ect does not vary significantly over a prolonged
period of
time. A pattern of drug accumulation following continuous administration of a
dosage
form at constant dosing intervals eventually achieves a "steady-state" where
the plasma
concentration peaks and plasma concentration troughs are essentially unchanged
for
each dosing interval.
[00056] Persons of skill in the art appreciate that plasma drug concentrations
obtained in individual subjects will vary due to intrapatient variability in
the many
parameters affecting drug absorption, distribution, metabolism and excretion.
For this
reason, unless otherwise indicated, mean values obtained from groups of
subjects are
16
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
used herein for purposes of comparing plasma drug concentration data and for
analyzing relationships between in vitYO dosage form dissolution rates and in
vivo
plasma drug concentrations.
VII. DETAILED DESCRIPTION OF THE INVENTION
AND ITS PREFERRED EMBODIMENTS
A. DOSAGE FORMS
[00057] The present invention can be practiced using a variety of techniques
known
in the art for producing controlled-release oral dosage forms. Non-limiting
examples of
such techniques include osmotic systems, diffusion systems such as reservoir
devices
and matrix devices, dissolution systems such as encapsulated dissolution
systems
(including, for example, "tiny time pills") and matrix dissolution systems,
combination
diffusion/dissolution systems and ion-exchange resin systems as described in
Remihgtof2's Plza~naaceutical Sciences, 1990 ed., pp. 1682-1685. Oxycodone
dosage
forms that operate in accord with any of these or other approaches are
encompassed by
the present invention to the extent that the drug release characteristics
and/or the
plasma oxycodone concentration characteristics of the appended claims are
achieved by
those dosage forms either literally or equivalently.
[00058] As illustrated by the examples set forth below, particularly preferred
dosage
forms for use in the practice of the invention are osmotic dosage forms.
Osmotic
dosage forms, in general, utilize osmotic pressure to generate a driving force
for
imbibing fluid into a compartment formed, at least in part, by a semipermeable
wall
that permits free diffusion of fluid but not drug or osmotic agent(s), if
present. A
significant advantage to osmotic systems is that operation is pH-independent
and thus
continues at the osmotically determined rate throughout an extended time
period even
as the dosage form transits the gastrointestinal tract and encounters
differing
microenvironments having significantly different pH values. A review of such
dosage
forms is found in Santus and Baker, "Osmotic drug delivery: a review of the
patent
literature," Journal of Controlled Release 35 (1995) 1-21, incorporated in its
entirety by
reference herein. In particular, the following U.S. Patents, owned by ALZA
Corporation and directed to osmotic dosage forms, are each incorporated in
their
entirety herein: Nos. 3,845,770; 3,916,899; 3,995,631; 4,008,719; 4,111,202;
4,160,020; 4,327,725; 4,519,801; 4,578,075; 4,681,583; 5,019,397; and
5,156,850.
17
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[00059] Figure 1 is a perspective view of one embodiment of a controlled
release
osmotic dosage form. Dosage form 10 comprises wall 20 that surrounds and
encloses
an internal compartment (not seen in Figure 1). The internal compartment
contains a
composition comprising oxycodone, and/or one or more of its pharmaceutically
acceptable acid addition salts. Wall 20 is provided with at least one drug
delivery exit
60 for connecting the internal compartment with the exterior environment of
use.
Accordingly, following oral ingestion of dosage form 10, fluid is imbibed
through wall
20 and oxycodone andlor one or more of its pharmaceutically acceptable acid
addition
salts is released through exit 60.
[00060] While the preferred geometrical embodiment in Figure 1 illustrates a
standard biconvex shaped tablet, the geometry may embrace a capsule shaped
caplet
and other oral dosage forms.
[00061 ] Figure 2 is a cutaway view of Figure 1 showing an embodiment of a
controlled release osmotic dosage form with internal compartment 15 containing
a
single component layer referred to herein as drug layer 30, comprising drug
31, i.e., at
least oxycodone and/or one or more of its pharmaceutically acceptable acid
addition
salts, in an admixture with selected excipients adapted to provide an osmotic
activity
gradient for driving fluid from an external environment through wall 20 and
for
forming a deliverable drug formulation upon imbibition of fluid. As described
in more
detail below, the excipients may include a suitable suspending agent, also
referred to
herein as drug can-ier 32, binder 33, lubricant 34 and an osmotically active
agent,
osmagent 35. In operation, following oral ingestion of dosage form 10, the
osmotic
activity gradient across wall 20 causes gastric fluid to be imbibed through
the wall 20
thereby forming a deliverable drug formulation, i.e., a solution or
suspension, within
the internal compartment. The deliverable drug formulation is released through
exit 60
as fluid continues to enter the internal compartment. As release of the drug
formulation
occurs, fluid continues to be imbibed thereby driving continued release. In
this manner,
the drug is released in a sustained and continuous manner over an extended
time period.
[00062] Figure 3 is a cutaway view of Figure 1 with an alternate embodiment of
internal compartment 15 having a bilayer configuration. In this embodiment,
internal
compartment 15 contains a bilayered-compressed core having a first component
drug
layer 30 and a second component push layer 40. Drug layer 30, as described
above
with reference to Figure 1, comprises at least oxycodone and/or one or more of
its
18
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
pharmaceutically acceptable acid addition salts in an admixture with selected
excipients.
[00063] As described in more detail below, second component push layer 40
comprises~osmotically active component(s), but does not contain any active
agent. The
components in push layer 40 typically comprise an osmagent 42 and one or more
osmopolymers 41 having relatively large molecular weights which exhibit
swelling as
fluid is imbibed such that release of these osmopolymers through the drug
delivery
orifice 60 does not occur. Additional excipients such as binder 43, lubricant
44, ,
antioxidant 45 and colorant 46 may also be included in push layer 40. The
second
component layer is referred to herein as an expandable or a push layer since,
as fluid is
imbibed, the osmopolymer(s) swell and push against the deliverable drug
formulation
of the first component drug layer to thereby facilitate release of the drug
formulation
from the dosage form.
[00064] In operation, following oral ingestion of the dosage form 10 as shown
in
Figure 3, the osmotic activity gradient across wall 20 causes gastric fluid to
be imbibed
through wall 20 thereby forming drug layer 30 into a deliverable formulation
and
concurrently swelling the osmopolymer(s) in push layer 40. The deliverable
drug layer
30 is released through exit 60 as fluid continues to enter internal
compartment 15 and
push layer 40 continues to swell. As release of drug layer 30 occurs, fluid
continues to
be imbibed and the push layer continues to swell thereby driving continued
release. In
this manner, drug is released in a sustained and continuous manner over an
extended
time period.
[00065] Drug layer 30, as described with reference to Figures 2 and 3,
comprises
oxycodone and/or one or more of its pharmaceutically acceptable acid addition
salts in
an admixture with selected excipients. Push layer 40, as described with
reference to
Figure 3, comprises osmotically active components) but does not contain any
active
agent.
[00066] Drug layer 30 comprises a composition formed of a pharmaceutically
effective amount of oxycodone drug 31, and/or one or more of its
pharmaceutically
acceptable salts, and a Garner 32. The drug oxycodone is comprised of 4, 5-
epoxy-14-
hydroxy-3-methoxy-17-methylmorphinan-6-one possessing analgesic therapy.
Oxycodone is known in the art. The Merck Index, 11th Ed., p. 1100 (1990).
19
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[00067] The oxycodone salts are, for example, represented by one or more
members
selected from the group consisting of the following: oxycodone sulfate,
oxycodone
hydrochloride, oxycodone trifluoracetate, oxycodone thiosemicarbazone
hydrochloride,
oxycodone pentafluoropropionate, oxycodone p-nitrophenylhydrozone, oxycodone o-
methyloxine, oxycodone thiosemicarbazone, oxycodone semicarbazone, oxycodone
phenylhydroazone, oxycodone hydrazone, oxycodone hydrobromide, oxycodone
mucate, oxycodone methylbromide, oxycodone oleate, oxycodone n-oxide,
oxycodone
acetate, oxycodone phosphate dibasic, oxycodone phosphate monobasic, oxycodone
inorganic salt, oxycodone organic salt, oxycodone acetate trihydrate,
oxycodone
bis(heptafluorobutyrate), oxycodone bis(methylcarbamate), oxycodone (bis-
pentafluoropropionate), oxycodone bis(pyridine-3-carboxylate), oxycodone
bis(trifluoroacetate), oxycodone bitartrate, oxycodone chlorohydrate and
oxycodone
sulfate pentahydrate.
[00068] The dosage form and the therapeutic composition in either manufacture
can
comprise 1 to 640 mg of oxycodone drug 31 and/or oxycodone drug 31
pharmaceutically acceptable salt. More typically, loading of compound in the
dosage
forms, whether using osmotic or other controlled-release technology, will
provide doses
of compound to the subject ranging from 10 mg to 160 mg and more usually 20 mg
to
80 mg per day. Generally, if a total drug dose of more than 160 mg per day is
required,
multiple units of the dosage form may be administered at the same time to
provide the
required amount of drug. Preferably, the once-a-day dosage forms of the
present
invention comprise a dose D of oxycodone and/or one or more of its
pharmaceutically
acceptable acid addition salts that is greater than or equal to about 10 mg
and less than
or equal to about 80 mg.
[00069] For reference, immediate release oxycodone is typically administered
at a
starting dose of about 10 mg, administered in two or three doses per day. The
effective
dose range has been determined to be generally 10 mg/day- 320 mg/day.
Observations of the patient's tolerability to side effects and the need for
additional
clinical effect over the starting dose often results in the dose being
increased in
increments of 5 mglday to 80 mg/day. Concurrently with these observations,
plasma
concentrations in a subject may be determined by clinical assay to determine a
correlation between side effect tolerability, clinical effect, and blood
plasma
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
concentrations of the drug. Oxycodone plasma concentrations may range from 0.1
ng/ml to 100 ng/ml (nanograms per milliliter), more typically 4 ng/ml to 40
ng/ml.
[00070] For some dosages administered by an osmotic dosage form, it is
desirable to
modulate the viscosity of the hydrated drug layer in operation by the addition
or
reduction of salt in the formulation. Traditional systems utilizing salt in a
drug
formulation dealt with compounds exhibiting a strong common ion effect. This
strong
common ion effect at high drug loading allowed the addition of salt to
modulate the
solubility of the compound, allowing more of the salt to be released earlier
in the
delivery cycle in order to produce a zero order release rate profile. These
systems
taught incorporation of salt in high drug loading systems with little or no
salt in low
drug loading systems where a salting out effect was unnecessary.
[00071 ] It has been found that oxycodone and other similar drugs that exhibit
a weak
common ion effect are not similarly affected by salts to modulate solubility
and affect
the release rate through a salting out effect. Specifically, it has been found
that
oxycodone does not benefit from the addition of salt at higher doses, but does
benefit
from the addition of salt in the low doses. It has also been found that this
addition of
salt to the lower doses can modulate the viscosity of the hydrated drug layer
to maintain
a proper delivery for the desired release rate profile.
[00072] The amount of salt iycorporated into the drug layer of the system is
from
about 25% if using a high molecular weight polymer and low doses of drug to
zero
percent if using low molecular weight polymer and higher doses of drug.
Representatives of a salt to be incorporated into the drug composition include
sodium
chloride, potassium chloride and the like. Most preferable is sodium chloride.
Preferably, the drug layer viscosity in operation is maintained between about
SOcps and
about 100cps. In this way, products containing lower drug concentrations (5-
15%) and
higher drug concentrations (15-40%) can essentially be produced such that they
have
equivalent release functionality.
[00073] The drug layer viscosity can be attained by using any of many
hydrophilic
polymers. Examples include water-soluble cellulose polymers such as NaCMC,
HPMC, etc. or polyethylene oxide polymers such as Polyox~ or water soluble
sugars,
such as maltodextrin, sucrose, mannitol. Any physical or chemical property of
the
polymer, which could be modified to achieve the desired viscosity, is also
included in
this description.
21
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[00074] Carrier 32 may comprise a hydrophilic polymer represented by
horizontal
dashes in Figures 2 and 3. The hydrophilic polymer provides a hydrophilic
polymer
particle in the drug composition that contributes to the controlled delivery
of active
agent. Representative examples of these polymers are poly(alkylene oxide) of
100,000
to 750,000 number-average molecular weight, including polyethylene oxide),
poly(methylene oxide), poly(butylene oxide) and poly(hexylene oxide); and a
poly(carboxymethylcellulose) of 40,000 to 400,000 number-average molecular
weight,
represented by poly(alkali carboxymethylcellulose), poly(sodium
carboxymethylcellulose), poly(potassium carboxymethylcellulose) and
poly(lithium
carboxymethylcellulose). The drug composition can comprise a
hydroxypropylalkylcellulose of 9,200 to 125,000 number-average molecular
weight for
enhancing the delivery properties of the dosage form as represented by
hydroxypropylethylcellulose, hydroxypropylmethylcellulose,
hydroxypropylbutylcellulose and hydroxypropylpentylcellulose; and a
poly(vinylpyrrolidone) of 7,000 to 75,000 number-average molecular weight for
enhancing the flow properties of the dosage form. Preferred among those
polymers are
the polyethylene oxide) of 100,000 - 300,000 number average molecular weight.
Garners that erode in the gastric environment, i.e., bioerodible Garners, are
especially
preferred.
[00075] Other carriers that may be incorporated into drug layer 30 include
carbohydrates that exhibit sufficient osmotic activity to be used alone or
with other
osmagents. Such carbohydrates comprise monosaccharide, disaccharides and
polysaccharides. Representative examples include maltodextrins (i.e., glucose
polymers produced by the hydrolysis of corn starch) and the sugars comprising
lactose,
glucose, raffmose, sucrose, mannitol, sorbitol, and the like. Preferred
maltodextrins are
those having a dextrose equivalence (DE) of 20 or less, preferably with a DE
ranging
from about 4 to about 20, and often 9-20. Maltodextrin having a DE of 9-12 has
been
found most useful.
[00076] Carbohydrates described above, preferably the maltodextrins, may be
used
in the drug layer 30 without the addition of an osmagent, and obtain the
desired release
of oxycodone and/or one or more of its pharmaceutically acceptable acid
addition salts
from the dosage form, while providing a therapeutic effect over a prolonged
period of
time and up to 24 hours with once-a-day dosing.
22
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[00077] The preferred molecular weight of the polymer carrier utilized in the
drug
layer range from 100,000 mw to 300,000 mw and more preferably about 200,000
mw.
[00078] Drug layer 30 may further comprise a therapeutically acceptable vinyl
polymer binder 33 represented by vertical dashes in Figure 2 and Figure 3. The
vinyl
polymer comprises a 5,000 to 350,000 average molecular weight, represented by
a
member selected from the group consisting of poly-n-vinylamide, poly-n-
vinylacetamide, polyvinyl pyrrolidone), also known as poly-n-vinylpyrrolidone,
poly-
n-vinylcaprolactone, poly-n-vinyl-5-methyl-2-pyrrolidone, and poly-n-
vinylpyrrolidone
copolymers with a member selected from the group consisting of vinyl acetate,
vinyl
alcohol, vinyl chloride, vinyl fluoride, vinyl butyrate, vinyl laureate, and
vinyl stearate.
Dosage form 10 and the therapeutic composition can comprise 0.01 to 25 mg of
the
binder or vinyl polymer that serves as a binder. Representative of other
binders include
acacia, starch and gelatin.
[00079] Dosage form 30 may further comprise lubricant 34 represented by a wavy
line in Figures 2 and 3. The lubricant is used during manufacture to prevent
sticking to
die walls or punch faces. Typical lubricants include magnesium stearate,
sodium
stearate, stearic acid, calcium stearate, magnesium oleate, oleic acid,
potassium oleate,
caprylic acid, sodium stearyl fumarate, and magnesium palmitate. The amount of
lubricant present in the therapeutic composition can be 0.01 to 10 mg.
[00080] Drug layer 30 typically will be a dry composition formed by
compression of
the carrier and the drug as one layer and the push composition as the other
layer in
contacting relation.
[00081 ] Drug layer 30 is formed as a mixture containing oxycodone and/or one
or
more of its pharmaceutically acceptable acid addition salts and the carrier
that when
contacted with biological fluids in the enviromnent of use provides a slurry,
solution or
suspension of the compound that may be-dispensed by the action of the push
layer. The
drug layer may be formed from particles by comminution that produces the size
of the
drug and the size of the accompanying polymer used in the fabrication of the
drug
layer. The means for producing particles include granulation, spray drying,
sieving,
lyophilization, crushing, grinding, jet milling, micronizing and chopping to
produce the
intended micron particle size. The process can be performed by size reduction
equipment, such as a micropulverizer mill, a fluid energy grinding mill, a
grinding mill,
a roller mill, a hammer mill, an attrition mill, a chaser mill, a ball mill, a
vibrating ball
23
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
mill, an impact pulverizes mill, a centrifugal pulverizes, a coarse crusher
and a fine
crusher. The size of the particle can be ascertained by screening, including a
grizzly
screen, a flat screen, a vibrating screen, a revolving screen, a shaking
screen, an
oscillating screen and a reciprocating screen. The processes and equipment for
preparing drug and carrier particles are disclosed in Pharmaceutical Sciences,
Remington, 17th Ed., pp. 1585-1594 (1985); Chemical Engineers Handbook, Perry,
6th
Ed., pp. 21-13 to 21-19 (1984); Journal of Pharmaceutical Sciences, Parrot,
Vol. 61,
No. 6, pp. 813-829 (1974); and Chemical Engineer, Hixon, pp. 94-103 (1990).
[00082] Drug layer 30 may further comprise surfactants and disintegrants.
Exemplary of the surfactants are those having an HLB value of between about 10
- 25,
such as polyethylene glycol 400 monostearate, polyoxyethylene-4-sorbitan
monolaurate, polyoxyethylene-20-sorbitan monooleate, polyoxyethylene-20-
sorbitan
monopalmitate, polyoxyethylene-20-monolaurate, polyoxyethylene-40 -stearate,
sodium oleate and the like. Disintegrants may be selected from starches,
clays,
celluloses, algins and gums and crosslinked starches, celluloses and polymers.
Representative disintegrants include corn starch, potato starch,
croscarmelose,
crospovidone, sodium starch glycolate, Veegum HV, methylcellulose, agar,
bentonite,
carboxymethylcellulose, alginic acid, guar gum and the like.
[00083] Push layer 40 comprises a displacement composition in contacting
layered
arrangement with the first component drug layer 30 as illustrated in Figure 3.
Push
layer 40 comprises osmopolymer 41 that imbibes an aqueous or biological fluid
and
swells to push the drug composition through the exit means of the device. A
polymer
having suitable imbibition properties may be referred to herein as an
osmopolymer.
The osmopolymers are swellable, hydrophilic polymers that interact with water
and
aqueous biological fluids and swell or expand to a lugh degree, typically
exhibiting a 2-
50 fold volume increase. The osmopolymer can be non-crosslinked or
crosslinked, but
in a preferred embodiment are at least lightly crosslinked to create a polymer
network
that is too large and entangled to exit the dosage form. Thus, in a preferred
embodiment, the expandable composition is retained within the dosage form
during its
operative lifetime.
[00084] Push layer 40 comprises 20 to 375 mg of osmopolymer 41, represented by
"V" in Figure 3. Osmopolymer 41 in layer 40 possesses a higher molecular
weight
than osmopolymer 32 in drug layer 20.
24
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[00085] Representatives of fluid-imbibing displacement polymers comprise
members selected from poly(alkylene oxide) of 1 million to 15 million number-
average
molecular weight, as represented by polyethylene oxide), and poly(alkali
carboxymethylcellulose) of 500,000 to 3,500,000 number-average molecular
weight,
wherein the alkali is sodium, potassium or lithium. Examples of additional
polymers
for the formulation of the push-displacement composition comprise osmopolymers
comprising polymers that form hydrogels, such as Carbopol~ acidic
carboxypolymer, a
polymer of acrylic cross-linked with a polyallyl sucrose, also known as
carboxypolymethylene, and carboxyvinyl polymer having a molecular weight of
250,000 to 4,000,000; Cyanamer polyacrylarnides; cross-linked water swellable
indenemaleic anhydride polymers; Good-rite~ polyacrylic acid having a
molecular
weight of 80,000 to 200,000; Aqua-Keeps~ acrylate polymer polysaccharides
composed of condensed glucose units, such as diester cross-linked polygluran;
and the
like. Representative polymers that form hydrogels are known to the prior art
in U.S.
Patent No. 3,865,108, issued to Hartop; U.S. Patent No. 4,002,173, issued to
Manning;
U.S. Patent No. 4,207,893, issued to Michaels; and in Handbook of Common
Pol~rs,
Scott and Roff, Chemical Rubber Co., Cleveland, OH.
[00086] Push layer 40 can comprise 0 to 75 mg, and presently 5 to 75 mg of an
osmotically effective compound, osmagent 42, represented by circles in Figure
3. The
osmotically effective compounds are known also as osmagents and as osmotically
effective solutes. Osmagent 42 that may be found in the drug layer and the
push layer
in the dosage form are those which exhibit an osmotic activity gradient across
the wall
20. Suitable osmagents comprise a member selected from the group consisting of
sodium chloride, potassium chloride, lithium chloride, magnesium sulfate,
magnesium
chloride, potassium sulfate, sodium sulfate, lithitun sulfate, potassium acid
phosphate,
mannitol, urea, inositol, magnesium succinate, tartaric acid, raffmose,
sucrose, glucose,
lactose, sorbitol, inorganic salts, organic salts and carbohydrates.
[00087] Push layer 40 may further comprise a therapeutically acceptable vinyl
polymer 43 represented by triangles in Figure 3. The vinyl polymer comprises a
5,000
to 350,000 viscosity-average molecular weight, represented by a member
selected from
the group consisting of poly-n-vinylamide, poly-n-vinylacetamide, polyvinyl
pyrrolidone), also lcnown as poly-n-vinylpyrrolidone, poly-n-
vinylcaprolactone, poly-n-
vinyl-5-methyl-2-pyrrolidone, and poly-n-vinylpyrrolidone copolymers with a
member
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
selected from the group consisting of vinyl acetate, vinyl alcohol, vinyl
chloride, vinyl
fluoride, vinyl butyrate, vinyl laureate, and vinyl steaxate. Push layer can
contain 0.01
to 25 mg of vinyl polymer.
[00088] Push layer 40 may further comprise 0 to 5 mg of a nontoxic colorant or
dye
46, identified by vertical wavy lines in Figure 3. Colorant 35 includes Food
and Drug
Administration Colorant (FD&C), such as FD&C No. 1 blue dye, FD&C No. 4 red
dye,
red ferric oxide, yellow ferric oxide, titanium dioxide, carbon black, and
indigo.
[00089] Push layer 40 may further comprise lubricant 44, identified by half
circles in
Figure 3. Typical lubricants comprise a member selected from the group
consisting of
sodium stearate, potassium stearate, magnesium stearate, stearic acid, calcium
stearate,
sodium oleate, calcium palmitate, sodium laurate, sodium ricinoleate and
potassium
linoleate. The concentration of lubricant can be 0.01 to 10 mg.
[00090] Push layer 40 may further comprise an antioxidant 45, represented by
slanted dashes in Figure 3 to inhibit the oxidation of ingredients comprising
expandable
formulation 40. Push layer 40 can comprise up to 5 mg of an antioxidant.
Representative antioxidants comprise a member selected from the group
consisting of
ascorbic acid, ascorbyl palmitate, butylated hydroxyanisole, a mixture of 2
and 3
tertiary-butyl-4-hydroxyanisole, butylated hydroxytoluene, sodium
isoascorbate,
dihydroguaretic acid, potassium sorbate, sodium bisulfate, sodium
metabisulfate, sbrbic
acid, potassium ascorbate, vitamin E, 4-chloro-2,6-ditertiary butylphenol,
alpha-
tocopherol, and propylgallate.
[00091 ] Figure 4 depicts a preferred embodiment of the present invention
comprising an overcoat 50 of drug 31 on the dosage form of Figure 3. Overcoat
50 can
be a therapeutic composition comprising 0.5 to 75 mg of oxycodone 31 and/or
one or
more of its pharmaceutically acceptable acid addition salts and 0.5 to 275 mg
of a
pharmaceutically acceptable Garner selected from the group consisting of
alkylcellulose, hydroxyalkylcellulose and hydroxypropylalkylcellulose. For
example,
the overcoat can contain methylcellulose, hydroxyethylcellulose,
hydroxybutylcellulose, hydroxypropylcellulose, hydroxypropylmethylcellulose,
hydroxypropylethylcellulose and hydroxypropylbutylcellulose. Overcoat 50
provides
therapy immediately as overcoat 50 dissolves or undergoes dissolution in the
presence
of gastrointestinal fluid and concurrently therewith delivers oxycodone drug
31 and/or
26
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
one or more of its pharmaceutically acceptable acid addition salts into the
gastrointestinal tract for immediate oxycodone therapy.
[00092] Exemplary solvents suitable for manufacturing the dosage form
components
comprise aqueous or inert organic solvents that do not adversely harm the
materials
used in the system. The solvents broadly include members selected from the
group
consisting of aqueous solvents, alcohols, ketones, esters, ethers, aliphatic
hydrocarbons,
halogenated solvents, cycloaliphatics, aromatics, heterocyclic solvents and
mixtures
thereof. Typical solvents include acetone, diacetone alcohol, methanol,
ethanol,
isopropyl alcohol, butyl alcohol, methyl acetate, ethyl acetate, isopropyl
acetate, n-
butyl acetate, methyl isobutyl ketone, methyl propyl ketone, n-hexane, n-
heptane,
ethylene glycol monoethyl ether, ethylene glycol monoethyl acetate, methylene
dichloride, ethylene dichloride, propylene dichloride, carbon
tet°rachloride nitroethane,
nitropropane tetrachloroethane, ethyl ether, isopropyl ether, cyclohexane,
cyclooctane,
benzene, toluene, naphtha, 1,4-dioxane, tetrahydrofuran, diglyme, water,
aqueous
solvents containing inorganic salts such as sodium chloride, calcium chloride,
and the
like, and mixtures thereof such as acetone and water, acetone and methanol,
acetone
and ethyl alcohol, methylene dichloride and methanol, and ethylene dichloride
and
methanol.
[00093] Wall 20 is formed to be permeable to the passage of an external fluid,
such
as water and biological fluids, and it is substantially impermeable to the
passage of
oxycodone and/or one or more of its pharmaceutically acceptable acid addition
salts,
osmagent, osmopolymer, and the like. As such, it is semipermeable. The
selectively
semipermeable compositions used for forming the wall are essentially
nonerodible and
they are substantially insoluble in biological fluids during the life of the
dosage form.
[00094] Representative polymers for forming wall 20 comprise semipermeable
homopolymers, semipermeable copolymers, and the like. Such materials comprise
cellulose esters, cellulose ethers and cellulose ester-ethers. The cellulosic
polymers
have a degree of substitution (DS) of their anhydroglucose unit of from
greater than 0
up to 3, inclusive. Degree of substitution (DS) means the average number of
hydroxyl
groups originally present on the anhydroglucose unit that are replaced by a
substituting
group or converted into another group. The anhydroglucose unit can be
partially or
completely substituted with groups such as acyl, alkanoyl, alkenoyl, amyl,
alkyl,
alkoxy, halogen, carboalkyl, allcylcarbamate, alkylcarbonate, alkylsulfonate,
27
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
alkysulfamate, semipermeable polymer forming groups, and the like, wherein the
organic moieties contain from one to twelve carbon atoms, and preferably from
one to
eight carbon atoms.
[00095] The semipermeable compositions typically include a member selected
from
the group consisting of cellulose acylate, cellulose diacylate, cellulose
triacylate,
cellulose acetate, cellulose diacetate, cellulose triacetate, mono-, di- and
tri-cellulose
alkanylates, mono-, di-, and tri-alkenylates, mono-, di-, and tri-aroylates,
and the like.
Exemplary polymers include cellulose acetate having a DS of 1.8 to 2.3 and an
acetyl
content of 32 to 39.9%; cellulose diacetate having a DS of 1 to 2 and an
acetyl content
of 21 to 35%; cellulose triacetate having a DS of 2 to 3 and an acetyl content
of 34 to
44.8%; and the like. More specific cellulosic polymers include cellulose
propionate
having a DS of 1.8 and a propionyl content of 38.5%; cellulose acetate
propionate
having an acetyl content of 1.5 to 7% and an acetyl content of 39 to 42%;
cellulose
acetate propionate having an acetyl content of 2.5 to 3%, an average propionyl
content
of 39.2 to 45%, and a hydroxyl content of 2.8 to 5.4%; cellulose acetate
butyrate having
a DS of 1.8, an acetyl content of 13 to 15%, and a butyryl content of 34 to
39%;
cellulose acetate butyrate having an acetyl content of 2 to 29%, a butyryl
content of 17
to 53%, and a hydroxyl content of 0.5 to 4.7%; cellulose triacylates having a
DS of 2.6
to 3, such as cellulose trivalerate, cellulose trilamate, cellulose
tripalmitate, cellulose
trioctanoate and cellulose tripropionate; cellulose diesters having a DS of
2.2 to 2.6,
such as cellulose disuccinate, cellulose dipalmitate, cellulose dioctanoate,
cellulose
dicaprylate, and the like; and mixed cellulose esters, such as cellulose
acetate valerate,
cellulose acetate succinate, cellulose propionate succinate, cellulose acetate
octanoate,
cellulose valerate palmitate, cellulose acetate heptanoate, and the like.
Semipermeable
polymers are lcnown in U.S. Patent No. 4,077,407, and they can be synthesized
by
procedures described in Encyclo~edia of Polymer Science and Technolo~y, Vol.
3, pp.
325-354 (1964), Interscience Publishers Inc., New York, NY.
[00096] Additional semipermeable polymers for forming the outer wall 20
comprise
cellulose acetaldehyde dimethyl acetate; cellulose acetate ethylcarbamate;
cellulose
acetate methyl carbamate; cellulose dimethylaminoacetate; semipermeable
polyamide;
semipermeable polyurethanes; semipermeable sulfonated polystyrenes; cross-
linked
selectively semipermeable polymers formed by the coprecipitation of an anion
and a
canon, as disclosed in U.S. Patents Nos. 3,173,876; 3,276,586; 3,541,005;
3,541,006
28
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
and 3,546,142; semipermeable polymers, as disclosed by Loeb, et al. in U.S.
Patent No.
3,133,132; semipermeable polystyrene derivatives; semipermeable poly(sodium
styrenesulfonate); semipermeable poly(vinylbenzyltrimethylammonium chloride);
and
semipermeable polymers exhibiting a fluid permeability of 10-5 to 10-2 (cc.
mil/cm
hr.atm), expressed as per atmosphere of hydrostatic or osmotic pressure
differences
across a semipermeable wall. The polymers are known to the art in U.S. Patents
Nos.
3,845,770; 3,916,899 and 4,160,020; and in Handbook of Common Pol mers, Scott
and
Roff (1971) CRC Press, Cleveland, OH.
[00097] Wall 20 may also comprise a flux-regulating agent. The flux regulating
agent is a compound added to assist in regulating the fluid permeability or
flux through
wall 20. The flux-regulating agent can be a flux-enhancing agent or a flux-
decreasing
agent. The agent can be preselected to increase or decrease the liquid flux.
Agents that
produce a marked increase in permeability to fluid such as water are often
essentially
hydrophilic, while those that produce a marked decrease to fluids such as
water are
essentially hydrophobic. The amount of regulator in the wall when incorporated
therein generally is from about 0.01 % to 20% by weight or more. The flux
regulator
agents may include polyhydric alcohols, polyalkylene glycols,
polyalkylenediols,
polyesters of alkylene glycols, and the like. Typical flux enhancers include
polyethylene glycol 300, 400, 600, 1500, 4000, 6000 and the like; low
molecular
weight glycols such as polypropylene glycol, polybutylene glycol and
polyamylene
glycol: the polyalkylenediols such as poly(1,3-propanediol), poly(1,4-
butanediol),
poly(1,6-hexanediol), and the like; aliphatic diols such as 1,3-butylene
glycol, 1,4-
pentamethylene glycol, 1,4-hexamethylene glycol, and the like; alkylene triols
such as
glycerine, 1,2,3-butanetriol, 1,2,4-hexanetriol, 1,3,6-hexanetriol and the
like; esters
such as ethylene glycol dipropionate, ethylene glycol butyrate, butylene
glycol
dipropionate, glycerol acetate esters, and the like. Presently preferred flux
enhancers
include the group of difunctional block-copolymer polyoxyalkylene derivatives
of
propylene glycol lcnown as pluronics (BASF). Representative flux-decreasing
agents
include phthalates substituted with an alkyl or alkoxy or with both an alkyl
and alkoxy
group such as diethyl phthalate, dimethoxyethyl phthalate, dimethyl phthalate,
and
[di(2-ethylhexyl) phthalate], aryl phthalates such as triphenyl phthalate, and
butyl
benzyl phthalate; polyvinyl acetates, triethyl citrate, eudragit; insoluble
salts such as
calcium sulfate, barium sulfate, calcium phosphate, and the like; insoluble
oxides such
29
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
as titanium oxide; polymers in powder, granule and like form such as
polystyrene,
polymethylmethacrylate, polycarbonate, and polysulfone; esters such as citric
acid
esters esterified with long chain alkyl groups; inert and substantially water
impermeable fillers; resins compatible with cellulose based wall forming
materials, and
the like.
[00098] Other materials may be included in the semipermeable wall material for
imparting flexibility and elongation properties, to make wall 20 less brittle
and to
render tear strength. Suitable materials include phthalate plasticizers such
as dibenzyl
phthalate, dihexyl phthalate, butyl octyl phthalate, straight chain phthalates
of six to
eleven carbons, di-isononyl phthalte, di-isodecyl phthalate, and the like. The
plasticizers include nonphthalates such as triacetin, dioctyl azelate,
epoxidized tallate,
tri-isoctyl trimellitate, tri-isononyl trimellitate, sucrose acetate
isobutyrate, epoxidized
soybean oil, and the like. The amount of plasticizes in a wall when
incorporated therein
is about 0.01 % to 20% weight, or higher.
[00099] Pan coating may be conveniently used to provide the completed dosage
form, except for the exit orifice. In the pan coating system, the wall-forming
composition for wall 20 is deposited by successive spraying of the appropriate
wall
composition onto the compressed single or bilayered core comprising the drug
layer for
the single layer core or the drug layer and the push layer for the bilayered
core,
accompanied by tumbling in a rotating pan. A pan coater is used because of its
availability at corrunercial scale. Other techniques can be used for coating
the
compressed core. Once coated, the wall can be dried in a forced-air oven or in
a
temperature and humidity controlled oven to free the dosage form of solvents)
used in
the manufacturing. Drying conditions will be conventionally chosen on the
basis of
available equipment, ambient conditions, solvents, coatings, coating
thickness, and the
like.
[000100] Other coating techniques can also be employed. For example, the wall
or
walls of the dosage form may be formed in one technique using the air-
suspension
procedure. This procedure consists of suspending and tumbling the compressed
single
or bilayer core in a current of air and the semipermeable wall forming
composition,
until the wall is applied to the core. The air-suspension procedure is well
suited for
independently forming the wall of the dosage form. The air-suspension
procedure is
described in U.S. Patent No. 2,799,241; in J. Am. Pharm. Assoc., Vol. 48, pp.
451-459
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
(1959); and, ibid., Vol. 49, pp. 82-84 (1960). The dosage form also can be
coated with
a Wurster air-suspension coater using, for example, methylene dichloride
methanol as
a cosolvent for the wall forming material. An Aeromatic air-suspension coater
can be
used employing a cosolvent.
[000101 ] Dosage forms in accord with the present invention are manufactured
by
standard techniques. For example, the dosage form may be manufactured by the
wet
granulation technique. In the wet granulation technique, the drug and carrier
are
blended using an organic solvent, such as denatured anhydrous ethanol, as the
granulation fluid. The remaining ingredients can be dissolved in a portion of
the
granulation fluid, such as the solvent described above, and this latter
prepared solution
is slowly added to the drug blend with continual mixing in the blender. The
granulating
fluid is added until a wet blend is produced, which wet mass blend is then
forced
through a predetermined screen onto oven trays. The blend is dried for 18 to
24 hours
at 24°C to 35°C in a forced-air oven. The dried granules are
then sized. Next,
magnesium stearate, or another suitable lubricant, is added to the drug
granulation, and
the granulation is put into milling jars and mixed on a jar mill for 10
minutes. The
composition is pressed into a layer, for example, in a Manesty~ press or a
Korsch LCT
press. For a bilayered core, the drug-containing layer is pressed and a
similarly
prepared wet blend of the push layer composition, if included, is pressed
against the
drug-containing layer. The intermediate compression typically takes place
under a
force of about 50-100 newtons. Final stage compression typically takes place
at a force
of 3500 newtons or greater, often 3500-5000 newtons. The single or bilayer
compressed cores are fed to a dry coater press, e.g., Kilian~ Dry Coater
press, and
subsequently coated with the wall materials as described above.
[000102] One or more exit orifices are drilled in the drug layer end of the
dosage
form, and optional water soluble overcoats, which may be colored (e.g., Opadry
colored coatings) or clear (e.g., Opadry Clear), may be coated on the dosage
form to
provide the finished dosage form.
[000103] In another manufacture the drug and other ingredients comprising the
drug
layer are blended and pressed into a solid layer. The layer possesses
dimensions that
correspond to the internal dimensions of the area the layer is to occupy in
the dosage
form, and it also possesses dimensions corresponding to the second push layer,
if
included, for forming a contacting arrangement therewith. The drug and other
31
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
ingredients can also be blended with a solvent and mixed into a solid or
semisolid form
by conventional methods, such as ballmilling, calendering, stirring or
rollmilling, and
then pressed into a preselected shape. Next, if included, a layer of
osmopolymer
composition is placed in contact with the layer of drug in a like manner. The
layering
of the drug formulation and the osmopolymer layer can be fabricated by
conventional
two-layer press techniques. The compressed cores then may be coated with the
semipermeable wall material as described above.
[000104] Another manufacturing process that can be used comprises blending the
powdered ingredients for each layer in a fluid bed granulator. After the
powdered
ingredients are dry blended in the granulator, a granulating fluid, for
example,
poly(vinylpyrrolidone) in water, is sprayed onto the powders. The coated
powders are
then dried in the granulator. This process granulates all the ingredients
present therein
while adding the granulating fluid. After the granules are dried, a lubricant,
such as
stearic acid or magnesitun stearate, is mixed into the granulation using a
blender e.g.,
V-blender or tote blender. The granules are then pressed in the manner
described
above.
[000105] Exit 60 is provided in each dosage form. Exit 60 cooperates with the
compressed core for the uniform release of drug from the dosage form. The exit
can be
provided during the manufacture of the dosage form or during drug delivery by
the
dosage form in a fluid environment of use.
[000106] Exit 60 may include an orifice that is formed or fonnable from a
substance
or polymer that erodes, dissolves or is leached from the outer wall to thereby
form an
exit orifice. The substance or polymer may include, for example, an erodible
poly(glycolic) acid or poly(lactic) acid in the semipermeable wall; a
gelatinous
filament; a water-removable polyvinyl alcohol); a teachable compound, such as
a fluid
removable pore-former selected from the group consisting of inorganic and
organic
salt, oxide and carbohydrate.
[000107] The exit, or a plurality of exits, can be formed by leaching a member
selected from the group consisting of sorbitol, lactose, fructose, glucose,
mannose,
galactose, talose, sodium chloride, potassium chloride, sodium citrate and
mannitol to
provide a uniform-release dimensioned pore-exit orifice.
[000108] The exit can have any shape, such as round, triangular, square,
elliptical and
the like for the uniform metered dose release of a drug from the dosage form.
The
32
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
dosage form can be constructed with one or more exits in spaced-apart relation
or one
or more surfaces of the dosage form.
[000109] Drilling, including mechanical and laser drilling, through the
semipermeable
wall can be used to form the exit orifice. Such exits and equipment for
forming such
exits are disclosed in U.S. Patents Nos. 3,916,899, by Theeuwes and Higuchi
and in
U.S. Patent No. 4,088,864, by Theeuwes, et al., each of which is incorporated
in its
entirety by reference herein. It is presently preferred to utilize a single
exit orifice.
[000110] Techniques corresponding to those described above for osmotic systems
are
used for dosage forms employing other controlled-release technologies. For
example,
matrix systems are described in various of the patents relating to Purdue
Pharma's
OXYCONTIN products. See, for example, US Pat. Nos. 4,861,598; 4,970,075;
5,226,331; 5,508,042; 5,549,912; and 5,656,295. Based on the present
disclosure,
persons skilled in the art will be readily able to adapt such other controlled-
release
technologies to produce the in vit~~o and in vivo profiles of the present
invention.
B. SINGLE DOSE C",aX VALUES
[000111 ] One of the advantages of the preferred embodiments of the invention
is the
production of single dose plasma profiles that have small CmaX values.
CmaX values that are large are known to be undesirable for a variety of
reasons. For
example, high oxycodone concentrations are known to be associated with
respiratory
depression and resulting high COZ levels in the blood. See Leino et al., "Time
course
of changes in breathing pattern in morphine- and oxycodone-induced respiratory
depression," Anaesthesia, 1999, 54:835-840.
[000112] Although specific studies have not been done with oxycodone, "liking"
studies have been performed using morphine and have shown higher "liking"
values for
higher plasma morphine concentrations. See Marsch et al., "Effects of Infusion
Rate of
Intravenously Administered Morphine on Physiological, Psychomotor, and Self
Reported Measures in Hurnans," Journal of Pharmacology and,Experimental
Therapeutics, 2001, 299:1056-1065. Marsch et al. summarized their findings in
this
regard at page 1063 of their article as follows: "These results suggest that
suggestive
measures of drug liking may depend on both the rapidity and magnitude of
changes in
blood levels of the drug...." Thus, in and of itself, reducing single dose
CmaX values
represents an important contribution to the art.
33
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[000113] As discussed above, the present invention provides substantially zero
order
(SZO) release profiles. The plasma oxycodone concentration profile for an oral
controlled-release dosage form with a constant release rate of R can be
modeled using
the following equation:
~(t) - k~ X R 1 (1 _ e-ka ) - 1 (1 _ ~-ka~ ) Eq. l
where ka is an absorption rate constant, ke is an elimination rate constant,
and Vd/F is
the mean apparent volume of distribution. ke can be derived as the ratio of
CL/F to
Va/F, where CL/F is the mean apparent clearance.
[000114] The plasma oxycodone concentration after a single administration of
oxycodone oral solution, 20 mg, has been previously modeled by Mandema, J. W.,
R.
F. Kaiko, B. Oshlack, R. F. Reder and D. R. Stanski (1996). "Characterization
and
validation of a pharmacokinetic model for controlled release oxycodone,"
British
journal of clinical pharmacology 42(747-756). The parameters used in this
article are
set forth in Table 1. Also included in Table 1 are corresponding parameter
values
derived from the pharmacokinetic data of Examples 5 and 6 below.
[000115] Using the data of Example 6 and Equation 1 above, a single dose
profile
was calculated for a substantially zero order release rate. The results are
shown in
Figure 5 by the curve 100. In addition, two other release profiles were
modeled, one
having a fast-followed-by-slow release rate and the other having a slow-
followed-by-
fast release rate. The specific release rates used are set forth in Table 2.
Each of these
release profiles, as well as the constant release profile used to produce
curve 100,
released the same amount of drug in 24 hours, i.e., 80 milligrams.
[000116] The results of the simulations for the fast-followed-by-slow and slow-
followed-by-fast release rates are shown in Figure 5 by curves 102 and 104,
respectively. As can be clearly seen in this figure, each of these curves have
higher
Cmax values than curve 100. The CmaX values for curves 102 and 104 are set
forth in
Table 2. For comparison, the CmaX value for curve 100 is 46.5, i.e., 18% lower
than the
curve 102 value and 24% lower than the curve 104 value.
[000117] Although not formally proved, it is believed that the results shown
in Figure
5 will be true of all other profiles, i.e., all profiles which administer the
same amount of
drug over 24 hours but do not have a constant release rate will have a CmaX
value larger
than that achieved with a constant release rate.
34
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[000118] In accordance with the first and seventh aspects of the invention
discussed
above, Cr,~,ax for a single dose is specified to be:
3.5 x 10-4 liter 1 < CmaX/D <_ 6.8 x 10-4 liter ' Eq. 2
where D is the dose.
[000119] The specified upper and lower limits on the CmaX to-dose ratio
(CmaX/D) in
Equation (2) are based on the mean C",~ value reported in Table 8 for SZO-24
oxycodone, plus and minus the reported standard deviation for CmaX.
(Similarly, the
upper and lower limits on the AUCo_4$-to-dose ratio (AUCo_48lD) of these
aspects of the
invention, as well as of the second, third, eighth, and ninth aspects, are
based on the
mean AUCo_4$ value for SZO-24 oxycodone reported in Table 8, plus and minus
its
reported standard deviation.)
[000120] Because the data of Table 8 is for a dosage form which had a
substantially
zero order release rate, based on the modeling of Figure 5, it is believed
that the range
for the C",~/D ratio specified in Equation (2) represents the lowest possible
range of
CmaX/D ratios achievable by any oral oxycodone formulation. The provision of
dosage
forms having such low Cmax/D ratios is one of the important contributions to
the art of
the present invention.
C. PROFILES
[000121 ] As discussed above, the present invention provides in vitro
dissolution/release profiles and ifZ vivo single dose and steady state plasma
profiles for
orally-administered oxycodone and/or one or more of its pharmaceutically-
acceptable
acid addition salts.
[000122] Based on how drugs are absorbed and eliminated by the body, the shape
of a
dosage form's steady state plasma profile is linked to the shape of its single
dose plasma
profile. In particular, for oxycodone, if one lowers the single dose CmaX
value while
keeping the single dose AUC value substantially the same, the result will be a
flatter
steady state plasma profile. In terms of the language of the above quoted
passage from
Benziger et al. 1997, this means that lowering CmaX while maintaining AUC will
result
in "comparatively constant blood levels" of oxycodone. Based on Purdue
Pharma's
teachings, such blood levels should be avoided because they run the risk of
tolerance
development.
[000123] The AUCo_48/D ratio specified in the first, seventh, and other
aspects of the
invention (i.e., the specification that 7.6 x 10-3 hour/liter _< AUCo_48/D <_
16.7 x 10-3
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
hour/liter) is characteristic of how the body absorbs and eliminates
oxycodone. Thus,
because OXYCONTIN administers its incorporated dose during such time as the
dosage form is in the body, its AUCo_48/D ratio is within the range for
AUCo_~$/D ratios
specified in the first and seventh aspects of the invention. Specifically, as
shown in
Table 8, OXYCONTIN has a mean AUCo_4$/D ratio of 12.6x10-3 hour/liter ((1007.3
hr-
ng/ml)/80 mg = 12.6x10-3 hour/liter), which is within the specified range of
7.6x10-3 to
16.7x 10-3 hour/liter.
[000124] While the specified AUCo_48/D values bracket the OXYCONTIN value, the
specified upper limit on the single dose CmaX/D value, i.e., 6.8 x 10-4 liter
1 is
significantly below that of OXYCONTIN. Specifically, in connection with the
pharmacokinetic study of Example 6, CmaX for a single dose of OXYCONTIN 40mg
was found to be 41.8 ng/mL. When divided by 40mg, the result is 10.5x10-4
/liter,
which is well above the specified upper limit of 6.8x10-4 /liter of the first
and seventh
aspects of the invention.
[000125] Thus, the first and seventh aspects of the invention specify a single
dose
AUC value which brackets OXYCONTIN, but a lower Cmax~ Based on the linkage
between single dose and steady state profiles discussed above, this means that
a steady
state profile is being specified that is generally flatter than that produced
by
OXYCONT1N. Figure 16D confirms that this is precisely what is observed. As can
be
seen in this figure, the SZO-24 steady state profile (curve 150) is almost
completely flat
while the OXYCONTIN profile (curve 152) clearly oscillates.
[000126] Based on the foregoing, it is evident that the single dose profiles
specified in
the first and seventh aspects of the invention call for a dosage form which is
exactly
opposite to what Purdue Pharma has taught, namely, that one should not use a
dosage
form which produces a flat steady state profile because of the risk of
tolerance. As
discussed fully below (see Example 8), experimentally it has been found that
notwithstanding Purdue Pharma's teachings, oxycodone tolerance levels
associated with
biphasic profiles (i.e., OXYCONTIN type profiles) and flat profiles (i.e., SZO-
24 type
profiles) are, in fact, not statistically different. This is plainly contrary
to what would
have been expected based on Purdue Pharma's warnings regarding "comparatively
constant blood levels" of oxycodone.
36
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[000127] With the foregoing as background, we now turn to a specific
discussion of
the preferred ifa vivo steady state, in vivo single dose, and in vitro release
profiles of the
invention.
1. IN VIYO STEADY STATE PLASMA PROFILES
S [000128] In accordance with certain aspects of the invention, it has been
found that
effective pain management can be achieved with steady state plasma profiles
that are
sufficiently flat. As used herein, a steady state plasma profile is
sufficiently flat to
achieve the pain management benefits of the invention if the ratio of the AUC
(area
under the curve) for each quartile for the profile to the AUC for the full
profile, i.e., the
full dosing period of 24-hours, is greater than 0.18 (such a profile is
hereinafter referred
to as a ">18%/quartile steady state profile").
[000129] As is conventional, the first quartile begins at 0 hours (i.e., the
time of
administration of the dosage form) and ends at 6 hours, the second quartile
begins at 6
hours and ends at 12 hours, the third quartile begins at 12 hours and ends at
18 hours,
and the fourth quartile begins at 18 hours and ends at 24 hours. As is also
conventional, the plasma profiles are mean profiles obtained from a study
population
and the AUC values for the quartiles and for the entire profile are obtained
using the
trapezoidal method. More particularly, the AUC ratios are determined for each
individual and then those values are averaged. Samples are taken from subjects
in
accordance with a sampling scheme selected to reflect the time course of the
plasma
profile, e.g., there may be more sampling points where the profile is changing
rapidly in
time.
[000130] Preferably, the ratio of the AUC for each quartile of the profile to
the AUC
for the full profile is greater than or equal to about 0.20. Even more
preferably, the
difference in ratios between any two adjacent quartiles is less than about
0.03 and/or
the difference in ratios between any two quartiles is less than about 0.05.
Most
preferably, both these criteria are satisfied, i.e., the difference in ratios
between any two
adjacent quartiles is less than about 0.03 and the difference in ratios
between any two
quartiles is less than about 0.05.
[000131 ] As the data present below demonstrates, it has been found that
>18%/quartile steady state profiles assure efficacy within each quartile, thus
reducing
the probability of brealcthrough pain which has been a long standing problem
in pain
management using controlled-release dosage forms.
37
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
2. IN VIVO SINGLE DOSE PLASMA PROFILES
[000132] In accordance with other aspects of the invention, it has been
further found
that such desirable >18%/quartile steady state profiles are related to single
dose plasma
profiles having certain preferred characteristics. One such preferred
characteristic of
the single dose plasma profile is a mean profile shape which increases
substantially
monotonically over a period of 24 hours or more.
[000133] In certain embodiments, such substantially monotonically increasing
mean
profile comprises a first rising phase and a second phase, where the slope of
the first
phase is greater than the magnitude of the slope of the second phase, where
the slope of
a phase is defined as the slope of a best fit straight line to the portion of
the mean
profile making up the phase. For example, the slope of the first phase can be
at least
approximately 10 times the magnitude of the slope of the second phase. In
other
embodiments, the first rising phase can include a first rising subphase
followed by a
second rising subphase, where the slope of the first rising subphase is
greater than the
slope of the second rising subphase, where slopes are defined in the same
manner as for
the first and second phases.
[000134] Generally, the transition from the first phase to the second phase
occurs at
about 14 hours, e.g., between about 12 hours and about 16 hours, while the
transition
from the first subphase to the second subphase occurs at about 2 hours, e.g.,
between
about 1 hour and about 3 hours.
[000135] The single dose plasma profiles also preferably have their maximum .
concentration values ~C,nax) at a time (TmaX) which is greater than about 17
hours, more
preferably greater than about 18 hours, and most preferably greater than about
19
hours.
[000136] The single dose plasma profiles also preferably have a 12-24 hour AUC
which is greater than their 0-12 hour AUC. In particular, the ratio of the 12-
24 hour
AUC to the 0-12 hour AUC is preferably greater than about 1.5, more preferably
greater than about 1.7, and most preferably about 2Ø
[000137] To reduce the probability of the dosage form having "liking"
problems, the
single dose plasma profile preferably has a CmaX/(TmaX x dose) ratio which is
less than
about 3x10-4 hour -1 liter 1, more preferably less than about 4x10-5 hour -1
liter 1, and
most preferably less than about 3x10-5 hour -1 liter 1. In this way, the user
of the dosage
form does not achieve an early, strong bolus of oxycodone and thus is less
likely to
38
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
experience the euphoria and other effects which can lead to a liking response.
For
comparison, the commercial OXYCONTIN product, which is known to suffer from a
liking, indeed, an abuse, problem, has a C",aX/(T",~ x dose) ratio of about
4x10-4 hour -1
liter 1 for its 40 mg dosage strength.
[000138] As with the steady state profiles, the single dose profiles are mean
profiles
obtained from a study population and the sampling scheme is selected to
reflect the
time course of the single dose plasma profile. As discussed above, the slopes
are
determined from the mean profiles. However, TmaX, CmaX, ~d CmaX/(TmaX x dose)
ratios
are obtained for individual subjects and then averaged.
3. IN VITRO RELEASE PROFILES
[000139] In accordance with other aspects of the invention, it has been
further found
that the desired >18%/quartile steady state profiles are related to the ifa
vitro
dissolution/release profile of the dosage form. In particular, the in vitro
dissolution/release profile preferably comprises an initial loading dose
component and a
controlled release component.
[000140] Preferably, the ratio of the amount of oxycodone in the initial
loading dose
to the total amount of oxycodone in the dosage form is less than 0.25, more
preferably
less than 0.10, and most preferably less than or equal to 0.05. The 0.25 upper
limit on
initial loading dose ensures that the dosage form does not generate plasma
concentrations above those produced by an immediate release dosage form
administered at an equivalent daily dose, and thus the probability of the
dosage form
having "liking" problems or other adverse side effects will be no worse than
for an
immediate release product. The 0.10 and 0.05 levels should make such "liking"
and
other problems even less.
(000141 ] The controlled release component preferably has a substantially
constant in
vitro dissolution/release rate so that when combined with the initial loading
dose, the
overall dosage form has substantially zero order ira vitro release kinetics,
i.e., the
overall ira vitro release rate is substantially constant over a 24 hour
period. Figures 9
and 10 are non-limiting examples of release profiles for dosage forms which
employ a
controlled-release component and an initial loading dose and exhibit
substantially zero
order ih vitro release kinetics, while Figure 8 is an example of a release
profile for a
dosage form which achieves those kinetics with only a controlled-release
component.
39
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[000142] Preferably, the dosage form releases 70% of the dosage form's label
dose
within a period (the T~o period) of between about 15 hours to about 18 hours.
More
particularly, the dosage form preferably has a delivery dose pattern of from
0% to 20%
in 0-2 hours, 30 to 65% (preferably 33 to 63%) in 0 to 12 hours, and 80 to
100% in 0 to
24 hours, as shown schematically in Figure 6.
[000143] As is conventional, mean in vitro dissolution/release profiles are
used which
are determined by testing a sample set of dosage forms using USP apparatus 1,
2, or 7,
or comparable apparatus which may be substituted in the future. Tao values,
however,
are an average of the Tao values for the individual dosage forms tested, and
similarly
the delivery dose pattern for a dosage form is determined by averaging the
results for
the individual dosage forms tested.
D. EXAMPLES
[000144] The following non-limiting examples illustrate various of the
features of the
invention.
EXAMPLE 1
Oxycodone Hydrochloride 17 m~ Osmotic Push Pull Systems (Fast and Slow)
[000145] A dosage form adapted, designed and shaped as an osmotic drug
delivery
device was manufactured as follows: Two granulations were made by the
following
procedure: 1479 g of oxycodone hydrochloride, USP and 7351 g of polyethylene
oxide
N80 with average molecular weight of 200,000 were added to a fluid bed
granulator
bowl. Next a binder solution was prepared by dissolving 500 g of
polyvinylpyrrolidone
identified as K.29-32 in 4500 g of water. The dry materials were fluid bed
granulated by
spraying with 1800 g of binder solution. Next, the wet granulation was dried
in the
granulator to an acceptable moisture content. The two granulations were then
sized by
passing through a 7-mesh screen into the same container. Next, the granulation
was
transferred to a blender and mixed with 3.53 g of butylated hydroxytoluene as
an
antioxidant and lubricated with 88 g of magnesium stearate.
[000146] Next, a push composition was prepared as follows: first, a binder
solution
was prepared. 27.3 kg of polyvinylpyrrolidone identified as K29-32 having an
average
molecular weight of 40,000 was dissolved in 182.7 kg of water. Then, 22.4 kg
of
sodium chloride and 1.12 kg of fernc oxide were sized using a Quadro Comil
with a
21-mesh screen. Then, the screened materials and 82.52 lcg of polyethylene
oxide
(approximately 2,000,000 molecular weight) were added to a fluid bed
granulator bowl.
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
The dry materials were fluidized and mixed while 43 kg of binder solution was
sprayed
from 3 nozzles onto the powder. The granulation was dried in the fluid-bed
chamber to
an acceptable moisture level. The granulation process was repeated four times
and the
granulations were blended together during sizing. The coated granules were
sized using
a Fluid Air mill with a 7-mesh screen. The granulations were transferred to a
tote
tumbler, mixed with 224 g of butylated hydroxytoluene and lubricated with 1.12
kg
stearic acid.
[000147] Next, the oxycodone hydrochloride drug composition and the push
composition were compressed into bilayer tablets. First, 113 mg of the
oxycodone
hydrochloride composition was added to the die cavity and pre-compressed;
then, 103
mg of the push composition was added and the layers were pressed into a 5/16"
diameter round, standard concave, bilayer arrangement.
[000148] The bilayered arrangements were coated with a semi-permeable wall.
The
wall forming composition comprised 99% cellulose acetate having a 39.8% acetyl
content and 1% polyethylene glycol comprising a 3.350 viscosity-average
molecular
weight. The wall-forming composition was dissolved in an acetone:water (95:5
wt:wt)
co solvent to make a 5% solids solution. The wall-forming composition was
sprayed
onto and around the bilayered arrangements in a pan coater until approximately
20 mg
of membrane was applied to each tablet to create "fast" systems. The coating
process
was repeated and approximately 30 mg of membrane was applied to each tablet to
create "slow" systems.
[000149] Next, one 25 mil (0.64 mm) exit passageway was laser drilled through
the
semi-permeable wall to connect the drug layer with the exterior of the dosage
system.
The residual solvent was removed by drying for 48 hours as 45°C and 45%
humidity
followed by 4 hours at 45°C to remove excess moisture.
[000150] The dosage forms produced by this manufacture were designed to
deliver
l7mg of oxycodone HCI, USP from the core containing 15.8% oxycodone
hydrochloride USP, 81.68% polyethylene oxide N80 possessing a 200,000
molecular
weight, 2% polyvinylpyrrolidone possessing a 40,000 molecular weight, 0.02%
butylated hydroxytoluene, and 0.5% magnesium stearate. The push composition
comprised 73.7% polyethylene oxide comprising a 7,000,000 molecular weight,
20%
sodium chloride, 5% polyvinylpyrrolidone possessing an average molecular
weight of
40,000, 1% ferric oxide, 0.05% butylated hydroxytoluene, and 0.25% magnesium
41
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
steaxate. The semi-permeable wall comprised 99% cellulose acetate of 39.8%
acetyl
content and 1% polyethylene glycol. The dosage forms comprised one passageway,
25
mils (0.64 mm) on the center of the drug side. The final dosage forms had a
mean
release rate of 1.35 mg oxycodone hydrochloride, USP per hour (7.95 %/hr) and
0.97
mg oxycodone hydrochloride USP per hour (5.70 %/hr) for the "fast" and "slow"
systems, respectively.
[000151 ] The formulation of this example is summarized in Table 3.
EXAMPLE 2
Oxycodone Hydrochloride 20 m~ Osmotic Push Pull System
[000152] A dosage form adapted, designed and shaped as an osmotic drug
delivery
device was manufactured as follows: 1933 g of oxycodone hydrochloride, USP ,
7803 g
of polyethylene oxide N80 with average molecular weight of 200,000, and 200 g
of
polyvinylpyrrolidone identified as K29-32 having an average molecular weight
of
40,000 were added to a fluid bed granulator bowl. Next a binder solution was
prepared
by dissolving 500 g of the same polyvinylpyrrolidone in 4500 g of water. The
dry
materials were fluid bed granulated by spraying with 2000 g of binder
solution. Next,
the wet granulation was dried in the granulator to an acceptable moisture
content, and
sized by passing through a 7-mesh screen. Next, the granulation was
transferred to a
blender and mixed with 2 g of butylated hydroxytoluene as an antioxidant and
lubricated with 25 g of magnesium stearate.
[000153] Next, a push composition was prepared as follows: first, a binder
solution
was prepared. 15.6 kg of polyvinylpyrrolidone identified as K29-32 having an
average
molecular weight of 40,000 was dissolved in 104.4 kg of water. Then, 24 kg of
sodium
chloride and 1.2 kg of fernc oxide were sized using a Quadro Comil with a 21-
mesh
screen. Then, the screened materials and 88.44 kg of polyethylene oxide
(approximately 2,000,000 molecular weight) were added to a fluid bed
granulator bowl.
The dry materials were fluidized and mixed while 46.2 lcg of binder solution
was
sprayed from 3 nozzles onto the powder. The granulation was dried in the fluid-
bed
chamber to an acceptable moisture level. The coated granules were sized using
a Fluid
Air mill with a 7-mesh screen. The granulation was transferred to a tote
tumbler, mixed
with 15 g of butylated hydroxytoluene and lubricated with 294 g magnesium
stearate.
[000154] Next, the oxycodone hydrochloride drug composition and the push
composition were compressed into bilayer tablets. First, 113 mg of the
oxycodone
42
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
hydrochloride composition was added to the die cavity and pre-compressed;
then, 103
mg of the push composition was added and the layers were pressed into a 5116"
diameter round, standard concave, bilayer arrangement.
[000155] The bilayered arrangements were coated with a semi-permeable wall.
The
wall forming composition comprised 99% cellulose acetate having a 39.8% acetyl
content and 1% polyethylene glycol comprising a 3.350 viscosity-average
molecular
weight. The wall-forming composition was dissolved in an acetone:water (95:5
wt:wt)
co solvent to make a 5% solids solution. The wall-forming composition was
sprayed
onto and around the bilayered arrangements in a pan coater until approximately
37 mg
of membrane was applied to each tablet.
[000156] Next, one 40 mil (1 mm) exit passageway was laser drilled through the
semi-permeable wall to connect the drug layer with the exterior of the dosage
system.
The residual solvent was removed by drying for 48 hours as 45°C. and
45% humidity.
After drilling, the osmotic systems were dried for 4 hours at 45°C. to
remove excess
moisture.
[000157] Next, the drilled and dried systems were coated with an immediate
release
drug overcoat. The drug overcoat was an 8% solids aqueous solution containing
157.5
g of oxycodone HCI, USP and 850 g of hydroxypropyl methylcellulose possessing
an
average molecular weight of 11,200. The drug overcoat solution was sprayed
onto the
dried coated cores until an average wet coated weight of approximately 8 mg
per system
was achieved.
[000158] Next, the drug-overcoated systems were color overcoated. The color
overcoat was a 12% solids suspension of Opadry in water. The color overcoat
suspension was sprayed onto the drug overcoated systems until an average wet
coated
weight of approximately 8 mg per system was achieved.
[000159] Next, the color-overcoated systems were clear coated. The clear coat
was a
5% solids solution of Opadry in water. The clear coat solution was sprayed
onto the
color coated cores until an average wet coated weight of approximately 3 mg
per
system was achieved. Next, clear-coated systems were coated with approximately
1 g
of Carnuaba wax by dispersing the wax over the systems as they tumbled in the
pan
coater.
[000160] The dosage form produced by this manufacture was designed to deliver
1
mg of oxycodone hydrochloride USP as an immediate release from an overcoat
43
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
comprised of 15% oxycodone HCI, USP and 85% hydroxypropyl methylcellulose
followed by the controlled delivery of 19 mg of oxycodone HCI, USP from the
core
containing 17.7% oxycodone hydrochloride USP, 78.03% polyethylene oxide
possessing a 200,000 molecular weight, 4% polyvinylpyrrolidone possessing a
40,000
molecular weight, 0.02% butylated hydroxytoluene, and 0.25% magnesium
stearate.
The push composition comprised 73.7% polyethylene oxide comprising a 7,000,000
molecular weight, 20% sodium chloride, 5% polyvinylpyrrolidone possessing an
average molecular weight of 40,000, 1% fernc oxide, 0.05% butylated
hydroxytoluene,
and 0.25% magnesium stearate. The semi permeable wall comprised 99% cellulose
acetate of 39.8% acetyl content and 1% polyethylene glycol. The dosage form
comprised one passageway, 40 mils (1 mm) on the center of the drug side. The
final
dosage form contained a color overcoat, a clear overcoat and a wax coat and
had a
mean release rate of 0.93 mg oxycodone hydrochloride, USP per hour (4.66
%/hr).
[000161 ] The formulation of this example is summarized in Table 4 and is
referred to
hereinafter as the "Example 2 SZO-24 dosage form."
EXAMPLE 3
Oxycodone Hydrochloride 80 m~ Osmotic Push Pull System
[000162] A dosage form adapted, designed and shaped as an osmotic drug
delivery
device was manufactured as follows: 34.36 kg of oxycodone hydrochloride, USP ,
63.7
kg of polyethylene oxide N150 with average molecular weight of 200,000, and
0.02 kg
of ferric oxide red, were added to a fluid bed granulator bowl. Next, a binder
solution
was prepared by dissolving 5.40 kg of polyvinylpyrrolidone identified as K29-
32
having an average molecular weight of 40,000 in 49.6 kg of water. The dry
materials
were fluid bed granulated by spraying with 33.3 kg of binder solution. Next,
the wet
granulation was dried in the granulator to an acceptable moisture content, and
sized by
passing through a 7-mesh screen. The granulation was then transferred to a
blender and
mixed with 0.02 kg of butylated hydroxytoluene as an antioxidant and
lubricated with
0.25 kg of magnesium stearate.
[000163] Next, a push composition was prepared as follows: First, a binder
solution
was prepared by dissolving 7.8 kg of polyvinylpyrrolidone identified as K29-32
having
an average molecular weight of 40,000 in 52.2 kg of water. Then, 24 kg of
sodium
chloride and 1.2 lcg of fernc oxide were sized using a Quadro Comil with a 21-
mesh
screen. The sized materials and 88.5 kg of polyethylene oxide (approximately
44
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
2,000,000 molecular weight) were added to a fluid bed granulator bowl. The dry
materials were fluidized and mixed while 46.2 kg of binder solution was
sprayed from
3 nozzles onto the powder. The granulation was dried in the fluid-bed chamber
to an
acceptable moisture level. The coated granules were sized using a Fluid Air
mill with a
7-mesh screen. The granulation was transferred to a tote tumbler, mixed with
24 g of
butylated hydroxytoluene and lubricated with 300 g magnesium stearate.
[000164] Next, the oxycodone hydrochloride drug composition and the push
composition were compressed into bilayer tablets. First, 250 mg of the
oxycodone
hydrochloride composition was added to the die cavity and pre-compressed, then
192
mg of the push composition was added and the layers were pressed into a 13/32"
(1.03
cm) diameter round, standard concave, bilayer arrangement.
[000165] The bilayered arrangements were coated with a semi-permeable wall.
The
wall forming composition comprised 99% cellulose acetate having a 39.8% acetyl
content and 1% polyethylene glycol comprising a 3.350 viscosity-average
molecular
weight. The wall-forming composition was dissolved in an acetone:water (95:5
wt:wt)
solvent mixture to make a 5 % solids solution. The wall-forming composition
was
sprayed onto and around the bilayered arrangements in a pan coater until
approximately
44 mg of membrane was applied to each tablet.
[000166] Next, two 40 mil (1 mm) exit passageways were laser drilled through
the
semi-permeable wall to connect the drug layer with the exterior of the dosage
system.
The residual solvent was removed by drying for 72 hours as 45°C and 45%
humidity
followed by 4 hours at 45°C to remove excess moisture.
[000167] Next, the drilled and dried systems were coated with an immediate
release
drug overcoat. The drug overcoat was a 12% solids aqueous solution containing
1.33
kg of oxycodone HCI, USP and 7.14 lcg of OpadryTM Clear. The drug overcoat
solution
was sprayed onto the coated systems until an average wet coated weight of
approximately 27 mg per system was achieved.
[000168] Next, the drug-overcoated systems were color overcoated. The color
overcoat was a 12% solids suspension of Opadry in water. The color overcoat
suspension was sprayed onto the drug overcoated systems until an average wet
coated
weight of approximately 8 mg per system was achieved.
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[000169] Next, the color-overcoated systems were coated with approximately 100
ppm of Carnuaba wax by dispersing the wax over the systems as they tumbled in
the
pan coater.
[000170] The dosage form produced by this manufacture was designed to deliver
4
mg of oxycodone hydrochloride USP as an immediate release from an overcoat
comprised of 15% oxycodone HCI, USP and 85% OpadryTM Clear followed by the
controlled delivery of 76 mg of oxycodone HCI, USP from the core containing
32%
oxycodone hydrochloride USP, 63.73% polyethylene oxide N150 possessing a
200,000
molecular weight, 4% polyvinylpyrrolidone possessing a 40,000 molecular
weight,
0.02% butylated hydroxytoluene, and 0.25% magnesium stearate. The push
composition comprised 73.7% polyethylene oxide comprising a 7,000,000
molecular
weight, 20% sodium chloride, 5% polyvinylpyrrolidone possessing an average
molecular weight of 40,000, 1% ferric oxide, 0.05% butylated hydroxytoluene,
and
0.25% magnesium stearate. The semi permeable wall comprised of 99% cellulose
acetate of 39.8% acetyl content and 1% polyethylene glycol. The dosage form
comprised two passageways, 40 mils (1 mm) equidistant on the center of the
drug side.
The final dosage form contained a color overcoat and a wax coat and had a mean
release rate of 3.94 mg oxycodone hydrochloride, USP per hour (4.93 %/hr).
[000171 ] The formulation of this example is summarized in Table 5 and is
referred to
hereinafter as the "Example 3 SZO-24 dosage form."
Example 4
Pharmacokinetics and Pharmacodynamics of Osmotic Oxycodone Hydrochloride
r
(Fast and Slow) and Immediate Release Oxycodone Hydrochloride in Healthy
Volunteers
[000172] This study investigated the pharmacokinetics and pharmacodynamics of
the
"fast" and "slow" osmotic oxycodone HCl systems of Example 1 and immediate
release
(1R) oxycodone HCl in healthy male volunteers. In particular, this single-
center,
randomized, three-treatment, three-period, single- and multiple-dose,
crossover,
pharmacolcinetic/pharmacodynamic study compared two osmotic oxycodone HCl
formulations and IR oxycodone HCl (Oxynorm~ capsule, 5 mg supplied by Napp
Pharmaceuticals, Cambridge Science Park, Milton Rd., Cambridge, United
Kingdom)
in healthy male subj ects over four days. The pharmacodynamic portion of the
study
was single blind and utilized a VAS pain score. Eighteen subjects enrolled and
15
46
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
completed all study periods. While operating, the fast-release and slow-
release osmotic
dosage forms released oxycodone in a zero-order fashion with different
durations and
neither dosage form had an immediate-release oxycodone overcoat.
[000173] Subjects each received three treatments according to a randomly
assigned
sequence:
one 17-mg dose of the fast-release dosage form (delivered over approximately
10
hours);
~ one 17-mg dose of the slow-release dosage form (delivered over approximately
20
hours);
~ four 5-mg doses of IR oxycodone HCl (one dose at hours 0, 6, 12, and 18 of
the
study period).
[000174] The fast-release formulation produced a larger reduction in the pain
score
than either the slow-release formulation or IR oxycodone HCI. The reduction in
pain
scores with the slow-release formulation were generally comparable to those
seen with
IR oxycodone HCI.
[000175] On average, the fast-release and the slow-release formulations were
105%
and 99% bioavailable, respectively, relative to IR oxycodone HCI. The plasma
oxycodone concentration profiles for the fast and slow formulations were
consistent
with their ifa vitro release rate data.
[000176] The mean plasma oxycodone concentration profiles after a single day
dosing
are shown in Figure 15A. After a single dose administration, the mean
CmaX/(TmaX*Dose) ratio was 7x10-5 (h*Liter)-1 and 4x10-5 (h*Liter)-1 for the
fast and
slow dosage forms, respectively. The mean plasma oxycodone concentration
profiles
after repeated dosing are shown in Figure 15B. The steady state quartile AUC
values
for the formulations are set forth in Table 6.
[000177] The steady-state plasma profiles for both the q6h regimen of the IR
product
and the once daily regimen of the slow formulation were of the >18% quartile
type while
that for the once daily regimen of the fast formulation was not. Based on the
findings of
this study, the osmotic dosage form was changed to have 5% of the labeled dose
in the
overcoat to enable rapid dissolution and absorption after ingestion, and 95%
of the
labeled dose in the core for slow release over the entire dosing interval,
i.e., 24 hours.
This modified design was evaluated in a Phase I
pharmacolcinetic/pharmacodynamic
47
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
study (Example 5) and in a Phase II dose-ranging study in osteoarthritis pain
(Example
7).
Example 5
Pilot Study to Evaluate SZO-24 Oxycodone Hydrochloride Pharmacodynamics
[000178] A single-center, randomized, three-treatment, double-blind, crossover
study
was performed to compare the Example 2 SZO-24 dosage form (2x20mg), IV
morphine
(10 mg), and placebo in healthy male subjects. This study was designed to
determine
the dose of oxycodone HCl when administered by the Example 2 SZO-24 dosage
form
that provides a statistically significant pharmacodynamic response as measured
by the
cold pain test.
[000179] Twelve male subjects enrolled and received all three treatments
according to
a randomly assigned sequence:
~ IV placebo and oral placebo;
~ IV morphine infusion (10 mg over 15 min) and oral placebo;
~ Example 2 SZO-24 dosage form (2x20 mg) and IV placebo (saline).
[000180] The treatment of IV morphine was intended to serve as a positive
control
due to the successful separation of this treatment from placebo as reported
previously
(Van and Rolan 1996), however, in this study, this treatment did not
statistically
separate from placebo as measured by the cold pain test. The pupil size
remained steady
over the study period for the placebo treatment, and the pupil size changes
for both the
IV morphine and the Example 2 SZO-24 dosage form were consistent with their
respective pharmacolcinetic profiles (see Figure 11).
[000181 ] The study generated single-dose plasma oxycodone, noroxycodone, and
oxymorphone concentration profiles for the Example 2 SZO-24 dosage form (2x20
mg)
(see Figure 12 and Table 7). The mean C",ax~(Tmax*Dose) ratio for oxycodone
for this
study was 2 x10-5 (h*Liter) -1
[000182] A pharmacokinetic model consisting of the in-vitro release rate for
the
Example 2 SZO-24 dosage form and a first-order absorption, first-order
elimination
disposition model was fitted to the plasma oxycodone concentration data using
NONMEM. As the data were not sensitive to the absorption rate constant, the
absorption rate constant was set to 6.48 h-1. The population mean appaxent
clearance
(Cl/F) was 67.7 L/h and the population mean apparent volume (V/F) was 556 L.
The
mean best-fit curve underestimated the mean data during the first few hours
after
48
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
dosing as shown in Figure 13. The expected pharmacokinetic profile for IR
oxycodone
HCI, 10 mg, given every 6 hours was also simulated and is included in Figure
13. The
simulated steady-state pharmacokinetic profiles for a q6h regimen of an IR
product, a
ql2h regimen of OXYCONTIN, and a qd regimen of the Example 2 SZO-24 dosage
form are presented in Figure 14. Based on the pharmacokinetic results, this
formulation
(5% of the labeled dose in the overcoat to enable rapid dissolution and
absorption after
ingestion, and 95% of the labeled dose in the core for slow release over the
entire
dosing interval, i.e., 24 hours, was further evaluated in a Phase II clinical
study
(Example 7).
Example 6
Single- and Multiple-Dose Pharmacokinetics of
SZO-24 Oxycodone Hydrochloride and OXYCONTIN
[000183] This study was a single-center, randomized, open-label, two-
treatment, two-
period, single- and multiple-dose crossover study in healthy subjects.
Subjects received
the following treatments:
~ Treatment A - a single dose of the Example 3 SZO-24 dosage form (80 mg)
followed 72 hours later by a QD regimen of the same dosage form (80 mg for 5
days);
~ Treatment B - two doses of OXYCONTIN~, 40 mg each dose, administered 12-
hours apart followed 72 hours later by a ql2h regimen of OXYCONTIN, 40 mg for
5 days.
[000184] All subjects took 50 mg naltrexone orally starting 14 hours before
dosing
and every 12 hours during the treatment periods and 24 hours after the last
dosing day
of oxycodone.
[000185] There was a minimum washout period of 5 days but not more than 14
days
between treatment periods.
[000186] The objectives of the study were:
~ To determine the plasma oxycodone concentration profile for a single dose of
the
Example 3 SZO-24 dosage form (80 mg) and the steady-state plasma oxycodone
hydrochloride concentration profile for a QD regimen of the dosage form;
~ To compare the steady-state plasma oxycodone concentration profile for a QD
regimen of the Example 3 SZO-24 dosage form (80 mg), and that for a ql2h
regimen of OXYCONTIN.
49
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
[000187] A total of 37 subjects completed the study. The mean plasma oxycodone
concentration profiles are given in Figure 16. The mean plasma oxycodone
concentration profile after the administration of the Example 3 SZO-24 dosage
form
(80 mg) can be found in Figure 16B and the same profile is plotted with the
mean
profile after the administration of two OXYCONTIN (40 mg each) separated by 12
hours in Figure 16C. From these figures and, in particular, the 12 hour data
point for
the Example 3 SZO-24 dosage form and the standard deviation for that data
point, it
can be seen that the single dose plasma profile for the dosage forms of the
invention
satisfies the relationship:
2.7 x 10-4 liter 1 <_ C12/D _< 5.7 x 10-4 liter 1.
[000188] For comparison, using the same 37 subjects, b.i.d. OXYCONTIN dosing
was found to produce a mean C12 concentration of 15.92 ng/ml (SD --'' 6.88
ng/ml).
Dividing this mean value by 80mg, the total OXYCONTIN dose over 24 hours,
gives
2.0 x 10-4 liters 1, which is substantially below the above range for the once-
a-day
dosage forms of the invention.
[000189] The steady-state plasma concentration profiles for a once daily
regimen of
the Example 3 SZO-24 dosage form (80 mg) and twice daily dosing of OXYCONTIN,
(40 mg each) can be found in Figure 16D. PK data are summarized in Tables 8
(single
dose) and 9 (steady state).
[000190] After the single administration of the Example 3 SZO-24 dosage form,
the
mean ratio of the area under the plasma concentration profile from 0 to 12
hour to
AUC;"f was 0.24(0.07) and the mean ratio of the area under the plasma drug
concentration profile from 12 to 24 hours to that from 0 to 12 hours was
1.94(0.49).
[000191 ] A comparison of plasma oxycodone concentrations at 72 (day 3), 96
(day 4),
and 120 (day 5) hours following start of dosing during the mufti-dose period
showed
that steady state had been reached by day 4 of dosing for both treatments.
[000192] A comparison of PK parameter AUC96_120 on day 5 of the mufti-dose
period
with AUC;"f following the single dose period for the Example 3 SZO-24 dosage
form
demonstrated time-invariant kinetics for this formulation (p = 0.9).
[000193] The bioavailability for the Example 3 SZO-24 dosage form was 92%
relative to OXYCONT1N as estimated by the AUC96-iao ratio. The 90% confidence
interval of this ratio falls within the 80-125% range for the bioequivalence
criteria.
Therefore, the amount of oxycodone provided by the Example 3 SZO-24 oxycodone
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
dosage form given once-daily is bioequivalent to that of OXYCONTIN given twice-
daily in the same total daily dose. The Cm;" value for the Example 3 SZO-24
dosage
form was 121% that for OXYCONTIN, while the Cmax value for the Example 3 SZO-
24 dosage form was 78% that for OXYCONTIN. The CmaX values were significantly
different (i.e., the ratio was significantly different from 1 (p < 0.001)).
These data
demonstrate that the oxycodone plasma profile is flatter following treatment
with the
Example 3 SZO-24 dosage form as compared to treatment with OXYCONTIN.
[000194] The steady state quartile AUC values for the Example 3 SZO-24 dosage
form and OXYCONTIN are set forth in Table 10. This data demonstrate that the
Example 3 SZO-24 dosage form given once daily and OXYCONT1N given twice daily
achieved >18%/quartile steady-state plasma oxycodone concentration profiles.
Once
daily dosing, however, is more convenient for patients and more likely to lead
to better
compliance. Also, as shown in Figure 16D, the Example 3 SZO-24 dosage form
produces a steady state profile that is clearly flatter than that produced by
OXYCONTIN, which clearly continues to be biphasic.
Example 7
Phase II Clinical Study of SZO-24 Oxycodone Hydrochloride
[000195] A Phase II, two-week, placebo-controlled study using the Example 2
SZO
24 dosage form (20 mg and 2x20 mg = 40 mg) in patients with osteoarthritis
pain of the
hips and/or knees was performed. In general, 40 mg showed statistically
significant
differences from placebo in pain assessments over the two-week treatment
period,
while 20 mg was superior to placebo over the first week of treatment but less
consistently so during the second week, despite the fact that the study was
not powered
to show a statistically significant difference between the 20 mg and placebo
in either
week. The results showed the general trend that 40 mg was more effective than
20 mg,
as expected, although the two dosage strengths did not show statistically
different
results in most cases. Scores of the Brief Pain Inventory (BPI), average pain
intensity,
showed significant results for both 20 mg (p = 0.042) and 40 mg (p = 0.010) at
the last
weelc on study medication.
[000196] Results from an analysis of the overall quality of sleep indicate
that for the
mg treatment, the mean increases from baseline to last week of treatment and
was
statistically superior to placebo (p=0.0360) in improving the quality of
sleep: 2.35 vs
1.21, on a scale of 0 (Very Poor) to 10 (Excellent).
51
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
Example 8
Rat Tolerance Study
[000197] This example reports the results of experiments performed to
determine the
effect of oxycodone input patterns on tolerance development in rats.
The specific obj ective of the study was to compare the degree of
antinociceptive
tolerance developed in rats administered oxycodone hydrochloride (HCl) for a
period
of three days, either by a biphasic dosing regimen (bolus/twice-a-day) or an
SZO
dosing regimen (substantially zero order/continuous). The biphasic dosing
regimen
used subcutaneous (SC) infusion, and the SZO regimen used subcutaneously-
implanted
ALZET~ osmotic pumps. The vehicle control for the study was 0.9% saline. The
test
solutions were oxycodone HCl dissolved in saline.
[000198] The rodent tail-flick assay was used to assess analgesia
(antinociception).
This assay is a well-characterized and standard method to assess
antinociception and
tolerance to opioid drugs (Cleary 1994, D'Annour & Smith 1941). In the assay,
rodents are briefly restrained and radiant heat is applied to the tip of the
tail. The time
it takes for the animal to flick its tail is recorded; a delay in this
response compared to
pre-dose readings is indicative of antinociception.
[000199] The tail flick latency methods used in the present study were similar
to those
described previously in the literature to assess antinociception (Duttaroy &
Yoburn
1995, Nielsen et al 2000) with slight modifications from the original method
described
by D'Armour and Smith (1941). An IITC Model 33 Tail Flick Analgesia Meter was
used to apply heat to the animal's tail (IITC Life Science, Woodland Hills,
CA). The
meter was programmed with the following conditions:
(1) Active Intensity: 75% (intensity of the stimulation light during the
test);
(2) Trigger Temperature: 30°C (this temperature allows pre-warming of
the
animal's tail to allow for more uniform measurements from day-to-day
and test-to-test);
(3) Cutoff Time: 10 seconds (i.e., the length of time from the start of the
test until the unit automatically ends the test to prevent tissue damage).
[000200] The animals were briefly restrained in plexiglass restrainers and
radiant heat
was applied to the tip of the animal's tail (approximately 1-2 cm from the
tip). After
the temperature reached 30°C, the meter increased the light intensity
providing a
noxious stimulus to the animal's tail. The time in seconds for the animal to
fliclc its tail
52
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
was recorded for each animal. If the animal did not flick its tail within 10
seconds
(cutoff time), the heat stimulus was removed in order to minimize injury to
the tail.
[000201] Three pre-dose readings were taken for each animal at intervals of
approximately 5-15 minutes. For the animals used in the study, these pre-dose
readings
did not vary by more than a second for an individual animal. The average pre-
dose
readings for animals within the same test group did not vary by more than
about two
second (range = 2.02 seconds). In this way, the variability of the
measurements was
decreased and thus the dynamic range of the assay was increased.
[000202] Tail flick latency values were converted to a percentage of the
maximum
possible effect (%MPE) using the following formula:
%MPE =100 x (OL/~Lr"aX)
where:
DL = Post-dose Latency - Pre-dose Latency, and
OLmaX = Cutoff Time - Pre-dose Latency.
[000203] For the biphasic dosing regimen, the animals were subcutaneously
infused
using a computer-controlled Harvard syringe pump. The STANPUMP computer
program (STANPUMP 1998) was used to drive an infusion device to administer
test or
control solutions as two boluses, approximately 12-hours apart. The animals
had
catheters implanted subcutaneously with approximately 7 cm of PE 10 tubing.
The
tubing was secured with sutures and sterile surgical skin glue to prevent
accidental
removal of the catheter. Prior to the start of infusion, the tubing was filled
with the
infusate (saline or oxycodone solutions).
[000204] During the treatment, the animals were connected to an Instech
tethering
system, which consisted of a Covance Infusion Harness and a stainless steel
dual
channel swivel mounted on a counter-balanced lever arm attached to an Instech
MTANK cage. This tethering system allowed the rats to roam freely in their
cages
while protecting the catheters. The rat tethering system was designed to
protect the
surgically implanted catheters while providing free mobility to the rat during
delivery.
During infusion, the animals were housed singly and had free access to food
and water.
After approximately 72 hours of infusion, the tether system was disassembled,
the
suture was cut, and the catheter removed.
[000205] For each 24-hour period, the infusion regimen produced a biphasic
profile,
with two peaks (Cmax) between 2 to 4 hours and 14 and 16 hours, and two
troughs
53
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
(Cmin) at approximately 12 hours and 24 hours. The ratio of Cmax to Cmin was
between three and four.
[000206] For the SZO dosing regimen, ALZET~ osmotic pumps (Model 2ML1) were
implanted subcutaneously in the animals. The pumps were primed overnight in
0.9%
saline in an oven at 37°C in order for the pump to have reached its
steady state
pumping rate at implantation (DURECT 2003). After approximately 72 hours, the
pumps were removed. For the SZO dosing, the rats were not tethered.
[000207] Male Sprague-Dawley (SD) rats obtained from Charles River (Hollister,
CA) and weighing at least 200g were used in the experiments. Extra animals
were
employed in the biphasic dosing regimen to take account of damaged catheters,
but
only enough animals were dosed on Day +3 to replace the animals with damaged
catheters. The study was performed in compliance with the animal welfare
regulations
of 9 CFR 1-3 and the Guide for the Care arid Use of Laboratory Animals
(National
Research Council 1996).
[000208] The animals were divided into four groups and on Day -1, each group
was
further divided into six subgroups and administered 0, 0.25, 0.5, 0.75, 1 or
1.5 mg/kg
oxycodone by subcutaneous (SC) injection, respectively. Animals were tested
for
antinociception (tail-flick latency) approximately 15 minutes after injection.
On Day 0,
animals in each group were treated in accordance with Table 11.
[000209] After approximately 72 hours, the pumps infusing vehicle or oxycodone
were stopped and the catheters were removed from the animals in Groups 1 and
2, and
the ALZET~ pumps were removed from the animals in Groups 3 and 4. Between six
to
eight hours after the end of infusion, each subgroup of Groups 1-4 was
administered 0,
0.25, 0.5, 0.75, 1 or 3 mg/kg oxycodone by subcutaneous (SC) injection,
respectively.
Animals were tested for antinociception (tail-flick latency) approximately 15
minutes
after injection. For both the biphasic and SZO dosing regimens, the dose of
oxycodone
over the 72 hour (3 day) test period was on average approximately 10 mg/kg~d,
i.e., a
total of approximately 30 mg/lcg was administered over the testing period.
[000210] The results of these experiments are shown in Tables 12A and 12B, and
in
Figures 17A and 17B, where Figure 17A plots all of the data of Tables 12A and
12B,
while Figure 17B plots the Day +3 data for tail flick testing doses of 0,
0.25, 0.5, 0.75,
and 1.0 mg/kg. The curve numbers in Figures 17A and 17B correspond to the
following:
54
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
curve 154a: SZO -- Day -1/Saline Group;
curve 154b SZO -- Day +3/Saline Group;
curve 156a: SZO -- Day -llOxycodone Group;
curve 156b: SZO -- Day +3/Oxycodone Group;
curve 158a: biphasic -- Day -1/Saline Group;
curve 158b biphasic -- Day +3/Saline Group;
curve 160a: biphasic -- Day -1/Oxycodone Group;
curve 160b: biphasic -- Day +3/Oxycodone Group.
[000211 ] As can be seen most clearly in Figure 17B, the groups that had been
treated
with oxycodone for 3 days (curves 156b and 160b) had generally smaller %MPE
values
for the same tail flick testing dose than the groups that had been treated
with saline for
3 days (curves 154b and 158b), i.e., the oxycodone-treated animals had become
tolerant
to oxycodone so that the same tail flick testing dose generally had a smaller
analgesic
effect and thus produced less latency before a tail flick occurred.
[000212] Examination of the dose-effect curves suggests that not all the
curves are
likely to be modeled by the same equation. Also, the curves representing Day
+3 data
do not increase monotonically, and all four of the Day +3 effects at the 1
mg/kg test
dose are below 50% of %MPE, thus making the estimation of ED50 difficult or
with
high uncertainty even with the much higher effect observed at 3 mg/kg.
[000213] Due to these modeling difficulties, an alternate approach was taken
to obtain
a statistical measure of the tolerance. The study design had each rat
receiving the same
testing dose of oxycodone on Day -1 and Day +3, except that the animals that
received
1.5 mg/kg on Day -1, received 3.0 mg/kg on Day +3. Intuitively, the difference
between the effect of the same test on Day +3 and Day -1 should be a direct
measure of
tolerance. The data collected from rats tested for responses at 0, 0.25, 0.5,
0.75, and 1
mg/kg were thus used to perform the statistical analysis.
[000214] For these rats, the overall study design followed a (2)x(2)x(5)
format, i.e.:
(2) SZO dosing regimen versus biphasic dosing regimen
(2) oxycodone treatment versus saline treatment
(5) 0 mg/kg versus 0.25 mg/kg versus 0.5 mg/kg versus 0.75 mg/kg versus
1.0 mg/lcg tail flick testing.
[000215] The total number of rats included in the analysis of the (2)x(2)x(5)
format
was 158. The data (difference between Day +3 and Day -1) were analyzed by the
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
analysis of variance (ANOVA) method. The full variance model consisted of the
three
primary factors, their first-order interaction terms and their second order
interaction
term, namely:
dosing regimen,
3-day treatment,
tail flick testing dose,
dosing regimen x 3-day treatment,
dosing regimen x tail flick testing dose,
3-day treatment x tail flick testing dose, and
dosing regiment x 3-day treatment x tail flick testing dose.
[000216] The ANOVA analysis was performed with SAS software. None of the four
interaction teens nor the dosing regimen term in the ANOVA model was
statistically
significant (with a critical a-value at 0.05). There was a statistically
significant effect of
the 3-day treatment (p=0.0039) and the tail flick testing dose (p<0.0001).
[000217] Therefore, the ANOVA analysis concluded that the tolerance was
statistically different between rats treated with oxycodone for 3 days versus
those
treated with saline, and different between rats tested at different tail flick
testing doses,
but not statistically significantly different between rats treated with the
SZO dosing
regimen versus the biphasic dosing regimen.
[000218] Due to the lack of statistically significant interaction terms in the
full
ANOVA model, the data were further analyzed using a reduced ANOVA model
containing only the primary design factors: dosing regiment, 3-day treatment,
tail flick
testing dose. This further analysis revealed the same conclusions as the
analysis with
the full ANOVA model. The tolerance was significantly different between
oxycodone
and saline treated rats (p=0.0035) and between rats tested with different
doses of
oxycodone (p<0.0001). The tolerance, however, was again not statistically
significantly different between rats treated with the SZO dosing regimen
versus the
biphasic dosing regimen. The estimated mean tolerance difference was -10.7
%MPE
between the oxycodone and saline treated rats and -3.2 %MPE between with the
SZO
dosing regimen and the biphasic dosing regimen. The -10.7 %MPE difference was
statistically different at an a-value of 0.05, but the -3.2 %MPE value was
not.
[000219] The laclc of a statistically significantly difference between rats
treated with a
SZO dosing regimen versus a biphasic dosing regimen is in direct contrast with
the
56
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
concerns expressed in the literature that substantially zero order dosing will
be more
likely to lead to tolerance than biphasic dosing (see Benziger et al. 1997 and
Kaiko
1997 discussed above). Based on this literature, one would have expected that
the rats
treated with the SZO dosing regimen would have exhibited more tolerance at a
statistically significant level than those treated with the biphasic dosing
regimen, but no
such statistically significant difference was found.
[000220] From the foregoing, it can be seen that the invention provides dosage
forms
suitable for providing once-daily dosing of oxycodone and/or one or more its
pharmaceutically-acceptable salts for relief of moderate to severe pain in
patients
requiring an opioid for more than a few days. Potential advantages for once-a-
day
dosing over current IR and CR oxycodone formulations include improved
convenience,
better compliance, a simpler dosing regimen, and more consistent pain relief
with fewer
adverse events over a 24-hour period.
[000221 ] Although specific embodiments of the invention have been described
and
illustrated, it is to be understood that a variety of modifications which do
not depart
from the scope and spirit of the invention will be evident to persons of
ordinary skill in
the art from the foregoing disclosure.
57
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
REFERENCES
Citations for various of the documents referred to above are set forth below.
The contents of these documents, as well as those referenced elsewhere in this
specification, are incorporated herein by reference.
Benzinger et al., "A PharmacokineticlPharmacodynamic Study of Controlled-
Release
Oxycodone," Journal of Pain and Symptom Management, 1997, 13:75-82
Cleary J, Mikus G, Somogyi A, Bochner F. The Influence of Pharmacogenetics on
Opioid Analgesia: Studies with Codeine and Oxycodone in the Sprague-
I)awley/dark Agouti Rat Model. J. Pharmacol Exp Ther 1994; 271:1528-1534
D'Armour FE, Smith DL. A Method for Determining Loss of Pain Sensation. J
Pharmacol Exp Ther 1941; 72:74-79.
DURECT Corporation, 2003. ALZET Osmotic Pump Model 2MLl, Instruction and
Specification Sheet.
Duttaroy A, Yoburn BC. The Effect of Intrinsic Efficacy on Opioid Tolerance.
Anesthesiology 1995; 82;1226-1236.
Ekblom M, Hammarlund-Udenaes M, Paalzow L. Modeling of Tolerance development
and rebound Effect During Different liztravenous Administrations of Morphine
to Rats. J Pharmacol Exp Ther 1993; 266(1):244-252.
Gardmark M, Ekblom,M, Bouw R, Hammarlund-Udenaes M.. Quantifiication of the
Effects Delay and Acute Tolerance Development to Morphine in the, Rat. J
Pharmacol Exp Therap 1993; 267(5):1061-1067.
Kaiko RF. Pharmacokinetics and Pharmacodynamics of Controlled Release Opioids
1997 Acta Anaesthiol Scand 1997; 41:166-174
Nielsen CK, Ross FB, Smith MT. Incomplete, Asymmetric, and Route-Dependent
Cross-Tolerance between Oxycodone and Morphine in the Darlc Agouti Rat. J
Pharmacol Exp Ther 2000; 295(1): 91-99.
Ouellet DM-C, Pollaclc GM. A Pharmacokinetic-Prarmacodynamic Model of
Tolerance to Morphine Analgesia During Infusion in Rats. J. Pharmacokinetics
Biopharmaceutics. 1995; 23(6):531-549
58
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
Ouellet DM-C, Pollack GM. Pharmacodynamics and Tolerance Development During
Multiple Intravenous Bolus Morphine Administration in Rats. J Pharmacol Exp
Ther 1997; 281(2):713-720.
Van, F. and P. E. Rolan. The utility of the cold pain test to measure
analgesia from
intravenous morphine. Br J Clin Pharmacol. 1996; 42: 663-4.
STANPUMP User's Manual 1998
http://anesthesia.Stanford.edulpkpd/Target%20Control%20Drug%20Delivery/
STANPUMP/Forms/AllItems.htm (August 2004)
National Research Council. Guide for the Care a~r.d Use of Labo~ato~y Animals.
Washington DC: National Academy Press 1996.
59
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
TABLE 1
Parameter Mean Value
Mandema et Example 5 Example 6
al.
1996
CL/F I h- 110 67.7 80
Vd/F I 593 556 431
ka h- 4.19 6.48 4.19
Ice h- 0.186 0.122 0.186
TABLE 2
Release Rate Cmax (ng/mL)
(mglh)
0-12 h 12-24 h
Fast-Slow 5 1.67 56.4
Slow-Fast 1.67 5 60.9
TABLE 3
Push Granulation
Material % mg
Polyethylene Oxide, NF, 7000K, TG 73.73% 75.94
Povidone, USP, Ph Eur, (ff29-32) 5.00% 5.15
Sodium Chloride, USP, Ph Eur, Powder20.00% 20.6
Ma nesium Stearate, NF, Ph Eur 0.25% 0.26
BHT, FCC, Ph Eur, Milled 0.02% 0.02
Iron Oxide, Green PB-1581 1.00% 1.03
ctive Granulations
Material % mg
Ox codone H drochloride, USP 15.80% 17.00
Pol eth lene Oxide N80, TG LEO 81.68% 92.30
Povidone, IJSP, Ph Eur, I<29-32 2.00% 2.26
Ma nesium Stearate, NF, Ph Eur 0.50% 0.57
BHT, FCC, Ph Eur, Milled 0.02% 0.02
Membrane Coat: fast slow
Material % mg mg
Cellulose Acetate, NF, 398-104.95% 19.80 29.70
Pol eth lene GI col 3350, 0.05% 0.20 0.30
NF, LEO
Acetone, NF, Bulk 90.25% - -
Purified Water, USP, Ph 4.75% - -
Eur
Unit Weights: fast slow
Dru La er Wei ht m 113 113
Push La er Wei ht m 103 103
Membrane Coating Wei m 20 30
ht
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
TABLE 4
Push Granulation
Material % mg
Pol eth lene Oxide, NF, 7000K,73.73% 75.94
TG
Povidone, USP, Ph Eur, K29-325.00% 5.15
Sodium Chloride, USP, Ph 20.00% 20.6
Eur, Powder
Ma nesium Stearate, NF, Ph 0.25% 0.26
Eur
BHT, FCC, Ph Eur, Milled 0.02% 0.02
Iron Oxide, Green PB-1581 1.00% ~ 1.03
~
Active Granulations:
Material % mg
Ox codone H drochloride, 17.70% 20.00
USP
Pol eth lene Oxide N80, TG 78.03% 88.17
LEO
Povidone, USP, Ph Eur, (K29-32)4.00% 4.52
esium Stearate, NF, Ph Eur 0.25% 0.28
Ma n
_ 0.02% 0.02
BHT, FCC, Ph Eur, Milled
Membrane Coat:
Material % mg
Cellulose Acetate, NF, 398-104.95% 37.62
Pol eth lene GI col 3350, 0.05% 0.38
NF, LEO
Acetone, NF, Bulk 90.25% -
Purified Water USP Ph Eur 4.75% ~ -
Drug Coat:
Material % mg
Ox codone H drochloride, 1.50% 1
USP
HPMC 2910, USP, Ph Eur, 3c 8.50% 6
s
Purified Water, USP, Ph Eur 90.00% -
~
Color Coat:
Material % mg
O adr ~, Gra TS-009525 12.00% 8
Purified Water, USP, Ph Eur 88.00% -
~
Clear Coat:
Material % mg
O adr ~, Clear YS-1-19025-A 5.00% 3.2
Purified Water, USP, Ph 95.00% -
Eur
Carnauba Wax, NF, Powder 0.01% 0.05
Unit Weights: 20mg
Dru La er Wei ht m 113
Push La er Wei ht m 103
Membrane Coatin Wei ht m 38
Dru Overcoat Wei ht m 7
Color Overcoat Wei ht m 8
Clear Overcoat Wei ht m 3.2
61
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
TABLE 5
Push Granulation:
Material % mg
Polyethylene Oxide, NF, 7000K,73.73 141.56
TG, LEO %
Povidone, USP, Ph Eur, (K29-32)5.00% 9.60
Sodium Chloride, USP, Ph Eur, 20.00% 38.40
(Powder)
Magnesium Stearate, NF, Ph 0.25% 0.48
Eur
BHT, FCC, Ph Eur, (Milled) 0.02% 0.04
Iron Oxide, Green PB-1581 I 1.00% I 1.92
Active Granulation:
Material % mg
Oxycodone Hydrochloride, USP 32.00% 80.00
Polyethylene Oxide N 150 FP 63.71 159.28
LEO %
Povidone, USP, Ph Eur, (K29-32)4.00% 10.00
Ferric Oxide, NF, (Red) 0.02% 0.05
Magnesium Stearate, NF, Ph 0.25% 0.63
Eur
BHT, FCC, Ph Eur, (Milled) 0.02% 0.05
I
Membrane Coat:
Material % mg
Cellulose Acetate, NF, (398-10)4.95% 43.56
PEG 3350 0.05% 0.44
Acetone, NF, (Bulk) 90.25%
Purified Water, USP, Ph Eur 4.75%
Drug Coat:
Material % mg
Oxycodone Hydrochloride, USP* 1.80% 4.00
Opadry Clear YS-1-19025-A 10.20% 22.67
Purified Water, USP, Ph Eur 88.00%
Color Coat:
Material % mg
Opadry~, Red (No. 03815632) 12.00% 8.00
'Carnauba Wax, NF, (Powder) 0.01 % trace
(
62
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
TABLE 5 (continued)
Unit Weights: 80mg
Drug Layer Weight (mg) 250
Push Layer Weight (mg) 192
Membrane Coating Weight (mg) 44
Drug Overcoat Weight* (mg) 26.7
Color Overcoat Weight (mg) 8
Formulation Characteristics: 80mg
Tablet Size (in) 13/32"
Core content* (mg) 80
Drug overcoat content (mg) 4
Total drug content (mg) 84
*including 5% system overage in core
TABLE 6
Mean (SD) Ratio of AUC For Each Quartile
to AUC For the Entire (0-24 hr) Steady-State Profile
0-6 h 6-12 h 12-18 h 18-24 h
IR 5 mg 0.29(0.03)0.27(0.03)0.19(0.03)0.24(0.03)
q6h
r=ast o ~~ro o s~rn n ~nrn n ~nrn
n:~~ n5~ n:~~ n:~~
Slow 0.2010.0310.2810.0310.30(0.0210.2310.041
63
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
TABLE 7
Single-Dose Plasma Concentrations for
40 ma (Oxycodone HCI) SZO-24 Dosage Form
Cmax (nglmL) AUCinf (hxng/mL)
Oxycodone 20.92 553.2
Noroxycodone 13.12 421.2
Oxymorphone 0.35 11.67
TABLE 8
Mean (SD) Oxycodone PK Parameters Following Single Dose
SZO-24 Oxycodone OXYCONTIN
80 mg 40 mg q12h
Cmax n /mL 41.2 13.1 57.5 18.6
max h 19.4 5.1 15.1 4.4
Cmax/ Tmax x Dose 4 x 10- 2 X 10- 4 x 10 3 x 10 a
h x Liter -
~,z h 5.4 0.9 5.1 0.6
UCo_48 n /mL.h 971.4 361.7 1007.3 330.2
UC;nf n /mL.h 989.2 376.1 Not done
aThis calculation used Cmax and Tmax during the first dosing interval (0 to 12
hr).
TABLE 9
Mean (SD) Oxycodone PK Parameters Following Multi-Dose
SZO-24 Oxycodone OXYCONTIN
80 mg 40 m 12h
~%max n /mL 53.2 15.3 67.3 19.5
max f1 105.1 8.6 104.8 6.6
Cmin n /mL 29.3 12.8 21.0 7.9
min h 109.3 9.5 106.6 7.1
UC96_~zo n /mL.h988.9 296.3 1063.7 338.0
64
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
TABLE 10
Mean (SD) Ratio of AUC for Each Quartile
to AUC for the Entire (0-24 hr) Steady-State Profile
0-6 h 6-12 h 12-18 18-24
h h
SZO-24 Oxycodone0.27(0.08)0.26(0.04)0.24(0.05)0.23(0.06)
OXYCONTIN 0.30(0.02)0.19(0.02)0.29(0.03)I 0.22(0.03)
TABLE 11
Group Treatment Route Oxycodone Number
Number Dose of
mg/ kg~d)a Animals
1 Vehicle SC Infusion 0 47
s rin a um
2 Oxycodone SC Infusion 10 47
s rin a um
3 Vehicle SC ALZET 0 48
4 Ox codone SC ALZET 10 48
a Doses calculated in terms of the hydrochloride salt.
b 0.9% saline.
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
TABLE 12A
SZO Dosing
TreatmentTail Flick
Testing Dose Results1
(mglkg) (% MPE)
Day -1 Day
+3
Saline 0 0.50 6.20 -2.377.58
Saline 0.25 13.69 g,g4 7.44 11.94
Saline 0.5 21.86 13.71 8.92 11.30
Saline 0.'l5 65.33 32.06 51.0946.28
Saline 1 90.33 19.58 35.5527,g4
Saline 1.5 100.00 0.00 100.000.00
Oxycodone0 0.33 5.14 -4.75g,51
Oxycodone0.25 14.72 16.16 -2.074.18
Oxycodone0.5 33.12 1 g,58 8.48 17.38
Oxycodone0.75 60.76 31.45 7.90 11.36
Oxycodone1 80.15 36.76 23.7525.96
Oxycodone1.5~ ~ 91.44 24.22 ~ 94.3511.48
~ mean ~ SD; n=8.
23.0 mg/kg for Day +3.
66
CA 02546691 2006-05-O1
WO 2005/041968 PCT/US2004/036132
TABLE 12B
Biphasic Dosing
TreatmentTail Flick
Testing Dose Resultsl
(mglkg) (%
MPE)
Day -1 Day
+3
Saline 0 3.20 7.23 1.71 4.11
Saline 0.25 13.82 7.87 9.25 12.00
Saline 0.5 26.59 22.79 15.2212.44
Saline 0.75 73.56 28.57 48.5939.89
Saline3 1 89.52 20.25 46.001 g,96
Saline 1.5 92.48 16.05 92.9715.65
Oxycodone0 0.47 1.06 1.55 4.70
Oxycodone0.25 4.75 6.80 0.48 2.28
Oxycodone0.5 34.19 28,28 10.647,72
Oxycodone0.75 49.32 32,g9 2.26 8,35
Oxycodone1 84.61 27.43 30.7319.05
Oxycodone~ 1.5 ~ 100.00 0.00 ~ 84.8126.06
mean ~ SD; n=8, except where indicated.
~ 3.0 mg/kg for Day +3.
n=7.
67