Note: Descriptions are shown in the official language in which they were submitted.
CA 02610436 2007-11-21
CARBOXAMIDE DERIVATIVES OF PYRROLIDINE, PIPERIDINE,
AND HEXAHYDROAZEPINE FOR THE TREATMENT OF
THROMBOSIS DISORDERS
BACKGROUND OF THE INVENTION
Platelet aggregation constitutes the initial hemostatic response to curtail
bieeding induced by vascular injury. However, pathological extension of
this normal hemostatic process can lead to thrombus formation. The final,
common pathway in platelet aggregation is the binding of fibrinogen to
activated, exposed platelet glycoprotein Ilb/Ilia (GPllb/111a). Agents which
interrupt binding of fibrinogen to GPIIb/Illa, therefore, inhibit platelet
aggregatiori_ These agents are, therefore, useful in treating platelet-
mediated thrombotic disorders such as arterial and venous thrombosis,
acute myocardial infarction, unstable angina, reocclusion following
thrombolytic therapy and angioplasty, inflammation, and a variety of vaso-
occiusive disorders. The fibrinogen receptor (GPIib/Illa) is activated by
stimuli such as ADP, collagen, and thrombin exposing binding domains to
two different peptide regions of fibrinogen: a-chain Arg-Gly-Asp (RGD) and
rchain His-His-Leu-Gly-Gly-Ala-Lys-Gln-Ala-Gly-Asp-Va!
(HHLGGAKQAGDV, y400-411). Since these peptide fragments themselves
have been shown to inhibit fibrinogen binding to GPllb/Ilia, a mimetic of
these fragments would also serve as an antagonist. In fact, prior to this
invention, potent RGD-based antagonists have been revealed which inhibit
both fibrinogen :binding ta GPilb/Illa and platelet aggregation e.g., Ro-
438857 (L. Alig, J. Med. Chem. 1992,35, 4393) has an ICsp of 0.094 M
against in vitrothrombin-induced. platelet aggregation. Some of these
agents have also shown in vivo efficacy as antithrombotic agents and, in
some cases, have been used in conjunction with fibrinolytic therapy e.g., t-
PA or streptokinase, as well (J. A. Zablocki, Current Pharmaceudcal Design
1995, 1, 533). As demonstrated by the results of the pharmacological
studies described hereinafter, the compounds of the present invention show
the ability to block fibrinogen binding to isolated,GPlib/tlla (IC5o's 0.0002-
1.39 M), inhibit platelet aggregation in vitro in the presence of a variety
of
.platelet stimuli (0.019-65.0 M vs. thrombin), and furthermore, inhibit ex
vivo
platelet aggregation in animal models; Additionally, these agents exhibit
CA 02610436 2007-11-21
efficacy in animal thrombosis models as their progenitors had shown. The
compounds of the present invention show efficacy as antithrombotic agents by
virtue of their ability to prevent platelet aggregation. Additionally, because
the
compounds of this invention inhibit integrin-mediated cell-cell or cell-matrix
adhesion, they may also be useful against inflammation, bone resorption, tumor
cell metastasis, etc. (D. Cox, Drug News&Perspectives 1995, 8, 197).
DISCLOSURE OF THE INVENTION
The present invention is directed to compounds as defined in claim 1. These
platelet aggregation inhibitors are useful in treating platelet-mediated
thrombotic
disorders such as arterial and venous thrombosis, acutemyocardic infarction,
reocclusion following thrombolytic therapy and angioplasty, inflammation,
unstable angina, and a variety of vaso-occlusive disorders. These compounds
are also useful as antithrombotics used in conjunction with fibrinolytic
therapy
(e.g., t-PA or streptokinase). Pharmaceutical compositions containing such
compounds are also part of the present invention.
According to one broad aspect, there is provided a process for preparing a
compound of the formula AG4
~N
~ ,
~ HO
Ph~ ~~, '
H~ OH
AG4
2
CA 02610436 2007-11-21
comprising treating a compound of the formula AG3
I " N
OH O
Ph
H OH
AG3
with penicillin amidase.
DETAILED DESCRIPTION OF THE INVENTION
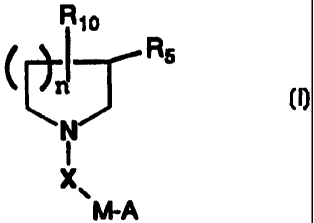
More particularly, the present invention is directed to compounds of the
following
formula (I) or (2):
~ra R~o
Rs Rs
N N
i I
X. X.
M-A MA
(1) (1)
wherein M is (CH2)m or piperidin-l-yl;
2a
CA 02610436 2007-11-21
wherein A is piperidin-2-yl, piperidin-3-yi, piperidi n-4-yl, piperazin- 1-yl,
pyrrolidi n-2-yi, pyrrolidi n-3-yi,
~
NHR2, or 81.1 wherein R9 is H, alkyl, CH(NH), CMe(NH) or acyl,
preferably R9 is hydrogen;
wherein R, is H or cycloalkyl;
wherein R2 is H, alkyl or acyl. Preferably, R 2 is hydrogen;
wherein Q is CH-heteroaryl, wherein heteroaryl is a pyridyl, thienyl,
furanyl or quinolinyl group optionally substituted with an alkyl group and R8
is H,
alkyl or aralkyl; preferably R8 is H.
wherein m is the integer 1, 2, or 3. Preferably m is 1 or 2;
wherein X is C(O), C(O)O. C(O)NH,CH2, or S02;
wherein n is the integer 1, 2, or 3;
wherein Y is CH(R3)(CH2) or (CH2)CH(R3)
wherein R3 is heteroaryl; (wherein heteroaryl is a pyridyl, thienyl, furanyl
or quinolinyl group, optionally substituted with an alkyl group);
wherein Z is CO2H, COZalkyl, SO3H, P03H2, or 5-tetrazole;
or the enantiomer or the pharmaceutically acceptable salt thereof.
Preferably, the group C(O)N(R' )YZ is attached to the ring carbon of the
central azacycie at the 3- or 4-position (4-position when larger than a five-
membered ring), and most preferably the 3-position.
3
CA 02610436 2007-11-21
As used herein, unless otherwise noted alkyl and alkoxy whether used
alone or as part of a substituent group, include straight and branched chains
having 1-8 carbons. For example, alkyl radicals include methyl, ethyl, propyl,
isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, n-pentyl, 3-(2-methyl)butyl,
2-
pentyl, 2-methylbutyl, neopentyl, n-hexyl, 2-hexyl and 2-methylpentyl. Alkoxy
radicals are oxygen ethers formed from the previously described straight or
branched chain alkyl groups. Cycloalkyl groups contain 5-8 ring carbons and
preferably 6-7 carbons.
The term "aryl", "heteroaryl" or "substituted heteroaryl" as used herein
alone or in combination with other terms indicates aromatic or heteroaromatic
groups such as phenyl,naphthyl, pyridyl, thienyl, furanyl, or quinolinyl
wherein
the substituent is an alkyl group. The term "aralkyl" means an alkyl group
substituted with an aryl group.
The term "acyl" as used herein means an organic radical having 2-6
carbon atoms derived from an organic acid by renioval of the hydroxyl group.
The compounds of the present invention may also be present in the form
of a pharmaceutically acceptable salt. The pharmaceutically acceptable salt
generally takes a form in which the nitrogen on the 1-piperidine (pyrrolidine,
piperazine) substituent is protonated with an inorganic or organic acid.
Representative organic or inorganic acids include hydrochloric, hydrobromic,
hydriodic, perchloric, sulfuric, nitric, phosphoric, acetic, propionic,
glycolic, lactic,
succinic, maleic, fumaric, malic, tartaric, citric, benzoic, mandelic,
methanesulfonic, hydroxyethanesulfonic, benezenesulfonic, oxalic, pamoic, 2-
naphthalenesulfonic, p-toluenesulfonic, cyclohexanesulfamic, salicylic,
saccharinic or trifluoroacetic.
Particularly preferred compounds of the present invention include those
compounds shown in Table I (compounds 1 to 9, 11 to 14, 16, 19, 20, 22 to 26,
28 to 30 and 32 are comparative), where "Subst" indicates the position of
4
CA 02610436 2007-11-21
attachment of the group C(O)N(R')YCO2H to the central azacycle and where the
letter "R" after the numeral "3" indicates the absolute configuration (Cahn-
ingold-
Prelog rules). Those numerals not having any configuration specified are
racemic mixtures.
CA 02610436 2007-11-21
R'
I
Q N, C02H
Y
C~)
N n
(CH2)m-Z N-R2
it ~uhs1 m Il X R1 fl2 Y. Z
1 3 2 2 C(O) H H CH(Ph)CH2 CH (comparative)
2 3 1 2 NHCO H H CH2CHMe CH (comparative)
3 3 1 2 OC(O) H H (R)-CH(CO2Me)CH2 CH (comparative)
4 3 2 1 C(O) H H CH(4-Me-Ph)CH2 CH (comparative)
5 4 2 2 C(O) H H CH(Me)CH2 cH (comparative)
6 4 2 2 c(O) H H- CH(4-CO2H-Ph)CH2 CH (comparative)
7 3 2 2 C(O) H Me CH2CH2 CH (comparative)
s See structure (comparative)
9 3 2 2 C(O) H H CH(Me3SI-ethynyl)CH2 CH (comparative)
10 See structure
11 3R .2 2 co H H CH2CH(OH) CI-i (comparative)
12 3 2 2 S02 H H CH2CH2 CH (comparative)
1.3 See structure (comparative)
1 4 3 2 2 co H Me CH(3,4-OCH20-Ph)CH2 N(Comparative)
15 3 2 2 co H Me CH(3-qulno(inyl)CH2 N
16 3R 2 2 co H H S-CH(3,4-OCH20-Ph)CH2 CH (comparative)
17 3 2 3 CO H H CH(3-quinolinyl)CH2 CH
18 3R 2 2 CO H H S-CH(3-quinolinyl)CH2 Cht
19 3R 2 2 co H H S-CH(t-butyfethynyqCH2 Ci-i (comparative)
20 3 2 2 CH2 H H S-CH(3,4-OCH2O-Ph)CH2 CH (comparative)
21 3R 2 2 co H H S-CH(3-pyridyl)CH2 C!-I
6
CA 02610436 2007-11-21
N
O o o O HO H
N H N - OH H2N N N
N H
H' o O OH
8 16
--\
O
0 0 N~., N O O I .i
H
r
rH
N H H N H Me O OH
13 14
O -1
Q
O O
H.~ ~ H
N N
N H
HO OH
16
The compounds of the invention wherein R10 is C(O)N(R1)YZ,
M is (CH2)m and A is piperidin-2-yl, piperidin-3-yl, piperidin-4-yl, piperazin-
1-yl, pyrrolidin-2-yi, pyrrolidin-3-yl or NHR2 may be prepared as shown in
Scheme AA. In this scheme nipecotic acid allyl ester (either the racemic
mixture or either separate enantiomer) may be treated with resin-bound 4-
piperidinepropionic acid in the presence of DIC/HOBT and a tertiary amine.
The allyl ester is then removed via palladium-mediated catalysis and the
iterative coupling process continued to give final product upon
saponification with potassium trimethylsilanolate (e.g., compound 1). By
analogy, urea and urethane-based replacements for the tertiary amide
(compounds 2 and 3) were prepared by reaction of solid-supported amine
(alcohol) with p-nitrophenylchloroformate and then ethyl nipecotate (S. M.
Hutchins, Tetrahedron Lett. 1994, 35, 4055).
7
CA 02610436 2007-11-21
Three-substituted 3-aminopropionic acid ester intermediates were
prepared utilizing a modified Knoevenagel procedure (Scheme AG; E. Profft,
J. Prakt. Chem. 1965, 30, 18) followed by Fischer esterification of the
carboxylic acid product (when ndt commercially-available). These
intermediates were prepared in enantiomerically-enriched form by penicillin
amidase resolution of racemic phenylacetamides such as intermediate AG3
(V. A. Soloshonok, Tetrahedron: Asymmetry 1995, 6, 1601). Here, the
undesired R-enantiomer is hydrolyzed by amidase while the desired S-
enantiomer retains the phenylacetyl group. Resolutions may also be
performed on the (-)-ephedrine salts of racemic three-substituted 3-N-Boc-
aminopropionic acids as published (J. A. Zablocki, J. Med Chem. 1995, 38,
2378). Ethyl nipecotate and ethyl isonipecotate are commercially-available
intermediates.
Synthesis of 5- and 7-membered ring analogues of nipecotamides (4
and 17, respectively) were prepared by solid-phase synthesis using methyl
pyrrolidine-3-carboxylate and methyl hexahydroazepine-3-carboxylate
intermediates for the analogous conversion of AA2 to AA3 (Scheme AA).
Methyl pyrrolidine-3-carboxylate and methyl hexahydroazepine-3-
carboxylate were prepared as published (H. Rapoport, J. Org. Chem. 1974,
39, 893). For example, N-benzyl hexahydroazepin-2-one was reacted with
lithium diisopropylamide/diethylcarbonate and this product then reduced
with lithium aluminum hydride to afford N-benzyl-3-hydroxymethyl-
hexahydroazepine. The benzyi group was removed by hydrogenolysis (H2,
Pd-C, MeOH), the nitrogen protected (di-t-butyldicarbonate/sodium
hydroxide), and the alcohol oxidized with chromium trioxide to give N-Boc-
hexahydroazepine-3-carboxylic acid. The Boc group was removed
concomitant with carboxylate esterification using HCI/MeOH to afford methyl
hexahydroazepine-3-carboxylate.
Piperazine analogs were prepared, as exemplified in Scheme AB, as
published (S. G. Gilbreath, J. Am. Chem. Soc. 1988, 110, 6172). Tetrazoles
(13) were prepared from the corresponding nitriles using
azidotrimethylsilane/dibutyitin oxide as published (Scheme AC; S. J.
Wittenberger, J. Org. Chem. 1993, 58, 4139). Here, the nitrile precursor
AC2 was prepared by standard amide bond coupling with 3-
aminopropionitrile, and reduced on the final synthetic step using platinum
8
CA 02610436 2007-11-21
dioxide-mediated hydrogenation (W. J. Hoekstra, J. Med. Chem. 1995, 38,
1582).
N-Methylpiperidine analogues can be prepared by Fmoc-based solid-
phase peptide synthesis techniques as shown in scheme AD (P. Sieber,
Tetrahedron Lett 1987, 28, 6147). The Fmoc protecting groups were
cleaved by 20% piperidine/DMF, couplings were effected using
DIC/HOBT/DMF, and final products were removed from the resin with 95%
TFA
Sulfonamide 12 was prepared as shown in Scheme AE. Intermediate
AE1 was isolated in two steps from 4-pyridineethanesulfonic acid by
hydrogenation/protection as described (J. I. DeGaw, J. Heterocyclic Chem.
1966, 3, 90), and then chlonnated using standard thionyl chloride
conditions (P. J. Hearst, Org. Syn. 1950, 30, 58) to give AE2. Intermediate
AE2 was then carried forward to final product using standard solution-phase
synthesis (W. J. Hoekstra, J. Med. Chem. 1995, 38, 1582).
Piperidinepropyl-nipecotamide 20 was prepared as shown in Scheme
AF. Ester AF1 was Boc-protected using standard Boc-ON conditions (D. S.
Tarbell, Proc. Natl. Acad. Sci. USA 1972, 69, 730), and then reduced to its
corresponding primary alcohol with DiBAL-HtTHF (E. Winterfeldt, Synthesis
1975, 617) to give intermediate AF2. This compound was converted to its
corresponding tosylate AF3 using p-TsCi (L. F. Awad, Bull. Chem. Soc. Jpn.
1986, 59, 1587). Ethyl nipecotate.was then alkylated with intermediate AF3
using standard conditions (benzene/heat; I. Seki, Chem. Pharm. Bull. Jpn.
1970, 18, 1104).
Enanttiomerically-enriched R-(-)-nipecotic acid ethyl ester was isolated
by chiral resolution of racemic material as its corresponding D-tartaric acid
salt (A. M. Akkerman, Rec. Trav. Chim. Pays-Bas 1951, 70, 899)
9
CA 02610436 2007-11-21
SCHEME AA
O O
Q~,/// (Ph3P)4Pd OH H-Nip-OC3H5
N TMSN3 N DIC/HOBT
D~ ~ DIEA, DMF
AA1 AA2
2-Chiorotrityl resin
O O O O
H O/~ / H D. ::)/N/I N (Ph3P)4Pd N OH
N TMSN3 r
DCE
AA3 AA4
~ ~
H~
H 1) -KOTMS
H2NCH(Ph)CH2CO2Me N N O M e TBF DIClHOBT N H 2) AcOH
DIEA, DMF ~ 3) TFA
AAS
O OH 0
H
N N OH
N H
H
CA 02610436 2007-11-21
'SCHEME A
1> H
C~
H H \\ ~ H N
, N O E t CH2CHCOCI N O E t Me
NMM, CH2CI2 EtOH
AB1 AB2 2) aq. NaOH
0 0
H 1) H2NCH(Ar)CHZCO2Mc
ID
C/HOBT, NMM
ON0H
M 8 2) aq. LiOH, THF
AB3
O"'\
O
0 0
~~ H H
N N H
N
Me 0 OH
14
10
=15
11
CA 02610436 2007-11-21
SCHEME AC
O O 0 0
H H
N O H H2N(CH2)2CN I~ N N/~C N
N / EDC/HOBT / H
AC1 AC2
0 0 N N=
H õ ~ N
1) n-Bu2SnO, TMSN3 N N/ "/ N
PhCH3 reflux N H
2) H2lPtO2 H
13
SCHEME AD
0 0
Fmoc, /\'~'/kO /3 piperidine /~O~ Fmoc-Nip-OH
H DMF H2N DIC/HOBT
DMF
AD1 AD2
Wang resin
0 0 0 0
Fmoc,
H ~~ H H
N N 0 piperidine = N N 0
H DMF H
AD3 AD4
1) CO2H 0 0 0
MeN
N H N/N\-'AOH
H
DIC/HOBT M~ N
DMF
2) TFA 7
12
CA 02610436 2007-11-21
SCHEME ' AE
0,0
O.,O
OH socr2 S% Ci
H-Nip-OEt
Z' N DCE reflux N Z NMM
AEI AE2
0=S'~ H O . ,O y O
1) aq. LiOH '
N OEt %N N
Z N 2) H-p-AIa-OBn . N H
EDC/HOBT Z BnO 0
AE3 AE4
O,"O O O
H
H2/Pd-C S' N H O H
aq. HCI
~N
H
IZ
20
13
CA 02610436 2007-11-21
SCHEME AF
0
1) BOC-ON
O Me Et3N O H p-TsC1
. N 2) DiBAL-H N pyridine
H THF Boc
AFI AF'Z
O
N O E t
O TS H-Nip-OEt -/"\ /,
Boc N C6H6 reflux Boc N
AF3 AF4
0
1) a.q. LiOH 0 (
H
:2c02Me ) H2NCH(Ar)CHN N ~
EDC/HOBT H ~
) aq. LiOH N
4) HC1, dioxane H. O H
5
15
14
CA 02610436 2007-11-21
SCHEME AG
N
C H 0 CH2(CO2H)2 H O PhCHZCOCI
N NH4OAc H2N OH E=sN, aq-acxt nc
EtOH reflux
AG1 AG2
I ~N ~N
/ penicillin amidase ~ /
oH o o ~ Ho
pH 7.5 (aq.) Ph~ ~V '
Ph~
N OH N OH
H H
AG3 AG4
N =2HC1
1) 6 N HCl rcflux
HO
2) HC1, MeOH ~
HzN OMe
AGS
Particulariy preferred compounds of the present invention include those
compounds shown in Tabie 1 (and Table 2), where the letter "R" after the
numeral "3" indicates the absolute configuration (Cahn-Ingoid-Prelog rules).
CA 02610436 2007-11-21
TABL.E Ii
O R2
H COzH
N
n H R
N 3
NRI
x n fli H2 BZ
22 2 H H NHCONH(3-MeOPh) (comparative)
23 2 H H NHCOOCHZPh (comparative)
24 2 H H NHCOOCHz(3-ClPh) (comparative)
2 5 2 H H NHSO2CH2Ph (comparative)
2 6 2 H H NHCONH(3,5-diMeOPh) (comparative)
27 See structure befow
2 8 2- H H NHCONH(2-naphthyl) (comparative)
29 See structure below (comparative)
3 0 2 H H NHCONHCHZCHZPh (comparative)
31 2 H 6-Me-3-pyridyt H
3 2 2 H S-Br-3-pyrictyl H (comparative)
3 3 2 CH(NH) 3-pyridyt H
CO2H
O
H ,,,H
Q o
_ U
. Ph
H H ~
N N/ :y N
~ N H H H
O N
O
NH NH
27 29
The diaminopropionic acid antagonists of the invention wherein R5 is
ONN,
C(0)NHQ(CHW)rC02R8,. R10 is H, M is piperidin-l-yl and A is Rg
16
CA 02610436 2007-11-21
may be prepared as shown in Scheme AH. Methyl N-a-Z-
diaminopropionate was acylated by HBTU-aetivated AH1, the Z group
removed by hydrogenolysis to afford AH2 (for 23 the Z group was retained),
and then the resultant primary amine reacted with the requisite isocyanate
(or alkyl chloroformate for 24, alkylsulfonyl chloride for 25) to give AH3.
The
Boc group of intermediate AH3 was removed with HCI and the resultant
secondary amine acylated with HBTU-activated AH4 to give AH5. This
material was saponified with lithium hydroxide and the Boc group removed
with HC! to give 22.
SCHEME AH
1)
CO2M e
H2N11~y O MeO NCO
NHZ
CO2H HBTUIHOBT H NCOzMe
c H H NHz
2) HZ/Pd(OH)I N NMM
Boc Boe
AH1 AH2
0
H COzMe
~ ~ 1) HCI, dioaane
H NH /
I
N ~ 2) HBTU/HOBT
8oc O H OMe C02H
AH3 BoC N AH4
O 0
H ~'COzMe 1) LiOH, aq. TI~ H NC02H
'~NH / H NH
N 2) HCI, dioxane N ~
ON OMe O N OMe
O H p H
N NH
AHS Boc 22
-15
17
CA 02610436 2007-11-21
The bipiperidine-urea based antagonists of the invention may be
prepared as shown in Scheme AJ. Intermediate AJ1 was prepared as
described in Scheme AG. AJ1 was acylated with p-nitrophenyl
chioroformate and then reacted with Boc-bipiperidine (for a synthesis, see
W. Bondinell, patent application WO 94/14776). The ester AJ2 was
saponified with lithium hydroxide and the Boc group removed with HCI to
afford 27. Substituted piperidine aldehyde intermediates such as AK2 were
prepared by lithium aluminum hydride reduction of their corresponding
nicotinic acid methyl esters (AK1) followed by oxidation with manganese
dioxide (Scheme AK). The aldehydes were then converted to p-amino acids
as shown in Scheme AG. Formamidine A1.3 was prepared as shown in
Scheme AL. Amine ALl was acylated with ethyl formimidate as described
by M. K. Scott (J. Med. Chem. 1983, 26, 534). The ester AL2 was
saponified with. 4 N HCI (RT, 20 h) to afford 33. Three-substituted P-amino
acid-type antagonists were synthesized as shown in Scheme AM. Resolved
6-methyl-pyridyi-o-amino ester was acylated with HBTU-activated AM1, and
the coupled product treated with HCI to afford amine AM2. The amine was
acylated with HBTU-activated AM4, the ester saponified, and the Boc group
removed with HCI to afford 31.
18
CA 02610436 2007-11-21
SCHEME AJ
o COzMe O CO2Me
H ,' H 1) 4-NO2PhOCOCl H %%' H
H N NMM H CN
H 2) Bocbipedine = 2 HC1 DMAP
O N
AJ1 AJZ
NBoc
O COzH
H ,%H
1) LiOH, ad. 'IBF N N
H J
2) HCI, dioxane N C
O/~N
Z~ NH
SCHEME AK
0 0
O M 1) LiAlH4, THF H
Me N 2) MnO2, CH2C12 Me N
AIC1 AK2
19
CA 02610436 2007-11-21
SCHEME AC
COz1Ule C OZR
O 0
N H
N HC(NH)OEt = HCl = N I~ N
EtOH
= HC1
O O
NH N H
X
ALl AL2 R= Me NH
33 R=H
SCHEME AM
CO2Me
1) HZN I O COZMe
H CO2H HBTU/HOBT N M8 H N N
H 1 ~
N 2) HCI, dioxane N Me
~
Boc H
AM1 AM2
1) HBTUlHOBT
02H 0 COZH
N H CJL
Boc' AM4 2) LiOH, aq. THF N Me
3) HCI, dioxane O
NH
31
CA 02610436 2007-11-21
To prepare the pharmaceutical compositions of this invention, one or more
compounds of formula (I) or satt thereof oTthe invention as the active
ingredient, is intimately admixed with a pharmaceutical carrier according to
conventional pharmaceutical compounding techniques, which carrier may
take a wide variety of forms depending of the form of preparation desired for
administration, e.g., oral or parenteral such as intramuscular. In preparing
the compositions in oral dosage form, any of the usual pharmaceutical
media may be employed. Thus, for liquid oral preparations, such as for
example, suspensions, elixirs and solutions, suitable carriers and additives
include water, glycols, oils, alcohols, flavoring agents, preservatives,
coioring agents and the like; for solid oral preparations such as, for
example,
powders, capsules, caplets, gelcaps and tablets, suitable carriers and
additives include starches, sugars, diluents, granulating agents, iubricants,
binders, disintegrating agents and the like. Because of their ease in
administration, tablets and capsules represent the most advantageous oral
dosage unit form, in which case solid pharrnaceuticai carriers are obviously
employed. If desired, tablets may be sugar coated or enteric coated by
standard techniques. For parente-rals, the carrier will usually comprise
sterile water, through other ingredients, for example, for purposes such as
aiding solubility or for preservation, may be included. Injectable
suspensions may also be prepared, in which case appropriate liquid
carriers, suspending agents and the like may be employed. The
pharmaceutical compositions herein will contain, per dosage unit, e.g.,
tablet, capsule, powder, injection, teaspoonful and the like, an amount of the
active ingredient necessary to deliver an effective dose as described above.
The pharmaceuticaf compositions herein wiif contain, per unit dosage unit,
e.g., tablet, capsule, powder, injection, suppository, teaspoonful arid the
like,
of from about 0.03 mg to 100 mg/kg (preferred 0.1-30 mg/kg) and may be
given at a dosage of from about 0.1-300 mg/kg/day (preferred 1-50
mg/kg/day). The dosages, however, may be varied depending upon the
requirement of the patients, the severity of the condition being treated and
the compound being employed. The use of either daily administration or
post-periodic dosing may be employed.
BIOLOGY
The compounds of the present invention interrupt binding of fibrinogen
to platelet glycoprotein llb/Illa (GPllb/Ilia) and thereby inhibit platelet
21
CA 02610436 2007-11-21
aggregation. Such compounds are, therefore, useful in treating piatefet-
mediated thrombotic disorders such as arteriai and venous thrombosis,
acute myocardial infarction, reocctusion following thrombolytic therapy and
angioplasty, and a variety of vaso-occiusive disorders. Because the final,
common pathway in normal platelet aggregation is the binding of fbrinogen
to activated, exposed GPlib/IIIa, inhibition- of this binding represents a
plausible antithrombotic approach. The receptor is activated by stimuli such
as ADP, collagen, and thrombin, exposing binding domains to two different
peptide regions of fibrinogen: a-chain Arg-Gly-Asp.(RGD) and ychain 400-
411. As demonstrated by the results of the pharmacological studies
described hereinafter, the compounds of the present invention show the
ability to block fibrinogen binding to isolated GPiIb/Ilia (IC5o-s 0.0002-1.39
M), inhibit platelet aggregation in vitro in the presence of a various of
platelet stimuli (0.019-65.0 M.vs. thrombin), and furthermore, inhibit ex
vivo
platelet aggregation in animal models.
IN VITRO SOLID PHASE PURIFIED GLYCOPROTEIN IIB/IlIA
BINDING ASSAY.
A 96 well lmmulon-2 microtiter plate (Dynatech-Immulon) is coated with
50 l/well of RGD-affinity purified GPltb/Ilta (effective range 0.5-10 g/mL)
in
10 mM HEPES, 150 mM NaCl, 1 mM MgCI2 at pH 7.4. The plate is covered and
incubated ovemight at 4 C. The GPIib/Illa solution is discarded and 150 lal of
5% BSA is added and incubated at RT for 1-3 h. The plate is washed
extensively with modified Tyrodes buffer. Biotiny{ated fibrinogen (25 41/well)
at 2 x final concentration is added to the wells that contain the test
compounds (25 l/wetl). The plate is covered and incubated at RT for 2-4 h.
Twenty minutes prior to incubation compfetion, one drop of Reagent A
(Vecta Stain ABC Horse Radish Peroxidase kit, Vector Laboratories, Inc.)
and one drop Reagent B are added with mixing to 5 mL modified Tyrodes
buffer mix and let stand. The ligand solution is discarded and the plate
washed (5 x 200 Uwell) with modified Tyrodes buffer. Vecta Stain HRP-
Biotin-Avidin reagent (50 l/welf, as prepared above) is added and
incubated at RT for 15 min. The Vecta Stain solution is discarded and the
wells washed (5 x 200 l/well) with modified Tyrodes buffer. Developing
buffer (10 mL of 50 mM citrate/phosphate buffer @ pH 5.3, 6 mg Q-
phenylenediamine, 6 l 30% H202; 50 Uwell) is added and incubated at
22
CA 02610436 2007-11-21
RT for 3-5 min, and then 2N H2SO4 (50 UweJi) is added. The absorbance
is read at 490 nM. The results are shown in Tables IIl and IV.
IN VITRO INHIBITION OF THROMBIN-INDUCED GEL-
FILTERED PLATELET AGGREGATION ASSAY.
The percentage of platelet aggregation iscaiculated as an increase in
light transmission of compound-treated platelet concentrate vs. control-
treated platelet concentrate. Human blood is obtained from drug free,
normal donors into tubes containing 0.13M sodium citrate. Platelet rich
plasma (PRP) is collected by centrifugation of whole blood at 200 x g for 10
min at 25 C. The PRP (5 mL) is gel filtered through Sepharose 2B (bed
voiume 50 mL), and the platelet count is adjusted to 2x107 platelets per
sample. The following constituents are added to a siliconized cuvette:
concentrated platelet filtrate and Tyrode's buffer (0.1 4M NaCI, 0.0027M KCI,
0.012M NaHC03, 0.76 mM Na2HPO4, 0.0055M glucose, 2 mg/mL BSA and
5.0mM HEPES @ pH 7.4) in an amount equal to 350 l, 50 i of 20 mM
calcium and 50 l of the test compound. Aggregation is monitored in a
BIODATA aggregometer for the 3 min following the addition of agonist
(thrombin 50 i of 1 unit/mL). The results are shown in Tables III and IV.
Trade-mark
23
CA 02610436 2007-11-21
j48LE !If
In Vitro -Aesutts
Fibrinogen Binding Platelet Aggregation'
Comnound # % Inh. (50 uM)~'~j~Q~ % Inh, (5OuM) 1G,; (uM)
1 95.0% 0.003 83.0% 3.6 (comparative)
2 93.0% 0.027 95.7% 54.0 (comparative)
3 81.0% NT 26.2% > 100 (comparative)
4 89.9% 0.121 81.0% 26.0 (comparative)
5 89.0% 0.012 100% 10.0 (comparative)
6 90.7 ' 0.197 71.2% 73.0 (comparative)
7 100% 0.006 75.6% 2.4 (comparative)
8 93.0% 0.332 94.8% 65.0 (comparative)
9 99.0% 0.002, 90.9% 0.37 (comparative)
10 91.3% 0.019 85.0% 1.6
11 79.6% 0.004 99.2% 1.55 (comparative)
12 97.0% 0.025 .88.0% 15.5 (comparative)
13 95.0% 1.39 67.0% 25.5 (comparative)
14 99.0% 0.004 91.0% 0.91 (comparative)
15 100% 0.0091 92.2% 1.9
16 .100% 0.0005 94.0% 0.028 (comparative)
17 96.0% 0.005 89.6% 0.45
18 100% 0.0002, 100% 0.019
19 99.0% 0.021 92.1% 0.079 (comparative)
20 99.0% 0.0007 89.7% 37.0 (comparative)
21 100% 0.0005 100% 0.060
Thrombin-induced aggregation of gel-fiitered platelets.
24
CA 02610436 2007-11-21
TABLE I V
In Vitro Results
Fibrinogen Bindtng Platelet Aggregation'
CQmpound # % Inh+S50 ldM1 j.Q_U (uMl % lnh, f50u 1 jQ_U fuMl
22 100% 0.0007 94.0% 0.046 (comparative)
2 3 100% 0.0003 97.0% 0.027 (comparative)
24 100% 0.0004 100% 0.018 (comparative)
25 100% 0.0003 97.0% 0.007 (comparative)
2 6 100% 0.0003 97.0% 0.016 (comparative)
27 100% 0.0006 100% 0.45
28 100% 0.0002 100% 0.17 (comparative)_
29 100% 0.068 100% 42 (comparative)
3 0 100% 0.0008 100% 0.19 (comparative)
31 100% 0.0003 100%p 0.045
3 2 100% 0.0004 100% 0.020 (comparative)
33 100% 0.0007 100% 0.30
' Thrornbin-induced aggregation of gel-filtered platelets.
EX VIVO DOG STUDY
Adult mongrel dogs (8-13 kg) were anesthetized with sodium' pentobarbitaJ (35
mg/kg, i.v.) and artificially respired. Arterial blood pressure and 'heart
rate were
measured using a Millar* catheter-tip pressure 'transducer inserted in a
femoral artery.
5 Another Millar transducer was placed in the left ventricle (LV) via a
carotid artery to
measure LV end diastolic pressure and indices of myocardial contractility. A
lead _I(
electrocardiogram was recorded from limb electrodes. Catheters were placed in
a
femoral artery.and vein to sample blood and infuse drugs, respectively.
Responses
were continuously monitored using a Modular Instruments data aquisition
system,
Arteri.al blood samples (5-9 ml) were withdrawn into tubes containing 3.8%
sodium citrate to prepare platelet rich plasma (PRP) and to determine effects
on
coagulation parameters: prothrombin time (PT) and activated partial
throrriboplastin
time (APTT). Separate blood samples (1.5 ml), were withdrawn in EDTA to
determine
hematocrit and cell counts (platelets, RBC's and white cells). Template
bleeding times
were obtained from the buccal surface using a symplate incision devise and
Whatman
filter paper.
CA 02610436 2007-11-21
Aggregation of PRP was performed using a BioData aggre,gometer. Aggregation
of whole blood used a Chronolog impedarToe aggregometer. PT and APTT were
determined on either a BioData or ACL 3000+ coagulation analyser. C',ells were
counted with a Sysmex K-1000. -
Compounds were solubilized in a small volume of dimethylformamide (DMF) and
diluted with saline to a final concentration of 10% DMF. Compounds were
administered by the intravenous route with a Harvard infusion pump. Doses was
administered over a 15 min interval at a constant rate of 0.33 rnl/min. Data
were
obtained after 'each dose and in 30 min intervals following the end of drug
administration. Oral doses were administered as aqueous solutions via synnge.
Compounds caused marked inhibition of ex vivo platelet aggregation responses.
Thus, in whole blood, the compounds inhibited collagen-stimulated (or ADP)
aggregation in doses of 0.1-10 mg/kg with marked inhibition of coliagen stimul-
ated
platelet ATP release. In PRP, the compounds also inhibited coliagen stimulated
platelet aggregaton with marked activity at 0.1-10 mg/kg. Compounds had no
measurable hemodynamic effect in doses up to 1 mg/kg, iv. The drugs produce an
increase in template bleeding time at 0.1-1 mg/kg with rapid recovery post
treatment.
No effects on coagulation (PT or APTT) were observed during treatment and
platelet,
white and R8C counts were unchanged at any dose of the compounds.
The results indicate that the compounds are broadly effective irihibitors of
platelet
aggregation ex vivo (antagonizing both collagen and ADP pathways) following iv
administration of doses ranging from 0.1-1 mg/kg or 1-10 mg/kg orally (Tables
V and
VI). The antiaggregatory effects are accompanied by increases in bleeding time
at the
higher doses. No other hemodynamic or hematologic effects are observed.
TABLE V
Ex Vlvo Dog Study Results
Intravenous Dosing Oral Dosing
Comnound #Dose Duration Qose Qrs ation'
15 1 mpk 30 min 10 mpk 120 min
16 0.1 mpk 60 min 1 mpk 60 min (comparative)
26
CA 02610436 2007-11-21
0.3 mpk NT 3 mpk >180 min
'1 a 01 mpk 30 rnin i-mpk 150 min
19 1 mpk 30 min 10 mpk 90 min (comparative)
21 0.3 mpk 150 min 1 mpk 180 min
' Indicates duration of >50% inhibition of coliagen- or ADP-induced ox
vivo platelet aggregation.
TABLE VI
Ex Vivo Dog Study Results
Intravenous Dosing Oral Dosing
~ Z= Durationf VILU Du[ation+
22 0.3 mpk 180 min 3 mpk 60 min (comparative)
23 0.1 mpk 60 min 1 mpk 180 min (comparative)
0.3 mpk NT 3 mpk 150 min
24 0.3 mpk 90 min 3 mpk 120 min (comparative)
2 5 0.3 mpk 30 min 3 mpk 60 min (comparative)
26 0.3 mpk NT 3 mpk 60 min (comparative)
2 7 0.3 mpk 60 min 3 mpk 120 min
28 0.3 mpk NT 3 mpk 120 min (comparative)
3 0 0.3 mpk 105 min 3 mpk 180 min (comparative)
3'1 0.3 mpk 120 min 3 mpk >180 min
31 0.3 mpk 60 min 3 mpk 180 min
" Indicates duration of >50% inhibition of collagen-induced ex vivo
platelet aggregation.
Compounds 1.6 and 18 have shown efficacy in a canine arteriovenous
shunt model of thrombosis in a dose-dependent fashion ( method (n
"Nipecotic Acid Derivatives As Antithrombotic Compounds," application
3 0 Serial No. 08/213772, filed March 16, 1994). For instance, compound 16
inhibits thrombus formation at 10, 30, and 100 g/kg/min cumulative doses
by iv infusion (75%, 37%, 12% of thrombus weight vs. vehicle control,
respectively). Compound 18 inhibits thrombus formation at 3, 10, and 30
g/kg/min cumulative doses by iv infusion (82%, 41%, 12% of thrombus
weight vs. vehicle control, respectively).
27
CA 02610436 2007-11-21
EXAMPLES
Protected amino acids were purchased from Aldrich Chemical or
Bachem Bioscience fnc. 2-Chlorotrityl resin and Wang resin were obtained
from Novabiochem Corp. Enantiomerically-enriched cycioalkylidene-3-
carboxyiic acid ethyl esters were isolated by chiral resolution of racemic
material as published (A. M. Akkerman, Rec. Trav. Chim. Pays-Bas 1951,
70, 899). All other chemicals were purchased from Aldrich Chemical
Company, Inc. Final product acid addition salts can be converted to free
bases by basic ion exchange chromatography. High field 1 H NMR spectra
*
were recorded on a BRUK8R AC-360 spectrometer at 360 MHz, and coupling
constants are given in Herz. Melting points were determined on a MEL-TEMPl,
!k melting point apparatus and are uncorrected. Microanalyses were :
performed at Robertson Microlit Laboratories, Inc., Madison, New Jersey. In
those cases where the product is obtained as a salt, the free base is
obtained by methods known to those skilled in the art, e.g. by basic ion
exchange purification. In the Examples and throughout this application, the
following abbreviations have the meanings recited hereinafter.
Bn or Bzl = BenZyi
Boc = t-Butoxycarbonyl
BOC-ON = 2-(t-Butoxycarbonyloxyimino)-2-phenylacetonitrile
BOP-Cl = Bis(2-oxo-3-oxazolidinyl)phosphinic chloride
CP = compound
DCE = 1,2-Dichioroethane
DCM = Dichloromethane
DIBAL-H = Diisobutyialuminum hydride
DIC = Diisopropyicarbodiimide
DIEA = Diisopropylethylamine
DMAP = 4-Dimethylaminopyhdine
DMF = N,. N-Dimethyiformamide
EDC = Ethyl dimethylaminopropylcarbodiimide
EDTA = Ethylenediaminetetraacetic acid
Et20 = Diethyl ether
HBTU = 2-(1 H-Benzotriazole-1=yl)-1,1,3,3-tetramethyluronium
hexafluorophosphate
HOBT = Hydroxybenzotriazole
i-Pr = Isopropyl
R
Trade-mark
28
CA 02610436 2007-11-21
KOTMS = Potassium trimethylsilanolate
NMM = N-Methylmorpholine
Nip = Nipecotyl (uniess noted otherwise, racemic at 3-position)
NT = not tested
PPT = precipitate
PTSA = p-Toluenesuffonic acid
RT = room temperature
TFA = Trifluoroacetic acid
TMSN3 = Azidotrimethylsilane
Z = Benzyloxycarbonyl
Allyl a-(4-pjperidine)propionate = HCI (AA1 nrecursor)
To a mixture of 3-(4-pyridine)acryiic acid (10.0 g, 0.066 mol) and aqueous
HCI (2.0 N, 50 mL) under a blanket of nitrogen was added platinum (tY)
oxide (0.54 g). This mixture was hydrogenated at 50 psi and RT for 21 h,
filtered through CeliteMand evaporated to give 3-(4-piperidine)propionic acid
= HCI as a white powder (12.9 g, 99%)._ This powder was treated with allyl
aicohol (50 mL) and warmed at 50 C for 2 h. This solution was cooled to RT,
evaporated to ca. 10 mL volume, and diluted with Et20 (250 mL). The
resultarit precipitate was collected and washed with Et20 to afford a white
powder (14.5 g, 94%): 1H NMR (DMSO-d6) S 8.7-9.1 (m, 2 H), 5.9 (m, 1- H),
5.25 (dd, J=7, 15, 2 H), 4.53 (d, J=4, 2 H), 3.21 (d, J=8, 2 H), 2.74 (t, J=7,
2
H), 2.35 (t, J=4, 2 H), 1.72 (d, J=8, 2 H), 1.5 (m, 3 H), 1.3 (m, 2 H); MS m/e
198 (Ml-i+).
Methyl (S)-3-amino-3-f3-ovridvll_arQRionate = 2HCI fAG51
Phenylacetamide intermediate AG3 was prepared using standard methods
as shown in Scheme AG (E. Profft, J. Rrakt Chem. 1965, 30, 18). A mixture
of AG1 (0.47 rnol), EtOH (100 mL), NH4OAc (0.47 mol), and malonic acid
(0.70 mol) was heated at reflux for 6 h, cooled, and filtered. The white solid
was washed with EtOH and MaOH and dried. This solid was dissolved in
2:1 acetone/water (360 mL), treated with triethylamine (0.72 mol) and
phenylacetyl chloride (0.36 mol), and stirred for 22 h. The mixture was
evaporated and the residue dissolved in water (500 mL) and adjusted to pH
29
CA 02610436 2007-11-21
12 (1 N NaOH). The aqueous layer was adjusted to pH 2 (conc. HC(),
extracted with EtZ0, and evaporated to a white foam. The foam was purified
by silica gel chromatography (10% MeOH/DCM) to give AG3. A solution of
compound AG3 (0.22 moi) in water (600 mL) at RT was adjusted to pH 7.5
using KOH (3.0 N) and treated with penicillin amidase (91520 unfts, Sigma).
This mixture was stirred for 47 h, acidified to pH 1 with HCf (conc), and the
resultant ppt filtered through Celite. The filtrate was extracted with EtZO
(3x300 mL), concentrated in vacuo, and treated with MeOH/conc. NH4OH
(9:1). This product-containing solution was purified by silica gel
chromatography (eluent DCM/MeOH/NH4OH, 78:18:4) to give (S)-3-
phenylacetamido-3-(3-pyridyl) propionic acid ammonium salt (19.5 g, 58%).
This product was treated with HCI (6.0 LL, 292 mL), heated at reflux for 5 h,
cooled to RT, and extracted with Et20 (3x200 mL). The aqueous layer was
adjusted to pH 12, concentrated in vacuo, and the resultant solid triturated
with MeOH (2x300 mL). This solution was evaporated to give ca. 14 g
sodium salt. This material was treated with MeOH (500 mL), 2,2-
dimethoxypropane (44 mL), and HCI (4 N in dioxane, 84 mL), and stirred for
90 h at RT. This mixture was filtered and the filtrate concentrated in vacuo.
The resultant off-white solid was triturated with Et20 (2 x 150 mL) and dried
to give compound AG5 (16.7 g, 96% ee) as a white, amorphous solid.
EXAMPLE 1 (comparative)
N-3-(4-Pli2eridinenroRionyJ)-nineco~f-(3-arpino-. - henyl) Qronionic acid -
TFA {11
A 25 mL sintered glass vessel under nitrogen was charged with 2-chlorotrityl
chloride resin (0.24 g, 0.36 mmol, Novabiochem) and DMF (5 mL). The
resin was agitated with nitrogen for 5 min to swell and the DMF removed.
The resin was treated with DMF (5 mL), DIEA (0.31 mL, 5 eq), and allyl 3-(4-
piperidine)propionate - HCI (0.20 g, 2.4 eq), sequentially, and agitated for 8
h. The resultant dark green solution was removed, and the resin washed
with DMF (3x5 mL), aqueous DMF (25%, 3x5 mL), THF (3x5 mL), DCM (3x5
mL), and Et2O (5 mL). The resin was swelled with DCE (5 mL) and treated
with a mixture of tetrabutylammonium fluoride hydrate (0.28 g, 3 eq),
azidotrimethylsilane (0.38 mL, 10 eq), tetrakis(triphenylphosphine)pailadium
(0.084 g, 20 mol %), and DCE (5 mL), The resin was agitated for 15 h and
in
CA 02610436 2007-11-21
the orange solution removed. Tiie resin was washed with DCM (3x5 mL),
DMF (3x5 mL), THF (3x5 mL), and Et20 T5 mL). The resin was swelled with
DMF (5 mL) and treated with DIEA (0.18 mL, 3 eq), allyl nipecotate = HCI
(0.17 g, 3 eq), DIC (0.17 mL, 3 eq), and HOBT (1 mg). The resin was
agitated for 15 h and then the reaction solution removed. The resin was
washed with DMF (3x5 mL), aqueous DMF (25%, 3x5 mL), THF (3x5 mL),
DCM (3x5 mL), and Et20 (5 mL). The resin was swelled with DCE (5 mL)
and treated with a mixture of tetrabutylammonium fluoride hydrate (0.28 g, 3
eq), azidotrimethylsilane (0.38 mL, 10 eq), tetrakis(triphenylphosphine)
palladium (0.084 g, 20 mol %), and DCE (5 mL). The resin was agitated for
h and the orange solution removed. The resin was washed with DCM
(3x5 mL), DMF (3x5 mL), THF (3x5 mL), and Et20 (5 mL). The resin was
swelled with DMF (5 mL) and treated with DIEA (0.18 mL, 3 eq), methyl D,L-
3-amino-3-phenylpropionate = HCI (0.23 g, 3 eq), DIC (0.17 mL, 3 eq), and
15 HOBT (1 mg). The resin was agitated for 17 h and then the reaction solution
removed. The resin was washed with DMF (3x5 mL), aqueous DMF (25%,
3x5 mL), THF (3x5 mL), DCM (3x5 mL), and Et20 (5 mL). The resin was
swelled with THF (5 mL) and treated with a solution of KOTMS (0.23 g, 10
eq) and THF (2 mL). The resin was agitated for 18 h and then the reaction
solution removed. The resin was washed with DMF (3x5 mL), acetic
acid/THF (1:1, twice), aqueous DMF (25%, 3x5 mL), THF (3x5 mL), DCM
(3x5 mL), and Et20 (5 mL). The resin was treated with TFA/DCM (1:1, 10
mL), agitated for 15 min, and the resultant red solution coliected. This
solution was evaporated and the resultant oil triturated with Et20 (3x5 mL)
and dried to afford compound 1 as a clear glass (0.11 g): 1 H NMR (DMSO-
d6) 5 8.6 (m, 1 H), 8.42 (d, J=7, 1 H), 8.2 (m, 1 H), 7.3 (m, 3 H), 7.2 (m, 2
H),
5.18 (d, J=6, 1 H), 4.3 (m, 1 H); 3.7 (m, 1 H), 3.2 (m, 3 H), 2.8 (m, 2 H),
2.6 (m,
2 H), 2.3 (m, 5 H), 1.1-1.9 (m, 11 H); MS m/e 416 (MH+).
~ 4 4
Using the same general solid phase synthesis technique as described
in Example 1, the compounds of indicated examples were made according
to Scheme AA as recited in the particular example.
31
CA 02610436 2007-11-21
EXAtiE.E 2 (comparative)
N_3L4-Pii2eridinemethvla ina carhonyj):njQgcotyl-(3-amino=2-methyl)
r ionjgacid - TFA (2)
Compound 2 was prepared as shown in Scheme AA. Resin-bound 4-
piperidinemethylamine (0.36 mmol) was swelled with DCE (5 mL), treated
with p-nitrophenylchloroformate (0,36 mmol) and DIEA (0.36 mmol), agitated
for 1 h, and the solvent removed. The resin was washed (see Example 1),
swelled with DCE (5 mL), treated with allyl nipecotate - HCI (0.36 mmof) and
DIEA (0.72 mmoL), and agitated for 16 h. The solvent was removed, the
resin washed (see Example 1), and the allyl ester cleaved to the
corresponding acid (see Example 1). The resin was swelled with DMF (5
rnL), the acid coupled with methyl 3-amino-2-methylpropionate (0.36 mmol),
and the synthesis completed as shown in Example 1. Compound 2 was
isolated as a clear glass (0.11 g): 1 H NMR (CD30D) S 3.9 (m, 2 H), 3.2 (m, 4
H), 3.10 (d, J=7, 2 H), 2.9 (m, 3 H), 2.6 (m, 2 H), 2.3 (m, 1 H), 1.9 (m, 4
H),
1.7-1.9 (m, 5 H), 1.3-1.5 (m, 5 H), 1.11 (d, J=7, 3 H); MS m/e 355 (MH+).
EXAMPLE 3 (comparafiive)
NtQ(4- ipp,ridin . pt lox,y.a rbonyl)-ni eco yL-D-asl2artic acid a-methy,l
est~.
-lFA (3}
Compound 3 was prepared as shown in Scheme AA. Resin-bound 4-
piperidinemethanol (0.36 mmol) was swelied with DCE (5 mL), treated with
p- nitrophenyichloroformate (0.36 mmol) and DIEA (0.36 mmol), agitated for
1 h, and the solvent removed. The resin was washed (see Example 1),
swelled with OCE (5 mL), treated with allyl nipecotate - HCI (0.36 mmol) and
DIEA (0.72 mmoL), and agitated for 16 h. The solvent was removed, the
resin washed (see Example 1), and the allyl ester cleaved to the
corresponding acid (see Example 1). The resin was swelled with DMF (5
mL), the acid coupled with H-D-Asp(OBn)-0Me (0.36 mmol), and the
synthesis completed as shown in Example I. Compound 3 was isolated as
a yellow glass (0.019 g): iH NMR (CD30D) 8 4.8 (m, 2 H), 3.9 (m, 3 H), 3.70
(d, J=9, 4 H), 3.39 (s, 3 H), 3.3 (m, 2 H), 2.9 (m, 4 H), 2.8 (m, 2 H), 1.9
(m, 4
H), 1.7 (m, 2 H), 1.4 (m, 4 H); MS m/e 400 (MH+).
32
CA 02610436 2007-11-21
EXAPNPLE 4 (comparative)
N-3-(4-Pineridineoronionyl) -o yl[!21i inQ43 - carbonyi-r3-a minQ; 3-(4-toIl
gm i~nic acid - TFA (41
Compound 3 was prepared as shown in Scheme AA. Intermediate AA2
(0.36 mmol) was swelled with DCE (5 mL), treated with methyl pyrrolidine-3-
carboxylate = HCI (0.36 mmoi), D(G (0.72 mmol), and DIEA (0.72 rnmoL), and
agitated for 1 6 h. The solvent vsras removed, the resin washed (see Example
1), and the methyl ester cleaved to the corresponding acid with KOTMS (see
Example 1). The resin was swelled with DMF (5 mL), the acid coupled witil
methyl 3-amino-3-(4-tolyl)propionate (0.36 mmol), and then the synthesis
completed as shown in Example 1. Compound 4 was isolated as a cfear
glass (0.081 g): 1H NMR (CD3OD) S 7.19 (d, J=5, 2 H), 7.10 (d, J=5, 2 H),
5.31 (dd, J=3, 10; 1 H) 3.6 (m, 4 H), 3.3 (m, 2 H), 2.9 (m, 4 H), 2.7 (m, 2
H),
2.3 (m, 2 H), 2.1 (m, 3. H), 1.9 (m, 4 H), 1.6 (m, 4. H), 1.3 (m, 4 H); MS m/e
416
(MH+).
_E XAMPLE 5 (comparative)
N-3-(4-F'i ridin .p roRio5Yl)-isonioecotvl _ja-a(gjro-3=methvll pronionic
acid,_
A(5)
Compound 5 was prepared as shown In Scheme AA. Intermediate AA2
(0.36 mmol) was swelled wfth DCE (5 mL), treated with ethyl isonipecotate
(0.36 mmol), DIC (0.72 mmol), and DIEA (0.72 mmoL), and agitated for 16 h.
The solvent was removed, the resin washed (see Example 1), and the ethyl
ester cleaved to the corresponding acid with KOTMS (see Example 1). The
resin was swelled with DMF (5 mL), the acid coupled with methyl 3-amino-3-
methylpropionate (0.36 mmol), and then the synthesis completed as shown
in Example 1. Compound 5 was isolated as a tan glass (0.033 g): 1H NMR
(CD3OD) S 4.5 (m, 1 H), 4.2 (m, 1 H), 3.9 (m,- 1 H), 3.3 (m, 2 H), 3.3 (m, 3
H),
3.1 (m, 1 H), 2.9 (m, 3 H), 2.7 (m, 2 H), 2.4 (m, 2 H), 2.0 (m, 2 H), 1.7 (m,
2 H),
1.5 (m, 6 H), 1.3 (m, 2 H), 1.15 (d, J=9, 3 H); MS m/e 354 (MH+).
33
CA 02610436 2007-11-21
EXAMPt_F 6 (comparative)
N-3-(4_pj12e[!dine proaionyl)-is b pcotyl j, -amino-3 44-carboxvDhenvl11
proniQnic acid- TFA (61
Compound 6 was prepared as shown in Scheme AA. Intermediate AA2
(0.36 mmol) was swe{led with DCE (5 mL), treated with ethyl isonipecotate
(0.36 mmol), D(C (0.72 mmol), and DIEA (0.72 mmoL), and agitated for 16 h.
The solvent was removed, the resin washed (see Example 1), and the ethyl
ester cleaved to the corresponding acid with KOTMS (see Example 1}. The
resin vvas swelled with DMF (5 mL), the acid coupled with methyl 3-amino-3-
(4-carboxymethyl-phenyl)propionate (0.36 mmol), and then the synthesis
completed as shown in Example 1. Compound 6 was isolated as a tan
glass (0.034 g): 1H NMR (CDaOD) S 7.9 (m, 3 H), 7.43 (d, J=5, 2 H), 5.4 (m, 1
H), 4.5 (m, 1 H), 4.0 (m, 1 H), 3.3 (m, 4 H), 3.1 (m, 1 H), 2.9 (m, 2 H), 2.7
(m. 2
H), 2.7 (m, 1 H), 2.5 (m, 4 H), 2.0 (m, 2.H), 1.2-1.9 (m, 10 H); MS mle 460
(MH+).
EXAMPLE 7 (comparative)
d-3-(4-N-Melhvl-ninPridineoroQiQOYl)-nio ,cQtyJ-3-aminoQronionic acid - TEA
Compound 7 was prepared as shown in Scheme AD. Resin-bound Fmoc-p-
Ala (1 mmol) was treated with 20% piperidine/DMF (10 mL), agitated for 2h,.
and the solvent removed. The resin was washed with DMF, swelled with
DMF (10 mL), and treated with Fmoc-nipecotic acid (1 mmol), DIC (2 mmol),
and DIEA (1 mmol). The resin was agitated for 16 h, the solvent removed,
and the resin washed with DMF and DCM. The resin was treated with' 20%
piperidine/DMF (10 mL) for 2h, the solvent removed, and the resin washed
with DMF. The resin was swelled with DMF (10 mL), treated with 4-N-
methylpiperidinepropionic acid (1 mmol), DIC (2 mmol), and DIEA (1 mmol),
and agitated for 16 h. The solvent was removed and the resin washed with
'DMF and DCM. The resin was cleaved with 95% TFA (10 mL) and the TFA
evaporated to afford 7 as a white powder (0.26 g): mp 172-177 C; 1 H NMR
(CDC13) 5 4.4 (m, 1 H), 3.7 (m, 1 H), 3.4 (m, 1 H), 3.2 (m, 1 H), 3.1 (m, 1
H),
34
CA 02610436 2007-11-21
2.7 (m, 2 H), 2.3 (m, 6 H), 2.21 (s, 3 H), 1.9 (m, 4 H), 1.3-1.8 (ni, 10 H);
MS
m/e 354 (MH+).
EXAMPLE 8 (comparative)
N:, -(4-PiRe dine l2ropionyl)- nit2e=ty1-4-oxonipecotic acid - TFA (89
Compound 8 was prepared as shown in Scheme AA. Intermediate AA2
(0.36 nimol) was swelled with DCE (5 mL), treated with ethyl nipecotate
(0.36 mmol), DIC (0.72 mmol), and DIEA (0.72 mmoL), and agitated for 16 h.
The solvent was removed, the resin washed (see Example 1), and the ethyl
ester c9eaved to the corresponding acid with KOTMS (see Example 1). The
resin was swelled with DMF (5 mL), the acid coupled with methyl 4-oxo-
nipecotate (0.36 mmol), and then the synthesis completed as shown in
Example 1. Compound 8 was isolated as a clear glass (0.04 g): 'H NMR
(DMSO-d6) S 8.5 (m, 1 H), 8.2 (m, 1 H), 6.5 (m, 1 H), 4.3 (m, 1 H), 3.4-3.8
(m,
4 H), 3.2 (m, 2 H), 3.0 (m, 1 H), 2.8 (m, 2 H), 2.2-2.6 (m, 6 H), 1.8 (m, 2-
H),
1.1-1.7 (m, 11 H); MS m/e. 394 (MH+).
);rXAMPLE .9 (comparative)
N-'3-(4-Pjp .r~idineploDionyj)-nipecojyl-f3-amjno-3-(2-
trimethylsilv!ethynvl)25 prQ nic,a0id - TFA (9)
Compound 9 was prepared as shown in Scheme AA. Intermediate AA2
(0.36 mmol) was swelled with DCE (5 mL), treated with ethyl nipecotate
(0.36 mmol), D1C (0.72 mmol), and DIEA (0.72 mmoL), and agitated for 16 li.
The solvent was removed, the resin washed (see Example 1), and the ethyl
ester cleaved to the corresponding acid with KOTMS (see Example 1). The
resin was swelled with DMF (5 mL), the acid coupled with methyl 3-amino-3-
(2-trimethylsilylethynyl)propionate (for a preparation, see J. Zablocki, J.
Med.
Chem. 1995, 38, 2378; 0.36 mmol), and then the synthesis completed as
shown in Example 1, Cornpound 9 was isolated as a yellow gfass (0.12 g):
1H NMR (CD3OD) fi 3.8 (m, 1 H), 3.2-3.4 (m, 4 H), 2.9 (m, 3 H), 2.7 (m, 2 H),
2.3-2.5 (m, 2 H), 1.9 (m, 4 H),- 1.1-1.9 (m, 13 H), 0.0=(s, 9 H); MS m/e 436
(MH+).
CA 02610436 2007-11-21
EXAMPLE 10
N-3-f6-Aminocaaroyll-niRecotvl-3-amino-3-(2-QVridvllQrogionic acid - 2TFA
LM
Compound 10 was prepared as shown in Scheme AA. Resin-bound 6-
aminocaproic acid (0.36 mmol) was swelled with DCE (5 mL), treated with
ethyl nipecotate (0.36 mmol), DIC (0.72 mmol), and DIEA (0.72 mmoL], and
agitated for 16 h. The solvent was removed, the resin washed (see Example
1), and the ethyl ester cleaved to the corresponding acid with KOTMS (see
Example 1). The resin was swelled with DMF (5 mL), the acid coupled with
methyl 3-amino-3-(3-pyridyl)propionate (0.36 mmol), and then the synthesis
completed as shown in Example 1. Compound 10 was isolated as a clear
glass (0.008 g): I H NMR (DMSO-ds) & 8.6 (m, 2 H), 8.1 (s, I H), 7.0-7.7 (m, 5
H), 5.15 (t, J=3, 1 H), 4.4 (m, 1 H), 4.1 (m, 1 H), 3.7 (m, 2 H), 3.1 (m, 1
H), 2.7
(rn, 4 H), 2.5 (m, 1 H), 2.3 (m, 2 H), 1.2-1.9 (m, 11 H); MS m/e 391 (MH~).
Anal. caicci. for CZpH3pN404 - 3TFA - 2H20 (768.60): C, 40.63; H, 4.85; N,
7.29; F, 22.25. Found: C, 40.81; H, 4.70; N, 6.12; F, 23.83.
EXAMPLE 11 (comparative)
N-3-(4-Pioeridinenrooionyl)-A-(-)-nin cotvl-(3-amino-2-hydroxy) proil1 anic~
acid = TFA (Y 1)
Compound 11 was prepared as shown in Scheme AA. Intermediate AA2
(0.36 mmol) was swelled with DCE (5 mL), treated with ethyl R-nipecotate
(0.36 mmol), D1C (0.72 mmol), and DIEA (0.72 mmoL), and agitated for 16 h.
The solvent was removed, the resin washed (see Example 1), and the ethyl
ester cleaved to the corresponding acid with KOTMS (see Example 1). The
resin was swelled with DMF (5 mL), the acid coupled with methyl 3-amino-2-
hydroxypropionate (0.36 mmol), and then the synthesis completed as shown
in ExaFnpie 1. Compound 11 was isolated as a pink glass (0.05 g): 1 H NMR
(DMSC?-d6) S 8.5 (m, 1 H), 8.2 (m, 1 H), 7.6 (m, 1 H), 4.0-4.4 (m, 2 H), 3.7
(r~,
1 H), 3.2 (rn, 3 H), 2.8 (m, 3 H), 2.6 (m, 1 H), 2.1-2.3 (m, 3 H), 1.8 (m, 4
H),
1.0-1,4 (m, 10 H); MS m/e 356 (MH+).
36
CA 02610436 2007-11-21
EXAMPLE 12 (comparative)
,t'I-3-(4-p1RPridineethanesuff o nx( -nipecoty,l-3-Omino0rop ic acid ~ HCI
Compound 12 was prepared as shown in Scheme AE. Intermediate AE1
was synthesized by the following procedure. 2-(4-Pyridine)ethanesulfonic
acid (3.0 g, 0.016 mol) was dissolved in aq. HCI (2.0 Lj. 12 mL) and this
solution treated with platinum dioxide (0.13 g) and hydrogenated at 50 psi
and RT for 18 h. This mixture was filtered through Calite and evaporated to
afford 2-(4-piperidine)ethanesulfonic acld - HCI (3.5 g, white powder). This
powder was dissolved in aq. THF (1:1, 70 mL) at RT and treated with NMM
(3.7 mL, 2.2 eq.) and benzyl chloroformate (2.2 mL, 1 eq.). This mixture was
stirred for 15 h, acidified with aq. citric acid,'and extracted with CHCI3
(2x100
mL). The organic layer was dried with NazSO, and evaporated to afford 2-
(4-N-Z-piperidine)ethanesulfonic acid (2.75 g, gold oil). This oil was
converted to final product 12 in five synthetic steps (Scheme AE, W. J.
Hoekstra, J. Med. Chem. 1995, 38, 1582) and isolated as a clear glass
(0.060 g): I H NMR (DMSO-d6) S 8.9 (m, 1 H), 8.6 (m, 1 H), 3.5 (m, 2 H), 3.1-
3.3 (m, 4 H), 3.0 (m, 2 H), 2.6-2.8 (m, 4 H), 2.3 (m, 3 H), 1.65-1.9 (m, 5 H),
1.6
(m, 3 H), 1.2-1.4 (m, 5 H); MS m/e 376 (MH+).
EXAMPLE 13 (comparative)
N-3-(4-Pi0 .eridinP,l2rol2ionyl~~cotyl-5 -(2-aminoethYllt tr zole = HCI (1 ~
Compound 13 was prepared as shown in Scheme AC. interinediate AC1
(prepared as in W. J. Hoekstra, J. Med Chem. 1995, 38, 1582; 1.9 mmol)
was dissolved in DCM (50 mL) and treated with BOP-CI (1.9 mmol), NMM
(1.9 mmol), and 3-aminopropionitrile (1.9 mmol). The reaction was stirred
for 18 h, diluted with sat'd NH.CI, and the layers separated. The organic
layer was evaporated and the product purified by silica gel chromatography
(10%EIOH/DCM) to give an oil. The oil was dissolved in toluene (10 mL),
treated with azidotrimethylsilane (2.4 mmol) and dibutyltin oxide (1.2 mmol),
and 'heated at reflux for 16 h. Cooling gave a brown ppt which was triturated
37
CA 02610436 2007-11-21
with EtzO. This solid was hydrogenated over platinum dioxide (0.08 g) in
MeOH (12 mL) at 50 psi for 15 h, fiitered, and evaporated to give 13 as a
yellow foam (0.065 g): 1 H NMR (DMSO-d6) & 8.9 (m, 1 H), 8.6 (m, 1 H), 8.13
(d, J-28., 1 H), 4.2 (m, 2 H), 3.2 (rn, 3 H), 3.0 (m, 4 H), 2.7 (m, 4 H), 2.31
(q,
J-8, 2 H), 1.7-1.9 (m, 3 H), 1.4-1.6 (m, 5 H), 1.1-1.3 (m, 4 H); MS m/e 364
(MHi ).
EXAMPLE 14 (comparative)
N-344-N-Methy(-12 iperazine12 rooionviL io~ ecotyj t3_amino-3-(3.4-
QtethylenedioxxRhenylaJnro ioni . cjd = Na (14)
Compound 14 was prepared as -shown in Scheme AB. Ethyl nipecotate (3
mmof) was dissolved in DCM (50 mL), treated with acryloyl chloride (3
mmol) and NMM (3 mmol), and stirred for 1 h. The. solvent was evaporated
and the residue dissolved in EtOH (50 mL) and treated with N-
methyipiperazine (3 mmol). The solution was warmed at 60 C for 15 h,
cooled to RT, and the solvent evaporated. The residue was partitioned
between DCM (100 mL) and water (10 mL), and the layers separated. The
organic layer was dried and evaporated to give a foam. The foam was
. dissolved in water, treated with NaOH (3 mmol), stirred for 1 h, and
evaporated to give A83-Na. The synthesis was completed as illustrated (W.
J. Hoekstra, J. Med. Chem. 1995, 38, 1582) using methyl 3-amino-3-(3,4-
methylenedioxyphenyl)propionate (2.5 rrirno!) to give 14 as a white,
amorphous solid (0.14 g): I H NMR (D20) S 6.8 (m, 3 H), 5.91 (s, 2 H), 5.0 (m,
1 H), 4.0 (m, 1 H), 3.7 (m, 1 H), 2.8-3.4 (m, 11 H), 2.69 (s, 3 H), 2.4-2.6
(m, 7
H), 1.9 (m, 1 H), 1.7 (m, 2 H), 1.5 (m, 1 H); MS m/e 475 (MH+). Anaf, calcd.
for C24H33N406 = Na - H20 (514.56): C, 56.02; H, 6.86; N, 10.89. Found: C,
55.72; H, 6.78; N, 10.52.
38
CA 02610436 2007-11-21
EXAMPLE 15
N; 3-(A-N-Methyl-pioerazinegrgpfonY()-nin, cotvLj3 -amino-3-l3-
qyjnQliByfltorogionic acid - 3TFA(1 )
Compound 15 was prepared as described in Example 14. The synthesis
was completed as illustrated (W. J. Hoekstra, J. Med Chem. 1995, 38,
1582) using methyl 3-amino-3-(3-quinoliny!)propionate (6 mmol) with AB3.
1'0 Compound 15 was isolated as a yellow powder (1.89 g): 'H NMR (DMSO-
d6) S 8.94 (s, 1 H), 8.12 (s, 1 H), 7.9 (m, 2 H), 7.6 (m, 2 H), 7.07 (d; J=4,
1 H),
5.2 (m, 1 H), 4.1 (m, 1 H), 3.7 (m, 1 H), 3.1-3.3 (m, 2 H), 2.9 (m, 2 H), 2.6
(m, 2
H), 2.43 (s, 3 H), 1.9-2.4 (m, 12 H), 1.2-1.5 (m, 4 H); MS m/e 482 (MH+).
EXAMPLE 16 (comparative)
N;Q-(4-Pioeri ineQro ionyl)-R-(-)-nioecotYl j(S --amino-3-f3.4-
methxfenedioxyl2 envij,lQro ionic acid = HCI (9 6)
To a cooled (5 C) solution of Bdc-R-nipecotic acid (9 mmol) and methyl (S)-
3-amino-3-(3,4-methylenedioxyphenyl)]propionate (see.AG5 example; 9
rnmol) in MeCN (100 mL) was added HBTU (9 mmol), HOBT (9 mmol), and
NMM (18 mmol). This mixture was stirred for 15 h, diluted with water (10
mL), and evaporated. The. residue was diluted with EtOAc (100mL) and the
organic layer dried and evaporated to give a white foam. The foam was
treated with HCI (2 N in dioxane, 20 mL), stirred for 3 h, and evaporated to a
foam. The foam was dissolved in MeCN (100 mL) and treated with Boc-
piperidinepropionic acid (7 mmol), HBTU (7 mmol), HOBT (7 mmol), and
NMM (14 mmol) with stirring for 6 h. The mixture was diluted with water (10
mL), evaporated, and diluted with EtOAc (100 mL). The organic layer was
dried, evaporated, and purified by silica gel chromatography (7%
EtOH/DCM) to give a foam. To a solution of the foam (4.6 mol) in THF cooled
in an ice bath was added LiOH=H20 (6.9 mmol dissolved in 30 mL water)
dropwise. This mixture was stirred for 1.5 h, acidified with 'AcOH (1.7 mL),
and warmed to RT. This solution was diluted with CHCI, (75 mL) and the
layers separated. The organic layer was dried (NaZSO4) and evaporated to
give a white foam. The foam was dissolved in dioxane (20 mL) and anisole
39
CA 02610436 2007-11-21
(0.3 mL), cooled in an ice bath, treated with HCI (15 mL, 4.0 N in dioxane),
and stirred for 3 h to give a ppt. The ppt was fiitered and washed with Et2O
(150 mL) and MeCN (20 mL) to give 16 as a white powder (1.78 g): mp 190-
200 C; iH NMR (DMSO-d6) S 8.9 (m, 1 H), 8.6 (m, 1 H), 8.4 (m, 1 H), 6.83 (d,
J=5, 1 H), 6.79 (d, J=5, 1 H), 6.7 (m, 1 H), 5.95 (s, 2 H), 5.08 (dd, J=5, 11,
1
H), 4.1-4.3 (m, 1 H), 3.7 (m, 1 H), 3.15 (d, J=10, 2 H), 3.0 (m, 1 H), 2.7 (m,
2
H), 2.6 (m, 3 H), 2.31 (d, J=7, 2 H), 1.81 (d, J=10, 2 H), 1.2-1.7 (m, 11 H);
MS
m!e 460 (MH+); (a124D -0.478 (c 1.00, MeOH).
EXAMPLE 17
N-3-(4-Piperidinepropionyl)-hexahydroazepine-3-carbonyl-f3-amino-3-(3-
guinolinyl)lpropionic acid = 2TFA (17)
Compound 17 was prepared as shown in Scheme AA. _ Intermediate AA2
(0.36 mmol) was swelled with DCE (5- mL), treated with methyl
hexahydroazepine-3-carboxylate - HCI (0.36 mmol), DIC (0.72 mmol), and
DIEA (0.72 mmoL), and agitated for 16 h. The solvent was removed, the
resin washed (see Example 1), and the methyl ester cleaved to the
corresponding acid with KOTMS (see Example 1). The resin was sweiled
with DMF (5 mL), the acid coupled with methyl 3-amino-3-(3-
quinolinyl)propionate (0.36 mmol), and theri the synthesis completed as
shown in Example 1. Compound 17 was isolated as a glass (0.10 g): 1 H
NMR (D20) S 9.06 (s, 1 H), 8.9 (m, 1 H), 8.2 (m, 1 H), 8.04 (s, 1 H), 8.0 (t,
J=4,
2 H), 7.8 (t, J=4, 2 H), 5.5 (m, 1 H), 3.8 (m, 1 H), 3.3 (m, 4 H), 3.0 (m, 2
H), 2.7
(m, 4 H), 2.0-2.4 (m, 6 H), 1.7-1.9 (m, 4 H), 1.1-1.6 (m, 8 H); MS m/e 481
(MH+)-
EXAMPLE 18
N-3-(4-Pioeridineotooionyj)-R-(-)-niaecotvt-f(S)-3-amino-3-(3.-
auinolinyl)]oroQionic acid - 2HCI (1 8)
Compound 18, prepared as described in Example 16 starting with Boc-R-
nipecotic acid (7.1 mmol) and methyl (S)-3-amino-3-(3-quinolinyl)propionate
(see example AG5; 7.1 mmol), was isotated as white flakes (1.11 g): mp 142-
144 C; MS m/e 467 (MH+); [a]24p -173 (c 0.1, MeOH). Anal. caicd. for
CA 02610436 2007-11-21
C26H34N404 = 2.25 HCI - H20 (566.64): C, 55.11; H, 6.80; N, 9.89; Cl, 14.08.
Found: C, 54.85; H, 6.62; N. 10.04; Cl, 13.68.
EXAM LE 19 (comparative)
N-3-(4-PiQeridineRro oionXj)-R-(-)-n 'necetxlõjfS1-3-amino-3-(2-t-
butylethynyju
~onionic acid = HCI (T 9)
Compound 19, prepared as described in Example 16 starting with Boc-R-
nipecotic acid (3.2 mmol) and methyl (S)-3-amino-3-(2-t-
butylethynyi)propionate (see J. A. Zablocki, J. Med. Chem. 1995, 38, 2378;
3.2 mmol), was isolated as a white powder (0.33 g): MS m/e 420 (MH+).
Anal. calcd. ior C2,3H37N304 = 1.07 HCI = 0.43 H20 (468.97): C, 59.21; H,
8.42; N, 8.96; Cl, 8.09. Found: C, 58.92; H, 8.58; N, 8.76; Cl, 7.82.
EXAMPLE 20 (comparative)
N=3-(4-Piperidi[]ej2rgpYl)-nil2ecgtyf-jj51-3-amino-3õ(0.4-
met y.lenedio gh eny rooiopic acid = 2TFA (2 0)
Compound 20 was prepared as shown in Scheme AF. Intermediate AF3
(2.8 mmol) was dissolved in benzene (50 mL), treated with ethyl nipecotate
(2.8 mmol), and heated at reflux for 7 h. The reaction was cooled,
partitioned between water (15 mL) and EtOAc (70 mL), and the layers
separated. The organic layer was dried and evaporated to give AF4. AF4
was converted to 20 as previously described (W. J. Hoekstra, J. Med. Chem.
1995, 38, 1582) and isolated as a white powder (0.33 g): 1 H NMR (CD30D)
S 8.6-8.8 (m, 3 H), 6.7-6.9 (m, 3 H), 5.91 (s, 2 H), 5.1-5.2 (m, 1 H), 3.3-3.5
(in,
4 H), 2.8-3.1 (m, 6 H), 2.6-2.7 (m, 3 H), 1.5-2.0 (m, 11 H), 1.2-1.4 (m, 4 H);
MS m/e 446 (MH+).
41
CA 02610436 2007-11-21
EXAMPL-E 21
N-3-(4-Pioeridnel2ropionYl)-R-(-1'-nioecntvl-ffSl-3-aminn-3-(3-Qvridvl)l
nroDionic ac{d - 2TFA (2 1)
Compound 21, prepared as described in Example 16 starting with Boc-R-
nipecotic acid (6.4 mmol) and methyl (S)-3-amino-3-(3-pyridyl)propionate
(see example AG5; 6.4 mmol), was isolated as a whtte amorphous solid
(1.60 g): mp 74-81 C; MS m/e.417 (MH+). Anal. catcd. for C22H32N404 - 2.1
10. C2HF302 - 0.7 H20 (668.58): C, 47.07; H, 5.35; N, 8.38; F, 17.90; KF,
1.89.
Found: C, 47.08; H, 5.31; N, 8.41; F, 17.68; KF, 2.00.
EXAMPLE 22 (comparative)
N-3-(4-Pineridineoro ionyjJ-R-(-lnioecoty(-[(S)-2-(3-
methoxyanilino).arbonylamino;3-aminoj~ropio nic ai~ad (221
Methyl Boc-R-nipecotyl-[(S)-2-Z-amino-3-amino)propionate (prepared from
methyl N-a-Z-L-diarninopropionate and oc-R-nipecotic acid as shown in
Example 16; 9.5 mmol) was dissolved in MeOH (40 mL) and hydrogenated
at 50 p.si over palladium hydroxide (0.4 g) for 24 h. The mixture was filtered
and evaporated to give white solid AH2. AH2 (9.1 mmol) was dissolved in
DCM (100 mL), cooled (5 C), treated with 3-rnethoxyphenylisocyanate (9.1
rnmol) and NMM (9.1 mmol), and stirred for 17 h. The solution was diluted
with sat'd NH4,CI (10 mL), the layers separated, and the organic layer dried,
evaporated to an oil, and purified by silica gel chromatography (4%
EiOH/DCM) to give AH3. Intermediate AH3 was converted to 22 in four
steps as in Example 16 to afford a white amorphous solid (1.35 g): mp 72-
'76 C; 1H NMR (DMSO-d6) S 8.7 (m, 3 H), 7.8 (m, 1 H), 7.1 (m, 2 H), 6.8 (d, 1
H), 6.5 (d, 2 H), 3.66 (s, 3 H), 3.4 (m, 2 H), 3.2 '(d, 2 H), 2.7 (dd, 4 H),
2.3 (m, 3
H), 1.6 -(m, 3 H), 1.1-1.7 (m, 11 H); MS m/e 504 (MH+). Anal. calcd. for
C25H37Ng0s - 1.2 HCI = 1..0 H20 (565.37): C, 53.11; H, 7.17; N, 12.39; Cl,
7.53. Found: C, 53.40; H, 7.44; N, 12.14; Cl, 7.66.
' = =
Using the same general synthesis technique as described in Example
22, the compounds of Examples 26, 28-30 were made according to
Scheme AH recited in the particuiar example. For carbamate analogues,
42
CA 02610436 2007-11-21
the acylating agent employed was the appropriate alkyl chloroformate
(analogous conversion of AH2 to AH3; one molar equivalent). For
sulfonamides, the sulfonating agent employed was the appropriate sulfonyl
chloride (one molar equivalent).-
EXAMPLE 23 (comparative)
.N-3-(4-PiReridine ro i~onyl)-R-(-)niRgr;otyl-f(S)-2-benzyloxyc3rbonylamino-
3-am~inolAro Alonirgcid = HCI (2 3)
Compound 23, prepared from methyl N-a-Z-L-diaminopropionate (8.8
mmol) and Boc-R-nipecotic acid (8.8 mmol) as shown in Example 16, was
isolated as a white powder (1.65 g): mp 110-113 C; MS m/e 489 (MH+).
15. Anal. calcd, for C25H36N406 = 1.15 HCI = 0.5 H20 = 0.5 Dioxane (583.57):
C,
55.56; H, 7.41; N, 9.60; Cl, 6.99. Found: C, 55.23; H, 7.79; N, 9.85; Cl,
7.01.
EXAMPLE 24 (comparative)
N-3-(4-Pi ridin . roniony1)-R-f- ninecotyl f(S)-2-(3-
chlorobenzyloxy)carbonylamino-3-a ino~r pionic acid = HCI (2 41
Compound 24, prepared by reacting 3-chlorobenzyloxycarbonyl chloride
(6.6 mmol) with AH2 (6.6 mrnol) as described in Example 22, , was isolated
as a white amorphous solid (1.33 g): mp 89-96 C; MS m/e 524 (MH+). Anal.
calad. for C25H35CIN406 = 1.25 HCI = 0.5 H20 - 1.0 Dioxane (637.20): C.
50.89; H, 7.08; N, 8.78; Cl, 12.52. Found: C, 51.10; H, 6.71; N, 8.38; Ci,
12.20.
E?CAMPLE 25 (comparative)
N- -(4-Pioeridineor ionyj)-R-(-)nioecotyl-r(S)-2-benzylsulfonviamino-3-
amino)~r pionic acid = HCI (Z 2)
Compound 25, prepared by reacting benzylsulfonyl chloride (5.2 mmol) with
AH2 (5.2 mmol) as shown in Example 22, was isolated as a white powder
.43
CA 02610436 2007-11-21
(0.87 g): mp 145-149 C; MS m/e 509 '(MH+). Anal. calcd. for C24H36N406S
= 1.3 HCI = 0.3 Dioxane (568.06): C, 50.75, H, 7.04; N, 9.86; Cl, 8.11. Found;
C, 51.03; H, 6.93; N, 9.46; Cl, 7.85.
EXAMPLE 26 (comparative)
N-3-(4-PiperidineprQpigny,j)-R-(-)nipecoty,u(S1-2,:(3.5-
imethoaanilinQ)carbonylaming-3-aminoloropionic acid - HCI (2 6)
Compound 26, prepared by reacting 3,5-dimethoxyphenylisocyanate (10.2
mmol) with AH2 (10.2 mmol) as shown in Example 22, was isolated as a
white powder (1.89 g): mp 190-193 C; MS nVe 534 (MH+). Anal. calcd. for
C26H39N507 - 1.2 HCI = 0.2 Dioxane (585.40): C, 53.35; H, 7.20; N, 11.96;
Cl, 7.27. Found: C, 53.48; H, 7.38; N, 12.05; Cl, 6.97.
EXAMPt. 27
N-((4.4'-QjRoeridin-1-yl-)cad~gnvfl-R-y-ni ecotv -[(;z)-3-amino-3-(3-RyndYl)1
pr~nionic acid = 3HC1 (2 7)
Intermediate AJ1 (5.5 mmol), prepared as shown in Example 16, was
dissolved in DCM (140 mL), cooied (5 C), treated with p-
nitrophenylchloroformate (5.5 mmol) and' (16.5 mmol), and stirred for 2 h.
The mixture was diluted with water (15 mL), the layers separated, and the
organic layer dried and evaporated to an oil. The oil was dissolved in MeCN
(70 mL), treated with N-8oc-4,4'-bipiparidine (7.5 mmol) and OMAP (5.5
rnmol), and heated at reflux for 24 h. The mixture was cooled, evaporated to
a solid, and partitioned between EtOAc (150 mL) and NaOH (1 N, 20 mL).
The layers were separated, and the organic layer dried, evaporated to a
solid, and purified by silica gel chromatography (8 10 ,EtOH/DCM) to give
green glass AJ2 (1.5 mmol). AJ2 was saponified and deprotected as
described in Example 16 to give 27 as a pale yellow powder (0.73 g): mp
:35 121-125 C; MS m/e 472 (MH+). Anal. calcd. for C25H37N504 = 3.6 HCl.= 1.0
Dioxane (690.98): C, 50.41; H, 7.09; N, 10.14; CI, 18.47. Found: C, 50.80; H,
7.31; N, 10.20; Cl, 18.78.
44
CA 02610436 2007-11-21
L-XAMPLE 2$ (comparative)
N-3-(3-Pi e0 ridine0ro io )-R-(-)niResotvl-((S)-2-(2~
naRhttiylg min o)carbonylgi min4-3-amino)propignic acid = HCI (28)
Compound 28, prepared by reacting 2-naphthyiisocyanate (8.5 mmol) with
AH2 (8.5 mmol) as shown in Example 22, was isolated as a white powder
(1.65 g): mp 187-193 C; MS m/e 524 (MH+). Anai, calcd. for C28H37N505 -
1.36"HCI = 0.72 Dioxane (602.07): C, 55.86; H, 7.39; N, 11.63; Cl, 8.01.
Found: C, 56.03; H, 7.11; N, 11.23; Cl, 7.97.
E C NipLE 29 (comparative)
N-3-(4-P ridinepronionyl)-R-(_)gjRecotylaminomethyl-5-(SI-(3-N-
benzyI)imidazQline-2.4-S,ijQne = HCI (29)
N-3-(4-Piperidi nepropionyf)-R-(-) nipecotyl-((S)-2-(2-
benzylamino)carbonylamino-3-amino]propionic acid hydrochiodde (0.15 g),
prepared from intermediate AH2 (4.4 mmol) and benzyiisocyanate (4.4
mmol) as described in Example 22, was dissolved in aq. HCI (3 M and
stirred for 18 h at RT. This solution was concentrated in vacuo to give a
white soiid. This solid was triturated and dried to give 29 as a white foam
(0.144 g) :1H NMR (DMSO-d6) 5 9.0 (m, 1 H), 8.6 (m, 1 H), 8.3 (m, 1 H), 7.2
(m, 5 H), 4.48 (s, 2 H), 4.2 (m, 2 H), 3.7 (m, 1 H), 3.4 (m, 1 H), 3.2 (d, 3
H), 2.7
(d, 3 H), 2.2 (m, 3 H), 1.7 (m, 3 H), 1.0-1.6 (m, 10 H); MS m/e 470 (MH+).'
FXAflRPLE 30 (comparative)
;'-3-(4-Pio ridieorooionyy-l3;(-)ni otyl-r(G)-2-(2;
nhengthylaminQ)carbony ~mino-3-arnino)praRonic acid = HCO= _t-(3 0)
Compound 30, prepared by reacting 2-phenethyfisocyanate (4.1 mmol
mmol) with AH2 (4.1 mmol) as shown in Example 22, was isolated as a tan
foam (0.41 g): mp 65-72 C; MS m/e 502 (MH+). An.al, calcd. for
CA 02610436 2007-11-21
C26Ha9N$05 - 1.2 HCOzH = 1.0 H20 (574.87): C, 56.83; H, 7.61; N, 12.18.
Found: C, 57.12; H. 7.80; N, 11.85.
6-Mettivl-3-Ryridine-carboxaldehv_de (AK21
Aidehyde precursor AK2 was 'prepared in two steps using standard
conditions. AK1 (0.066 mol) was dissolved in THF (100 mL), cooled (-78 C),
treated with LiAIH4 (0.066 mol), and stirred for 4 h, The reaction was
quenched with sat'd NH4C1, warmed, filtered with CHCI3 washes (250 mL),
and the layers separated. The organic layer was dried and evaporated to.
give a clear oil (0.054 mol). The oil was dissolved in DCM (200 mL); treated
with MnQ2 (70 g), and heated at reflux for 6 h. The mixture was cooled,
filtered, and the solvent evaporated to give AK2 (0.052 mol) as a brown oil.
EXAMPLE 31
N-3-(A-Pio eridineRrooionyl)-R-(-)nil2pco yJ-ffSl-3-amino-3-(6-methyl-3-
py-pdyl)j12ronionic acid - 2HCI (311
Compound 31, prepared as described in Example 16 starting with Boc-R-
nipecotic acid (6.9 mmol) and methyl (S)-3-amino-3-(6-methyl-3-
pyridyl)propionate (see examples AK5, AG5; 6.9 mmol). Compound 31 was
isolated as a white foam (1.20 g): mp 99-105 C; MS m/e 431 (MH+). Anal.
calcd. for C23H34N404 - 2.24 HCI - 1.0 H20 = 0.24 Acetonitnle (534.33): C,
51.70; H, 7.35; N, 11.11; Ci, 14.82. Found: C, 51.32; H, 7.45; N, 11.23; Cl,
14.42.
EXAMPLE 32 (comparative)
IL3-(4-Pi ridin .oroflionyl)-R-(-)nip ecoYyl-f(5)-3-2 mino-3-(5-bromo-3-
Wdylll rlrogionic acid - 2FICI (3 2)
Compound 32, prepared as described in Example 16 starting with Boc-R-
nipecotic acid (4.8 mmol) and methyl 3-S-amino-3-(5-bromo-3-
pyridyl)propionate (see examples AK5, AG5; 4.8 mmol), was isolated as a
white 9oam (1.24 g): mp 98-101 C; MS m/e 496 (MH+). Anal. calcd. 'for
46
CA 02610436 2007-11-21
C22H31 BrN~O4 = 2.2 HCI = 11.0 F#10 (593.67): C, 44.51; H, 5.98; N, 9.44; Cl,
13.14. Found: C, 44.17; H, 6.37; N, 9.81; Cl, 13.10.
EXAMPLE 33
N-3-(4-Forma midinopi endin l2rQ io I)-R ;(-)njl2ecQtyl-((S?-3-amino-3-!3-
t~y~v#)~ ~QQionic acid = 2HCI Q3}
Formamidine 33 was prepared according to the procedure of M. K. Scott (J.
Med. Chem. 1983, 26, 534) as shown in Scheme AL. Intermediate ALl
(see Example 21; 2.3 mmol) was dissolved in EtOH (20 mL), treated with
ethyl formimidate-HCI (3.7 mmol), stirred for 22 h, and filtered. The filtrate
was treated with EtZO (40 mL), cooled in an ice bath, and filtered to give
glassy AL2. AL2 was dissolved in aq. HCI (4 N, 15 mL), stirred for 28 h, and
evaporated to give 33 as a white foam (0.75 g): mp 49-55 C. 1H NMR
(DMSO-ds) S 9.35 (s, 1 H), 9.1 (m, 2 H), 8.8 (m, 2 H), 8.70 (d, 1 H), 8.5 (m,
1
H), 7.8 (m, 2 H), 5.2 (dd, 1 H), 4.2'(m, 1 H), 3.8 (m, 2 H), 3.2 (m, 2 H), 2.8
(m,
2 H), 2.6 (m, 1 H), 2.3 (m, 2 H), 1.8 (m, 3 H), 1.0-1.7 (m, 12 H); MS m/e 444
(MH+).
47