Note: Descriptions are shown in the official language in which they were submitted.
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
TARGET MATERIAL REMOVAL USING RARE EARTH METALS
CROSS REFERENCE TO RELATED APPLICATION
The present application claims the benefits of U.S. Provisional Application
Serial
No. 61/113,435, filed November 11, 2008, entitled "Arsenic Removal Using Rare
Earth
Metals"; U.S. Provisional Application Serial No. 61/179,622, filed May 19,
2009, entitled
"Arsenic Removal Using Rare Earth Metals"; U.S. Provisional Application Serial
No.
61/186,258, filed June 11, 2009, entitled "Arsenic Removal Using Rare Earth
Metals";
U.S. Provisional Application Serial No. 61/186,662, filed June 12, 2009,
entitled "Arsenic
Removal Using Rare Earth Metals"; U.S. Provisional Application Serial No.
61/223,222,
filed July 6, 2009, entitled "Arsenic Removal Using Rare Earth Metals"; U.S.
Provisional
Application Serial No. 61/223,608, filed July 7, 2009, entitled "Arsenic
Removal Using
Rare Earth Metals"; U.S. Provisional Application Serial No. 61/240,867, filed
September
9, 2009, entitled "Arsenic Removal Using Rare Earth Metals"; U.S. Provisional
Application Serial No. 61/224,316, filed July 9, 2009, entitled "Removal of
Soluble
Arsenic from a Sulfide Waste Stream"; U.S. Provisional Application Serial No.
61/232,702, filed August 10, 2009, entitled "Lanthanide-Based Compound for
Arsenic
Removal in Sulfide Waste Stream"; and U.S. Provisional Application Serial No.
61/232,703, filed August 10, 2009, entitled "Aluminum-Induced Precipitation
for Arsenic
Removal in Sulfide Waste Stream"; which are all incorporated herein by this
reference in
their entirety.
Cross reference is made to U.S. Patent Application Serial Nos. 11/958,602,
filed
December 18, 2007; 11/958,644, filed December 18, 2007; and 11/958,968, filed
December 18, 2007, each of which is incorporated herein by this reference in
its entirety.
FIELD
The invention relates generally to removal, using rare earth metals, of target
materials and particularly to removal and stabilization, using rare earth
metals, of arsenic.
BACKGROUND
Harmful metals, such as arsenic, oxyanions of heavy metals, and their
radioactive
isotopes, naturally occur in a variety of combined forms in the earth. Their
presence in
natural waters may originate, for example, from geochemical reactions,
industrial waste
discharges (including those generated by nuclear, oil, and/or coal fired power
plants), or
agricultural, industrial, and/or home uses of pesticides, herbicides,
insecticides, and
1
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
rodenticides, and other sources. Because the presence of high levels of
certain harmful
metals, particularly arsenic, may have carcinogenic and other deleterious
effects on living
organisms, the U.S. Environmental Protection Agency ("EPA") and the World
Health
Organization have set the maximum contaminant level ("MCL") for various
harmful
metals in drinking water. Harmful metal concentrations in wastewaters, ground
waters,
surface waters, subterranean waters, and geothermal waters frequently exceed
this level.
Thus, the current MCL and any future decreases create the need for new
techniques to
economically and effectively remove arsenic from drinking, well, and
industrial waters.
Many of the harmful metals have multiple oxidation states, which can
complicate
their removal. For example, under normal conditions, arsenic is found
dissolved in
aqueous or aquatic systems in the +3 and +5 oxidation states, usually in the
form of
arsenite (As02-1) and arsenate (As04-3). The removal of arsenic by adsorption
or
precipitation technologies requires the arsenic to be in the arsenate form.
Arsenite, in
which the arsenic exists in the +3 oxidation state, is only partially removed
by adsorption
and precipitation technologies because the predominate form of arsenite is
arsenious acid
(HAsO2). Arsenious acid is a weak acid and maintains a neutral charge (that
is, contains
minimal, if any, arsenite (As02-1)) at a pH between pH 5 and pH 8 where
adsorption takes
place most effectively.
Various other technologies have been used to remove harmful metals from
aqueous
systems. Examples of such technologies include adsorption on high surface area
materials, such as alumina and activated carbon, ion exchange with anion
exchange resins,
co-precipitation optionally using flocculants, and electrodialysis. Most
technologies for
harmful metal removal are hindered by the difficulty of removing a number of
these
metals.
Harmful metal removal may be further complicated by co-occurrence with
valuable metals. In many industrial processes, contaminated process solids and
solutions
contain not only harmful metals, such as arsenic, but also valuable metals,
such as copper,
nickel, cobalt, and/or precious metals. Arsenic is often dissolved selectively
from the
solid wastes and isolated from streams using a co-precipitation process. This
process uses
iron reagents to precipitate arsenic as ferric arsenate. This precipitation
method requires a
series of pH adjustments to form and, in many applications, produces an
excessively large
volume of, the ferric arsenate precipitate.
2
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
Precipitation using rare earth metals is a newly invented technology that has
shown promise removing harmful and/or valuable metals from contaminated waste
streams. Cerium, in particular, has been used to remove oxyanions of various
harmful
metals, such as arsenic, antimony, molybdenum, tungsten, vanadium, and
uranium.
There is a need for a process to remove harmful and/or valuable metals
effectively
from solids and/or liquid streams.
SUMMARY
These and other needs are addressed by the various embodiments and
configurations of the present invention. This disclosure relates generally to
target material
removal from fluids and stabilization of the removed target material.
In one embodiment, a process is provided that includes the steps of:
(a) contacting a process stream (which may be a liquid, gas, slurry, and the
like)
comprising a target material other than arsenic with a soluble fixing agent,
the soluble
fixing agent comprising a rare earth, to form an insoluble target material-
containing
composition comprising the target material and the rare earth; and
(b) removing the insoluble target material-containing composition from the
process
stream to form a purified process stream.
The insoluble target material-containing composition is typically in the form
of
precipitate that can be removed as a solid. Preferably, the insoluble target
material-
containing composition has at least about 0.01 wt. %, even more preferably at
least about
0.1 wt. %, and even more preferably ranges from about 5 to about 50 wt. % of
the target
material. The target material is commonly in the form of an oxygen-containing
anion with
an oxyanion being illustrative. The soluble fixing agent, or precipitant, can
be supported
by a suitable carrier or be unsupported. The ability to form the insoluble
target material-
containing composition in the form of a solid comprising a relatively high
concentration of
the target material can greatly reduce the volume of the insoluble target
material-
containing composition requiring disposal, thereby reducing disposal costs.
In another embodiment, a process is provided that includes the steps:
(a) providing an arsenic and a valuable metal-containing solid material;
(b) contacting the solid material with a leaching agent to form a leach stream
comprising dissolved arsenic and an arsenic depleted solid, the dissolved
arsenic
comprising most of the arsenic contained in the solid material and the arsenic
depleted
solid comprising most of the valuable product contained in the solid material;
3
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
(c) contacting the leach stream with a soluble fixing agent to form a target
material-containing composition comprising most of the arsenic in the leach
stream and
the soluble fixing agent; and
(d) removing most of the target material-containing composition from the leach
stream, wherein the soluble fixing agent comprises a rare earth.
The fixing agent can be in any suitable form, such as a solid, a coating, a
particle, a
nano-particle, a sub-micron particle, a dissolved rare earth species, and/or
powder. The
rare earth can be in the form of a solid, or the solid may be supported by a
polymeric
binder interconnecting particles of the rare earth-containing compound. The
coating can
be on any suitable carrier. In one application the fixing agent is a
lanthanoid, particularly
cerium. The cerium is typically in the form of a cerium (IV) oxide or a
dissolved cerium
species, which, for example, can be a cerium (III) and/or (IV) salt solution.
The valuable product can be any metal or metalloid, with a transition metal,
aluminum, tin, and lead being typical and titanium, chromium, manganese, iron,
cobalt,
nickel, copper, zinc, molybdenum, a platinum group metal, a precious metal,
and mixtures
thereof being even more typical.
In another embodiment, a solid-phase composition is provided that includes:
(a) a target material;
(b) oxygen;
(c) water; and
(d) a rare earth.
The lattice structure of the crystalline phase is believed to belong to a
trigonal
space group.
In the case of arsenic as the target material, the chemical formula of the
composition is believed to be:
REAsO4 = (H2O), where 0 < X < 10 and "RE" refers to a rare earth element.
The composition is substantially crystalline, with the arsenic, oxygen, rare
earth
element, and water of hydration forming a crystal lattice.
In another embodiment, a method includes the steps of:
(a) providing a target material-containing stream;
(b) contacting the target material-containing stream with one or both of the
following:
4
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
(i) a rare earth salt additive, the rare earth salt additive comprising a rare
earth in the +3 oxidation state and a non-rare earth metal in the +3 oxidation
state;
and
(ii) a non-rare earth salt additive, the non-rare earth salt additive
comprising
a non-rare earth metal in the +3 oxidation state and being substantially free
of a
rare earth; and
(c) forming a precipitate between the target material and at least one of the
rare
earth and non-rare earth salt additives.
The non-rare earth metal can be any non-rare earth metal in the +3 oxidation
state,
with transition metals, boron, aluminum, gallium, indium, thallium, and
bismuth being
preferred, and the transition metals and aluminum being particularly
preferred. Preferred
transition metals include the elements having atomic numbers 22-29, 40-45, 47,
72-77,
and 79.
The first salt additive is, in one formulation, a bimetallic, lanthanide-based
salt
solution. In a preferred formulation, the first salt additive includes cerium
in the +3
oxidation state and aluminum in the +3 oxidation state.
The second salt additive, in a preferred formulation, contains aluminum in the
+3
oxidation state. The first and second salt additives can provide significant
reductions in
the amount of rare earths required to remove selected target materials,
particularly arsenic.
In another embodiment, a process is provided that includes the steps of:
(a) providing a feed stream comprising a target material;
(b) contacting the feed stream with an insoluble fixing agent to form a target
material-loaded insoluble fixing agent, the insoluble fixing agent comprising
at least one
of yttrium, scandium, and a lanthanoid, and the target material-loaded
insoluble fixing
agent comprising most of the target material in the feed stream, whereby the
target
material, in the target material-loaded insoluble fixing agent, forms a
composition with the
insoluble fixing agent;
(c) contacting the target material-loaded insoluble fixing agent with a
stripping
solution to dissolve, solubilize, or otherwise displace most of the target
material in the
target material-loaded insoluble fixing agent to form a loaded stripping
solution and barren
insoluble fixing agent; and
(d) removing at least most of the dissolved target material from the loaded
stripping solution.
5
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
In one process configuration, first and second fixing agents are used. In a
first
step, the feed comprises a target material-bearing aqueous solution, having a
first
concentration of target material. The target-bearing aqueous solution is
contacted with an
insoluble first fixing agent, such as an adsorbent or absorbent, to produce a
target material-
bearing first fixing agent. The first step removes most, if not all, of the
target material
from the target material-bearing aqueous solution. In a second step, the
target material-
bearing first fixing agent is contacted with an alkaline stripping solution
("release agent")
to produce an intermediate target material-rich solution having a second
concentration of
the target material. The second concentration of target material may exceed
the first
concentration of target material. The alkaline stripping solution can be or
include, for
example, the leaching agent discussed above. Commonly, the second
concentration of
target material is a concentration about equal to the solubility limit of the
target material
(at the process conditions of the second step). More commonly the second
concentration
of the target material is between about 0.1 and about 2,500 g/L, even more
commonly
between about 0.1 and about 1,000 g/L, and even more commonly between about
0.25 g/L
and about 500 g/L. Finally, a soluble or dissolved second fixing agent is
contacted with
the intermediate target material-rich solution in an amount sufficient to
precipitate most, if
not all, of the target material as a target material-bearing solid. The target
material-
bearing solid may be separated from the intermediate solution by any suitable
solid/liquid
separation technique to produce a separated solid for disposal and a stripping
solution for
recycle to the second step.
The insoluble first fixing agent is commonly a particulate solid. The first
fixing
agent preferably is an insoluble rare earth metal compound, preferably an
insoluble rare
earth oxide comprising an insoluble rare earth compound, such as hydrous or
anhydrous
rare earth oxides, fluorides, carbonates, fluorocarbonates, silicates, and the
like. A
particularly preferred first fixing agent is CeO2. The first fixing agent is
particularly
effective in removing arsenic having an oxidation state of +3 or +5.
The soluble second fixing agent typically has an oxidation state lower than
the
oxidation state of the first fixing agent. Preferably, the oxidation state of
the second fixing
agent is one of +3 or +4. The soluble fixing agent preferably is a soluble
rare earth metal
compound and more preferably includes salts comprising rare earth compounds,
such as
bromides, nitrates, phosphites, chlorides, chlorites, chlorates, nitrates, and
the like. More
preferably, the soluble fixing agent is a rare earth (III) chloride.
6
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
In some embodiments, the target material will be present in a reduced
oxidation
state and this condition might be undesirable. In such cases, an oxidant may
be contacted
with the solution to increase the target material oxidation state. Using
arsenic as an
example, the presence of arsenite might favor the use of an oxidant before the
fixing agent
is applied.
The intermediate solution can include a residual valuable product. The
valuable
product is commonly any metal of interest, more commonly includes one or more
of the
transition metals and even more commonly includes a metal selected from the
group of
metals consisting of copper, nickel, cobalt, lead, precious metals, and
mixtures thereof.
All or a portion of the residual valuable product may be recovered from the
intermediate
solution.
In yet another embodiment, a method is provided that includes the steps of:
(a) receiving a target material-containing stream, the target material-
containing
stream comprising an interferor, the interferor adversely impacting (e.g.,
impairing the
level, extent, and/or degree of) rare earth precipitation of the target
material;
(b) removing at least most of the interferor from the target material-
containing
stream to form a treated stream comprising at least most of the target
material; and
(c) thereafter contacting the treated stream with a rare earth fixing agent to
precipitate most of the target material from the treated solution.
It has been discovered that interferors, particularly phosphates, fluorides,
carbonates, silicates, and vanadate can readily form compositions with or
otherwise
impede target material removal by the rare earth fixing agent, thereby
consuming
unnecessarily the fixing agent when it is desired to remove target materials,
such as
arsenic. Removing the competing or otherwise obstructing oxyanion interferors
prior to
fixing agent contact with the target material can reduce fixing agent
consumption.
In a further embodiment, a method is provided that includes the steps:
(a) providing a target material-containing stream comprising a dissolved
target
material and dissolved valuable product, the target material being in the form
of an
oxyanion and the valuable product being at least one of a transition metal,
aluminum, tin,
and lead and in a form other than an oxyanion;
(b) contacting the target material-containing stream with a rare earth fixing
agent
to precipitate at least most of the dissolved target material as a target
material-containing
7
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
precipitate while leaving at least most of the valuable product dissolved in a
treated
stream; and
(c) separating at least most of the target material-containing precipitate
from the
treated stream.
The present invention can include a number of advantages depending on the
particular configuration. The process of the present invention can remove
variable
amounts of target materials as needed to comply with application and process
requirements. For example, the target material removal process can remove high
concentrations of target materials to produce a treated solution having no
more than about
500 ppm, in some cases no more than about 100 ppm, in other cases no more than
about
50 ppm, in still other cases no more than about 20 ppb, and in still other
cases no more
than about 1 ppb target material. The insoluble rare earth/target material
product can be
qualified as non-hazardous waste. The target material removal process can be
relatively
insensitive to pH. The disclosed process can effectively fix target materials,
particularly
arsenic, from solutions over a wide range of pH levels, as well as at
extremely high and
low pH values. In contrast to many conventional target material removal
technologies,
this capability can eliminate the need to alter and/or maintain the pH of the
solution within
a narrow range when removing the target material. Moreover, where the aqueous
solution
is produced from the remediation of an arsenic-bearing material, it adds
flexibility because
the selection of materials and processes for leaching arsenic from an arsenic-
bearing
material can be made without significant concern for the pH of the resulting
arsenic-
containing solution. Further still, elimination of the need to adjust and
maintain pH while
fixing arsenic from an arsenic-containing solution can provide significant
cost advantages.
The target material removal process can also be relatively insensitive to
target material
concentration. The process can remove relatively low and high levels of target
materials,
particularly arsenic, from aqueous streams. The process can be a robust,
versatile process.
These and other advantages will be apparent from the disclosure of the
invention(s)
contained herein.
As used herein, the term "a" or "an" entity refers to one or more of that
entity. As
such, the terms "a" (or "an"), "one or more" and "at least one" can be used
interchangeably
herein. It is also to be noted that the terms "comprising", "including", and
"having" can
be used interchangeably.
8
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
As used herein, "absorption" refers to the penetration of one substance into
the
inner structure of another, as distinguished from adsorption.
As used herein, "adsorption" refers to the adherence of atoms, ions,
molecules,
polyatomic ions, or other substances of a gas or liquid to the surface of
another substance,
called the adsorbent. The attractive force for adsorption can be, for example,
ionic forces
such as covalent, or electrostatic forces, such as van der Waals and/or
London's forces.
As used herein, "at least one", "one or more", and "and/or" are open-ended
expressions that are both conjunctive and disjunctive in operation. For
example, each of
the expressions "at least one of A, B and C", "at least one of A, B, or C",
"one or more of
A, B, and C", "one or more of A, B, or C" and "A, B, and/or C" means A alone,
B alone, C
alone, A and B together, A and C together, B and C together, or A, B and C
together.
As used herein, a "composition" refers to one or more chemical units composed
of
one or more atoms, such as a molecule, polyatomic ion, chemical compound,
coordination
complex, coordination compound, and the like. As will be appreciated, a
composition can
be held together by various types of bonds and/or forces, such as covalent
bonds, metallic
bonds, coordination bonds, ionic bonds, hydrogen bonds, electrostatic forces
(e.g., van der
Waal's forces and London's forces), and the like.
As used herein, "insoluble" refers to materials that are intended to be and/or
remain as solids in water and are able to be retained in a device, such as a
column, or be
readily recovered from a batch reaction using physical means, such as
filtration. Insoluble
materials should be capable of prolonged exposure to water, over weeks or
months, with
little (< 5%) loss of mass.
As used herein, "oxyanion" or oxoanion is a chemical compound with the generic
formula AxOy' (where A represents a chemical element other than oxygen and 0
represents an oxygen atom). In target material-containing oxyanions, "A"
represents
metal, metalloid, and/or Se (which is a non-metal), atoms. Examples for metal-
based
oxyanions include chromate, tungstate, molybdate, aluminates, zirconate, etc.
Examples
of metalloid-based oxyanions include arsenate, arsenite, antimonate,
germanate, silicate,
etc.
As used herein, "particle" refers to a solid or microencapsulated liquid
having a
size that ranges from less than one micron to greater than 100 microns, with
no limitation
in shape.
9
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
As used herein, "precipitation" refers not only to the removal of target
material-
containing ions in the form of insoluble species but also to the
immobilization of
contaminant-containing ions on or in insoluble particles. For example,
"precipitation"
includes processes, such as adsorption and absorption.
As used herein, "rare earth" refers to one or more of yttrium, scandium,
lanthanum,
cerium, praseodymium, neodymium, samarium, europium, gadolinium, terbium,
dysprosium, holmium erbium, thulium, ytterbium, and lutetium. As will be
appreciated,
lanthanum, cerium, praseodymium, neodymium, samarium, europium, gadolinium,
terbium, dysprosium, holmium erbium, thulium, ytterbium, and lutetium are
known as
lanthanoids.
As used herein, "soluble" refers to materials that readily dissolve in water.
For
purposes of this invention, it is anticipated that the dissolution of a
soluble compound
would necessarily occur on a time scale of minutes rather than days. For the
compound to
be considered to be soluble, it is necessary that it has a significantly high
solubility
product such that upwards of 5 g/L of the compound will be stable in solution.
As used herein, "sorb" refers to adsorption and/or absorption.
The preceding is a simplified summary of the invention to provide an
understanding of some aspects of the invention. This summary is neither an
extensive nor
exhaustive overview of the invention and its various embodiments. It is
intended neither
to identify key or critical elements of the invention nor to delineate the
scope of the
invention but to present selected concepts of the invention in a simplified
form as an
introduction to the more detailed description presented below. As will be
appreciated,
other embodiments of the invention are possible utilizing, alone or in
combination, one or
more of the features set forth above or described in detail below.
BRIEF DESCRIPTION OF THE DRAWINGS
The accompanying drawings are incorporated into and form a part of the
specification to illustrate several examples of the present invention(s).
These drawings,
together with the description, explain the principles of the invention(s). The
drawings
simply illustrate preferred and alternative examples of how the invention(s)
can be made
and used and are not to be construed as limiting the invention(s) to only the
illustrated and
described examples.
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
Further features and advantages will become apparent from the following, more
detailed, description of the various embodiments of the invention(s), as
illustrated by the
drawings referenced below.
Figs. IA and B depict a process flow chart according to a first embodiment;
Fig. 2 depicts a process flow chart according to a second embodiment;
Fig. 3 is a plot of loading capacity (mg/g) (vertical axis) versus arsenic
concentration (g/L) (horizontal axis);
Fig. 4 is a plot of final arsenic concentration (mg/L) (vertical axis) versus
molar
ratio of cerium:arsenic (horizontal axis);
Fig. 5 is a plot of final arsenic concentration (mg/L) (vertical axis) versus
molar
ratio of cerium to arsenic (horizontal axis);
Fig. 6 is a series of XRD patterns for precipitates formed upon addition of Cc
(III)
or Cc (IV) solutions to sulfide-arsenite solutions and sulfate-arsenate
solutions;
Fig. 7 is a plot of arsenic sequestered (micromoles) (vertical axis) and
cerium
added (micromoles) (horizontal axis);
Fig. 8 is a series of XRD patterns exhibiting the structural differences
between
gasparite (CeAsO4) and the novel trigonal phase CeAsO4 = (H20)X;
Fig. 9 is a plot of residual arsenic concentration (mg/L) (vertical axis)
versus molar
ratio Ce/As (horizontal axis); and
Fig. 10 is a plot of loading capacity (As mg Ce02 g) (vertical axis) versus
molar
ratio (Ce/As) (horizontal axis);
Fig. 11 is a plot of residual arsenic concentration (mg/L) (vertical axis)
versus
molar ratio (horizontal axis); and
Fig. 12 is a series of XRD patterns exhibiting the structural differences
among
trigonal CeAsO4 = (H2O) trigonal CeAsO4 = (H2O) and
trigonal BiPO4 = (H20)0 67 (simulated).
DETAILED DESCRIPTION
In one aspect, the present invention uses an insoluble or soluble fixing agent
or
both to remove selected target materials from an aqueous solution. The fixing
agent,
whether soluble or insoluble, preferably includes a rare earth. Specific
examples of such
materials that have been described as removing arsenic include lanthanum (III)
11
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
compounds, soluble lanthanum metal salts, lanthanum oxide, cerium dioxide, and
soluble
cerium salts.
The particular target materials removed depend on whether the fixing agent is
insoluble or soluble in an aqueous process, particularly under standard
conditions (e.g.,
Standard Temperature and Pressure "STP"). While not wishing to be bound by any
theory, it is believed, using arsenic and cerium as an example, that insoluble
cerium fixing
agents remove effectively arsenic, when part of a complex multi-atomic unit
having an
oxidation state preferably of +3 or higher and even more preferably a
oxidation state from
+3 to +5, while soluble cerium fixing agents remove effectively arsenic, when
part of a
complex multi-atomic unit, having an oxidation state of +5. "Target
materials", as used
herein, preferably includes not only arsenic but also elements having an
atomic number
selected from the group of consisting of atomic numbers 5, 9, 13, 14, 22 to
25, 31, 32, 33,
34, 35, 40 to 42, 44, 45, 49 to 53, 72 to 75, 77, 78, 80, 81, 82, 83, 85, 92,
94, 95, and 96
and even more preferably from the group consisting of atomic numbers 5, 13,
14, 22 to 25,
31, 32, 33, 34, 40 to 42, 44, 45, 49 to 52, 72 to 75, 77, 78, 80, 81, 82, 83,
92, 94, 95, and
96. These atomic numbers include the elements of arsenic, aluminum, astatine,
bromine,
boron, fluorine, iodine, silicon, titanium, vanadium, chromium, manganese,
gallium,
thallium, germanium, selenium, mercury, zirconium, niobium, molybdenum,
ruthenium,
rhodium, indium, tin, antimony, tellurium, hafnium, tantalum, tungsten,
rhenium, iridium,
platinum, lead, uranium, plutonium, americium, curium, and bismuth. Uranium
with an
atomic number of 92 is an example of a target material having radioactive
isotope.
Examples of target materials amenable to removal and stabilization by the
insoluble fixing
agent include, without limitation, target materials in the form of complex
anions, such as
metal, metalloid, and selenium oxyanions.
In one configuration, the fixing agent reacts with an aqueous solution
comprising
one or more target material-containing oxyanions to form a purified aqueous
stream. The
fixing agent can be soluble or in the aqueous solution under standard
conditions (e.g.,
STP). In some instances, the fixing agent can comprise a mixture of fixing
agents, the
mixture comprising soluble or insoluble fixing agents. The fixing agent reacts
with one or
more of the target material-containing oxyanions, oxyanion radioactive
isotopes, or other
toxic elements in an aqueous feed to form insoluble species with the fixing
agent. The
insoluble species are immobilized, for example, by precipitation, thereby
yielding a treated
and substantially purified aqueous stream.
12
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
Although the disclosure is discussed primarily with reference to arsenic and
arsenic-containing species, such as the arsenate and arsenite oxyanions, it is
to be
understood that the teachings of this disclosure apply equally to the other
arsenic-
containing compounds and the non-arsenic elements and compounds listed above.
Referring to a first embodiment in Fig. IA, the target material-containing
solid 100
includes one or more target materials and, optionally, a valuable product
(which may itself
fall within the definition of a target material), such as a transition metal
(such as nickel,
cobalt, copper, a precious metal (such as, gold, and silver) and/or a platinum
group metal
(such as, ruthenium, rhodium, palladium, osmium, iridium, and platinum,
aluminum, tin,
and lead). The target material and valuable product can be present as an
element or
compound. Examples of the solid 100 include products, byproducts and waste
materials
from industries such as: mining; metal refining; steel manufacturing; glass
manufacturing;
metal working processing and/and manufacturing; chemical and petrochemical
production,
processing and manufacturing; as well as contaminated soil, wastewater sludge,
and
process stream remediation and the like. Specific examples of target material-
bearing
solids 100 include ores, mine or mill tailings, concentrates, calcines, slag,
and mattes, and
spent catalysts.
In one application, the target material-containing solid 100 is derived from
an
electrolyte, stripping solution, or leach solution containing dissolved nickel
in a
concentration of from about 5 mg/L to about 1,000 g/L nickel, chlorine in a
concentration
of from about 5 mg/L to about 1,000 g/L, sulfate in a concentration of from
about 5 mg/L
to about 5,000 g/L, arsenic (III) in a concentration of from about 1 to about
1,500 mg/L,
cobalt in a concentration of from about 5 to about 5,000 mg/L, copper in a
concentration
of from about 0.1 to about 1,500 mg/L, sodium in a concentration of from about
1 to about
1,500 mg/L, and lead in a concentration of from about 10 mg/L to about 1,500
g/L. The
target material-containing solid 100 is derived by contacting, such as by
sparging, a
reductant, preferably H2S, through the solution. The resulting target material-
containing
solid 100 typically includes from about 1 to about 10 wt. % As2S3, from about
25 to about
75 wt. % CuS, from about 0.1 to about 2.5 wt. % lead, and from about 1 to
about 25 wt. %
NiS.
In step 104, the target material-containing solid 100 is contacted with an
aqueous
leaching agent to dissolve the target material (and optionally the valuable
product) and
form a target material-containing stream 108. The aqueous leaching agent can
be any
13
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
acidic (e.g., pH less than about pH 7) or alkaline (e.g., pH more than about
pH 7) leach
solution that is capable of dissolving, from the target material-containing
solid 100, at least
most of the target material. Examples of leaching agents include inorganic
salts (e.g.,
alkali and alkaline earth metal phosphates, chlorides, nitrates, sulfates, and
chlorates),
inorganic acids (e.g., mineral acids such as sulfuric acid, hydrochloric acid,
nitric acid, and
phosphoric acid), organic salts (e.g., citrate and acetate), organic acids
(e.g., citric acid and
acetic acids), and alkaline agents (e.g., hydroxide, cyanide, thiosulfate, and
thiourea).
Preferably, the leaching agent is a base, such as a carbonate (XCO3),
bicarbonate
(XHCO3), hydroxide (XOH), and other metal oxides and compounds of oxygen,
nitrogen,
and sulfur with nonbonded electron pairs. X is normally an alkali or alkaline
earth metal.
More preferably, the alkaline solution includes a leaching agent in an amount
of less than
about 25 % by wt, even more preferably less than about 20 wt. %, and even more
preferably ranges from about 1 to about 15 wt. %, with about 5 wt. % being
preferred.
When the target material is arsenic, the aqueous leaching agent selectively
dissolves most
of the arsenic while leaving most of the valuable product in the solid
material. While not
wishing to be bound by any theory, it is believed that the caustic leaching
agent
metastasizes with copper to form soluble arsenic compounds.
When the aqueous leaching agent is a carbonate and is contacted with the
target
material-containing solid 100 discussed above, the arsenic-containing aqueous
leaching
agent commonly includes from about 15 to about 25 g/L Na2CO3, 1 to about 30
g/L
arsenic (III), from about 1 to about 10 g/L sulfur (e.g., as sulfide, sulfate,
and/or sulfite),
no more than about 5 g/L chlorine, no more than about 10 mg/L nickel, and no
more than
about 5 mg/L copper. The pH of the resulting solution is typically about pH 9
or higher
and even more typically ranges from about pH 9 to about pH 12.
The target material-containing stream 108 is separated from the target
material-
depleted solid by any well known liquid/solid separation technique.
Solid/liquid
separation is commonly performed by a number of techniques, including
filtering,
hydrocycloning, screening, centrifuging and gravity separating techniques,
such as by
counter current decantation and settling.
The target material-containing stream 108 typically contains a concentration
of
dissolved or otherwise solubilized target material ranging from about 0.1 g/L
up to the
solubility limit of the material in the stream under the conditions of the
stream. More
typically, the target material-containing stream concentration ranges from
about 0.1 g/L to
14
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
about 1,000 g/L, even more typically the target material concentration ranges
from about
0.1 to about 500 g/L, and even more typically the target material
concentration ranges
from about 0.1 g/L to about 100 g/L.
Optional step 112 adjusts the charge of most and even more preferably of about
75% or more of the target material or a composition incorporating the target
material to a
selected charge. For example, when the target material is arsenic the
preferred oxidation
state may be +5 because the soluble fixing agent may not form a precipitate
with the
arsenic at other arsenic oxidation states, specifically -3 (arsenides) and +3
(arsenites). The
oxidation state can be adjusted by any suitable oxidation and/or reduction
technique and/or
using any suitable oxidant and/or reductant. A non-limiting example of a
preferred
oxidant is a molecular oxygen-containing gas. The molecular oxygen-containing
gas is
normally sparged through the target material-containing stream.
Although not shown, the concentration of the target material in the target
material-
containing stream may be increased by suitable techniques, such as through
water
removal. Water may be removed, for example, by evaporation, distillation,
and/or
filtration techniques (such as, membrane filtration). Other techniques include
precipitation
and redissolution, absorption or adsorption followed by stripping, ion
exchange followed
by stripping, and the like of the target material.
In one application, most of the interferors (which interfere with removal of
the
target material), particularly fluorides, phosphates, carbonates, silicates,
and vanadium
oxides, are removed from the target material-containing stream by suitable
techniques,
such as ion exchange, membrane filtration, precipitation, a complexing agent,
and the like.
Interferors can compete with other target materials, particularly arsenic, for
available
fixing agents, thereby increasing fixing agent consumption and/or lowering
levels of target
material removal. In this application, the concentration of interferors is
maintained
preferably at a concentration of no more than about 300 ppm/interferor species
and even
more preferably no more than about 10 ppm/interferor species.
In step 116, the target material-containing stream 100 is contacted with a
soluble
fixing agent to form a precipitate-containing solution 120 containing a target
metal-
containing precipitate 128. Preferably, most and even more preferably 75% or
more of the
target material or a composition incorporating the target material forms, with
the soluble
fixing agent, the target material-containing precipitate 128. The soluble
fixing agent is
preferably one or more of scandium, yttrium, and a lanthanoid and is in a form
that is
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
soluble in water and/or the aqueous leaching agent. When the soluble fixing
agent
comprises cerium, it typically has an oxidation state of +4 or less. The
fixing agent can
be, without limitation, a soluble salt, such as bromides, nitrates,
phosphites, chlorides,
chlorites, chlorates, and the like of scandium, yttrium, or a lanthanoid, with
a chloride of
cerium (III) or cerium (IV) being preferred. While not wishing to be bound by
any theory,
it is believed that soluble forms of cerium (IV) can form nanocrystalline
cerium dioxide,
which then sorbs target materials or a composition incorporating the target
material. The
soluble fixing agent is added, commonly as a separate aqueous solution, to the
target
material-containing stream preferably in an amount to produce an average molar
ratio of
fixing agent to target material in solution of less than about 8:1 and more
preferably
ranging from about 0.5:1 to about 5:1.
During step 116, the pH of the target material-containing stream preferably
ranges
from about pH 4 to about pH 9 and even more preferably from about pH 5.5 to
about pH
8. In some instances, a pH adjustment may be required before step 116. The pH,
when
too high or too low, can cause the soluble fixing agent (discussed below) to
precipitate out
of solution (e.g., when the pH is too high, the fixing agent can precipitate
out of solution
as a carbonate or hydroxide and when the pH is too low the fixing agent can
precipitate
out of solution as a sulfate).
A chelating agent can be added to the soluble fixing agent aqueous solution to
increase the solubility of the fixing agent in the aqueous solution. A typical
chelating
agent is a chemical compound containing at least two nonmetal entities capable
of binding
to a metal atom and/or ion. While not wishing to be bound by any theory,
chelating agents
function by making several chemical bonds with metal ions. Exemplary chelating
agents
include ethylene diamine tetra acetic acid (EDTA), dimercaprol (BAL),
dimercaptosuccinic acid (DMSA), 2,3-dimercapto-l-propanesulfonic acid (DMPS),
and
alpha lipoic acid (ALA), aminophenoxyethane-tetraacetic acid (BAPTA),
deferasirox,
deferiprone, deferoxamine, diethylene triamine pentaacetic acid (DTPA),
dimercapto-
propane sulfonate (DMPS), dimercaptosuccinic acid (DMSA), ethylenediamine
tetraacetic
acid (calcium disodium versante) (CaNa2-EDTA), ethylene glycol tetraacetic
acid
(EGTA), D-penicillamine, methanesulfonic acid, methanephosphonic acid, and
mixtures
thereof.
The soluble fixing agent can further include an organic or inorganic additive.
Preferably, the additive is one or more of a flocculent, coagulant, and
thickener, to induce
16
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
flocculation, settling, and/or formation of the precipitated solids. Examples
of such
additives include lime, alum, ferric chloride, ferric sulfate, ferrous
sulfate, aluminum
sulfate, sodium aluminate, polyaluminum chloride, aluminum trichloride,
polyelectrolytes,
polyacrylamides, polyacrylate, and the like.
In one process configuration, the target material-containing stream includes,
in
addition to the target material, a dissolved valuable product as a dissociated
or dissolved
cation. In other words, the dissolved valuable product is not, under the
conditions of the
stream, in the form of an oxyanion but occurs as a positively charged metal
ion. When the
soluble fixing agent is added, most of the target material is precipitated
while most of the
valuable product remains dissolved in the precipitate-containing solution.
In step 124, at least most of the target material-containing precipitate in
the
resulting slurry is separated from the aqueous leaching agent (which may be
recycled to
step 104) to form the separated target material-containing precipitate 128
(which includes
most of the target material) and treated stream 140 (which includes most of
the leaching
agent). After step 124, the treated stream 140 typically contains no more than
about 500
ppm and even more typically no more than about 50 ppm dissolved target
material.
Residual soluble fixing agent dissolved in the aqueous leaching agent can be
removed by adding a salt, such as mineral acid salt (e.g., NaCl) or a halide
(e.g., an alkali
metal or alkaline earth metal fluoride), or selected oxyanion, such as
phosphate or sulfate,
to the aqueous leaching agent. Alternatively, the soluble rare earth can be
oxidized, such
as by sparging with oxygen, to a higher oxidation state, optionally followed
by pH
adjustment to a higher pH, to precipitate the rare earth as an insoluble
compound, such as
a rare earth oxide. In another technique, the pH of the aqueous leaching agent
is
increased, preferably to a pH of at least about pH 7 and even more preferably
to a pH of at
least about pH 10 to precipitate out the residual soluble fixing agent. The
removal of
excess soluble fixing agent can occur before or after step 124.
The separated target material-containing precipitate 128 is dewatered in step
132 to
form the dewatered precipitate 136. Preferably, dewatering is performed for a
time and at
a temperature sufficient to remove at least about 50% and even more preferably
at least
about 75% of the water contained within the separated target material-
containing
precipitate 128. Typically, the separated target material-containing
precipitate 128 will be
dewatered for a time ranging from about 0.1 to about 24 hours at a temperature
ranging
from about 0 to about 250 C, with about 8 hours at about 100 C being even more
17
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
preferred. The dewatered precipitate 136 is typically a low surface area
agglomerate
having a high bulk density and low solubility in the aqueous leaching agent.
When arsenic
(V) is the target material and cerium (III) the soluble fixing agent, it is
believed that the
precipitate is predominantly gasparite (cerium arsenate). Typically, the
dewatered
material includes at least about 5 wt. %, even more preferably at least about
10 wt. %, and
even more preferably at least about 20 wt. % of the target material. The
dewatered
precipitates 136 contains preferably at least most and even more preferably at
least about
85% of the dissolved target material in the target material-containing stream
108 while the
treated stream 140 contains typically no more than about 25% of the dissolved
target
material in the target material-containing stream 108.
In one configuration, step 116 is performed using a concentrated and acidic
rare
earth salt solution added at a relatively rapid rate to produce a precipitate
that sequesters
more arsenic for a given amount of rare earth than is anticipated based on
theoretical
"best-case" calculations (which is for a rare earth: arsenic molar ratio of
1:1). The
preferred rare earth salt concentration in the salt solution is preferably at
least about 50
g/L, even more preferably from about 100 to about 400 g/L, and even more
preferably
from about 300 to about 400 g/L. The preferred pH of the salt solution is no
more than
about pH 2 and even more preferably no more than about pH 0. A particularly
preferred
formulation includes a solution of cerium in the +3 and/or +4 oxidation state
comprising
chloride and/or nitrate counter ions. The resulting precipitate has a low
density and is gel-
like. The precipitate is substantially free of any crystalline phases of
arsenic and rare earth
solids. For each mole of rare earth (e.g., cerium (III) or (IV)) in the gel,
there is typically
more than one mole of arsenic, more typically at least about 1.1 moles of
arsenic, and even
more typically at least about 1.25 moles of arsenic.
In another configuration, a novel rare earth - target material precipitate is
produced. In one process, the rare earth is cerium in the +3 oxidation state,
and the target
material is arsenic in the +5 oxidation state. Preferably, the cerium is in
the form of
cerium chloride (CeC13) and/or cerium nitrate (Ce(NO3)3). The target material-
containing
stream 108 commonly has an acidic pH and even more commonly a pH of no more
than
about pH 5. After or concurrently with the addition of the rare earth-
containing soluble
fixing agent, the pH of the target material-containing stream 108 is raised to
a second pH,
preferably of at least about pH 6 and even more preferably in the range of
about pH 6 to
about pH 10. The pH of the target material-containing stream 108 is raised
with a strong
18
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
base, such as an alkali metal hydroxide and group I salt of ammonia, amides,
and primary,
secondary, tertiary, or quaternary amines, with alkali metal hydroxides being
more
preferred, and alkali metal hydroxides being even more preferred. The
precipitate has a
crystal structure different from gasparite. Gasparite (CeAsO4) has a
monoclinic space
group with a monazite-type structure. While not wishing to be bound by any
theory, it is
believed that the crystal structure of the precipitate belongs to a trigonal
space group, such
as that of an apparently structurally analogous compound, BiPO4 (H2O)0.67 with
space
group P3121. The PDF card number for trigonal hydrated BiPO4 is 01-080-0208.
It is
further believed that the formula of the precipitate is REAsO4 = (H2O), 0 < X
< 10
and "RE" is a rare earth element. The water molecules are believed to occupy
lattice
positions, or are believed to be packed, in the crystalline structure.
In another configuration, the soluble fixing agent is combined with other
arsenic
removal agents to form a mixed salt additive. For example, the soluble fixing
agent(s) are
combined with one or more non-rare-earths having a +3 oxidation state,
particularly a
transition metal or metal from Groups 13 of the Periodic Table of the
Elements, with
aluminum or iron in the +3 oxidation state being preferred. Preferably, the
soluble fixing
agent is a rare earth metal in the +3 oxidation state, and the soluble fixing
agent and non-
rare-earth metal are each in the form of water dissociable salts. For example,
a double salt
mixture is formed by mixing cerium (III) chloride with aluminum (III)
chloride. In
another example, the double salt mixture is formed by mixing lanthanum (III)
chloride
with aluminum (III) chloride. In another example, the double salt mixture is
formed by
mixing lanthanum (III) chloride with iron (III) chloride. In a preferred
formulation, at
least one mole of the non-rare-earth is present for each mole of the rare
earth soluble
fixing agent. In a more preferred formulation, at least 3 moles of the non-
rare-earth are
present for each mole of the rare earth soluble fixing agent. In an even more
preferred
formulation, at least one mole of the non-rare-earth having an oxidation state
of +3 is
present for each mole of the rare earth soluble fixing agent having an
oxidation state of +3.
In a yet even more preferred formulation, at least 3 moles of the non-rare-
earth having an
oxidation state of +3 are present for each mole of the rare earth soluble
fixing agent having
an oxidation state of +3.
The contacting conditions for the mixed salt additive and target material-
containing stream 108 depend on the application. The mixed salt additive can
have any
19
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
pH; that is, the mixed salt can have an acidic, neutral, or basic pH.
Preferably, the mixed
salt additive has a pH less, that is more acidic, than the target material-
containing stream
108 pH. More preferably, the mixed salt additive has an acidic pH,
particularly when the
pH of the target material-containing stream 108 is basic. The mixed salt
additive, which is
typically a bimetallic lanthanide-based salt solution, is contacted with the
target material-
containing stream 108 at standard or higher temperature. The pH of the target
material-
containing stream 108, before and after mixed salt addition, can range from
about pH 0 to
about pH 14. More preferably, the pH of the mixed solution ranges from about
pH 8.5 to
about pH 13.5. The mixed salt solution can be contacted with the target
material-
containing stream over a wide temperature range, preferably from about the
freezing point
of the stream to about the boiling point of the target material-containing
stream.
The contacting of the bimetallic lanthanide-based salt and the target material-
containing stream 108 produces a precipitate. When the target material is
arsenic, the
precipitate is, for example, believed to be arsenoflorencite-(RE)
[(RE)Al3(As04)2(OH)6]
when the mixed salt additive comprises a rare earth ("RE") and aluminum (III),
and
graulichite-(RE) [(RE)Fe3(As04)2(OH)6] when the mixed salt additive comprises
a rare
earth and iron (III).
The separated liquid phase of the precipitate-containing solution 120
(hereinafter
the treated stream 140) retains most, if not all, of the dissolved sulfides,
while the target
material-containing precipitate 128 contains most, if not all, of the target
material, rare
earth, and the non-rare-earth(s) contained in the mixed salt additive. As can
be seen from
the above mineral formulas, the ratio of rare earth to arsenic is at least
about 1:2, which
represents a significant reduction in rare earth consumption relative to the
configurations
noted above in which rare earth arsenates, [REAsO4], are precipitated.
In another configuration, the soluble fixing agent is a non-rare-earth salt
additive
that does not include, or is substantially free of, rare earth metals. For
example, the non-
rare-earth is particularly a transition metal or metal from Groups 13 of the
Periodic Table
of the Elements, with aluminum in the +3 oxidation state being preferred.
Preferably, the
soluble fixing agent is the form of a non-rare earth (in a +3 oxidation state)
salt which
substantially dissociates in water under standard conditions (e.g., STP).
The contacting conditions for the mixed salt additive and target material-
containing stream 108 are not critical. Although the pH of the salt additive
solution can
be acidic or basic, the preferred pH is acidic, particularly when the pH of
the target
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
material-containing stream 108 is basic. The salt additive solution is
contacted with the
target material-containing stream 108 at standard (e.g., STP) or higher
temperature. The
pH of the target material-containing stream 108, before and after salt
addition, can range
from about pH 0 to about pH 14. More preferably, the pH of the mixed solution
ranges
from about pH 8.5 to about pH 13.5. The mixed salt solution can be added to
the target
material-containing stream over a wide temperature range, preferably from
about the
freezing point to about the boiling point of the target material-containing
stream.
A precipitate forms from the contacting of the salt additive solution with the
target
material-containing stream 108. When the target material is arsenic, the
precipitate is
believed to be alarsite [AlAsO4] or mansfieldite [AlAsO4 = H20] when the mixed
salt
additive comprises aluminum (III), and scorodite [FeAsO4] when the mixed salt
additive
comprises iron (III).
The separated liquid phase of the precipitate-containing solution 120 (or
treated
stream 140) retains most, if not all, of the dissolved sulfides while most, if
not all, of the
target material and non-rare-earth metal in the +3 oxidation state are
contained in the
target material-containing precipitate 128.
In one process configuration described above, the target material-containing
stream
includes dissolved valuable product(s) in a form other than as oxyanions. The
stream is
subjected to steps 112 (optional), 120, 124 and 128 to form a treated solution
and an target
material-containing precipitate. At least most, if not all, of the dissolved
valuable
product(s) remain in solution for recovery by any suitable technique. While
not wishing to
be bound by any theory, it is believed that soluble and insoluble rare earth
fixing agents
commonly do not remove metal and metalloid dissociated cations from solution.
This can
permit metal and metalloid oxyanions to be removed selectively from a solution
containing both metal and metalloid oxyanions and dissociated cations.
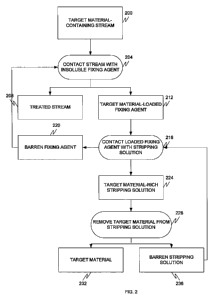
A second embodiment will now be discussed with reference to Fig. 2.
A target material-containing stream 200 is provided. The target material-
containing stream 200 can be the stream 108 or any other process stream,
byproduct and
waste stream from industries such as: mining; metal refining; steel
manufacturing; glass
manufacturing; metal manufacturing, working and/or processing; chemical and
petrochemical production, processing and manufacturing; streams produced from
treating
and/or remediating a contaminated soil, a wastewater sludge, and the like.
Specific
examples of target material-bearing streams include pregnant or barren leach
solutions
21
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
and/or other effluent streams, such as contaminated water. While portions of
the
disclosure herein refer to the removal of arsenic from mining tailings and
residues from
hydrometallurgical operations, such references are illustrative and should not
be construed
as limiting. As will be appreciated by one of ordinary skill in the art, the
teachings are
applicable to other target materials.
The target material concentration in the stream 200 is typically the same as
the
target material concentration in the target material-containing stream 108.
Arsenic, for
example, can be present in concentrations of more than about 20 ppb arsenic
and even
more than 1,000 ppb arsenic. The stream 200 can include other dissolved
components,
such as sulfides and/or sulfates in concentrations noted elsewhere in this
disclosure. The
pH of the stream 200 can be acidic, neutral, or basic, depending on the
application. The
stream 200 can also include dissolved solids with a common total dissolved
solid ("TDS")
level being at least about 5g/L, more commonly at least about 20 g/L, and even
more
commonly at least about 100 g/L.
In step 204, the stream 200 is contacted with an insoluble fixing agent to
form a
target material-loaded insoluble fixing agent 212 and a treated stream 208.
Preferably
most, and even more preferably about 75% or more, of the target material is
loaded on the
insoluble fixing agent. The target material forms a composition with the
insoluble fixing
agent. The affinity of the insoluble fixing agent for specific target
materials is believed to
be a function of pH and/or target material concentration. The insoluble fixing
agent is
commonly used as a particulate in a fixed or fluidized bed and, in certain
configurations,
may be desirable for use in a stirred tank reactor. In one configuration, the
insoluble
fixing agent is contained in one or more columns arranged in series or
parallel. In one
configuration, the insoluble fixing agent includes a flocculent and/or
dispersing agent,
such as those discussed above, to maintain a substantially uniform particle
distribution in
the bed.
For some insoluble fixing agents, step 204 may be preceded by an oxidation
step
112 to oxidize the target material for better target material removal
efficiency and/or
affinity of the target material for the insoluble fixing agent.
The insoluble fixing agent is preferably a rare earth and is in a form that is
substantially insoluble in water. The insoluble fixing agent can be, for
example, a hydrous
or anhydrous rare earth oxide, fluoride, carbonate, fluorocarbonate, or
silicate of
scandium, yttrium, or a lanthanoid, with an oxide of cerium being preferred
and cerium
22
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
(IV) oxide even more preferred. The insoluble fixing agent is preferably a
finely divided
solid having an average surface area of between about 25 m2/g and about 500
m2/g, more
preferably between about 70 m2/g and about 400 m2/g, and even more preferably
between
about 90 m2/g and about 300 m2/g.
The insoluble fixing agent can be blended with or include other components,
such
as ion-exchange materials (e.g., synthetic ion exchange resins), porous carbon
such as
activated carbon, metal oxides (e.g., alumina, silica, silica-alumina, gamma-
alumina,
activated alumina, acidified alumina, and titania), metal oxides containing
labile metal
anions (such as aluminum oxychloride), non-oxide refractories (e.g., titanium
nitride,
silicon nitride, and silicon carbide), diatomaceous earth, mullite, porous
polymeric
materials, crystalline aluminosilicates such as zeolites (synthetic or
naturally occurring),
amorphous silica-alumina, minerals and clays (e.g., bentonite, smectite,
kaolin, dolomite,
montmorillinite, and their derivatives), ion exchange resins, porous ceramics
metal silicate
materials and minerals (e.g., one of the phosphate and oxide classes), ferric
salts, and
fibrous materials (including synthetic (for example, without limitation,
polyolefins,
polyesters, polyamides, polyacrylates, and combinations thereof) and natural
(such as,
without limitation, plant-based fibers, animal-based fibers, inorganic-based
fibers,
cellulosic, cotton, paper, glass and combinations thereof).
The insoluble fixing agent may be derived from precipitation of a rare earth
metal
salt or from thermal decomposition of, for example, a rare earth metal
carbonate or oxalate
at a temperature preferably between about 100 to about 700 and even more
preferably
between about 180 and 350 C in a furnace in the presence of an oxidant, such
as air.
Formation of the insoluble fixing agent is further discussed in copending U.S.
Application
Serial No. 11/932,837, filed October 31, 2007, which is incorporated herein by
this
reference.
Although the preferred insoluble fixing agent comprises a rare earth compound,
other fixing agents may be employed. Any fixing agent, whether solid, liquid,
gaseous, or
gel, that is effective at fixing the target material in solution through
precipitation ion
exchange, or some other mechanism may be used. Examples of other fixing agents
include at least those set forth above.
In one configuration, the insoluble fixing agent is an aggregated particulate
having
a mean surface area of at least about 1 m2/g. Depending on the application,
higher surface
areas may be desired. For example, the aggregated particulates can have a
surface area of
23
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
at least about 5 m2/g; in other cases, more than about 10 m2/g; and, in still
other cases,
more than about 25 m2/g. Where higher surface areas are desired, the
particulates can
have a surface area of more than about 70 m2/g; in other cases, more than
about 85 m2/g;
in still other cases, more than about 115 m2/g; and, in still other cases,
more than about
160 m2/g. The aggregated particulates can include a polymer binder, such as
thermosetting polymers, thermoplastic polymers, elastomeric polymers,
cellulosic
polymers, and glasses, to at least one of bind, affix, and/or attract the
insoluble fixing
agent constituents into particulates having one or more of desired size,
structure, density,
porosity, and fluid properties.
The insoluble fixing agent can include one or more flow aids, with or without
a
binder. Flow aids can improve the fluid dynamics of a fluid over and/or
through the
insoluble fixing agent to prevent separation of slurry components, prevent the
settling of
fines, and, in some cases, hold the fixing agent and other components in
place.
The process 200 operational conditions should be controlled. When arsenic is
the
target material, for example, the insoluble fixing agent, under proper process
conditions,
selectively removes at least most of the arsenic while leaving at least most
of the valuable
product as dissolved (cationic) species in solution. Although the insoluble
fixing agent
can effectively fix arsenic from solutions over a wide range of pH levels, the
pH of the
target material-containing stream preferably is no more than about pH 6 and
even more
preferably ranges from about pH 2 to about pH 5 to adsorb both arsenic (V) and
arsenic
(III). Arsenic (III) sorbs onto the insoluble fixing agent over a broad pH
range while
arsenic (V) is preferably sorbed by the insoluble fixing agent at lower pH
levels. The
aqueous solution may contain dissolved solids, with a total dissolved solid
content of at
least about 50 g/L being typical.
The treated stream 208 has, relative to the stream 200, a reduced
concentration of
the target material. Commonly, most, even more commonly about 75% or more, and
even
more commonly about 95% or more of the target material in the stream 200 is
loaded onto
the insoluble fixing agent. In one application, the treated stream 208
preferably has no
more than about 1,000 ppm, even more preferably no more than about 500 ppm,
even
more preferably no more than about 50 ppm, and even more preferably no more
than
about 5 ppm of the target material.
When a pre-selected degree of target material loading occurs, the target
material-
loaded fixing agent 212 is contacted, in step 216, with a stripping solution,
or release agent
24
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
238, to unload, or dissolve, preferably most and even more preferably about
95% or more
of the agent and form a barren fixing agent 220 (which is recycled to step
204) and a target
material-rich stripping solution 224. Any acidic, neutral, or basic stripping
solution or
release agent may be employed. The desorption process of the rare earth loaded
insoluble
fixing agent is believed to be a result of a -one or more of. 1) a stronger
affinity by the rare
earth for the release agent than the sorbed target material or its composition
and 2) an
upward or downward adjustment of the oxidation state of the rare earth on the
surface of
the fixing agent 212 and/or of the sorbed target material and/or the sorbed
target material-
containing oxyanion.
In one application, the stripping solution is alkaline and comprises a strong
base,
including the strong bases discussed above. While not wishing to be bound by
any theory,
it is believed that, at high concentrations, hydroxide ions compete with, and
displace,
oxyanions from the surface of the insoluble fixing agent. In one formulation,
the stripping
solution includes a caustic compound in an amount preferably ranging from
about 1 to
about 15 wt. %, even more preferably from about 1 to about 10 wt. %, and even
more
preferably from about 2.5 to about 7.5 wt. %, with about 5 wt. % being even
more
preferred.
The preferred pH of the stripping solution 238 is preferably greater (e.g.,
more
basic) than the pH at which the target material was loaded onto the fixing
agent 212. The
stripping solution 238 pH is preferably at least about pH 10, even more
preferably at least
about pH 12, and even more preferably at least about pH 14.
In another application, the (first) stripping solution comprises an oxalate or
ethanedioate, which, relative to target material-containing oxyanions, is
preferentially
sorbed, over a broad pH range, by the insoluble fixing agent. In one process
variation to
desorb oxalate ions, the insoluble fixing agent is contacted with a second
stripping
solution (not shown) having a preferred pH of at least about pH 9 and even
more
preferably of at least about pH 11 to desorb oxalate and/or ethanedioate ions
in favor of
hydroxide ions. A strong base is preferred for the second stripping solution
(not shown).
Alternatively, the sorbed oxalate and/or ethanedioate anions can be heated to
a preferred
temperature of at least about 500 C to thermally decompose the sorbed oxalate
and/or
ethanedioate ions and remove them from the insoluble fixing agent.
In another application, the (first) stripping solution 238 includes a strongly
adsorbing exchange oxyanion, such as phosphate, carbonate, silicate, vanadium
oxide, or
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
fluoride, to displace the sorbed target material-containing oxyanion. The
first stripping
solution has a relatively high concentration of the exchange oxyanion.
Desorption of the
exchange oxyanion is at done at a different (higher) pH and/or exchange
oxyanion
concentration than the first stripping solution. For example, desorption can
be by a second
stripping solution (not shown) which includes a strong base and has a lower
concentration
of the exchange oxyanion than the oxyanion concentration in the first
stripping solution.
Alternatively, the exchange oxyanion can be thermally decomposed to regenerate
the
insoluble fixing agent. Alternatively, the exchange oxyanion can be desorbed
by
oxidation or reduction of the insoluble fixing agent or exchange oxyanion.
In another application, the stripping solution includes a reductant or
reducing
agent, such as ferrous ion, lithium aluminum hydride, nascent hydrogen, sodium
amalgam,
sodium borohydride, stannous ion, sulfite compounds, hydrazine (Wolff-Kishner
reduction), zinc-mercury amalgam, diisobutylaluminum hydride, lindlar
catalyst, oxalic
acid, formic acid, and a carboxylic acid (e.g., a sugar acid, such as ascorbic
acid), to
reduce the rare earth, sorbed target material, and/or sorbed target material-
containing
oxyanion. While not wishing to be bound by any theory nor by way of example,
surface
reduction of the insoluble fixing agent will reduce cerium (IV) to cerium
(III), which may
interact less strongly with target materials and oxyanions. Following or
concurrently with
surface reduction of the insoluble fixing agent, the pH is increased to desorb
the sorbed
target material or its oxyanion.
In another application, the stripping solution includes an oxidant or
oxidizing
agent, e.g., peroxygen compounds (e.g., peroxide, permanganate, persulfate,
etc.), ozone,
chlorine, hypochlorite, Fenton's reagent, molecular oxygen, phosphate, sulfur
dioxide, and
the like, that oxidizes the sorbed target material and/or its oxyanion to a
higher oxidation
state, e.g., arsenic (III) to arsenic (V), followed by a pH adjustment and a
desorption
process. Desorption of arsenic (V) from insoluble rare earth compounds, for
example,
typically occurs at a pH of at least about pH 12 and even more typically at
least about pH
14.
Regardless of the precise stripping mechanism, a first concentration of the
target
material in the target material-containing stream 200 is typically less than a
second target
material concentration in the target material-rich stripping solution 224.
Commonly, the
first concentration of the target material is no more than about 75% of the
second
concentration and even more commonly no more than about 50% of the second
26
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
concentration. By way of example, a first concentration of the arsenic is
between about
0.1 mg/L to about 5 g/L, and the second concentration of arsenic is between
about 0.25
g/L and about 7.5 g/L.
In step 228, the target material is removed from the rich stripping solution
224 by a
suitable technique to form a target material 232 and a barren stripping
solution 236 (which
is recycled to step 216). Removal may be effected by any suitable technique
including
precipitation (such as using a sulfide (for transition metals), an alkaline
earth metal
carbonate (for fluoride), and a rare earth or iron salt (for arsenic)),
adsorption, absorption,
electrolysis, cementation, amalgamation, and the like. In one configuration,
the target
material is precipitated using a soluble rare earth fixing agent as noted
above.
In another embodiment, the target material-containing stream 200 is the target
material-containing stream 104 (Fig. IA) and steps 204 and 216 are performed
immediately before step 112 or after step 112 and before step 116 to increase
the
concentration of the target material in the solution prior to step 116. This
can provide
benefits, such as handling reduced volumes of aqueous solutions in step 116.
Additionally, when arsenic is the target material and the solution to be
treated contains
dissolved metals, performing arsenic removal using an insoluble fixing agent
before
precipitation by the soluble fixing agent can isolate and selectively remove
arsenic from
the other metals. As will be appreciated, the soluble fixing agent may not
exclusively
precipitate arsenic and may depress/remove dissolved metals too.
EXPERIMENTAL
The following examples are provided to illustrate certain embodiments of the
invention and are not to be construed as limitations on the invention, as set
forth in the
appended claims. All parts and percentages are by weight unless otherwise
specified.
EXAMPLE 1
A set of tests were conducted to determine a maximum arsenic loading capacity
of
soluble cerium (III) chloride CeC13 in an arsenic-containing stream 108 to
reduce the
arsenic concentration to less than 50 ppm. As shown by Table 1, the arsenic-
containing
streams 108 (hereinafter alkaline leach solutions) tested had the following
compositions:
27
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
Table 1
Test Volume Na2CO3 Na2SO4 Na2HAsO4-7H20 As g/L
Number of DI (g) (g) (g)
mL
1 500 10 8.875 1.041 0.5
2 500 10 8.875 2.082 1
3 500 10 8.875 4.164 2
4 500 10 8.875 6.247 3
500 10 8.875 8.329 4
6 500 10 8.875 10.411 5
7 500 10 8.875 12.493 6
The initial pH of the seven alkaline leach solutions was approximately pH 11,
the
5 temperatures of the solutions were approximately 70 to 80 C, and the
reaction times were
approximately 30 minutes.
Seven alkaline leach solutions were made with varying arsenic (V)
concentrations,
which can be seen in Table 1 above. Each solution contained the same amount of
sodium
carbonate (20 g/L) and sodium sulfate (17.75 g/L). In a first series of tests,
3.44 mL of
cerium chloride (CeC13) were added to every isotherm and equates to 0.918 g
CeO2
(approximately 0.05 mole Cc) In a second series of tests, 6.88 mL of cerium
chloride was
added to every test and equates to 1.836 g CeO2 (approximately 0.1 mole Ce).
Below is
the guideline on how each isotherm test was performed.
In a first step, 200 mL of solution were measured out by weight and
transferred
into a 400 mL Pyrex beaker. The beaker was then placed on hot/stir plate and
heated to
70-80 C while being stirred.
In a second step, 3.44 mL of cerium chloride were measured out, by weight, and
poured into the mixing beaker of hot alkaline leach solution. Upon the
addition of cerium
chloride, a white precipitate formed instantaneously. To ensure that the white
precipitate
was not cerium carbonate [Ce2(CO3)3 = xH2O], step three was performed.
In the third step, 4.8 mL of concentrated HC1 were slowly added dropwise.
Fizzing was observed. The solution continued to mix for 30 minutes and was
then
allowed to cool for 4 hours before sampling.
28
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
The results are shown in Table 2:
Analysis using ICP-AES
Approximate Molar Final As Arsenic Loading Percent
Moles of Arsenic Ratio Concentration Removed Capacity Arsenic
Cerium (g/L) (Ce/As) (mg/L) (mg) (mg/g) Removed
Added
0.5 4.2 0 100 104 100
1.0 2.1 8 199 206 99
2.0 1.0 159 367 380 92
0.005 3.0 0.7 903 412 426 69
4.0 0.5 1884 408 422 51
5.0 0.4 2663 445 461 45
6.0 0.4 3805 409 422 34
0.5 8.3 0 102 53 100
1.0 4.2 0 201 104 100
2.0 2.1 55 388 201 97
0.01 3.0 1.4 109 577 299 96
4.0 1.1 435 709 367 89
5.0 0.8 1149 759 392 76
6.0 0.7 1861 810 419 67
Fig. 3 shows that the loading capacity begins to level off at the theoretical
capacity
of 436 mg/g if cerium arsenate (CeAsO4) was formed, leading one to believe it
was
formed. Fig. 4 displays that the molar ratio of cerium to arsenic required to
bring down
the arsenic concentration to less than 50 ppm lies somewhere between a 1 molar
and 2
molar ratio. However, at a 2 molar ratio a loading capacity of 217 was
achieved. Fig. 5
shows very similar results (essentially double the addition of CeC13); at a
molar ratio
between 1 and 2, the dissolved arsenic concentration can be below 50 ppm. This
capacity
may be improved with a lower molar ratio and tighter pH control.
EXAMPLE 2
In another experiment, 40 grams of cerium (IV) dioxide particles were loaded
into
a 1-inch column giving a bed volume of approximately 50 ml. The cerium dioxide
bed
had an arsenic-containing process stream [75% As(V), 25% As (III)] flowed
through the
bed and successfully loaded the media with approximately 44 mg of arsenic per
gram
CeO2 or with approximately 1,700 mg of arsenic total added to the column.
Following
this, the arsenic loaded cerium dioxide bed had the equivalent of six bed
volumes of 5%
NaOH solution passed through the bed, at a flow rate of 2 mL/min. This
solution released
29
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
approximately 80% of the 44 mg of arsenic per gram CeO2. Subsequently, the
same
cerium media was then treated again with the arsenic contaminated process
stream [75%
As(V), 25% As(III)], loading the media with another 25 mg of arsenic per gram
CeO2 or
with another 1,000 mg of arsenic. This experiment demonstrates how to
regenerate, and
thereby prolong the life of, the insoluble fixing agent and shows that the pH
of the arsenic-
containing solution can be important to determining the performance of the
insoluble
fixing agent.
EXAMPLE 3
A test was performed to remove residual rare earth fixing agents from an
alkaline
leach solution.
Fifteen grams of table salt (NaCl) were added to 150 mL of alkaline leach
solution
that contained residual cerium from cerium nitrate addition. Table 6 shows the
beginning
(control) and post-salt concentrations in the alkaline leach solution:
Table 3
Sample As Cc
(ppm) (ppm)
Control 220 4700
Salt Addition 250 270
As can be seen from this Table 3, 94% of the residual cerium has been removed.
EXAMPLE 4
In this example, the product of cerium and arsenic was shown to contain more
arsenic than would be anticipated based upon the stoichiometry of gasparite,
the
anticipated product of cerium and arsenic. Furthermore, the X-ray diffraction
pattern
suggests that the product is amorphous or nanocrystalline and is consistent
with ceria or,
possibly, gasparite. The amorphous or nanocrystalline phase not only permits
the
recycling of process water after arsenic sequestration but does so with a far
greater arsenic
removal capacity than is observed from other forms of cerium addition,
decreasing
treatment costs and limiting environmental hazards.
Eight 50 mL centrifuge tubes were filled with 25 mL each of a fully oxidized
solution of arsenate/sulfate/NaOH while another eight 50 mL centrifuge tubes
were filled
with 25 mL each of a fully reduced solution of arsenite/sulfide/NaOH that had
been
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
sparged with molecular oxygen for 2 hours. Both solutions contained 24 g/L
arsenic, 25
g/L NaOH, and the equivalent of 80 g/L sulfide. Each sample was then treated
with either
cerium (IV) nitrate or cerium (III) chloride. The cerium salt solutions were
added in doses
of 1, 2, 3, or 5 mL. No pH adjustments were made, and no attempt was made to
adjust the
temperature from ambient 22 C.
Fifteen of sixteen test samples showed the rapid formation of a precipitate
that
occupied the entire - 25 mL volume. The reaction between the two concentrated
solutions
took place almost immediately, filling the entire solution volume with a gel-
like
precipitate. The sixteenth sample, containing 5 mL of cerium (IV) remained
bright yellow
until an additional 5 mL of 50% NaOH was added, at which point a purple solid
formed.
Solids formed from the reaction of cerium and arsenic were given an hour to
settle
with little clarification observed. The samples were then centrifuged at 50%
speed for 5
minutes. At this point, the total volume of the solution and the volume of
settled solids
were recorded, and a 5 mL sample was collected for analysis. Since little more
than 5 mL
of supernatant solution was available (the concentration of arsenic was 24
g/L, meaning
that the concentration of cerium was also quite elevated), the samples were
filtered using
0.45 micron papers. The four samples with 5 mL of cerium salt added were not
filtered.
The supernatant solutions were collected and the volume recorded.
The filter cake from the reaction was left over the weekend in plastic weight
boats
atop a drying oven. Seventy-two hours later, the content of each boat was
weighed, and it
was determined that the pellets were still very moist (more mass present than
was added to
the sample as dissolved solids). The semi-dry solids of the samples with 2 mL
of cerium
salt solution were transferred to a 130 C drying oven for one hour, then
analyzed by XRD.
The XRD results are shown in Fig. 6. XRD results are presented for gasparite
(the
expected product) and the various systems that were present during the
experiments., with
"ceria" corresponding to cerium dioxide. As can be seen from Fig. 6, the XRD
analysis
did not detect any crystalline peaks or phases of arsenic and cerium solids in
the various
systems. The only crystalline material present was identified as either NaCl,
NaNO3
(introduced with the rare earth solutions) or Na2SO4 that was present in the
samples
prepared from Na2SO4. However, the broad diffraction peaks at about 29, 49,
and 57
degrees 2-Theta could be indicative of very small particles of ceria or,
possibly, gasparite.
The arsenic content of supernatant solutions was measured using ICP-AES. It
was
observed that both cerium (IV) and cerium (III) effectively removed arsenic
from the
31
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
system to about the same extent. As can be seen from Table 4 below and Fig. 7,
a greater
difference in arsenic removal was found between the fully oxidized system, and
the
system which was fully reduced before molecular oxygen sparging. Fig. 7 shows
a plot
for arsenic micromoles removed in an "oxidized" system staring with arsenate
and a
"molecular oxygen sparged" system starting with arsenite, which was
subsequently
oxidized to arsenate through molecular oxygen sparging.
Table 4
Arsenite/sulfide/NaOH + 02 Arsenate/sulfate/NaOH
Cerium Additive mL Cc CeO2 As ppm As capacity As ppm As
(g) (mg/g) capacity
(mg/g)
cerium (III) chloride 1 0.33 21200 242 20000 276
2 0.65 18800 271 8700 576
3 0.98 11200 324 1000 596
cerium (IV) nitrate 1 0.26 21600 265 19200 429
2 0.52 18800 237 8000 764
3 0.77 13600 322 3200 672
control 0 0.0 25200 24400
Fig. 7 shows the amount of arsenic consumed by the formation of precipitated
solids, plotted as a function of the amount of cerium added. The resultant
soluble arsenic
concentrations from this experiment can be divided into two groups: samples
containing
fully oxidized arsenate and sulfate and samples containing arsenite and
sulfite that was
sparged with molecular oxygen. The oxidation state of the cerium used as the
soluble
fixing agent had considerably less impact on the efficacy of the process,
allowing both
Ce(III) and Ce(IV) data to be fit with a single regression line for each test
solution. In the
case of the fully oxidized solution, arsenic sequestration with the solids
increases in an
arsenic to cerium molar ratio of 1:3, potentially making a product with a
stoichiometry of
Ce3AS4.
EXAMPLE 5
A series of experiments were performed, which successfully synthesized a novel
Ce-As compound. The experiments embody the precipitation of arsenic, in the As
(V)
32
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
state, from a highly concentrated waste stream of pH less than pH 2 by the
addition of a
soluble cerium salt in the Cc (III) state followed by a titration with sodium
hydroxide
(NaOH) solution to a range of between pH 6 and pH 10.
In a first test, a 400 mL solution containing 33.5 mL of a 0.07125 mol/L
solution
of NaH2AsO4 was stirred in a beaker at room temperature. The pH was adjusted
to
roughly pH 1.5 by the addition of 4.0 mol/L HN03, after which 1.05 g of
Ce(NO3)3 = 6
H2O was added. No change in color or any precipitate was observed upon the
addition of
the cerium (III) salt. NaOH (1.0 mol/L) was added to the stirred solution at a
dropwise
pace to bring the pH to pH 10.1. The pH was held at pH 10.2 0.2 for a period
of 1.5
hours under magnetic stir. After the reaction, the solution was removed from
the stir plate
and allowed to settle undisturbed for 12 to 18 hours. The supernatant was
decanted off
and saved for ICP-MS analysis of Cc and As. The solids were filtered through a
0.4 gm
cellulose membrane and washed thoroughly with 500 to 800 mL of de-ionized
water. The
solids were air-dried and analyzed by X-ray diffraction.
In a second test, a simulated waste stream solution was prepared with the
following
components: As (1,200 ppm), F (650 ppm), Fe (120 ppm), S (80 ppm), Si (50
ppm), Ca
(35 ppm), Mg (25 ppm), Zn (10 ppm), and less than 10 ppm of Al, K, and Cu. The
pH of
the solution was titrated down to pH 0.4 with concentrated HC1(12.1 mol/L),
and the
solution was heated to 70 C. A solution of CeC13 (6.3 mL, 1.194 mol/L) was
added to the
hot solution, and the pH was slowly increased to pH 7.5 by dropwise addition
of NaOH
(20 wt. %, 6.2 mol/L). The solution was then allowed to age at 70 C under
magnetic
stirring for 1.5 hours, holding pH at pH 7.5 0.2. The solution was then
removed from
the heat and allowed to settle undisturbed for 12 to 18 hours. The supernatant
was
decanted off and saved for ICP-MS analysis of Cc and As. The precipitated
solids were
centrifuged and washed twice before being filtered through a 0.4 gm cellulose
membrane
and washed thoroughly with 500 to 800 mL of de-ionized water. The solids were
air-dried
and analyzed by X-ray diffraction.
In a third test, solid powders of the novel Ce-As compound were tested for
stability
in a low-pH leach test. 0.5 g of the novel Ce-As compound were added to 10 mL
of an
acetic acid solution with a pH of either pH 2.9 or pH 5Ø The container was
sealed and
rotated for 18 2 hours at 30 2 revolutions per minute at an ambient
temperature in the
range of 22 5 C. After the required rotation time, the solution was filtered
through a 0.2
33
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
micron filter and analyzed by ICP-MS for Cc and As which may have been leached
from
the solid. Less than 1 ppm of As was detected by ICP-MS.
Fig. 8 compares the X-Ray Diffraction ("XRD") results for the novel Ce-As
compound (shown as trigonal CeAs 04 = (H20)X (both experimental and simulated)
and
gasparite (both experimental and simulated). Fig. 12 compares the XRD results
for
trigonal CeAs 04 = (H20)X (both experimental and simulated) and trigonal BiP
04
(H20)0.67 (simulated). The XRD results show that the precipitated crystalline
compound is
structurally different from gasparite (CeAsO4), which crystallizes in a
monoclinic space
group with a monazite-type structure, and is quite similar to trigonal BiP 04
= (H20)0.67-
Experiments with different oxidation states of Cc and As demonstrate that the
novel Cc - As compound requires cerium in the Cc (III) state and arsenic in
the As(V)
state. pH titration with a strong base, such as sodium hydroxide, seems to be
necessary.
As pH titration with sodium carbonate produces either gasparite, a known and
naturally
occurring compound or a combination of gasparite and trigonal CeAsO4 = (H20)X.
The use
of cerium chloride and cerium nitrate both successfully demonstrated the
successful
synthesis of the novel compound. The presence of other metal species, such as
magnesium, aluminum, silicon, calcium, iron, copper, and zinc, have not been
shown to
inhibit the synthesis of the novel compound. The presence of fluoride will
compete with
arsenic removal and produce an insoluble CeF3 precipitate. Solutions
containing only
arsenic and cerium show that a Ce:As atomic ratio of 1:1 is preferable for
forming the
novel compound, and solutions containing excess cerium have produced a cerium
oxide
(CeO2) precipitate in addition to the novel compound. Additionally, the novel
compound
appears to be quite stable when challenged with a leach test requiring less
than 1 ppm
arsenic dissolution in solution of pH 2.9 and pH 5Ø
EXAMPLE 6
In a first test, 50 mL of synthetic waste water containing 24 g/L arsenic, 25
g/L
sodium hydroxide, and 80 g/L sodium sulfide were added to a flask and heated
to 70 C
under magnetic stir. Initial solution pH was found to be pH 12Ø Dropwise
addition of
19.6 g of cerium-aluminum chloride solution (83.7 g/L Ce, 54.0 g/L Al, D=1.29
g/L)
yielded a flaky, white solid precipitate. Sodium hydroxide solution (NaOH,
20%) was
added as needed to maintain a solution pH of pH 10.0 or higher during addition
of the
bimetallic lanthanide-based salt solution. After complete addition of the
bimetallic
34
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
lanthanide-based salt solution, the solution is aged at 70 C under magnetic
stir for 30
minutes. After cooling, the final solution pH is pH 10.4. The solid
precipitate was filtered
through a 0.4 m membrane and dried. ICP-AES analysis of the feed and treated
solutions indicates that the arsenic concentration was decreased from 23,800
ppm to 4,300
ppm. This is an 82% removal rate at a capacity of 730 mg arsenic/gram of Ce02.
In a second test, 30 mL of synthetic waste water containing 24 g/L arsenic, 25
g/L
sodium hydroxide, and 80 g/L sodium sulfide were added to a flask at 22 C
under
magnetic stir. Initial solution pH was found to be pH 13Ø Dropwise addition
of 11.9 g
of cerium-aluminum chloride solution (83.7 g/L Ce, 54.0 g/L Al, D=1.29 g/L)
yielded a
flaky, white solid precipitate. Sodium hydroxide solution (NaOH, 20%) was
added as
needed to maintain a solution pH of pH 10.0 or higher during addition of the
bimetallic
lanthanide-based salt solution. After complete addition of the bimetallic
lanthanide-based
salt solution, the solution is heated to 70 C under magnetic stir and aged for
60 minutes.
After cooling, the final solution pH is pH 11Ø The solid precipitate was
centrifuged and
washed with water two times, then dried. ICP-AES analysis of the feed and
treated
solutions indicates that the arsenic concentration was decreased from 23,800
ppm to 2,750
ppm. This is an 89% removal rate at a capacity of 770 mg arsenic/gram of Ce02.
EXAMPLE 7
In a first test, 30 mL of synthetic waste water containing 24 g/L arsenic, 25
g/L
sodium hydroxide, and 80 g/L sodium sulfide were added to a flask and heated
to 70 C
under magnetic stir. Initial solution pH was found to be pH 12.8. Dropwise
addition of
17.3 g of aluminum chloride solution (54.0 g/L Al, D=1.20 g/L) yielded a
flaky, white
solid precipitate. Sodium hydroxide solution (NaOH, 20%) was added as needed
to
maintain a solution pH of pH 10.0 or higher during addition of the aluminum
chloride
solution. After complete addition of the aluminum-based salt solution, the
solution is aged
at 70 C under magnetic stir for 30 minutes. After cooling, the final solution
pH is pH
10.3. The solid precipitate was centrifuged and washed with water two times,
then air
dried. ICP-AES analysis of the feed and treated solutions indicates that the
arsenic
concentration was decreased from 23,800 ppm to 6,830 ppm. This is a 73%
removal rate
at a capacity of 200 mg arsenic/gram of A1203.
In a second test, 30 mL of synthetic waste water containing 24 g/L arsenic, 25
g/L
sodium hydroxide, and 80 g/L sodium sulfide was added to a flask and heated to
70 C
under magnetic stir. Initial solution pH was found to be pH 12.5. Dropwise
addition of
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
17.3 g of aluminum chloride solution (54.0 g/L Al, D=1.20 g/L) yielded a
flaky, white
solid precipitate. Sodium hydroxide solution (NaOH, 20%) was added as needed
to
maintain a solution pH of pH 9.0 or higher during addition of the aluminum
salt solution.
After complete addition of the aluminum salt solution, the solution is heated
to 70 C under
magnetic stir and aged for 30 minutes. After cooling, the final solution pH is
pH 9.2. The
solid precipitate was centrifuged and washed with water two times, then air
dried. ICP-
AES analysis of the feed and treated solutions indicates that the arsenic
concentration was
decreased from 23,800 ppm to 3,120 ppm. This is an 87.5% removal rate at a
capacity of
245 mg arsenic/gram of A1203-
EXAMPLE 8
A number of tests were undertaken to evaluate solution phase cerium ion
precipitations.
Test 1:
Solutions containing 250 ppm of Se(IV) or Se(VI) were amended with either
Ce(III) chloride or Ce(IV) nitrate at concentrations sufficient to produce a
2:1 mole ratio
of Se:Ce. Solids formation was observed within seconds in the reactions
between Cc and
Se(IV) and also when Ce(IV) was reacted with Se(IV). However, no solids were
observed
when Ce(III) reacted with Se(VI).
Aliquots of these samples were filtered with 0.45 micron syringe filters and
analyzed using ICP-AES. The remaining samples were adjusted to pH 3 when
Ce(IV) was
added, and to pH 5 when Ce(III) was added. The filtered solutions indicated
that Ce(III)
did not significantly decrease the concentration of Se(VI). However, Ce(IV)
decreased the
concentration of soluble Se(VI) from 250 ppm to 60 ppm. Although Ce(IV) did
not
initially decrease the concentration of Se(IV) at the initial system pH of
1.5, after
increasing to pH 3 >99% of the Se was filtered from the sample. Ce(III)
decreased the
concentration of Se(IV) from 250 ppm to 75 ppm upon addition and adjustment to
pH 5.
Test 2:
Solutions containing 250 ppm of Cr(VI) were amended with a molar equivalent of
cerium supplied as either Ce(III) chloride or Ce(IV) nitrate. The addition of
Ce(III) to
chromate had no immediate visible affect on the solution, however 24 hours
later there
appeared to be a fine precipitate of dark solids. In contrast, the addition of
Ce(IV) led to
the immediate formation of a large amount of solids.
As with the previous example, aliquots were filtered, and the pH adjusted to
pH 3
36
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
for Ce(IV) and pH 5 for Ce(III). The addition of Ce(III) had a negligible
impact on Cr
solubility, however Ce(IV) removed nearly 90% of the Cr from solution at pH 3.
Test 3:
Solutions containing 250 ppm of fluoride were amended with cerium in 1:3 molar
ratio of cerium: fluoride. Again the cerium was supplied as either Ce(III)
chloride or
Ce(IV) nitrate. While Ce(IV) immediately formed a solid precipitate with the
fluoride, Cc
(III) did not produce any visible fluoride solids in the pH range 3 - 4.5.
Test 4:
Solutions containing 50 ppm of molybdenum Spex ICP standard, presumably
molybdate, were amended with a molar equivalent of Ce(III) chloride. As with
previous
samples, a solid was observed after the cerium addition and an aliquot was
filtered through
a 0.45 micron syringe filter for ICP analysis. At pH 3, nearly 30 ppm Mo
remained in
solution, but as pH was increased to 5, the Mo concentration dropped to 20
ppm, and near
pH 7 the Mo concentration was shown to be only 10 ppm.
Test 5:
Solutions containing 50 ppm of phosphate were amended with a molar equivalent
of Ce(III) chloride. The addition caused the immediate precipitation of a
solid. The
phosphate concentration, as measured by ion chromatography, dropped to 20-25
ppm in
the pH range 3-6.
EXAMPLE 9
A series of tests were performed to determine if certain halogens,
particularly
fluoride, interfere with arsenic and other target material removal when using
cerium
chloride (CeC13). This will be determined by doing a comparison study between
a stock
solution containing fluoride and one without fluoride. For materials used
were: CeC13
(1.194 M Cc or 205.43 g/L REO) and 400 mL of the stock. The constituents of
the stock
solution are shown in Tables 5-6:
Table 5. Amount of Reagents Added
37
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
Amount of Amount of
Compound Reagent Added Reagent Added to
to 3.5L (g) 3.5L (g) No
Fluoride
NaF 5.13 0
AICI3. 6H20 0.13 0.13
CaC12.2 H2O 0.46 0.46
CuSO4. 5H2O 0.06 0.06
FeSO4. 7H2O 2.17 2.16
KCI 0.16 0.15
MgC12. 6H20 0.73 0.74
Na2S'03.9H2O 1.76 1.76
ZnS04. 7H2O 0.17 0.17
Na2HAsO4. 7H2018.53 18.53
Table 6. Calculated Analyte Concentrations
Theoretical Theoretical
Element Concentration Concentration
(mg/L) (mg/L) No
Fluoride
Cl 19032 15090
Na 1664 862
K 24 22
Cu 4 4
Fe 125 124
Zn 11 11
As 1271 1271
Mg 25 20
Ca 36 36
Al 16 16
Si 50 50
S 79 79
F 663 0
The initial pH of the stock solution was pH -0-1. The temperature of the stock
solution was elevated to 70 C. The reaction or residence time was
approximately 90
minutes.
The procedure for precipitating cerium arsenate with and without the presence
of
fluorine is as follows:
Step 1:
Two 3.5L synthetic stock solutions were prepared, one without fluorine and one
with fluorine. Both solutions contained the above listed constituents.
Step 2:
38
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
400 mL of synthetic stock solution was measured gravimetrically (402.41 g) and
transferred into a 600 mL Pyrex beaker. The beaker was then placed on hot/stir
plate and
was heated to 70 C while being stirred.
Step 3:
Enough cerium chloride was added to the stock solution to meet a predetermined
molar ratio of cerium to arsenic. For example, to achieve a molar ratio of one
ceria mole
to one mole of arsenic 5.68 mL of cerium chloride was measure gravimetrically
(7.17g)
and added to the stirring BHP solution. Upon addition of cerium chloride a
yellow/white
precipitate formed instantaneously, and the pH dropped due to the normality of
the cerium
chloride solution being 0.22. The pH was adjusted to approximately 7 using 20%
sodium
hydroxide.
Step 4:
Once the cerium chloride was added to the 70 C BHP solution, it was allowed
to
react for 90 minutes before being sampled.
Step 5:
Repeat steps 2-4 for all desired molar ratios for BHP solution containing
fluoride
and without flouride.
The results are presented in Table 7 and Figures 10-11.
Table 7. The residual arsenic concentration in supernatant solution after
precipitation with
cerium chloride solution
Molar Ratio Residual As Concentration w/ Residual As Concentration no
Fluoride Present (mg/L) Fluoride Present (mg/L)
1.00 578 0
1.10 425 0
1.20 286 0
1.30 158.2 0
1.40 58.1 0
1.50 13.68 0
1.60 3.162 0
1.71 0 0
1.81 10.2 0
1.90 0 0
2.01 0 0
39
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
A comparison of loading capacities for solutions containing or lacking
fluoride
suggest a benefit in eliminating the fluoride before the addition of cerium.
Figure 10
shows that the loading capacities (which is defined as mg of As per gram of
CeO2) for
solutions lacking fluoride are considerably higher at low molar ratios of
cerium to arsenic.
Steps should be taken to determine a method for the sequestration of fluoride
from future
stock solutions.
Solutions with a cerium to arsenic molar ratio of approximately 1.4 to 1 or
greater
had a negligible difference in the loading capacities between solution that
contained F- and
not having F-. This leads one to believe that an extra 40% cerium was needed
to sequester
the F-; then the remaining cerium could react with the arsenic.
These results confirm that the presence of fluoride is interfering with the
sequestration of arsenic. The interference comes from the competing reaction
forming
CeF3; this reaction has a much more favorable Ksp. A method for pretreatment
of
fluoride should be considered and developed in order to achieve more efficient
use of the
cerium.
Accordingly, a fluoride free solution gives better arsenic removal when using
lower cerium to arsenic molar ratios, in effect giving higher loading
capacities.
EXAMPLE 10
40.00 g of cerium was added to 1.00 liter of solution containing either 2.02
grams
of As(III) or 1.89 grams of As(V). The suspension was shaken periodically,
about 5 times
over the course of 24 hours. The suspensions were filtered and the
concentration of
arsenic in the filtrate was measured. For As(III), the arsenic concentration
had dropped to
11 ppm. For As(V), the arsenic concentration was still around 1 g/L, so the pH
was
adjusted by the addition of 3 mL of cone HCL
Both suspensions were entirely filtered using a vacuum filter with a 0.45
micron
track-etched polycarbonate membrane. The final or residual concentration of
arsenic in
solution was measured by ICP-AES. The solids were retained quantitatively, and
resuspended in 250 mL of DI water for about 15 minutes. The rinse suspensions
were
filtered as before for arsenic analysis and the filtered solids were
transferred to a weigh
boat and left on the benchtop for 4 hours.
The filtered solids were weighed and divided into eight portions accounting
for the
calculated moisture such that each sample was expected to contain 5 g of
solids and 3.5 g
of moisture (and adsorbed salts). One sample of each arsenic laden solid
(As(III) or
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
As(V) was weighed out and transferred to a drying oven for 24 hours, then re-
weighed to
determine the moisture content.
Arsenic-laden ceria samples were weighed out and transferred to 50 mL
centrifuge
tubes containing extraction solution (Table 8). The solution (except for H202)
had a 20
hour contact time, but with only occasional mixing via shaking. Hydrogen
peroxide
contacted the arsenic-laden solids for two hours and was microwaved to 50 deg
C to
accelerate the reaction.
A control sample was prepared wherein the 8.5 g arsenic-laden ceria samples
were
placed in 45 mL of DI water for the same duration as other extraction tests.
The first extraction test used 45 mL of freshly prepared 1 N NaOH. To increase
the chances of forcing off arsenic, a 20% NaOH solution was also examined. To
investigate competition reactions, 10% oxalic acid, 0.25 M phosphate, and 1
g/L carbonate
were used as extracting solutions. To test a reduction pathway 5 g of arsenic-
laden ceria
was added to 45 mL of 0.1 M ascorbic acid. Alternatively an oxidation pathway
was
considered using 2 mL 30% H202 added with 30 mL of DI water
After enough time elapsed for the selected desorption reactions to occur, the
samples were each centrifuged and the supernatant solution was removed and
filtered
using 0.45 micron syringe filters. The filtered solutions were analyzed for
arsenic content.
Litmus paper was used to get an approximation of pH in the filtered solutions.
Because the reactions based upon redox changes did not show a great deal of
arsenic release, the still arsenic-laden solids were rinsed with 15 ML of 1 N
NaOH and 10
mL of DI water for 1 hour, then re-centrifuged, filtered, and analyzed.
The results of these desorption experiments can be seen in Table 8. In short,
it
appears that the desorption of As(III) occurs to a minimal extent. In
contrast, As(V)
adsorption exhibits an acute sensitivity to pH, meaning that As(V) can be
desorbed by
raising the pH above a value of 11 or 12. As(V) adsorption is also susceptible
to
competition for surface sites from other strongly adsorbing anions present at
elevated
concentratons.
Using hydrogen peroxide to convert As(III) to As(V) appeared to be relatively
successful, in that a large amount of arsenic was recovered when the pH was
raised using
NaOH after the treatment with H202. However, until the NaOH was added, little
arsenic
desorbed.
41
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
While ascorbate did cause a dramatic color change in the loaded media, it was
unsuccessful in removing either As(III) or As(V) from the surface of ceria. In
contrast,
oxalate released a detectable amount of adsorbed As(III) and considerably
greater amounts
of As(V).
In experiments with other adsorbates:
These experiments examined the adsorption and desorption of a series of non-
arsenic anions using methods analogous to those established for the arsenic
testing.
Permanganate:
Two experiments were performed. In the first experiment, 40 g of ceria powder
were added to 250 mL of 550 ppm KMnO4 solution. In the second experiment, 20 g
of
ceria powder were added to 250 mL of 500 ppm KMnO4 solution and pH was lowered
with 1.5 mL of 4 N HC1. Lowering the slurry pH increased the Mn loading on
ceria four
fold.
In both experiments the ceria was contacted with permanganate for 18 hours,
then
filtered to retain solids. The filtrate solutions were analyzed for Mn using
ICP-AES, and
the solids were washed with 250 mL of DI water. The non-pH adjusted solids
were
washed a second time.
Filtered and washed Mn-contacted solids were weighed and divided into a series
of
three extraction tests and a control. These tests examined the extent to which
manganese
could be recovered from the ceria surface when contacted with 1 N NaOH, 10%
oxalic
acid, or 1 M phosphate, in comparison to the effect of DI water under the same
conditions.
The sample of permanganate-loaded ceria powder contacted with water as a
control exhibited the release of less than 5% of the Mn. As with arsenate,
NaOH
effectively promoted desorption of permanganate from the ceria surface. In the
case of the
second experiment, where pH was lowered, the effect of NaOH was greater than
in the
first case where the permanganate adsorbed under higher pH conditions.
Phosphate was far more effective at inducing permanganate desorption than it
was
at inducing arsenate desorption. Phosphate was the most effective desorption
promoter we
examined with permanganate.
Oxalic acid caused a significant color change in the permanganate solution,
indicating that the Mn(VII) was reduced, possibly to Mn(II) or Mn(IV), wherein
the
formation of MnO or Mn02 precipitates would prevent the detection of
additional Mn that
may or may not be removed from the ceria. In the sample that received no pH
adjustment,
42
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
no desorbed Mn was detected. However, in the sample prepared from acidifying
the
slurry slightly a significant amount of Mn was recovered from the ceria
surface.
Chromate
250 mL of solution was prepared using 0.6 g sodium dichromate, and the
solution
was contacted with 20 g of cerium powder for 18 hours without pH adjustment.
The
slurry was filtered and the solids were washed with DI water then divided into
50 mL
centrifuge tubes to test the ability of three solutions to extract chromium
from the ceria
surface.
Ceria capacity for chromate was significant and a loading of > 20 mg Cr / g
ceria
was achieved without any adjustments to pH or system optimization (pH of
filtrate was
approximately 8). Likewise, the extraction of adsorbed chromate was also
readily
accomplished. Raising the pH of the slurry containing chromate-laden ceria
using 1 N
NaOH was the most effective method of desorbing chromium that was tested.
Considerably less chromate was desorbed using phosphate and even less was
desorbed
using oxalic acid. In the control sample, only 5% of the chromate was
recovered when the
loaded solid was contacted with distilled water.
Selenite
A liter of selenite solution was prepared using 1 g of Na2SeO2. The pH was
lowered using 2 mL of 4 M HC1. 40 g of ceria was added to create a slurry that
was
provided 18 hours to contact. The slurry was filtered and the Se-loaded ceria
was retained,
weighed, and divided into 50 mL centrifuge tubes for extraction.
Ceria was loaded with > 6 mg/g of Se. While the solids from this reaction were
not washed in the preparation stages, the control extraction using DI water
exhibited less
than 2% selenium release. The extent of selenium adsorption was diminished by
adding 1
N NaOH to the loaded ceria, but the effect was not as dramatic as has been
seen for other
oxyanions. However, by using hydrogen peroxide to oxidize the Se(IV) to Se(VI)
the
adsorbed selenium was readily released from the ceria surface and recovered.
Oxalic acid
had no noticeable impact on the extent of selenium adsorption.
Antimony
The solubility of antimony is rather low and these reactions were limited by
the
amount of antimony that could be dissolved. In this case, 100 mg of antimony
(III) oxide
was placed into 1 L of distilled water with 10 mL concentrated HC1, allowed
several days
to equilibrate, and was filtered through a 0.8 micron polycarbonate membrane
to remove
43
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
undissolved antimony. The liter of antimony solution was contacted with 16 g
of ceria
powder, which was effective removing antimony from solution, but had too
little Sb(III)
available to generate a high loading on the surface. In part due to the low
surface
coverage and strong surface-anion interactions, the extraction tests revealed
little Sb
recovery. Even the use of hydrogen peroxide, which would be expected to
convert Sb(III)
to a less readily adsorbed species of Sb(V), did not result in significant
amounts of Sb
recovery.
Tables 8-11 show the test parameters and results.
Table 8: Loading of cerium oxide surface with arsenate and arsenite for the
demonstration
of arsenic desorbing technologies.
A B C D E F G H I J K L M
[As] Mass pH Resid As- We We Dry % Rins Rinse Final
(g/L) Ce02 [As] loadi t t (g) Soli e Vol [As] [As]
(g) (ppm) ng Ma ma ds (mL) (ppm (mg/g)
(mg/ ss ss )
As( 2.02 40.0 9.5 0 50.5 68 7.4 4.63 61.9 250 0 50.5
III 8
As( 1.89 40.0 5 149 43.5 69 8.8 5.33 60.2 250 163 42.5
V 6
Table 9: Loading of cerium oxide surface with arsenate and arsenite for the
demonstration
of arsenic desorbing technologies.
[As] Residual As-loading Rinse [As] Final [As]
(g/L) pH [As] (ppm) (mg/g) (ppm) (mg/g)
As(III) 2.02 9.5 0 50.5 0 50.5
As(V) 1.89 5 149 43.5 163 42.5
Table 10: Arsenic extraction from the ceria surface using redox and
competition reactions
Extractant pH % As(III) recovered % As(V) recovered
Water 7 0.0 1.7
1 N NaOH 13 0.2 60.5
20% NaOH 14 2.1 51.8
44
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
0.25 P04 8 0.4 15.0
g/L C03 10 2.0 7.7
10% oxalate 2.5 3.0 16.5
30% H202 6 2.0 1.5
H202/NaOH 13 25.2 31.0
0.1 M ascorbate 4 0.0 0.0
Table 11: Loading and extraction of other adsorbed elements from the ceria
surface
(extraction is shown for each method as the `percent loaded that is recovered)
5
Per- Per-
chromate antimony selenite manganate manganate
loading pH 8 2 6 6 11
loading (mg/g) 20 1 6 4 0.7
water (% rec) 5.1 <2 1.6 2.6 3.4
1 N NaOH (% rec) 83 <2 40.8 49.9 17.8
10% oxalic (% rec) 25.8 2.3 0.2 22.8 < 3
0.5 M P04 (% rec) 60.7 78.6 45.8
30% H202 (% rec) 2.3 71.9
EXAMPLE 11
Experiments were performed to determine whether cerium (IV) solutions can be
used
10 to remove arsenic from storage pond process waters, and accordingly
determine the
loading capacity of ceria used. In these trials the storage pond solutions
will be diluted
with DI water, since previous test work has confirmed that this yields a
better arsenic
removal capability. The soluble cerium (IV) species used are Ceric Sulfate -*
0.1 M
Ce(S04)2 and Ceric Nitrate -* Ce(N03)4. The pond solution used has an arsenic
split
between 27% As (III) and 73% As (V), with a of ph 2. Additional components in
the pond
solution are presented in Table 12:
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
Additional Sol'n Components:
Analyte As B Ce Cl Co Cu Fe Na Ni Pb S Si
(PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM)
Tailings
Pond 2500 270 4 1100 140 2400 130 4800 19500 9 15000 870
Solution
Test 1:
50 mL of storage pond solution was diluted to 350 mL using DI water, a seven
fold
dilution. The diluted pond solution was heated to a boil and 50 mL of 0.1M
Ce(S04)4 was
added and mixed for 15 minutes while still at a boil. A yellow/white
precipitate formed.
This was filtered using a Buchner funnel and 40 Whatman paper. The precipitate
was
dried at 110 C overnight, and was weighed at 0.5 g. The filtrate was sampled
and filtered
using a 0.2p filter. A full assay was performed on the filtrate using ICP-AES.
Test 2:
200 mL storage pond solution was diluted to 300 mL using DDI water. The
solution was heated to a boil and 8.95 mL of 2.22 Ce(N03)4 was added. The
solution
boiled for 15 minutes, and a yellow/white precipitate formed. This was
filtered using a
Buchner funnel and 40 Whatman paper. The precipitate was dried at 110 C
overnight,
and was weighed at 2.46 g. The filtrate was sampled and filtered using a 0.2p
filter. A
full assay was performed on the filtrate using ICP-AES.
The results are presented in Tables 11-12 below:
Table 13:
Analyte As B Ce Cl Co Cu Fe Na Ni Pb S Si
(PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM) (PPM)
Storage
Pond 2500 270 4 1100 140 2400 130 4800 19500 9 15000 870
Solutio
n
Test 1 364 273 850 N/A 133 2240 126 5250 14700 7 N/A 840
7 FD
Test4 639 254 2900 N/A 99 2464 94 4620 18480 9 N/A 601
1.54 FD
*Note: FD denotes "fold dilution" and the dilution has been factored for the
reported
concentrations
Table 14: Calculated Capacities
Test As CeO2 Capacity Percent Percent
# Removed Used (g) ~ (mg As/ As Cc still in
46
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
(mg) CeO2) Removed solution
1 107 0.86 124 85 42
2 372 3.44 108 74 32
Tables 13 and 14 demonstrate that the cerium (IV) solutions have a
preferential
affinity for the arsenic. When examining the data closer, it appears that some
of the other
metals fluctuate in concentrations i.e., nickel. According to the dilution
scheme used and
the limitations of the instrument, there could be up to 15% error in the
reported
concentrations, explaining some of the fluctuations. Moving onto to table 12,
it shows that
tests 1 and 2 removed 85% and 74% of the arsenic respectively.
A number of variations and modifications of the invention can be used. It
would
be possible to provide for some features of the invention without providing
others.
While the various processes are discussed with reference to liquids, it is to
be
appreciated that the processes can be applies to other fluids, such as gases.
Examples of
arsenic-containing gases include smelter and roaster off-gases and utility
flue gas.
The present invention, in various embodiments, configurations, or aspects,
includes
components, methods, processes, systems and/or apparatus substantially as
depicted and
described herein, including various embodiments, configurations, aspects,
subcombinations, and subsets thereof. Those of skill in the art will
understand how to
make and use the present invention after understanding the present disclosure.
The
present invention, in various embodiments, configurations, and aspects,
includes providing
devices and processes in the absence of items not depicted and/or described
herein or in
various embodiments, configurations, or aspects hereof, including in the
absence of such
items as may have been used in previous devices or processes, e.g., for
improving
performance, achieving ease and\or reducing cost of implementation.
The foregoing discussion of the invention has been presented for purposes of
illustration and description. The foregoing is not intended to limit the
invention to the form
or forms disclosed herein. In the foregoing Detailed Description for example,
various
features of the invention are grouped together in one or more embodiments,
configurations, or aspects for the purpose of streamlining the disclosure. The
features of
the embodiments, configurations, or aspects of the invention may be combined
in alternate
embodiments, configurations, or aspects other than those discussed above. This
method
of disclosure is not to be interpreted as reflecting an intention that the
claimed invention
requires more features than are expressly recited in each claim. Rather, as
the following
47
CA 02743304 2011-05-10
WO 2010/056742 PCT/US2009/064023
claims reflect, inventive aspects lie in less than all features of a single
foregoing disclosed
embodiment, configuration, or aspect. Thus, the following claims are hereby
incorporated
into this Detailed Description, with each claim standing on its own as a
separate preferred
embodiment of the invention.
Moreover, though the description of the invention has included description of
one
or more embodiments, configurations, or aspects and certain variations and
modifications,
other variations, combinations, and modifications are within the scope of the
invention,
e.g., as may be within the skill and knowledge of those in the art, after
understanding the
present disclosure. It is intended to obtain rights which include alternative
embodiments,
configurations, or aspects to the extent permitted, including alternate,
interchangeable
and/or equivalent structures, functions, ranges or steps to those claimed,
whether or not
such alternate, interchangeable and/or equivalent structures, functions,
ranges or steps are
disclosed herein, and without intending to publicly dedicate any patentable
subject matter.
48