Note: Descriptions are shown in the official language in which they were submitted.
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
SELECTIVE CATALYTIC REDUCTION VIA ELECTROLYSIS OF UREA
FIELD OF INVENTION
[0001] The present invention relates to methods and devices for treating
exhaust gases.
BACKGROUND
[0002] There is concern over the environmental impact of emissions from
fossil fuel
combustion sources. For example, the exhaust gas of vehicles powered with
diesel fuel
contains chemical pollutants such as nitrogen oxides ("NOx") and sulfur oxides
("S0x"), as
well as particulates. Selective catalytic reduction (SCR) and selective non-
catalytic reduction
(SNCR) are means for converting nitrogen oxides (NOõ) into diatomic nitrogen,
N2, and
water, H20. In SCR, a catalyst is used in combination with a gaseous
reductant, which is
added to a stream of flue or exhaust gas and is absorbed onto the catalyst. In
SCNR, the
reductant is injected into the flue gas in a furnace within an appropriate
temperature window.
Additionally, flue gas conditioning with a gaseous reductant can also enhance
electrostatic
precipitator performance for removing fly ash. In SCR, SNCR, and fly ash
removal systems,
the reductant is typically ammonia or urea.
[0003] The NO, reduction reaction takes place as the gases pass through the
catalyst
chamber. Before entering the catalyst chamber the ammonia, or other reductant,
such as urea,
is injected and mixed with the gases. The chemical equations for using either
anhydrous or
aqueous ammonia for a selective catalytic reduction process are:
4N0 + 4NH3 + 02 ¨> 4N2 + 6H20 (Equation 1)
2NO2 + 4NH3 + 02 ¨> 3N2 + 6H20 (Equation 2)
NO + NO2 + 2NH3 ¨> 2N2 + 3H20 (Equation 3)
The reaction for urea as a reductant instead of ammonia is:
4N0 + 2(NH2)2C0 + 02 ¨> 4N2 + 4H20 + 2CO2 (Equation 4)
[0004] Compared to urea, ammonia is more reactive, is more easily dispersed
uniformly
into the flue gas stream, and is active over a broader temperature range, as
well as being more
efficient. Urea, as such, while also an effective reductant, forms unwanted
byproducts, such
as carbon monoxide (CO) and nitrous oxide (N20), both of which are now under
critical
scrutiny by environmental authorities.
1
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
[0005] Commonly urea is thermally hydrolyzed to form ammonia for exhaust gas
treatment applications. The hydrolysis of urea to form ammonia can be broken
down into
two distinct reactions. The first reaction is a mildly exothermic reaction,
wherein heat is
given off as urea hydrolyzes to form ammonium carbamate. The second reaction,
in which
the ammonium carbamate is converted to ammonia and carbon dioxide, is strongly
endothermic, which overall dominates the thermodynamics of the conversion of
urea to
ammonia and carbon dioxide, i.e., the overall reaction is endothermic.
Therefore, the
hydrolysis of urea requires a substantial amount of heat and quickly stops
when the supply of
heat is withdrawn. For example, the liberation of ammonia commences at around
110 C and
becomes rapid at around 150 C to 160 C, with or without catalytic assistance.
H2O + (NH2)2C0 ¨> (NH2)CO2-NH4+ + NH3 + heat (Equation 5)
(NH2)CO2-NH4+ + heat ¨> 2NH3 + CO2 (Equation 6)
Excess water promotes the hydrolysis reaction, the overall reaction for which
is as follows:
(x+1)H20 + (NH2)2C0 + heat ¨> 2NH3 + CO2 + (x)H20 (Equation 7)
[0006] However, under the reaction conditions necessary to affect useful
throughput, the
water quality is important. For example, in a conventional thermal hydrolysis
of urea to
ammonia for an SCR system, an aqueous solution of urea is atomized through a
spray nozzle
into a heated vaporization chamber. As such, the excess water is also
vaporized during the
hydrolysis of urea to ammonia, thereby leaving behind any non-volatile
substances such as
minerals. Minerals and other non-volatile substances can adhere to equipment
surfaces, such
as spray nozzles and the vaporization chamber walls, and build up over time,
which may lead
to blockage of the spray nozzle or reduced heat transfer efficiency to the
vaporization
chamber. Thus, the water used in thermal hydrolysis systems needs to be
demineralized.
Further, the thermal hydrolysis of urea method is also sensitive to the
quality of the urea. For
example, formaldehyde present in urea can negatively affect the performance of
an SCR
system in a way similar to that of using demineralized water.
[0007] In view of the foregoing, the hydrolysis of urea requires an external
heat source to
initiate the reaction, even when coupled with combustion engines, and also is
sensitive to the
extent of demineralization of the water, and the quality of urea used in the
hydrolysis.
Therefore, more efficient and/or safer methods for generating ammonia for
exhaust gas
treatment applications are needed.
2
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
SUMMARY OF THE INVENTION
[0008] The present invention is premised on the realization that ammonia can
be
produced from the electrolysis of urea to supply exhaust gas treatment
applications, such as
selective catalytic reduction (SCR) systems, selective non-catalytic reduction
(SNCR)
systems, and/or flue gas conditioning systems.
[0009] According to one embodiment of the present invention, a method for
supplying
NH3 to an exhaust gas treatment system is provided. The method includes
supplying urea to
an electrolytic flow cell that includes an inlet, an outlet, a cathode having
a first conducting
component, an anode having a second conducting component, an alkaline
electrolyte
composition in electrical communication with the anode and the cathode, and a
recirculation
system operatively coupled to the inlet and the outlet of the electrolytic
cell; producing
ammonia by the electrolytic hydrolysis of urea by applying a voltage
difference to the
electrolytic flow cell, wherein the voltage difference is applied across a
cathode and an
anode, wherein the voltage difference is sufficient to effect the electrolytic
hydrolysis of urea
to produce at least NH3; recovering at least a portion of the NH3;
transferring the at least a
portion of the NH3 to the exhaust gas treatment system; and recirculating at
least a portion of
the alkaline electrolyte composition. The alkaline electrolyte composition has
a hydroxide
concentration of at least 0.01 M or a pH of at least 8, and the recirculation
system contains at
least a portion of the alkaline electrolyte composition.
[0010] According to another embodiment of the invention, an exhaust gas
treatment
system for a combustion engine is provided. The exhaust gas treatment system
includes at
least one of a selective catalytic reduction system, a selective non-catalytic
reduction system,
or a flue gas conditioning system; and an ammonia generator. The ammonia
generator
includes an electrolytic flow cell having an inlet, at least one outlet, a
cathode having a first
conducting component, an anode having a second conducting component, an
alkaline
electrolyte composition in electrical communication with the anode and the
cathode, where
the alkaline electrolyte composition has a hydroxide concentration of at least
0.01 M or a pH
of at least 8, and a recirculation system operatively coupled to the inlet and
the at least one
outlet of the electrolytic cell. The at least one outlet from the ammonia
generator is further in
communication with the at least one of the selective catalytic reduction
system, the selective
non-catalytic reduction system, or the flue gas conditioning system.
3
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
[0011] The invention will be further appreciated in light of the following
detailed
description and drawings in which:
BRIEF DESCRIPTION OF THE DRAWINGS
[0012] The accompanying drawings, which are incorporated in and constitute a
part of
this specification, illustrate embodiments of the invention and, together with
the general
description of the invention given above, and the detailed description given
below, serve to
describe the invention.
[0013] FIG. 1 is a schematic representation of a method to produce ammonia
from urea;
[0014] FIG 2 is a diagrammatical view of a simplified electrolytic cell
coupled to
exhausted combustion gases;
[0015] FIG. 3 is a diagrammatical view of a method to purify exhaust gases
from a
combustion engine;
[0016] FIG. 4 is a plot of current density at a constant voltage (1.4 V) in
the
electrochemical cell over time;
[0017] FIG. 5 is a plot of current density at a constant voltage (1.33 V) in
the
electrochemical cell over time;
[0018] FIG. 6 is a diagrammatic depiction of an electrolytic ammonia generator
system
according to an embodiment of the present invention;
[0019] FIG. 7 is exploded view of an electrolytic flow cell according to an
embodiment
of the present invention;
[0020] FIG. 8 is a diagrammatic depiction of an electrolytic ammonia generator
system
according to another embodiment of the present invention; and
[0021] FIG. 9 is a diagrammatic depiction of an electrolytic ammonia generator
system
according to yet another embodiment of the present invention.
DETAILED DESCRIPTION
[0022] The treatment of combustion exhaust gas is facilitated by the
electrolysis-induced
hydrolysis of urea and is described herein. Advantageously, the electrolytic
cell conditions
may be modified to additionally generate hydrogen, which may be injected to
increase fuel
efficiency, to provide heat into the electrolytic cell, or to provide
electricity into the
electrolytic cell.
4
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
[0023] Referring now to FIG. 1, urea may be subjected to electrolysis-induced
hydrolysis
in an electrolytic device. The electrolytic device may comprise a cell or
multiple cells that
each contains an anode and a cathode. The electrolytic cell can operate in
batch mode,
continuous mode, semi-continuous, and with recirculation, as needed to provide
on demand
and controlled injection of ammonia into a process gas stream such as a
combustion gas
exhaust. At the anode, the working electrode of the cell, urea is hydrolyzed
to ammonia. The
overall hydrolysis reaction is provided in Equation 8 below.
[0024] (NH2)2C0 + H20 ¨> NH3 i + c02 i (Equation 8)
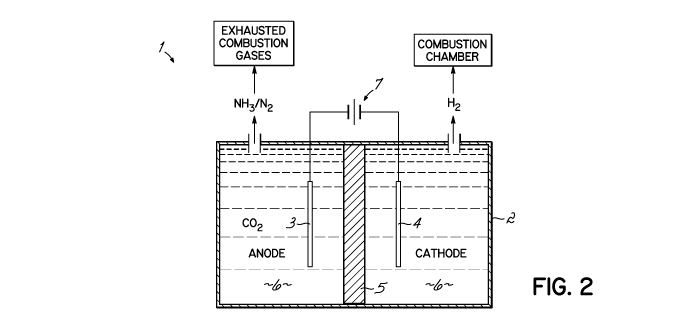
[0025] Referring more particularly to FIG. 2, a simplified electrolytic cell
1 representing
a single batch-type arrangement comprises a tank 2, which may be made of light
gauge iron,
steel, TEFLON , or other material not attacked by an alkaline electrolyte
composition. An
electrode assembly comprising two electrodes, an anode 3 and a cathode 4, is
suspended
within an alkaline electrolyte composition 6 contained in tank 2. Optionally,
a separator 5
may be positioned between the anode and cathode. In this single batch-type
arrangement, the
alkaline electrolyte composition 6 includes an effective amount of urea as
described below.
The anode 3 and cathode 4 are electrically connected to a voltage source 7,
which provides
the electrical energy for the electrolysis of urea contained in the alkaline
electrolyte
composition 6. In a batch-type arrangement, the alkaline electrolyte
composition may be
stirred to facilitate mass transfer. It will be readily apparent to one of
ordinary skill in the art
that the above cell is readily adaptable to a continuous flow cell
configuration, semi-
continuous, and with recirculation of the alkaline electrolyte composition, as
discussed in
detail below.
[0026] The electrodes comprise a conductor or a support which can be coated
with one or
more active conducting components. Exemplary conductors include, but are not
limited to,
metals such as nickel and platinum, alloys such as carbon steel or stainless
steel, or other
materials capable of conducting electricity such as carbon or graphite.
Exemplary electrode
support materials may be chosen from many known supports, such as foils,
meshes, sponges,
and beads, for example. The support materials may include, but are not limited
to, Ni foils,
Ti foils, graphite, carbon fibers, carbon paper, glassy carbon, carbon
nanofibers, and carbon
nanotubes. Aside from these specific support materials listed, other suitable
supports will be
recognized by those of ordinary skill in the art.
5
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
[0027] Accordingly, the cathode may comprise a conductor that is inert to an
alkaline
electrolyte composition. Additionally, the cathode may further include a
support material
that is inert to the alkaline electrolyte compositions and coated with one or
more active
conducting components. For example, the conducting component of the cathode
may include
carbon, cobalt, copper, iridium, iron, nickel, palladium, platinum, rhodium,
ruthenium, or
mixtures or alloys thereof. Exemplary conducting components include carbon
steel and
stainless steel.
[0028] The anode may comprise a conductor that is inert to the alkaline
electrolyte
composition. Additionally, the anode may further include a support material
that is inert to
the alkaline electrolyte compositions and coated with one or more active
conducting
components. According to embodiments of the present invention, the reaction of
urea
hydrolysis occurs at the conducting component of the anode. Therefore, the
conductor and/or
the conducting component at the anode is one or more metals active toward
electrolytic
hydrolysis of urea. Active metals may include cobalt, copper, iridium, iron,
platinum, nickel,
rhodium, ruthenium, or mixtures or alloys thereof, for example, and in
particular, nickel. The
active metals may be in an oxidized form, such as nickel oxyhydroxide.
[0029] The structure of the anode is not limited to any specific shape or
form. For
example, the active metal may be formed as foil, wire, gauze, bead, or coated
onto a support.
[0030] Exemplary working electrodes include, nickel electrodeposited on a
carbon
support, such as carbon fibers, carbon paper, glassy carbon, carbon
nanofibers, or carbon
nanotubes, and nickel formed into beads and suspended in a nickel gauze.
[0031] One electrode found to be favorable to the electrolysis-induced
hydrolysis of urea
is an activated nickel oxyhydroxide modified nickel electrode (NOMN) on
different 4 cm2-
metallic substrates (Ni foil, Ni gauze, Ti foil and Ti gauze) that have been
electroplated with
0.1 mg of Ni using a Watts bath. Specifically, the plated nickel electrode is
activated by
immersed in a solution containing nickel sulfate, sodium acetate, and sodium
hydroxide at
33 C. Stainless steel is used as counter electrode. The plated nickel
electrode may be used
as the anode and cathode by manual polarity switching at 6.25 A/m2 for four 1-
minute cycles
and two 2-minute cycles. Finally, the electrode is kept as the anode at the
same current and
maintained thereat for two hours. The activated electrode yields higher
current densities than
those of M/Ni, where M represents a metallic substrate.
6
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
[0032] The separator 5 compartmentalizes the anode and cathode. Separators
should be
constructed from materials chemically resistant to the alkaline electrolyte
composition. Many
polymers are suitable for constructing separators, such as Teflon and
polypropylene.
Separators are not required for simple batch-type arrangements, but may be
advantageous for
continuous flow electrochemical cells or fuel cells. Separators may include
ion exchange
membranes, solid electrolytes or electrolytic gels, for example. Separators
may be
permeable, semi-permeable or impermeable to gases or liquids.
[0033] According to the present invention, the electrolyte composition is
alkaline and has
a hydroxide ion concentration of at least 0.01 M or a pH of at least 8.
According to one
example, the alkaline electrolyte composition has a hydroxide concentration of
at least 0.01
M and a pH of at least 8. As such, the alkaline electrolyte composition may
include a
sufficient quantity of any suitable hydroxide salt, carbonate salt or
bicarbonate salt to provide
an electrolyte composition with a hydroxide ion concentration of at least
0.01M and/or a pH
of at least 8. An alkali metal hydroxide or alkaline earth metal hydroxide
salt, such as lithium
hydroxide, rubidium hydroxide, cesium hydroxide, barium hydroxide, strontium
hydroxide,
potassium hydroxide, sodium hydroxide, magnesium hydroxide, calcium hydroxide,
and
mixtures thereof may be used. In particular, the alkaline electrolyte
composition includes
potassium hydroxide. Advantageously, the sequestration of CO2 gas, shown in
Equation 1,
may be realized by the reaction of CO2 with hydroxide to form carbonate, which
may be
retained in the alkaline electrolyte composition. Similarly, alkali metal
carbonates or
bicarbonate salts or alkaline earth metal carbonates or bicarbonate salts are
also suitable
electrolytes.
[0034] The concentration of the hydroxide, carbonate, or bicarbonate salts
may vary
according to embodiments of the invention. For example, according to one
embodiment, the
concentration of the hydroxide, carbonate, or bicarbonate salts may be from
about 0.01 M to
about 8 M. In another example, the concentrations of potassium hydroxide,
potassium
carbonate, potassium bicarbonate, sodium hydroxide, sodium carbonate, or
sodium
bicarbonate from about 2 M to about 8 M and from about 4 M to about 8 M, are
particularly
effective.
[0035] The alkaline electrolyte composition may comprise a gel, such as a
solid polymer
electrolyte. Suitable gels include, but are not limited to, those containing
polyacrylic acid,
polyacrylates, polymethacrylates, polyacrylamides and similar polymers and
copolymers.
7
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
[0036] The electrolytic gel may be prepared using any suitable method. One
method
includes forming a polymer and then injecting a hydroxide, a carbonate or a
bicarbonate salt
electrolyte into the polymer to form a polymeric mixture. In another method,
the monomer
may be polymerized in the presence of a hydroxide, a carbonate, or bicarbonate
salt
electrolyte.
[0037] According to one embodiment, the electrodes are separated by the
electrolyte gel
which contains an effective hydroxide, carbonate, or bicarbonate ion
concentration. The
anode is contacted with a urea solution as the feed stock. The cathode is then
contacted with
a suitable aqueous solution, such as water or a hydroxide, carbonate, or
bicarbonate solution,
for example.
[0038] Alternatively, the gel electrolyte is not fixed and can flow through
an electrolytic
cell. According to another embodiment, urea may be contained within the gel or
an aqueous
solution comprising urea may flow within the gel electrolyte.
[0039] In the cell shown in FIG. 2, the electrolyte composition 6 includes
urea, which
may vary from trace amounts up to about a saturated solution, which is
approximately 12 M
at standard temperature and pressure. Advantageously, the specific source and
purity of the
urea is not particularly limited.
[0040] Moreover, for the formation of aqueous solutions of urea, the specific
source and
purity of the water used in making the aqueous solution is not particularly
limited or critical.
One reason for this advantage is that, according to embodiments of the present
invention, the
entire aqueous solution comprising urea is not volatilized to thereby leave
behind trace
minerals and other non-volatile materials. Instead, the majority of the water
remains in the
liquid form, which substantially maintains the trace minerals in solution.
Additionally, after
the electrolytic hydrolysis of at least a portion of the urea within the
electrolytic cell, the
aqueous solution or the alkaline electrolyte composition being discharged from
the
electrochemical cell may be recirculated.
[0041] Voltage source 7 may be any available source, such as batteries, fuel
cells, power
from the grid, and renewable energy sources, such as a solar cell or a wind-
turbine generator,
for example. When the electrolytic cell is coupled with an SCR system on a
motor vehicle,
the electric source may be from an alternator. In order to attain desired
efficiencies, a voltage
sufficient to initiate the electrolytic hydrolysis of urea is required. But it
is preferable that the
voltage not be so high as to significantly electrolyze water. Generally, the
minimum voltage
8
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
required to electrolyze or electrolytically-hydrolyze urea is about 0.85
volts. The voltage
required to electrolyze water is greater than 1.7 volts with a platinum
electrode at standard
conditions, but the rate of electrolysis and/or electrolysis-induced
hydrolysis depends on
other factors, such as temperature and ionic strength/conductivity. Based on
the foregoing,
the voltage range applied to the electrolytic cell to electrolytically-
hydrolyze urea may be
from about 0.85 volts to less than about 1.7 volts. The voltage range may be
from about 1.2
volts to about 1.6 volts. Typically, the electrolytic cell will be operated at
a constant voltage
within these ranges.
[0042] Additionally, the rate of producing ammonia and/or hydrogen from urea
may be
controlled by varying the voltage within different regions of the electrolytic
cell. For
example, in a packed-bed type electrolytic cell, the voltage within the packed-
bed of an
anodic catalyst material can be adjusted along the catalyst bed to control the
rate of ammonia
production and/or injection into an SCR or SNCR device. As such, different
regions in the
catalyst bed may have different potentials to control the rate of ammonia
production. For
example, a packed bed column configuration may include a plurality of anodes
with each
being electrically insulated from the other anodes and capable of having
voltage controlled
separately thereto, such as that represented in FIG. 6. For a given maximum
production of
ammonia, the totality of the anodes may be polarized. However, when a lower
amount of
ammonia is desired, then less than all of the anodes are polarized.
[0043] Amperage or current density may affect the performance of an
electrolytic cell, as
well. Pure water has poor electrical conductivity and, as such, electrolysis
in pure water is
very slow and essentially occurs due to the self-ionization of water.
Generally, the rate of
electrolysis increases by adding an electrolyte, such as a salt, an acid or a
base. Therefore,
the presence of an added hydroxide ion, a carbonate ion or a bicarbonate ion
and its
respective counter ion in the alkaline electrolyte composition enhances the
conduction of
electrical current. The current density of the electrolytic cell described
herein ranges from
about 1 mA/cm2 to about 500 mA/cm2. In some embodiments, the current density
range may
be from about 50 mA/cm2 to about 400 mA/cm2. The current density range may be
from
about 200 mA/cm2 to about 300 mA/cm2. Overall, it is only necessary to provide
a sufficient
amount of current to induce the active form of the active metal, which
comprises the anode,
to cause the hydrolysis of urea. Typically, the electrolytic cell will be
operated at a constant
current or current density within these ranges.
9
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
[0044] The electrical current may also be used to control the production of
ammonia from
the electrolytic hydrolysis of urea and therefore control the rate of
injecting ammonia into an
exhaust gas treatment system. For example, a given electrical current may be
required to
induce the active form of the active metal in all the regions of the anode to
maximize the
production of ammonia. The applied current may be lowered when the need for
ammonia
decreases.
[0045] Electrolytic cells may operate over varying ranges of pressure and
temperature.
The operating pressure may be about atmospheric pressure or ambient pressure
with no upper
pressure limit other than the physical limits of the reaction vessel. If
desired, the operating
pressure of the electrolytic cell may be varied to control the rate of ammonia
that is injected
into an exhaust gas. The operating temperature range may be from about 0 C to
about
100 C. An acceptable operating temperature range may be from about 40 C to
about 80 C.
More specifically, an operating temperature range from about 60 C to about 70
C is
particularly useful.
[0046] The temperature in the electrolytic cell may be controlled with any
available
source. For example, the electrolytic cell may further include a heater
apparatus operatively
coupled to electrolytic cell, and/or a recirculation system operatively
coupled to the
electrolytic cell, wherein the recirculation system contains at least a
portion of the alkaline
electrolyte composition. Exemplary heating apparatus include heating jackets
that surround
the electrolytic cell, from which heat may be supplied by external sources,
such as steam,
heated water, or other heated fluids. Other possible heating sources can
include, but are not
limited to, electric heaters or combustion gases. Alternatively, or in
addition, the
recirculation system may also include a heating apparatus for increasing the
temperature of
the alkaline electrolyte composition at a point external to the electrolytic
cell. The desired
heating source may depend on the availability and/or compatibility with the
system. For
example, electric heat may be the most convenient way to provide the heat to
achieve a
desired operating temperature for the use of the electrolytic cell in an
automobile SCR
system, especially during cold start and during extreme weather conditions.
Accordingly, the
electrolytic cell may have temperature control that is independent of the
temperature of the
engine.
[0047] It will be readily apparent to one of ordinary skill in the art that
the above-
described electrolytic cell is readily adaptable to a continuous flow cell
configuration, semi-
continuous, and with recirculation of the alkaline electrolyte composition.
For example, an
10
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
exemplary system for the continuous generation of sufficient quantities of
ammonia to
adequately supply the needs of a coal fired power plant on a continuous basis
is shown in
FIG. 6. From a urea storage container 10, urea prill is supplied via a rotary
feed valve 12 to a
mix tank 14 where the urea prill is mixed with water from a water supply 16 to
form a urea
solution. The mix tank 14 includes a discharge line 18, which supplies the
urea solution to a
urea solution feed pump 20 to transfer the urea solution to a urea electrolyte
tank 24 through
the urea electrolyte tank inlet 22. A urea solution recirculation line 26
permits continuous
operation of the urea solution feed pump 20. According to this embodiment, the
urea
electrolyte composition is formed by mixing the urea solution from the mix
tank 14 with an
alkaline electrolyte composition including hydroxide, carbonate, or
bicarbonate salts of alkali
metals or alkaline earth metal, or combinations thereof. The urea electrolyte
tank 24 includes
a discharge line 28, which supplies the urea electrolyte solution to a urea
electrolyte solution
feed pump 30 to transfer the urea electrolyte solution through an electrolytic
cell inlet 32 to
an electrolytic cell 34. A urea electrolyte solution recirculation line 36
permits continuous
operation of the urea electrolyte solution feed pump 30, and may also
participate in control of
the volume or level of urea electrolyte solution within the electrolytic cell
34. The
electrolytic cell 34 includes a heating jacket 38 having an inlet line 40 and
an outlet line 42
for recirculating heating fluids therethrough.
[0048] One typical flow cell design is that of a packed-bed type of
electrolytic flow cell,
which enables the voltage and/or the current within the packed bed of anodic
catalyst material
to be varied along the catalyst bed and thereby control the rate of ammonia
evolution. A
packed-bed type flow cell is depicted in FIG. 6 with V1-V6 representing the
variable voltage
capability of the electrolytic cell 34, where the insulating materials between
the electrically
insulated regions of the packed anodic catalyst bed are not shown. This
configuration is also
adaptable for controlling the amount of urea being hydrolyzed based on the
level or volume
of urea electrolyte solution covering the catalyst bed. In other words,
varying an area
percentage of a total area of the anodic catalyst bed in contact with a urea
solution will vary
the rate of ammonia production. As such, increasing the amount of urea
electrolyte solution
covering the available catalyst bed will increase the rate of ammonia
production.
[0049] During operation, the urea electrolyte solution flows through the
electrolytic cell
34 and thereby contacting the electrodes. Accordingly, the generated ammonia
from the
electrolytic hydrolysis of urea is supplied to an exhaust gas treatment system
through an
ammonia discharge line 44. Depending on the electrolytic cell operating
conditions,
11
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
hydrogen may also be produced and supplied to auxiliary systems through a
hydrogen gas
discharge line 46. The urea electrolyte solution, after having been depleted
of at least a
portion of its urea, is returned to the urea electrolyte tank 24 though urea
electrolyte return
line 48.
[0050] According to one aspect of this embodiment, the alkaline electrolyte
composition
is concentrated to a desired level prior to being returned to the urea
electrolyte tank 24. In
one example, the urea electrolyte return line 48 delivers the discharge of the
electrolytic cell
34 to an evaporator 50 wherein excess water is removed via a water discharge
line 52 to
achieve a desired concentration. Advantageously, any non-hydrolyzed urea is
principally
retained in solution and is therefore also concentrated therein. The
concentrated electrolyte
solution is then transferred to the urea electrolyte tank 24 through a
concentrated electrolyte
discharge line 54. The rate of excess water removal can be controlled by
modifying various
parameters, such as temperature and pressure. Accordingly, the operating
temperature of the
evaporator 50 may range from about 120 C to about 90 C, depending on the
relative
operating pressure of the evaporator. For example, the evaporator 50 may be
operated at
about 110 C and near or below atmospheric pressure. The source of heat for the
evaporator
50 is not particularly constrained to any source.
[0051] Other flow cell designs are also amenable to the instant embodiment. As
shown
in FIG. 7, a flow cell 60 may include a jacketed containment vessel 62 having
a tubular
cathode 64, a tubular anode 66 and a vessel lid 68. The jacketed containment
vessel 62 may
be thermally controlled by any suitable method. The jacketed vessel 62 further
includes inlet
70. When present, a tubular separator 72 compartmentalizes the tubular cathode
64 and the
tubular anode 66, which permits separation of the effluents therefrom.
Accordingly, each
electrode chamber may have its own discharge port, whereby the vessel lid 68
is configured
to accommodate a cathode connector tubing 74 and an anode connector tubing 76.
For
example, the cathode connector tubing 74 may be hollow and include a conductor
to thereby
provide both a discharge flow path from the proximity of the tubular cathode
64 and an
electrical connection. Similarly, the anode connector tubing 76 may be hollow
and include a
conductor to thereby provide both a discharge flow path from the proximity of
the tubular
anode 66 and an electrical connection.
[0052] According to another embodiment shown in FIG. 8, as an alternative to
supplying
urea from prill, which needs to be dissolved prior to electrolysis-induced
hydrolysis of the
urea, the urea may be supplied to the electrolytic flow cell 90 as a pre-
dissolved, concentrated
12
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
aqueous solution. Many conventional SCR systems use to treat diesel exhaust
utilize diesel
exhaust fluid (DEF), which can be, for example, about 32 wt% urea dissolved in
deionized
water. The exhaust gas treatment systems in accord with the principles of the
present
invention can include an ammonia generator 80, which further includes a DEF
tank 82.
Pump 20, via inlet line 84 and discharge line 85, transfers the DEF from the
DEF tank 82 to a
three way feed valve 86, which mixes the DEF and alkaline electrolyte
composition in the
desired portions to achieve a target urea concentration therein. If the DEF
tank 82 is kept
pressurized, for example, about 20 psig, a pump 20 will not be needed for the
system. This
may allow a more compact system for transportation applications.
Advantageously, the
alkaline electrolyte composition is supplied via recirculating alkaline
electrolyte composition
supply line 87. The urea electrolyte solution is transferred through an
electrolytic cell inlet
88 to the electrolytic cell 90. Accordingly, the generated ammonia from the
electrolytic
hydrolysis of urea is supplied to an exhaust gas treatment system through an
ammonia
discharge line 92 through valve 93. Depending on the electrolytic cell
operating conditions,
hydrogen may also be produced and supplied to auxiliary systems through a
hydrogen gas
discharge line 94 through valve 95. Further, after electrolysis-induced
hydrolysis of at least a
portion of the urea, the discharge of the electrolytic cell is directed to an
evaporator 96 via
pump 20, wherein the alkaline electrolyte composition is concentrated via
removal of water
via water discharge line 98. The alkaline electrolyte composition may be
concentrated near
to the saturation point for the given alkaline electrolyte(s), which elevates
the freezing point
of the solution.
[0053] As shown in FIG. 9, in yet another embodiment the ammonia generator 110
may
include an electrolytic cell 130 having a supply of solid urea in a urea
cartridge 120. The
urea may be transferred from the cartridge 120 via supply line 122 and mixed
with water in a
rotary mixing valve 12 prior to being added to the electrolytic cell 130 via
inlet 128. In this
embodiment, in the absence of physical losses, water need only be introduced
into the system
from water supply tank 124 via water supply line 126 over time to make up for
the water
consumed during the hydrolysis of urea (see Equation 8). For example, the
water may be
introduced at regularly scheduled time intervals and/or based on one or more
operating
parameter. The water can be added directly in the electrolytic cell 130 at
regularly schedule
time intervals to decrease freezing point due its mixture with the
electrolyte. The ammonia
and/or hydrogen, which can be transported via discharge lines 132, 136 and
valves 133, 137,
respectively, are utilized for the treatment of exhaust combustion gas as
described above.
13
CA 02809092 2013-02-21
WO 2012/027368 PCT/US2011/048809
According to another aspect, the urea cartridge 120 may be contained within
the electrolytic
cell 130. According to another aspect, the electrolyte may be contained within
the
electrolytic cell and may be stored in gel form. Although not shown, the
electrolyte can be
recirculated within the electrolytic cell 130 to facilitate improved mixing
with the urea
cartridge 120.
[0054] The present invention will be further appreciated in view of the
following
examples.
EXAMPLE 1
[0055] Two closed cells (1000 mL) were assembled. Each cell was filled with
200 mL of
7 M KOH and 0.33 M urea solution, and stirred at 120 rpm. Voltage (1.4 V) was
applied to
cell B (supplied with Arbin Industries MSTAT) using a Rh-Ni anode (0.15 mg/cm2
Rh on Ni
foil, 10 cm2) and platinum foil (10 cm2) as the cathode. Samples were taken
via liquid
sampling ports and analyzed for ammonia concentration periodically by
extracting 10 mL and
diluting 1:100 with distilled water. A 50 mL aliquot of this analyte was added
to a flask, 1
mL of pH adjusting solution was added with stirring, and the solution was
analyzed using an
ion selective electrode. After two hours of constant voltage operation, Cell A
and B
contained aqueous ammonia concentrations of 3600 and 4700 ppm, respectively
(Table 1).
After 3 hours of operation, cell A increased to 3800 ppm while cell B
increased to 6450 ppm,
which provided that the cell with applied potential had 41% higher conversion
of urea to
ammonia. Cell B averaged about 25 mA/cm2 during the first two hours, which
decreased to
near 8 mA/cm2 for the third hour (Figure 4). These results show that the lower
current
density was more effective in converting the urea to ammonia.
[0056] TABLE 1. Urea hydrolysis via electrolysis samples.
Test Time Cell ppm NH3 Avg. Current % increase w/
(total hrs) (liquid Phase) (mA) electrolysis
A 3637
2 B 4715 98 23
A 3800
3 B 6450 30 41
[0057] Application of 1.4 V to cell B resulted in a 41% higher conversion
after 3 hours of
operation, indicating that the urea to ammonia reaction is in fact enhanced by
electrolysis.
Electrolysis at a low voltage contributes to kinetics of the urea to ammonia
conversion.
14
CA 02809092 2013-02-21
WO 2012/027368 PCT/US2011/048809
EXAMPLE 2
[0058] Two closed cells (1000 mL) were assembled with Rh-Ni anodes (8 cm2
each; cell
A: 0.05 mg/cm2, cell B: 0.15 mg/cm2) and platinum foil cathodes (15 cm2),
filled with 7 M
KOH and 0.33 M urea, and heated to 70 C. Liquid sampling ports were included
for
monitoring aqueous ammonia concentration ex-situ by ISE throughout the
duration of the
experiment. Voltage (1.33 V) was applied to both cells A and B (supplied with
Arbin
Industries MSTAT) with 120 rpm stirring. A lower voltage was chosen as
compared to
Example 1 above because it was postulated that a lower voltage, which will
provide a lower
current density, was needed to affect the Ni0OH catalyzed reaction to ammonia.
[0059] Samples were taken and analyzed for ammonia concentration periodically
by
extracting 10 mL and diluting 1:100 with distilled water. A 50 mL aliquot of
this analyte was
added to a flask with stirrer and ISE electrode and 1 mL of pH adjusting
solution, as
described in Experiment 1. After two hours of constant voltage operation, Cell
A and B
contained aqueous ammonia concentrations of 4890 and 6470 ppm, respectively
(Table 2).
These concentrations did not increase after the third hour of operation. The
average current
in each cell for the first two hours was 1.5 and 2.0 mA/cm2 for cell A and B,
respectively
(Figure 5). It is postulated that the apparent stoppage in urea conversion to
ammonia after the
first sample period is likely the result of the current density dropping to
around 1 mA/cm2
after 2 hours, which may be below the level necessary to affect the reaction.
It was observed
that a black precipitate formed on the platinum cathode in both cells. Most of
the conversion
affected by applied potential probably took place within the first hour where
average current
was 2-3 mA/cm2. Otherwise, leakage from the liquid sampling ports could
explain the lack
of increase in conversion.
[0060] TABLE 2: Urea hydrolysis via electrolysis samples.
Time Cell A ppm NH3 Cell B ppm NH3
2 hrs 4890 6470
3 hrs 4580 6400
[0061] Based on these results, the effect of current density on the conversion
of urea to
ammonia and the effect of catalyst loading Cell B exhibited a higher
conversion than cell A,
probably because it had an anode with higher loading of rhodium and operated
under a
slightly higher average current density. Again, these results show that
electrolysis at a low
voltage can contribute to favorable kinetics of the urea to ammonia
conversion.
15
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
[0062] For example, for a Diesel truck application, providing 0.5 Kg of
ammonia per
hour to an SCR unit at a current of 6.25 amps and a cell voltage of 1.33
volts, would
correspond to 8.31 watts of power. The thermal energy consumed would be 1,980
kilojoules.
Additionally, under these conditions, approximately 0.23 g/hour of hydrogen
may be
generated, which equates to about 33 kilojoules of thermal energy, and may be
injected into
the combustion engine of the diesel truck to minimize carbon dioxide emissions
and increase
fuel efficiency.
[0063] In another example, for a 500 MW coal-fired power plant, providing 200
Kg of
ammonia per hour to an SCR unit at a current of 2,500 amps and a cell voltage
of 1.33 volts,
would correspond to 3.325 kilowatts of power. The thermal energy consumed
would be
792,000 kilojoules. Additionally, under these conditions, approximately 93.3
g/hour of
hydrogen may be generated, which equates to about 13,228 kilojoules of thermal
energy.
EXAMPLE 3
[0064] Electrolytic Hydrolysis of Urea: A cell containing 7 M KOH/0.33 M urea
solution at atmospheric pressure was subjected to electrolysis-induced
hydrolysis. A cell
voltage of 1.4 volts was applied to a 2x2.5 cm2 carbon-paper anode deposited
with Ni, and a
5x5 cm2 Pt foil cathode. Under these conditions, the presence of ammonia was
detected from
the conversion of urea into ammonia and carbon dioxide. The hydrolysis pathway
becomes
favorable with increasing hydroxide salt concentration and increasing
temperatures. For
example, urea samples contained in 0 M, 1 M, 5 M and 7 M KOH at 50 C for 89
hours
produced 0.7%, 4.2%, 27.4% and 36.7% hydrolysis, respectively. A 7 M KOH
sample of
urea at 70 C for 24 hours provided over 95% hydrolysis.
EXAMPLE 4
[0065] Flow Cell Hydrolysis of Urea: In a sandwich-style urea electrolytic
cell that
compartmentalized the anode and cathode, a polypropylene membrane was used as
a
separator. The anode was constructed of a 5 cm2 carbon-paper support, on which
was
electrodeposited Ni. The cathode was constructed of a 5 cm2 carbon paper
support, on which
was electrodeposited Pt. The electrodes were immersed in 5M KOH/0.33 M urea at
70 C. A
cell voltage of 1.33 volts was applied and ammonia evolved from the anode. It
was noted
that a small amount of hydrogen was produced from the cathode. The respective
gases were
analyzed using a MG2 SRI 8610C gas chromatograph with a thermal conductivity
detector
(TCD), Haysep column, and a molecular sieve column. Pure hydrogen was observed
at the
16
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
cathode, while ammonia, N2 and small amounts H2 were observed from the anode
in gas
phase. The hydrogen on the anode side of the separator is believed to arise
from hydrogen
passing through the polypropylene membrane. Ammonia was further detected in
the liquid
phase using an Orion ammonia selective electrode (ISE). No carbon species were
detected in
gas phase. It is postulated that any CO2 that may have been generated was
quickly
transformed into potassium carbonate.
EXAMPLE 5
[0066] Electrolysis of Urea: A cell containing 5 M KOH/0.33 M urea solution
at 25 C
and atmospheric pressure was subjected to electrolysis. A cell voltage of 1.4
volts was
applied to a 2x2.5 cm2 carbon-paper anode deposited with Ni, and a 5x5 cm2 Pt
foil cathode.
It was determined by gas chromatography that the electrolysis of urea produced
nitrogen at
the anode of this electrolytic cell, whereas hydrogen was produced at the
cathode. Ammonia,
which is presumably derived from the electrolysis-induced hydrolysis of urea,
was detected
in the electrolyzed solution using an Orion ammonia selective electrode (ISE).
No carbon
species were detected in the gas phase. It is postulated that the generated
CO2 was quickly
transformed into potassium carbonate by reacting with potassium hydroxide in
the alkaline
electrolyte composition.
[0067] Therefore, at the anode, urea may be oxidized to nitrogen and carbon
dioxide. At
the cathode, the counter electrode, hydrogen may be produced, as shown in the
following
reaction:
(NH2)2C0+ H20 ¨> N2 i + c02 i + 3142 i (Overall Electrolysis Reaction)
[0068] Therefore, in addition to the electrolysis-induced hydrolysis of urea
to supply the
requisite ammonia reductant to an exhaust gas treatment system, under the
appropriate
conditions, the foregoing electrolysis of urea may provide hydrogen, which may
be injected
into a combustion chamber that is attached to the exhaust gas treatment
system, as shown in
FIG. 3. Thus, adding hydrogen to the combustion chamber may facilitate
improved fuel
combustion efficiency, as well as reducing unwanted emission by-products.
EXAMPLE 6
[0069] The model system in accordance with the embodiment represented in
FIG. 6 has
been designed with an electrolytic cell having a total volume of 825 liter,
with 660 liters of an
anodic bed providing 1,247 m2 of active metal surface. Extrapolating the
experimental data
obtained from a batch-type configuration, operation mass transfer parameters
were calculated
17
CA 02809092 2013-02-21
WO 2012/027368 PCT/US2011/048809
for the foregoing system. Additionally, a comparison was made between the
inventive
electrolytic hydrolysis method (EU2A) and the commonly-used chemical
hydrolysis. As
shown in Table 4 below, the electrolytic urea to ammonia (EU2A) hydrolysis
method
provides an ammonia stream which is predominantly (e.g., 64 molar %) comprised
of
ammonia. The calculated parameters and comparison data are shown in Tables 3
and 4,
respectively.
[0070] TABLE 3: Calculated operating parameters.
Stream Rate (kg/hr) Composition (%)
1: Prill urea 352.9 100.0
2: Water 119.4 100.0
3: Concentrated urea 472.3 Urea: 74.7
H2 = 0. 25.3
4: Urea electrolyte reactor feed 1138.0 Urea: 31.0
K2CO3: 22'5
H2 = 0. 46.5
5: Electrolyte recycle 665.7 K2CO3: 38'4
H2 = 0. 61.6
6: NH3 to SCR (@ 70 C; 30 472.2 NH3: 42.4
psig) CO2: 54.7
H20: 2.9
7: Hydrogen to fuel cell 0.1 100.0
8: Saturated steam (150 psig) 394.8 100.0
9: Exhausted steam (150 psig) 394.8 100.0
[0071] TABLE 4: Comparison of methods.
Description Chemical hydrolysis EU2A
Volume of reactor (liters) 7,250 825
SCR Ammonia Reagent 200 kg/hr 200 kg/hr
Dry Urea Flow Rate 352 352
Reagent Concentration 50 % wt 40-60 % wt
DI water 375 kg/hr 119 kg/hr
Steam heating (150 psig) 840 kg/hr 395 kg/hr
Power N/A 1.8 kW
Gas Molar Composition
NH3 (%) 22.8 64.0
CO2(%) 11.4 32.0
H20 (%) 65.8 4.0
** Reusing the hydrogen in a fuel cell with 50% efficiency.
18
CA 02809092 2013-02-21
WO 2012/027368 PCT/US2011/048809
EXAMPLE 7
[0072] The model system in accordance with the embodiment represented in
FIG. 8 was
calculated for an electrolytic cell having a volume of 2 liters. A comparison
was made
between the inventive electrolytic hydrolysis method (EU2A) and the commonly-
used DEF-
SCR systems used on diesel trucks (which are based on chemical hydrolysis). As
shown in
Table 5 below, the electrolytic urea to ammonia (EU2A) hydrolysis method
provides an
ammonia source that is substantially more volume and weight efficient compared
to that of
DEF-SCR chemical hydrolysis systems.
[0073] TABLE 5: Comparison of methods.
Service life (miles) 12,000
Volume of reactor (liters) 2
Volume electrolyte recovery (liters) 2
Total volume of system (liters) 4*
Fraction of volume compare to a DEF full tank (30 gal) 3.5%
Weight of reactor (kg) 10
Weight electrolyte and recovery vessel (kg) 3
Total system weight (kg) 13*
Fraction of weight compare to a DEF full tank (123 kg) 10.5%
*Tubing, valves, and fittings are not included in these calculations.
EXAMPLE 8
[0074] The model system in accordance with the embodiment represented in
FIG. 9 was
calculated for an electrolytic cell having a volume of 2 liters. A comparison
was made
between the inventive electrolytic hydrolysis method (EU2A) using solid urea
versus the
conventional DEF-SCR system. As shown in Table 6 below, the electrolytic urea
to
ammonia (EU2A) hydrolysis method provides an ammonia source that is
substantially more
volume efficient that chemical hydrolysis systems.
19
CA 02809092 2013-02-21
WO 2012/027368
PCT/US2011/048809
[0075] TABLE 6: Comparison of methods.
Service life (miles) 12,000 60,000
Volume of reactor with contained electrolyte (liters) 2 2
Volume of Urea (liters) 62 82
Water consumed (liters) 25 123
Total volume of system (liters) 87* 205*
Fraction of volume compared to a DEF full tank (30 gal or 76% N/A
113 liters)
Weight of reactor with contained electrolyte (kg) 12 12
Weight urea (kg) 82 410
Weight of water consumed 25 123
Total system weight (kg) 107* 533*
Fraction of weight compare to a DEF full tank (123 kg) 87% N/A
*Tubing, valves, and fittings are not included in these calculations.
[0076] One issue commonly encountered in electrolytic cells, is the slow
deactivation of
the one or both of the electrodes. In some instances, the deactivation may be
attributed to the
attachment of an oxidized film on the anode and/or the attachment of scale on
the surface of
the cathode. This deactivation process deteriorates the electrolytic
efficiency of the cell. For
example, as this deactivation occurs, the current density can, in some
instances, decrease for a
constant applied voltage, thereby reducing the rate of electro-oxidation.
Alternatively, the
current density sometimes can be sustained by increasing the applied voltage.
In either
instance, energy is wasted and the overall efficiency of the cell is
diminished.
[0077] From an operational perspective, regeneration of the electrodes by
reversing the
applied voltage for a period of time can be useful. The reversed voltage may
be the same or
different as the operating voltage. The reversal voltage may range from about
0.5 volts to
about 2.0 volts. Another suitable reversal voltage may range from about 1.4
volts to about
1.6 volts.
[0078] During regeneration, the period of time for applying a reversed
voltage may vary
from just a few minutes to tens of hours. For example, the first and second
conducting
components may both include one or more metals active toward electrochemical
oxidation of
urea, therefore either electrode may function as a cathode and produce
hydrogen. As such,
reversing the voltage is effectively an uninterrupted process, thereby
allowing the reversed
voltage to be applied for an indefinite period of time or until deactivation
is again
encountered. According to the operating conditions of the electrochemical cell
described
herein, electrodes may be operated for about 5 hours to about 20 hours before
losing activity
and requiring activation.
20
WO 2012/027368 CA 02809092 2013-02-21 PCT/US2011/048809
[0079] Conversely, if the anode's conducting component is comprised of a metal
inactive
toward electrochemical oxidation of urea, the regeneration may be achieved in
about 1
minute to about 20 minutes at about 1.4 volts. In some instances, reactivation
can be
achieved in about 6 minutes at 1.4 volts.
[0080] For SCR applications, the SCR unit is not particularly limited to any
specific
configuration or catalyst. For example, plate, honeycomb, pellet, bead, fiber
or corrugated
configurations are suitable for use. Moreover, the catalyst is not limited to
any species or
form. For example, traditional catalysts based on vanadium, titanium, or iron
or copper-
promoted zeolite catalysts are suitable for use. Additionally, newer SCR
catalysts, such as
those disclosed in U.S. Patent No. 7,527,776 by Golden et al. may be used.
Similarly, for
SNCR applications and/or gas flue conditioning applications, the SNCR unit and
/or the
particle precipitator are not particularly limited to any specific design.
[0081] Accordingly, the electrolytic cells according to embodiments of the
present
invention may be adapted to couple with commercially available SCR or SNCR
units or flue
gas conditioning systems. For example, the electrolytic cell may be adapted to
work with
existing ammonia generators that thermally hydrolyze urea, or the electrolytic
cell may be
designed to be the lone source of ammonia for the exhaust gas treatment
systems.
Alternatively, the cell and the exhaust gas treatment system, such as an SCR
or an SNCR
system, may be designed as a combined unit.
[0082] The ammonia may normally be introduced into the exhaust gas prior to an
electrostatic precipitator, an SNCR unit, or prior to contacting a catalyst
within an SCR unit.
The exhaust gas and the ammonia as a reducing agent may be contacted with the
catalyst,
thereby reducing the nitrogen oxides in the exhaust gas. The optimization of
temperatures,
pressures, flow rates and the like can readily be achieved by one having
ordinary skill in the
art of exhaust gas treatment technology.
[0083] As used herein and in the appended claims, the singular forms "a",
"an", and "the"
include plural reference unless the context clearly dictates otherwise. As
well, the terms "a"
(or "an"), "one or more" and "at least one" can be used interchangeably
herein. It is also to be
noted that the terms "comprising", "including", "characterized by" and
"having" can be used
interchangeably.
[0084] While the present invention has been illustrated by the description of
one or more
embodiments thereof, and while the embodiments have been described in
considerable detail,
21
WO 2012/027368 CA 02809092 2013-02-21PCT/US2011/048809
they are not intended to restrict or in any way limit the scope of the
appended claims to such
detail. The various features of exemplary embodiments described herein may be
used in any
combination. Additional advantages and modifications will readily appear to
those skilled in
the art. The invention in its broader aspects is therefore not limited to the
specific details,
representative product and method and illustrative examples shown and
described.
Accordingly, departures may be made from such details without departing from
the scope of
the general inventive concept.
22