Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02093203 2002-03-13
21489-8646 (S)
-1-
Pyrimidine derivatives and processes for the preparation thereof
The invention relates to N-phenyl-2-pyrimidine-amine derivatives, to processes
for the
preparation thereof, to medicaments comprising those compounds, and to the use
thereof in the
preparation of pharmaceutical compositions for the therapeutic treatment of
warm-blooded
animals.
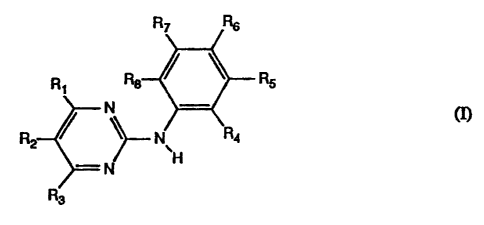
The invention relates to N-phenyl-2-pyrimidine-amine derivatives of formulal
R R ~ ~ Rs
N CI).
R / ~~N R4
~H
'N
R3
wherein
R, is pyrazinyl, 1-methyl-1 H-pyrrolyl, amino- or amino-lower alkyl-
substituted phenyl
wherein the amino group in each case is free, alkylated or acylated, 1 H-
indolyl or 1 H-imidaiolyl
bonded at a five-membered ring carbon atom, or unsubstituted or lower alkyl-
substituted pyridyl
bonded at a ring carbon atom and unsubstituted or substituted at the nitrogen
atom by oxygen,
R2 and R3 are each independently of the other hydrogen or lower alkyl,
one or two of the radicals R4, R5, Rs, R, and R8 are each vitro, fluoro-
substituted lower alkoxy or
a radical of formulall
-N~Rs)'C~=X)-(Y)n-R~o
wherein
R9 is hydrogen or lower alkyl,
X is oxo, thio, imino, N-lower alkyl-imino, hydroximino or O-lower alkyl-
hydroximino,
CA 02093203 2002-05-10
21489-8646 (S)
-2-
Y is oxygen or the group NH,
n is 0 or 1 and
Rlo is an aliphatic radical having at least 5 carbon atoms, or an aromatic,
aromatic-
aliphatic, cycloaliphatic, cycloaliphatic-aliphatic, heterocyclic or
heterocyclic-
aliphatic radical,
and the remaining radicals R4, R5, Rs, R~ and Rg are each independently of the
others
hydrogen, lower alkyl that is unsubstituted or substituted by free or
alkylated amino,
piperazinyl, piperidinyl, pyrrolidinyl or by morpholinyl, or lower alkanoyl,
trifluoro-
methyl, free, etherified or esterifed hydroxy, free, alkylated or acylated
amino or
free or esterified carboxy,
and to salts of such compounds having at least one salt-forming group.
1-Methyl-1H-pyrrolyl is preferably 1-methyl-1H-pyrrol-2-yl or 1-methyl-1H-
pyrnol-3-yl.
Amino- or amino-lower alkyl-substituted phenyl Rl wherein the amino group in
each case
is free, alkylated or acylated is phenyl substituted in any desired position
(ortho, meta or
para) wherein an alkylated amino group is preferably mono- or di-lower
alkylamino, for
example dimethylamino, and the lower alkyl moiety of amino-lower alkyl is
preferably
linear Ct-C3alkyl, such as especially methyl or ethyl.
1H-Indolyl bonded at a carbon atom of the five-membered ring is 1H-indol-2-yl
or
1H-indol-3-yl.
Unsubstituted or lower alkyl-substituted pyridyl bonded at a ring carbon atom
is lower
alkyl-substituted or preferably unsubstituted 2-, 4- or preferably 3-pyridyl,
for example
3-pyridyl, 2-methyl-3-pyridyl or 4-methyl-3-pyridyl. Pyridyl substituted at
the nitrogen
atom by oxygen is a radical derived from pyridine N-oxide, i.e. N-oxido-
pyridyl.
Fluoro-substituted lower alkoxy is lower alkoxy carrying at least one, but
preferably
several, fluoro substituents, especially trifluoromethoxy or 1,1,2,2-
tetrafluoro-ethoxy.
When X is oxo, thio, imino, N-lower alkyl-imino, hydroximino or O-lower alkyl-
hydrox-
imino, the group C=X is, in the above order, a radical Cue, Cue, C=N-H, C=N-
lower
alkyl, C=N-0H or C=N-O-lower alkyl, respectively. X is preferably oxo.
~~1fi~1 /~ 5,~
f.J a ~ V r~1
-3-
n is preferably 0, i.e. the group Y is not present.
Y, if present, is preferably the group 1V13.
The term "lower" within the scope of this text denotes radicals having up to
and including
7, preferably up to and including 4 carbon atoms.
bower alkyl Rl, Rz, R3 and R9 is preferably methyl or ethyl.
An aliphatic radical Rto having at least 5 carbon atoms preferably has not
more than 22
carbon atoms, generally not more than 10 carbon atoms, and is such a
substituted or
preferably unsubstituted aliphatic hydrocarbon radical, that is to say such a
substituted or
preferably unsubstituteri alkynyl, alkenyl or preferably alkyl radical, such
as CS-C~alkyl,
for example n-pentyi. An aromatic radical Rlo has up to 20 carbon atoms and is
unsubstituted or substituted, for example in each case unsubstituted or
substituted
naphthyl, such as especially 2-naphthyl, or preferably phenyl, the
substituents preferably
being selected from cyano, unsubstituted or hydroxy-, amino- or 4-methyl-
piperazinyl-
substituted lower alkyl, such as especially methyl, trifluoromethyl, free,
etherified or
esterified hydroxy, free, alkylated or acylated amino and free or esterified
carboxy. In an
aromatic-aliphatic radical Rto the aromatic moiety is as defined above and the
aliphatic
moiety is preferably lower alkyl, such as especially Ct-C2alkyl, which is
substituted or
preferably unsubstituted, for example benzyl. A cycloaliphatic radical Rto has
especially
up to 30, more especially up to 20, and most especially up to 10 carbon atoms,
is mono- or
poly-cyclic and is substituted or preferably unsubstituted, for example such a
cycloalkyl
radical, especially such a S- or 6-membered cycloalkyl radical, such as
preferably cyclo-
hexyl. In a cycloaliphatic-aliphatic radical Rto the cycloaliphatic moiety is
as defined
above and the aliphatic moiety is preferably lower alkyl, such as especially
Ct-C.zalkyl,
which is substituted or preferably unsubstituted. A heterocyclic radical Rtp
contains
especially up to 20 carbon atoms and is preferably a saturated or unsaturated
monocyclic
radical having 5 or 6 ring members and 1-~ hetero atoms which are preferably
selected
from nitrogen, oxygen and sulfur, especially, for example, thienyl or 2-, 3-
or 4-pyridyl, or
a bi- or tri-cyclic radical wherein, for example, one or two benzene radicals
are annellated
(fused) to the mentioned monocyclic radical. In a heterocyclic-aliphatic
radical Rtp the
heterocyclic moiety is as defined above and the aliphatic moiety is preferably
lower alkyl,
such as especially Ct-Czalkyl, which is substituted or preferably
unsubstituted.
fix, i1. ~ j
~a . e.9 r'~-t !,~ =~
<;
-4-
Etherified hydroxy is preferably lower alkoxy. Esterified hydroxy is
preferably hydroxy
esterified by an organic carboxylic acid, such as a lower alkanoic acid, or a
mineral acid,
such as a hydrohalic acid, for example lower alkanoyloxy or especially
halogen, such as
iodine, bromine or especially fluorine or chlorine.
Alkylated amino is, for example, lower alkylamino, such as methylamino, or di-
lower
alkylamino, such as dimethylamino. Acylated amino is, for example, lower
alkanoyl-
amino or benzaylamino.
Esterified carboxy is, for example, lower alkoxycarhonyl, such as
methoxycarbonyl.
A substituted phenyl radical may carry up to 5 substituents, such as fluorine,
but
especially in the case of relatively large substituents is generally
substituted by only from
1 to 3 substituents. Examples of substituted phenyl that may be given special
mention are
4-chloro-phenyl, pentafluora-phenyl, 2-carboxy-phenyl, 2-methoxy-phenyl, 4-
fluoro-
phenyl, 4-cyano-phenyl and 4-methyl-phenyl.
Salt-forming groups in a compound of formula I are groups or radicals having
basic or
acidic properties. Compounds having at least one basic group or at least one
basic radical,
for example a free amino group, a pyrazinyl radical or a pyridyl radical, may
form acid
addition salts, for example with inorganic acids, such as hydrochloric acid,
sulfuric acid or
a phosphoric acid, or with suitable organic carboxylic or sulfonic acids, for
example
aliphatic mono- or di-carboxylic acids, such as trifluoroacetic acid, acetic
acid, propionic
acid, glycolic acid, succinic acid, malefic acid, fumaric acid, hydroxymaleic
acid, malic
acid, tartaric acid, citric acid or oxalic acid, or amino acids such as
arginine or lysine,
aromatic carboxylic acids, such as benzoic acid, 2-phenaxy-benzoic acid, 2-
acetoxy-
benzoic acid, salicylic acid, 4-aminosalicylic acid, aromatic-aliphatic
carboxylic acids,
such as mandelic acid or cinnamic acid, heteroaromatic carboxylic acids, such
as nicotinic
acid or isonicotinic acid, aliphatic sulfonic acids, such as methane-, ethane-
or 2-hydroxy-
ethane-sulfonic acid, or aromatic sulfonic acids, for example benzene-, p-
toluene- or
naphthalene-2-sulfonic acid. When several basic groups are present mono- or
poly-acid
addition salts may be formed.
Compounds of formula I having acidic groups, for example a free carboxy group
in the
radical Rto, may form metal or ammonium salts, such as alkali metal or
alkaline earth
metal salts, for example sodium, poeassium, magnesium or calcium salts, or
ammonium
2 ~~ a
~~L~W~~3
-5-
salts with ammonia or suitable organic amines, such as tertiary monoamines,
far example
triethylamine or tri-(2-hydroxyethyl)-amine, or heterocyclic bases, for
example N-ethyl-
piperidine or N,N'-dimethyl-piperazine.
Compounds of formula I having both acidic and basic groups can form internal
salts.
For the purposes of isolation or purification, as well as in the case of
compounds that are
used further as intermediates, it is also possible to use pharmaceutically
unacceptable
salts. Only pharmaceutically acceptable, non-toxic salts are used for
therapeutic purposes,
however, and those salts are therefore preferred.
Owing to the close relationship between the novel compounds in free form and
in the form
of their salts, including those salts that can be used as intermediates, fox
example in the
purification of the novel compounds or for the identification thereof,
hereinbefore and
hereinafter any reference to the free compounds should be understood as
including the
corresponding salts, where appropriate and expedient.
The compounds of formula I have valuable pharrmacological properties and can
be used,
for example, as anti-tumoral drugs and as drugs against atherosclerosis.
The phosphorylation of proteins has long been known as an important step in
the differ-
entiatian and proliferation of cells. The phosphorylation is catalysed by
protein kinases
which are divided into serine/threonine kinases and tyrosine kinases. The
serine/threonine
kinases include protein kinase C and the tyrosine kinases the PDGF (platelet-
derived
growth factor)-receptor tyrosine kinase.
The compounds of formula I wherein R4 and Rg are hydrogen selectively inhibit
the
enzyme protein kinase C.
Several species of protein kinase C, which is dependent on phospholipids and
calcium,
occur within cells (distribution of the species is tissue-specific); protein
kinase C partici-
pates in various fundamental processes, such as signal transmission,
proliferation and
differentiation, as well as the release of hormones and neurotransmitters. The
enzyme is
activated either by means of receptor-mediated hydrolysis of phospholipids of
the cell
membrane or by direct interaction with certain tumour-promoting agents.
Cellular
functions that are controlled with the aid of protein kinase C can be
influenced by
ct f. ~1 ~~. s~ ~~F v"r
E,, ~, ~ ~ a i7
-6-
modulation of the enzyme activity of protein kinase C.
To determine protein kinase C-inhibiting activity, protein kinase C from pig
brain purified
in accordance with the procedure described by T. Uchida and C.R. Filbrwrr in
J. Biol.
Chem. 259, 12311-4 (I984) is used. The protein- kinase C-inhibiting activity
of the
compounds of formula I is determined by the method of D. Fabro et al., Arch.
Biochem.
Biophys. 239, 1U2-111 (1985). In that test the compounds of formula I inhibit
protein
kinase C at a concentration ICSO of as low as approximately from U.1 to 10
~,mol/litre,
especially approximately from 0.05 to 5 ~,mol/litre. On the other hand, the
compounds of
formula I inhibit other enzymes, for example protein kinase A, phosphorylase
protein
kinase and certain types of tyrasine protein kinase, for example the tyrosine
protein kinase
of EGF (epidermal growth factor) receptors, only at a far higher
concentration, for
example 100 times higher. That is an indication of the selectivity of the
compounds of
formula I. With a view to reducing undesired side effects, it is important for
the protein
kinase C-inhibitors to be as selective as possible, i.e. inter _alia to have
as little effect as
possible on other enzymes, especially when the effect of the activity of those
other
enzymes has no equivalent or synergistic effect on the disease to be treated.
Owing to their inhibiting activity towards protein kinase C, the compounds of
formula I
wherein R4 and Rx are hydrogen, and their pharmaceutically acceptable salts,
can be used
as tumour-inhibiting, immunornodulating and anti-bacterial active ingredients
and, further,
as drugs against atherosclerosis, the immunode~ciency disorder AIDS, and
diseases of the
cardiovascular system and the central nervous system.
As might already be expected on the basis of the inhibiting action on protein
kinase C
described above, the compounds of formula I wherein R~ and Rs are hydrogen,
and their
pharmaceutically acceptable salts, have anti-proliferative properties which
can be
demonstrated directly in the following, different test. In that test the
inhibiting action of
compounds of formula I on the growth of human T24 bladder carcinoma cells is
determined. Those cells are incubated in "Eagle's minimal essential medium",
to which
% (v/v) foetal calf serum has been added, in a humidified incubator at
37°C and with
5 percent by volume C02 in the air. The carcinoma cells (1000-1500) are
transferred to
96-well rnicrotitre plates and incubated overnight under the above-mentioned
conditions.
The test compound is added in serial dilutions on day 1. The plates are
incubated for
5 days under the above-mentioned conditions. During that period the control
cultures
undergo at least 4 cell divisions. After the incubation, the cells am fixed
with 3.3 0l0 (g/v)
~~ t ~ e3 ~e'~ ~.~
_7_
aqueous glutaraldehyde solution, washed with water and stained with 0.05 %
(weight/-
volume) aqueous methylene blue solution. After washing, the stain is eluted
with 3 %
(g/v) aqueous hydrochloric acid. Then the optical density (OD) per well, which
is directly
proportional to the number of cells, is measured using a photometer (Titertek
multiskan) at
665 nm. The ICSp values are calculated by means of a computer system, using
the formula
OD665 (test) minus OD665 (sty) x 100
~D665 (~Oritr01) mlnuS OD665 (Start)
The ICSo values are defined as that concentration of active ingredient at
which the number
of cells per well at the end of the incubation period is only 50 % of the
number of cells in
the control cultures. The ICSO values thus determined are, for the compounds
of formula I,
approximately from 0.1 to 10 pmol/litre.
Owing to the properties described, the compounds of formula I wherein R4 and
R8 are
hydrogen can be used especially as tumour-inhibiting active ingredients, for
example for
the treatment of tumours of the bladder. In addition, they are suitable fnr
the further
applications mentioned above for protein kinase C-modulators and can be used
especially
in the treatment of diseases that respond to inhibition of protein kitiase C.
Some of the compounds of formula I wherein R4 and Rg are hydrogen inhibit not
only
protein kinase C but, at a concentration ICSp as low as approximately from
0.01 to
~.mol/litre, especially approximately from 0.05 to 1 umol/litre, also certain
tyrosine
kinases, such as especially PDGF-receptor kinase or abl-kinase, for example v-
abl-kinase.
Compounds of formula I wherein at least one of the radicals R4 and R8 is other
than
hydrogen and is, for example, lower alkyl, such as methyl, are especially
selective for the
above-mentioned PDGF-receptor and abl-tyrosine kinases and inhibit protein
kinase C
virtually not at all.
PDGF (platelet-derived growth factor) is a very frequently occurring growth
factor which
plays an important role both in normal growth and in pathological cell
proliferation, such
as in carcinogenesis and disorders of the smooth muscle cells of blood
vessels, for
example in atherosclerosis and thrombosis.
The inhibition of protein kinase C and of PDGF-receptor kinase has in this
sense a
~; a'~ ~°s '..> '~'
~v ~t; ~s ~3 ws ~~ e~
_g_
virtually synergistic effect in the same direction vrith regard to the
regulation of cell
growth.
The inhibition of PDGF-stimulated receptor tyrosine kinase activity in vitro
is measured in
PDGF receptor immunocomplexes of BALB/c 3T3 cells, analogously to the method
described by E. Andrejauskas-Buchdunger and U. Regenass in Cancer Research 52,
5353-5358 (1992). The compounds of formula I described in detail above inhibit
PDGF-
dependent cell-free receptor phosphorylation at concentrations of frnm 0.005
to 5 ltmol/-
litre, especially from 0.01 to 1.0, more especially from 0.01 to 0.1
pmol/litre. The
inhibition of PDGF-receptor tyrosine kinase in the intact cell is detected by
means of
Western Blot Analysis, likewise analogously to the method described by
E. Andrejauskas-Buchdunger and U. Regenass in Cancer Research 52, 5353-5358
(1992).
In that test the inhibition of ligand-stimulated PDGF-receptor
autophosphorylation in
BALB/c mouse cells is measured with the aid of anti-phosphotyrosine
antibodies. The
compounds of formula I described in detail above inhibit the tyrosine kinase
activity of the
PDGF receptor at concentrations of from 0.005 to 5 ~.mol/litre, especially
from 0.01 to 1.0
and more especially from 0.01 to 0.1 p.mol/litre. At concentrations below 1.0
p,mo1/litre,
those compounds also inhibit the cell growth of a PDGF-dependent cell Line,
namely
BALB/c 3T3 mouse fibroblasts.
The above-mentioned inhibition of v-abl-tyrosine kinase is determined in
accordance with
the methods of I~1. Lydon et al., Oncogene Research 5, 161-173 (1990) and J.
F. Geissler
et al., Cancer Research 52, 4492-4498 (1992). In those methods [Vats]-
angiotensin_ II and
[T s2P]-ATP are used as substrates.
Owing to the properties described, compounds of formula I can be used not only
as
tumour-inhibiting active ingredients but also as drugs against non-malignant
proliferative
diseases, e.g. atherosclerosis, thrombosis, psoriasis, sclerodemtitis and
fibrosis. They are
also suitable for the further applications mentioned above for protein kinase
C-modulators
and can be used especially in the treatment of diseases that respond to the
inhibition of
PDGF-receptor kinase.
In addition, the compounds of formula I prevent the development of resistance
(multi-drug
resistance) in cancer treatment with other chemotherapeutic drugs or remove
existing
resistance to other chemotherapeutic drugs.
CA 02093203 2002-03-13
.21489-8645(S)
_g_
Preference is given to compounds of formula I wherein
one or two of the radicals R4, R5, Rs, R, and R8 are each nitro or a radical
of formula II
wherein
R9 is hydrogen or lower alkyl,
X is oxo, thio, imino, N-lower alkyl-imino, hydroximino or O-lower alkyl-
hydroximino,
Y is oxygen or the group NH,
n is 0 or 1 and
R,o is an aliphatic radical having at least 5 carbon atoms or an aromatic,
aromatic-aliphatic,
cycloaliphatic, cycloaliphatic-aliphatic, heterocyclic or heterocyclic-
aliphatic radical,
and the remaining radicals R4, R5, RB, R~ and Re are each independently of the
others hydrogen,
lower alkyl that is unsubstituted or substituted by free or alkylated amino,
piperazinyl,
piperidinyl, pyrrolidinyl or by morpholinyl, or lower alkanoyl,
trifluoromethyl, free,
etherified or esterified hydroxy, free, atkylated or acylated amino or free or
esterified
carboxy,
and the remaining substituents are as defined above,
and to salts of such compounds having at least one salt-forming group.
Preference is given especially to compounds of formula I wherein
R, is pyrazinyl, 1-methyl-1 H-pyrrolyl, amino- or amino-lower alkyl-
substituted phenyl
wherein the amino group in each case is tree, alkylated by one or two lower
alkyl
radicals or acylated by lower alkanoyl or by benzoyl, 1 H-indolyl or 1 H-
imidazolyl bonded
at a five-membered ring carbon atom, or unsubstituted or lower alkyl-
substituted pyridyl
bonded at a ring carbon atom and unsubstituted or substituted at the nitrogen
atom by
oxygen, .
R2 and R3 are each independently of the other hydrogen or lower alkyl,
one or two of the radicals R4, R5, RB, R~ and Rg are each nitro, fluoro-
substituted lower alkoxy or
a radical of formula II wherein
R9 is hydrogen or lower alkyl,
X is oxo, thio, imino, N-lower alkyl-imino, hydroximino or O-lower alkyl-
hydroximino,
Y is oxygen pr the group NH,
n is 0 or 1 and
Rio is an aliphatic hydrocarbon radical having 5-22 carbon atoms, a phenyl or
naphthyt radical
each of which is unsubstituted or substituted by cyano, lower alkyl,
-10- ~i)~W ~~
hydroxy-lower alkyl, amino-lower alkyl, (4-methyl-piperazinyl)-lower alkyl,
tri-
fluoromethyl, hydroxy, lower alkoxy, lower alkanoyloxy, halogen, amino, lower
alkylamino, di-lower alkylamino, lower alkanoylamino, benzoylamino, carboxy or
by lower aikoxycarbonyl, or phenyl-lower alkyl wherein the phenyl radical is
unsub-
stituted or substituted as indicated above, a cycloalkyl or cycloalkenyl
radica:
having up to 30 carbon atoms, cycloalkyl-lower alkyl or cycloalkenyl-lower
alkyl
each having up to 30 carbon atoms in the cycloalkyl or cycloalkenyl moiety, a
monocyclic radical having 5 or 6 ring members and 1-3 ring hetero atoms
selected
from nitrogen, oxygen and sulfur, to which radical one or two benzene radicals
may
be fused, or lower alkyl substituted by such a monocyclic radical,
and the remaining radicals R4, R5, R6, R~ and Rg are each independently of the
others
hydrogen, lower alkyl that is unsubstituted or substituted by amino, lower
alkyl-
amino, di-lower alkylamino, piperazinyl, piperidinyl, pyrrolidinyl or by
morpho-
linyl, or lower alkanoyl, trifluoromethyl, hydroxy, lower alkoxy, lower
alkanoyloxy,
halogen, amino, lower alkylamino, di-lower alkylamino, lower alkanoylamino,
benzoylamino, carboxy or lower allcoxycarbonyl,
and to salts of such compounds having at least one salt-forming group.
Special preference is given to compounds of formula I wherein
Rt is pyridyl bonded at a ring carbon atom and unsubstituted or substituted at
the
nitrogen atom by oxygen,
RZ and R3 are each hydrogen,
R4 is hydrogen or lower alkyl,
RS is hydrogen, lower alkyl or fluoro-substituted lower alkoxy,
R6 is hydrogen,
R~ is vitro, fluoro-substituted lower alkoxy or a radical of formula II
wherein
R9 is hydrogen,
X is oxo,
n is 0 and
Rto is an aliphatic hydrocarbon radical having 5-22 carbon atoms, a phenyl
radical that
is unsubstituted or substituted by cyano, lower alkyl, (4-methyl-piperazinyl)-
lower
alkyl, lower alkoxy, halogen or by carboxy; a cycloalkyl radical having up to
30
carbon atoms or a monocyclic radical having 5 or 6 ring members and 1-3 sulfur
ring atoms, and
Rg is hydrogen,
and to pharmaceutically acceptable salts of such compounds having at least one
salt-
b c fiz ~ '~ ~~ ~v
fd Y.~s,~ x~ ø,e ~.~ r~
-11-
forming group.
Special preference is given especially to compounds of formula I wherein
RI is pyridyl or N-oxido-pyridyl each of which is bonded at a carbon atom,
R2 and R3 are each hydrogen,
R4 is hydrogen or lower alkyl,
RS is hydrogen, lower alkyl or trifluoromethyl,
R6 is hydrogen,
R~ is nitro, fluoro-substituted lower alkoxy or a radical of formula II
wherein
R9 is hydrogen,
X is oxo,
n is the number 0 and
Rlp is pyridyl bonded at a carbon atom, phenyl that is unsubstituted or
substituted by
halogen, cyano, lower alkoxy, carboxy, lower alkyl or by 4.-methyl-piperazinyl-
methyl, or CS-C~alkyl, thienyl, 2-naphthyl or cyclohexyl, and
Rg is hydrogen,
and to pharmaceutically acceptable salts of such compounds having at least one
salt-
forming graup.
Preference is given above all to compounds of formula I wherein R4 and R8 are
each
hydrogen or wherein at least one of the radicals R4 and Rs is lower alkyl, and
the other of
the radicals R4 and Rx and the remaining radicals are as defined above, and to
pharmaceut-
ically acceptable salts of such compounds having at least one salt-forming
group.
Preference is given above all especially to compounds of formula I wherein
Rl is pyridyl bonded at a carbon atom,
R2, R3, Rn, R5, R6 and Rg are each hydrogen and
R~ is nitro or a radical of formula II wherein
R9 is hydrogen,
X is oxo,
n is the number 0 and
Rtp is pyridyl bonded at a carbon atom, phenyl that is unsubstituted or
substituted by
fluorine, chlorine, cyano, lower al.koxy, carboxy, lower alkyl or by 4-methyl-
piper-
azinyl-methyl, or CS-C~alkyl, thienyl or cyclohexyl,
and to pharmaceutically acceptable salts thereof.
~~t.~~~~;3
_ 1~ _
Most especially preferred are the compounds of formula I described in the
Examples and
pharmaceutically acceptable salts of such compounds having at least one salt-
forming
group.
In view of their inhibition of protein kinase C, greatest preference is given
to those above-
mentioned compounds of formula I wherein R4 and R8 are each hydrogen and the
remaining substituents are as defined above, and to pharmaceutically
acceptable salts of
such compounds having at least one salt-forming group.
The compounds of fornmla I and salts of such compounds having at least one
salt-forming
group are prepared in accordance with processes known per se. The process
according to
the invention is as follows:
a) a compound of formula III
Rt
O
R2
NrRtt
R3 Rtz
wherein Rlt and R12 are each independently of the other lower alkyl and Rt, R~
and R3 are
as defined above, functional groups present in a compound of formula III, with
the
exception of the groups participating in the reaction, being if necessary in
protected form,
or a salt of such a compound, is reacted with a compound of formula IV
R5
NH (IV),
NHz H
wherein the substituents are as defined above, functional groups present in a
compound of
>'t . ~i r3 s"' ''~ " ~
~r ''~' 3 e3 !~ ~~ '~'
-13-
formula IV, with ih; exception of the guanidine group participating in the
reaction, being
if necessary in protected form, or with a salt of such a compound, and any
protecting
groups present are removed, or
b) for the preparation of a compound of formula I wherein the radicals Rh, R5,
R6, R7 and
Rg are as defined above with the exception of nitre and fluc~ro-substituted
lower alkoxy, a
compound of formula V
Rts Rts
R' Rt ~ ~ Rt4
N (V)~
R2 ~ ~ Ns Rya
N
- N
R3
wherein one or two of the radicals Rlg, Rt4, R15, R16 ~d Rc7 ~'e each amino
and the
remaining radicals Rt3, Rt4, R15, Rts and Rt7 are each independently of the
others
hydrogen, lower alkyl that is unsubstituted ar substituted by free or
alkylated amino,
piperazinyl, piperidinyl, pyrrolidinyl or by morphalinyl, or lower alkanoyl,
trifluaro-
methyl, free, etherified or esterified hydroxy, free, alkylated or acylated
amino or free or
esterifted carboxy, and the remaining substituents are as defined above,
functional groups
present in a compound of formula V, with the exception of the amino groups)
participating in the reaction, being if necessary in protected foam, is
reacted with a
compound of formula VI
I-IO-~:(=X)-('Y)n Rto (VI),
wherein the substituents and symbols are as defined above, functional groups
present in a
compound of formula VI, with the exception of the I-IO-C(=X) group
participating in the
reaction, being if necessary in protected form, or with a reactive derivative
of a compound
of formula VI, and any protecting groups present are removed, or
c) for the preparation of a compound of formula I wherein Rt is pyridyl
substituted at the
nitrogen atom by oxygen, and wherein the other substituents and symbols are as
defined
14-
above, a compound of formula I wherein l~l is pyridyl is converted into the N-
oxido
compound with a suitable oxidising agent,
and, if desired, a compound of formula I obtainable by any one of processes a
to c is
converted into its salt, or an obtainable salt of a compound of formula I is
converted into
the free compound.
The procedure for the above-mentioned process variants is explained in detail
below:
General notes:
The end products of formula I may contain substituents that can also be used
as protecting
groups in starting materials for the preparation of other end products of
formula I. Thus,
within the scope of this text, only a readily removable group that is not a
constituent of the
particular desired end product of formula I is designated a "protecting
group", unless the
context indicates otherwise.
Protecting groups, and the manner in which they are introduced and removed are
described, for example, in "Protective Groups in Organic Chemistry", Plenum
Press,
London, New York 1973, and in "lVlethoden der organischen Chemie", Houben-
Weyl, 4th
edition, Vol. ill, Georg-Thieme-Verlag, Stuttgart 1974 and in Theodore W.
Greene,
"Protective Groups in Organic Synthesis", John Whey & Sons, New York 191. E1
characteristic of protecting groups is that they can be removed readily, i.e.
without the
occurrence of undesired secondary reactions, for example by solvolysis,
reduction,
photolysis or alternatively under physiological conditions.
Hydroxy-protecting groups are, for example, acyl radicals, such as
unsubstituted or
substituted, for example halo-substituted, lower alkanoyl, such as 2,2-
dichloroacetyl, or
acyl radicals of carbonic acid semiesters, especially tert-butoxycarbonyl,
unsubstituted or
substituted benzylaxycarbonyl, for example 4-nitrobenzyloxycarbonyl, or
diphenyl-
rnethoxycarbonyl, or 2-halo-lower alkoxycarbonyl, such as 2,2,2-
trichloroethoxycarbonyl,
also trityl or formyl, or organic silyl or stannyl radicals, and also readily
removable etheri-
fying groups, such as tent-lower alkyl, for example tent-butyl, 2-oxa- or 2-
thia-aliphatic or
2-oxa- or 2-thia-cycloaliphatic hydrocarbon radicals, especially 1-lower
alkoxy-lower
alkyl or 1-lower alkylthio-lower alkyl, for example methoxymethyl, 1-methoxy-
ethyl,
1-ethoxy-ethyl, methylthiomethyl, 1-methylthioethyl or 1-ethylthioethyl, or 2-
oxa- or
2-thiacycloalkyl having ~ or 6 ring atoms, for example tetrahydrofuryl or 2-
tetrahydro-
pyranyl or corresponding this analogues, and unsubstituted or substituted 1-
phenyl-lower
., p' ~ S 3 ' 3
e9Fa~
-15-
alkyl, such as unsubstituted or substituted benzyi or diphenylmethyl, suitable
substituents
of the phenyl radicals being, for example, halogen, such as chlorine, lower
alkoxy, such as
methoxy, and/or vitro.
A protected amino group may be, for example, in the form of a readily
cleavable acyl-
amino, arylmethylamino, etherified mercaptoamino, 2-acyl-lower alk-1-en-yl-
amino, silyl-
amino or stannylamino group or in the form of an azido group.
In a corresponding acylamino group, aryl is, for example, the aryl radical of
an organic
carboxylic acid having, for example, up to 18 carbon atoms, especially an
alkanecarb-
oxylic acid that is unsubstituted or substituted, for example, by halogen or
by aryl, or of a
benzoic acid that is unsubstituted or substituted, for example, by halogen,
lower alkoxy or
by vitro, or of a carbonic acid semiester. Such acyl groups are, for example,
lower
alkanoyl, such as formyl, acetyl or propionyl, halo-lower alkanoyl, such as 2-
haloacetyl,
especially 2-chloro-, 2-brorno-, 2-iodo-, 2,2,2-trifluoro- or 2,2,2-trichloro-
acetyl, benzoyl
that is unsubstituted or substituted, for example, by halogen, lower alkoxy or
by vitro, for
example benzoyl, 4-chlorobenzoyl, 4-methoxybenzoyl or 4-nitrobenzoyl, or lower
allcoxy-
carbonyl that is branched in the 1-position of the lower alkyl radical or
suitably substituted
in the 1- or 2-position, especially tart-lower alkoxycarbonyl, for example
tent-butoxy-
carbonyl, arylmethoxycarbonyl having one or two aryl radicals which are
preferably
phenyl that is unsubstituted or mono- or poly-substituted, for example, by
lower alkyl,
especially tart-lower alkyl, such as tart-butyl, lower alkoxy, such as
methoxy, hydroxy,
halogen, for example chlorine, and/or by vitro, such as unsubstituted or
substituted benzyl-
oxycarbonyl, for example 4-vitro-benzyloxycarbonyl, or substituted
diphenylmethoxy-
carbonyl, for example benzhydryloxycarbonyl or di(4-
methoxyphenyl)methoxycarbonyl,
aroylmethoxycarbonyl wherein the amyl group is preferably benzoyl that is
unsubstituted
or substituted, for example, by halogen, such as bromine, for example
phenacyloxy-
carbonyl, 2-halo-lower alkoxycarbonyl, for example 2,2,2-
trichloroethoxycarbonyl,
2-bromoethoxycarbonyl or 2-iodoethoxycarbonyl, or 2-(trisubstituted silyl)-
ethoxy-
carbonyl wherein each of the substituents, independently of the others, is an
aliphatic,
araliphatic, cycloaliphatic or aromatic hydrocarbon radical having up to 15
carbon atoms
that is unsubstituted ar substituted, for example, by lower alkyl, lower
alkoxy, aryl,
halogen or by vitro, such as corresponding unsubstituted or substituted lower
alkyl,
phenyl-lower alkyl, cycloalkyl or phenyl, for example 2-tri-lower
alkylsilylethoxy-
carbonyl, such as 2-trimethylsilylethoxycarbonyl or 2-(di-n-butyl-methyl-
silyl)-ethoxy-
carbonyl, or 2-triarylsilylethoxycarbonyl, such as 2-
triphenylsilylethoxycarbonyl.
G xa~
2~ »je
- 16-
Other acyl radicals that are suitable as amino-protecting groups are
corresponding radicals
of organic phosphoric, phosphoric or phosphinic acids, such as di-lower
alkylphosphoryl,
for example dimethylphosphoryl, diethylphosphoryl, di-n-propylphosphoryl or
diiso-
propylphosphoryl, dicycloalkylphosphoryl, for example dicyclohexylphosphoryl,
unsubstituted or substituted diphenylphosphoryl, for example
diphenylphosphoryl,
unsubstituted or substituted, for example vitro-substituted, di(phenyl-lower
alkyl)-
phosphoryl, for example dibenzylphosphoryl or di(4-nitrobenzyl)phosphoryl,
unsubstituted or substituted phenoxy-phenyl-phosphonyl, for example
phenoxyphenyl-
phosphonyl, di-lower alkylphosphinyl, for example diethylphosphinyl, or
unsubstituted or
substituted diphenylphosphinyl, for example diphenylphosphinyl.
In an arylmethylamino group that is a mono-, di- or, especially, tri-
arylmethylamino
group, the aryl radicals are especially unsubstituted or substituted phenyl
radicals. Such
groups are, for example, benzyl-, diphenylmethyl- and especially trityl-amino.
An etherified mercapto group in an amino group protected by such a radical is
especially
arylthio or aryl-lower alkylthio, wherein aryl is especially phenyl that is
unsubstituted or
substituted, for example, by lower alkyl, such as methyl or tent-butyl, lower
alkoxy, such
as methoxy, halogen, such as chlorine, and/or by vitro. A corresponding amino-
protecting
group is, for example, 4-nitrophenylthio.
In a 2-acyl-lower alk-1-en-1-yl radical that can be used as amino-protecting
group, aryl is,
for example, the corresponding radical of a lower alkanecarboxylic acid, of a
benzoic acid
that is unsubstituted or substituted, for example, by lower alkyl, such as
methyl or
tert-butyl, lower alkoxy, such as methoxy, halogen, such as chlorine, and/or
by vitro, or
especially of a carbonic acid semiester, such as a carbonic acid lower alkyl
semiester.
Corresponding protecting groups are especially 1-lower alkanoyl-prop-1-en-2-
yl, for
example 1-acetyl-prop-1-en-2-yl, or 1-lower alkoxycarbonyl-prop-1-en-2-yl, for
example
1-edioxycarbonyl-prop-1-en-2-yl.
JPreferred amino-protecting groups are acyl radicals of carbonic acid
semiesters, especially
tert-butoxycarbonyl, benzyloxycarbonyl that is unsubstituted or substituted,
for example,
as indicated, for example 4-vitro-benzyloxycarbonyl, or
diphenylmethoxycarbonyl, or
2-halo-lower alkoxycarbonyl, such as 2,2,2-trichloroetlioxycarbonyl, also
trityl or formyl.
The removal of the protecting groups that are not constituents of the desired
end product
c ~; "~ ~~ ~~ ~ '~~
I~ 'tJ ~ r.~ ra "vi :3
-17-
of formula I is effected in a manner known per se, for example by means of
solvolysis,
especially hydrolysis, alcoholysis or acidolysis, or by means of reduction,
especially
hydrogenolysis or chemical reduction, as desired stepwise or simultaneously.
A protected amino group is freed in a manner known her se and, according to
the nature of
the protecting groups, in various ways, preferably by means of solvolysis or
reduction..
2-Halo-lower alkoxycarbonylamino (optionally after conversion of a 2-bromo-
lower
alkoxycarbonylamino group into a 2-iodo-lower alkoxycarbonylamino group),
aroyl-
rnethoxycarbonylamino or 4-nitrobenzyloxycarbonylamino can be cleaved, for
example,
by treatment with a suitable chemical reducing agent, such as zinc in the
presence of a
suitable carboxylic acid, such as aqueous acetic acid.
Aroylmethoxycarbonylamino can be
cleaved also by treatment with a nucleophilic, preferably salt-forming,
reagent, such as
sodium thiophenolate, and ~1-vitro-benzyloxycarbonylamino can be cleaved also
by
treatment with an alkali metal dithionite, for example sodium dithionite.
~Tnsubstituted or
substituted diphenylmethoxycarbonylamino, tert-lower alkoxycarbonylamino ar 2-
tri-
substituted silylethoxycarbonylamino can be cleaved by treatment with a
suitable acid, for
example formic or trifluoroacetic acid; unsubstituted or substituted
benzyloxycarbonyl-
amino can be cleaved, for example, by means of hydrogenolysis, that is to say
by
treatment with hydrogen in the presence of a suitable hydrogenation catalyst,
such as a
palladium catalyst; unsubstituted or substituted triarylmethylamino or
formylamino can be
cleaved, for example, by ~eatment with an acid, such as a mineral acid, for
example
hydrochloric acid, or an organic acid, for example formic, acetic or
trifluoroacetic acid,
optionally in the presence of water, and an amino group protected by an
organic silyl
group can be freed, for example, by means of hydrolysis or alcoholysis. An
amino group
protected by ~,-haloacetyl, for example 2-chloroacetyl, can be freed by
treatment with
thiourea in the presence of a base, or with a thiolate salt, such as an alkali
metal thiolate,
of thiourea and subsequent solvolysis, such as alcoholysis or hydrolysis, of
the resulting
condensation product. An amino group protected by 2-substituted
silylethoxycarbonyl can
also be converted into the free amino group by treatment with a salt of
hydrofluoric acid
yielding fluoride anions.
A hydroxy group protected by a suitable acyl group, an organic silyl group ar
by
unsubstituted or substituted 1-phenyl-lower alkyl is freed analogously to a
corresponding-
ly protected amino group. Hydroxy protected by unsubstituted or substituted 1-
phenyl-
lower alkyl, for example benzyl, is preferably freed by catalytic
hydrogenation, for
example in the presence of a palladium-on-carbon catalyst. A hydroxy group
protected by
~~i~~i~~v~i
-18-
2,2-dichloroacetyl is freed, for example, by basic hydrolysis, and a hydroxy
group
etherified by tert-lower alkyl or by a 2-oxa- or 2-thia-aliphatic or 2-oxa- or
2-this-cyclo-
aliphatic hydrocarbon radical is freed by acidolysis, for example by treatment
with a
mineral acid or a strong carboxylic acid, for example trifluoroacetic acid.
Hydroxy
etherified by an organic silyl radical, for example trimethylsilyl, can also
be freed by a salt
of hydrofluoric acid yielding fluoride anions, for example tetrabutylammonium
fluoride.
Process a:
Preferably Rtt and R~2 are each methyl.
Free functional groups in a compound of formula III that are advantageously
protected by
readily removable protecting groups are especially amino groups in the radical
Rt and the
imino group of 1H-indolyl. The latter can be protected, for example, by
benzyl.
Free functional groups in a compound of formula IV that are advantageously
protected by
readily removable protecting groups are especially amino groups, but also
hydroxy and
carboxy groups.
A salt of a compound of formula IV is preferably an acid addition halt, for
example a
nitrate or one of the acid. addition salts mentioned for the end products of
formula I.
The reaction is carried out in a suitable solvent or dispersing agent, for
example a suitable
alcohol, such as 2-methoxy-ethanol, or a suitable lower alkanol, for example
isopropanol,
at a temperature of from room temperature (approx. 20°C) to
150°C, for example under
reflux. Especially when the compound of formula IV is used in the form of a
salt, that salt
is converted into the free compound, preferably in situ, by the addition of a
suitable base,
such as an alkali metal hydroxide, for example sodium hydroxide.
It is preferable to use as starting materials compounds of formula IV wherein
one or two
of the radicals R4, Rs, R6, R~ and Rs are each nitre and the remaining
radicals Rn, R5, R6,
R~ and R8 are each independently of the others hydrogen, lower alkyl that is
unsubstituted
or substituted by free or alkylated amino, piperazinyl, piperidinyl,
pyrrolidinyl or by
morpholinyl, or lower alkanoyl, trifluoromethyl, free, etherified or
esterified hydroxy,
free, alkylated or acylated amino or free or esterified carboxy.
The starting material of formula III is obtained by reacting a compound of
formula VII
,1,,'ta~e~~'"~~
-19-
R~
R (v~>
2
wherein the substituents are as defined above, with a compound of formula VIII
OR~9 ~ it
RtBO ~ \ (VIII),
R3 R12
wherein RtR and Rt9 are each lower alkyl and the remaining substituents are as
defined
above, in a manner analogous to that described in the European Patent
Application having
the Publication No. 233461. Typical representatives of a compound of formula
VIII are
N,N-dimethylformamide-dimethylacetal and N,N-dimethylacetamide-dimethylacetal.
The
reaction is carried out with heating of the reactants of formulae VII and VIII
for several
hours, for example for from 4 to 24 hours, at a temperature of approximately
from 50°C to
15a°C, in the absence or, if necessary, in the presence of a solvent.
The starting material of formula III is alternatively obtained by reacting a
compound of
formula VII with an ester of the formula R3-C(=O)-O-CH2-CH3 wherein R3 is as
def°med
above, and reacting the resulting product with an amine of the formula H-
N(Rtt)-Rt2
wherein the substituents are as def'med above.
The starting material of formula IV is obtained in the fozm of an acid
addition salt by
reacting a compound of formula IX
R7 Rs
R5 (1X)>
N .Rd
i
HI
wherein the substituents are as defined above, with cyanamide (NC-NI-I2). The
reaction is
~~~~'~e~.3w3~~
r~ ~t
-20-
carried out in a suitable solvent or dispersing agent, for example a suitable
alcahol, for
example a suitable lower alkanol, such as ethanol, in the presence of
equimolar amounts
of the salt-forming acid at a temperature of from room temperature to
150°C, for example
under reflux.
Process b:
Free functional groups in a compound of formula V or VI that are
advantageously
protected by readily removable protecting groups are especially amino groups,
but also
hydroxy and carboxy groups, that are not intended to participate in the
desired reaction,
for example amino in the radical Rt.
A reactive derivative of a compound of formula VI wherein X is oxo is
especially a
reactive (activated) ester, a reactive anhydride or a reactive cyclic amide.
The same is true
for the derivatives wherein X has one of the other definitions given above.
Reactive (activated) esters of an acid of formula VI are especially esters
unsaturated at the
linking carbon atom of the esterifying radical, for example esters of the
vinyl ester type,
such as actual vinyl esters (obtainable, for example, by transesterification
of a corres-
ponding ester with vinyl acetate; activated vinyl ester method),
carbamoylvinyl esters
(obtainable, for example, by treatment of the corresponding acid with an
isoxazolium
reagent; 1,2-oxazolium or Woodward method), or l-lower alkoxyvinyl esters
(obtainable,
for example, by treatment of the corresponding acid with a lower
alkoxyacetylene; ethoxy-
acetylene method), or esters of the amidino type, such as N,N'-disubstituted
amidino
esters (obtainable, for example, by treatment of the corresponding acid with a
suitable
N,N'-disubstituted carbodiimide, for example N,N'-dicyclohexylcarbodiimide;
carbodi-
imide method), or N,N-disubstituted amidino esters (obtainable, for example,
by treatment
of the corresponding acid with an N,N-disubstituted cyanamide; cyanamide
method),
suitable aryl esters, especially phenyl esters suitably substituted by
electron-attracting
substituents (obtainable, for example, by treatment of the corresponding acid
with a
suitably substituted phenol, for example 4-nitrophenol, 4-methylsulfonyl-
phenol, 2,4,5-tri-
chlorophenol, 2,3,4,5,6-pentachloro-phenol or 4-phenyldiazophenol, in the
presence of a
condensation agent, such as N,N'-dicyclohexylcarbodiimide; activated aryl
esters
method), cyanomethyl esters (obtainable, for example, by treatment of the
corresponding
acid with chloroacetonitrile in the presence of a base; cyanomethyl esters
method), thio
esters, especially unsubstituted or substituted, for example nitro-
substituted, phenylthio
esters (obtainable, for example, by treatment of the corresponding acid with
unsubsdtuted
i~e ~.' ; ~ ei '~ t>
-21-
or substituted, for example nitro-substituted, thiophenols, inter alia by the
anhydride or
carbodiimide method; activated thiol esters method), amino or amido esters
(obtainable,
for example, by treatment of the corresponding acid with an N-hydroxy-amino or
N-hydroxy-amido compound, for example N-hydroxy-succinirraide, N-hydroxy-
piperidine,
N-hydroxy-phthalimide or 1-hydroxy-benzotriazole, for example by the anhydride
or
carbodiimide method; activated N-hydroxy esters method), or silyl esters
(which are
obtainable, for example, by treatment of the corresponding acid with a
silylating agent, for
example hexamethyl disilazane, and react readily ~.vith hydroxy groups but not
with amino
groups).
Anhydrides of an acid of formula VI may be symmetric or preferably mixed
anhydrides of
that acid, for example anhydrides with inorganic acids, such as acid halides,
especially
acid chlorides (obtainable, for example, by treatment of the corresponding
acid with
thionyl chloride, phosphorus pentachloride or oxalyl chloride; acid chloride
method),
azides (obtainable, for example, from a corresponding acid ester via the
corresponding
hydrazide and treatment thereof with nitrous acid; azide method), anhydrides
with
carbonic acid semiderivatives, such as corresponding esters, for example
carbonic acid
lower alkyl semiesters (obtainable, for example, by treatment of the
corresponding acid
with haloformic, such as chloroformic, acid lower alkyl esters or with a 1-
lower alkoxy-
carbonyl-2-lower alkoxy-1,2-dihydroquinoline, for example 1-lower
alkoxycarbonyl-
2-ethoxy-1,2-dihydroquinoline; mixed O-alkylcarbonic acid anhydrides method),
or
anhydrides with dihalogenated, especially dichlorinated, phosphoric acid
(obtainable, for
example, by treatment of the corresponding acid with phosphorus oxychloride;
phosphorus
oxychloride method), or anhydrides with organic acids, such as mixed
anhydrides with
organic carboxylic acids (obtainable, for example, by treatment of the
corresponding acid
with an unsubstituted or substituted lower alkane- or phenylalkane-carboxylic
acid halide,
for example phenylacetic acid chloride, pivalic acid chloride or
trifluoroacetic acid
chloride; mixed carboxylic acid anhydrides method) or with organic sulfonic
acids
(obtainable, for example, by treatment of a salt, such as an alkali metal
salt, of the corres-
ponding acid, with a suitable organic sulfonic acid halide, such as lower
alkane- or aryl-,
for example methane- or p-toluene-sulfonic acid chloride; mixed sulfonic acid
anhydrides
method), and symmetric anhydrides (obtainable, for example, by condensation of
the
corresponding acid in the presence of a carbodiimide or of 1-
diethylaminopropyne;
symmetric anhydrides method).
Suitable cyclic amides are especially amides with five-membered diazacycles of
aromatic
9
~"v8
-22-
character, such as amides with imidazoles, for example imidazole (obtainable,
for
example, by treatment of the corresponding acid with N,N'-carbonyldiimidazole;
imidazolide method), or pyra.zoles, for example 3,5-dimethyl-pyrazole
(obtainable, for
example, by way of the acid hydrazide by treatment with acetylacetone;
pyrazalide
method).
Derivatives of acids of formula VI that can be used as acylating agents can
also be formed
in situ. For example, N,N'-disubstituted amidina esters can be formed in situ
by reacting
a mixture of the starting material of formula V and the acid used as acylating
agent in the
presence of a suitable N,N-disubstituted carbodiimide, for example N,N'-
dicyclohexyl-
carbodiimide. In addition, amino or amido esters of the acids used as
acylating agents can
be formed in the presence of the starting material of formula V to be
acylated, by reacting
a mixture of the corresponding acid and amino starting materials in the
presence of an
N,N'-disubstituted carbodiimide, for example N,N'-dicyclohexyl-carbodiimide,
and an
N-hydroxy-amine or N-hydroxy-amide, for example N-hydroxysuccinimide, where
appropriate in the presence of a suitable base, for example 4-dimethylamino-
pyridine.
The reaction is preferably carried out by reacting a xeactive carboxylic acid
derivative of a
compound of formula VI with a compound of formula V wherein ltte amino group
or
hydroxy group participating in the reaction is in free form.
The reaction can be carried out in a manner known her se, the reaction
conditions being
dependent especially on whether, and if so how, the carboxy group of the
acylating agent
has been activated, usually in the presence of a suitable solvent or diluent
or of a mixture
thereof and, if necessary, in the presence of a condensation agent, which, for
example
when the carboxy group participating in the reaction is in the form of an
anhydride, may
also be an acid-binding agent, with cooling or heating, for example in a
temperature range
from approximately -30°C to approximately +150°C, especially
approximately from 0°C
to +100°C, preferably from room temperature (approx. +20°C) to
+70°C, in an open or
closed reaction vessel and/or in the atmosphere of an inert gas, for example
nitrogen.
Customary condensation agents are, for example, carbodiimides, for example
N,N'-diethyl-, N,N'-dipropyl-, N;N'-dicyclohexyl- or N-ethyl-N'-(3-
dimethylamino-
propyl)-carbodiimide, suitable carbonyl compounds, for example
carbonyidiimidazole, or
1,2-oxazoliurn compounds, for example 2-ethyl-5-phenyl-1,2-oxazolium 3'-
sulfonate and
2-tert-butyl-5-methyl-isoxazolium perchlorate, or a suitable acylamino
compound, for
example 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroduinoline. Customary acid-binding
a ; r1 ':7 '~ ';'~ ~3
~a'~ ~ tJ'i'~e''
-23-
condensation agents are, far example, alkali metal carbonates or hydrogen
carbonates, for
example sodium or potassium carbonate or hydrogen carbonate (customarily
together with
a sulfate), or organic bases, such as, customarily, pyridine or sterically
hindered tri-lower
alkylamines, for example N,N-diisopropyl-N-ethyl-amine.
The starting material of formula V is obtained by reduction of the nitro
groups) in a
compound of formula I wherein one or two of the radicals R4, R5, R6, R~ and Rg
are each
nitro. That reduction can be carried out, for example, by catalytic
hydrogenation in a
suitable solvent, such as a suitable acyclic or cyclic ether, such as in
tetrahydrofuran.
There is preferably used as hydrogenation catalyst palladium on active carbon
(S °/a) and
in that case the hydrogenation is preferably carried out under normal
pressure.
Process c:
A suitable oxidising agent for converting a compound of formula I wherein Rt
is pyridyl
into the N-oxido compound is preferably a suitable peracid, for example a
suitable
perbenzoic acid, such as especially m-chloro-perbenzoic acid. The reaction is
carried out
in an inert solvent, for example a halogenated hydrocarbon, such as preferably
methylene
chloride, at temperatures of approximately from -20°C to +1S0°C,
especially approx-
imately from 0°C to the boiling point of the solvent in question,
in.general below +100°C,
and preferably at room temperature or at slightly elevated temperature
(20°C-70°C).
Acid addition salts of compounds of formula I are obtained in customary
manner, for
example by treatment with an acid or a suitable anion exchange reagent.
Acid addition salts can be converted into the free compounds in customary
manner, for
example by treatment with a suitable basic agent.
Mixtures of isomers can be separated into the individual isomers in a manner
known
her se, for example by fractional crystallisation, chromatography, etc..
The processes described above, including the processes for removing protecting
groups
and the additional process steps, are, unless otherwise indicated, carried out
in a manner
known per se, for example in the presence or absence of preferably inert
solvents and
diluents, if necessary in the presence of condensation agents or catalysts, at
reduced or
elevated temperature, for example in a temperature range of from approximately
-20°C to
approximately 1S0°C, especially from approximately 0°C to
approximately +70°C,
tf~~~~fr~~5;~'i
Ca ~.: :.s ~ ~a "tD z~
-24-
preferably from approximately +10°C to approximately +50°C, and
more especially at
room temperature, in a suitable vessel and if necessary in an inert gas
atmosphere, for
example a nitrogen atmosphere.
In those process steps, taking account of all the substituents in the
molecule, if necessary,
for example when readily hydrolysable radicals are present, especially mild
reaction
conditions should be used, such as short reaction times, the use of mild
acidic or basic
agents at low concentrations, stoichiometric quantity ratios, and the
selection of suitable
catalysts, solvents, temperature and/or pressure conditions.
Z'he invention relates also to those forms of the process in which a compound
obtainable
as intermediate at any stage of the process is used as starting material and
the remaining
steps are carried out or the process is interrupted at any stage or a starting
material is
formed under the reaction conditions or is used in the form of a reactive
derivative or salt.
It is preferable to begin with those starting materials which in accordance
with the process
result in the compounds described above as being especially valuable.
The present invention relates also to novel starting materials and/or
intermediates and to
processes for the preparation thereof. The starting materials used and the
reaction
conditions chosen are preferably those which result in the compounds described
in this
Application as being especially preferred.
The invention relates also to a method of treating warm-blooded animals
suffering from a
tumoral disease, which comprises administering to warm-blooded animals
requiring such
treatment an effective, tumotar-inhibiting amount of a compound of formula I
or of a
pharmaceutically acceptable salt thereof. The invention relates further to the
use of a
compound of formula I or of a pharmaceutically acceptable salt thereof for
inhibiting
PDGF-receptor kinase or to the use of a compound of formula I wherein R4 and
R8 are
each hydrogen, or of a pharmaceutically acceptable salt thereof, for
inhibiting protein
kinase C in warm-blooded animals or for preparing pharmaceutical compositions
for use
in the therapeutic treatment of the human or animal body. Effective doses, for
example
daily doses of approximately from t to 1000 mg, especially from 50 to 500 mg,
are
administered to a warm-blooded animal of approximately 70 kg body weight
according to
species, age, individual condition, mode of administration and the individual
syndrome.
The invention relates also to pharmaceutical compositions comprising an
effective
_~5_ ~'e~~~~~
amount, especially an amount effective in the prevention or therapy of one of
the above-
mentioned diseases, of the active ingredient together with pharmaceutically
acceptable
carriers that are suitable for topical, enteral, for example oral or rectal,
or parenteral
administration, and may be inorganic or organic, solid or liquid. For oral
administration
there are used especially tablets or gelatin capsules comprising the active
ingredient
together with diluents, for example lactose, dextrose, sucrose, mannitol,
sorbitol, cellulose
and/or glycerol, and/or lubricants, for example silicic acid, talc, stearic
acid or salts
thereof, such as magnesium or calcium stearate, and/or polyethylene glycol.
Tablets may
also comprise binders, for example magnesium aluminium silicate, starches,
such as corn,
wheat or rice starch, gelatin, methylcellulose, sodium carboxymethylcellulose
and/or
polyvinylpyrrolidone, and, if desired, disintegrators, for example starches,
agar, alginic
acid or a salt thereof, such as sodium alginate, and/or effervescent mixtures,
or adsorbents,
dyes, flavourings and sweeteners. The pharmacologically active compounds of
the
present invention can also be used in the form of parenterally administrable
compositions
or in the form of infusion solutions. Such solutions are preferably isotonic
aqueous
solutions or suspensions, which, for example in the case of lyophilised
compositions that
comprise the active ingredient alone or together with a carrier, for example
mannitol, can
be prepared before use. The pharmaceutical compositions may be sterilised
and/or may
comprise excipients, for example preservatives, stabilisers, wetting agents
and/or
emulsifiers, solubilisers, salts for regulating the osmotic pressure and/or
buffers. The
present pharmaceutical compositions which, if desired, may comprise further
pharmaco-
logically active substances, such as antibiotics, are prepared in a manner
known per se, for
example by means of conventional mixing, granulating, confectioning,
dissolving or
lyophilising processes, and comprise approximately from 1 % to 100 %,
especially from
approximately 1 % to approximately 20 %, active ingredient(s).
The following Examples illustrate the invention but do not limit the invention
in any way.
The Ftt values are determined on silica gel thin-layer plates (Merck,
Darmstadt, Germany).
The ratio to one another of the eluants in the eluant mixtures used is given
in proportions
by volume (v/v), and temperatures are given in degrees Celsius.
Abbreviations:
HV: high vacuum
n: normal (straight-chain)
Example 1: 41.3 g (0.17 mol) of 3-nitrophenyl-guanidine nitrate, made into a
slurry in
-26-
50 ml of isopropanol, are added to a solution of 30 g (0.17 mol) of 3-
dimethylamino-1-(3-
pyridyl)-2-propen-1-one described in EP-A-0 233 461] in 250 ml of isopropanol.
After
the addition of 7.49 g (0.19 mol) of sodium hydroxide, the yellow suspension
is boiled at
reflex for 8 hours. After cooling to 0°, the mixture is filtered and
washed with 200 ml of
isopropanol. The filtration residue is made into a slurry in 300 ml of water
and stirred for
30 minutes, filtered and washed with 200 m1 of water. After again making into
a slurry in
200 ml of ethanol and washing with 200 ml of ethanol/diethyl ether (1:l) there
is obtained
N-(3-nitrophenyl)-4-(3-pyridyl)-2-pyrimidine-amine; m.p. 212-213°, Rf =
0.75 (chloro-
form:methanol = 9:1).
The starting material is obtained as follows:
Step 1.1: 42 ml (0.6 mol) of nitric acid (65 %) are added dropwise to a yellow
suspension
of 82.88 g (0.6 mol) of 3-nitroaniline in 200 ml of ethanol. After the
exothermic reaction
has subsided, 75.7 g (0.9 mol) of cyanamide (50 % in water) are added and the
reaction
mixture is boiled at reflex for 21 hours. After cooling to 0°, the
mixture is filtered and
washed six times with ethanol/diethyl ether (1:1). Drying under HV at
40° yields 3-nitro-
phenyl-guanidine nitrate; m.p. 205-207°.
Step 1-2: 8 g (0.35 mol) of sodium are placed in 260 ml of toluene and at
100° made into a
suspension using a vibromixer. After cooling to 0°, 17 ml (0.42 mol) of
methanol are
added dropwise, with cooling, and the mixture is then stirred for 45 minutes
at 75°. At 25°
and with ice-cooling, a solurion of 38.5 ml (0.35 mol) of 3-acetylpyridine and
28 ml (0.35
mol) of ethyl formate in 300 ml oftoluene are added dropwise in the course of
45 minutes.
The yellow suspension is stirred for 16 hours at 25° and then 23.7 g
(0.52 mol) of
dimethylamine are added. After the addition of 100 rnl of toluene, the mixture
is stirred
for 45 minutes at 25°, and then at 0° a solution of 20 rnl of
acetic acid in 150 ml of toluene
is added dropwise in the course of 30 minutes and the mixture is then boiled
at reflex for
1 hour. After cooling to 25°, the mixture is filtered and washed with
500 ml of toluene/-
hexane (1:1) and the filtrate is concentrated until crystallisation begins.
Cooling to 0°,
filtration and drying at 80° under HV yield 3-dimethylamino-1-(3-
pyridyl)-2-propen-1-
one; m.p. 81-82°.
Example 2: 100 mg (0.38 mmol) of N-(3-aminophenyl)-4-(3-pyridyl)-2-pyrimidine-
amine
are dissolved in 5 xnl of pyridine; 58.5 p.1 (0.46 mmol) of 4-chlorobenzoyl
chloride are
added and the mixture is stirred at room temperature for 24 hours. 10 ml of
water are
-27-
added to the reaction mixture which is then cooled to 0° and filtered.
Washing with water
and drying yield N-[3-(4-chlorobenzoylamido)-phenyl]-4-(3-pyridyl)-2-
pyrimidine-amine;
m.p. 238-240°, Rf = 0.66 (chloroform:methanol = 9:1).
The starting material is obtained as follows:
Step 2.1: A suspension of 17.0 g (0.058 mol) of N-(3-nitrophenyl)-4-(3-
pyridyl)-2-
pyrimidine-amine in 1700 ml of tetrahydrofuran is stirred with 1.7 g of
palladium tin
active carbon (5 %) under a hydrogen atmosphere at normal pressure fox 21
hours. The
suspension is filtered and the filtrate is concentrated in a rotary
evaporator. The yellow
solid product that remains behind is stirred overnight in 200 ml of methylene
chloride.
Filtration and drying yield N-(3-aminophenyl)-4-(3-pyridyl)-2-pyrimidine-
amine;
m.p. 89-90°, Rp = 0.38 (chloroform:methanol = 9:1 ).
Example 3: 53 p,1 (0.46 mmol) of benzoyl chloride are added to a solution of
100 mg
(0.38 mmol) of N-(3-aminophenyl)-4-(3-pyridyl)-2-pyrimidine-amine in 5 ml of
pyridine
and the mixture is stirred under a nitrogen atmosphere for 24 hours at room
temperattue.
ml of water are added to the reaction mixture which is then cooled to
0°, filtered and
washed with water. Drying under HV yields N-(3-benzoylamidophenyl)-4-(3-
pyridyl)-2-
pyritnidine-amine; m.p. 207-209°, Rf = 0.53 (chloroform:methanol =
9:1).
Example 4: A solution of 100 mg (0.38 mmol) of N-(3-aminophenyl)-4-(3-pyridyl)-
2-
pyrimidine-amine and 59 mg (0.46 mmol) of 2-pyridinecarboxylic acid chloride
in 5 ml of
pyridine is stirred under nitrogen for 24 hours at room temperature. After the
addition of
30 mg (0.23 mmol) of 2-pyridine-carboxylic acid chloride, the mixture is
stirred for
18 hours and then a further 25 rng (0.19 mmol) of 2-pyridinecarboxylic acid
chloride are
added and the mixture is stirred for 72 hours at 25°. After the
addition of 10 ml of water
and cooling to 0°, the mixture is filtered and washed with water.
Separation by chromato-
graphy (silica gel, CH(Cl~/MeOf-I = 9:1) yields N-[3-(2-pyridyl)-carboxamido-
phenyl]-
4-(3-pyridyl)-2-pyrimidine-amine; m.p. 18 7-190°, Rf = 0.58
(chloroform:methanol = 9:1).
Example 5: Analogously to Example 4, N-[3-(3-pyridyl)-carboxamido-phenyl]-4-
(3-pyridyl)-2-pyrimidine-amine is prepared from 3-pyridinecarboxylic acid
chloride;
m.p. 217-220°, Rf = U.29 (chloroform:methanol = 9:1).
Example 6: Analogously to Example 4, N-[3-(4-pyridyl)-carboxamido-phenyl]-4-(3-
~i~~~J~~e~3
-28-
pyridyl)-2-pyrimidine-amine is synthesised from 4-pyridinecarboxylic acid
chloride;
m.p. 224-226°, Rf = 0.29 (chloroform:methanol = 9:1).
Example 7: 63 w1 (0.46 mmol) of pentafluorobenzoyl chloride are added to a
solution of
100 mg (0.38 mmol) of N-(3-aminophenyl)-4-(3-pyridyl~2-pyrimidine-amine in 5
rnl of
pyridine and the mixture is stirred under nitrogen at room temperature for 17
hours. 10 ml
of water are added to the brown reaction solution which is then cooled to
0° and filtered.
The residue is recrystallised from ethanol/acetone and yields the crystalline
product N-(3-
pentafluorobenzoylamido-phenyl)-4-(3-pyridyl)-2-pyrimidine-amine; m.p. 234-
244°, Rp =
0.41 (chloroform:metha~no19:1).
Example 8: 28 mg (0.19 nunol) of phthalic acid anhydride are added to a
solution of
50 mg (0.19 mmol) of N-(3-aminophenyl)-4-(3-pyridyl)-2-pyrimidine-amine in 1
ml of
pyridine. After 2.5 hours, a further 14 rng (0.095 mmol) of phthalic acid
anhydride are
added to the yellow reaction solution and the mixture is stirred for 20 hours
at 25°. The
suspension is f°lltered and washed with a small amount of cold
pyridine. The residue is
digested with 2 x 2.5 ml of absolute ethanol and yields N-[3-(2-
carboxybenzoylamido)-
phenyl]-4-(3-pyridyl)-2-pyrimidine-amine; m.p. 206-209°, Rp = 0.07
(chloroform:-
methanol = 9:1).
Example 9: A solution of 100 mg (0.38 rnmol) of N-(3-aminophenyl)-4-(3-
pyridinyl)-2-
pyrimidine-amine and 105 p.1 (0.46 mmol) of caproic acid anhydride in 5 ml of
pyridine is
stirred under a nitrogen atmosphere for 24 hours at 25° and then
concentrated in a rotary
evaporator. The residue is purified by flash chromatography (silica gel,
chloroform:-
methanol 95:5), yielding N-(3-n-hexanoylamido-phenyl)-4-(3-pyridyl)-2-
pyrimidine-
amine; m.p. 180-184°, Rf = 0.78 (chloroform:methanol = 9:1).
Example 10: 1 g (5.68 rnmol) of 3-dimethylamino-1-(2-pyridyl)-2-propen-1-one
[EP-A-
233 461] is dissolved in 8 ml of isopropanol, and 1.38 g (5.68 mmol) of 3-
nianphenyl-
guanidine nitrate are added. After the addition of 0.25 g (6.24 mmol) of
sodium
hydroxide, the yellow suspension is heated at reflux for 20 hours, then cooled
to 0°,
filtered and washed with 30 rnl of isopropanol. The filtration residue is
stirred in 15 ml of
ethanol for 20 minutes, filtered and washed with a small amount of cold
ethanol, yielding
N-(3-nitrophenyl)-4-(2-pyridyl)-2-pyrimidine-amine; m.p. 213-219°.
Example 11: 1.38 g (5.68 mmol) of 3-nitrophenyl-guanidine nitrate and 0.25 g
-29-
(6.24 mmol) of sodium hydroxide are added to a solution of 1 g (5.68 mmol) of
3-dimethylamino-1-(4-pyridyl)-2-propen-1-one [LJS Patent 4 281 000] in 8 ml of
iso-
propanol. The yellow suspensian is heated at reflux for 20 hours and then
cooled to 0°.
After w4shing with 30 ml of isopropanol the filtration residue is made into a
slurry in
successian in 15 ml of ethanol and then in I5 ml of water and filtered each
time. Drying
under HV yields N-(3-nitrophenyl)-4-(4-pyridyl)-2-pyrimidine-amine; rn.p. 282-
284°.
Example 12: Analogously to Example 2, N-[3-(2--methoxybenzoylamido)-phenyl]-4-
(3-
pyridyl)-2-pyrimidine-amine is prepared from 2-methoxybenwyl chloride; m.p.
115-117°,
Rf = 0.76 (chloroforrn:methanol = 9:1).
Example I3: Analogously to Example 2, N-[3-(4-fluorobenzoylamido)-phenyl]-4-(3-
pyridyl)-2-pyrimidine-amine is prepared from 4-fluorobenzoyl chloride; m.p.
215-216°,
Rr = 0.34 (chloroform:methanol = 9:1 ).
Example 14: Analogously to Example 2, N-[3-(4-cyanobenzaylamido)-phenyl]-4-(3-
pyridyl)-2-pyrimidine-amine is prepared from 4-cyanobenwyl chloride; m.p. 220-
222°,
Rf = 0.31 (chloroform:methanol = 9:1).
Example 15: Analogously to Example 2, N-[3-(2-thienylcarboxamido)-phenyl]-4-(3-
pyridyl)-2-pyrimidine-amine is prepared from 2-thiophenecarboxylic acid
chloride;
m.p. 139-141°, Rf = 0.35 (chloroform:methanol = 9:1).
Example 16: Analogously to Example 2, N-(3-cyclohexyl-carboxamido-phenyl)-4-(3-
pyridyl)-2-pyrimidine-amine is prepared from cyclohexanecarboxylic acid
chloride;
m.p. 205-206°, R f = 0.36 (chloroform:methanol = 9:1 ).
Example 17: Analogously to Example 2, N-[3-(4-methylbenwylamido)-phenyl]-4-(3-
pyridyl)-2-pyrimidine-amine is prepared from 4-methylbenwyl chloride; m.p. 214-
216°,
R f = 0.64 (chloroform:rnethanol = 9:1).
Example 18: Analogously to Example 2, N-[3-(4-chloro-benzoylamido)-phenyl]-4-
(4-
pyridyl)-2-pyrimidine-amine is prepared by treatment of 100 mg (0.38 rnmol) of
N-(3-
aminophenyl)-4-(4-pyridyl)-2-pyrirnidine-amine with 58 ltl (0.46 mmol) of 4-
chloro-
benzoyl chloride; m.p. 258-261 °; R f = 0.37 (CHCl3:methanol = 9:1 ).
I' e~,
-30- ~~,}~~~~~4:3
The starting material is obtained as follows:
Step 18.1: Analogously to Step 2.1, N-(3-aminophenyl)-4-(4-pyridyl)-2-
pyrimidine-amine
is obtained by treatment of 3~ mg (1.0 mmol) of N-(3-nitrophenyl)-4-(4-
pyridyl)-2-
pyrimidine-amine (see Example 11) under a hydrogen atmosphere; m.p. 200-
202°,
Rf = 0.27 (CHCl3:methanol = 95:5).
Example 19: Analogously to Example 2, N-{3-{4-(4-methylpiperazinomethyl)-
benzoyl-
amido]-phenyl}-4-(3-pyridyl)-2-pyrimidine-amine is prepared from 98 mg (0.3
mmol) of
4-(4-methyl-piperazinomethyl)-benzoyl chloride; m.p. 198-201°.
Example 20: A solution of 8.0 g (28.85 mrnol) of N-(5-amino-2-methylphenyl)-4-
(3-
pyridyl)-2-pyrimidine-amine and 4.0 rnl (34.6 mmol) of benzoyl chloride in 320
ml of
pyridine are stirred under nitrogen at room temperature for 23 hours. The
reaction mixture
is concentrated under HV; 200 ml of water are added and, after cooling to
0°, the mixture
is filtered. After drying at 80° under HV, the crude product is made
into a slurry with
CH2Ch/methanol (95:5) and filtered, yielding N-(5-benzoylamido-2-methylphenyl)-
4-(3-pyridyl)-2-pyrimidine-amine. After separation by chromatography there are
obtained
further amounts of that product; m.p. 173-176°, Rf = 0.65
(CHCl3:methanol = 9:1).
The starting material is obtained as follows:
Step 20.1: 9.1 ml (0.13 mol) of 65 % nitric acid are added dropwise in the
course of 5
minutes to a yellow suspension of 20.0 g (0.13 mol) of 2-amino-4-nitrotoluene
in 50 ml of
absolute ethanol. When the exothermic reaction has subsided, 8.32 g (0.198
mol) of
cyanamide dissolved in 8.3 ml of water are added. The brown reaction mixture
is boiled
at reflex for 25 hours, cooled to 0° and filtered. Washing with 4 x 100
ml of ethanol/-
diethyl ether (1:1) and drying yield 2-methyl-5-nitrophenyl-guanidine nitrate;
m.p. 219-226°.
Step 20.2: 24$.2 g (0.96 mol) of 2-methyl-5-nitrophenylguanidine nitrate ~~re
added to a
solution of 170 g (0.96 mol) of 3-dimethylamino-1-(3-pyridyl)-2-propen-1-one
in 2.0 litres
of isopropanol. After the addition of 42.5 g of sodium hydroxide, the reddish
suspension
is boiled at reflex for 12 horars. After cooling to 0°, filtration,
washing with 2.0 litres of
isopropanol and 3 x 400 ml of methanol and drying, there is obtained N-(2-
methyl-5-nitro-
phenyl)-4-(3-pyridyl)-2-pyrirnidine-amine, m.p. 195-198°, Rf = 0.68
(methylene chloride:-
-31-
methanol = 9.1).
Step 20.3: A suspension of 143.0 g (0.46 mol) of N-(2-methyl-5-nitrophenyl)-4-
(3-
pyridyl)-2-pyrimidine-amine in 7.15 litres of ethyl acetate is stirred with
14.3 g of
palladium on active carbon (10 % Pd) under a hydrogen atmosphere at normal
pressure for
6.5 hours. The suspension is filtered and the filtrate is concentrated in a
rotary evaporator.
The crude product is recrystallised from methylene chloride, yielding N-(5-
amino-2-
methylphenyl)-4-(3-pyridyl)-2-pyrimidine-amine; m.p. I38-140°, Rf =
0.36 (methylene
chloride:methanol = 9:1).
Example 21: Analogously to Example 20, N-{ 5-[4-(4-methyl-piperazino-methyl)-
benzoyl-
amido]-2-methylphenyl)-4-(3-pyridyl)-2-pyrimidine-amine is prepared from 10.68
g
(32.8 mmol) of 4-(4-methyl-piperazinomethyl)-benzoyl chloride; m.p. 211-
213°, Rf= 0.33
(methylene chloride:methanol:25 % aqueous ammonia solution = 95:5:1).
Example 22: Analogously to Example 20, N-[5-(4-methyl-benzoylamido)-2-methyl-
phenyl]-4-(3-pyridyl)-2-pyrimidine-amine is prepared from 0.23 ml ( 1.7 mrnol)
of
p-toluoyl chloride (p-toluyl chloride); m.p. 102-106°, Rf = 0.4
(methylene chloride:-
methanol = 9:1 ).
Example 23: Analogously to Example 20, N-[5-(2-naphthoylamido)-2-methylphenyl]-
4-(3-pyridyl)-2-pyrimidine-amine is prepared from 330 mg (1.73 mmol) of 2-
naphthoyl
chloride; m.p. 97-101°, Rf = 0.45 (methylene chloride:methanol = 9:1).
Example 24: Analogously to Example 20, N-[5-(4-chloro-benzoylamido)-2-methyl-
phenyl]-4-(3-pyridyl)-2-pyrimidine-amine is synthesised from 0.22 ml (1.73
mmol) of
4-chlorobenzoyl chloride; m.p. 216-219°, R f = 0.39 (methylene
chloride:methanol = 9:1).
Example 25: Analogously to Example 20, N-[5-(2-methoxy-benzoylamido)-2-methyl-
phenyl]-4-(3-pyridyl)-2-pyrimidine-amine is prepared from 0.28 ml (1.87 mmol)
of
2-methoxybenzoyl chloride; m.p. 88-92°, R f = 0.45 (methylene
chloride:methanol = 9:1 ).
Example 26: Analogously to Example 1, N-(3-trifluoromethoxy-phenyl)-4-(3-
pyridyl)-2-
pyrimidine-amine is obtained from 1.0 g (5.68 mmol) of 3-dimethylamino-1-(3-
pyridyl)-
2-propen-1-one and 1.53 g (5.68 mmol) of 3-trifluoromethoxyphenyl-guanidine
nitrate;
Rf = 0.7 (chloroform:methanol = 9:1).
x~ r.) #et ~ c~
-32-
The starting material is obtained as follows:
Step 26.1: Analogously to Step 1.1, 3-trifluoromethoxy-phenyl-guanidine
nitrate is
prepared from 2.0 g (11.3 mmol) of 3-trifluoromethoxy-aniline and 1.4 g (16.6
mol) of
cyanamide (50 % in water); Rf = 0.1 (methylene chloride:methano1:25 % aqueous
ammonia solution = 150:10:1 ).
Example 27: Analogously to Example 1, N-(3-[1,1,2,2-tetrafluoroethoxy]-phenyl)-
4-(3-
pyridyl)-2-pyrirnidine-amine is obtained from 1.0 g (5.68 mmol) of 3-
dimethylamino-
1-(3-pyridyl)-2-propen-1-one and 1.78 g (5.68 mmol) of 3-(1,1,2,2-
tetrafluoroethoxy)-
phenyl-guanidine nitrate; Rf= 0.75 (chloroform:methanol = 9:1).
The starting material is obtained as follows:
Step 27.1: Analogously to Step 1.1, 3-(1,1,2,2-tetrafluoro-ethoxy)-phenyl-
guanidine
nitrate is prepared from 2.09 g (10 mmol) ef 3-(1,1,2,2-tetrafluoroethoxy)-
aniline and
1.26 g (15 mol) of cyanaznide (50 % in water); Rf = 0.15 (methylene
chloride:methanol:-
25 % aqueous ammonia solution = 150:10:1).
Example 28: Analogously to Example 1, N-(3-nitro-5-methyl-phenyl)-4-(3-
pyridyl)-2-
pyrimidine-amine is obtained from 1.0 g (5.68 mmol) of 3-dimethylamino-l-(3-
pyridyl)-
2-propen-1-one and 1.46 g (5.68 mmol) of 3-nitro-5-methylphenyl-guanidine
nitrate;
Rf = 0.72 (chloroform:methanol = 9:1).
The starting material is obtained as follows:
Step 28.1: Analogously to Step, 1.1, 3-nitro-5-methyl-phenyl-guanidine nitrate
is prepared
from 1.52 g (10 mrnol) of 3-nitro-5-methylaniline and 1.26 g (15 mot) of
cyanamide (50
in water); Rp = 0.1 (methylene chloride:methano1:25 % aqueous ammonia solution
=
150:10:1).
Example 29: Analogously to Example 1, N-(3-nitro-S-trifluoromethylphenyl)-4-(3-
pyridyl)-2-pyrimidine-amine is obtained from 1.0 g (5.68 rnmol) of 3-
dimethylamino-
1-(3-pyridyl)-2-propen-1-one and 1.76 g (5.68 mmol) of 3-nitro-5-
trifluoromethylphenyl-
guanidine nitrate; R f = 0.8 (chloroform:methanol = 9:1).
-33-
The starting material is obtained as follows:
Step 29.1: Analogously to Step 1.1, 3-vitro-5-trifluoromethylphenyl-guanidine
nitrate is
prepared from 2.06 g (10 mmol) of 3-vitro-5-trifluoromethylaniline and 1.26 g
(15 rnol) of
cyanamide (50 % in water); Rp = 0.2 (methylene chloride:methano1:25 % aqueous
ammonia solution = 150:10:1).
Example 30: 200 mg (0.6$ mmol) of N-(3-nitrophenyl)-4-(3-pyridyl)-2-pyrimidine-
amine
are suspended in 5 ml of methylene chloride, and 225 mg (0.71 mmol) of 3-
chloroper-
benzoic acid are added. After 2 hours a further 10 ml of methylene chloride
are added.
The suspension is stirred for a further 20 hours at room temperature.
Filtration and flash
chromatography of the residue (methylene chloride:methanol:25 % aqueous
ammonia
solution = 90:10:1) yield N-(3-vitro-phenyl)-4-(N-oxido-3-pyridyl)-2-
pyrimidine-amine;
Rf = 0.4 (methylene chloride:methanol:25 % aqueous ammonia solution = 90:10:1
),
m.p. 252-25$°.
Example 31: 150 mg (0.39 mmol) of N-(3-benzoylamido-5-methylphenyl)-4-(3-
pyridyl)-
2-pyrimidine-amine are suspended in 6 ml of methylene chloride, and 129 mg
(0.41 mmol) of 3-chloroperbenzoic acid are added. After 22 hours, the mixture
is filtered
and the residue is purified by flash chromatography (methylene
chloride:methano1:25 %
aqueous ammonia solution = 90:10:1), yielding N-(3-benzoylamido-5-
methylphenyl)-
4-(N-oxido-3-pyridyl)-2-pyrimidine-amine; Rf = 0.3 (methylene
chloride:methano1:25 %
aqueous ammonia solution = 90:10:1), m.p. 295-300°.
Example 32: Tablets comprising 20 mg of active ingredient, for example one of
the
compounds of formula I described in Examples 1 to 31, and having the following
composition are prepared in customary manner:
-34-
Composition:
active ingredient 20 mg
wheat starch 60 mg
lactose 50 mg
colloidal silicic acid 5 mg
talc 9 mg
magnesium stearate 1 mg
145 mg
Preparation: The active ingredient is mixed with a portion of the wheat
starch, with the
lactose and the colloidal silicic acid and the mixture is forced through a
sieve. A further
portion of the wheat starch is made into a paste, on a water bath, with five
times the
amount of water and the powder mixture is kneaded with the paste until a
slightly plastic
mass is obtained.
The plastic mass is pressed through a sieve of about 3 mm mesh size and dried,
and the
resulting dry granules are again forced through a sieve. Then the remainder of
the wheat
starch, the talc and the magnesium stearate are mixed in and the mixture is
compressed to
form tablets weighing 145 mg and having a breaking notch.
Example 33: Tablets comprising 1 mg of active ingredient, for example one of
the
compounds of formula I described in Examples 1 to 31, and having the following
composition are prepared in customary manner:
Composition:
active ingredient 1 mg
wheat starch 60 mg
lactose 50 mg
colloidal silicic acid 5 mg
talc 9 mg
magnesium stearate 1 mg
126 mg
Preparation: The active ingredient is mixed with a portion of the wheat
starch, with the
~~9~~~J
-35-
lactose and the colloidal silicic acid and the mixture is forced through a
sieve. A further
portion of the wheat starch is made into a paste, on a water bath, with five
times the
amount of water and the powder mixture is kneaded with the paste until a
slightly plastic
mass is obtained.
The plastic mass is pressed through a sieve of about 3 mm mesh size and dried,
and the
resulting dry granules are again forced through a sieve. Then the remainder of
the wheat
starch, the talc and the magnesium stearate are mixed in and the mixture is
compressed to
form tablets weighing 126 rng and having a breaking notch.
Example 34: Capsules comprising 10 mg of active ingredient, for example one of
the
compounds of formula I described in Examples 1 to 31, are prepared in
customary manner
as follows:
Composition:
active ingredient 2500 mg
talc 200 mg
colloidal silicic acid 50 mg
Preparation: The active ingredient is intimately mixed with the talc and the
colloidal
silicic acid and the mixture is forced through a sieve of 0.5 mm mesh size and
then
introduced in 11 mg portions into hard gelatin capsules of a suitable size.