Note: Descriptions are shown in the official language in which they were submitted.
IM 50 314
~ e and
imidaz -thienopyridine comPounds
The invention relates to new imidazo-isoquinoline
and imidazo-thienopyridine compoundls, as well as
to processes for their preparation and pharmaceutical
compositions containing them.
GB-A-1594907 describes compounds o~ general
formula (A)
.
RO
RO ~ ~ Y (A)
A B
wherein, for example, R represents hydrogen or
Cl_4 alkyl,
Y represents alkyl~
8 represents a lone pair of electrons and
A is nitrile or carbamoyl.
According to the description in GB-A-1594907
these compounds are effective as strong broncho-
; 15 dilating agents and may therefore be used ~as active
~ substances or antiasthmatic pharmaceutical prepara-
- ~ tions. However compounds in which Y represents
alkyl, A represents carbamoyl and B represents
a lone pair of electrons are neither men~ioned
nor exemplified.
It has now been found that cer~ain imidazo-
,,
~- isoquinolines an~ imidazo-thienopyridines possess
pharmacologically interesting cardioprotective
~properties.
~,
:~ ,
5~5
-- 2 --
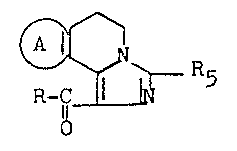
Thus according to one aspect of the invention
we provide compounds of general formula (I3
~ ~ ~ 5 (I)
and the acid addition salts thereof, wherein
A represents a group which, together wi~h the 2
carbon atoms of the pyridyl system to which it
is bonded, forms-a mono- or polysubstituted phenyl
group or a mono- or polysubstituted thieno group,
the substituent or substituents being selected
from Cl 3 alkoxy, hydroxy and methanesulphonyloxy
groups;
R5 represents a Cl ~ alkyl group; and
R represents
(i) a hydroxy group;
: : 15 (ii) a saturated or unsaturated, branched
or unbranched Cl 5 alkoxy group;
(iii) a group -NRlR2, wherein Rl and R2 in p
dently of each other represent
. a~ a hydrogen atom;
b) a saturated or unsaturated, branched
or unbranched Cl 8 hydrocarbon moiety
~optionally mono- or polysubstituted
: : by halogen atoms or by hydroxy, methoxy,
amino, methylamino, anilino, dimethylamino,
~:~ : 25 ~ C3 7 cycloalkyl or phenyl groups (said
phenyl groups optionally being mono-
: : or polysubstituted by halogen atoms
~; or methoxy groups~ or ~y a heterocyclic
~: :: 5- to 7-membered ring which contains
~ :
~,:
,
~ ''"'
~ Z~25~5
one or two identical or different hetero-
atoms selected from N~ S and O and
which is optionally substituted by
a ~Cl 3) alkyl group);
c) a C3 7 cycloalkyl group;
d) a phenyl group, optionally mono
or polysubstituted by halogen atoms
or methoxy groups;
e~ a heterocyclic 5-7 membered ring
which contains one or two identical
or different heteroatoms selected from
N, S and O;
f) an amino group substituted by a
heterocyclic 5-7 membered ring which
contains one or two identical or different
heteroatoms selected ~rom N, S and
O, said ring optionally being mono-
or polysubstituted by halogen atoms;
or Rl and R2 together with the nitrogen
:~ 20 atom to which they are both attached
~: form a 5- or 6-membered ring which
optionally contains an oxygen atom
:~ or a further nitrogen atom as a ring
heteroatom and which ring may be substi-
tuted by a 5 or 6 membered homocyclic
.
: or heterocyclic ring, which, when hetero-
~: cyclic, contains one or two identical
or different heteroatoms selected from
N, S and O.
:
The term "halogen" indicates a fluorine,
chlorine, bromine or iodine atom.
The preferred compounds are compounds of
general formula (II)
:
,
, ~ '; ,
.
' :
,. - ,: .
'a45
-- 4 --
R~ CH~ (II)
wherein
R3 and R4 independently of each other represent
hydrogen atoms or hydroxy, methoxy or methanesul~honyl
oxy groups and R-is as hereinbefore defined,
and the acid addition salts thereof.
In the compounds of general formula (II) the
following combinations of R3 and ~4 are preferred:
R3 = R4 = methoxy
~10 R3 = methoxy, R4 = hydroxy or vice versa,
:~R3 = methoxy, R4 = methanesulphonyloxy or vice
versa,
R3 = methoxy, R4 = hydrogen or vice versa~
Particular mention should also be made of
compounds of general formulae (III) and ~IV)
S
~ ~ CH~ (IIIj :
: ~ R ~ L --N
CH3 (IV)
R-~ N
~: :
,
,
-- 5 --
and the acid addition salts thereof.
If R in formula (I) is a Cl_5 alkoxy group
or a -~RlR2 group, this is preferably methoxy~
ethoxy or an -NR1R2 group wherein:
Rl = a hydrogen atom, and
R2 = a Cl 5 alkyl, C3_s alkenyl~ C3_5 alkynYl~
C3 h cycloalkyl ~Cl 5) alkyl or C3 6 cycloalkyl
group, e.g.: a methylS ethyl, n-propyl, n-butyl,
n-pentyl, isopropyl~ isobutyl, isopentyl, allyl,
propynyl, cyclopropyl, cyclohexyl or cyclohexylmethyl
group;
or R1 and R2 éach represent a Cl 2 alkyl group;
or Rl = a hydrogen atom, and
R2 = a C~ 5 alkyl group substituted by
a) an unsaturated heterocyclic 5 or 6 membered
ring containing at least one heteroatom selected
from among N, S and O, e.g. thienyl, imidazoly],
furyl or pyridinyl; or
b) a saturated heterocyclic 5- or 6-membered
; ring having a nitrogen atom as a ring hetroatomr
said nitrogen atom optionally being substituted
by a methyl group, said ring also optionally containing
an O atom as a further heteroatomt e.g. morpholinyl,
pyrrolidinyl or N-methyl-pyrrolidin-2-yl;
or Rl = a hydrogen atom,
R2 = a phenyl or halophenyl group, e.g.
4-fluorophenyl or 3-chlorophenyl,
or Rl = a hydrogen atom,
R2 = a Cl_5 alkyl group substituted by a
halogen atom or a C1_2 alkoxy, hydroxy, di(Cl 2
alkyl) amino or anilino group, e.g. a methoxy
(C2 or C3~ alkyl, 2-hydroxyethyl, 3-hydroxy-
` .'
. :
-- 6 --
n-propyl, 2-chloroethyl, 3-chloro-n-propyl,
2-dimethylamino-ethyl, 2-anillno-ethyl or
3-dimethylamino-n-propyl group;
or Rl = a hydrogen atom
R2 = an unsaturated heterocyclic 5 or 6
membered ring which contains one or two identical
or different heteroatoms selected from N,
S or O;
or Rl and R2 together with the nitrogen atom to
which they are both attached form a piperazine
group which is substituted by a heterocyclic 6-
membered ring ccntaining one or two N atoms, e~g.
4-(2-pyridillyl)-piperazinyl,
4-(2-pyrazinyl)-piperazinyl or
lS 4-(2-pyrimidinyl)-piperazinyl;
or
` Rl and R2 together with the nitrogen atom to which
: they are both attached form a saturated heterocyclic
5 or 6 membered ring which optionally contains
an N or O atom as further heteroatom.
Thus the class of compounds within formula
(I) as defined above corresponding to formula (VIII)
b (VIII)
and the acid addition salts thereof, wherein R3'
: 25 and ~4' independently of each other represent hydroxy
,
,' ,
,
,
~: .
~ _ 7 _
or methoxy groups and R5 is a Cl 4 alkyl group,
Eall within general formula (A) above of GB-A-1594907
although no compounds of formula (VIII) are disclosed
specifically by Gs A-1594907.
The compounds of formula (VIII) are effective
in the treatment of the indications mentioned herein-
below. There is no suggest-on in Gs-A-l5949o7
of any pharmacological activity for the compounds
of formula (A) other than as bronch~lytics, and
the cardioprotective and other properties of the
compounds of the present invention are thus surprisinq~
The compounds according to the invention
have a significant cardioprotective activity.
~s is well known; the myocardiac Ca2~ content is
a measurement of hypoxic heart damage or heart
damage caused by toxic catecholamine doses (Higgins
et. al. Mol. Cell, Cardiol. 10, 427-438, 1984;
Nakanischi et. al. Am. J. Physiol. 242, 437-439,
1982, Fleckenstein A., Vortrage der Erlanger Physiol.
Tagung 1970, Edit. Keidel, Springer Verlag, Berlin,
Heidelberg, New York, 1971). Conversely, the inhibition
of hypoxic or isoprenalin-induced myocardiac calcium
uptake is a measurement of the cardioprotective
effectiveness of calcium antagonists (Fleckenstein,
see above), of calmodulin inhibitors (Higgins loc.
cit~) and other drugs, e.g. ~-andrenolytics (Arndts,
Arzneimittelforschung 25, 1279-1284, 1975).
The card;oprotective activity of the compounds
according to the invention was tested on conscious
rats after subcutaneous or oral administration
of the active substance using the me~hod described
by Arndts (loc. cit.) and the degree of activity
of the test subs~ance was calculated as the H50
value; this value corresponds to the dosage which
results in a 50~ inhibition in the m~ocardiac radio-
calcium uptake induced by the administration of
30 mg/kg s.c. of isoprenalin.
, '
-
~
.
:: : ,
. .
5'~
-- 8
The new compounds tested proved to be upto five times more effective than the known commercial
product propranolol.
In vitro tests on smooth muscle (strips of
aorta) showed that the compounds according to the
invention are calcium antagonists with a new mechanism
of activity.
In view of these findings, the compounds
of general formula ~I~ or the acid addition salts
thereof should be effective as active substances
in drugs to combat coronary diseases, particularly
angina pectoris or to improve the ischaemia tolerance
in the myocardium.
In addition, the substances according to
the ;nvention promote blood flow through the tissues
and aid the ox~gen supply to the tissues, particularly
in the central ner~ous system. In tests ;n which
the short-term memory is restricted by the admini~
stration of the muscarinic-cholinergic antagonist
scopolamine (0.6 mg/kg i.p., Psychopharmacology
78, page 104 111 (1982)) the compounds are capable
of counteracting or remedying this pharmacologically
induced cerebral insufficiency.
Thus according to a further aspect of the
invention we provide a me~hod of treatment of coronary
disease or conditions of restricted cerebral perfor-
mance, which comprises administering to a human
or non-human animal subject an effeetive amount
of a compound of general formula (I) as herein
defined or a phys;ologically acceptable acid addition
salt thereof.
In tests on the survival of animals in a
sealed chamber (hypoxia tolerance test) through
which a m;xture consisting of 96.5~ nitrogen and
3.5~ oxygen was passed, the animals which had been
previously treated with the substances according
to the invention sho~ed a statistieally highly
. ~
.
-
'
~2~ 5
g
significant increase in survival compared withcontrol animals or animals which had been pretreated
with diltiazem, verapamil or nifedipine.
The protective effect of these substances
on the brain tested by this method was marked even
at a dosage of 5 mg/kg p.o. Thus, the compounds
according to the invention are clea~rly superior
to the known substances mentioned both in terms
of the effective dose and also in the improvement
in performance achieved in animal experiments.
In view of these findinys, the compounds
of general formula ~I) and the acid addition salts
thereof are suitable as active substances for pharma-
ceutical compositions to treat cerebral metabolic
disorders or organic brain psychosyndromes as well
as post-traumatic and alcoholic brain damage.
Thus according to a yet further aspect Oe
the invention we provide a pharmaceutical composition
containing as active substance at least one compound
~ 20 of general formula (I) as herein defined or a physio-
i logically acceptable acid addition sàlt thereof.
The new compounds may be administered on
their own or in conjunction with other active sub-
stances according to the invention, optionally
in combination with other pharmacologically active
substances, e.g. other cerebroactivators. Suitable
forms for admin;stration include tablets, capsules,
suppositories, solutions, syrups, emulsions and
dispersible powders. Tablets may be produced,
for example, by mixing the active substance or
substances with known excipients, e~g. inert diluents
such as calc;um carbonate, calcium phosphate or
lactose, disintegrants such as corn starch or alginic
-~ acid, binders such as starch or gelatine, lubricants
such as magnesium stearate or talc and/or agents
for obtaining delayed release such as carboxypoly-
~ ' , .' ` ' ,
: ;
:: ' ~. .
s
-- 10 --
methylene, carboxymethylcellulose, cellulose acetate-
phthalate or polyvinylacetate. The tablets may
also consist of several layers.
Coated tablets may be produced accordingly
by coating cores prepared analogously to the tablets
with the agents conventionally used for tablet
coatings, e.g. collidone or shellac, gum arabic,
; talc, titanium dioxide or sugar~ To obtain delayed
release or avoid incompatibilities, the core may
also consist of several layers. Similarly, the
tablet coating may consist of several layers in
order to obtain delayed release, using the active
substances mentioned above for the tablets.
Syrups of the active substances or combinations
of active substances according to the invention
may additionally contain a sweetener such as saccharin,
cyclamate, glycerol or sugar and a ~lavour-improving
agent, e.g. flavourings such as vanillin or orange
extract. They may also contain suspension adjuvants
or thickeners such as sodium carboxymethylcellulose,
wetting agents, e.g. condensation products of fatty
alcohols with ethylene oxide, or preservatives
such as p-hydroxyben~oates.
In~ection solutions may be prepared in the
usual way, e.g. by adding preservatives, such as
p-hydroxybenzoa~es, or stabilisers such as alkali
metal salts of ethylenediamine tetraacetic acid
and then transferred into injection vials or ampoules~
Capsules containing one or more active substances
or combinations of active substances may be produced~
~ for example, by mixing the active substances with
;; inert vehicles such as lactose or sorbitol and
encapsulating the mix~ure in ~elatine capsules.
Suitable suppos;tories may be produced by
3S mixing with the appropriate vehicles such as neutral
fats or polyethylene glycol or derivatives thereof.
The compounds may be administered by both
enteral and parenteral routes. The proposed dosage
~ `
-- 11 --
for oral administration is from O.l to lO mg/kg,
pre~erably from 0.2 to 0O5 mg/kg o active substance
per dosage, whilst for parenteral administration
it is from O.Ol to 0.5 mg/kg, preferably 0.05 to
0.2 mg/kg.
The compounds according to the invention
may be prepared by various methods as described
below, which form a yet still further aspect of
the invention:
Process A
(to prepare compounds of general formula (I) wherein
R does not represent -N~2~.
A compound of qeneral formula (V)
O O
Il 11
Ar-CH2(~H2-NII-C-C~EI-C-R (V)
F~N-~-R5
o
(wherein ~r represents an unsubstitu~ed or a mono-
or polysubstituted phenyl group or an unsubstituted
or mono- or disubstituted thieno group, with the
proviso that the phenyl or thieno group possesses
a -CH= group adjacent to the point of attachment,
i.e. possesses an ~ hydrogen atom, and the or
each substituent is selected from Cl 3 alkoxy,
hydroxy and methylsulphonyloxy groups, and R and
R5 are as hereinbefore defined1, is cyclised, convenien-
tly in the presence of a condensing agent.
Suitable condensing agents incIude Lewis
acids such as phosphorus oxychloride, phosphorus
pentachlorider tin tetrachloride and also inorqanic
acids such as polyphosphoric acid, siloxane-polyphos-
; phoric acid mixtures or solutions of p2Os in methane-
sulphonic acid. They are generally used in excess.
Preferred condensing aqents are phosphorus oxychloride,
:
'~. " -
.
,, ,
~ ~2~;~5
- 12 -
polyphosphoric acid and methanesulphonic acid/P2O5
mixtures containing about lO~ by weight of P2O5.
The cyclisation may be effected in the presence
or absence of a solvent. Any inert solvent is
suitable provided that it has sufficient solubility
for the reactants and a sufficiently high boiling
point, e.g. benzene, alkyl benzenes, chloroform,
methylene chloride and acetonitrile.
If desired, the condens;ng agent, e.g. phosphorus
oxychloride, polyphosphoric acid or a methanesulphonic
acid/P2O5 mixture, may be used as solvent.
The reaction may be carried out within a
wide temperature ~ange, preferably with heating
up to about the boiling point of the solvent.
].5
Process B
(to prepare compounds o general ormula (I) wherein
R is -NRlR2).
A compound of general formula (VI)
~ ~ ~ (VI~
(wherein R5 and A are as hereinbefore defined) or a Cl 5
alkyl ester or an activated derivative thereof
is reacted with a compound of general formula (VII)
HNRlR2 (VII)
~'
(wherein Rl and R~ are as hereinbefore defined).
Examples of suitable activated derivatives
of the comPound of formula Vl include ncid halides,
- 13 -
azides or mixed anhydrides (e.g~ with an aromatic
or aliphatic carboxylic acid, alkyl carbonic acid
or dialkylphosphoric acid, etc.~, also amides (for
example with imidazole, 4~substituted im;dazole,
dimethylpyrazole, triazole or tetrazole), or activated
esters, e.g. cyanomethyl, methoxymethyl, vinyl,
propargyl or p-nitrophenyl esters or esters with,
for example, dimethylhydroxylamine, l-hydroxysuccin-
imide or dicyclohexylurea. The active imidazolides
and the Cl 5 alkyl esters are preferred.
The amide formation is generally carried
out in an inert solvent such as dioxan, acetonitrile,
dimethylformamide or a mixture of one or more solvents,
optionall~ in the presence of an inorganic or organic
base as acid acceptor. However, it is also possible
to carry out the reaction without a solvent with
an excess of the amine.
Depending on the starting materials used,
the reaction temperature may vary within wide limits
and may range between about 0C and the boiling
temperature of the reaction mixture.
The end products of the reactions described
; above are generally bases and may be converted
into acid addition salts, generally physiologically
acceptable acid addition salts, in the usual way
with inorganic or organic acids. If salts are
obtained these can be converted into the free bases.
Acids suitable for salt formation include,
for example, hydrochloric, hydrobromic, sulphuric,
phosphoric, nitric, acetic, propionic, butyric,
oxalic, malonic, succinic, maleic, fumaric, lactic,
tartaric, citric, malic~ benzoic, cinnamic, ascorbic
and methanesulphonic acids, etc.
~ome of the starting compounds of general
; 3~ formula (V) are new. They may be prepared by reacting
a suitable reactive derivat;ve o~ an N-acylamino-
malonic acid with a correspondinq amine or, on
.
25~5
~ 14 -
the other hand, first a monoamide of the corresponding
N-acylamino malonic acid is prepared and this is
reacted with another amine to form a diamide of
general formula (V) either as ;t is or after being
converted into an active form.
The N-acyl-malon;c acid derivatives of general
formula (V) may be prepared using the same methods
as those described hereinbefore for preparing the
carboxamides of ~eneral formula ~
Again, methods involving the use of active
imidazolides are preferred.
The invention is further illustrated by the
following non-limiting Examples in which percentages
and ratios are by weight unless otherwise indicated:
,
~ .
;
.,
s~
- 15 -
_xample 1
Ethyl 5,6-dihydro-8,9-dimethoxy-3-methyl-imidazo-
[1,5-a]-isoquinoline-1-carboxylate
50 g of ethyl 2-(3,4-dimethoxyphenyl)-ethylamino
carbonyl-N-acetyl-glycine are added with stirring
to 100 g of a methanesulphonic acid/P2O5 mixture
(100:10), previously heated to 100C, and left
at about 120C for 30 minutes. After the reaction
has ended the reaction mixture is successively
poured onto ice, neutralised with sodium carbonate
solution, extracted with CH2C12~ dried over Na2SO4,
concentrated by evaporation to about 50 ml, mixed
with ethanolic hydrochloric acid and the hydrochloride
(m.p. 214C) is precipitated with the addition
of diethylether.
Yield: 38.2 g (85% of theory)
Example 2
5,6-Dihy~ro-8,9-dimethoxy-3-methyl-imidazo-~1,5-a~-
isoquinoline l-carboxylic acid
25.9 ~ of ethyl S,6-dihydro-8,9-dimethoxy-
3-methyl-imidazo-[1,5-a]-isoquinoline-1-carboxylate
are saponified in a solution of 5~0 g of KOH in
ethanol at about 60C. After the reaction has
ended the reaction mixture is successively neutralised
with ethanolic HCl, cooled to 0C, suction filtered
to remove precipitated KCl and the filtrate is
` evaporated. The resulting hydrochloride (m.p.
214C) is crystallised from ethanol.
Yield: 22.6 g (96~ of theory)
-
~2~
- 16 -
Example 3
5,6-Dihydro-8,9-dimethoxy-3-methyl-imidazo-~1,5-a~-
isoquinoline-l-(N-methyl)-carboxamide
: 5
V iant _
A mixture of 10 g of 2-(3,4-dimethoxyphenyll-
ethylamino-carbonyl-N-acetyl-glycine N-methylamide,
100 g of acetonitrile and 30 ml of phosphorus oxychloride
is refluxed for 1 to 2 hours. After the reaction
has ended the reaction mixture is concentrated
by evaporation in vacuo, made alkaline with saturated
soda solution, the reaction product is isolated
in the usual way, purified on silica gel (CH2C12:MeOH
100:5) and crystallised in the form of the hydrochloride
(m.p. 208C) from ethanol.
Yield: 4.2 g (40% of theory)
Variant B
A mixture of 16 g of 2-(3,4-dimethoxyphenyl)-
ethylaminocarbonyl-N-acetylglycine-N-methylamide
and 60 g of polyphosphoric acid is heated to 130C
for about half an hour. After the reaction has
ended the reaction mixture is left to cool to ambient
temperature, poured onto ice and made alkaline
with concentrated NH3 solution. The reaction product
is isolated by shaking with CH2C12, purified over
silica gel (eluent: CH2C12/MeQH, 100:5~ and converted
into the hydrochloride (m.p. 208-209C).
Yield: 7.2 g ~50% of theory)
Variant C
5 g of 5,6-dihydro-8,9 dimethoxy-3~methyl-
imidazo-~1,5-a]-isoquinoline-1-carboxylic acid
: 35 hydrochloride are stirred in 40 ml of anhydrous
dimethylformamide at ambient temperature with 3.1 g
of N,N'-carbonyldiimidazole for 30 minutes and
5~5
- 17 -
then mixed with 10 ml of a saturated solution of
methylamine in CHC13. The resulting solution is
stirred for 1 hour, the solvent is drawn off in
vacuo, the product is distributed b~etween CH2C12
and water and the organic phase is dried over Na2SO~
and concentrated by evaporation in vacuo. In order
to form the hydrochloride, the residue is dissolved
in a little anhydrous ethanol, mixed with ethanolic
hydrochloric acid and then the hydrochloride (m.p.
209C) is crystallised by the addition of ether.
Yield 5.4 g (93% of theory~
Variant D
10 g of methyl 5,6-dihydro-8,9 dimethoxy-
3 methyl-lmidazo-[1,5-a]-isoquinoline carboxylate
are st;rred ;n 200 ml o~ a chloroform solution
saturatecl with methylamine or 24 hours at ambient
temperature. When the reaction has ended the reaction
mixture is evaporated in vacuo and the reaction
product is converted into the hydrochloride (m.p.
208C).
Yield: 8.0 g (81~ of theor~).
The following end products were also syntheslsed
analogously to the methods given in Examples 1
to 3:
'' ''
;.. ;,
5~5
- 18 -
Table 1
CH30
~ CH . HCl
; CH~ ~ ~ 3
R-C
Example R MpC Yield ~ of
theory
4 -OCH3 197-202 77
-NH2 215 ~ 85
6 -NHC2H5 . 180-183 79
7 -NH(CH2)2-CH3 219-222 6a
; -NH(CH2)3-CH3 212-214 70
9 -NH(CH2)4CH3 204 71
-NH-CH(C~3)2 >250 66
11 -NH-CH2-CH(CH3)2 : 228-229 64
12 -NH(cH2~2-ca(cH3)2 208-220 71
,~ :
.
: .,
- . .
S4~
-- 19 --
Example R MpC Yield % of
theory
13 2 2 Z 16 67
14 -NHCH2C_C~ 189 57
-NH-CH2-~ 239-241 61
16 -NHCH2-O . 228-230 72
17 -NH-~ 208-210 69
18 -NH~ 237-238 61
19 -tlH~(CH2)20CH3 219-224 74
-N~- (CH2) 30C~3 Z03 67
21 ( 2) 2 155 68
...
22 -NH(cH2)3-oH. 164-165 S4
` 23 -NH~CH2)2C1 217 46
: 24 -NH~cH2)3cl 218 43
, ~ 25 -NH(CH2)Z-N(CH3)2 215 68
26 -NH(CHZ)2-NH-~ 214 63
:~ :
: 27 -NH(CHz)2-~ 239-240 70
Z8 -NH-CH2-~ 226 73
: ; 29 -N~-(CHz)2~ Z70-273 68
`
s~s
- 2Q -
Example R MpC Yield % of
theory
-NH-~C~2)2- ~ 244-246 ~3
31 -NH-(CH2)2- ~ 155 (Decomposed) 64
CH3
32~ -NH-(CH2)2-N ~ 96 67
33 -NH-CH2 ~ 221 63
34 -NH ~ 176-179 59
-NH ~ F 242-243 67
36 ~ Cl 212 ~eco~posecl) 70
37 -N(CH3)2 244-246 59
38 -N ~ 228-230 53
. 39 -N ~ 262-263 55
; ~40 -NH(CH2~3-N(CH3)2 239-241 67
41 -N ~ ~ 175 63
42 -N ~ ~ 153-154 65
: 43 -N ~ ~ 210-212 r 67
44 -NH- ~ : Z60 56
-NH-~H- ~ -Cl 219-220 57
, N.N
* The compound of Example 32 ~was obtained as a
ree base~ M.p. and yie~ld apply to the base.
: ~ -
,: ~
~ ~ .
~ ~2~15
-- 21 --
Table 2
__
3 ~
H0 /~ CH~; . HCl
Bxample R M pC Yield ~
of theory
46 -OC2~5 238-239 62
47 -~H-(CH2)2-OCH3 >250 59
48 -NH(CH2)2~3 >250 61
'
:
' :
~,
. . .
. ' ,~`: :
` . '` '
,.
5~5
_ 22 _
Table 3
H0
CH30~ CH3 . HCI
.
Example R MpC Yield % of
theory
49 -NK-(CH~) ~OCH3 l9q-199 56
;
;
'
:
:
:
'`
"
~ - 23 -
Table 4
.
C~30 ~
H~C02S0 ~ ~ `I ~ _CH3 . HCl
Example - R MpC Yield %
of theor~
-OC2~5 Z10-211 67
, .
Table S
::
'
CH30 ~ H3
~':
. . .
~ , ~
~ ~Z~i45
- 24 -
Example R MpC Yield ~
of theory
51 2 5 201-203 ~7
~ -N~-(CH2)~- ~ 148-149 65
52(a) ~NHC~2CH(CH3)2 184-1~6 --__
Table 6
N ~ CH3 . HCl
R~
~, .
Example R MpC Yield ~
of theory
53 -OC H 198-199 78
: : 54 -NH(cH2)2ocH3 156-15~ : 63
'' ~
~, :
.
.. . .. .....
; : .. ~ . -.
. ~
i2~
-- 25 --
Table 7
CH3
R-
Example E'~ M'p. C Y:ield 9~
o:E theory
-OC2~5 20~-210 67
5 6 ( 2 ) 2 3 2 0 ~ 6 2
57 -NH(CH2 ) ;~3 >250 61
58 -NH(CH2)z~ 245-ZS0 58
59 -NM-CH2-CH=CH2 274 73
-NH-(CH2)2~7 226 229 75
;;.. :.
.'~, ;
- ,
~6;~5~5
- 26 -
Examples of pharmaceutical formulations
a) Tablets per tablet
Active substance 25 mg
Lactose 140 mg
5 Corn starch 240 mg
Polyvinylpyrrolidone 15 mg
Magnesium stearate ~
425 mg
The finely ground active substance, lactose
and some of the corn starch are mixed together.
The mixture is screened then moistened with a solution
of polyvinylpyrrolidone in water, kneaded, granulated
whilst wet and drled. The granulate, the remaining
corn starch and the magnesium stearate are screened
and mixed together. The mixture is compressed
to form tablets o~E suitable size and shape.
b) Tablets per tablet
Active substance 15 mg
Corn starch 190 mg
20 Lactose 55 mg
Microcrystalline cellulose 35 mg
Polyvinylpyrrolidone 15 mg
Sodium carboxymethyl starch 23 mg
Magnesium stearate 3 mg
335 mg
The finely ground active substance, some
of the corn starch, lactose, microcrystalline cellulose
and polyvinylpyrrolidone are mixed together, the
mixture is screened and combined with the remaining
corn starch and water to form a granulate which
is dried and screened. The sodium carboxymethyl
starch and the magnesium stearate are added and
mixed in and the mixture is compressed to form
. ~ .
2~5
- 27 -
tablets of sui.table size.
c) Ampoules
Active substance 10.0 mg
5 Sodium chloride lO.O mg
Doubly distilled water q.s. acl 1.0 ml
The active substance and sodium chloride
are dissolved in doubly distilled water and the
solution is transferred into ampoules under sterile
10 conditions.
d) Drops
Active substance 1.0 mg
Methyl p-hydroxybenzoate 0.1 mg
Propyl p-hydroxybenzoate 0.1 m~
15 Demineralised water q.s. ad lOO.O ml
The active substance and preservatives are
; dissolved in demineralised water and the solution
is filtere~ and transferred into lOO ml vials.
.
'
:
: :~
,
:. :
. ~ . .
,