Note: Descriptions are shown in the official language in which they were submitted.
~;2~
Process for the preparation of novel thieno(2,3-d)-
imidazole derivatives and pharmaceutically acceptable
acid addition salts thereof
.. . . _ . . _ ~ .
Description:
The invention relates to a process -for the pre-
paration of novel derivatives of thieno(2,3-d)imidazoles
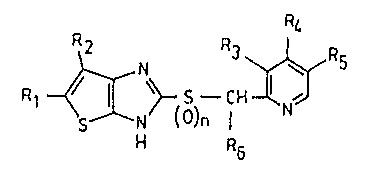
of the general formula
Rl ~ \~ S -CH
H R6
in which R1 denotes hydrogen, lower alkyl, chlorine,
bromine, acetyl, cyclopropylcarbonyl, methoxycarbonyl or
ethoxycarbonyl, R2 denotes hydrogen or lower alkyl, R3,
Rs and R6 independently of one another denote hydrogen
or lower aLkyl, R4 denotes hydrogen or lower alkoxy and
n denotes 0 or 1, and of pharmaceutically acceptable acid
addition salts ~hereof; the thieno(2,3-d)imida~ole deriva-
tives of the general formula I and their salts can be
prepared accord;ng to the invent;on by a process which
comprises
a) reacting a compound of the general formula
~ N
wherein R1 and R2 have the meaning given in claim 1,
with a cornpound of the general formula
'~
7~.~
2 -
X lC ~ ~
R6
wherein X represents chlorine or bromine and R3, R4, Rs
and R6 have the meaning given in the case of formula I,
in the presence of at least 2 equivalents of a strong
base, and then
b) if appropriate reacting the compounds thus obtained,
of the general formula I wherein n denotes 0, with equiva-
lent amounts of an organic peracid or hydrogen peroxide
to give a compound o-f the general formula I in which n
denotes the number 1, and
c) if desired converting a compound of the general formula
I obtained in stage a) or b) into a pharmaceutically
acceptable acid addition salt.
The term "lower alkyl" used in this description
means straight-chain or branched saturated hydrocarbon
groups with 1 - 4 carbon atoms, such as, for example,
methyl, ethyL, propyl, isopropyl, butyl and tert.-butyl~
The term "lower alkoxy" relates to hydrocarbonoxy groups
with 1 - 4 carbon atoms, for example methoxy, ethoxy,
propyloxy, isopropyloxy, butyloxy and tert~-butyloxy.
In a preferred group of compounds of the formula
I, R1 denotes hydrogen, acetyl, methoxycarbonyl or
ethoxycarbonyl, hydrogen or acetyl being particularly
preferred. R2 is preferably hydrogen. R3, Rs and
R6 preferably represent hydrogen or methyl. R4 prefer-
ably represents hydrogen or methoxy.
A particularly preferred yroup within the com-
pounds of the general formula I is that in which n denotes
the number 1~
The reaction according to process step a) is
advantageously carried out by a procedure in which the
compounds of the formulae II and lII are suspended in an
-- 3
inert organic solvent, for example a low-boiling aliphatic
alcohol, 5uch as methanol, ethanol, isopropanol and the
like, preferably in methanol, the compound of the formula
II advantageously being used in excess for reasons of
easier working up, and at least 2 equivalents of a strong
base, preferably NaOH or KOH, dissolved in a little water,
are then added dropwise at room temperature. The reaction
time is 2 - 4 hours.
According to process step b), the sulfoxide com-
pounds in which n in the general formula I denotes thenumber 1 can be obtained from the sulfide compounds ob-
tained by process step a), where n denotes 0, by partial
oxidation with suitable oxidizing agents. Such oxidizing
agents which are preferably used are equivalent amounts
of an organic peracid, such as peracetic acid, perbenzoic
acid or m-chloroperbenzoic acid, in a solvent which is
inert in the reaction, for example methylene chloride or
chloroform, at temperatures between - 5C and -50C, or
equivalent amounts of 30 per cent strength H22 in glacial
acetic acid at room temperature.
The compounds of the general formula I are subject
to tautomerism and can therefore also exist in all the
forms tautomeric to formula I~
The starting compounds of the general formula III
are known. The starting compounds of the general formula
II used for the process are either known or can be pre-
pared in a manner which is known per se, starting from
known products. In particular, they can be synthesized in
accordance with the following equations and in accordance
with the specific information in the examples. The start-
ing compounds of the formulae IV a, IV b, V and VI shown
in the e~uations are known from the literature. Equation
I relates to the preparation of compounds of the formula
II in which R1 denotes hydrogen, lower alkyl, chlorine
or bromine and R2 denotes hydrogen or lower alkyl, and
equation II relates to the preparation of compounds of the
formula II in which R1 denotes acetyl, cyclopropyl-
carbonyl, methoxycarbonyl or ethoxycarbonyl and R2 again
~enotes hydro~en or lower alkyl
Eauat ion I
R2 ~,,~COOCH3 R2 \~COOCH3
Rl S NH2 S NH2
:E a ( Rl = H,A~ky I ) E b
~ (Ac)2o ~ 2 SO2C12 ,Br2
2; ~ COOCH3 R2~COOCH3
Rl S NtlAc Rl S NHAc
~2 ( Rl- H, Alkyl ) . ~[b ( R1= Cl, Br )
/ O
R2~COOH R2~3~C o
Rl S NHAc Rl S N CH3
( Rl=H, Alkyt; Cl, Br ) ~
H ¦ 11
R2 ~ ~N R2~ --C-N3
c=o ~ 11 11
Rl S N Rl~\Sf~NHAc
Ac
:~ ~.
R
2~ C=O ~ Ir
R 5 N/
H ( Rl = H, Alkyl, Cl, Br )
~' ( R2=H,Alkyl )
~6~7;~
Eauat ion I I
R2`1~ Cl COOH s~\CI
vr
I C~. COCI / I 1. SOC12
~ COCI / .3 1 2. CH30H / C2 H50H
Rl ~\a R l~`CI
a( Rl~COCH3, CO <~ b ( R1=COOCH3,COOC2H5 )
~ '~
R2 \, NO2
R S Cl
~ ( Rl=COCH3, CO ~, COOCH3, COOC2H5 )
R2,~No2
R 1 S NH2
~:
R2~NH2
Rl 5 NH2
r
( Rl =COCH3 ,CO <1 ~ COOCH3 COOC2H5 )
( R2 =H, Alkyl )
7~
-- 6
~~he compounds of the general formula I have
strongly basic properties~ They can therefore easily be
converted, in accordance with process step c, into crystal-
line~ pharmaceutically acceptable acid addition salts,
which, like, for example, the hydrochlorides, can readily
be purified by recrystallization. For this, the crude
base is advantageously dissolved in a suitable solvent,
for example a lower alcohol, an equivalent amount of pro-
ton acid is added, the solvent is evaporated off in vacuo
and the residue is recrystallized from methanol or etha-
nol, if appropriate with the add;tion of ether.
Suitable examples of such pharmaceutically accept-
able salts are, in addition to the salt of hydrochloric
acid, for example, the salts of sulfuric acid, nitric
acid, phosphoric acid, sulfonic acids, benzoic acid,
maleic acid, tartaric acid or citric acid.
The compounds of the general formula I and their
pharmaceutically acceptable acid addition salts exhibit
useful pharmacological properties in animal experiments.
In particular, they effect a blockade of (H~ + K ) -
ATP-ase and can therefore be used, for examPle, in human
medicine for the treatment or prophylaxis of gastric and
duodenal ulcers and other diseases caused by increased ~astric
secretion. The following test methods were used to in-
vestigate the pharmacological properties:
The substances were dissolved in 20% strengthdimethylsulfoxide and administered orally to conscious
female rats (strain: Charles River CD) in increasing doses
of 125, 25û and 500 ~mol/kg with a uniform dosage volume
of 20 ml/kg. 60 minutes later, the animals were subjected
to a pylorus ligature. 4 hours after the pylorus ligature
had been applied, the acid concentration and total acid
content in the gastric volume of the animals were deter-
mined. The control group received ~0 ml/kg of dimethyl-
sulfoxide or 20 ml/kg of distilled waterO
ln this standard test, compounds of the generalformula I, such as, for example, 5-acetyl-2-((4-methoxy-
3,5-dimethyl-2-pyridyl)-methylsulfinyl)-3H-thienot2,3-d)-
7imidazo~e (compound A) or 2-((4-methoxy-2-pyridyl)methyl-
sulfiny~-3H-thieno(2,3-d)imidazole (compound ~), effect a
significant, dose-dependent inhibition of gastric secre-
tion (Tables I and II).
Table I:
Gastric juice secretion in rats with pylorus ligature
following administration of compounds of the general
formula I
AmountAcid Total acid
(ml)concentration (meq)
(meq/ml)
Water 4.7+0.7 0.12+0.005 0.56+0.09
(control)
DMSO 3.8~0.5 0.08+0.008 0.34~0.07
(carrier)
Compound A
125 ~mol/kg 4.7+0.4 0.08+0.008 0.37+0.06
250 ~mol/kg 3.4+0.3 0.04+0.006 0.16+0.03
500 ~mol/kg 3.1+0.4 0.04+0.005 0.13+0.03
Table II:
Gastric juice secretion in rats with pylorus ligature
following administration of compounds of the general
formula I
AmountAcid Total acid
(ml)concentration (meq)
(meq/ml)
Water 4.9+0.6 0.13+0.003 0.62+0.08
(control)
30 DMSO 5.1+0.4 0~10+0aO06 0.50+0.05
(carrier)
Compound ~
125 ~mol/kg 4.5+0.6 0.08+û.007 0.37+0.07
25û ,umol/kg 5.0+0.o 0.05+0.004 0.27+0.05
35 500 ,umol/kg 4.5+0.4 0.03+0.004 0.14+0.02
~ 2~
-- 8
The compounds of the general formula I and salt
thereof can be used as medicines, for example in the form
of pharmaceutical products which contain the compounds
according to the invention mixed with a pharmaceutical
organic or inorganic excipient suitable for enteral or
parenteral administration, for example water, gelatin, gum
arabic, lactose, starch, magnesium stearate, talc, veg-
etable oils, polyalkylene glycols, vaseline or the like.
The pharmaceutical products can be in a solid form, for
examPle as tablets, coated tablets, supPositories or cap-
sules, or in a liquid form, for example as solutions,
suspensions or emulsions~ If appropriate, they are
sterilized and/or contain auxiliaries, such as preserva-
tives, stabilizers or emulsifiers, salts for modifying
the osmotic pressure or buffers. They can also be admin-
istered in combination ~ith other therapeuticalLy useful
substances~
The following examples illustrate the in-
vention.
Example 1:
5-Methyl-2-(2-pyridylmethylthio)-3H-thieno(2,3-d)imidazole
(formula I: R1 = CH3, R2 - R6 = H, n = O)
1.56 g (9.162 mmol) of 1,3-dihydro-5-methyl-
thieno~2,3-d)imidazole-2-thione (formula II: R1 = CH3,
R2 = H) and 1.33 9 (8.108 mmol) of 2-chloromethylpyridine
hydrochloride are suspended ;n 40 ml of methanol, and
8.64 ml of 2N NaOH (17.28 mmol) are slowly added dropwise
in the course of 20 minutes, while stirring. The tempera-
ture thereby rises up to 29C and a clear solution forms,
from which the product already precipitates after about
30 minutesn The mixture is stirred at room temperature
for a forther 2~5 hours, the solvent is then largely
removed in vacuo, the residue is taken up in 30 ml of
water and a pH of 4.5 is established by addition of
0.5 ml of glacial acetic acid. The mixture is extracted
four times by shaking with 25 ml of chloroform each time
and the combined organic phases are dried over sodium
sulfate and evaporated. The crude product (2.0 g, 94% of
'73~
theory) is recrystalli~ed from acetonitrile~
Yield: 1.56 9 of colorless crystals (73~6% of theory)
Melting point = 171 - 17ZC (CH3CN)
The starting material can be prepared as follows:
Methyl 2-acetylamino-S-methyl-3-thiophenecarboxylate
(XIa: R1 = CH3, R2 = H)
284.0 9 (1.659 moles) of methyl 2-amino-5-methyl-
3-thiophenecarboxylate (IVa, known from the literature)
are stirred in portions into 600 ml of acetic anhydride,
the temperature being kept between 18 and 25C by inter-
mediate cooling with ice. After stirring for 2.5 hours,
the mixture is evaporated in vacuo and the brownish crys-
talline residue is recrystallized from d;isopropyl ether.
Yield: 277.5 9 of yellowish crystals (78.5% of theory)
Melting point = 100 - 101C (diisopropyl ether)
2-Acetylamino-5-methyl-3-thiophenecarboxylic acid
(XII: R1 = CH3, R2 = H)
200.0 g (0.938 mole) of methyl 2-acetylamino-5-methyl-
3-thiophenecarboxylate are stirred into 1,400 ml of
methanol at 50C. A solution of 37.6 9 (0.940 mmol) of
NaOH in 970 ml of water is added dropwise at the boiling
point in the course of 2.5 hours, with mechanical stirring.
The mixture is heated under reflux for a further 30 min-
utes and, after cooling, the solution is largely evapo-
rated~ The residue which remains is taken up in 1,800 ml
of water and 200 ml of saturated sodium bicarbonate solu-
tion and the mixture is extracted three times with Z50 ml
of methylene chloride each time. The aqueous phase is
acidified to pH 1 with concentrated hydrochloric acid and
the precipitate which has separated out is filtered off
with suction, washed three times with water and dried at
70C in a circulating air drying cabinet for 3 hours.
Yield: 121.8 g of colorless crystals (65.2% of theory)
Melting point = 197 - 198C (MeOH)
3,6-3imethyl-4H-thieno(2,3-d)-(1,3~-oxazin-4-one
(XlII: R1 = CH3, R2 - H)
170.0 g (0.853 mole) of 2-acetylamino-S-methyl-3-
thiophenecarboxylic acid (XII) are suspended in 800 ml of
3~
-- 10 -
acetic anhydride and the suspension is heated under reflux
for 2.5 hours. The solution is then cooled and evaporated
comDlete~y in vacuo~ The residue which remains is re-
crystallized from carbon tetrachloride.
Yield: 128~0 g of colorless crystals ~82.8% of theory)
Melting point = 140C (diisopropyl ether)
2-Acetylamino-5-methyl-3-thiophenecarboxylic acid azide
(XIV: R1 = CH3, R2 = H)
72.0 9 (0.397 mole) of 3,6-dimethyl-4H-thieno-
(2,3-d)-(1,3~-oxazin-4-one are d;ssolved in 1,100 ml of
dioxane, and a solution of 51.7 g (0.795 mole) of sodium
azide in 170 ml of water is introduced in a layer below
the solution. The mixture is stirred at a lower speed at
room temperature for 50 hours and a total of 23.8 ml
(0.397 mole) of glacial acetic acid is added in 4 portions
at intervals of 8 - 12 hours.
The phases are then separated and the organic
phase is evaporated at a bath temperature of 40C. The
dark oil which remains is taken up in 600 ml of methylene
chloride and the mixture is combined with the aqueous
phase and 150 ml of saturated sodium bicarbonate solution.
The phases are separated and the aqueous phase is extrac-
ted twice more by shaking with 200 ml of methylene chlor-
ide each time. The combined organic extracts are dried
over sodium sulfate and evaporated.
Yield: 72.0 9 of yellow crystals (80.8% of theory)
The crude product can be recrystallized from
methanol.
Melting point = 78 - 79C (MeOH)
3-Acetyl-1,3-dihydro-S-methyl-thieno(2,3-d)imidazol-2-one
(XV: R1 = CH3, R = H)
16.3 9 (72.7 mmol) of 2-acetylamino-5-methyl-3-
thiophenecarboxylic acid azide are dissolved in 250 ml of
absolute dioxane and the solution is heated at 100C for
4.5 hours. The dark solution is left to cool overnight,
the precipitate which has separated out is filtered off
with suction, the mother liquor is concentrated to half
the volume and left to crystallize and the crystals are
filtered off with suction again.
Yield: 9 9Z g of colorless crystals (69.6% of theory)
Melting point = about 200C, decomposition (dioxane)
1,3-Dihydro-5-methyl-thieno(2,3-d)imidazol-2-one
(XVI: Rl = CH3, R2 = H)
34.7 g (0.177 mole) of 2-acetyl-1,3-dihydro-5-
methyl-thieno(2,3-d)-imidazol-2-one are introduced into
420 ml of 2N NaOH at room temperature and, after the solid
has dissolved completely, the solution is stirred for a
further 15 minutes. The deep red solution is acidified
to pH 2.5 by adding concentrated HCl dropwise and is
cooled briefly in an ice-bath, and the precipitate which
has separated out is filtered off with suction and washed
twice with cold water. The crystals are dried at 70C/
20 mbar.
Yield: 26.5 9 of colorless crystals (97.2% of theory)
Melting point = 275 - 278C, decomposition.
1,3-Dihydro-5-methyl-thieno(2,3-d)imidazole~2-thione
(II: R1 = CH3, R2 = H)
25.0 9 (0.162 mole) of 1,3-dihydro-5-methyl-
thieno(2,3-d)imidazol-2-one and 27.0 9 (0.122 mole) of
P25s are added to 700 ml of absolute pyridine at room
temperature, with mechanical stirringO The suspension
is warrned in an oil bath under nitrogen, whereupon a clear
red solution forms at about 100Co The solution is
heated under reflux for 3 hours.
The mixture is then cooled and the solvent ;s
distilled off in vacuo. The dark viscous-oily residue is
dissolved in 300 ml of 2N NaO~I and this solut;on ;s poured
slowly into a vigorously stirred m;xture of 1,100 ml of
ethyl acetate and 400 ml of 3N HCl. The m;xture is
stirred for a further 10 minutes and filtered with suction
over a filtration auxiliary, and the phases are separated.
The aqueous phase is extracted four more times by shaking
~ith a total of ~00 ml of ethyl acetate. The combined
organic phases are dried over sodium sulfate/active char-
coal and evaporated. The dark residue (12.4 9; 45~ of
theory) is dissolved in 90 ml of 2N NaOH, the solution is
.. ,
filtered with active charcoal and the clear red solution
is acidified to pH 2 with concentrated HCl~ The crystals
which have precipitated are filtered off with suction,
washed twice with water and dried in vacuo at 60Cn
Yield: 9.8 9 of pale brown crystals (35.5% of theory)
Melting point = greater than 330C, continuous decomposi-
tion.
~xample 2:
5-Methy~-2-~4-methoxy-2-Pyridyl)methylthio)-3H-thieno-
~2,3-d)imidazole
~I: Rl = CH3, R2, R3, Rs and R6 = H, R4 = 0CH3,
n = 0)
3.46 9 ~20.3 mmol) of 1,3-dihydro-5-methyl-
thieno~2,3)imidazole-2-thione and 3.35 9 (17.3 mmol) of
2-chloromethyl-4-methoxypyridine hydrochloride are suspencd-
ded in 70 ml of ethanol and the susPension is slowly added
dropwise to 18.8 ml (37.6 mmol) of 2N NaOH, while stirring.
The temperature rises up to 29C and a clear solution
forms. The solution is stirred at room temperature for a
further 2 hours and then largely evaporated in vacuo. The
residue which remains is taken up in 70 ml of water and
a pH of 4.5 is established by addition of about 1 ml of
glacial acetic acid. The mixture is extracted four times
by shaking with a total of 300 ml of chloroform and the
combined organic phases are dried over sodium sulfate and
evaporated.
The oily residue which remains is triturated with
acetonitrile and dissolved in 280 ml of acetonitrile at
the boiling point. After addition of active charcoal, the
mixture is filtered hot, the solution is concentrated to
60 ml, the concentrate is left to crystallize overnight
in a refrigerator and the crystals are filtered off with
suction and washed with cold acetonitrile.
Yield: 3.62 9 of pale brown crystals (71.9% of theory)
Melting point = 173C (CH3CN)
. ..
~ 7
- 13-
Example 3:
2-(2-Pyridyl)methylthio-3H-thieno(Z,3-d)imidazole
(I: R1 - R6 = H, n = O)
1.20 9 (7.68 mmol) of 1,3-dihydro-thieno(2,3-d)-
imidazole-2-thione (II: R1 and R2 = H) and 1.13 9
(6.91 mmol) of 2-chloromethylpyridine hydrochloride are
dissolved in 15 ml of methanol, and 7.4 ml (14.8 mmol) of
2N NaOH are added dropwise in the course of 20 minutes,
with stirring. The mixture is stirred at room temperature
for a further 1.5 hours and then concentrated on a Rota-
vapor~ The res;due is partitioned between 60 ml of water
and 80 ml of methylene chloride. For phase separation,
the mixture is filtered over a filtration auxiliary and
the aqueous phase is extracted three more times with 70 ml
of methylene chloride each time. The comb;ned organic
phases are dried over sodium sulfate and evaporated.
The crude product (1.52 9 of brown crystals; 92.4%
of theory) is recrystallized from acetonitrile with the
addition of active charcoal.
Yield: 1.10 9 of colorless crystals (64.3% of theory)
Melting point = 155 - 156C (CH3CN)
The starting material can be prepared as follows:
1,3 Dihydro-thieno(2,3-d)imidazole-2-thione
(II: R1 and R2 = H)
20.0 9 (0.143 mole) of 1,3-dihydro-thienot2,3-d)-
imidazol-2-one (known from the literature) are introduced
into 500 ml of absolute pyridine at room temperature, with
mechanical stirring. Thereafter, 23.8 9 (0.107 mole) of
P2S5 are added and the mixture is heated under reflux
for 5.5 hours. It is cooled and the solvent is largely
distilled off in vacuo. The dark residue is dissolved
in 300 ml of 2N NaOH and the solution is emptied into a
mixture of 370 ml of 2N HCl and 400 ml of ethyl acetate,
with vigorous stirring. The two-phase mixture is f;ltered
off with suction over a f;ltration auxiliary and the
phases are separated~ The aqueous phase is extracted six
more times with 150 ml of ethyl acetate each time and the
combined organic phases are dried over sodium sulfate and
. . ,
a~
~ 14 ~
evaporated.
Yield: 8.81 9 of brown crystals (39.5% of theory)
Melting point = continuous decomposition
Example 4:
2-((4-Methoxy-2-pyridyl)methylthio)-3H-thieno(2,3-d)imidazole
(I: R1, R2, R3, R5 and R6 = H, R4 = OCH3,
n = 0)
4~65 g (29.76 mmol) of 1,3-dihydro-thieno(2,3-d)-
imidazole-2-thione and 4.91 9 (25.30 mmol) of 4-methoxy-
2-chloromethylpyridine hydrochloride are suspended in
60 ml of methanol, and 27.6 ml (55.2 mmol) of 2N KOH are
slowly added dropwise in the course of 25 minutes, with
stirring. The temperature thereby rises to 32C. When
the addition has ended, the mixture is stirred at room
temperature 'for a further 4 hours. The reaction mixture
is then evaporated in vacuo, the residue is taken up in
50 ml of water and a pH of 4.5 is established by dropwise
addition of glacial acetic acid. The precipitate which
has separated out is dissolved in 80 ml of chloroform and
the mixture is shaken vigorously and, for phase separa-
tion, filtered with suction over a filtration auxiliary.
The phases are seParated and the aqueous phase is ex-
tracted three more times with 40 ml of chloroform each
time. The combined organic phases are dried over sodium
sulfate and evaporated. The crude product (5.8 g; 82%
of theory) is dissolved in 400 ml of acetonitrile at the
boiling point, the solution is filtéred hot w;th active
charcoal, the filtrate is concentrated to 150 ml and the
concentrate is allowed to crystallize out.
Yield: 4.34 9 of brownish crystals (61.8% of theory)
Melting point = 140 - 141C (CH3CN)
Example 5:
S-Acetyl-Z-((4-methoxy-3,5-dimethyl-2-pyridyl)-methylthio)-
3H-thieno(2,3-d)imidazole
(I: R1 ~ COCH3, R2 and R6 = H, R3 and R5 = CH3~ ~4 =
OCH3, n = 0)
5.64 9 (28.4 mmol) of 5-acetyl-1,3-dihydro-thieno-
(2,3-d)imidazole-2-thione and 6.00 9 (27.0 mmol) of 2-
, . ,
chloromethyl-4-methoxy-3,5-dimethylpyridine hydrochloride
are suspended in 250 ml of methanol, and 28 ml of 2N NaOH
(56.0 mmol) are added dropwise in the course of 15 minutes,
while stirring, whereupon the temperature rises up to
27C. The product already precipitates from the result-
ing solution after a short time.
The reaction mixture is subsequently stirred at
room temperature for a total of 4 hours and the largely
evaporated. The residue is taken up in 200 ml of water
and a pH of 4.5 is established by addition of about 2 ml
of glacial acetic acid. The suspension is extracted four
times by shaking with a total of 350 ml of chloroform and
the combined organic phases are dried over sodium sulfate/
active charcoal and evaporated.
The dark oil which remains is crystallized with
80 ml of acetonitrile, the mixture is cooled and the pre-
cipitate is filtered off with suction. It is washed three
times with cold acetonitr;le and dried at 40C in vacuo.
Yield: 8.92 9 of slightly yellowish crystals S95.0% of
theory)
Melting point = 177 - 180C (CH3CN)
The starting material can be prepared as follows:
1-(5-Chloro-4-nitro-2-thienyl/-1-ethanone
(VIIIo R1 = COCH3, R2 = H)
30.0 g (0.18b8 mole) of finely ground 1-(5-chloro-
2-thienyl)-1-ethanone (VI~ a: R1 = COCH3, R2 = H) are
introduced into 180 ml of concentrated H2SO4 at -20C,
with stirring. A nitrating mixture of 7~8 ml of fuming
HNO3 (0.1868 mole) and 30 ml of concentrated H2504 is
added dropwise to the red-colored solution at a tempera-
ture between -20C and -25C in the course of 35 minutes~
The mixture is subsequently stirred at -20C for
a further 30 minutes and the reaction mixture is then
emptied onto 1,400 ml of ice-waterO The product which has
precipitated is taken up in 500 ml of methylene chloride
and the aqueous phase is extracted twice more by shaking
with 250 ml of methylene chloride each time. The combined
organic phases are washed once with 300 ml of water, dried
, ~,
- 16 -
over sodium sulfate and evaporated.
The crude product (36.0 9; 93.8% of theory) con-
tains about 10% of an impurity (runs higher in the ~hin
layer chromatogram), which does not interfere in the
further reaction but can be removed by recrystallization
from ethanol.
rield: 26.0 9 of slightly yellowish crystals (67.7% of
theory)
Melting point = 80C (EtOH)
(5-Amino-4-nitro-2-thienyl)-1-ethanone
(IX: R1 = COCtl3, R2 = H)
Dry NH3 gas is passed into a solution of 22.0 9
(0.108 mole) of 1-(5-Chloro-4-n1tro-2-thienyl)-1-ethanone
in 250 ml of dry dioxane, whereupon the temperature rises
up to 45C. When the exothermic reaction has subsided,
the temperature is kept at 50C by warming on a waterbath
and further NH3 gas is passed in for another 1.5 hours.
A precipitate which has separated out is then
filtered off with suction and is discarded. The ~iltrate
is stirred with active charcoal at 50C, -filtered again
and evaporated. The residue which remains is recrystal-
lized from acetonitrile.
Yield: 11.9 9 of yellow needles (5q.2% of theory)
Melting point = 224 - 225C (CH3CN)
5-Acetyl-1,3-dihydro-thieno(2,3-d)imidazole-2-thione
(II: R1 = COCH3, R2 = H)
10.0 g (0.0537 mole) of 1-(5-amino-4-nitro-2-thi-
enyl)-1-ethanone (IX) are dissolved in a mixture of 150 ml
of methanol and 150 ml of dioxane and hydrogenated with
3 spoon~uls of W2 Raney nickel, as a catalyst, in a PARR
medium pressure hydrogenation apparatus at room tempera-
ture in the course of 2~5 hours, until the calculated
amount of hydrogen has been taken up. The catalyst is
filtered off with suction over a filtration auxiliary and
washed thoroughly with methanol and dioxane.
8.39 9 (0.0644 mole) of sodium methyl xanthogenate
are added to the filtrate (consisting of X: R1 = COCH3,
R2 = H), and the mixture is heated with stirring and
,,
t7.~
under reflux (temperature about 70C) for 4 hours.
After cooling, ~he mixture is largely evaporated and the
residue is dissoLved in 250 ml of water. The solution is
stirred with two teaspoonfuls of active charcoal for 10
minutes and then filtered with suction over a filtration
auxiliary. The filtrate is slowly acidified to pH 1 with
concentrated HCl, with stirring, the mixture is further
stirred briefly and the yellow precipitate which has
separated out is filtered off with suction. The crystals
are dried at 90C/20 mbar for 2 hours.
Yield: 8.2 9 of crystals (77.0% of theory)
Melting point = 302C (decomposition)
Example 6:
Methyl 2-((4-methoxy-3,5-dimethyl-2-pyridyl)-methylth;o)-
3H-thieno(2,3-d)imidazole-5-carboxylate
(I: R1 = COOCH3, R2 and R6 = H, R3 = CH3~ R4 = OCH3
n = 0)
The abovementioned compound of the general formula
I was prepared by a working method analogous to that des-
cribed in Example 5 by reacting methyl 1,3-dihydro-thieno-
(2,3-d)imidazole-2-thione-5-carboxylate with 2-chloro-
methyl-4-methoxy-3,5-dimethyl-pyridine hydrochloride.
Yield: 89% ot theory
Melting point = 173 - 174.5C (CH3CN)
The starting material was prepared by the reaction
sequence described in Example 5 from methyl 5-chloro-4-
nitro-thiophene-2-carboxylate (VIII: R1 = COOCH3, R2 =
H, known from the literature) via methyl 5-amino-4-nitro-
thiophene-2-carboxylate (IX: R1 = COOCH3, R2 = H, melting
point = 271C, decomposition), reduction to give methyl
4,5-diamino-thiophene-2-carboxylate (X: R1 = CO0CH3,
Rz = H) and reaction of this compound with sodium methyl
xanthogenateO Methyl 1,3-dihydro-thieno-(2,3-d)imida~ole-
2-thione-5-carboxylate (II: R1 = COOCH3, R2 = H; melting
point = 27qC, decomposition) is obtained in this manner.
~ 2
- 18 -
Examp~e 7:
S-Acetyl-2((4-methoxy-2-pyridyl)methylthiol)-3H-thieno-
(2,3-d)imiclazole
(I: R1 = CCH3, R2, R3, Rs and R6 = H, R4 = OCH3,
n = O)
1.30 9 (6.567 mmol) of 5-acetyl-1,3-dihydro-
thieno(2,3-d)imidazole-2-thione and 1.08 g (5.565 mmol)
of 4-methoxy-2-chloromethylpyridine hydrochloride are
suspended in 15 ml of methanol, and 6.2 ml of 2N Na~H
(12.4 mmol) are added dropwise in the course of 10 minutes,
with stirring, whereupon the temperature rises up to
27C. The mixture is stirred at room temperature for a
further 1.5 hours.
The reaction m;xture is then largely evaporated,
the residue is taken up in 5û ml of water and the mixture
is brought to pH 4 by addition of about 1 ml of glacial
acetic acid~ The mixture is extracted three times by
shaking with 40 ml of chloroform each time and the com-
bined organic phases are drierJ over sod;um sulfate and
evaporated.
The crude product (1.52 9 of a brown oil) is
triturated with acetonitrile and recrystallized from
acetonitrile with the acidition of active charcoal.
Yield: 0.76 g of colorless crystals (42.8% of theory)
Melting point = 192 - 194C (CH3Cn)
Example 8:
5-Acetyl-2-(2-pyridyl)methylthio-3H-thieno(2,3-d)imidazole
(I: R1 = COCH3, R2 ~ R6 = ~I~ n O)
1.50 9 (7.565 mmol) of S-acetyl-1,3-dihydro-
thieno(2,3-d)imidazole-2-thione and 1.12 g (6.809 mmol)
of 2-chloromethylpyridine hydrochloride are suspended in
60 ml of methanol, and 7.2 ml of ZN NaOH (14.4 mmol) are
added dropwise in the course of 8 minutes, with stirring,
whereupon the temperature rises up to 26C. The mixture
is stirred at room temperature for a further 2 hoursO
The clear solution formed is largely evaporated,
the residue is taken up in 80 ml of water and a pH of 4.5
is established by addition of 1.2 ml of glacial acetic
~2~
- 19
acid. The mi~ture is extracted three times with a total
of 150 m~ of chloroform and the organic phase is dried
over sodium sulfate and evaporated. The crystals which
remain are recrystallized from acetonitrile.
Yield: 1.85 g of colorless crystals (84.5% of theory)
Melting point = 171 - 173C (CH3CN)
Example 9:
5-Methyl-2-(2-pyridyl)-methylsulfinyl-3H-thieno(2,3-d)-
imidazole
(I: R1 = CH3, R2 ~ R6 = H~ n 1)
1.0 g (3.826 mmol) of 5-methyl-1-(2-pyridyl)-
methylthio-3H-thieno(2,3-d)imidazole is dissolved in 20
ml of chloroform at room temperature and the solution is
cooled to -10C. A solution of 0.76 g (3~749 mmol) of
85 per cent strength 3-chloroperbenzoic acid in 10 ml of
chloroform is added dropwise at a temperature between
-12C and -9C in the`course of 10 minutes, with stirring.
When the addition has ended, the mixture is stirred at
-1ûC for a further 10 minutes.
The reaction mixture is extracted twice by shaking
with 10 ml of saturated sodium bicarbonate solution each
time and the combined organic phases are dried over sodium
sulfate and evaporated.
The crude product (0.96 9; 90.5% of theory) is
recrystallized from acetonitrile.
Yield: 0.76 9 of colorless crystals t71.c6% of theory)
Melting point = 175 - 176C, decomposition (CH3CN)
Example 10:
5-Methyl-2-((4-methoxy-2-pyridyl)methylsulfinyl)-3H-
thieno-(2,3-d)imidazole
(I R1 = C~l3, R2, R3, Rs and R6 = H,
R4 = OCH3~ n = 1)
6.0 9 (20.59 mmol) of 5-methyl-2-((4-methoxy-2-
pyridyl)methylthio)-3H-thieno(2,3-d)imidazole are dissolved
in 120 ml of chloroform at room temperature. The solution
is cooled to -12C and a solution of 4.10 g (Z0.1~ mmol)
of 85 per cent strength 3-chloroperbenzoic acid in 60 ml
of chloroform is added dropwise at a temperature of bet-
- 20 -
~een -12C and -80C in the course of 20 minutes~ while
stirring. When the addition has ended, the mixture is
stirred at -10C for a further 10 minutes. It is then
extracted twice with 25 ml of saturated sodium bicarbonate
S solution each time, the aqueous phases are washed twice
with a little chloroform and the combined organic phases
are dried over sodium sulfate and evaporated.
The residue which remains (about 6 9 of a reddish
oil) i5 crystallized with a little acetonitrile and dis-
solved in 40 ml of dimethylformamide at 80C. The solu-
tion is filtered hot with active charcoal, the filtrate
is emptied into 220 ml of warm acetonitrile at 60C and
the mixture is allowed to cool slowly. Crystallization
is brought to completion overnight in a refrigerator.
The crystals are then -filtered off with suction and washed
with cold acetonitrile.
Yieldo 4.81 9 of colorless crystals (76% of theory)
Melting point = 183 - 184C (dimethylformamide/CH3CN)
Example 11:
2-(2-Pyridyl)methylsulfinyl-3H-thieno(2,3-d)imidazole
(I: R1 ~ R6 = H~ n = 1)
0.45 9 (1.82 mmol) of 2-(2-pyridyl)methylthio-3H-
thieno(2,3-d)imidazole is dissolved in 10 ml of chloroform,
and a solution of 0.37 9 (1.82 mmol) of 85 per cent
strength 3-chloroperbenzoic acid in 5 ml of chloroform is
added dropwise at a temperature between -11 and -6C,
with stirring. The mixture is stirred at a temperature
below 0C for a further 30 minutes and the solution is
diluted with a little methylene chloride and washed twice
with 5 ml of saturated sodium b;carbonate solution each
time.
The aqueous phase is extracted twice with S ml of
methylene chloride each time and the combined organic
phases are dried over sodium sulfate and evaPorated.
The residue (0.42 9; 87.7% of theory), which is
crystalline after trituration with acetonitrile~ is re-
crystallized from acetonitrile/active charcoalu
Yield: 0.33 9 of colorless crystals (68.9~ of theory)
~%~
Melting point = 151 - 152C, decomposition (CH3CN)
Example 12:
2-((4-Methoxy-2-pyridyl~methylsulfinyl)-3H-thieno(2,3-d)-
imidazole
(1: R1 ~ R3, R5 and R6 = H, R4 = OCH3, n = 1)
4.30 9 (15.50 mmol) of 2-((4-methoxy-2-pyridyl)-
methylthio)-3H-thieno(2,3-d)imidazole are dissolved in
90 ml of chloroform at room temperature and the solution
is cooled to -10C. Thereafter, a solution of 3.08 9
(15.19 mmol) of 85 per cent strength 3-chloroperbenzoic
acid in 35 ml of chloroform is added dropwise at a tem-
perature between -11 and -8C in the course of 30
minutes, while stirring. The mixture is then stirred at
-10C for a further 15 minutes.
The solution is extracted twice with a total of
30 ml of saturated sodium bicarbonate solution, the aque-
ous phase is extracted three times by shaking with 20 ml
of chloroform each time and the combined organic phases
are dried over sodium sulfate and evaporated.
The crude product (5.2 9 of .3 red oil) is tritura-
ted with a little acetonitrile and the brownish crystals
are dissolved in 22 ml of dimethylformamide at 80C.
Active charcoal is added, the mixture is filtered hot and
the filtrate is emptied into 130 ml of warm aceton;trile
Z5 at 60C. The mixture is allowed to cool slowly and crys-
tallization is brought to completion in a refrigerator
overnight.
Yield: 4.03 9 of colorless crystals t88.6% of theory)
Melting point = 157 - 9C, decomposition tCH3CN)
Example 13:
5-Acetyl-2-(t~-methoxy-3,5-dimethyl-2-pyridyl)-methyl-
sulfinyl)-3H-thienot2,3-d)imidazole
(1: R1 = COCH3, R2 and R6 = H, R3 and Rs = CH3,
R4 = OCH3, n = 1)
4.5 9 (1Z.95 mmol) of 5-acetyl-2-(4-methoxy-3,5-
dimethyl-2-pyridyl)-methylthio)-3H-thieno(2,3-d)imidazole
are almost completely dissolved in 120 ml of chloroform
at room temperature and the solution is cooled to -10C.
2~3~
- 22 -
A solution of 2.58 9 (12.69 mmol) of 85 per cent strength
3-chloroperbenzoic ac;d in 40 ml of chloroform is added
dropwise at a temperature between -8C and -10C in the
course of 25 minutes, with stirring.
The mixture is stirred at -10C for a further 10
minutes and the now completely clear solution is extracted
three times with a total of 60 ml of saturated sodium bi-
carbonate solution. The aqueous phase is washed three
times with a little chloroform and the combined organic
phases are dried over sodium sulfate and evaporated.
The residue which remains (about S g of a red oil)
is dissolved in 50 ml of dimethylformamide at 80C and
the solution is filtered with active charcoalO The solu-
tion is emptied into 300 ml of warm acetonitrile at 60C,
the mixture is allowed to cool slowly and crystallization
is brought to completion overnight in 3 refrigerator. The
crystals are filtered off with suction and washed three
times with acetonitrileO
Yield: 3.30 g of colorless crystals (70.1~ of theory)
Melting point = 190 - 191C (dimethylformamide/CH3CN)
Example 14:
5-Acetyl-2~ -methoxy-2-pyridyl)methylsulfinyl)-3H-
thieno-(2,3-d)imidazole
(I: R1 = CCH3, R2, R3, Rs and R6 = H, R4 =
OCH3, n = 1)
0.53 g (1.628 mmol) of 5~acetyl-2-(4-methoxy-2-
pyridyl)methylthio)-3H-thieno(2,3-d)imidazole is suspended
in 15 ml of chloroform and a solution of 0.33 g (1n628
mmol) of 85 per cent strength 3-chloroperbenzoic acid in
6 ml of chloroform is added dropwise at a temperature
between -12 and -8C in the course of 7 minutes, with
stirring, whereupon a clear solution formsO The mixture
is stirred at -10C for a further 10 minutes. It is
then extracted twice with 6 ml of saturated sodium bi-
carbonate solution each tirne, the aqueous phases areextracted twice by shaking with S ml of chloroform each
time and the combined organic phases are dried over sodium
sulfate and evaporated. The oily crude product is re--
~2~
-- 23 -
crysta~lized from acetonitrile/active charcoal.
Yield: 0.37 9 of colorless crystals t67.8~ of theory)
Melting point = 159 - 162C, decomposition (CH3CN)
Example 15:
5-Acetyl-2-((4-methoxy-2-pyridyl)methylsulfinyl)-3H-
thieno(2,3-d)imidazole
(I: R1 = COCH3, R2, R3, R5 and R6 = ~l, R4 = OCH3
n = 1~
0.53 9 (1.628 mmol) of S-acetyl-2-(4-methoxy-2-
pyridyl)methylthio)-3H-thieno(2,3-d)imidazole is taken up
in 15 ml of glacial acetic acid, and a solution of 181 mg
(1.6Z8 mmol) of 30 per cent strength H22 is slowly added
at 5 to 10C, whereupon a clear solution forms. The mix-
ture is stirred at room temperature for a further 20
minutes. It is then diluted with 100 ml of water and ex-
tracted three times by shaking with 20 ml of chloroform
each time, and the combined organic phases are washed
neutral with sodium bicarbonate solution, dried over
sodium sulfate and concentrated. The oily crucde product
is recrystallized from acetonitrile/active charcoal.
Yield: 0.28 9 of colorless crystals (51.3% of theory)
Melting point = 159 - 162C~ decomposition (CH3CN)
Example 16:
5-Acetyl-2-(2-pyridyl)methylsulfinyl-3H-thieno(2,3-d)-
imidazole
(I: R1 = COCH3, R2 - R6 = ~, n = 1)
1.0 9 (3.46 mmol) of 5-acetyl-2-(2-pyridyl)methyl-
thio-3H-thieno-(2,3-d)imidazole is almost completely dis-
solved in 15 ml of chloroform at room temperature and the
30 solution is cooled to -12C. A solution of 0.69 g
(3.39 mmol) of 85 per cent strength 3-chloroperbenzoic
acid in 10 ml of chloroform is added dropwise at a tem-
perature betweeri -14 and -9C in the course of 10
minutes.
The mixture is stirred at -10C for a further 10
minutes and the solution, which has meanwhile become com-
pletely clear, is diluted with a little chloroform and
extracted twice with 7 ml of saturated sodium bicarbonate
7.~:~
- 2~
solution each time. The organic phase is dried over
sodium sulfate and evaporated. The residue which remains
is crysta~lized with acetonitrile and the crystals are
recrystallized from acetonitrile with the addition of
active charcoal~
Yield: 0.78 g of colorless crystals (73.9% of theory)
Melting point = 194 - 197C (CH3CN)
Example 17:
Methyl 2-((4-methoxy-3,5-dimethyl-2-pyridyl)-methyl-
sulfinyl)-3H-thieno(2,3-d)imidazole~5-carboxylate
(1: Rl = COOCH3, R2 and R6 = H, R3 and R4 = OCH3,
n = 1)
The abovementioned compound of the general formula
I is obtained by a procedure analogous to that described
in Example 16 by partial oxidation of methyl 2-((4-methoxy-
3,5-dimethyl-2-pyridyl)-methylthio)-3H-thieno(2,3-d)-
imidazole-5-carboxylate with perbenzoic acid in methylene
chloride as the solvent.
Yield: 72.5% of theory
Melting point = 179 - 179.5C (CH3CN)