Note: Descriptions are shown in the official language in which they were submitted.
663s~ llog-7281D
This Application is a Divisional Application from
Application 455,398, filed May 30th, 1984.
Background o~ the Invention
This invention relates to polymers including isopropenyl
benzyl amine compounds as monomers.
In Canadian Patent Application Serial No. 432,926,
filed July 21, 1983, there is disclosed a method for production
of tertiary aralkyl isocyanates, such as tetramethylxylylene diiso-
cyanates (TMXDI), by thermal cracking of corresponding urethanes
formed by addition to corresponding olefins of carbamic acid esters
at moderate temperatures and in the presence of acid catalyst.
Such process is particularly useful in producing the meta- and
para-isomers of TMXDI and as the by-product thereof, substantial
amounts of the corresponding meta~isopropenyl-~,~-dimethylbenzyl-
isocyanate (m-TMI) and para-isopropenyl~ dimethylbenzylisocyan-
ate (p-TMI), respectively~ are formed.
The m-TMI or p-TMI by-product in such systems may be
recycled within the process to improve the overall yield of~ ~ DI
therein, but has substantial utility per se as a separate product
due to its difunctLonal character, viz., the presence of reactive
isocyanato (-NCO) and isopropenyl groups.
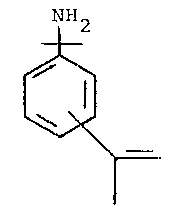
The present invention is directed to polymers of com-
pounds which are derivable from TMI, specifically to meta- or
;~ para- compounds of the formula:
+ 2
~ .
"; '''-~'" "' : ' '
: ` ~
'
- 2 ~ 635~ 1109-7281D
These monomers are the subject of Application 455,398,
which also discloses acid salts of these monomers, of the formula:
NH2-A
\\~
wherein A is a mineral acid or a carboxylic acid. Preferably A
represents acetic, nitric, or sulfuric acid.
Application 455,398 also discloses a method of making
meta~ or para-isopropenyl-~,~-dimethylbenzylamine, comprising:
(a) hydrolyzing the corresponding meta- or para-isomer of
isopropenyl-~,~-dimethylbenzylisocyanate in the presence of hydro-
chloric acid to form isopropenyl-~,~-dimethylbenzylamine hydro-
chloride; and
b) reacting the isopropenyl-~,~-dimethylbenzylamine hydro-
chloride with a stoichiometric excess of sodium hydroxide form the
isopropenyl-~,~-dimethylbenzylamine.
In this process the acid used in step (a) is preferably
at a strength of less than 20N.
An alternative method for making m-isopropenyl-~,~-
dimethylbenzylamine also disclosed in Application 455,398 comprises:
(a)~ heating a first solution of m-isopropenyl-~,~-dimethyl-
~20 benzylisocyanate in methanol for sufficient time to form methyl
N-~3-isopropenyl-~,~-dimethylbenzyl)carbamate (TMU) in said first
solution;
(b) mixing sald first solution containing TMU with potassium
,: , .: -.
. ' .-- ' ' ' ' ''': '
_ 3 _ ~26~3s~ 1109-7281D
hydroxide at a molar ratio of TMU: potassium hydroxide of from
about 0.25 to about 0.50 and in sufficient amount of 2-methoxy-
ethanol to form a second solution therewith;
(c) heating said second solution for sufficient time and at
sufficient temperature to convert said TMU to m-isopropenyl-~,~-
dimethylbenzylamine; and
(d) recovering m-isopropenyl-~,~-dimethylbenzylamine.
In this process, step (a) preferably is conducted at the
boiling point of the solution, and for a period of about 2 to
about 5 hours.
This invention relates to poly m-isopropenyl-~,~-dimethyl-
benzylamine), poly(p-isopropenyl-~,~-dimethylbenzylamine), and
to copolymers of m- or p-isopropenyl-~,~-dimethylbenzylamine and
at least other ethylenically unsaturated comonomer.
m-Isopropenyl-~,~-dimethylbenzylamine and p-isopropenyl-
~,~-dimethylbenzylamine (hereinafter m-TMA and p-TMA, respectively)
can be polymerized to give dimeric or oligomeric cationic products,
and are copolymerizable with other ethylenically unsaturated co-
monomers to yield copolymers, and m-TMA and p-TMA are also homo-
polymerizable to form high molecular weight products. Meta- and
para-TM~ and their polymer products, as well as the acid salts
thereof have potential utility in sizing of paper, as flocculants
and in the formation of water-soluble cationic polymers for water
~ clarification and hair spray end-use applications.
-~; p-TMI, as for example is produced by the above-described
~ method disclosed in Canada Serial No. 432,926 is readily reacted
:
.: . ~.~ , . :
.. , .: . . -
.: ~
. .. .
.~: . .~ :
: . ::: ~ ~ ,
3a 12~6~0 1109~7281D
to form the hydrochloric acid salt of p-TMA by hydrolysis in a
reaction volume comprising p-TMI, dilute aqueous hydrochlorlc acid
and acetone. Such reaction mixture may be reacted at room tempera-
ture in the presence of agitation to form a resulting fine suspen-
sion of reaction product. Acetone may be evaporated from the
reaction product, following which filtration of the acetone-
depleted reaction product yields the crude acid salt, isopropenyl-
~::
,. ,.~ , ,
,, ~, . -,. ..
:, :. : ,
4 ~61~35lL~
-dimethylbenzylamine hydrochloride (TMA-HCl). From the
crude acid salt product, TMA-HCl may be recovered, as for
example by recrystallization from water. The free base
(TMA) then may be obtained by mixing the acid salt with a
stoichiometric excess of a strong base, such as sodium hy-
droxide, and extraction of the resulting reaction product
with a solvent for TMA, such as hexane.
Although the foregoing procedure may be employed
to prepare the acid salts and pure compounds of TMA, in
preferred practice such procedure is employed for the pre-
paration of the para acid salt and pure compound. An al-
ternative procedure, which is highly efficient in the prep-
aration of both isomers, and is the preferred preparation
method for meta-isomers of the TMA acid salt and pure TMA.
In the alternative procedure, TMU may be prepared
for example by heating a methanol solution of TMI as for
example Erom six to about twenty hours at reflex without
the presence of any catalyst. Alternatively, TMU may be
prepared as disclosed in Canada Patent Application Serial
No. 444,794~ filed January 6, 1984 in the names of B. Singh
et al., in accordance with which N-tertiary aralkyl ure-
thane esters are prepared by reaction of carbamic acid
esters of an alkyl or aralkyl alcohol and a tertiary ar-
alkyl diol. If the stoichiometric amount of the carbamateis limited in this reaction system, a selective conversion
of the diol to mono-urethane takes place.
A solution of TMU then may be hydrolyzed by
heating in the presence of base such as potassium hydroxide
in an alcohol solvent such as 2-methoxyethanol at the boil-
ing point of the solvent for a period of from about 1 to
about 3 hours. The boiled solution is then cooled and
mixed with methylene chloride to give a solution from which
the 2-methoxyethanol is extractec] using repeated treatmenls
with water ~ollowing this the methylene chloricle contain-
in~ product TMA may be dr;ed as ~or example with molecular
sieves or anhydrous potassium carbonate and distilled to
; recover TMA.
. ~ . . . . .
:' ~,..... ~ , ,", .:
. .
.. ..
:: . . :
-,: , . .
~rz6~ 35~
Polymers of TMA conveniently are prepared from polymers of
TMI which may be homopolymerized or oligomerized as de-
scribed in copending patent application Canada Serial No.
455,396, filed May 30, 1984, in the name of F. C. Schaefer
or TMA copolymers may be formed from TMI copolymers, the
latter being prepared for example as disclosed in copending
Canada Application Serial Nos. 455,399 and 455,400, filed
May 30, 1984, with the respective TMI homopolymers or
copolymers being hydrolyzed under acid catalyzed conditions
to convert the isocyanato groups in the TMI homo or co-
polymer to amino groups.
In such manner, poly(m-ixopropenyl-~ ~-dimethyl-
benzylamine), poly(p-isopropenyl-~,~-dimethylbenzylamine)
and copolymers of m- or p-isopropenyl-d,~-dimethylbenzyl-
amine and at least one other ethylenically unsaturatedcomonomer polymerizable therewith, may be formed.
The invention is more fully illustrated by the
following non-limiting examples hereinafter set forth,
wherein all parts and percentages are by weigh~-, un~ess
otherwise specified.
:
: :: : :
~ : :
::: :
:
::: ~ :
35~3
-- 6 --
EXAMPLE I
Polymers of the hydrochloric acid salt of m-TMA,
poly(m-TMA^HCl), were obtained by hydrolysis of poly(m-TMI)
by the following procedures:
~C:H2~ ~--CE~
0 12NHC1 ~ 2 - HC
A mixture of 43 cc. of 12N NCl (0.51 equiv.) and 400 cc. of
toluene was preheated to 60 and with vigorous stirring 170
g. of 60.5% solution of poly-m-TMI (0.41 equiv.) in toluene
was added in 75 min. The poly-m-TMI used in this experiment
had^ a molecular weight range of 900-22000, peaking at 2900
(GPC analysis) and showed 3.99 meq-NCO/g. GLC analysis show-
ed ~8% residual monomeric m-TMI. Carbon dioxide was evolved
smoothly, and a granular suspension of the polyamine salt
was produced. After another 90 minutes at 60-65, the mix-
ture was cooled and filtered. The granular, very slightly
caked solid was washed with toluene and with methyIene chlor-
ide and was air-dried to constant weight. There was no aque-
ous phase in the filtrate. The product (108 g.) contained
7.2% water and on a dry basis showed a C.E.Q. of 3.25; i.e.
3.25 meq. of amine per g. Total chloride was 4.31 meq/g.
The IR spectrum showed evidence for a minor amount of urea
structure, in line with the reduced amine content relative
to the in;tial isocyanate content of the polymer. However,
the principal cause of this decrease was removal of monomer-
ic m-TMi as m~TMA-HCl (soluble in CH2C12/toluene).
In several similar preparations, up to a 10-fold
excess of 12N NCl was used. Even at this level essentially
no polyamine hydrochloride dissolved although removal of
impurities which are water-soluble probably resulted. If
~; the reaction temperature was a~ove ~70C, the lower molecu-
.
. .
, .. .. .
~ 3
-- 7
lar weight polymers tended to form a gum~y mass which great-
ly impeded hydrolysis and separation and purification of the
desired product.
Poly-_-TMA-HCl was soluble in water to at least
the 10% level although with high molecular weight material
dissolution was quite slow. The T~ polymeric salt was rea-
dily soluble in wet acetone, which therefore could not be
used to wash out the residual hydrochloric acid.
EXAMPLE II
The polyamine base poly(TMA) was prepared by
treat~ent of the hydrochloride polymer obtained in Example II
in water with excess NaOH and extraction with toluene or meth-
ylene chloride. The extract was dried over solid K2CO3 and
evaporated to recover the polyamine as a syrup or glass, de-
pending on its molecular weight. A typical polyamine pre-
pared from 2900 peak M.W.-poly-m-TMI had 5.57% N (~3.98
meq/g.) and -NH2 content of 4.03 meq/g by conventional analy-
sis.
EXAMPLE III
p-TMI (40 g.) gave no apparent reaction when mix-
ed with 4N hydrochloric acid (75 cc) at room temperature.
Acetone (75 cc) was then added to viev some compatability
to the mixture and the mixture was stirred overnight. Dur-
ing this time the upper liqu-id phase disappeared, and a fine
suspension was produced. Filtration gave 9.0 g. of crude
p-TMA-HCL, mp ~220, and another 22 g, mp 216-218 was ob-
tained after most of the acetone was evaporated from the
filtrate. Recrystallization from water or acetonitrile gave
a material with mp, 223-225 at a ~80% yield. The mother
liquors contained a substantial amount of N,N'-bis(4-iso-
propenyl~ -dimethylbenzyl)urea.
EXAMPLE IV
p-TMA-HCl (9.5 g.) was added to 40 cc conc.~sul-
furic acid. A deep red color developed with a mild exo-
therm and hydrogen chloride was evolved. The reaction
~ mixture was heated on the steam bath for l hour, poured on
;~ ice, basified with sodium hydroxide, and extracted with
.
. : , , .................. , . ~ . ~ :
: ;
': ~ :, .: - :
~ 3
-- 8 --
CH2C12. The extract was dried with solid H20H and then
evaporated. Dilution of the residual syrup with hexane
caused separation of a small amount of a solid which proved
to be an aromatic sulfonate salt. Re-evaporation of the
hexane solution left 7.3 g. of syrup. There was essentially
no change in the infrared spectrum after distillation of
this product; the b.p. was ~ 195/0.5 min. The NMR spectrum
was indicative of an indane dimeric TMA structure shown be-
low. The mass spectrum confirmed the empirical formula,
and as chromatography showed 97.9 area % of this compound.
EXAMPLE V
The indane diamine
l-[4~ Amino-l-Methylethyl)phenyl]-6-(l-amino-l-meth
ethyl)-1,3,3-trimethylindane : _
~
NH2
.~
t NH2
of Example IV was also prepared by hydrolysis/dimerization
of p-TMI in 7G% H2S04 in a single step. ~-TMI (10 g) in 10
cc of toluene was added dropwise to 98 g. of preheated 70%
H2S04 at 60+5. After carbon dioxide evaluation had subsi-
ded, the mixture was heated at 80 for 90 minutes. It was
then cooled and poured on ice. This caused crystallization
of the diamine sulfate which was filtered and pumped as
dry as possible. The cold toluene/dil. H2S0~ filtrate was
determined to contain virtually no dissolved amine salt.
The collected amine salt (soluble in acetone or in water)
,.. .. ... ..
~ 5~
was dissolved in water and basified with NaOH. The precipi-
tated gum was extracted with CH2C12, the extract was dried
over K2CO3, and the product was recovered by evaporation of
the solvent. The infrared spectrum showed this to be equi-
valent to the diamine prepraed from p-TMI-HCl by the proce-
dure of Example IV.
EXAMPLE VI
In contrast to ~-TMIoHCl, the meta isomer of TMA
was difficult to prepare satisfactorily by hydrolysis of
_-TMI with hydrcchloric acid, since linear oligomeric pro-
ducts tended to predominate in the reaction product.
m-TMA base was therefore prepared by alkaline hy-
drolysis of methyl N-(3-isopropenyl-~,~-dimethylbenzyl)car-
bamate(m-TMU) as follows.
~ ~Ic CU3 ~O~ + ~2co~
2 0 m-TMU m-T~A
m-TMU was prepared by heating a methanol solution of _-TMI
overnight at reflux without catalyst. The pure compound
25 had a boiling point of 12610.5 mm., and is an extremely
viscous liquid which eventually crystallizes at room temper-
ature if stabilzied with t-butylcatechol. In the absence of
a stabilizer, m-TMI polymerizes slowly at room temperature,
e.g., 20% after one month at ambient conditions.
A solution of 250 g m-TMU (91%, ~ 1.0 mole) and
165 g 85% KOH (2.S moles) in 460 cc of 2-methoxyethanol
was heated at the boil for 2.5 hours. It then was cooled
and mixed with 500 cc of H2O and 700 cc of CH2C12. The
amine-containing CH2C12 phase was separated and washed ~e
peatedly with 500 cc portions of water until its volume re-
mained cons~ant, thus extracting residual 2-methoxyethanol.
.
.
:, ;: ; ;,, ~ i ~. ` ' .
'
" ' ;' `
3S~
-
- 10 -
The CH2C12 solution was then dried with K2C03 and distilled.
m-TMA was recovered in 93% purity, b.p. 106/2 mm. The
yield was 128 g, 73% of theory. A non-separable by-product
was found by capillary GLC which may have constituted an
S additional 2% yield of p-TMA. About 4-5% of unconverted _-
TMU was recovered, indicating that the hydrolysis might
have been conducted somewhat longer, to advantage. Two by~
products found in the higher boiling fractions proved to be
_-TMXDA from the slight amount of m-TMXDU in the starting
material and the transesterified urethane:
~ ~ X NHCOOCH2CH20CH3
~OJ
~ .
which would have been converted to _-TMA on longer hyd-roly-
sis.
EXAMPLE VI I I
1-[3-(1-amino-1-methylethyl~phenyl]-5-(1-amino-1-
methylethyl)-1,3,3-trimethylindane:
~ ~
NH2
~ . ~
~" ' '' ~ - " ' '`
i3~
- 11 -
was produced by dimerization of m-TMA according to the pro-
cedure set forth. Several experiments in which m-TMA oligo-
merization was studied lead to the conclusion that relative-
ly dilute acid (e.g. 37% HCl) yielded only linear oligomers
while more concentrated acid (e.g. 85% H2SO4) produced cy-
clized (indane) dimer even at room temperature.
A solution of 28.9 g of m-TMA (93%; ~170 meq. base)
in 150 cc. of hexane was chilled in ice and treated dropwise
with 8 cc 18 N H2SO4 with vigorous stirring. This resulted
in a creamy suspension of crystallized amine salt. To this
was added gradually 30 cc. of 36 N H2SO4. A red-violet
color developed at once and reaction was mildly exothermic,
but the temperature was held at 20-30 by ice-bath cooling.
the amine salt rapidly dissolved in the viscous acid phase.
After 15 minutes the reaction mixture was warmed to 50C
briefly to ensure completion of the reaction. This caused
degradation of the bright color to tan. After addition of
200 g. of ice, the mixture was basified with excess s.trong
NaOH $olution ane the hexane phase was separated. This was
dried over solid K~CO3, evaporated to a syrup (19.2 G.) and
analyzed by GLC which showed ~90 wt. % with less than 5 area
% impurities. The IR spectrum showed only this structure
present. Some hexane insoluble gum in the reaction mixture
largely dissolved on further extraction with CX2C12, leaving
a little flocculant insoluble material. The CH2C12 extract
yielded another 8.6 g. of syrup showing 72 wt % of the
dimeric cyclic indane but containing a significant amount
of much less volatile higher oligomers as well as minor
linear dimers. The total yield of the cyclized indane dimer
was approximately 86% based on starting real _-TMA (GLC an-
alysis).
~XAMPL~ V~II
; In a larger run carried out in the same manner as
Example VII, but with toluene instead of hexane, alkylation
of the solvent was a serious side reaction:
: :
.
'
- . ~ : , . ~ ,
.: .-, ;, ~ : ~
. . .... . .
,. ~., ~ , , .
~L2~i~35~
- 12 -
5 ~ ~> ~H3
N~2 . NH2
2- [ m~ amir,o-I-metnylethyl)phenyl]-
2-p- tolylpropane
Distillation of the crude product from 78 g of m-TMA gave a
71% yield of the above alkylation product in sev-eral frac-
tions. About 10% of the product was essentially non-vol-
atile polyamine. Fractionation of m-indanediamine from the
less volatile diamine was inefficient because of the very
high boiling points of these compounds. A fraction boiling
at 172-181 at 1 mm. contained 36% monoamine and 61% of the
cyclized indane dimer. The best product, b.p. 186/1 mm
was 96.8 area % m-indanediamine with 0.4 area % monoamine
and 1% each of three slightly higher boiling by-products.
On analysis, this material was determined to
contain 5.74 meq NH2/g.: calc~, 5.71. It was a very vis-
cous syrup at room temperature.
Successful dimerization of _-TMA in strong sul-
furic acid required initial conversion to amine salt (ei-
ther hydrochloride or sulfate) in less concentrated acid at
low temperature to avoid violent localized reaction. Pre-
ferably the amine salt then should be added to strong sul-
furic acid (e.g., 85%) to minimize formation of higher
oligomers under weaker acid conditions.
; 30 EXAMPLE IX
The monoamine shown in Example VIII was also
~prepared from m-TMI by the ~ollowing procedure.
; ~ A mixture of 200 cc. of`;36N H2S04~and lOO cc. of
heptane~was preheated to 65 and to it gradually was added
during a two-hour period a solution of 252 g. o~ m-TMI
(1.24~mole~ in 250 cc. of heptane. The reacti~on temperature
was held at 70-80 C;by gentle heating, and the rate of
~: :
~ ' , " ' . . ' ' . ' ':
:, , :, , ,.,. ' ;. . : ~, . :-
. . ,,, .,.. : .. .: ~ ~
: : ., - . .;. ;. . ~ .
- ~ . .
~ 35~
tion was controlled to keep evolution of carbon dioxide vi-
gorous. The reaction mixture was a viscous red/violet syrup
throughout this operation. After carbon dioxide evolution
had stopped, the mixture was allowed to cool and was then
basified (at 50-60C) by cautious addition of strong aque-
ous sodium hydroxide (8 moles). The heptane phase then was
separated and the aqueous phase re-extracted with a small
amount of additional heptane. The combined heptane solution
was dried with solid K2CO3 and evaporated to obtain 219.5
g. of crude amine (theory, 217 g).
The crude indane diamine was distilled through a
simple Vigreaux apparatus, yielding 154 g., b.p. ~ 165C/5 mm.,
approx. 97 area % pure by GLC (71% yield). Approximately
23 g. (11% yield) of much higher boiling material was also
obtained, presumably a trimer mixture of at least four prin-
cipal components (GLC analysis), b.p. 220-230l0.5 mm.,
amine equivalent wt. 207 (theory 175). 13C-NMR shows indane
structure in this fraction at a level of about one indane
unit to four phenyl groups. Additional less volatile pro-
duct was obtained as a distillation residue. Two minor com-
ponents appeared to be chromatographically identical to the
linear dimers obtained in 12 N HCl in Example X.
EXAMPLE X
Attempts to hydrolyze _-TMI to m-TMA-`Hcl using 12
N HCl in acetone (at 25C) or toluene (65C) did ~ot give a
crystalline salt as with the ~-isomer. Apparently the hydro-
chloride which remained dissolved in the acid solution or
aqueous phase underwent some degree of oligomerization. In
the 12 N HCl/acetone system about 15% could be recovered as
m-TMA by basification after 20 hours at room temperature.
Another 15% of the TMI was converted to l-(l-amino~l-methyl-
ethyl)-3(1-hydroxy-1-methylethyl~benzene,
.
" '-',
:. .
- ~., ~ ' , i
i35
- 14 -
(b.p. 150C/1 mm, f.p. 86C). GLC analysis of the undis-
tilled higher boiling residue showed no indane diamine of
the type shown in Example VII.
The 12 N HCl/toluene system at 65C (90 min.)
gave slight amounts of _-TMA and hydroxyamine with a trace
of III. About half of the base produced was distillable at
230-380C/l mm., apparently a mixture of the same linear
dimers and trimers as obtained at lower temperature. No
indication of indane structure was found by 13C-NMR.
EXAMPLE XI
A copolymer of TMA and ~-methylstyrene was formed
by hydrolysis of a 70 mole % m-TMI/30 mole % ff-methylstyrene
copolymer by the following procedure.
Sixty-five grams of 70 mole % m-TMI/30 mole %
o~-methylstyrene copolymer were dissolved in 200 g. of anhy-
drous toluene and placed in a dropping funnel. During a
ten minute period, the copolymer solution was added to 215
ml. of 12N hydrochloric acid vigorously stirred in an 85C
; ~ oil bath. Precipitation of white solid produced a thick
mixture, which was stirred for an additional hour at 90-
95C. and then cooled to room temperature.
The product was filtere~d to obtain finely-divided
white solid. After drying at 25C/100 Mm Hg, the solid
weighed 125.3 g. ~ `
Sixty grams of the solid was shaken with a~mixture
of 100 ml. of water, 250 ml. of methylene chloride, and
38.4 g. of 33.3% sodium~hydroxide solution~ The solid
dissolved to form a cloudy solut~ion of the polyamine in
methylene chloride. The remaining 65 g. of solid was sim-
:
.:
. ~ ., . , .--
. : . . :
. , ~,,, : ,
, ... . . ..
635~
ilarly treated.
The two methylene chloride solutions were com-
bined, the solvent was evaporated under vacuum, and the
residue was dried at 60C/ 1 mm Hg to g:ive 48.2 g. of off-
white solid. The infrared spectrum of the product wasconsistent with the expected m-TMA/G~-methylstyrene co-
polymer structure.
~ .
- - .
'`' ~ "' ~. ' `
.,. :.
.. :.
.