Note: Descriptions are shown in the official language in which they were submitted.
6-OXYGENATED-1,3,4,5-TETRAHYDROBENZ~ cd~ INDOL-4-AMINES
This invention relates to new 6-oxygenated-
1,3,4,5-tetrahydrobenz~cd]indol-4~amines, to pharma-
ceutical compositions containing these compounds and to
methods of modulating serotonergic activity by
administering them. The invention also relates to a
process for preparing ~hese compounds and to indol-4-one
intermediates.
The compounds of this invention are selective
serotonin 15-HT) receptor antagonists. They bind
selectively to 5-HTl (vs 5-HT2) serotonin receptors.
~; Such selective 5-HTl receptor antagonists are useful, for
example~ in cardiovascular, gastrointestinal and renal
;~ disease s~ates, as well as in the treatment of other
2~ disorders, such as depression, in which serotonin
modulation is important. The compounds have
`~ antihypertensive activity.
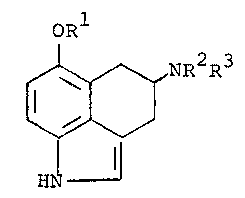
The 6-oxygenated-1,3,4,5-tetrahydrobenz[cd]indol-
4-amine compounds of this invention are represented by the
following formula (I):
QRl 2 3
: ~ NR R
HN
~ ` ~
~: `
::
~`'`
~' '`~ : ' '~ ' '
,` in which.
l is hydrogen or Cl 5 al'~yl; and
R2 and R3, being the same or different, are
hydrogen, Cl ~ alkyl, phenyl(CH2)n where n is 0-3,
C5 ~ cycloalkyl or C3 5 alkenyl;
or a pharmaceutically acceptable acid addition salt thereof.
Particular compounds of formula (I) are those in
which Rl is hydrogen or methyl.
Other particular compounds of for~ula (I) are
those in which R2 and R3 are both hydrogen or are both
methyl.
Specific compounds of formula (I) are:
6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4 amine
6-hydroxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine
N,N-dimethyl-6-methoxy-1,3,4,5-tetrahydrobenz-
[cd]indol-4-amine.
The compounds of formula (I) are prepared by the
following procedures.
`:~
~` 20 ~ethod A
. O,R . oRl
NH2
HN HN
(II)
,;
~ 30
. `i.~ 1
R is de~ined as in formula (I).
According to the procedure of Method A, a 4-nitro-
1,3,4,5-tetrahydrobenz[cd]indole (II) is hydrogenated using
a noble ~etal catalyst such as palladium, or prererably,
platinum oxide.
:
~ ~ ,
: : .. : .- '. . :'
;' ' ' ~:
.., ::
;7~`
3 - _~
Method B
.
R4 ~R4
~0 ~\~ NR2R3
W ~
H HN
(III)
O~I
¢~,NR~R3
R4 is Cl 5 alkyl and R2 and R3 are as defined in
formula (I).
. The first step in Method B, which is a reductive
amination method, is also included in this invention.
-~ The reductive amination is carried out with sodium
cyanoborohydride and an amine of the formula:
R2R3NH
:~ and an inorganic acid addition salt, such as a
hydrochloride salt, of said amine~ The process is
preferably carried out with the inorganic acid addition
~ 30 salt of the amine and the basic amine present in a ratio of
:~ about 10:1 to 10:3.
According to the second step of Method B,
compounds of ormula (I) where ~ is hydrogen are
~;~ prepared by dealkylating the 6-alkoxy compounds using, for
~; 35 example, boron tribromide, hydrobromic acid or pyridine
.~ hydrochloride.
: ,.
:
-
. :
~ -
. .
. .
.
-- 4
The compouncls of formula (I) where R2 and R3
are other than hydrogen may be prepared by alkylation of
the unsubstituted amino compounds when Rl is Cl 5
alkyl. However, these compounds, particularly where R2
and R3 are both other than hydrogen, are preferably
prepared by the reductive amination process of Method B.
The secondary amine compounds of formula (I),
i.e. where only one of R2 or R3 is other than hydrogen,
may be prepared by acylating the primary amine with an
acylating agent such as an acid halide, anhydride or
activated ester such as a p-nitrophenyl or dinitro-
phenyl ester, and then reducing, for example with an alkali
metal hydride such as lithium (or sodium) aluminum hydride,
or other reducing agent such as borane or borane/
tetrahydrofuran or borane/dimethylsulfide, to give the
N-substituted amines of formula (I).
The compounds of formula (I) form pharmaceutically
acceptable acid addition salts with organic or inorganic
acids. Examples of these acids are hydrochloric,
hydrobromic, sulfuric, phosphoric, acetic, tartaric,
citric, maleic, lactic, oxalic, succinic, methanesulfonic,
and benzenesulfonic acids. The salts are formed according
to methods known to the art. If the product is isolated as
acid addition salt, it may be treated with base such as
aqueous sodium hydroxide and converted to the corresponding
base. The base can then be treated with an appropriate
acid for example in an aqueous miscible solvent, such as a
lower alkanol preferably methanol or ethanol, to give the
desired salt.
~ 35
; :`
.
,~
' ' . ',:
. - 5 -
The indol-4~one intermediates of this invention
are represented by the following formula (IV):
R5
. 5 ~ o
(IV)
HN~
in which R5 is hydrogen or Cl 5 alkoxy.
The compounds of formula (IV) in which R5 is
Cl 5 alkoxy are intermediates of formula (III) in
Method B. Those compounds in which R5 is hydrogen are
: intermediates, for example, for the preparation of the
NIN-dipropyl-1,3,4,~-tetrahydrobenz~cd]indol-4-amine of
~ U.S. 4,110,339 by reductive amination.
:~ The 4-nitro-tetrahydrobenz[cd]indole
intermediates of formula (II) in Method ~ are prepared by
the following process:
~:
-:
,~
. :~
: :` :
. ;~
~ :,
. ~.
~: ~
. .',:
~: ~ . . . , . ; .
... .
-,
: ~ ' .
6 -- -~
OH ~ ~
H COOR COOR COOH
1 1
CuCN
\
~R4 R4 loR4
~HO ~ CE=NNHCONH2 ~,~N
HN HN HN
:'' \~,
~ R~ oR4 oR4
~ H=CHN02 ~H2CH2N02 ~[~3,No2
:~ 25 /
:` QH OR
~-N2 < ~`J2
; ` '
R is methyl or ethyl and R4 is Cl 5 alkyl. This
process is illustrated in examples 1 and 2.
., .:, .
; .,
,. . .
.
,
,. ..
The 4-keto intermediates of formula (III) are
prèpared by treating the 4-nitro compounds (~ormula II
where R is Cl 5 alkyl) with solid sodium methoxide in
methanol, then with aqueous titanium trichloride. This
process is illustrated in example 3 herebelow.
I The selective 5-HTl receptor antagonist
activity of the compounds of formula (~) is demonstrated by
the displacement of [ H~5-HT from rat frontal cerebral
cortex preferably over the displacement of
[3H]spiroperidol which binds preferentially with 5-H~2
receptors. Peroutka et al, Molecular Pharmacology
16:687-699 ~1979) describe this test procedure and
designate as 5-HTl and 5-HT2 those serotonin receptors
that are labeled by ~3H]5-HT and [3H]spiroperidol,
respectively. The XC50 (concentration required to
displace 50~ of [3~]5-HT binding from rat frontal
cerebral cortex) for compounds of examples 1-3 is about
40-7OnM.
;~ Also, selective 5-HTl receptor antagonist
activity of the compounds of formula (I) may be shown by
the ability to antagonize the effect of serotonin in an
in vitro dog basilar artery preparation, Peroutka et al,
Braln Research 259:327-330 (1983). The compound of
example 1 herebelow showed selective 5-HTl activity at
KBY4.63 x lO 7M.
~; Inhibition of the activity of serotonin (i.e.
5-hydroxytryptamine) is an activity known to the art; in
;~ U.S. 4,435,405, certain quinoline compounds are described
as 5-hydroxytryptamine antagonists which demonstrated
binding a~ 5-HTl and 5-HT2 receptors.
Antihypertensive activity of compounds of formula
`~ (I) is demons~rated by administration ~o spontaneously
hypertensive rats (S~R) at doses of about 0.4-l.O mg/kg,
i.v. In this test procedure, diastolic blood pressure and
heart rate are reduced.
~:`
,. . ... .
.: ~ . ,,........ ~ .
: - . , , ~.
.
-
8 -
This invention also includes pharmaceutical
compositions having 5-HTl receptor antagonist activity
comprising a compound of formula (I) or a pharmaceutically
I acceptable acid addition salt thereof and a pharmaceutically
acceptable carrier.
The pharmacologically active compounds of formula
(I) can be administered orally or parenterally.
Preferably, these compounds are administered in
conventional dosage unit forms prepared by combining an
appropriate dose of the compound with standard
pharmaceutical carriers. The dosage units will contain the
active ingredient in an effective amount selected from
about 0.01 mg. to about 500 mg., preferably 1 mg. to 50 mg.
; The pharmaceutical carrier employed may be, for
example, either a solid or li~uid. Exemplary of solid
carriers are lactose, terra alba, sucrose, talc, gelatin,
agar, pectin, acacia, magnesium stearate, stearic acid and
the like. Exemplary of liquid carriers are syrup, peanut
oil, olive oil, water and the like. Similarly, the carrier
or diluent can include any time delay material well known
to the art r such as glyceryl monostearate or glyceryl
distearate alone or with a wax
A wide variety o pharmaceutical forms can be
employed. Thus, if a solid carrier is used the preparation
can be tableted, placed in hard gelatin capsule in powder
or pellet form or in the form of a trouche or lozenge. The
amount of solid carrier will vary widely but preferably
will be from about 25 mgO ~o about 1 g. If a li~uid
carrier is used, the preparation will be in the form or a
; 30 syrup, emulsion, soft gelatin capsule, sterile injectable
liquid such as an ampul or an aqueous or nonaqueous liquid
suspension.
'~
,
.
'
- .
The pharmaceutical compositions are prepared by
conventional techniques involving procedures such as
mixing, granulating and compressing when`necessary or
variously mixing and dissolving the lngredients as
appropriate to the desired composition.
The method of inhibiting the action o serotonin
at the S-HTl receptors in accordance with this invention
comprises administering internally to a subject in need of
said activity a compound of formula (I) or a
pharmaceutically acceptable acid addition salt thereof, in
an amount sufficient to produce said activity.
Also, included in this invention is a method of
producing antihypertensive activity which comprises
administering internally to a subject in need of said
activity a compound of formula (I) or a pharmaceutically
acceptable acid addition salt thereof, in an amount
sufficient to produce said activity.
The compound will preferably be administered in a
- dosage unit form orally or parenterally. Advantageously
equal doses will be administered one to four times daily
with the daily dosage regimen being from about 1 mg. to
about 250 mg~, preferably from 1 mg. to 10 mg. When the
method described above is carried out, the action of
serotonin at the 5-HTl receptors is inhibited.
; 25 One skilled in the art will recognize that in
determining the amounts of the compound needed to produce
the desired pharmacological effect without toxic side
effects, the activity of the particular compound as well as
the size of the host animal must be considered.
3Q The following examples illustrate the invention
but are not to be construed as limiting the scope thereof.
Temperatures are in degrees Centigrade unless otherwise
stated.
.
'~`
~' .
:
~ ;.
: ~,
` ' ~ I - . .. :
.:
j, -- 10 --
}
A mixture of ethyl 5-methoxy-(lH)indole-2-
carboxylate (186 g, 0.85 mol) and glacial acetic acid
(4.2 L) was stirred and warmed on a hot plate until only a
small amount of indole remained undissolved. The warm
solution was stirred vigorously during the dropwise
addition (15 min) of bromine (43.4 mL, 135.4 g, 0.85 mol),
then allowed to stand at ambient temperature for 24 hours.
The resulting mixture was filtered, the crystalline product
was washed sequentially with acetic acid (2 x 150 mL),
hexane (2 x 200 m~), and then dried at 65C to give ethyl
4-bromo-5-methoxy-(lH)indole-2-carboxylate m.p. 176-178C
(softens at 166C).
A 5-L flask was charged with sodium hydroxide
(60 g, 1.5 mol) and water (0.5 L), and the contents were
stirred until homogeneous. Ethanol (l L), tetrahydrofuran
(0.25 L), and ethyl 4-bromo-5 methoxy-(lH)indole-2-
carboxylate (180 g, 0.6 mol) were added and the mixture was
heated at re1ux for l hour, then cooled to ambient
temperature. The solution was stirred vigorously as
concentrated hydrochloric acid (0.25 L, 3 mol) and water
(2.5 L) were added. The resulting thick yellow paste was
cooled at 0C overnight, filtered/ and the crystalline
product was washed with water (3 x 400 mL), and dried at
60C to give 4-bromo-5-methoxy-(lH)indole-2-carboxylic acid
(161 g, 99.3%), m.p. 263C(d).
A l-L flask was charged with freshly distilled
N,N'-dimethylacetamide (450 mL) and the solvent was
degassed (15 min) with a vigorous stream of argon.
4-Bromo-5-methoxy-~lH)indole-2-carboxylic acid (75 g/ 0.28
~ mol) and copper (I) cyanide (75 g, 0.84 mole) were added,
;~ the solution was stirred and heated at reflux for 28 hours,
cooled to ambient temperature, and poured into a mixture or
water (500 mL) and ethyl acetate (500 mL). The mixture was
~ 35 filtered through diatomaceous earth, the precipitate was
.: `~
'
- -
'~
' `' ' '
.
6~
- 11 - .
~ washed with ethyl acetate (250 mL), the combined ethyl
; acetate ~iltrates were separated from the aqueous layer,
and washed with water (5 x 250 mL) and brine. The solution
was dried and concentrated, and the residue was
recrystallized from ethyl acetate/hexane to give
5-methoxy-(lH)indole-2-carbonitrile (21 g, 44~), m.p.
139-141 C.
A 450-mL Parr hydrogenation bottle was charged
with sodium acetate (12 g, 0.146 mol), semicarbazide
hydrochloride (16 g, 0.144 mol) and water (45 mL), and the
~ mixture was heated u~til homogeneous. A heaping tablespoon
l~ of Raney nickel catalyst was added to the cooled Parr
j bottle followed by a solution of ~-methoxy-(lH)indole-4-
carbonitrile (20 g, 0.116 mol) in hot methanol (185 mL).
1 15 The mixture was hydrogenated at 50 p5i until the
theoretical uptake of hydrogen was complete (18-24 hours).
The mixture was heated to near boiling on a steam bath,
filtered hot, and the precipitate was washed with hot
N,N-dimethylformamide until the catalyst was free o
product. The filtrate was concentrated and traces of
I solvent were removed from the residue by further
¦ ~ concentration using a dry ice cold trap to ~ive a solid
residue. The residue was triturated with a mixture of
~ ethanol (60 mL) and water (180 mL), filtered, washed with
i` ~ 25 water and dried at 60C to yield 5-methoxy-(lH)indole-4-
carboxaldehyde semicarbazone (18.89 g, 70%), m.p.
219-221C(d).
A mixture of water (0.46 L), glacial acetic acid
(0.91 L), freshly distilled pyruvic acid (33.23 g, 0.378
mol), and anhydrous sodium acetate (3~.16 g, 0.416 mol) was
stirred un~il homogeneous, then 5-methoxy (lH)indole-
4-carboxaldehyde semicarbazone (17.5 g, 0.0754 mol) was
added and the mixture was stirred for 16 hours. The
~; resulting solution was concentrated with a dry-ice cold
trap (water bath temperature ~25C), the residue was taken
, .
.
,~
,
`:
6~
_,
- 12 -
up in ethyl acetate (0.5 L) and filtered, and the filtrate
was washed sequentially with water (2 x 250 mL), 5% aqueous
sodium carbonate (3 x 250 mL), brine, dried, and warmed
briefly with decolorizing charcoal before concentration.
The crude product was dissolved in a little ethyl acetate
and applied to a silica gel (2S0 g) column; elution witn
2:1 ethyl acetate~hexane and concentration of the first
1.0 L of eluate gave 5-methoxy-(lH)indole-4-carboxaldehyde
(9.8 g, 74.2%), mOp. 134-136C (softens at 125C).
A solution of 5-methoxy-(lH)indole-4-
carboxaldehyde (8.61 g, 49.2 mmol) and ammonium acetate
(l S g) in nitromethane (70 mL) was heated on a steam bath
for 3 hours, and diluted with ethyl acetate (150 mL). The
solution was washed twice with water, once with brine,
dried, and concentrated. The solid product was
recrystallized from ethyl acetate/hexane. The
crystallization filtrates were concentrated, purified by
1ash chromatography using 1:1 hexane-ethyl acetate as
eluant, recrystallized from ethyl acetate/hexane, and
combined with the first crop of product to give
5-methoxy-4-(2-nitroethenyl)-(lH)indole (9.15 9, 85%), m.p.
175-176C.
A 2-L flask was charged with methanol (0.42 L) and
,
S-methoxy-4-(2-nitroethenyl)-(lH)indole (9.1 g, 41.7 mmol),
and the resulting mixture was stirred during the slow (20
min) addi~ion of sodium borohydride (7.0 g, 0.185 mol).
During the addition, vigorous hydrogen evolution occurred
and the mixture became warm. The solution was stirred for
20 minutes after the addition was completed, g~acial acetic
acid (15 mL) was added to a pH of 6, and the solution was
concentrated. The solid residue was partitioned between
water and ethyl acetate. The aqueous layer was washed
twice with ethyl acetate and the combined ethyl acetate
phases were washed with water, twice with 10~ aqueous
3S sodium carbonate, brine, then dried, and concentrated. The
. ~
~; .
~' .
'' , '
~i6~
13 -
resulting oil was purified by flash chromatograph~ using
40~ ethyl acetate~hexane as eluant to give S-methox~-4-~2-
nitroethyl)-(lH)indole (8.38 g, 91%), m.p. 77-79C.
N,N-Dimethyl~ormamide (20 mL) was stirred under
argon in a 500 mL flask during the addition (over 2
minutes) of phosphorus oxychloride (3.45 mL, 37.1 mmol),
then the flask was cooled in a cold water bath during the
addition (over 2 minutes) of 5-methoxy-4-(2-nitroethyl)-
(lH)indole (7.15 g, 32.5 mmol). After the addition was
completed, the cooling bath was removed, stirring was
continued for 15 min at ambient temperature, a mixture of
water (6.5 mL, 0.36 mol) and N,N-dimethylformamide (26 mL)
was added, and the resulting solution was stirred for 10
min. A mixture of triethylamine (45.5 mL, 0.33 mol) and
methanol (130 mL) was added, the solution was heated at
reflux for 30 min, water (75 mL) was added slowly ~o the
hot solution and the mixture was cooled at 20C for 2
hours. The mixture was filtered and the precipitate was
washed sequentially with water, 2:1 methanol-water
20 (2 x 25 mL), ether (3 x 50 mL), and then dried at 60C to
yield 6-methoxy-4-nitro-1,5-dihydrobenz[cd]indole (6.8 g,
91%), m.p. 350C(d).
A mixture of 6-methoxy-4-nitro~1,5-dihydro-
benz[cd~indole (7.26 g, 31 6 mmol) and methanol (0.5 L) was
stirred vigorously in a 2-L flask during the slow (20 min)
addition of sodium borohydride (18.25 g, 0.48 mol). During
the addition, vigorous hydrogen evolution occurred and the
mixture became quite warm. After stirring an additional
10 min, glacial acetic acid (45 mL) was added to a final pH
of 5-6. The solution was concentrated, partitioned between
water and ethyl acetate, the aqueous layer was extracted
twice with ethyl acetate, and the combined oryanic layers
were washed sequentially with water, 5% aqueous sodium
carbonate, and brine. The solution was dried and
concentrated, and the solid residue was purified by flash
'; `
` ' .
, .:
:. . ~ ..
:. ., . .:
.. , :
. . .
~266~7S
- 14 -
` chromatography using 2:1 hexane-ethyl acetate as eluant to
give 6 methoxy-4-nitro 1,3,4,5-tetrahydrobenz[cd]indole
(6.5 g, 89~), m.p. 134-136C.
~, Platinum oxide (300 mg) was reduced under 60 psi
hydrogen pressure in methanol (30 mL) for 30 min.
6~Methoxy-4-nitro~1,3,4,5-tetrahydrobenz[cd]indole (1.0 g,
4.31 mmol) was added and the resulting solution was
hydrogenated under 60 psi hydrogen pressure until hydrogen
uptake ceased (about 2 houxs). The mixture was filtered,
the filtrate was concentrated, and the residue was
dissolved in chloroform and decolorized with activated
carbon. The resulting mixture was filtered and the
filtrate was concentrated. The residue, which contained
6-methoxy-4-nitro-1,3,4,5-tetrahydrobenz[cd]indole-4-amine,
lS was dissolved in absolute ethanol (8 mL) and the solution
was stirred and heated to near reflux as a solution of
oxalic acid dihydrate (540 mg, 4.31 mmol) in absolute
ethanol (S mL) was added slowly. The mixture was diluted
with acetone (10 mL), cooled at 0C, filtered, and the
precipitate was washed with acetone (2 x 10 mL), hot
methanol (2 x 10 mL), and dried to yield 6-methoxy-1,3,4,5-
tetrahydrobenæ[cd]indole-4-amine hemioxalate (0.74 g, 70%),
;~ m.p. 260-265C(d).
Exam~le 2
6-Methoxy-4-nitro-1,3,4,5-tetrahydrobenz[cd]indQle
(0.210 g) was dissolved in 30 ml of anhydrous methylene
chloride under nitrogen in a flame dried flask. The
mixture was cooled ~o 0C and 4 mL o 1 molar boron
~` tribromide-in methylene chloride was added. The mixture
was stirred at 0C or 6 hours. The reaction was quenched
with 5% aqueous sodium bicarbonate and extracted twice with
ethyl acetate. The organic extracts were dried and the
solvent removed. The residue was chromatographed over
silica using 60~40 hexane/ethyl acetate to give 6-hydroxy-
4-nitro-1,3,4,5-tetrahydrobenz[cd]indole.
~"
: "
: : .. :
:: .:::, ,
': . .':, ...
', ': ::
~L~6~7~
- 15 -
6~Hydroxy-4-nitro 1,3,4,5-tetrahydrobenz~cd]indole
I (52 mg) was dissolved in 10 mL of methanol. The solution
i was added to 50 mg of platinum oxide which had been reduced
with hydLogen at 50 psi in 10 mL of methanol for 3 hours,
then filtered under argon into a flask containing 21.5 mg
(1 equiv.) of oxalic acid in 2 mL of methanol. The
solution was concentrated to about 10 mL volume and 100 mL
of diethyl ether was added. After 1 hour at -20C, the
solid material was filtered, washed with diethyl ether and
dried to yield 31 mg of 6-hydroxy-1,3,4,5-tetrahydro-
benz[cd]indol-4-amine oxalate, m.p. 218-220C(d).
. .
Exam~le 3
A mixture of 6-methoxy-4-nitro-1,3,4,5-tetrahydro-
benz[cd]indole (1.16 g, 5 mmol) and methanol (25 mL) was
treated with solid sodium methoxide (0.3 g, 5.5 mmol) and
stirred under argon until homogeneous. A solution made by
mixing 20% aqueous titanium trichloride (17.5 mL) with
ammonium acetate (10.5 g) dissolved in water (35 mL) was
added, and the resulting suspension was stirred for 1 hour.
The mixture was shaken vigorously and aecanted with diethyl
ether (8 x 50 mL). The diethyl ether extracts were washed
se~uentially with water (3 x 100 mL), 5% aqueous sodium
carbonate (2 x 100 mL) and brine, then dried and
concentrated to give 6-methoxy-1,5-dihydrobenz[cd]indol-
; 25 4(3H)-one (0.82 g, 81~) m.p. 130-132C(d).
A solution of anhydrous dimethylamine (0.26 g,
5.76 mmol), dimethylamine hydrochloride (2.45 g, 30.0
mmol), and sodium cyanoborohydride (1.88 g, 30.0 mmol) in
methanol (30 mL) was prepared. 6-~ethoxy-1,5-dihydro-
benæCcd]indol-4(3H)-one (0.60 g, 3.0 mmol) in methanol (30
;~ mL) was added. The reaction mixture was stirred at ambient
~ temperature for 1 hour, then poured into 5~ aqueous sodium
;; bicarbonate solution (100 mL). The resulting mixture was
;~ extracted with ethyl acetate (3 x 50 mL) and the combined
organic extracts were washed with 5% a~ueous hydrochloric
:;`
:, ~
, ~
:
: . . .
,.:.
.
'~2~
- 16 -
acid (3 x 30 mL). The acidic extracts were made basic with
20% aqueous sodium hydroxide and extracted with ethyl
/, acetate (3 x 50 mL), the combined organic extracts were
washed with 10% aqueous sodium sulfate, dried and
concentrated to yield a product which was purified by 1ash
chromatography using 5% diisopropylamine-ethyl acetate as
eluant to give N,N-dimethyl-6-methoxy-1,3,4,5-
tetrahydrobenz[cd]indol-4-amine as an oil. This free base
was dissolved in methanol (lOmL) and treated with a
solution of oxalic acid (0.156 g, 1.73 mmol) in methanol
(5 mL). The crystalline product was collected and dried
(25~C, 0.1 torr) to yield N,N-dimethyl-6-methoxy-1,3,4,5-
tetrahydrobenz[cd]indol-4-amine oxalate.
Example 4
By the procedure o~ Example 3, using in place of
dimethylamine the following amines:
diethylamine
dipropylamine
dibutylamine
dibenzylamine
- dicyclohexylamine
diallylamine
aniline
: allylamine
the products are:
N,N-diethyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine
N,N-dipropyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine
: N,N-dibutyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amineN,N-dibenzyl-6-methoxy-1,3,4,5 tetrahydrobenz L cd]indol-4-amine
N,N-dicyclohexyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-
amine
N,N-diallyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine
N-phenyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine
N-allyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine
: ~;
.', .
::
~'
': '
.,, ~ .
, .
~æ~
- 17 -
Similarly, using N-butyl-~1-propylamine in place
of dimethylamine, the product is N-butyl-N-propyl-6-
methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine.
A mixture of 6-methoxy-1,3,4,5-tetrahydrobenz~cd]-
indol-4-amine (2.0 g, 0.01 mol), benzoyl chloride (2.1 g,
0.015 mol) and 10 ml of pyridine is stirred at room
temperature for 10 minutes, then poured into water.
Extracting with ethyl acètate and then concentrating the
extracts gives N-benzoyl-6 methoxy-1,3,4,5-tetrahydrobenz-
[cd]indol-4-amine as the eesidue.
One gram o~ this N-benzoyl compound in 20 ml of
tetrahydrofuran is added to one gram of lithium aluminum
hydride in 20 ml of tetrahydrofuran and the resulting
mixture is stirred at room temperature overnight, then
quenched with water, filtered and concentrated to give
N-benzyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine.
~y the same procedure, using 6-hydroxy-1,3,4,5-
tetrahydrobenz[cd]indol-4-amine in place of the 6-methoxy
; 20 compound, the product is N-benzyl-6-hydroxy-1,3,4,5-tetra-
~-~ hydrobenz[cd]indol-4-amine.
Example 6
A mixture of 6~methoxy-1,3,4,5-tetrahydrobenz-
~cd]indol-4 amine (2.0 g, 0.01 mol) and p-nitrophenyl
formate (1.7 g, 0.01 mol) in 25 ml of ethyl acetate and
10 ml of pyridine is stirred overnight at room temperature,
`~ then washed with water and concentrated to giYe
N-formyl-6-methoxy-1,3,4,5-tetrahydrobenz~cd]indol-4-amine.
The above prepared N-formyl compound is reduced
with lithium aluminum hydride in tetrahydrofuran and the
resulting mixture is worked up by the procedure of
Example 5 to give N-methyl-6-methoxy-1,3,4,5-tetrahydro-
benz~cd]indol-4-amine.
'
: ::
.
' ~ ' ` . -,.
.. .. , ~ .
~, ~ . `: . - .
.
:
6~
18 -
.
By the same procedure, using 0.01 mol of acetic
anhydride ln ethyl acetate, the product is N-acetyl-6-
methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine. Reducing
with lithium aluminum hydride in tetrahydrofuran by the
procedure of Example 5 gives N-ethyl-6 methoxy-1,3,4,5-
tetrahydrob-enz[cd]indol-4-amine.
Using 6-hydroxy-1,3,4,5-tetrahydrobenz[cd~indol-
4-amine in place of the corresponding 6-methoxy compound in
the above procedures~ the products are:
N-methyl-6-hydroxy-1,3,4,5 tetrahydrobenz[cd]indol-
4-amine and N-ethyl-6-hydroxy-1,3,4,5-tetrahydrobsnz[cd]-
indol-4-amine.
Exarnple 7
By the procedure of Example 2, the following
6-methoxy compounds of Example 4 are reacted with boron
tribromide:
N,N-dimethyl-6-methox~-1,3,4,5-tetrahydrobenz[cd]indol-4-amine
N,N-diethyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4 amine
N,N-dibenzyl-6-methoxy-1,3,4,5-tetrahydrobenz[cd]indol-4-amine
N,N~diallyl-6-methoxy-1,3,4,~-tetrahydxobenz[cd]indol-4-amine
N-phenyl-6-methoxy-1~3,4,5-tetrahydrobenz~cd]indol-4-amine
to give the corresponding 6-hydroxy compounds.
~ .
. ~
6-Methoxy-1,3,4,5-tetrahydrobenz[cdlindol-4-amine
~S (50 mg) i9 mixed with 100 mg of lactose and 3 mg of
magnesium stearate. The resulting mixture is filled into a
hard gelatin oapsule.
:
~ .
:,,
~: '
~';
'
'
,,
.. ..
.
, . ., ,
: ~ :,. :
~: .
.