Note: Descriptions are shown in the official language in which they were submitted.
9~
X-6711A -1-
4-SUBSTITUTED DIAZOLIDINONES
This invention is directed to 4-substituted
diazolidinone compounds which are intermediates for
7-substituted bicyclic pyrazolidinone antimicrobials.
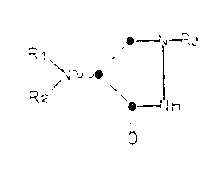
The present invention embraces compounds of
the Formu~a I:
R1 , _~-R3
1 0 ~N~V~
R2 ~9 2 IH
wherein R1, R2 and R3 are as defined below.
The ring system of the compound of Formula I
is a 4-(substituted amino)-3-oxo-1,2-diazolidine, which
for brevity's sake will be referred to as a "diazoli-
dinone" compound. In the above Formula I, the undulat-
ing line between position 4 of the diazolidinone ring
and the protected amino group indicates that the instant
diazolidinone compounds exist either as a mixture of
varying proportions of enantiomers or as the pure
4~(R) or the pure 4-(S) enantiomer.
In the above Formula I, R1 and R2 are
a) taken together to form a phthalimido
group; or
b) either R1 or R2 is hydrogen and -the other
of R1 or R2 is an amino-protecting group;
or an acid-addition salt thereof.
R3 in the above Formula I is either hydrogen
or trifluoroacetyl.
i~i9~U~i
X-6711A -2-
The terms "amino-protecting group" and "pro-
tected amino" as used in the specification refer to
substituents of the amino group commonly employed to
block or protect the amino functionality while carrying
out reactions at other functional groups on the compound.
Examples of such amino protecting groups include the
formyl group, the trityl group, the phthalimido group,
the trichloroacetyl group, the chloroacetyl, bromoacetyl
and iodoacetyl groups, urethane-type blocking groups
such as benzyloxycarbonyl, 4-phenylbenzyloxycarbonyl,
2-methylbenz.yloxycarbonyl, 4-methoxybenzyloxycarbonyl,
4-fluorobenzyloxycarbonyl, 4-chlorobenzyloxycarbonyl,
3-chlorobenzyloxycarbonyl, 2-chlorobenzyloxycarbonyl,
2,4-dichlorobenzyloxycarbonyl, 4-bromobenzyloxyca.rbonyl,
3-bromobenzyloxycarbonyl, 4-nitrobenzyloxycarbonyl,
4-cyanobenzyloxycarbonyl, 2-(4-xenyl)iso-propoxycar-
bonyl, 1,1-diphenyleth-1-yloxycarbonyl, 1,1-diphenyl-
prop-1-yloxycarbonyl, 2-phenylprop-2-yloxycarbonyl,
2-(p-toluyl)prop-2-yloxycarbonyl, cyclopentanyloxy-
carbonyl, l-methylcyclopentanyloxycarbonyl, cyclo-
hexanyloxycarbonyl, l-methylcychexanyloxycarbonyl,
2-methylcyclohexanyloxycarbonyl, 2-(4-toluylsulfonyl)-
ethoxycarbonyl, 2-(methylsulfonyl)ethoxycarbonyl,
2-(triphenylphosphino)ethoxycarbonyl, 9-fluorenyl-
methoxycarbonyl ("FMOC"), 2-(trimethylsilyl)ethoxy-
carbonyl, allyloxycarbonyl, l-(trimethylsilylmethyl)-
prop-l-enyloxycarbonyl, 5-benzisoxalylmethoxycarbonyl,
4-acetoxybenzyloxycarbonyl, 2,2,2-trichloroethoxycar-
bonyl, 2-ethynyl-2-propoxycarbonyl, cyclopropylmethoxy-
carbonyl, 4-(decyloxy)benzyloxycarbonyl, isobornyloxy-
carbonyl, 1-piperidyloxycarbonyl and the like, the
~i9.~0
X-6711A -3-
benzoylmethylsulfonyl group, the 2-(nitro)phenylsul-
fenyl group, the diphenylphosphine oxide group and like
amino protecting groups. The species of amino-protect-
ing group employed is not critical so long as the deriva-
tized amino group is stable to the condition of subse-
quent reaction(s) on other positions of the diaæolidinone
molecule and can be removed at the appropriate point in
the synthesis of 7~substituted bicyclic pyraæolidinone
antimicrobials without disrupting the remainder of the
molecule.
In particular, it is important not to subject
the amino-substituted diazolidinone molecule (wherein
R3 is hydrogen) to strong nucleophilic bases or reductive
conditions employing highly activated metal catalysts
such as Raney nickel.
Preferred amino-protecting groups are the
allyloxycarbonyl, the t-butoxycarbonyl and the trityl
groups. Similar amino-protecting groups used in the
cephalosporin, penicillin and peptide art are also em-
braced by the above terms. Further examples of groupsreferred to by the above terms are described by J.W.
Barton, "Protective Groups In Organic Chemistry", J.G.W.
McOmie, Ed., Plenum Press, New York, N.Y., 1973,
Chapter 2, and T.W. Greene, "Protective Groups in
Organic Synthesis", John Wiley and Sons, New Yor~, N.Y.,
19~1, Chapter 7.
The term "acid addition salt" encompasses
those salts formed by standard acid-base reactions with
amino groups and organic or inorganic acids. Such acids
include hydrochloric, sulfuric, phosphoric, acetic, suc-
cinic, citric, lactic, maleic, fumaric, palmitic,
i9~
X-671].A -4-
cholic, pamoic, mucic, D-glutamic, d-camphoric, glutaric,
phthalic, tartaric, lauric, stearic, salicyclic, d-10-
camphorsulfonic, methanesulfonic, benzenesulfonic,
para-toluenesulfonic, sorbic, picric, benzoic, cinnamic
and like acids.
The compounds of Formula I also embrace the
correspondlng crystalline solvates. Thus, diazoli-
dinones that crystallize with any number of (or any
fraction thereof) of molecules of the mother liquor
solvent are a part of the instant invention. The mother
li~uor solvent can be water or an organic solvent.
Examples of the compounds of Formula I include:
4-(R,S)-(t-butoxycarbonylamino)-3-oxo-1,2-
diazolidine,
4-(S)-(t-butoxycarbonylamino)-3-oxo-1,2-
diazolidine,
4-(R)-(t-butoxycarbonylamino)-3-oxo-1,2-
diazolidine,
4-(R,S)-(i-butoxycarbonylamino)-3-oxo-1,2-
diazolidine p-toluenesulfonate salt,
4-(S)-(allyloxycarbonylamino)-3-oxo-1,2-
diazolidine,
4-(S)-(trityiamino)-3-oxo-1,2-diazolidine,
4-(S)-(benzyloxycarbonylamino)-3-oxo-1,2-
diazolidine,
4-(R,S)-(allyloxycarbonylamino)~3-oxo-1,2-
diaæolidine d-10-camphorsulfonate salt,
4-~R,S)-(tritylamino)-3-oxo-1,2-diazolidine,
4-(R,S)-(trichloroacetylamino)-3-oxo-1,2-
diazolidine,
4-(R,S)-(benzolycarbonylamino)-3-oxo-1,2-
diazolidine,
910~3
X-6711A -5-
4-(R,S)-(chloroacetylamino)-3-oxo-1,2-
diazolidine,
4-(R,S)-(4-methoxybenzyloxycarbonylamino)-3-
oxo-1,2-diazolidine,
4-(R,S)-(cyclohexanoyloxycarbonylamino)-3-
oxo-1,2-diazolidine,
4-(R,S)-(4-nitrobenzyloxycarbonylamino)-3-
oxo-1,2-diazolidine,
4-(R,S)-(l,1-diphenylethoxycarbonylamino)-3-
oxo-1,2-diazolidine,
4-(R,S)-(2-(methylsulfonyl)ethyloxycarbonyl-
amino)-3-oxo-1,2-diazolidine,
4-(R,S)-(2-(trimethylsilyl)ethyloxycarbonyl-
amino)-3~oxo-1,2-diazolidine,
4-(R,S)-(2,2,2-trichloroethoxycarbonylamino)-
3-oxo-1,2-diazolidine,
4-(R,S)-(benzoylmethylsulfonylamino)-3-oxo-
1,2-diazolidine,
4-(R,S)-(t-butoxycarbonylamino) l-(trifluoro-
acetyl)-3-oxo-1,2-diazolidine,
4-(R,S)-(allyloxycarbonylamino)-1-(trifluoro-
acetyl)-3-oxo-1,2-diazolidine),
4-(R,S)-(trimethylsilylamino)-1-(trifluoro-
acetyl)-3-oxo-1,2-diazolidine),
4-(R,S)-(tritylamino)-1-trifluoroacetyl)-3-
oxo-1,2-diazolidine,
4-(R,S)-benzyloxycarbonylamino~-3-oxo-1,2-
diazolidine,
4-(R,S)-(trichloroacetylamino)-3-oxo-1,2-
diazolidine,
0~
X-6711A -6-
4-(S)-(t-butoxycarbonylamino)-l-(trifluoro-
acetyl)~3-oxo-1,2-diazolidine,
4-(S)-(allyloxycarbonylamino)-1-(trifluoro-
acetyl)-3-oxo-1,2-diazolidine,
4 (S)-(trimethylsilylamino)-1-~trifluoroacetyl)-
3-oxo-1,2-diazolidine, or
4-(S)-(tritylamino)-1-(trifluoroacetyl)-3-
oxo-1,2-diazolidine;
and the p-toluenesulfonic acid addition salt of the
above non-salt examples.
A preferred group of compounds of Formula I
is the p-toluenesulfonic acid addition salt. A second
preferred group of compounds of Formula I is when R1 or
R2 is hydrogen and the other is t-butoxycarbonyl, and
the corresponding p-toluenesulfonic acid addition salt.
A third preferred group of compounds occurs
when the C4 carbon of the diazolidinone ring is in the
S configuration. Preferred compounds with the third
preferred group have either R1 or R2 as hydrogen and
the other as t-butoxycarbonyl.
X-6711A -7-
The synthesis of enantiomeric mixtures of
diazolidinones of Formula I is outlined below in
Scheme I.
Scheme 1
CHaO f OH
NH-(t-Boc) protected serine
1) 1 TsCI
0~,
15 CH30/ ~/ O--Ts tosyl serine
NH-(t-~oc)
2 ) ~ NH2NH2
H ~ NH
(t-Boc)-N~,~a
race~ic
~ -NH diazolidine
The above Scheme depicts the synthesis of
4-(t-butoxycarbonylamino) diazolidinone compounds.
Diazolidinone compounds with different amino protecting
groups are obtained from serine derivatized with an
amino-protecting group other than t-butoxycarbonyl.
~i9:1~3~i
X-6711A -8-
The tosylate group (OTs) can be replaced by
any other appropriate leaving group such as mesylate.
Similarly, those skilled in the art will appreciate that
amino protecting groups other than -t-butoxycarbonyl
(t-Boc) can be utilized. The first step in the syn-
thesis o~ l-(unsubstituted)diazolidinones, represented
by Reaction 1 in the above Scheme, is the tosylization
of the hydroxy group of the protected serine derivative.
The tosylization is carried out in methylene chloride
with p-toluenesulfonyl chloride in the presence of a
catalytic amount of 4-dimethylaminopyridine and greater
than one equivalent of pyridine. The reaction mixture
is stirred at room temperature overnight.
The tosylated serine obtained is reacted with
97% hydrazine to give enantiomeric mixtures of 1-
(unsubstituted)diazolidinones, as depicted in Reaction 2.
Reaction 2 should be carried in polar solvents such as
chlorinated hydrocarbons, cyclic or acyclic ethers or
C1 to C~ alcohols. A preferred group of solvents for
the reaction is dichloromethane, methanol and chloro-
form, with dichloromethane being more preferred.
The temperature for Reaction 2 is not criti-
cal. It is preferred that the reaction be carried out
between about room temperature to about the freezing
temperature of the solvent. A more preferred tempera-
ture is approximately room temperature.
The reaction usually requires a period of
about one to about forty-eight hours. The optimal reac-
tion time can be determined by monitoring the progress
of the reaction by conventional means such as chromato-
graphic techni~ues (thin layer chromatography, high per-
91~3~.3
X-6711A -9-
formance liquid chromatography, or column chromatography)
and spectroscopic methods, alone or in conjuction with
chromatographic techniques, such as infrared spectros-
copy, nuclear magnetic resonance spectrometry and mass
spectrometry. A preferred time period is from between
about five to about sixteen hours.
The usual stoichiometry for Reaction 2 in the
above Scheme 1 is a 4:1 ratio of hydrazine to tosyl
serine reagent. Of course, a 1:1 ratio of reagents is
permissible. It is preferred that the hydrazine reagent
be present in excess, and especially preferred that the
hydrazine be present in a 4:1 excess. Furthermore, the
order of addition of either reagent is not critical.
The stereospecific synthesis of chiral dia-
lS zolidinones of Formula I is diagrammed below in Scheme 2.
9~
X-6711A -10~
Scheme 2
~ Protected serine
H0/ \~/ NH-NH~ acyl hydrazide
NH-(t-Boc)
l 3) ET-TFA
~ ~ g N-(Trifluoroacetyl)
H ~ 1 NH-NH CF3 acyl hydrazide
NH-(t-Boc)
l ~) DEAD, TPP
H ,~ ~ CFo Chiral
t-60cN~c~ -(Trifluoroacetyl)
~-NH diazolidine
l 5) hydroxide ion
t-BocN~ Chiral
~-NH
10~
X-6711A -ll-
The above Scheme depicts the sy~thesis of
4~(S)-(t-butoxycarbonylamino)diazolidine compounds.
Diazolidine compounds with the 4-(R) configuration are
synthesized by starting with the protected D-serine
acyl hydrazide instead of the L-isomer depicted above.
Chiral diazolidines with amino-protecting groups other
than t-butoxycarbonyl are synthesized starting with a
serine acyl hydrazide derivatized with an amino-protecting
group other than t-butoxycarbonyl.
The protected serine acyl hydrazide precursor
of Scheme 2 is synthesized in a procedure analogous
to B. Iselin and R. Schwyzer, Helv. Chim. Acta, 44, -
p 169 (1961). The precursor is acylated with the tri-
fluoroacetyl moiety, as set forth in Reaction 3 in the
Scheme. The acylation is carried out in ethanol with an
excess of ethylthio trifluorothioacetate ("ET-TFA").
The reaction mixture is stirred at room temperature for
65 hours.
The l-(trifluoroacetyl)acyl hydrazide obtained
from Reaction 3 is reacted with triphenylphosphine
("TPP") and diethyl azodicarboxylate ("DEAD"), as
depicted above in Reaction 4. (Although the above
Scheme depicts only the use of DEAD, the reaction will
also proceed if either dimethyl azodicarboxylate or
di(iso-propyl)azodicarboxylate are substituted in the
reaction.) In addition, the racemic serine acyl
hydrazide can be utilized as a starting material, and
other trihaloacetyl derivatives similarly provided can
be ring closed to yield racemic trihaloacetyl diazolidines.
As one skilled in the art will appreciate,
any trihaloacetyl group which serves as an effective
electron-withdrawing group (on the acyl hydrazide) would
3~
~-6711A -12-
be efficacious. Such other trihaloacetyl derivatives
can be prepared by using ethylthio trihalothioacetates
as the acylating agent in Scheme 2.
The stoichiometry of the process of Reaction 4
has the N-(trifluoroacetyl)acyl hydrazide, phosphine and
diethyl azodicarboxylate reagent present in at least
approximately a 1:1:1 molar ratio. The reaction will
proceed in the presence of molar excesses above this
ratio of any of the reagents or of the starting material.
The reaction is initiated by first combining
(in any order) the solvent, the l-(trifluoroacetyl)acyl
hydrazide and the phosphine, and secondly adding the
azodicarboxylate reagent.
The reaction temperature of Reaction 4 is
a not critical parameter. The process can be carried
out from approximately the freezing point to approx-
imately the reflux temperature of the solvent. The
preferred temperature is approximately room temperature.
The duration of Reaction 4 can be from
approximately five minutes to approximately twenty four
hours. The progress of the process can be monitored by
standard methods (for example, thin layer chromatography,
high performance liquid chromatography, etc.) The
process is stopped when the monitoring method demon-
strates that the reaction is substantially complete.
The solvents for Reaction 4 are aromatic hydro-
carbon solvents such as benzene, toluene, xylenes,
etc.; ethers such as diethyl ether, tetrahydrofuran,
or 1,4-dioxane; chlorinated hydrocarbons such as
methylene chloride, chloroform, carbon tetrachloride,
dichloroethane, or chlorobenzene; amides such as dimethyl-
formamide and dimethylacetamidei and other solvents such
as hexamethylphosphoramide. Tetrahydrofuran is the
9'1(3~
X-6711A -13-
preferred solvent. It is also desirable, but not
essential, to dry and deoxygenate the solvent before use
in the process.
The chiral l-(trifluoroacetyl)diazolidine
S obtained from Reaction 4 is deacylated with dilute
sodium hydroxide solution to yield the chiral l-
(unsubstituted)diazolidine. The deacylation reaction is
represented as Reaction 5 in Scheme 2. The Reaction
entails generally suspending the chiral l-(trifluoro-
acetyl)diazolidine in water then adding at least twoequivalents of dilute aqueous sodium hydroxide solution.
(For instance, a two-fold excess of lM sodium hydroxide
can be used. Preferably, sufficient sodium hydroxide
solution is added to give the reaction solution an
initial pH of from between about 11 to about 12). The
resultant solution stirred from about 10 minutes to
about 3 hours at a temperature from about 10C to 25C.
When the reaction is substantially complete the reaction
solution is neutralized by the addition of dilute acid,
such as lN hydrochloric acid.
The optimal reaction time for Reaction 5 can
be determined by monitoring the progress of the reaction
by conventional means such as chromatographic techniques
(thin layer chromatography, high performance liquid
chromatography, or column chromatography) and/or spectro-
scopic methods, such as infrared spectroscopy, nuclear
magnetic resonance spectrometry and mass spectrometry.
A preferred reaction time period is from between about
30 minutes to about 1.5 hours. The above chiral
synthesis can be modified to yield racemic product, if
desired.
~2~
X-6711A -14-
The diazolidinones of Formula I are intermedi-
ates to pyrazolidinium ylides of the Formula II:
~ frR4
R1> /5 ' ~Rs
R~ ~3 2~ II
In the above Formula II,
R1 and R2 are
1) taken together and form a
phthalimido group; or
2) either R1 or R2 is hydrogen and
the other of R1 or R2 is an amino
protecting groupi and
R4 and R5 are the same or different and are
hydrogen, C1 to C6 alkyl, C1 to C6 substituted alkyl,
C~ to C12 arylalkyl, C7 to C12 substituted arylalkyl,
phenyl, substituted phenyl or a group of the formula
-COOR6
wherein R6 is C1 to C6 alkyl, C1 to C6 substitu-ted
alkyl, C~ to C1 2 arylalkyl, C~ to C1 2 substituted
2~ arylalkyl, phenyl, substituted phenyl, a carboxy
protecting group, or a non-toxic, metabolically-labile
ester-forming group.
The ylides of Formula II are synthesized by
condensing a ketone or aldehyde with a 1-(unsubsti-
tuted)diazolidine of Formula I.
1~i9~
X-6711A -15-
As a useful alternative procedure, the ketal
of the ketone may be condensed with the diazolidine in
the presence of an acid. For example, the diazolidine
reagent is combined with acetone dimethyl acetal in
methanol and then the solution is treated with d-10
camphorsulfonic acid. The mixture is refluxed for 1.5
hours to give the dimethyl ylide (i.e., R4 and R5 are
methyl). The unsubstituted ylide (when R4 and R5 are
hydrogen) is synthesized by combining the diazolidine
reagent and 37% aqueous formaldehyde in methanol and
stirring the mixture for between about twenty minutes to
about 1.5 hours at room temperature. When R4 and R5 are
different those skilled in the art will recognize that
this final reaction will produce a mixture of E and Z
isomers.
Chiral pyrazolidinium ylide intermediates
(wherein C~ is either in the (R) or (S) configuration)
are syntheslzed from the corresponding chiral l-(unsub-
stituted)diazolidines of Formula I using the above-
described conditions.
The synthesis of the above pyrazolidiniumylide intermediates are further described by L.N.
Jungheim and R.E. Holmes, Canadian Patent Application No .
507,782, filed Aprll 28, 1986.
~i9~
X-6711A -16-
The ylides of Formula II are intermediates
to 7-substituted bicyclic pyrazolidinone antimicrobials
of Formula III
R5 R4
R2~
In Formula III, R4 and R5 include the substituents for
R4 and Rs of the ylides of Formula II plus a carboxylic
acid or a carboxylate salt. R1 and R2 of Formula III
include all the substituents of the corresponding terms
in Formula II plus substituents when either R1 or R2
is hydrogen and the other is an acyl group derived from
a C1 to C30 carboxylic acid. (Examples of such acyl
groups are the acyl groups bonded to the 6- and 7-amino
groups of penicillins and cephalosporins, respectively).
R7 and R8 in Formula III can be a variety of substitu-
ents, including a group of the formula
-COORg
wherein R~ includes the substituents of the above R6
plus, for example, hydrogen or an organic or inorganic
cation. Further examples of substituents at R1, R2, R3,
R4, R5, R7 and R8 in Formula III can be found in L.N.
Jungheim, S.K. Sigmund, C.J. Barnett, R.E. Holmes and
R.J. Ternansky, Canadian Patent Application No. 507,777,
30 filed April 28, 1986.
91~
X-6711A -17
The 7-substituted bicyclic pyrazolidinones
of Formula III are synthesized, for example, by various
1,3-dipolar cycloaddition reactions with the ylides of
Formula II. One method of cycloaddition reaction,
(the addition of an ylide and a substituted acetylene)
is represented below in Scheme 3:
Scheme 3
Rs
Rz\ ~ R~ I R~
ylide (II)acetylene III
In the above Scheme 3, for brevity's sake,
Formula III indicates only one of the two possible 2,3-
regioisomer products of the reaction. The reaction
represented by Scheme 3 can also produce the opposite
2,3-regioisomer as well as a mixture of the regioisomers.
In the above Scheme R1, R2, R4 and Rs are as
defined above for Formula II, R7 and R8 are as defined
for Formula III and either R1o or R11 is an amino pro-
tecting group and the other of R1o or R11 is hydrogen.
When carrying out the reaction it is preferable to
s''
X-6711A -18-
derivatize with protecting groups any of the acidic
groups represented by R4, R5, R7 or R8. Examples of
such acidic groups are the carboxylic acid group and the
hydroxyimino group. It is especially preferred that any
carboxylic acid groups be protected.
The reaction should be carried out in ~protic
solvents. Examples of such solvents are the chlorinated
hydrocarbons, the aromatic hydrocarbons and alkyl or
aromatic cyano solvents. The preferred solvents for the
above reaction are dichloromethane, acetonitrile, and
1,2-dichloroethane.
The temperature for the reaction is not criti-
cal. It is preferred that the reaction be carried out
between about room temperature to about the reflux tem-
perature of the solvent.
The reaction usually requires a period ofabout 1 to about 168 hours. The optimal reaction time
can be determined by monitoring the progress of the
reaction by conventional means such as chromatographic
techniques (thin layer chromatography, high performance
liquid chromatography, or column chromatography) and
spectroscopic methods (alone or in conjuction with
chromatographic techniques), such as infrared spec-
troscopy, nuclear magnetic resonance spectrometry and
mass spectrometry.
The usual stoichiometry for the reaction is
a 1:1 ratio of ylide to acetylene reagent. Of course,
an excess of either reagent is permissible. It is
preferred that the acetylene reagent be present in
excess, and especially preferred that the acetylene be
present in a 2:1 excess. Furthermore, the order of
addition of either reagent is not critical.
~2~
X-6711A -19-
The regiospecificity of the cycloaadition in
Scheme 2 is unpredictable. The stereochemical and
electronic properties of the ylide and acetylene and
the various reaction conditions have as yet yielded no
clearly discernable regiospecific trends. Usually the
reaction yields widely varying mixtures of 2,3-regio-
isomer products.
The stereospecificity of the cycloaddition
of Scheme 3 at the C~ position of the bicyclic
pyrazolidinone product is determined by the stereo-
chemistry at C4 of the ylide starting material. Thus,
i~ the ylide is chiral (either 4-(R) or 4-(S)) then the
cycloaddition product will be chiral (7-(~) or 7-(S),
respectively). Similarly, a C4 enantiomeric mixture of
ylide starting materials will yield a C7 enantiomeric
mixture of cycloaddition products.
The compounds produced by Scheme 3 above are
the 7-(protected amino) derivatives of Formula'III.
In order to enhance the antimicrobial activity of the
bicyclic pyrazolidinone compounds, it is desira~le to
replace the amino-protecting group with an acyl group
derived from a C1 to C3 o carboxylic acid. As discussed
above, the acyl groups employed are typically those used
to achieve the same purpose when bonded to the 6-amino
group of a penicillin or a 7-amino group of a cephalo-
sporin.
The first step for the acylation of a 7-(pro-
tected amino) bicyclic pyrazolidinone compound ("7-pro-
tected amino nucleus") is the removal of the amino pro-
tecting group. The conditions for the removal of thesegroups are well known in the cephalosporin and peni-
1~i9~
X-6711A -20-
cillin arts. For example, the trimethylsilyl protecting
group is removed by simple hydrolysis, the t-butoxy-
carbonyl group is removed by acidic hydrolysis (either
trifluoroacetic acid or a mixture of hydrochloric acid
in glacial acetic acid), and the allyloxycarbonyl group
is removed as a palladium complex.
Removal of the acid-labile amino protecting
groups usually yields the 7-amino nucleus as a salt.
The salt of the nucleus is neutralized by conventional
procedures before acylation. For instance, the removal
of the t-butoxycarbonyl group with trifluoroacetic acid
leaves the trifluoroacetate salt of the resultant
7-amino compound. The salt is taken up in tetrahydro-
furan and bis(trimethylsilyl)trifluoroacetamide was
added to yield the corresponding 7-amino compound.
The (neutralized) 7-amino compound can be isolated then
acylated or acylated ln situ. Similarly, the removal of
the t-butoxycarbonyl group with a mixture of hydro-
chloric acid in acetic acid leaves the hydrochloride
salt. The hydrochloride salt is neutralized with a base
such as N-methylmorpholine and generally acylated ln
SltU
-
The methods fGr the acylation of the 7-amino
bicyclic pyrazolidinone compounds with the acyl side
chain are similar to the methods for the acylation of 6-
aminopenicillanic acid, 7-aminodesacetoxycephalosporanic
acid and 7-aminocephalosporanic acid. One method is to
simply combine the 7-amino nucleus wi-th an acid chloride
or acid bromide. The acid chloride or acid bromide may
be formed in situ. Another method is to combine the
7-amino nucleus with the free carboxylic acid form of
X-6711A -21-
the side chain (or its acid salt) and a condensing
agent. Suitable condensing agents include N,N'-disub-
stituted carbodiimides such as N,N'-dicyclohexylcarbodi-
imide, N,N'-diethylcarbodiimide, N,N'-di-(n-propyl)-
carbodiimide, N,N'-di-(iso-propyl)carbodiimide, N,N'-
diallylcarbodiimlde, N,N'-bis(p-dimethylaminophenyl)-
carbodiimide, N-ethyl-N'-(4''-ethylmorpholinyl)carbodi-
imide and the like. Other suitable carbodiimides are
disclosed by Sheehan in U.S. Paten-t No. 2,938,892 and by
Hofmann et al. in U.S. Patent No. 3,065,224. Azolides,
such as N,N'-carbonyldiimidazole and N,N'-thionyldi-
imidazole may also be used. Dehydrating agents such as
phosphorus oxychloride, alkoxyacetylenes and 2-halogeno-
pyridinium salts (such as 2-chloropyridinium methyl
iodide, 2-fluoropyridinium methyl iodide, and the like)
may be used to couple the free acid or its acid salt
with the 7-amino nucleus.
Another acylation method entails first con-
verting the free carboxylic acid form (or the corre-
sponding salt) of the acyl side chain to the activeester derivative which is in turn used to acylate the
nucleus. The active ester derivative is formed by
esterifying the free acid form with groups such as
p-nitrophenol, 2,4-dinitrophenol, trichlorophenol,
pentachlorophenol, N-chlorosuccinimide, N-chloro maleic
imide, N-chlorophthalimide, 2-chloro-4,6-dimethoxy-
triazene, 1-hydroxy-lH-benzotriazole or l-hydroxy-6-
chloro-lH-benzotriazole. The active ester derivatives
can also be mixed anhydrides, formed with groups such as
methoxycarbonyl, ethoxycarbonyl, iso-butoxycarbonyl,
trichloromethylcarbonyl, and iso-but-2-ylcarbonyl and
~X~i91C)~i
X-6711A -22-
the carboxylic acid of the side chain. The mixed
anhydrides are synthesized by acylating the carboxylic
acid of the acyl side chain.
Alternatively, the 7-amino nucleus can be
acylated with the N-ethoxycarbonyl-2-ethoxy-1,2-dihydro-
quinoline (EEDQ) derivative of the acyl side chain. In
general, the free acid form of the acyl side chain and
EEDQ are reacted in an inert, polar organic solvent
(e.g. tetrahydrofuran, acetonitrile, etc.). The resul-
tant EEDQ derivative is used ln situ to acylate the7-amino nucleus.
Once the bicyclic pyrazolidinones are acylated
with the appropriate acyl group derived from a Cl to
C30 carboxylic acid, they are converted to the corre-
sponding antimicrobial final product form by removingany remaining amino, hydroxy and/or carboxy protecting
groups on the molecules. As discussed above, such re-
moval methods are well known in the cephalosporin,
penicillin and peptide arts. Once the carboxy groups
are deprotected, the oral ester may be put on the
desired carboxy group(s) at R4, R5, R7 and R8. The
methods for making the oral ester derivatives are well
known in the cephalosporin and penicillin art.
The antimicrobial compounds of Formula III
inhibit the growth of certain organisms pathogenic to
man and animals. The antimicrobial compounds are com-
pounds wherein the various amino, hydroxy and/or carboxy
protecting groups have been removed. The antimicrobial
activity can be demonstrated in vitro using standard
tube-dilution techniques. The ln vitro tests demon-
strate that the 7-(S) antimicrobial compounds are
~2~i~10~
X-6711A -23-
more active than either a mixture of corresponding C7
enantiomers or the corresponding 7-(R) compounds.
Representative pathogens which are sensitive to the
antimicrobial compounds of Formula III include
Staphylococcus aureus Xl.l, Streptococcus pYogenes C203,
Streptococcus pneumoniae Park, Hemophilus influenzae 76
(ampicillin reslstant), Escherichia coll N10,
Escherichia coli EC14, Escherichia coli TEM (b-lactamase
producer), Klebsiella pneumoniae X26, Klebsiella
pneumoniae KAE (~-lactamase producer), Klebsiella
pneumoniae X68, Enterobacter aerogenes C32,
_
Enterobacter aerogenes EB17, Enterobacter cloacae EB5
(non-~-lactamase producer), Salmonella typhi X514,
Salmonella typhi B35, Serratia marcescens X99, Serratia
marcescens SE3, Proteus morganii PR15, Proteus
-
inconstans PR33, Proteus rettgeri C24, Citroboaeter
freundii CF17, and the like.
The antimicrobial compounds for which the
diazolidinones of this invention are intermediates are
useful for the therapeutic or prophylactic treatment of
infections in warm-blooded animals caused by both
gram-positive, gram-negative and acid-fast bacteria.
The antimicrobial compounds can be admin-
istered orally, parenterally (e.g. intravenously,
intramuscularly or subcutaneously) or as a topical
ointment or solution in treating bacterial infections of
warm-blooded animals.
Further description of the synthesis and the
properties of the bicyclic pyrazolidinones of Formula
III are found in L.N. Jungheim, S.K. Sigmund, C.J.
Barnett, R.E. Holmes and R.J. Ternansby, Canadian Patent
~2~ 0~
X-6711A -24-
Application No. 50~,777, filed April 28, 1986.
The following Examples are provided to further
illustrate the invention. It is not intended that the
invention be limited in scope by reason of any of the
following Preparations or Examples.
In the following Preparations and Examples,
the terms melting point, nuclear magnetic resonance
spectra, field desorption mass spectra, electron impact
mass spectra, infra-red spectra, ultraviolet spectra,
elemental analysis, high performance liquid chroma-
tography and thin layer chromatography are abbreviated
m.p., n.m.r., f.d.m.s., m.s., i.r., u.v., anal., HPLC
and TLC, respectively. In addition, the adsorption
maxima listed for the i.r. spectra are only those of
interest and not all of the maxima observed.
The abbreviations THF, TFA and BSTFA stand
for tetrahydrofuran, trifluoroacetate and N,O-bis-
(trimethylsilyl)trifluoroacetamide, respectively.
In conjunction with the n.m.r. spectra, the
following abbreviations are used: "s" is singlet, "d"
is doublet, "dd" is doublet of doublets, "t" is triplet,
"q" is quartet, "m" is multiplet, "dm" is a doublet of
multiplets and "br. s", "br. d" and "br. t" stand for
broad singlet, doublet and triplet, respectively. "J"
indicates the coupling constant in Hertz. "DMSO/d6" is
dimethyl sulfoxide where all protons have been replaced
with deuterium.
1~9~1U~ -
X-6711A -25-
The n.m.r. spectra were obtained on a Varian
Associates EM-390 90 MHz, on a Jeol FX-9OQ 90 MHz
instrument, or on a ~eneral Electric QE-300 MHz instru-
ment. The chemical shifts are expressed in ~ values
(parts per million downfield from tetramethylsilane).
The field desorption mass spectra were taken on a
Varian-MAT 731 Spectrometer using carbon dendrite
emitters. Electron Impact Mass Spectra were obtained on
a CEC 21-110 instrument from Consolidated Electrodynamics
Corporation. Infrared spectra were obtained on a
Perkin-Elmer Model 281 instrument. Ultraviolet Spectra
were obtained on a Cary Model 118 instrument. Specific
rotations were obtained on a Perkin-Elmer Model Q-41
instrument. Thin layer chromatography was carried out
on E. Merck silica gel plates. Melting points reported
are uncorrected.
Preparation 1
Methyl 3-(p-Toluenesulfonate)-2-(S)-(t-
Butoxycarbonylamino~Propionate
Methyl (3-hydroxy)-2-(S)-(t-buto~ycarbonyl-
amino)propionate (58 g, 196 mmol), dry methylene chlo-
ride (150 ml), p-toluenesulfonyl chloride (43.35 g,
227.4 mmol), 4-(dimethylamino)pyridine (2.4 g, 19.6
mmol) and pyridine (30 ml, 371 mmol) were combined and
stirred at room temperature overnight. The reaction
solution was concentrated in vacuo to a pale yellow oil.
The oil was stored ln vacuo overnight, then the white
solid that formed was isolated to give 75.33 g of crude
91~
X-6711A -26-
product. The product was triturated in petroleum ether
(approximately 200 ml) to yield methyl 3-(p-toluenesulfo-
nate)-2-(S)-~t-butoxycarbonylamino)propionate: n.m.r.:
(CDCl3, 90 MHz): ~ 7.72, 7.31 (2x dd, 4, aromatic protons),
5.26 (m, 1, nitrogen proton), 4.48 (m, 1, C-2 proton),
4.32 (m, 2, C-3 protons), 3.68 (s, 3, methyl protons of
methyl ester), 2.44 (s, 3, methyl protons of toluene
moiety), 1.40 (s, 9, protons of t-butyl moiety); i.r.
(CHCl3): 3435, 3019, 1753, 1711, 1502, 1369, 1351, 1250,
10 1215, 1190, 1177 cm~1; m.s.: 279, 210, 172, 91, 41;
Anal. Calcd. for C16H23NO7S:
Theory: C, 51.19; H, 6.71; N, 3.73; S, 8.54.
Found: C, 51.05; H, 6.50; N, 3.63; S, 8.13.
Exam_le l
4-(R,S)-(t-Butoxycarbonylamino)-3-Oxo-1,2-
Diazolidine
Under a nitrogen atmosphere, dry methylene
chloride (50 ml) was cooled in an ice bath and anhydrous
hydrazine (11.0 g, 333 mmole), (97%) was added. The ice
bath was removed and the solution was stirred until it
warmed to room temperature. At this time a solution of
methyl 3-(p-toluenesulfonate)-2-(S)-(t-butoxycarbonyl-
amino)propionate (20.0 g, 53.6 mmole) in dry methylene
chloride (50 ml) was gradually added. The reaction
solution was stirred under nitrogen at room temperature
for 5 hours. The solution was then concentrated under
reduced pressure and the concentrate was taken up in
saturated aqueous sodium bicarbonate solution. The
~X~i910~;
X-6711A -27-
aqueous solution was continuously extracted for 14 hours
with methylene chloride (700 ml). The methylene chlo-
ride solution was dried over sodium sulfate, filtered
and concentrated under reduced pressure to yield approxi-
mately 5.15 g, 48% of 4-(R,S)-(t-butoxycarbonylamino)-
3-oxo-1,2-diazolidine: n.m.r. (CDC13, 90 MHz): ~ 7.04
(m, 1), 5.12 (m, 1), 4~28 (m, 1, C-4 proton), 3.94 (m,
1, C-5 proton), 3.20 (m, 1, C-5 proton), 1.45 (s, 9,
t-butyl protons); i.r. (CHC13): 3430, 3250, 3019, 2983,
1702, 1545, 1503, 1370, 1297, 1241, 1215, 1165 cm~l;
f.d.m.s.: M = 201;
Anal. Calcd. for C8H15N3O3:
Theory: C, 47.75; H, 7.51; N, 20.88.
Found: C, 47.80; ~, 7.56; N, 20.61.
Example 2
4-(R,S)-(t-Butoxyczrbonylamino)-3-Oxo-1,2-
Diazolidine p-Toluenesulfonate Salt
4-(R,S)-(t-Butoxycarbonylamino)-3-oxo-1,2-
diazolidine (1.7 g, 8.45 mmol) was slurried in methylene
chloride (50 ml). p-Toluenesulfonic acid hydrate (1.6 g,
8.45 mmol) was added to the slurry. After 20 minutes
the resultant solid material was collected then dried
ln vacuo for approximately 48 hours to yield 2.95 g of
colorless 4-(R,S)-(t-butoxycarbonylamino)-3-oxo-1,2-
diazolidine p-toluenesulfonate salt: n.m.r. (90 MHz,
DMSO-d6): ~ 7.5 (d, 2, J = 8), 7.1 (d, 2, J = 8), 4.32
(m, 1), 3.9 (m, 1), 3.4 (m, 1) 2.3 (s, 3), 1.4 (s, 9);
i.r. (KBr): 1742, 1704, 1537 cm
~ 3
X-6711A -28-
Preparation 2
4~(R,S)-(t-Butoxycarbonylamino)-3-Oxo-1-
(Methylene)-1,2-Pyrazolidinium Ylide
4-(R,s)-t-(sutoxycarbon~lamino)-3-oxo-1,2-
diazolidine (4.02 g, 20 mmol) was dissolved in dry
methanol (50 ml). 37% Aqueous formaldehyde (1.62 g,
20 mmol) was added, the mixture was stirred for 20
minutes at room temperature then concentrated in vacuo.
The solvent was removed by azeotropic distillation with
methanol in vacuo at 40C. The resultant residue was
dried ln vacuo at 40C overnight to yield 4-(R,S)-(t-
butoxycarbonylamino)-3-oxo-1-(methylene)-1,2-pyra-
zolidinium ylide: n.m.r. (90 MHz, CDCl3): ~ 6.1-5.3
(m, 2), 4.9-4.2 (m, 6), 4.0-3.6 (m, 2), 3.5-3.1 (m, 2),
1.4 (s, 18); i.r. (KBr): 3379, 2980, 2930, 1705, 1524,
1519, 1504, 1455, 1393, 1368, 1297, 1252, 1166 cm~l;
f.d.m.s.: M = 213.
Preparation 3
2,3-di(Allyl Carboxylate)-7-(R,S)-(t-Butoxy-
carbonylamino)-8-Oxo-1,5-Diazabicyclo[3.3.0]Octa-2-ene
4-(R,S)-(t-Butoxycarbonylamino)-3-oxo-1-
(methylene)-1,2-pyrazolidinium ylide from Preparation 2
above was dissolved in dry acetonitrile (50 ml) and
diallyl butynedioate (3.88 g, 20 mmol) was added. The
mixture was heated to reflux for 3 hours then concen-
trated _ vacuo. The resultant solid was chromato-
X-6711A -29-
graphed by HPLC on silica gel eluted with 2:1 hexane:-
ethyl acetate, to yield 2.67 g, 32.8% yield of 2,3-
di(allyl carboxylate)-7-(R,S)-(t-butoxycarbonylamino)-
8-oxo-1,5-diazabicyclo[3.3.0]octa-2-ene: n.m.r. (90 MHz,
CDCl3~: ~ 6.20-5.70 (m, 2, unsaturated protons on allyl
groups), 5.52-5.0 (m, 5, C-7 proton and unsaturated
protons in allyl group), 4.82 (dm, 2, ~ = 6, unsaturated
protons on allyl group on C-2 carboxylate), 4.64 (dm,
2, J = 6, saturated protons on allyl group on C-3 car-
10 boxylate group), 4.38 (d, l, J = 13, C-4 proton), 4.04
(t, 1, J = 8, C-6 proton), 3.92 (d, 1, J = 13, C-4
proton), 2.88 (dd, 1, J = 8, 12, C-6 proton), 1.45 (s,
9, protons of t~butyl group); u.v. (methanol): AmaX =
345 ( = 8500); i.r. (CHC13): 3019, 1750, 1736, 1709,
15 1384, 1370, 1278, 1234, 1215, 1162 cm~1;
Anal. Calcd. for C1gH25O7N3:
Theory: C, 56.01; H, 6.19; N, 10.31.
Found: C, 56.24; H, 6.35; N, 10.10.
PreParation 4
2,3-di(Allyl Carboxylate)-7-(R,S)-[2-(Thien-2-
yl)Acetamido]-8-Oxo-1,5-Diazabicyclo[3.3.0]0cta-2-ene
A. Removal of Amino Protecting Group and
Formation of TFA Salt
2,3-di(Allyl carboxylate)-7-(R,S) (t-butoxy-
carbonylamino)-8-oxo-1,5-diazabicyclo[3.3.0~octa-2-ene
(407 mg, 1 mmol) was dissolved in trifluoroacetic acid
(2 ml) and the solution was stirred for 5 minutes then
concentrated ln vacuo.
o~i
X-6711A -30-
~. Neutralization of TFA salt
The residue from Step A was taken up in THF
(5 ml) and BSTF~ (1.5 ml) was added while the mixture
was being cooled to 0C.
C. Acylation of Nucleus
A THF solution (1 ml) of 2-(thien-2-yl)acetyl
0 chloride (176 mg, 1.1 mmol) was added to the solution
from Step B and the resultant mixture was stirred at
0C for 20 minutes. The reaction mixture was then poured
into ethyl acetate and the resulting organic mixture was
washed with saturated sodium bicarbonate solution, 0.2N
hydrochloric acid, brine, dried over magnesium sulfate
and filtered. The filtrate was concentrated ln vacuo to
give 700 mg of crude oily residue. The residue was
chromatographed on a silica gel preparatory-scale TLC
plate eluted with 1:1 hexane:ethyl acetate solution to
20 give 270 mg, 62% yield of 2,3-di(allyl carboxylate)-7-
(R,S)-[2-(thien-2-yl)acetamido]-8-oxo-1,5-diazabicyclo-
[3.3.0]octa-2-ene: n.m.r. (90 MHz, CDC13): ~ 7.22 (m,
1, C-5 proton of thienyl group), 6.96 (m, 2, C-3 and C-4
protons of thienyl group), 6.56 (br. d, 1, J = 6, amido
25 proton), 6.20-5.60 (m, 2, C-2 proton of allyl groups),
5.60-5.10 (m, 4, C-3 (unsaturated) protons of allyl
groups), 5.0 (m, 1, C-7 proton), 4.80 (dm, 2, J = 6, C-1
protons of allyl group on C-2 carboxylate group), 4.64
(dm, 2, J = 6, C-1 protons on allyl group on C-3 car
30 boxylate group), 4.36 (d, 1, J = 12, C-4 proton), 4.08
(t, 1, J = 8, C-6 proton), 3.92 (d, 1, J = 12, C-4 proton),
- - .
l~i9~
X-6711A -31-
3.80 (s, 2, methylene protons of acetamido group), 2.86
(dd, 1, J = 8, 12, C-6 proton); u.v. (methanol): AmaX =
340 ( = 6850), 230 ( - 12,500); m.s.: M~ = 431; i.r.
(CHCl3): 1750, 1705 cm
Anal. Calcd. for C20H22N3O6S:
Theory: C, 55.68; H, 4.91; N, 9.74; s, 7.43.
Found: C, 55.97; H, 5.21; N, 9.52; S, 7.23.
_reparation 5
2,3-di(Carboxylic Acid)-7-(R,S)-[2-(Thien-2-
yl)Acetamido]-8-Oxo-1,5-Diazabicyclo[3.3.0]Octa-2-ene
Triphenylphosphine (35 mg, 0.13 mmol) was
added to a solution of palladium(II) acetate (6 mg,
0.026 mmol) in acetone (3 ml). The mixture was stirred
until a white precipitate formed (10 minutes). An
acetone solution (3 ml) of 2,3-di(allyl carboxylate)-
7-(S)-[2-(thien-2-yl)acetamido]-8-oxo-1,5-diazabicyclo-
[3.3.0]octa-2-ene (200 mg, 0.46 mmol) was added to the
mixture. After the resultant mixture became homo-
geneous, it was cooled to 0C and tri(n-butyl)tin hydride
(0.27 ml, 1 mmol) was added. The solution was stirred
at 0C for 30 minutes. lN Hydrochloric acid (l ml)
was added and the solution was stirred for an additional
10 minutes. The solution was filtered, diluted with
water (30 ml), then extracted with he~ane (4 X, 50 ml).
The aqueous phase was separated and freeze-dried to give
170 mg of yellow powder. The powder was triturated
with ethyl acetate, sonicated, centrifuged, and the
recovered solid was dried ln vacuo to give 2,3-di(car-
X-6711A -32-
boxylic acid)-7-(R,S)-[2-(thien-2-yl)acetamidol-8-oxo-
1,5-diazabicyclo[3.3.0]octa-2-ene: n.m.r. (90 MHz,
acetone-d6): ~ 7.20 (m, 1, C-5 proton of thienyl group),
6.94 (m, 2, C-3 and C-4 protons of thienyl group),
5.2-4.6 (m, 2, acetamido nitrogen proton and C-7 proton),
4.~4 (d, l, J = 13, c-4 proton), 4.0-3.8 (m, 2, side
chain methylene proton), 3.80 (s, 2, a C-6 proton and a
C-4 proton), 3.0 (dd, 1, J = 8, 12, a C~6 proton); u.v.
(methanol): AmaX = 345 ( = 4000), 226 ( = 7000)i
f.d.m.s.: (M~1)+ = 352; i.r. (KBr): 1730, 1699, 1533,
1438, 1405, 1377, 1338, 1246, 1209, 1188 cm 1.
Preparation 6
N-(t-Butoxycarbonyl) (L)-Serine Trifluoroacetyl
Acyl Hydrazide
N-(t-Butoxycarbonyl) (L)-serine acyl hydrazide
(32.85 g, 150 mmol) was suspended in ethanol (400 ml).
Ethylthio trifluorothioacetate (30 ml, 37.02 g, 234.3
mmol) was added to the suspension and the resultant
mixture was stirred at room temperature for 65 hours.
The solvent was removed ln vacuo and the residue was
dissolved in diethyl ether (160 ml). A seed crystal was
added to the diethyl ether solution and the resultant
crystals were collected by filtration (approx. 27 g).
The filtrate was evaporated ln vacuo and diethyl ether
(50 ml) was added to the residue. The solids that
formed on standing were removed by filtration to yield
approximately 16.5 g of additional product. The two
batches of solids collected by filtration were combined
12tj9~0~
X-6711A -33-
and recrystallized from diethyl ether (3 liters). After
effecting solution, the solution was reduced to approx-
imately 450 ml to yield (after a second crop) 41.04 g,
87% yieid of N-(t-butoxycarbonyl) (L)-serine trifluoro-
acetyl acyl hyrdrazide: n.m.r. (300 MHz, DMSO-d6): ~
11.5 (br. s, 1), 10.33 (s, 1), 6.84 (d, 1, J=9), 4.9 (t,
1, J=7, (OH)), 4.1 ~m, 1), 3.59 (br. m, 2), 1.4 (s, 9);
specific rotation: [~]D5 = ~ 25.87 (10.05 mg/ml in
methanol); m.p. 143-144C (first crop), 142-144C
(second crop).
Anal. Calcd. for C1oH16N3O5F3:
Theory: C, 38.10; H, 5.12; N, 13.33;
Found: C, 38.34; H, 4.89; N, 13.16.
Example 3
4-(S)-(t-Butoxycarbonylamino)-1-(Trifluoro-
acetyl)-3-Oxo-1,2-Diazolidine
N-(t-Butoxycarbonyl) (L)-serine trifluoro-
acetyl acyl hydrazide (3.78 g, 12 mmol) and triphenyl-
phosphine (3.46 g, 13.2 mmol) were dissolved in THF (50
ml). To the solution was added a THF solution (10 ml)
of 95% diethyl azodicarboxylate (2.42 g, 2.19 ml, 13.2
mmol). The resultant mixture was stirred at room
temperature for six hours and then the solvent was
removed ln vacuo. The residue was dissolved in ethyl
acetate (100 ml) and then the solution was washed with
aqueous sodium bicarbonate solution (33 ml, 3X). The
sodium bicarbonate extracts were combined, aqueous
saturated brine solution (70 ml) was added and the
;91~i
X-6711A -34-
resultant mixture was extracted with ethyl acetate (120
ml, 3X). The sodium bicarbonate solution was then
layered with additional ethyl acetate (200 ml) and lN
hydrochloric acid (approx. 80 ml) was added until the
sodium bicarbonate solution had a pH of 2.5. The ethyl
acetate layer was separated and the aqueous layer was
extracted with additional ethyl acetate (4X, 125 ml).
The ethyl acetate extracts were combined, washed with
saturated aqueous brine (125 ml, 2X), dried over sodium
sulfate, filtered, and taken to dryness 1n vacuo. The
resultant residue was dissolved in acetonitrile (100 ml)
then the acetonitrile was removed ln vacuo. Treatment
of the residue with acetonitrile was repeated to yield
3.06 g, 96% yield of 4-(S)-(t-butoxycarbonylamino)-1-
(trifluoroacetyl)-3-oxo-1,2-diazolidine: n.m.r. (300
MHz, CDCl3): ~ 5.25 (d, 1, J=6), 4.81 (t, 1), m 4.6 (m,
1), 4.06 (t, 1), 1.46 (s, 9); i.r. (CHC13): 1722, 1682,
1518 cm 1; (f.d.m.s.) (m/e): M = 297; specific
rotation: [~]D5 = -88.14 (10.03 mg/ml in methanol);
Anal. Calcd for C1oH14N3O4F3:
Theory: C, 40.41; H, 4.75; N, 14.14.
Found: C, 40.58; H, 5.01; N, 13.92.
Example 4
4-(S)-(t-Butoxycarbonylamino)-3-Oxo-1,2
Diazolidine
4-(S)-(t-butoxycarbonylamino)-1-(trifluoro-
acetyl)-3-oxo-1,2-diazolidine (2.97 g, 10 mmol) was
310~
X-671lA -35-
suspended in water (30 ml), lN sodium hydroxide solution
(20 ml, 0.8 g, 20 mmol, pH 12.2) was added and the
resultant mixture was stirred for one hour at room
temperature. The pH of the mixture was adjusted to
7.2 by the addition of lN hydrochloric acid (10 ml).
Sodium chloride (13 g) was added to the solution and the
mixture was extracted with chloroform (50 ml, 8X). The
chloroform extracts were combined, washed with saturated
aqueous sodium chloride solution (75 ml), dried over
sodium sulfate, filtered, and evaporated to dryness
1n vacuo. Diethyl ether (100 ml) was added to the
residue and then the ether was removed ln vacuo to yield
0.798 g of a white solid of 4-(S)-(t-butoxycarbonyl-
amino)-3-oxo-1,2-diazolidine: n.m.r. (300 MHz,
DMSO-d6): ~ 9.23 (s, 1), 7.04 (d, 1, J=9), 5.24 (br. s,
1,), 4.24 (m, 1), 3.41 (t, 1), 2.88 (t, 1), 1.38 (s,
9); specific rotation: [~]D5 = ~ 74.16 (10.06 mg/ml
in methanol); (the compound was dried overnight at 80C
before analysis):
Anal. Calcd. for C8H15N3O3:
Theory: C, 47.75; H, 7.51; N, 20.88.
Found: C, 47.75; H, 7.46; N, 20.62.
Procedure 8
2-(Allyl Carboxylate)-3-(Methyl Carboxylate)-
7~(S)-(t-Butoxycarbonylamino)-8-Oxo-1,5-Diazabicyclo-
[3.3.0]Octa-2-ene
1~6910~
X-6711A -36-
Step 1
Formation of Pyrazolidinium Ylide
4-(S)-(t-butoxycarbonylamino)-3-oxo-1,2-
diazolidine (20.1 g, 100 mmol) was suspended in 1,2-
dichloroethane (400 ml), 37% aqueous formaldehyde
solution (8.51 ml, 3.15 g, 105 mmol) was added and
the resultant mixture was stirred at room temperature
for 1.5 hours.
Step 2
15 Cycloaddition of Acetylene
Allyl methyl butynedioate (18.48 g, 110 mmol)
was added to the mixture from Step 1 and the resultant
mixture was refluxed for 6.5 hours. The volume of the
reaction mixture was reduced by half in a flask fitted
with a Dean-Stark trap. Hexane (200 ml) was added and
the mixture was allowed to stand until an oil formed.
The solvent was decanted, the oil was dissolved in ethyl
acetate (300 ml) and the solution was taken to dryness
ln vacuo to yield 17.3 g of a foam. The foam was
chromatographed using preparatory-scale high performance
liquid chromatography using a silica column eluted with
a gradient of 0 to 40% ethyl acetate in isooctane (8
liters). The product-containing fractions were combined
to yield 1.456 g of a yellow solid. The solid was
recrystallized from a mixture of ethyl acetate and
1~910~
X-6711A -37-
hexane to yield 0.55 g of 2-(allyl carboxylate)-3-
(methyl carboxylate)-7-(S)-(t-butoxycarbonylamino)-8-
oxo-1,5-diazabicyclo[3.3.0]octa-2-ene: n.m.r. (300 MHz,
CDC13): ~ 6.00 (m, 1), 5.38 (m, 2), 5.1 (br. d, J=6),
4.86 (d, 2), 4.74 (m, 1), 4.37 (d, 1, J=13), 4.08 (t,
1), 3.91 (d, 1, J=13), 3.77 (s, 3), 2.86 (t, 1), 1.46
(s, 9); i.r. (KBr): 1751, 1710, 1687 cm 1; u.v.
(ethanol): AmaX = 346 (~max = 8489); f-d-m-s- (m/e)
M = 381; specific rotation: [~]Ds = _ 481.92 (10.01
mg/ml in methanol); m.p. 111-113~C; Anal. Calcd for
C17H23N3 7
Theory: C, 53.54; H, 6.08; N, 11.02.
Found: C, 53.83; H, 6.06; N, 10.77
Procedure 9
2-(Allyl Carboxylate)-3-(Methyl Carboxylate)-
7-(S)-Amino-8-Oxo-1,5-Diazabicyclo[3.3.0]0cta-2-ene
Hydrochloride Salt
2-(Allyl carboxylate)-3-(methyl carboxylate)-
7-(S)-(t-butoxycarbonylamino)-8-oxo-1,5-diazabicyclo-
[3.3.0]octa-2-ene (0.1905 g, 0.5 mmol) was added to 3 M
hydrochloric acid in glacial acetic acid (7 ml) and the
resultant mixture was stirred at room temperature for
five minutes then taken to dryness ln vacuo. The
resultant yellow solid was dissolved in methylene
chloride (20 ml) and the mixture was sonicated and
evaporated ln vacuo. The methylene chloride/sonication
procedure was repeated two more times. The solid was
dried ln vacuo for 1.5 hours to yield to 2-(allyl
carboxylate)-3-(methyl carboxylate)-7-(S)-amino-8-oxo-
1,5-diazabicyclo[3.3.0]octa-2-ene hydrochloride salt.
X-6711A -38-
Procedure 10
2-(Allyl Carboxylate)-3-(Methyl Carboxylate)~
7-(S)-[2-(2-(Allyloxycarbonylamino)Thiazol-4-yl-2-~Z)-
(Methoxyiminoacetamido)]-8-Oxo-1,5-Diazabicyclo
[3.3.0]Octa-2-ene
Under a nitrogen atmosphere, 2-[2-(N-allyloxy-
carbonylamino~thiazolo-4-yl]-2-(z)-methoxyiminoacetic
acid (0.1425 g, 0.5 mmol) was suspended in dried
methylene chloride (5 ml). The suspension was cooled
to 0C then 6-chloro-2,4-dimethoxy-1,3,5-triazine (0.088
g, 0.5 mmol) and N-methylmorpholine (0.0505 g, 0.5 mmol)
were added. The resultant solution was stirred at 0C
for forty minutes. Additional N-methylmorpholine
(0.0505 g, 0.5 mmol) and then a methylene chloride
suspension (5 ml) of 2-(allyl carboxy)-3-(methyl
carboxylate)-7-(S)-amino-8-oxo-1,5-diazabicyclo[3.3.0]-
octa-2-ene hydrochloride salt (0.5 mmol) were added.
After all the solid dissolved, the solution was stirred
at room temperature for 20 hours then evaporated to
dryness in vacuo. The residue was dissolved in ethyl
acetate ~70 ml) and water (15 ml), the layers were
separated, and the ethyl acetate was extracted sequen-
tially with 0.1N hydrochloric acid (10 ml, 3X), satu-
rated aqueous sodium bicarbonate solution (20 ml, 3X),
brine solution (20 ml, 3X), dried over sodium sulfate,
filtered, and evaporated to dryness ln vacuo to yield
280 mg of a yellow solid. The solid was recrystal-
lized from a mixture of methylene chloride and di(iso-
propyl) ether to ~ield 136 mg of the 2-(allyl
;9~
X-6711A -39-
carboxylate)-3-(methyl carboxylate)-7-(S)-L2-(2-(t-allyl-
oxycarbonylamino)thiazol-4-yl)-2-(Z)-methoxyimino~
acetamido)]-8-oxo-1,5-diazabicyclo[3.3.0]octa-2-ene:
n.m.r. (300 MHz, DMSO-d6): ~ 12.1 (s, 1), 9.32 (d, 1,
J=9), 7.43 (s, 1), 5.94 (m, 2), 5.34 (m, 4), 5.09 (m,
1), 4.83 (d, 2, J=6), 4.7 (d, 2, J=6), 4.31 (d, l,
J=13), 4.02 (d, 1, J=13), 3.88 (overlapping t and s, 4),
3.69 (s, 3), 3.18 (t, 1); u.v. (ethanol)i ~max = 342
(~max = 8680), 264 (13,626), 209 (25,137); f.d.m.s.
(m/e): M = 548, 490; specific rotation: [~]D5 =
-351.45 (10.01 mg/ml in methanol).
Anal. Calcd for C22H24N6OgS:
Theory: C, 48.17; H, 4.41; N, 15.32.
Found: C, 48.09; H, 4.41; N, 15.02.
Procedure 11
2-(Carboxylic Acid)-3-(Methyl Carboxylate)-7-
(S)-[2-(2-Aminothiazol-4-yl)-2-(Z)-Methoxyiminoacetamido]-
8-Oxo-1,5-Diazabicyclo[3.3.0]0cta-2-ene Hydrate
Palladium(II) acetate (18 mg, 0.08 mmol) was
suspended in acetone (4 ml). Triphenylphosphine
(105 mg, 0.4 mmol) was washed into the suspension with
additional acetone (2 ml) and the resultant mixture was
stirred at room temperature for 20 minutes. 2-(Allyl
carboxylate)-3-(methyl carboxylate)-7-(S)-[2-(2-(allyloxy-
carbonylamino)thiazol-4-yl)-2-(Z)-methoxyiminoacetimido]-
8-oxo-1,5-diazabicyclo[3.3.0]octa-2-ene (0.497 g, 0.9096
mmol) was suspended in a mixture of acetone (45 ml) and
1~i9 1V~
X-6711A -40-
acetonitrile (15 ml~ was then added to the reaction
suspension. The suspension was stirrea at room tem-
perture for 35 minutes then cooled to 0C. Tri(n-
butyl)tin hydride (0.548 g, 1.81 mmol, 0.506 ml) was
slowly added to the cooled suspension and the mixture
was stirred at 0C for 30 minutes then at room tem-
perature for 50 minutes. The mixture was cooled to
0C then lN hydrochloric acid (1.82 ml, 1.81 mmol) was
added. The resultant mixture was stirred at 0C for
10 minutes then at room temperature for 5 minutes. The
mixture was filtered, water (130 ml) was added to the
filtrate, and the resultant mixture was filtered through
a pad of CeliteTM. The filtrate was extracted with
hexane (4X, 40 ml), and the aqueous layer was filtered
through a pad of Celite~ then reduced ln vacuo to about
75% volume. The resultant yellow solid was recovered by
filtration through a pad of Celite~ and the filtrate was
extracted with ether (2X, 75 ml), concentrated ln vacuo
to remove any residual ether and the resultant yellow
solution was lyophilized. The lyophilized solid was
dissolved in water (75 ml), filtered and chromatographed
on a preparatory-scale high performance liquid chromato-
graph using a C18 reverse phase column eluted with a
gradient of 0 to 10% methanol/0.5% acetic acid/water (8
liters) then a gradient of 10 to 25% methanol/0.5%
acetic acid/water (8 liters) to yield 91.5 mg of 2-
(carboxylic acid)-3-(methyl carboxylate)-7-(S)-[2-(2-
aminothiazol-4-yl)-2-(Z)-methoxyiminoacetamido]-8-
oxo-1,5-diazabicyclo[3.3.0~octa-2-ene: n.m.r. (300
MHz, DMSO-d6): ~ 9.18 (d, 1, J=lO), 7.24 (br- s, 2),
6.94 (s, l), 5.02 (m, l), 4.23 (d, 1, J=13), 3.9 (d, 1,
X-6711A 41-
J=13), 3.8 (overlapping t and s, 4), 3.1 (t, 1); i.r.
(KBr): 1726, 1688, 1670.5 cm 1; u.v. (ethanol~: AmaX=328
(maX=10,950~, 233 (16,013); f.d.m.s. (m/e): M
425; specific rotation: [~]Ds = -326.35 (9.83 mg/ml
in methanol);
Anal. Calcd f r 15 16 6 7 2
Theory: C, 40.72; H, 4.10; N, 19.00
Found: C, 40.81; H, 3.70; N, 19.03.