Note: Descriptions are shown in the official language in which they were submitted.
~7~ 8
~A3~3
--1--
S The pres~nt invention relat~s to 5,6-
epo~y-7-oxabicycloheptane subs~ituted pxo~taglandin
analogs which are cardiovascular agents useful, for
example, in the treatment of thrombotic disease.
:~ These compounds hav~ th~ structural fonmula
*~ 2~ 2 )m-C02R
~ A-~H-Rl
~ 15 o *\ I 0~
O
and incl~ading all stereoisomers thereof, wherein
m is 1 to 5; R is hydros~en, lower alkyl,
: 20 alkali metal sallt or E~olyhydroxylamine salt; A is
-C~=~ or -(C:EI2)2-; and Rl is lower alkyl, aryl,
;~ aralkyl, cycloal~yl, cycloalkylalkyl or lowex
~ alXenyl.
:
,
.
: '
,
: -.
-
~ .
~L~;:71198
-2- HA363
The present invention also includes
intermediates for preparing the above compounds of
the invention which have the structure
IA ~ CH2-C~O
\ I, ~ C~2-
and
IB ~ C~2 c~=C~-(C~2)m-C02alkYI
O I X
/~
wherein X is CH2 ~ ~ , -CH2OH, -CH0 or
-C~=CH-C R1 wherein m and Rl are as defined
above~
The term "lower alkyl" or l'alkyll' as employed
h~r~in by i~self or as par~ of anothex group
i~cludes both str~ight and branched chain radicals
of up to 12 carbons, preferably 1 to 8 carbons,
such as methyl, ethyl, propyl, isopropyl, butyl,
t-butyl, isobutyl, pentyl, hexyl, heptyl,
4,4-dimethylpentyl, octyl, 2,2,4-trimethylpentyl,
nonyl, decyl, undecyl, dodecyl, the various
,
' '
.
.
7~3~
HA363
--3--
branched chain isomers thereof, and the like as
well as such groups including a halo-sukstituent,
such as F, Br, Cl or I or CF3, an alkoxy substi-
tuent, an aryl substituent, an alkyl~aryl substi-
tuent, a haloaryl substituen~, a cycloalkyl sub-
s~ituent, an alkylcycloalkyl substituent, hydroxy,
an alkylamino substituent, a nitro substitue~t, an
amino subs~itue~, a cyano substi~uent, a ~hiol
substituen~ or an alkylthio substituent~
: 10 The term "cycloalkyl" by itself or as part
o~ arlother group includes saturate~ cyclic
hydrocarbon groups containing 3 to i 12 carbons,
preferably 3 to 8 carbons, which include cyclo-
propyl, cyclobutyl, cyclopentyl, cyclohexyl,
cycloheptyl, cyclooctyl, cyclodecyl and cyclodo-
decyl, any of which groups may be substituted with
1 or 2 halogens, 1 or 2 lower alkyl groups and/or
: lower alkoxy groups, an aryl group, 1 or 2 hydroxy
groups, 1 or 2 alkylamino groups, 1 or 2 amino
groups, 1 or 2 nitro groups, 1 or 2 cyano groups, 1
or 2 thiol groups or 1 or 2 alkylthio groups.
Th~ term "aryl" or 'IAr" as employed herein
by itself or as part of another group refers
to monocyclic or bicyclic aromatic groups
containing from 6 to 10 carbons in the ringportion, such as phenyl, naphthyl, substltuted
phenyl or substituted naphthyl wherein th~
substituen~ on either the phenyl or naph~hyl may be
1 or 2 lower alkyl groups, 1 or 2 haloyens (Cl,
~r or F), an aryl group, 1 or 2 lower alkoxy
: groups, 1 or 2 hydroxy groups, 1 or 2 alkylamino
groups,.1 or 2 amino groups, 1 or 2 nitro groups, 1
'
'
:, .
.
HA363
_ ~ "
or 2 cyano groups, 1 or 2 thiol groups or 1 or 2
alkylthio groups.
The term "aralkyl", "aryl-alkyl" or
"aryl-lower alkyl" as used herein by itself or as
part of another group refers to lower alkyl groups
as discus~ed above having an aryl substitue~t, such
as benzyl.
The term "lower alkenyl" or "alkenyl"
: includes straight or branched chain radicals of
fxom 2 to 12 carbons, preferably 2 to 6 carbons in
the normal chain, which includ~ one double bond in
the normal chain, such as ethenyl,~2-propenyl,
3-butenyl, 2-butenyl, 1-pentenyl, 3-pentenyl,
2-hexenyl, 3-h~xenyl, ~-heptenyl, 3-heptenyl,
4-h~ptenyl, 3-oc~enyl, 3-nonenyl, 4 decenyl,
3-undecenyl, 4 dodecenyl and the like.
The ter~ "lowar alkoxy", "alkoxy" or
"aralkoxy" include~ any of the above lower alkyl,
alkyl or ar~lkyl groups li~ked to a~ oxygen atom.
The term "halog~n" or "halol' as used herein
refers to chlorine, bromine, fluorine or iodine
~ with chlorine being preferred. ~-
`: The term "polyhydroxylamine" ref~rs to
gluca~ine salt, tri(hydroxyme~hyl)aminomethane
salt and the like.
The term 1l (C~2)ml~ includes a straight
or branched chain radicals having from 1 to
S carbon~ in the normal chain and may
contain on~ or more lower alkyl or halo
subs~ituents. Examples of (C~2)m
groups include CH2, CH2CH~, (CH2~3, (CH2)4,
`~ ,
-
~7~
~5- ~363
~H3 ~H3 IH3 ~H3
C- CH~ CwCH2-, -C-C~2 ~ (CH2~5~ ( 2 6
2 7' ( ~2~2 tH , _I_CH_, C~2-FH ~ C~ CH2~,
C~3 CH3 C~3 CH3
-C~ H~CH2-C~I-, a~d the like.
~H3 ~3
. Preferred are those compound~ of formula I
wherein A i~ CH~CH, and m is 2 or 4, ~
is ~, and R1 is lower alkyl, aryl,lsuch as phenyl,
or ~ralkyl such as b~nzyl, or b~zyl-l~methyl or
cycloalkyl, such as cyclohexyl.
The various compounds of the invention may
be prepared as outlined below.
The co~pounds of fonmula I of the invention
may be pxepared as describ~d below.
The startlng compounds of the invention II
may be prepared as follows.
Dione-A having the structure
~ O O
: 30
that is, 7-oxabicyclo[2.2.1]-5-hepten~-2,3-
- dicarboxylic anhydride [Ber. 62, 554 (1929); Ann.
; 460, 98 (19283]~ is reduced, for exampl~, by
.
.~ .
. .
. ~ .
:: '
7~
HA363
reacting with lithium aluminum hydride or
diisobutyl aluminum hydride in the presence of
an inext organic solvent such as tetrahydrofuran,
ether or toluene at reduced temperatures of from
S about -78C to about 67C to fonm diol B of the
structure
10- ~
¦ CH~OH
O
The diol B is subjected to a chloroformylation
reaction by reacting B dissolvad in an inert organic
solvent as descri~ed above, with phos~ene in the
presence of a solvent such as tetrahydrofuran,
toluene, benzene or xyl~ne, ~o form an alcohol of
the structure
~f H2-0CC
\ I CEI20
o
The alcohol _ is dissolved in an inert organic
solvent such as methylene chloride, tetrahydrofuran
or ether and then reacted with an organic base,
; such as pyridine, triethylamine, N,N-dimethyl-
aminopyridine or diazabicycloundecane (DBU) at
.
~A363
-7-
reduced temperatures of from about -78C to about
25 C, to form cyclic carbonate D
.
D ~ C~2
o
1~
The cyclic caxbona~e D i~ then subj!ected ~o
alcoholysis by reacti~g D with a~ alkanol
(alkyl-OH) having from 1 to 12 carbons, such as
ethanol, n-propa~ol, isopropanol, butanol,
pentanol, hexanol, heptanol, octanol, no~enol or
decanol, including all the Yariou~ isomers thereof,
pr~ferably isopropyl alcohol, ~mploying a molar
ratio of D:alkanol of within the range of from
about 1:10 to about 1:100 to form hydroxycarbo~ate
E (which itself is a novel compound)
~ ~
:. ~
12-OIlOalkyl
o o
~; 30 (wherein alkyl contains 1 to 12 carbons as defined
herein).
Thereafter, the hydroxy carbonate E is
tos~lated (or otherwise protected) by reacting E
, ~ . . .
', - ' ' .
~8- HA363
(dissolved in methylene chloride, and a basic
solvent such as pyridine, triethylamine or
dimethyl~linopyridine) with tosyl chloride or other
protecting agent, such as methane sulfonyl chloride
(mesyl chloride) and trifluorometh~nesulfonic
anhydride, to form the tosylate F or other
protected compound
~ CH2-OTs (or other protecting group)
F ~/ ~
CH2-OCO-alkyl
O O
Then, the tosylate F dissolved in an inert solvent
such as dimethylsulfoxide, or dimethylformamide is
cya~ated by reacting F with an alkali m~tal
cyanide such as NaCN or KCN employing a molar ratio
of IV:cyanide of within the range of from about 1:1
to about 10:1, at elevated temperatures of from
about 80C to about 130C, in an inert atmosphere,
such a~ an argon atmosphere, to form the cyano-
: carbonate G (which itself is a new compound3
: 25
C~2-CN
\ ¦ C~-0~0-alkyl
,
~,
,
7~ 8
_g_ HA363
CyaIlocarbonate G is dissolved in an alcohol
such as methanol or ethanol and treated with
agueous alkali metal carbonate such as potassium
carbonate at reduced temperature to form
cyano-alcohol H
~ ~ C~2-S:N
~ .
\ ~ CH2 OEI
O
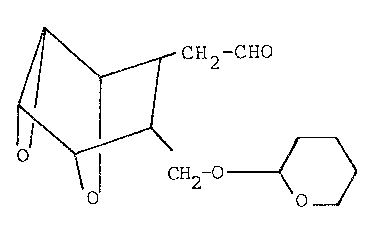
which is made to underyo tetrahydropyranyl ether
fonmation by reacting cyano alcohol H with
dihydropyran in ~he presence of an inert organic
I solvent ~uch as methylene chloride or ether and
catalytic amount of p-toluene sulfonic acid at
reduced tempera~ures of from about O~C to about
10C, to form the tetrahydropyranyl ether of
formula J
C~CN
\O
~- ~
Compound J is then mad~ to undergo epoxide
formation by treating a solution o J in methylene
. .
. ' , .
: .
~_ ~D ~ L ~ 3 ~
HA363
--10--
chloride or other appropriate solvent with
m-chloxoperoxybenzoic acid at reduced temperatures
to form epoxy nitrile II (which itself is a novel
compound)
~ IC~;2-C~
10 Ot~\C~2-0 ~
O O
(which is a novel compound).
ThA compounds of formula I of the invention
may be formed starting with compound II in
accordance wi~h the following reaction sequence.
~ . .
.
.
.
:
363
Ul ~' ~
.~
o
:s:
I H N
u :c C' I~
~ 0
~
~
~\--O
~ oU
a B ~ ~ ~ ~}
:~
~ ~ u
$ ~+=~ 3'`~ ~ ~
C o
~ H
.
.,
e71~
,.12- HA363
C
,,
.x
~6 'e
.~ ' .
: '
'
7~
. 13- HA36 3 `
~.
U1~ ~ H
U I H t.) ~' 2
^~, g ~ y~o
U
~ ~
ns
5 U=O
h Z
a
~ ,
O=
O= ',~
_
C~
H
`
, , . ' '
"~,' '~ ' ' '
.. ' ' ' '
~ . ~,.
: `, ' . ' '
, . . .
~:7~
_ 14- HA363
. 8
~`:
H
O
~
`
r~
:~ ~ O
:~
Il H
. H O X
H
o
, . .
~:,
'
~ ' ' ' ' .
.
. ..
3~3
-15~ H~36~
As seen from the reaction seq~ence set out
above, compounds of the invention may be formed by
treating I I with diisobutyl aluminum hydride
( DIBAL ) in the pre~ence of an inert solvent such
5 as toluene or tetrahydrofuran at reduced
temperatures of from abou~ -70 ~o about -~5C to
form epoxy aldehyde III (which itself is a new
compound )
III ~CE~2-C~O
O I C~I20 ~
0 ~
Epoxy aldehyde I I I in appropriate solvent
such a~ te~xahydrofuran is then react~d wi~h a
susp0nsis:~n ~ormed by mixing dry carboxyalkyltri-
20 phenylphosphonium halide K
.
K ( C H ) P+ B ~ ( (::~I ) COOH.~ :
in tetrahydrofuran with potassium t-a;nylate
25 in toluen~ at reduc:ed temperature and the reaction
produclt treated with ethereal diazoalkane to form
th~ ester IV (which also is a novel compound )
. ,
: .. . .
~ a3 7~ 3~3
~IA3 63
lV f~ 2 CE C}~-(C~2)m-COOR
O ~ OE~2 ~>
O o
(wherein R is lower alkyl )
Compound IV is dissol~ed i~ Ime~thanol and is
the~ hydrolyzed by ~reat~en~ with ~trong acid such
as E~Cl, ~mberlyst r~si~ or ac~tic acid ~o ~orm
al~ohol V ~which also is a ~ovel compound~
2 CEI-(C52)~1~-C02R
O I C~12-0
-.: O
(wherein R is lower alkyl )
2S which also i5 a nov~l co~pou~d.
The e~o:cy e ter V containiIlg the hydroxy-
lue!thyl grouE~ is used to form epoxy aldehyde VI
by subjee~ing epoxy ~s~er V to Collins oxidation,
for ~ aple, by ~e~cting V with chromium oxide in
30 pyridine.
Aldeh~d~ VI of the st~ ture
~ !
* Trade Mark
. ~
.- : -
: -
3L~7 ~L~Le~3
HA3 63
--17--
VI ~ H2-CH=C~- ( CH~ )m-COOR
5 O/\~CHO
wherein R is lower ~lkyl (which also is a novel
10 compound3 is reacted with a dialkoxy phosphonate,
such as of the s~ructure
O O
L ( 30 ) 2P-CH2-~-R
employin5~ a mol2lr ra~io of V:L of within ~e
raIlge of fro~ about 1:1 to about 0 . 5 :1, under
h~sic: cotlditions, such as triethylamine,
diazabicyclo u~decene (DBu) in t::he presen~e of
20 anhydrous lithi~Ln bromide and an inert organic
~olvent, such as methylene chloride or acetonitrile
to orm ~po~cy compound V~I o~ the structure
,~ CH2-CEl-CEI-~C~I2)m-COOR
~5 VII ~
O ~ C~I=C~ R
O, O
. (wherein R is lower alkyl )
~ ' , .
.
i ~ ` , ' -
: .
~7~
H~363
-la-
which also is a novel compound.
Compound VII may then be reduced by two
different ways as outlined above to form compounds
of the invention VIII or IX
~ CE12-C~=C~(CE12) -COOR
Ol ~ A-CH-R
O OH
(wherein R is lower alkyl)
VIII - A is (~2)2
IX - A is -C~=CH-
or compound~ o th~ invention of th~ general
formula X
X ~ 12 C~ CEI-(C~2)~-COOR
: /
O \ I A-CE-Rl
~' ~ .
- ~wherein ~ i~ lower alkyl3
Compounds of formula I
~;
,
.
,
~A363
--19--
~ CH2-CH=C~I-(CH2)m-COOR
IA / ~
\ I A~CH-R
O 1H
(wherein R is hydrogen3
may be prepared by hydrolyzing ester YIII or IX by
trea~ment wi~h a strong base such as sodium
hydroxide or lithium hydroxide in the ~resence o~
an inert solve~t such as tetrahydrofuran, methanol
or dimetho~ye~hane~water to form ~he corresponding
alkali metal salt which i~ then tr~ated wi~h
strong acid such as HCl to form ~he acid compound
of the inv~ntion IF.
The compounds of this invention have seven
cent~rs of asymmetry as indicated by the asterisks
in formula I. ~owever, it will be apparent that
: each of the formulae set out ahove which do not
include asterisks still represent all of the
possible stere~iso~ers thereof. ~11 o~ the v~rious
~ 25 stereoiso~ ric forms are within the scop~ of ~he
inv~nti~n.
The various s~ereoisomeric forms of th~
compou~ds of the invention, namely, cis-exo,
cis-endo and all trans forms ~nd st~reoisomeric
pairs may be prepared as shown in ~he working
Exampl~s which follow and by ~mploying starting
materials a~d following the procedures a outlined
~L~7~1~3~3
~A3 63
-20
in U. 5 ~ Patent No. 4, 143, 054. Ex~nples of such
stereoisomers axe set out below.
Ia TH2~CH=CH~(CH2 )m~C2R
O I t
( cis-endo )
Ib ~ - ~ C~2-cE~=cEi- ( C~2 )I C2
10 ~ '
O~ A-~I R
` OE~
~ ~ ( cis-axo )
.
.
, '
~ ' .
98
HA3 63
--21~
Ic ~L--cH2-cH=cH~(c~2)m C2R
A-~
O ~
( trans )
Id ~E~2 C~-CH-(C~2 )m-C02R
~}I
O A-~ R~
OH
( trans )
The wavy line ( ~ ) in the absve formulae
indica~es that ~he hydroxy ~roup in each of the
compc~unds of fo~ulae Ia-Id is either R(~ 3 or S~a ) .
Th~ nucleus in each of the compounds of the
invention is depi ::ted as
. ~ .
:~;' ' ' , . : -
7~19~3
EIA353
S o~
for matter of con~eni~nce; it will al~o b~
appreciated that the rlucleus in the compounds of
10 the invention may be depicted as
The compounds of this invention are
~: cardiovascula~ agents useful as platelet aggre-
gation inhibitors, suc:h a~ inhibiting aras:hidonic:
20 acid-indu ed platelet aggregation ( e . g ., for
trea~nent of thrombotic disease, such as coronary
or cer~bral thromboses) and iI~ inhibiting bronc:ho-
- . constrictiorl as induced by asl:hma. They are also
selectiv~ ~hromboxane ~ receptor antagonists and
25 ~ he~ase ir~hibi~ors, e.g., having a vasodilatory
ef~ct for ~r~a~en~ of myocardial ischemic
diseas~, such ~s angina pectoris.
The compou~d~ o this invention may also be
u~ed in combination wi~h a cyclic AMP phosphodi-
30 es~era~ (PD13) ir~ibitor such s theophylline or
~- ~ papaverine in the preparation and storage of
platelet concentrates.
.
:
.
:
3~3
EIA3 63
23 -
The compounds of the invention can be
administered orally or paxenterally to various
~nammalian species known to be subject to such
maladies, e.g., humans, cat5, dogs, and the like in
S an effective amount within the dosage range of
about 1 to 100 mg/kg, preferably about 1 to 50
mg/kg and especially abc: ut 2 to 25 mg/kg on a
regim~an in single or 2 to 4 divided daily doses.
The compounds of the inventiorl may also be
10 admini~tered topic:ally to any of the above
mammalian species in amounts of ~rom about 0.1 to
10 mg/kg in single or 2 to 4 divided daily doses.
The artive substance can be utilized in a
compo~ition such as ta3~1et, capsule, solution or
15 suspenslon containing about 5 ~o about 500 mg per
unit of dosage of a compound or mixture o~
compounds of formula I. They may be compounded in
conve~tional matter with a phy iologically
accsptable vehicle or carrier, excipient, binder,
20 preservati~re, stabilizer, flavor, etc. as called
for by accepted phannaceutical practice. Also as
indi ::ated in the discussio~ above, certain members
additionally s~rve as intennediates for other
members of the yroup.
, . .
.~. ~ . - ' .
~7~
H~363
-24-
The following Examples represent preferred
embodiments of the invention. Unless otherwise
indicated, all temperatures are expressed in
degrees Centigrade.
[1~,2~(5Z),3~(1E,3S),4a,5~,6a3-7-~5,6 Epo~y-3-(3-
hydroxy-1-oc~enyl~-7-oxabicyclo r 2.2.1~hept~2-yl]
A. 7-O~abicyclo[2.2.1~.5-hepten-2,3-
To a ~usl?ension of 6 . 84 g of lit:hium
alun~inum hydride ( ï80 mmol ) in 200 ml of freshly
distilled ~, cooled in an ica-water bath was
15 added dropwise, a solution o~ 20 g of
7-oxabicyclo [ 2 . 2 . l J -5-heptene 2, 3 -dicarboxylic
anhydride (120 mmol) in 150 ml o:E dry q~F, over a
period o 1 hour. Ater the addition, the cooliny
:i bath was r~moved and the reac~ion mixture was
20 stirred at room ~emperature for 2~ hours. The t
reaction mixtur~ was now cooled in an ice-water
ba~ a~d excess of hydride was destroyed by slow
addition of freshly prepared saturated sodium
sulfate solution. Addition was corltinued until all
25 th~ organic salts were pres:ipitat~d as white
gr~ular solids. Anhydrous r~agne~ium sulfate was
added to the.reac~ion mix~ure and it was filtered.
:: Th~ residue was thoroughly washed with methylene
chloxide. The residu~ was taken up in 500 ml of
10% acetoni~rile in ethyl aceta~e, stirred for 30
minutes and fina}ly was filtered. The combined
filtrate was concentrated under reduced pressure.
The crude residue was chromatographed on a silica
~' ' , ...
,
:
:~ ' ' ' '
,' ~ ' .
HA363
-25-
gel column. Elution with 50% ethyl acetate in
hexane followed by ethyl a~etate and finally with
10% methanol in ethyl acetate afforded 17.25 g of
title diol as a colorless viscous oil.
B. 7-Ox~bicy~1O[2.2.1]-5-h~ptene-2,3
To a solutio~ of ~6.73 g of Part A diol
(107.4 mmole) in 200 ml of freshly distilled T~F,
cool~d in an ic~-water ba~h was added dxopwise 90
ml of a 12.5% by weight solution o~ phosgene in
toluene (112.5 mmol), over a period of 45 minutes.
Th~ reac~ion ~ixture was s~irred for an additional
15 minut~s, wh~reupon argon was bubbled thxough to
r~move excess of phosgene and hydrogen chloride
formed during the reaction. The reaction mi~ture
was ~ow con¢e~trated u~der reduced pressure. The
crude monochloroformate was now dissolved in 250 ml
of m~thylen~ chloride a~d cooled at 50C in a dry
ice-acetone bath. A solution of 25 ml of pyridine
in 50 ml of methylene chloride was now adde~
dropwise ~ver a period of 20 minutes. An immediate
~ white precipitate was ormed upon addition. .The
- reatio~ ~ixture was left at -50C for a~ additional
30 minutes, whexeupon ~he cooling ba~h was remo~ed
and the reac~io~ mixture wa~ washed thoroughly
: . with water. Th~ me~hylene chloride layer was
~ drie~ ov~r ~nhydrous magnesium sul~ate, filtered
: and the filtrate was concentrated u~der reduçed
:~ 30 pr~ssurs. The crude residue was triturated with
e~her, cooled at 0C and the precipitated title
carbo~at~ was filtered off. 15.25 g of white
- crystalline title carbonate was obtained.
~ .
.`.~' , .
.
:', , . , :
- .
' ' . '
.
'' ' . . ,
:
~L~7~
H~363
-26~
C. 2-Hydrox~methyl-3-isopropyloxycarbonyl-
oxymeth~l-7-oxabicyclo[2.2.11heptene
To a suspension o~ 15.25 g of Part B cyclic
carbonate (83.8 mmole) in 200 ml of isopropyl
S alcohol was added wi~h stirring 1 g of p-toluene
sulfonic acid. The reaction mix~ure was hea~ed
und~r re1ux for 8 hours whereupon it was cooled
and isopropanol wa~ remov~d by distillation under
reduced pressur~. The crude r~sidue was dissol.ved
in methylene chloride and washed with aqueous
sodium bicarbonate solution. The ~queou~ layer
was e~tracted several time~ wi~h m~thylen~
chloride. The combined methylene chloride extract
was dried over anhydrous ~agnesium sulfate and was
then co~centra~ed under reduced pressure ~o obtain
22.53 g of title i~opropyloxycarbonate as a
viscous oilO
:~ .
D. 2~p~Toluene~ulfonyloxyme~hyl-3-isopro~
pyloxycarbonyloxyme~hyl-7 oxabicyclo-
~_ ,.;~
- To a solution of 22.53 g of P~rt C isopropyl-
` oxycarbonate (~4 ~ole) in 190 ~1 of pyridine wa~
added with stirri~g 19.2 g of p-toluene sulfonyl
chloride (101 mmole) at 0-5C. The reaction
~ix~ure wa~ s~irred at xoom ~emp~rature for 24
hours, whereupoM it was dilu~ed with methylene
chloride and wash~d ~horou~hly with water,
saturated copper sulfate solu~ion and finally with
water~ The combined aqueous layer was extracted
with two 200 ml portions of methylene chloride.
The combined methylene chloride ~xtract was dried
over anhydrous magnesium sulfate and finally was
~ '
7~
~A3~3
-~7-
concentrated under reduced pressure. The crude
residue was triturated with ethex, cooled at 0C
and th~ precipitate title tosylate (28.3 g) was
filtered off. The mother liquor was con~entrated
and chromatographed on a silica gel column to
obtain addi~ional S.~ g of crystalline title
tosylate ~eluting solvent 15-30% ethyl acetate in
hexane).
E. 2-Cyano~ethyl 3 isopropylo~ycarbonyl-
To a solution of 5.3 g o.f Part D tosylate
(12.99 ~ol~) in S0 ml o dry dimethylsulfoxide
: was added with ~tirring 1.28 g of powdered sodium
cya~ide (26 mmole). The reaction mixture was
placed on a~ oil bath (bath temperature 90-95C)
and heated ~or 2 hours. It wa~ now cooled and
diluted with 200 ~1 of ether. The rea~tion
: mixtur~ was now thoroug~ly washed with water~ The
combined aqueous extract was extracted with two
150 ml of ether. The e~her layer was now dried
over anhydrous magnesiu~ sulate and concentrated
under reduced pr~s~ure. The crude residue was
chromatoyraphed o~ a silica gel column. Elution
25 with 25-50% ehtyl aceta~e i~ hexane aforded 2.~8
g of title cyano-carbonate.
F. 2-Cyanomethyl,-3Dhyroxymeth~1-7-oxabi
To a ~olution of 1 g of jotassium carbonate
. in 25 ml of watex a~d 75 ml of methanol, cooled in
-` an ice-water ba~h wa~ added with stirring a
;~ solution of 2.58 g of Part E cyano~carbonate (9.8
' , ' ' , '.
' ' ' .
'
~'7~
~A363
~28-
mmol) in lO ml of methanol. After 15 minutes, the
cooling bath was removed and the reaction mixture
was allowed to stand at room temperature for
additional 6 hours, whereupon it was cidified
with lN aqueous hydrochloric acid solution. Most
of me~hanol was now removed by dis~illation under
red~ced pressuxe. The residue was now
exhaustively extracted with methylene chloride
SXl2~ ~ater saturating it with sodium chloride).
The combined organic extract was dried over
anhydrous magn~sium ~ulfate and co~centrated under
reduced pressure. The crude resid~e was
chromatographed on a silica gel column and eluted
with 25-50% ethyl acetate in hexane, followed by
ethyl acetate to obtain 1.23 g of ~itle cyano
alcohol.
G. 2-Cyanomethyl-3-tetrahydropyranyloxy-
A solution of 1.23 g of Part F
:~ cyano-alcohol (7.36 mmole) in 20 ml of dry
methylene chloride was tr~ated with 800 ml of
dihydropyra~ (8.89 mmole) and catalytic amou~t of
p-toluene sulfonic acid at 0-5C. After 4 hours,
~h~ reaction ~ixture was diluted with e~her and
washed wi~h aqu~ous sodium bicarbonate solution.
- Th~ agueous layer was reextracted twice with
ether. Th~ co~bined organic ex~rac~ was dried
~; over anhydrou~ maqnesium sulfate and conce~tra~ed
under reduced pressure. The crude residu~ was
cAro~atogxaphed on.a silica gel column and eluted
with 20-25% ethyl acetate i~ hesane to obtain 1.61
g of title tetr~hydropyranyl ether.
:
. ~ ,
HA363
-29-
. 5,6-Epoxy-2 cyanomethyl-3-tetrahydro-
pyranyloxymethyl-7-oxabicycls[2.2.1]-
A solution of 1.61 g of P~rt G cyano ether
(6.4 mmole~ i~ 20 ml of dry m~thylene chloride wastreated with 1.66 g of ~0% pur~ m-chloroperoxy-
benzoic acid (9.6 l~ole) at 0-5~C~ Af~er a few
mi~utes, ~he cooling-bath was re~oved and ~he
reactioD mi~ure wa~ l~t ~tand at roo~ temperature
10 for 6 hours. The reaction mix~ure was now diluted
wi~h e~her and e~cess of peracid w~s de~omposed by
addition of ~queous sodiu~ meta-bi~ulfits
solution. After stirring for 30 minute~, the
~ organic layer was ~eparated and the agueous layer
.~ 15 wa~ e~tracted t~ir~ with methylene chloride. The
co~bined organic extract was dried over anhydrous
~agne~iu~ sulfa~e and conc~trated under reduced
pressure. Purifica~io~ by chromatography o~ a
silica gel colu~n (eluting solve~t 25 67% e~hyl
acetate i~ hexane) ~forded 1.57 g of title epoxide.
.
~ J. 5,6-~poxy-2-formyIme~hyl-3-tetrahydro-
;~ ~ pyranylo~ymethyl-7-oxabicyclo[2.2.1]-
: 25 To a solution o Par~ Y epoxy-ni~rile (1.57
g, 5.88 m~ole) in 25 ml of toluene, cooled at
-78C i~ a d~y ic~-acetone bath was added with
tirring, 6.a ~l o a 25% by wei~h~ ~olution of
~` diisobu~ylaluminum hydrid~ in ~oluene (~12 mmole),
dropwi~e over a period of 5 minutes. After 4
:~ hour~ a~ -78C, exces of hydride was destxoyed by
dropwise addition of 1 ml of glacial acetlc acid.
~ The cooling bath was removed and 20 g of silica
:'
.
- .: . . . .
. ' :. '
, - :
: -
, , .
HA363
~30-
gel was added to the reaction mixture with
stirring, followed by 1.5 ml of water dropwise.
Stirring was sontinued for 30 minutes, whereupon
the reaction mixture was filtered and the residual
S silica gel was washed successively with THF, 5%
acetonitrile in ethyl acetate and finally with
acetone. The combined filtrate was conce~trated
under reduced pressure and the crude residue was
chromatographed on a silica gel column. Elution
with 50% e~hyl aceta~e in hexane, followed by
e~hyl acata~e afforded 1.16 g of title
epoxyaldehyde which crys~allized on s~anding at
_20C .
K. [la,2~(5Z~,3~,4a,5a,6a]-7-[5,6-Epoxy-3-
~tetrahydropyranyloxymethyl)-7-oxabi~
cyclo[2.2.1~hept-2-yl]-5-heptenoic
acid, methyl ester
,~
A suspension of 5.77 g of freshly dried
carboxybutyltriphenylphosphonium bromide (13.03
mmol), in 50 ml of freshly distilled THF, cooled
in a~ ice~water ba~h was ~reated dropwise with 12
ml of a l.S M solution of K-t-amylate in toluene
(19.2 mmol~). The yellow-orange suspe~sion was
: 25 s~irred at 0C ~or 30 ~inute~ and finally at room
temperature for 1 hour, whereupon it was cooled
to -20C and a solution.of 2.33 g of Part J epoxy
ald~hyde (8.69 mmole) in 10 ml of dry T~F was
: added dropwise over a p~riod of several minutes.
An instant discolorization of the ylide solution
was observed. The reaction mi~ture was stirred at
-20C for 2 hours, whereupon it was warmed to 0C
and left for 15 minutes, prior to addition of
, ~:
HA3 63
--31--
glacial acetic acid. The rea ::tion mixture was now
diluted with e ther and washed with water . The
ether extract was washed several times with
saturated ~odium bicarbonate solution. The
S combined aqueous extract was now washed with ether
~X2 ) . The aqueous layer was now care~ully
acidified with lN aqueous hydrochloric acid to pH
2. It was now extracted with ether and then with
me~thylerle chloride. Th~ combined ether alld
methylene chloride extract was dried over
anhydrous magnesium sulfate and co~centatad under
reduced pressure. The crude residue was diluted
with 75 ml of ether, cooled in an ice-water bath
and an etheral diazometharle solution was added
dropwise until 'che color persisted. After 30
minutes, exces diazomethane was remov~d by
bubbling argon through th~ reaction mixture. It
was ~ow concentrated arld the crude residue was
chromatographed on a silica gel column. Elution
with 15-40% ethyl acetate in hexane afforded 1.27
q of title 5Z~ester (contaminated with 10-15% of
~` undesired 5E ester).
Lo [1~,2~l5Z),3~,4a,5a,6a3-7~[5,6~
Epoxy-3-hydroxymethyl 7-oxabicyclo-
~2.2.1]hept-2-yl~-5-heptcnoic acid,
: m ~
To a solution of 1.27 g of Part K tetrahydro-
pyranyl ether ( 3 . 46 mmol~ 30 ml of methanol
was added with stirri~g 250 mg of powdered and
dried Amberlyst-15. After 6 hours at room
temperature, the reaction mixture was diluted with
ether and anhydrous ma~nesium sulfate was added.
~dE
.,' ~ ,'-' ' ~ ' ~ '
~ A363
-3~-
It w~s ncw filtered and the residual solid was
washed ~horoughly wi~ ether. The combined
organic extract was dried over anhydrous magnesium
sulfate and conc~ntrated undar reduced pressure.
The crude residue was c.~romatographed on a siLica
gel colu~n a~d eluted with 50-75~ e~yl ~cetat~ in
hexa~ to obtain 892 mg of title alcohol ester.
M. ~la,2~(5Z),3~,4a,5a,6a~-7-[5,6-
a Fpoxy ~ 3 ~ for~yl-7-~abi~ycLo[2.2.1~-
he~t-2-yl]-5 hept~oic acid, methyl
To a ~uspe~sion of 325 mg of pyridinium
chlorochromate and 325 mg of Celite in 20 ml of
d~y methylene chloride was added with stirri~g a
s~lution o~ 211 mg Part ~ alcohol ester (0.75
mmole) in ~ ml of ~eth~l~n~ chlorida. A ter 4
hours at room temperature, ~he r~action mi~ture
wa~ diluted with 100 ml of ether and filter~d
through a pad of Florisil. Florisil was washed
~everal times with ether and ethyl acotat~. The
combined organic e~tract was washed with water,
d~ied over ~nhydrous m~qnesium sulfate and w~s
then co~centrated under reduced pressure to obtain
2S 174 mg of title aldehyda.
N. [la,2~(5Z),3~(1E~,4~,5,6a]-7-~5,6-
~poxy-3~(3-~xo-l-octe~yl~-7-
oxabicyclo~2.2.1]hept~2-yl]-5-
To a suspe~sion o~ 90 mg of dry li~ium
:~: bromide in 5 ml of dry methylene chloride was
added with stirring 140 ~1 of triethylamine,
* Trade Mark
:
.
.~ .
7~
HA363
-33-
followed by a solution of 222 mg dimethyl-l2-
oxohep~yl) phosphonate (1 mmole) in 1 ml of
methylene chloride. After stirring for 15 minutes
at room temperature, a solution of 174 mg Part M
5 aldehyde (O.62 mmole) in 3 ml of methylene
chloride was added ~ropwise. The reaction was
stirred overnight, whereupon it was diluted with
ether and washed with water. The aqueous layer
was extracted with ether ~X2). The co~bined ether
extract was dried over a~hydrous magnesium sulfat~
and concentrated under reduced preslsure. The
crude oily residue was chromatographed on a silic~
gel column and eluted with 15~30% ekhyl acetate in
hexane to obtain 175 mg of titla enone.
O. ~1~,2~(5Z),3~(1E,3S),4~,5a,6~]-7-[5,6-
Epoxy-3-(3~hydroxy-1-octenyl) -7oxabi-
cyclo[2.2.1~hept-2-yl]-5-heptenoic
and
P. {1~,2~5Z),3~(1E,3R),4~,5a,6~]-7~[5,6~
Epoxy~3-~3-hydroxy-1-oct~nyl)-7-oxabi-
cyclo~2.2.13hept 2-yl~-5-heptenoic
To a solutio~ of 170 ~g of Part N enone
:~ (0.45 mmole) in 5 ml of methanol and THF each, was
added wi~h s~irring I70 mg of ceric chloride
hydrate. After 10 minutes at room temperablre,
the homogeneous solution was cooled ~o ~50C in a
- 30 dry ice-acetone bath and 20 mg of solid sodium boro-
hydride (O.S mmole) was added. The reaction
mixture was stirred at -50C for 1 hour, whereupon
t was treated with aqueous ammonium chloride
- :'
HA3~3
-34-
solution. The cooling bath was removed and the
reaction mixture was diluted with ether. The
organic layer was separated and the aqueous layer
was reextracted successively with ether and
methylene ~loride. The combi~ed or~anic extract
was dried over anhydrous magnesium sulfate and
concentrated. Purification by chromatography on a
~ilica gel colu~n and ~lution with 30-50% ethyl
acetate in hexane afforded 130 mg of title
fast-moving alcohol epimer and 40 mg of
slow-moving isomer.
Example 2
[1~,2~(5Z),3~ ,3S),4~,5u,6~ 7-[5,6-~poxy-3-(3-
: 15 hydroxy-1-octenyl~-7 oxabicyclot2.2.13hept-~-yl]-
A solution of 130 mg of Example 1
:~ fast-moving alcohol epimer (0.35 mmole) in 5 ml of
distilled lnF was treated with 2 ml of a lN
. 20 aqueou~ lithium hydroxide solution. After B hours
; at room temperatur~, the reaction mixture was
diluted with ether and acidified to pH 1 by
additio~ of lN agueous hydrochloric acid
~ solution. The ether layer was separated and the
- 25 aqueous layer was extracted with methylene
chloride (X2). The combined organic extract was
dri~d over anhydrous magnesium sulf~te a~d
conce~trated under reduced pressure to obtain 120
.~ mg of crude acid (contaminated with 10-15% of
io presumably 5E-isomer~. Chroma~ography on a silica
gel col~unn and elution with 2-3% me~hanol in
me~hylene chloride af~orded 80 mg of pure title
.~ acid.
. .
~ .
', ~ .
. . : . . .
7~1~8
HA363
-35~
Anal calcd for C21~325 C~ 69-20; ~ 8-85
Found: C, 69.24; ~, 8.84
xam~le 3
[la,2~(5Z~,3~(1E,3R),4a,5a,6a]~7-[5,6-Epoxy-3-(3-
hydroxy-1-octenyl)-7-oxabicyclo[2.2.1]hept-2-yl]
5-heE~tenoic acid (s _ _ _
To a sslution of 40 mg ~la,2~(5Z),3~(1E,3S),-
4a,5a,~a] 7-[5,6-epoxy 3-~3-hydroxy-oct~nyl)-
7-oxabicyclo~2.2.1]hept~2-yl]-5-heptenoic acid,
me~hyl ester slow-~oving alcohol ep~mer (prepared
a~; described i~ ~xample 1 Paxt P) (0.11 m~ole) in
3 ml of distilled THF was added with stirring 1 ml
of a lN aqueous lithium hydroxide solution. After
8 hours at room temperature, the reaction mixture
was diluted with e~her and acidified with lN
agueou~ hydrochloric acid solution of p~ 1.
The organic layer was separated and the aqueous
layer was ~x~racted with methylene chloxide. The
; 20 combined organic extract was dried with anhydrous
magnesium sulfat~ and then concentrated under
reduced pressure to obtain 33 mg of title acid as
- an oil.
25Anal Calcd for C21H325~ 0-96 mole of ~2
; C, 66~06; ~, 8.95
~ Found: C, 66.06~ .47
~ .
[1~,2~(5Z),3~(1E,3S),4a,5a,6a]-7-[5,6-Epoxy-3~(3-
cyclohexyl-3-hydroxy-1-propenyl)-7-oxabicyclo-
[2.2.1]hept-2-yl]-5-hept~noic acid (fast moving
isomer)
- --
~7~1~8
~A3 63
--36--
A. [la,2~(5Z),3~(1E),4a,5a,6a]-7-[5,6-
Epoxy-3-(3-oxo-3-~yclohexyl-1-propenyl)-
7~oxabicyclo[2.2.1]hept 2-yl]-5-
heptenoic acid, meth~l ester
To a suspension of 135 mg of dxy lithium
bromide in 5 ml of methylene chloxide was added
with stirring 200 ~l of triethylamine, followed by
a solution of 350 mg 2-oxo-cyclohexyl~dimethyl)-
phospho~ate ~1.5 mmole) in 2 ml of methylene
chloxide. After stirring for 15 minutes at room
temperature, a solution of 220 mg of ~1~,2~(5Z~,-
3~(1E,3S),4~,5a,6a]-7-~5,6-epoxy-3+formyl-7-
oxybicyclo[2.2.1]hep~ 2-yl]~5-heptenoic acid,
methyl ester prepared as described in Example 1
Part M in 3 ml of methylene chloride was added
dropwise~ Th~ reaction mixture was stirred at
room tamperature for 3 hours whereupon it was
diluted wi~h ether and washqd wi~h water. The
aqueous layer was extracted with e~her (X~). The
combined e~her extract was dried over anhydrous
ma~nesium sulfat~ and was then concentrated under
reduced pressure. Chromatographic pur.ification on
; a silica gel col~mn (eluting sol~ent 10 30% ethyl
acetate i~ he~an23 gave 235 mg of desired title
eno~e.
B. [la,2~(52),3~(1E,3S),4a,5a,Sa]-7-[5,6-
Epoxy-3-~3 cy~lohe~yl-3-hydxoxy~
:~ propenyl3-7-o~abicyclo[2.2.1]hept-2 yl]-
:~ ~ 30 5-he~tenoic acid, methyl ester (fast
movina isomerl
and
::
- ~ .
' : : ' . '. .
.
~ '
~7~
~A363
37-
C. [la,2~(5Z),3~(1E,3R),4a,5~,6a]-7 [5,6-
Epoxy-3-~3-cyclohexyl-3-hydroxy-1-
propenyl)-7-oxabicyclo~2.2.1]hept-2-yl]-
5-heptenoic acid, methyl ester (slow
To a solu~ion of 235 mg of Part A enone
(O.61 mmole) ~n 5 ml of methanol and T~F was
added with stirring 235 mg ceric (111~ chloride
hydra~e. Aftar ctirring or lO minut~s at room
temperature, the reactio~ mixture was cooled to
-50C and 25 mg of solid sodium borohydride (O.66
mmole) was added. After 1 hour at! 50C, the
reaction mixture was quenched b.y addition of
aqueous ammonium chlorid solution. It was now
warmed to room temper~ture and diluted with
ether. The organic layer was separated and the
agueous layex was extracted with methylene
chloride. The combined organic extract was dried
over anhyd~ous magnesium sulfate and concentrated
: 20 und~r r~duced pr~ssure. The crude residue was
~ chromatograph~d on a silica gel column and eluted
: with 30-67% ethyl acetate in hexane to obtain 17S
mg of title B fast-moving aIcohol epimer and 35 mg
of title C slow-moving alcohol epimer.
: 25
: [la,2~(5Z),3~(1E,35~,4~,5a,6a]-7-[5,6-Epoxy-3~(3-
cyclohexyl-3-hydroxy-1-prope~yl)-7-oxabicyclo-
[2.2.1]he~t-2-yl]-5 h~ptenoic acid (fast moving
~
A solution of 175 mg of E~ample 4, Part B,
alcohol-ester (fast moving isomer) in ~ ml of dry
T~F was trea~ed with 2 ml of lN aqueous lithium
~ ' ' ' .
:
,, ,
. ' , ::
~' ' ' '
~ ~7~
HA363
-38-
hydroxide solution. The reaction mixture was
stirred at room temperature for 8 hours, whereupon
it was acidified with lN aqueous hydrochloric acid
solution. It was then diluted with ether and the
organic layer was separated. The aqueous layer
was extracted with methylene chloride twice. The
combined organic ex~rac~ was dried over anhydrous
mag~esium sulfate and was then concentrated under
reduced pressure to obtain 163 mg of crude acid.
Chromatography on a silica gel column and elution
with 3-5~ methanol in methylene chloride afforded
110 mg.of title acid.
for C22H3205: C, 70.18; H, 8.57
15 Found: C, 70.03; H, 8~59
Exam~le 6
[la,2~(5Z3,3~(1E,3R),4a,5a,6a]-7-[5,6-Epoxy-3-(3-
cyclohe~yl-3-hydroxy-1-propenyl)~7-o~abicyclo-
2Q ~2.2.1]hept-~yl]-5-hept~noic acid ~slow moving
isomer 2 ,.. ,
: To a ~olution of 35 mg Example 4 Part ~
slow-moving alcohol epimer (O.09 mmole) in 3 ml of
distilled THF was added wi~h stirring 1 ml of a lN
aqueous li~hium hydroxide solution. After 8 hours
at room temperature, the reactio~ mi~ture was
dilu~ed with ether a~d acidifiad to p~ 1 with
: lN aqueous hydrochloric acid solution. Th~
organlc layer was separated and th~ agueous layer
was extracted wi~h methylene chlorideO The
combined organic extract was dried over anhydrous
: magnesium sulfate and finally was concentrated
. ~ .
H~363
-39-
under reduced pressure ~o obtain 2a mg of title
acid.
A~al Calcd for C22H3205, 0.27 mole of water:
S C, 69.28; H, 8.60
Fou~d: C, 69.28; H, 8.71
~la,2~(5Z),3~(1E,3R,4S),4a,5~,6a]-7-[5,6~Epoxy-
: 10 3 ~3-hydroxy-4-phenyl-1-pentenyl)-7-oxabi yclo-
. [1~,2~(5Z),3~(1E,3R,4S),i~,5a,6a]-7-
[5,6-Epoxy-[3-~3-oxo-4-phenyl-1-
pent~nyl~-7-oxabicyclo[2.2.1]hept-2
~ ~
To a suspension of 90 my of anhydrous
li~hi~ bromide (1 mmole) in 5 ml of dry methyle~e
chloride was added with stirring 140 ~l triethyl-
amine (1 mmol~). 256 mg of S+) dimethyl(2-oxo-3-
methyl~3-phen~lj propyl phosphonate was then added
dropwise. After 15 minutes at room temperature, a }
solution of Example 1 Part ~ 5,6-exo epo~y aldehyde
(170 mg, 0.62 mmole) in 3 ml of methylene chloride
was add~d slowly. The reaction mixture was stirred
at room te~peratuxe overni~ht, whereupon it was
diluted wi~h e~her and washed with wa~er. The
org~nic layer ~as dried over anhydrou~ ~agnesium
sul~ate, filtered and concentratred und~x reduced
pressure. The crude oily residue was chromato-
: 30 graphed on a silica gel column and eluted with
15-30% ethyl acetate in hexane to obtain 177 mg o
title enone.
~2 ~
HA363
~40-
B. [1~,2~(5Z),3~(1E,3R,4S),4~,5a,6a]-7-
[5,6-Epoxy-3~ hydroxy-4-phenyl-1-
pentenyl)-7-oxabicyclo[2.2.1]hept-2-yl]-
5-heptenoic acid, methyl ester
To a solution of 177 mg of Part A e~one in
5 ml of dry me~hanol and 5 ml of distilled THF was
added wi~h stirring 175 mg of ceric (III) chloride
hydrate. After 10 minutes at room temperature,
the homogeneous solution was cooled to -50C in
dry iceacetone bath and 20 mg of solid sodium boro-
hydride was added with stirring. ~fter 1 hour at
-50C, the reac~io~ mixture was quënched by
addition of aqueous ammonium chloride solution,
wa~med to room temperature and w~s then diluted
with e~her. The organic lay~r was separated and
the agueous layer was ex~racted twice with ether
and ~wice with methyl~ne chloride. The combined
organic extract was dried over anhydrous magnesium
sulfate and concentrated under reduced pressure.
: 20 The cru~e residue was chromatographed on a silica
gel column and eluted with 20-50% ethyl ac~tate in
hexane to obtain 133 mg of title alcohol-ester as
an oil.
Ex~mç~
[la,2~(5Z),3~1E,3R,4S),4a,5a,6a]-7-[5,6-Epoxy-3-
(3-hydroxy-4-phe~yl-1-p~ntenyl)-7-oxabicyclo-
A solutio~ o~ 133 mq Example 7 alcohol-ester
(0.32 mmole) in 5 ml of distilled THF was ~reated
with 2 ml of lN aqueous lithium hydro~ide solution.
The reaction mixture was stirred at room tempera-
- ture for 16 hours, whereupon it was carefully
: .,
.. . .
` '
:' .
.
7~
HA363
-41-
acidified to pH 1 by addition of lN aqueous
hydrochloric acid solution. It was now diluted
with ether and the organic layer was separated.
The aqueous layer was e~tracted with methylene
chloride (X2). The combined organic extract was
dried over anhydrous magnesium sulfate and
co~centrated under reduced pres~ure to obtain 124
mg crude acid, contaminated with ~15% of ~-side
chain ol~fin isomer. Chromatography on a silica gel
column and elution with 1-3% m~hanol in methylene
chloride afforded 51 mg of title afid as an oil.
Anal Calcd for C24H305 C~ 72 33; ~ 7-59
Found: C, 72.32; H, 7.57
: 15
Exam~le 9
~1~,2~(5Z) 3~(1E,3S~,4a,5a,6a]-7-[5,6-Epoxy-
3-~3-hydro~y3-phenyl-l~propenyl)-7-oxabicyclo-
Following the procedure of Examples 7 and 8
excep~ substituting dimethyl(2-oxo-~-phenyl)-
ethyl phosphonate for (+)dimethyl(~-o~o 3-methyl-
3-phenyl)propyl phosphonate, acid, the title
compound is obtained.
tl~,2~(5Z~,3~(113,35),4a,5a,6a]]-7-[5,6-Epoxy-
3 (3-hydroxy-4-phenyl-1-butenyl)-7-oxabicyclo
Following the procedure of Ex~mples 7 and 8
except substituting dimethyl(2-oxo-4-phenyl)-
butyl phosphona~e or (+)dimethyl(2-oxo-3-
.
: :`
. .
HA363
-42-
methyl-3-phenyl)propyl phosphonate, the title
compound i5 obtained.
~e~
[la,2~(5æ~,3~(1E,3S~4~,5~,6~]-7-[5,6 Epoxy-
3-(3-hydro~y-5-phe~yl-1-pe~tenyl)-7-oxabi-
cyclo~2.2.1~hept-2-yl]-5-heptenoic acid,
m ~
A. ~la,2~(5Z),3~(1E),4~,5a,6a~-7-[5,6-
Epo~y-3-~3-oxo~5-phenyl-1-pentenyl)-
7-oxabicyclo[2~2.13heptT2-yl]-5-
To a suspension of 135 mg anhydrous li~hium
bxomide (1.56 mmole) in 3 ml of dry methylene
chloride was added wi~h stirring 198 ~l
triethylamin~ ~1.42 mmol~. 386 mg dimethyl
(2-oxo-4-phenyl)butyl phosphona~e (1.51 mmole) in
1 ml o me~hylene chloride wa~ now add~d
dropwise. After 30 minutes at room temperature, a
solution of Example 1 Part M 5~6-exo~epoxy
aldehyde (200 mg, 0.7 mmole) in 3 ml of methylene
chloride was added dropwise. The reaction mixture
: wa~ s~irred at room tempera~ure overnight,
whereupo~ it was diluted with ether and washed
wi~h water. The organic layer was dried over
anhydrou~ ~agnei~um sulate and concentrated under
re~uced pres~ure. The crude residue was
chroma~ographed on a silica gel col~mn a~d elu~ed
; wi~h 40% ethyl acetate in h~xane to obtain 208 mg
of title e~one.
. :
, ~ :
.
H~363
-43-
B. [la,2~(5Z),3~(1E,3S),4a,5~,6a]-7-[5,6-
Epo~y-3-~3-hydroxy-5-phenyl-1-pentenyl)-
7 oxabicyclo~2.~ hept-2-yl]~5-
heptenoic acid, meth~l ester
~o a solution of 20~ mg of Part ~ enone
(0~5 mmole) in 1 ml of methanol and 1 ml of
methylene chloride was added with stirring 124 mg
ceric chloride hydrate. After 10 minu~es at room
temperatur , the homogeneous solution was cooled
to -50C and 19 mg of solid sodium borohydride
(0.5 mmole) was added. The reacti~n mixture was
let stand at -50C ~or 3 hours, wh~reupon it was
treated with aqueous ammonium chloride solution.
The cooling bath was removed and the reaction
mix~ure was diluted with ether. The organic layer
was separated and the aqueous layer W~8 extracted
~ucce~sively with ether and me~hylene chloride.
The combined organic extract was dxied over
anhydrous magnesium sulfate and co~centrat~d. The
~ 20 crude residue was chromatographed on a silica gel
:; column and eluted with 40% ethyl acetat~ in hexane
to o~tain 11~ mg of ~itle fast moving alcohol
epimer and 40 mg of slow movi~g isomer.
`~ 25 ~
[1~,2~(5Z~,3~(1E,3S),4a,5~,6a]~7-~5,6-Epoxy-3-(3-
~ . hydroxy-5~phenyl-1-pentenyl3-7 o~abicyclo[2.2.1]-
; . . ~ solution of 114 mg of ~xam~le 11 ester
30 (0.27 mmole) in 1 ml o f THF and 1 ml o f lN aqueous
hium hydroxide was stirred at 25C for 2 hours.
The reaction mix~ure was concen~rated and ~h4n
:~.
. ~
: .
$~3~
H~363
-44~
acidified with oxalic acid solution to pH 3. It
was now e~tracted with ether (X3). The combined
ether extract was dried over anhydrous magnesium
sulfate and concentrated under reduced pressure to
give 100 mg crude oil. Purifications on
preparative ilica gel plates (elu~ing solven~ 10%
in methylene chloride) gave 37 mg of title acid.
alcd for C24H305 C~ 72-33; ~ 7.59
Fo~lnd: C, 72.10; ~, 7.5g
~ i
r~ 2~(5z3~3~(lE~3s)4a~5a~6a]-7-[5~6oEpoxy-
3-(3-hydroxy-4-cyclope~tyl-1-butenyl)-7-oxabi-
cyclor~2.Z~Ih ~ _
Following the procedure of Exa~ples 7 and 8
~xcept substituting dimethyl(2~oxo-3-cyclo-
pentyl)propyl phosphonate for (~dimethyl(2~oxo-3-
methyl-3 phe~yl)propyl phosphonate, the title
29 compound i5 obtained.
[1~,2~(5Z),3~(1E,35~4a,5a,6a]-7-[5,6-Epo~y-
3-(3-hydroxy-1,5-hexadienyl)-7-oxabicyclot2.2.1]-
Following ~he pxocedur~ of Examples 7 and B
except subs~i~uting di~ethyl(2-oxo4-pentenyl)-
phosphonate for (~)dime~hyl(2-oxo-3-methylQ3-
phe~yl)propyl phosphonate, the title compound is
obtained.
HA363
-~5-
[la,2~(5Z),3~(1E,3S)4~,5a,6a]-7-~5,6-Epoxy-
3 (3-hydroxy-1 nonenyl)-7-o~abicyclo[2.2.1]hept-
Following the procedure of Ex~mples 7 and 8
e~cept substitu~i~g dimethyl(2 oxo-octyl)phos
phona~e for (~dime~hyl(2-oxo=-3-~ethyl-3-phenyl)~
propyl phosphonate, ~he title compou~d is obtained.
~
[la,2~(5Z),3~1E,3S)4a,5a,6a]-7-[5,6-Epoxy-
3-(3-hy~roxy1-pentenyl)~7-oxabicy~10[2.2.1~hept-
2~ 5-heptenols acid
Following the procedure of Examples 7 and 8
except substituti~g dimethyl(2-oxo-butyl)phos-
pho~ate for (~)dimethyl(2-oxo-3-methyl 3-phenyl)
propyl phosphonat~, the titl~ co~pound is obtained.
Example 1~
[la,2~5Z~,3~3R,4S),4a,5a,6a] 7-[5,6-Epoxy-3-
(3-hydro~y-4-phenyl-1-pentyl)-7-oxabicyclo[2.2.1]-
~' _
. A. [la,2~(5Z),3~(3R,4S),4~,5~,6a]-7-[5,~-
~ Epoxy3-(3-hydroxy-4~phenyl-1-pentyl)-
:~ 25 7-oxabicy~10[2~2.1Jhept-2-yl]-5-
,~:
:~; To a suspension of 686 mg of purified
cuprous bromide (4.8 mmole) in 12 r~l of dry TH~,
~: cooled at 0-5C is added with ~tirring 1.35 ml of
3û a 3 . 5 M solution of red-A1 ( sodium bis ( 2-methoxy-
ethoxy)aluminum hydride) in toluene dropwise. The
;~ solution is stirred at 0-5C for 30 minutes,
whereupon it is c::ooled to 78C and 2 ml of
butanol ( 18 mmole ) ls added rapidly~ followed
35 by A solutliDn of 672 mg of Example 7 Part A enone
.
.
HA363
-~6-
t2 mmole) in 4 ml of dry THF. After 10 minutes at
-78C, the reaction mixture is warmed to -20C and
left for an additional 1 hour. The reaction
mixture is quenched by addition of 70 ml of water
and then poured into saturated ammonium chloride
solution and e~tracted with e~her ~X3). The ether
extract is dried over anhydrous magnesium sulfate,
filtered and the filtrat~ is co~centrated under
reduced presæure. 675 ~g of desired title keton~
0 i5 obtained.
To a solution of 338 mg of ketone
~1 mmol~) (prepared as described a~ove) in 2 ml of
me~thanol and 2 ml of dry T~F is added with stirring
400 mg of ceric (III) chloride hydrate (1 mmole).
After stirring at room temperature or 10 minutes,
~he reaction mixture is cooled to -50C a~d 38 mg
: of solid sodiu~ borohydride (~1 mmole~ is added to
the reaction ~ixture. The reactio~ mixture is
stirred at ~50C for 45 minutes, whereupon 5 ml of
acetone is added to destroy excess of borohydride.
The mixture is stirred for an additional S minutes
a~ -50C. The cooling bath is removed and the
reaction mix~ure is evaporated to dryness. The
; crude residue is diluted wi~h ether and washed
with 1 N aqueous hydrochloric acid solution. The
ether ex~rac~ is dried over anhydrous MgS04 and
concen~rated under reduced pressure. The crude
residu~ is chromatographed on a silica gel column
and eluted with ethyl acetate (30-50%) in hexane
to obtain the desired title alcohol.
B. [1~,2~(5Z),3~(3R,4S),4a,5a,6u]-7~
~5,6-Epoxy-3~(3-hydxoxy-4-phenyl-1-
pentyl)-7~o~abicyclo[~.2.1]hept-2-yl]-
~
.
,
HA363
-47
Following the procedure of Example 8 except
substituting the above Part A alcohol ester for
the Example 7 alcohol ester, the title compound is
obtained.
~1~,2~(5Z),3~(3S),4~,5~,6~]-7-~5,6-Epoxy-3-(3- -
hydroxy-3 phenyl-1-propyl)-7 oxabicyclo[2.2ul]-
~ L~L-~L~ acid
Following the proc~dure of ~xample 16 and
E~amples 7 and 8 exc~pt substituti~g benzoic acid
for 2-phenylpro~ionic acid, the title compound is
obtai~ed.
~ .
{la,2~(5Z),3~(3S~,4~,5a,6a~-7-~5,6-Epoxy-3-(3-
hydroxy~-phenyl1-buty})-7-oxabicyclo[2.2.1J-
Follswing the procedure of Example 16 and
E~mples 7 and 8 except substituting phenylacetic
acid for 2-ph~nylpropionic acid, the title compound
~: is obtained.
~;
2~ ~3~Ee~
[1~,2~(5Z~,3~(3S),4~,5a,6a] 7-[5,6-Epoxy-3-~3-
~y~ro~y 3-cyd ohexyl-1-propyl) 7-oxabicyclo[2.2.1~-
- Following the procedure o~ Exampl~ 16 and
Examples 7 aIld 8 exc:ept ~ubstitutirlg cyclohexyl-
car}:~oxylic acid for 2-phenylpropionic acid, the
title compound is obtained.
.
: '
,~ .
~,~
,
~. :
,
lX7~1"3B
HA363
-48-
Examples 20 to 29
Following the proceduxe of Examples 7 and 8
(where A is C~=CH~ and Example 16 (where A is
(CH2)2)~ except ~ubstituting for carboxybutyl-
triphenylphosphonium bromide, the compound shown inColumn I of Table I set out below and substituting
for (~3dim~thyl(2-oxo-3-methyl-3-phenyl)propyl
phospho~ate, the compound shown in Column II,
the compound of the invention shown in Column III
is obtained~
' ~,
, - , '
~, .. .
~7~L9f~
~49- HP.363
o
a ~ u~ ~ ,,
- ~ X~ X~ J ~ ~
~ S
~ C~ _~
C`l ~ ~ ~ ~ U'~
o ::
~ o I ~ S~
~" Oel~
W ~ ~1
~¦ ~ o= IL~ ~ t~ 11 ~ O
o
D U ~
a
5:
C~
'~ ~ C~
o ~
~ U-
. _ .
. ~ .
.
X ~ C: ~ C~l ~ ~ U~ ~ r~
~ .
:`
:.
~.~7~19~
HA363
--SO--
::
~ ::~
~ ~1
ot,
~: 3: ,_
W ~,
~ ~1
~ i~l~
~ ~ '
. .
:4 ~ X ~
' ' ` C`t ~ ,
,~
.
'
.
'. '
' - ' '
-..'.
~7~C~
~A363
-51
Example 30
[la,2~(5Z),3~(1E),4a,5a,6a]-7-[5,6-Epoxy-3-[3-
hydroxy-3~ methylcyclohexyl)-1-propenyl]-
7-oxabicyclo[2.2.1~hept-2-yl];5-heptenoic acid,
S m
A. [la,2~5Z),3~(1E),4u,5a,6a]-7-[5,6-
Epoxy-3 [3-oxo-3-(1-me~hylcyclohexyl)-
1-propenyl3 7-oxabicyclo[2.2.1]hept-2-
y~lL5~ noic acid~ tky~_c~e~
To a slurry of 135 mg of lithium bromide
~1.56 mmole, 2.2 egui~.) in 3 ml of dry methylene
chloxide at 25C was added a solu~ion of 293 mg of
dimethyl [2~ methylcyclohexyl)]-2-oxo ethyl
phosphonate (loS mmole, 2.1 equiv.) i~ 1 ml of
methylene chloride ~nd 198 ml of triethylamine
(1.42 ~mole, 2.01 equiv.). After stirring for 30
minutes, a solution of ca. 0.70 mmole of [la,2~-
' ~ (5Z),3~(1E),4a,5a,6a]-7-t5,6~epoxy-3-formyl-
7-o~abicyclo[2.2.1]hept~2-yl]-5-h~pt~noic acid,
~ 20 methyl ester (prepared as describ~d in Example 1
-~ Part M) in 1 ml of methylene chloride was added.
~: The stirring was continued a~ 2SC for 18 hours.
The reaction mixture was then treated with 5 ml of
lM Na~2P04 solution and diluted with 30 ml of
~25 ether. The layers were separated. The organic
: layer was was~ed wi~h 10 ml of a saturated K~C03
solution, lO ~l f ~2 and 10 ml of brine. The
organic layer was then dried (MgS04) and
concentrated.
The residue wa~ purifi~d on a silica gel
.~ coIumn. Elution wi~h 25% EtOAc/hexane gave 170 mg
of title enone as a clear oil.
~' ~ ' ".
~ , :
.~
127~ g~3
.
HA363
-52~
B. [1~,2~5Z),3~(1E,3S),4a,5a,6a]-7-[5,6-
Epoxy-3-[3-hydroxy~3-(1-methylcyclo-
hexyl)-l propenyl]~7~oxabicyclo-
f2.2.1]hept 2-yl~-5-heptenoic acid,
m~th l ester
To a solution of 170 mg of Part A enone
(0.42 mmole) in 1 ml of me~hanol and 1 ml of THF
a~ 25C was added 102.9 mg of c~rium trichloride
(0.42 ~nole, 1 equiv.). ~f~er stirring at 25C
for 15 minutes, the mixture was cooled to -50C
a~d 15.9 mg of sodium borohydride ~.42 mmole, 4
equi~. was added. The mix~ure waslstirred at
-50C for 3 hours, then poured into 30 ml of a
saturated solution of ammonium chloride. The
agueous solution was extracted with three 15 ml
portlons of ether. The combined extract was
wsahed with 10 ml f ~2~ dried (MgSO4) and
concentrated. The residue was purified on a
silica gel column. Elution with 25% of
EtOAc/hexane gave 12a mg of title alcohol ester.
: 1~,2~(5Z~,3~(1E,35),4~,5a,6a]-7-[5,6-Epoxy-3-[3-
hydroxyw3-~l methylcyclohexyl3-1-propenyl3-
~
A mix~ure of 128 mg of Exampl~ 30 alcohol
:~ e~ter (O.3~ mmol~), 1 ml of lN LiO~ ~1.0 mmole, 3
eguiv.) in 1 ml o~ T~F was stirred at 25~C for 2
hours and ~hen concentrat~d. Th~ residu~ was
dilut~d with 5 ml of H20, acidifyiny to p~ 3 with
a saturat~d solution o~ oxalic acid, then
extract0d with three 15 ml portions of ether. The
combined ethereal e~tract was washed with 15 ml of
:
- . . . . .
.
' - . : ,
7 ~
HA363
-53-
H~0, dried (MgSO4) and concentrated to give 112 mg
of a crude oil.
This oil was purified on a silica gel
preparative plate (50 mg batchPs, O.5 mm silica
gel plate, 10% MeOH/CH2C12~ to yield a total of
49.6 mg of clean acid product.
TLC: silica gel; 10% MeOH/C~2C12; R~ ~ 0.60
Anal Calcd fo~ C23H345~5 ~2
~'l 8.82
Found: C, 69.10; H,' 8.51
[la,2~(5Z),3~(1E,3S),4u,5~,6~]-7-[5,6-Epoxy 3-(3-
hydroxyw4,4-dimethyl-l-octenyl)-7-oxabicyclo[2.2.1J-
A. [la,2~(5Z),3~(1E3,4~,5a,6~]~7~[5,6-
: Epoxy-3-(3-oxo-4,4-dimethyl-1-oc~enyl)-
7-oxabicyclo[2.2.1]hept-2-yl]-5-
e~e~noic acid, methyl ester
To a slurry of 102 mg of lithium bromide
; - (1.17 mmol, 2.2 equiv.) in 3 ml of dry CH2C12 at
25C was added a solution of 293 mg of
~:~ 25 3,3-dimethyl-2-oxo-heptyldimethylphosphonate
(1.11 mmole, 2.1 equiv.) in 1 ml of C~2C12 and 148
~1 o~ triethylamine (1.06 ~mol~, 2.01 eguiv.).
~f~er ætirring a~ 25C for 30 minutes, a solution
-~ of c~. 0.53 mmole of [1~,2~(5Z~,3~(1E),4~,5a,6a]-
~ 30 7-~5,6-epoxy-3-formyl 7-oxabicyclo[2.2.1]hept-2-
:~ yl]-5-heptenoic acid, methyl ester (prepared as
described in Example ~ Part M~ in 1 ml of CH~C12
~as added. The stirring was contlnued at 25C for
~ ~ ' ' , ` .
~ ~ 7~
.
HA363
-54-
18 hours. The reaction m1xture was then treated
with 5 ml of lM NaH2PO4 solution, and diluted with
30 ml of ether. The layers were separated. The
oxganic lay~r was washed with 1 ml of a saturated
KHCO3 solution, 10 ml of H2O and 10 ml of brine.
The organic layer was th~ dried (MgS04) and
co~c~ntrated.
The residue was purified on a silica gel
column. Elution with 25% EtOAc/hexane gave 130 mg
of title enone as a clear oil.
B. ~la,2~(5Z),3~(1E,3S),4aJ5a,6a]-7-[5,6-
Epoxy~3-(3-hydroxy 4,4-dimethyl-1-
octe~yl3 7-oxabicyclo[2.2.1]hept-2-yl]-
~
To a solution of 130 ~g o Part A enone
(0.32.mmole) in 1 ml of methanol and 1 ml o~ THF
at 25UC was added 78.4 mg of cerium trich~oride
0~32 mmole, 1 ~quiv.~. After stirring at 25~C for
15 minutes, ~he mixture was cooled to -50C and
12.1 mg of sodium borohydride (O.32 mmole, 4
equiv.) was added. ~he mi~ture was s~irred at
50C for 3 hours, ~hen poured into 30 ml of a
saturated ~olutio~ of ammonium chloride. The
aqueous solution was extracted with three 15 ml
. portio~s of e~her. The combined ex~ract was
washed with 10 ml of ~2~ dried (MgS04) and
co~centrated. The residue wa~ purified on a
silica gel col~m~. ~lu~ion with 25% EtOAc/hexane
gave 96 ng of title alcohol ester.
.
. .
.. ..
7~
~A363
-55~
Example 33
[la,2~(5Z~,3~(1E,3S),4a,5a,6a]~7-[5,6-Epoxy-3~3-
hydroxy-4,4-dimethyl~l octenyl)-7-oxabicyclo-
[2.2.1~hept-2-yll-5-heptenolc acld _
A mixtuxe of 96 mg of Example 31 alcohol
ester (O.23 mmole), 1 ml of lN LioH (1.O mmol~l 4
eguiv.) in 1 ~l of T~F was stirred at 25C for 2
hours, the~ concentrated. The residue was diluted
with 5 ml of H2O, acidifying to p~ 3 with a
saturated solution of oxalic acid, the~ extracted
with three 15 ml portions of etherl The combined
ethereal extract was washed with lg ml f ~2~
dried (MgSO4) and concentrated to give 75 mg of a
crude oil.
This oil was purified on silica gel
preparative plates (50 mg batches; 0.5 mm pla~es;
10% MeO~ 2C12) to yield a ~o~al of 49.8 mg of
clean acid product.
20 TLG: silica gel; 10% MeOH/CH2C~2; Rf ~ 0.55
1 Calcd for C23~36O5: C, 70.37; H, g.24
Found: C, 70.34; H, 9.43
:
.~' .
`~.