Note: Descriptions are shown in the official language in which they were submitted.
~O'ECULAR I~IG~T DISTP~IBUTIO;~ ~ODIFICATIO~
I~' TUBULAR REACTOR
Eeck~round of the Invention
The present invention relates to no~el co-
poly~ers o_ alpha-olefins. More specifically, it re-
12;es to novel copolymers of e~hylene with other
Glrha-olefins which have a polymodal molecular weight
etctribution wherein indiv.dual nodes comprising t~e`~
po'y~er have narrow molecular weight distributions.
For convenience, certain ter~s that are re-
peeeed throughout the present specification are defined.
below:
(a) Inter-CD de~ines eompositional variation,
in terms of ethylene content, among poly~er chains. It
-s expressed as the minimu~ deviction tanalogous to a
stendard deviation) in terQs of weight percent ethylene
~rom the average ethylene composition for a given co-
po'~-~er sa~ple needed to inclu~e 2 given ~eight percent
of the total copolymer sample -~hich is obtained by ex-
cluding equal weigh~ fraetions from both ends of the
distribution. ~he deviation need not be symmetrical.
~nen expressed as a single n~ber, for example, 15Z
Inter-CD, it shall mean the larger of the positive or
ne~ative ~eviations. For :e~,e;;~ple, ~or a Gaussian corl~po-
sitional distribution, 95.5Z of the polymer is within 20
~t~ ethylene o~ the ~ean if ~he standard deviation is
10~. The Inter-CD for 95~5 wtZ of the polymer is 20 wt%
e~hylene for such a s2rople.
(b) Intra-CD is the co~positional variation~
in terms oS ethylene, ~ithi~ a copolymer chain. It is
.
,
.
7~
--2--
expressed as the minimum difference in weight (wt) Z
ethylene ehat exists between two portions of a single
copolymer chain, each portion comprising at least 5 wtZ
of the chain~
(c) Molecular weight distribution (I~) is a
measure of the range of molecular weights within a given
copoly~er sample. It is characterized in terms of at
least one of the ra~ios of weight average to number av-
erage moleculax weigh~, MW/Mn, and Z average to weight
average molecular weight, Mz/ ~ , where: Mw = ~NiMi2
Mn = NiMi, and
M = ~ NiMi3, and
Z ~ iMi2
Ni is the number of molecules of weight Mi.
Ethylene-propylene copolymers, particularly
elastomers, are important commercial products. Two
basic types of ethylene-propylene copolymers are co~mer-
cially available; ethylene propylene copolymers and
ethylene propylene terpolymers~ Ethylene-propylene co-
polyners ~EPM) are saturated compounds requiring vulcan-
ization with free radical generators such as organic
peroxides. Ethylene-propylene terpol~ers (EPDM) con-
tain a small amount of non-conjugated diolefin, such as
dicyclopentadiene; 1 ,4-hexadiene or ethylidene nor-
bornene, which provides sufficient unsaturation to per-
mit w lcanization with sulfur. Such ethylene-propylene
poly~erc that include ae least two monomers, i.e., EP~
and EPDM, will be hereinaf~er collectively referred to
as et~ylene-propylene copolymers.
These copolymers ~ave outstanding resistance
to weathering, good heat aging properties and the abil-
ity to be compounded with large quantities of fillers
~ , . . .
. : :
.
~ ,. . ,' - :
-
-3-
and plasticizers- resulting in low cost compounds which
are particularly useful in automotive and industrial
mechanical goods applications. Typical automotive uses
2re tire sidewalls, inner tubes, radiator and heater
hose, vacuum tubing 9 weather stripping, sponge doorseals
and Viscosity Index (V.I.) improvers for lubricating oil
compositions. Typical mechanical goods uses are for ap-
pliance, industrial and garden hoses, botk molded and
e~cruded sponge parts, gaskets and seals and conveyor
belt covers. These copolymers also find use in adhe-
sives, appliance parts as in hoses and gaskets, wire and
cable and plastics blending.
As can be seen from the above, based on their
respective properties, EPM and EPDM find many, varied
uses. It is known that the properties of such copoly-
mers which make them useful in a particular application
are, in turn, determined by their composition and struc-
ture. For example, the ultimate properties of an EPM
and EPDM copolymer are determined by such factors as
co~position, compositional distribution, sequence dis-
` tribution, molecular weight, and molecular weight dis-
tribution (~D).
The efficiency of peroxide curing depends on
composition. As the ethylene level increases, it ~an be
shown that the "ch~mical" crosslinks per peroxide mole-
cule increases. Ethylene content also influences the
rheological and processing properties, because crystal-
linity, which acts as p~ysical crosslinks, can be intro-
duced. The crystallinity present at very high ethylene
contents may hinder processability and may make the
cured product too "hard" at temperatures below the crys-
talline melting point to be useful as a rubber.
Milling behavior of EPM or EPDM copolymers
varies radically with MW~. Narrow ~D copol~mers crum-
ble on a mill, whereas broad M~ materials will band un-
der conditions encountered in normal processing opera-
tions. At the shear rates encountered in processing
- ' :
-- 4
equipment, broader MWD copolymer has a substantially
lower viscosity than narrower MWD copolymer of the same
weight average molecular weight or low strain rate vis-
cosity. Thus, there exists a continuing need ~or dis-
covering polymers with unique properties and compositions.
For elastomer applications the processability
is often measured by the Mooney viscosity. The lower
this quantity the easier the elastomer is to mix and
fabricate. It is desirable to have low Mooney yet to
maintain a high number average molecular weight, Mn, so
that the polymers form good rubber networks upon cross-
linking. For EP and EPDM, narrowing the molecular
weight distrib-~tion results in the production of polymer
with higher number average molecular weight a-t a given
Mooney than the broader distribution polymer. In cer-
tain cases, the poor milling, calendering or extrusion
behavior that results from the narrow MWD must be ame-
liorated. _Rather than perform a MWD broadening which
includes low molecular weight components which reduce
Mn~ it is possible to broaden the MWD without dispropor-
tionately lowering Mn. This is done by superposing one
or more narrow MWD modes, i.e., different Mooney compo-
nents, each of which contains no appreciable amount of
low molecular weight polymer. The result is a polymodal
molecular weight distribution comprised of narrow ~WD
polymer fractions of different molecular weights.
The present invention is drawn to a novel co-
polymer of ethylene and at least one other alpha-olefin
monomer which copolymer is composed of several such MWD
components each of which is very narrow. It is well
known that the breadth of the MWD can be characterized
by the ratios of various molecular weight averages. For
example, an indication of the narrow MWD of each compo-
nent in accordance with the present invention is that
the ratio of weight to number average molecular weight
(MW/M ) is less than 2. Alternatively, a ratio of the
~;1
. - -
--, ~ ` : ' '
.
:
.
7~ ~ ~6
--5--
Z-average molecular weight ~o the weight average molecu-
lar weight (Mz/ ~ ) of less than 1.8 typifies a narrow
in accordance with the present invention. It is
known that the property advantages of copolymers in ac-
cordance with the present invention are rela~ed to these
ratios. Smail weight fractions of material can dispro-
portionately influence these ratios while not signifi-
cantly al~ering the property advantages which depend on
them. For instance, the presence of a small weight
~raction (e.g.~ 2Z) of low molecular weight copolymer
can depress Mn, and thereby raise ~IW/Mn above 2 while
maintaining Mz/ ~ less than 1.8. Therefore, the compo-
nent polymers, in accordance with the present invention,
are characterized by having at least one of two charac-
teristics; ~ /M~ less than 2 and Mæ/ ~ less than 1.8;
To o~tain naxrow M~, the copolymers in accordance with
the present invention are preferably made in a tubular
reactor. It has been discovered that to produce such
copolymers requires the use of numerous heretofore un-
disclosed method steps con~ucted within heretofore un-
disclosed preferred ranges. Accordingly, the present
invention is also drawn to a method for making th~ novel
copolymers of the present invention.
.
. . .: : .
.' ~ ~ ' ..
--6--
Des ~
Representative prior art dealing with tubular
reactors to make copolymers are as follows:
In "Polymerization o ethylene and propylene
to amorphous copolymers with catalysts of vanadium oxy-
chloride and alkyl aluminum halides"; E. Junghanns, A.
~umboldt and G. Bier; Makro~ol. Chem, v. 58 ~12/12/62~:
18-42, the use of a tubular reactor to produce ethylene-
propylene copolymer is disclosed in which the composi-
tion varies along the chain length. ~ore specifically~
~his reference discloses the production in a tubular
reactor of amorphous ethylene-propylene copolymers using
Ziegler catalysts prepared from vanadium compound and
aluminum alkyl. It is disclosed ~hat at the beginning
of the tube ethylene is preferentially polymerized9 and
if no additional ma~e-up of the nonomer mixture is made
during the polymerization the concentra~ion of monomers
changes in favor of propylene along the tube. It is
Lurther disclosed that since these changes in concentra-
tion take place during chain propagation, eopolymer
chains are produced which contain more ethylene on one
end than at the other end. It is also disclosed that
copolymers made in a tube are chemically non-uniform,
but fairly uniform with respect to molecular weight dis-
~ribu~ion. Using t~e data reported in their Figure 17
Cor copolymer prepared in the tube, it ~-as estimated
~hat the ~ /Mn ratio for this copolymer was 1.6.
"Laminar Flow Polymerization of EPD~ Polymer";
J.F. Wehner; ACS Symposium Serîes 65 (1978); pp 140-152
discloses th~ results of co~puter simulation work under-
taken to determine the effect of tubular reactor solu~
tion polymerization with Ziegler eatalysts on the molec-
ular weight distribution o~ the polymer produc~. The
speci~ic polymer simulated was a~ eiastomeric terpolymer
of ethylene-propylene-l, 4-hexadiene. On page 149, it
is stated that sinee the ~onomers have dirferent reac~
tivities, a pol~mer of varying composition is obtained
- - ~ ; , . ..
.
.
~ ~ 7
'~
--7--
as the monomers are depleted. However, whether the com-
position varies inter- or intramolecularly is not dis-
tinguished. In Table III on page 148, various polymers
having ~ /Mn f about 1.3 are predictcdO In the third
paragraph on page 144, it is stated that as the tube di-
aneter increases, the polymer molecular weight is too
low to be of prac~ical interes~, and i~ is predicted
that the reactor will plug. It is implied in the fixst
paragraph on page 149 that the compositional dispersity
produced in a tube would be detrimental to product qual-
ity.
U.S. 3,681,306 is drawn to a process for
producing ethylene/higher alpha-olefin copolymers having
good processability, by polymerization in at least two
consecuti~e reactor stages. According to this refer-
ence, ~his two-stage process provides a simple poly~eri-
z~tion process that permits tailor-making ethylene/
alpha-ole~in copolymers having predetermined properties,
particularly those contributing to processability in
co~mercial applications such as cold-flow, high green
strength and millability. Allegedly, the disclosed pro-
cess is par~icularly adapted ~or producing elastomeric
copolymers, such as ethylene/ propylene/5-ethylidene-2-
norbornene, using any of the coordination catalysts use-
~ul for making EP~I. The preferred process uses one
tubular reactor followed by one pot reactor. However,
it is also disclosed that one tubular reactor could be
used, but operated at diferent reaction conditions to
si~ulate two stages. As is seen from the disclosure at
column 2~ lines 14-20, the process makes polymers of
broader r~D than those made in a single sta~e reac~or.
Al~hough intermediate polymer from the first (pipeline)
reactor is disclosed as having a ratio of M~tMn o~ about
2 (column 5, lines 54-57) the ~inal polymers produced by
the process have an Mw/ ~ of 2.4 ~o 5.
U. S. 3, 6?5, 658 to Closon discloses a closed
circui~ tubular reactor apparatus with high
7~
--8--
recirculation ra~es of the reactants which can be used
to make elastomers of ethylene and propylene. With par-
ticular reference to E'ig. 1 of th~ patent, a hinged sup-
port lO for vertical leg 1 of the reactor allows for
horizontal expansion of ~he bottom leg thereof and pre-
vents harmful de~ormations due to ther~al expansions,
particularly those experienced during reactor clean out.
U~S. 4,065,520 ~o Bailey et al. discloses thP
use of a batch reactor to make ethylene copolymers, in-
cluding elastomers, having broad composi~ional distribu-
~ions. Several feed tanks for the reactor are arranged
in series, with the feed to each being varied to make
the polymer. The products made have crystalline to
se~i-crystalline to amorphous regions and gradient
ch2n~es in between. The catalyst sys~em can comprisë
vanadium compounds alone or in combination with titanium
coEpounds and is exemplified by vanadium oxy-tri-chlo-
ride and diisobutyl aluminum chloride. In all of the
examples, titanium compounds are used. In several exam-
ples, hydrogen and diethyl zinc, known transfer agen~s,
are used. The polymer chains produced have a cor;posi-
tionally disperse first length and uniform second
length. Subsequent lengths have various other CoQposi-
~ional distributions.
` In "Estimation of Long-Chain Branching in
Ethylene-Propylene Terpolymers from Infinite-Dilution
Viscoelastic Properties"; Y. Mitsuda, J. Schrag, and J.
Ferry; J. Appl~ Pol. Scl.~ 18, 193 (1974) narrow ~ co~
polymers of ethylene-propylene are disclosed. For exam-
ple, in TABLE II on page 198, EPDM copolymers are dis-
closed which have ~ /~n of from l.lg to 1.32.
In "The Effect o~ Molecular Weight and Molecu~
lar Ueight Distribution on the Non-~ewtonian Behavior of
~tkylene-Propyle~e-Diene Polymers; Trans. SocO Rheol.,
14, 83 (1970); C.K. Shih, a ~hole series of composi~ion-
ally homogeneous fractions were prepared and disclosedO
For example, the d~ta in TABLE I discloses polymer
.
.
J ~27~
_9_
S2-~ple B as naving a high de~ree of homogeneity. Also,
based on the reported data, the ~ of the sample is
very narrow. Howe-Jer, the poly~ers are not disclosed as
heting intraDolecular dispersity.
Molecular weight ~is~ribution (~I~) is a very
i ?ortant characteris~ic of ethylene-propylene copoly-
~e-s and terpolymers. Favorable distributions result in
polymers which can have both f2ster cures and better
processing characteristics. An opti~um coDbination of
t~.ese properties is achieved ~here the polymers have a
polymodal molecular weight distribution and a polymodal
compositional distribution.
A significant amount of effort ~2S been ex-
pe-.ded by the polyuer industry in an atte~pt to produce
su^h poly~odal ethylene-propylene polymers. Generally,
th.ese efforts have been directed toward physical blends
of poly~ers having different ~;~ or by sequential poly-
merization in a multiple reactor system. For example~ a
poly~erization is carried out in a first reaction stzge
to produce a polymer of a particular 2~WD and composition
~-ith a subsequent polymerization in a second reactor
stêge to produce a poly~er of a different ~.~ from that
o the first stage and, if desired, of a ~ifferent mono-
~e- composition. Representêtive prior 2rt dealing with
t~.e preparation of bimodal ~.~ ethylene-propylene co-
po1y~ers are as follows:
British Patent ~o. 1,233,599 is illustrative
o~ t~o stage polymeri2ation processes. I~ile copolymers
of ethylene are incidently disclosed, the exa~ples and
disclosure are directed tow2rd polyethylene homopolymers
2nd crystalline copoly~ers, e.g.~ 95~ ethylene. The
p-eferred catalysts are vanadium compounds such as
vcr~yl halide, van2dium tetrachloride or vanadium
tr-s-(aeetyi-acetonate) in conjunction ~ith an aluminum
~c=pound, e.g., Br2AlCH Br2. The different ~s are ob-
te_ned by using differing ~mounts of h~drogen in the
~-rst and second stage poly~erization.
: : ` .''
-10-
UOS. Patent No. 4,078,131 discloses an ethyl-
ene-propylene rubber ~omposition having a bimodal dis-
tribution in molecular weights comprising ~wo polymer
fractions each having a wide distribution of molecular
weig~ts and a monomer composition different from that of
the other principal fractions. The polymers are further
characterized in ~ha~ they are formed of: (a~ a first
principal fraction ~omprising from about 30Z to about
85Z ~by weight referred to the total weight of elasto-
mers) of molecular weight fractions having an intrinsic
viscosity dis~ribution of from about 0.2 to about 3, and
average intrinsic viscosity between about 0.8 to about
1.5 t an average propylene content between abou~ 36 to
about 52% by weight, and a termonomer content of between
0% and about 5Z, and of (b) a second fraction comprising
about 70% to about 15Z by weight of molecular weight
fractions having an intrinsic viscosity distribution
~rom about 3 to about 15, an average intrinsic viscosity
of about 3.5 to about 7, and average propylene content
of between about 26~ to about 32~ by weight and a ter-
monomer content of about 0 to about 5%.
The polymers are prepared by carrying out
polymerization in two separate reactors connected in se-
ries. The catalyst systems utilized include organic and
inorganic component of a transition metal of Group 4A to
8A o~ the Mendeleev periodic table of the elements,
e.g., VOC13, VCl4, vanadium esters and acetyl aceton-
ates. Co-catalysts include organoaluminum compounds or
mixtures of compounds, e.g., aluminum alkyls.
U.S. Patent 3,681,306 discloses a two stage
polymerization process for the preparation of ethylene-
propylene co~and terpolymers. In one embodiment the
first stage is a "pipe reactor" and t~e second stage is
a bac~-mixed pot reactor. The polymerization is carried
out so that the average ethylene/alpha olefin ratio in
one state is at least 1.3 ti~es the avera~e ratio o~ the
other s~age. Any of the coordination ca~alysts know ~o
.
~ .
J ~ ~ 7~
be useful in producing EPDM polymers is said to be ef
fective for the process.
U.S. Patent No. 4,~59,468 discloses a broad
~olecular weight ethylene-propylene-diene rubber pre-
pared using as a catalyst (a) the alcohol reaction prod-
uct of vanadium oxytrichloride and (b) a mixture of alu-
minum sesquichloride and ethylaluminum dichloride. The
polymer is characterized in that the higher molecular
weight fraction con~ains a larger proportion of the
diene than does the lower molecular weight fraction.
The polymer has an intrinsic viscosi~y of about 1.0 to
about 6.0 dl/g and a weight average molecular weight/
number ra~io of about 3 ~o about 15.
U.S. Patent No. 4,306,401 discloses a method
of manufacture of EPDM type terpolymers ~hich utilizes a
two stage polymerization process. Substantially all of
the non-conjugated diene monomer is fed to the first
stege thereby producing a poly~er having a non-uniform
diene content.
Brief Description of the Drawin~s
The accompanying drawings depict, for illus-
tration purposes only, processes e~bodied by the present
invention, ~herein:
Fig. 1 is a schematic representation of a pro-
cess ~or producing polymer in accordance with the pre-
sent invention.
Fig. 2 schematically illustrates a polymodal
~WD poly~er comprising narrow MWD polymers for each
mode,
Fig. 3 is a graphical illustration of a tech~
nique for determining Intra-CD of a copolymerD
Fig. 4 graphically ~llustrates various copoly-
~er structures that can be a~tained USillg processes in
accordance with the present invention.
- ~ . .
~2 7~
12-
Deta~led Descr~tion of the Inven~ion
The instant invention relates to a noYel co-
polymer of ethylene and at least one other alpha-olefin
monomer, whi~h copolymer is a superposition of t~o or
~ore copolymers, each of which has a 1,~ characterized
by having at least one of t~o characteristics; an ~Iw/Mn
of less than 2 and Mz/ ~ of less than 1.8.
As already noted, copolymers in accordance
~i~h the present invention are comprised of ethylene and
~t least one other alpha-olefin. Such alpha-olefins can
include those containing 3 to 18 carbon atoms. Alpha-
olefins of 3 to 6 carbons are preferred beca~se o~ eco-
no-~c considerations.
Illustrative non-limiting examples of alpha
ole ins userul in the practice of this invention are
pro~ylene, butene-l, pentene-l, hexene-l, h~ptene-l,
oc ene-l, dodecene-l, etc. The ~st preferred copoly-
mc s in accordance with the present in~ention are those
co-2rised of ethylene and propylene or ethylene, propyl-
ene and non-conjuga~ed diene.
As is ~ell known to those skilled in the art,
co?olymers of ethylene and higher alph2-olefins such as
propylene often include other polymerizable monomers.
T~ical of these other monomers can be non-conjugated
GieneS. Illustrative non-l~iting exa~ples of such
no~-conjugated dienes are:
a. straight chain acyclic dienes such as:
1l4-hexadiene; 1,6-octadiene;
b. branched chain acyclic dienes such as:
5-methyl-1, 4-hexadiene; 3,7 dimethyl-l, 6-
`octadiene; 3,7-di~thyl-1,7-octadiene and the
mixed isomers of dihydro-myrcene;
c. single ring 21icyclic diene~ such as:
1,4-cyclohexadiene; 1,5-cyclooctadiene; and
1,5-cyclododecadiene;
~ ' .
~: ' ' `
. ~ , . ,
~ ;
-13-
d. m~lti-ring alicyclic fused and bridg~d
ring dienes such as: tetrahydroindene;
~ethyltetra~ydroindene; dic~clopent~diene; bi-
cyclo-(2,2,1)-hepta-2,5-diene; alkenyl, alkyl-
idene, cycloalkenyl 2nd cycloalkylidene nor-
bornenes such es 5-methylene-2-norbornene
~ , 5-ethylidene-2-norbornene (E~B~, S-pro-
pylidene-2-norbornene, 5-isopropy~idene-2-norbor-
nener 5-(4-cyclopentenyl)-2-norbornene; S-cyclo-
he~ylidene-2-nor~ornene.
Of t'ne non-conjug2ted dienes typica ly used to
pr~p2re these copolymers, dienes containing at least one
of the double bonds in a strained rin~ zre preferred.
The ~ost prelerred diene is 5-ethylidene-2-norbornene
(E`~). The a~ount of diene (wt. basis) in the copolymer
ca~ be about 0% to 20Z with OZ to 15% being-preferred.
The nost pre~erred range is OZ to 10~.
As already noted, the ~ost preferred copolymer
in cccordance with the present invention is ethylene-
pr~p~lene or ethylene-prop;~lene non-conJugated diene.
In either event, the avera&e et~ylene content of each
co=ponent of these copolymers can be 2S low as about la%
on a ~eight basis. The prererred ~inimum ethylene ~on-
te~ is about 25%. A more preferred ~ini~um is abou~
30n~ The maxim~m ethylene oontent can b~ about 90% on a
weight basis. The preferred maximum is abou~ 85~, with
the ~ost pre.erred being about 80~
The molecular weight of the component copoly-
~er ~ade in accordance witb the present invention can
~2'~ over a wide range. The weight average molecular
~e_ght ~ ) can be as low as about 2~000O The ~referred
mi..i~um is about 10,000. The most preferred minimum is
cbout 20,000. The maximu~ weight a~erage molecular
~e_ght can ~e as high as 2bout 12,000,000. The pre-
ferred maximum is about 1,~00,000. The most preferred
~:i~um i~ about 75G~QOOo
.
-~.
,
- . . .
: . . . - ' ' ~ ~ ' . ', .
'
72
-14-
Another feature of the copolymers made in ac-
co-2ance with the present invention is that the ~olecu-
ler weight distribution (~D) of each component is very
n~rrow, as characterized by having at least one of two
c..eracteristics; a ratio of ~W/Mn of less than 2 2nd a
r~tio of Mzt~ of less thzn 1.8. The ~/1~n ratio for
t~ hole copolymer can range fro~ about 1 to about 50.
Tr.e ~w and 1~ of the copol~er is controlled by adjust-
the ~ and weight fraction of polymer that make uptr.e individual narrow ~ ~ components. In a preferred
e-.~odiment, the Mw of any t~o adjzeent ~h~ modes should
di~fer by at least 50% and any one mode ~hould com-
p-ise at least 10 wtZ of the total copolymer. As it re-
lc es to EPM and EPDM, a t~pical advantage of such co-
pcl~ners conposed of several Dodes having narrow ~ is
t~t when co~pounded and t~lcznized, faster cure and
b~.~er physical properties result than ~hen copolymers
hê~ing lower Mn for a given ~ooney are used.
Processes in accordance with the present in-
~ntion produce copolymer by polymerization of a reac-
.ion mixture comprised of catalyst, ethylene, at least
o..e additional alpha-ole~in ~onomer, and optionally, a
nc~- conju~ated diene. Solution polymerizations are
preferred.
Any kno~ solvent for the reaction mixture
t~.ct is effective for the purpose can be used in con-
d~cting solution polymerizations in accordance with the
p-esent invention. For exa~ple, suitable solvents are
h;;~rocarbon solvents such 2S aliphatic, cycloaliphatic
2nd aromat;c hydrocarbon solvents, or halogenated ana-
lc~s of such solvents. The preferred solvents are C4 to
Cl~, straight chain o~ branched chain, saturated hydro
cc-bons, C5 to Cg saturated alic~clic or aro~atic hydro-
cæ-bons or C2 to C6 halogenated hydrocarbons. Most pre-
ferred are C6 to C12~ straight chain or branched c~ain
h;~rocarbons, p~rticularly hexane. Nonlimiting illus-
t2ctive examples of such solvents are butane, pent2ne,
.
,
'' : ' ' `
2~
-15-
hexane, heptane, cyclopentane, cyclohexane, cyclohep-
tane, methyl cyclopentane, methyl cyclohexane, isooc-
tane t benzene, toluene, x~lene, chloroform, chloro-
benzenes~ tetrachloroethylene, dichloroethane and
trichloroethane.
A number of processes can be used to prepare
the copolymer products of this invention. These pro-
cesses are based on carrying out the copoly~erization in
a batch or tubular reactor. As described in our co-
pending patent application, Serial Number ~04~582, co-
polymers of narrow MWD with MW/Mn less than 2.0 or Mz/Mw
less than 1.8 can be obtained by operating such reactors
at certain specified conditions. Firstly, in the course
of the polymerization, substantially no ~ixing must
occur between polymer chains that have been initiated at
di~ferent times. This condition is defined as "mix
~ree." Tubular reactors are well known and are designed
to ~inimize mixing of the reactants in the direction o
flow. As a result, reactant concentration will vary
along the reactor length. In contrast, the reaction
mixture in a continuous flow stirred tank reactor
(CFSTR) is blended with the incoming feed to produce a
solution of essentially uniform composition everywhere
in the reactor. Consequently, the growing chains in a
portion of the reaction mixture will have a variety of
ages and thus a single CFS~R is not suitable for the
process of this invention. However, it is well known
that 3 or more stirred tanks in series with all of the
catalyst fed to the first reactor can approximate the
per~ormance of a tubular reactor. Accordingly, such
tanks in series are considered to be in accordance with
the present inven~ion.
A batch reactor is a suitable reactio~ vessel
in which to c2rry out the process o this invention,
preferably equipped with adequate agitation. The cata-
lyst, solvent, and monomer are added to the reactor at
the start of the po'ymerization. The charge of
., ~
'' ~ " '' ' :
.
~1 .
reactants is then left to polymerize for a time long
enough to produce the desired product. For economic
reasons, a tubular reactor is preferred to a batch reac-
tor for carrying out the processes of this invention.
~ n addition to the i~portance of the reactor
s~stem to make narrow ~'D cc=2onent copolymers the poly-
merization should be conducted in 2 menner such that for
each component or mode in the h~:
a. the catalyst system produces essentially
one active catalyst species,
b. the reaction ~i~;ture is essentially free
of chain transfer egents, and
c- for each mode the polymer chains are es-
sentially all initiated simultaneously, which~
is at the sa~e ti~e for a batch reactor or at
the same point along the length of the tube
for a tubular reactor.
The desired poly~er can also be obtained if
additional solvent and reactants ~e.g., at least one of
the ethylene, alpha-olefin and diene) are added either
along the length of a tub~lar rezctor or during the
cc~rse of polymerization in a batch reactor. Operating
in this f2shion can be decirable in certain cireum-
stences to control the pol.~Prization r2te or pol~er
cc-?osition. However, it is necessary to add the eata-
l~st at the inlet or specific locetions of the tube or
at the onset of or ae specific times in batch reactor
operation to meet the require~ent tha~ for each mode
essentially all ~olymer chains are initiated simultaneously.
Accordingly, narrou ~.1~ co~ponent copolymers
are produced by carrying out a pol~erization reaction:
(a) in a least one mix free reactor,
- (b) us;ng catalyst syste~s such ~hat each com-
po-.ent or ~ode in the I.~ is produced by essentially one
active catalyst species,
(c) using at least one reaction mixture which
is essentially transfer ager~-free, and
,'
. ' ~ . ' .
' ' , ,
.
-17-
(d) in such a manner and under conditions suf-
ficient to initiate propag2tion of essentially all poly-
mer ~hains made with a p2rticular ca~alyst species
siuultaneously~
To produce the mul~imodal MWD poly~er of this
in~ention, these polymeriz~tion conditions are used to
generate each of the narro~ ~WD modes that comprise the
final polymer product. A number of techniques are
availablP for achieving this: ~
1) In a single mux free reactor operated as
described above, portions o. the polymer product can be
~i.hdrawn after varying ti~es in a batch reactor or at
rying distances along a tubular reactor representing
ci rPrent average molecular weights and these portions
cc~ be blended. -
2) Mix free reactors can be operated either in~e-allel or sequentially and ~he products blended.
3) Two or more cetalysts that form narrow MWD
pcl}~er of difrering molecular weight can be added at
the onset of polymerization in a mixfree reactor. Each
ca~21yst must meet the requirements of minimizing chain
trcnsfer and initiating sir~ltaneous propagation of 211
the chains produced by that catalyst.
4) A catalys~ system that generates mul~iple
~c;ive catalyst species can be added at the start of the
poly~erization. Each cat21yst species produced must
gi~e simult2neous ch~in initiation and mini~ize chain
transfer .
5) Add;tional catalyst and monomer, if
desired, can be added at varying lengt~s along a tubular
reector o~ times in a batch reactor to initiate the for-
~aLion of additional M~.~ ~odes. The catalysts can ~e
the same or different, as long as chains are initiated
si-ultaneously and chain tr2nsfer is minimized.
6~ For c talyst system that show a decay i~
activity as a function of ~i~e due to deac~ivation, ca~
alys~ reactivator can be added during the course of the
:. . :, . . -
- . ' -
.
~7~
-18-
polymerizatiGn to regenerate the dead catalyst and form
a new mode o~ narrow ~.~ copolymer~
Catalyst reactivators are well known in the
ar~ for increasing the productivity of vanadium Ziegler
ca~alysts. These materials rejuvenate catalyst sites
~hat have become inert due to termination reactions and
thereby cause reinitiation of polymer chain growth.
~en added to a reactor operating according to the pro-
cess of this invention, catalyst reactivators have an
ef~ect similar to that of ad~ing a second catalyst feed.
~any reactivators are known, and examples of suitable
materials can be found in U.S. Patents 3,622,548,
3,629,212, 3,723,348, 4,168,358, 4,181,790 and
4,361,686. Esters of chlorinated organic acids are pre-
ferred reactivators for use with the vanadium catalyst
systems of t~is invention. Especially preferred is
buLyl perchlorocrotanate.
In ~he processes of this invention that uti-
lize multiple catalysts or multiple catalyst additions
during the course of polymerization the mix free condi-
tion of the reactor refers to the polymer chains of each
individual mode of the MWD and not to the polymer as a
whole.
A preferred multiple catalyst system comprises
t'C14 combined with VOCl3 and an alkyl aluminum sesqui-
~alide as a cocatalyst. The resultant polymer is a
bi~odal M~ polymer.
Since the present invention is considered to
be most preferred in the context of ethylene-propylene
(EPM) or ethylene-propylene-diene (EPDM) copolymers, it
will be d~scribed in detail in the context of EPM and/or
EPDM.
Copolymer in accordance with the present in-
veneion is preferably made in a tubular reactor. When
produced in a tubular reactor with monomer feed only at
the tube inlet, it is known that at the beginn~ng o the
~u~ular reactor ethylene, due to its high reacti~ity,
.
. . .
.. '
-l
will be pr~ferentially polymerized. However, the con-
centration of monomers changes along the tube in favor
of propylene as the ethyl~ne is depleted. The result i5
copolymer chains which are higher in ethylene concen-
tration in the chain segments grown near the reactor in-
let (as defined at the point at which the polymerization
reaction commences), and higher in propylene concen-
tration in the chain segments formed near ~he reactor
outlet. An illustrative copolymer chain of ethylene-
pr¢pylene is schematically presented below the E repre-
senting ethylene constituents and P representing
propylene constituents in the chain:
1 2 3 4
S gment E-E-E-E-P-E-E-E-P-P-E-E-P-P-P-E-P-P-P-P
As can be seen from this illustrativP schemat-
ic chain, the far left-hand segment (1) thereof repre-
sents that portion of the chain formed at the reactor
inlet where the reaction mixture is proportionately
richer in the more reactive constituent ethylene. This
segment comprises four ethylene molecules and one propy-
lene molecule. However, as subsequent segments are
~ormed from left to right with the more reactive ethy-
lene baing depleted and the reaction mixture proportion-
ately increasing in propylene concentration, the subse-
quent chain segments become more concentrated in
propylene. The resulting chain is intramolecularly het-
erogeneous .
In the event that more than two monomers are
used, e.g., in the production of EPDM using a diene ter-
monomer, for purposes of describing the present in~en-
tion all prop~rties related to homogeneity and hetero~
geneity will refer to the relative ratio of ethylene to
the o~her monomers in the chain~ ,The property, of the
copolymer discussed herein, related to intramolecular
compositional dispe~sity (compositional variation within
a chain) shall be referred to as Intra-CD, and that
re'ated to intermolecular compositional dispersi~y
.
- . .
.
7~
-20-
(compositional variation between chains) shall be re
ferred to as Inter~CD.
For copolymers in accordance with the present
in~ention, composition can vary between chains as well
2S along the length of the chain. In one embodiment of
~his invention, the Inter-CD can be characterized by the
difference in composition between some raction o~ the
copolymer and the average composition, as well as by the
total difference in composition between the copolymer
rrections containing the highest and lowest quantity of
ethylene. Techniques for measuring the breadth of ~he
In~er-CD are known as illustrated by Junghanns, et al.,
wherein a p-xylene-dimethylformamide solvent/non solvent
~as used to fractionate copolymer into fractions of dif-
ering intermolecular co~position. Other solvent/non-
solvent systems can be used9 sucb as hexane-2-propanol,
as will be discussed in more detail below.
In one embodiment of this invention, the-
Inter-CD of the individual component copolymers in ac-
cordance with the present invention is such that 95 wt%
of the copolymer chains have an ethylene composition
thet differs from the average component weight per~ent
ethylene composition by 15 wt% or less. The preferred
Inter-CD is about 13% or less, with the most preferred
being about 10% or less. In comparison, Junghanns, et
al., found that their tubular reactor copolymer had an
Inter-CD of greater than 15 wt%. Broadly, in one embod-
i~ent of this invention, the Intra-CD of copoly~er in
accordance with the present invention is such that at
least two portions o~ an individual component intramo-
lecularly heterogeneous chain, each portion comprising
at least 5 wt~ of the chain, differ in composition from
one another by at least 5 wt~ ethylene. Unless other-
wise indicated, this property of Intra-CD as referred to
he~ein is based upon at least two 5 wt~ portions of
copolymer chain. The Intra-CD of copolymer in accor-
dance with the present lnvention can be such that at
~, . : . . -.
.
.
, - : :
. - ~,
,~,,
,
-21-
least two portions of copolymer chain differ by at least
10 wt% ~thylene. Differences of at least 20 wt%, as
~ell as of at least 40 wt% ethylene are also considered
to be in accordance with the present inventionO
The experimental procedure for determining
Intra-CD is as follows. First, the Inter-CD is estab-
lished as described below, then the polymer chain is
broken into fragments along its contour and the Inter-CD
of the fragments is determined. The difference in the
~o results is due to Intra-CD as can be seen in the il-
lustrative example below.
Consider a heterogeneous sample polymer con-
taining 30 monomer units. It consists of 3 molecules
designated A, B, C.
A EEEEPEEEPEEEPPEEPPEPPPEPPPPPPP
B EEEEEPEEEPEEEPPEEEPPPEPPPEEPPP
C EEPEEEPEEEPEEEPEEEPPEEPPPEEPPP
~ Iolecule A is 36.8 wtZ et~ylene, B is 46.6%,
and C is 50% ethylene. The average ethylene content for
the mixture is 44.3Z. For ~his sample the Inter-CD is
sucb that the highest ethylene polymer contains 5.7%
more ethylene than the average while the lowest ethylene
content polymer contains 7.5~ less ethylene than the av-
er~ge. Or, in other words, 100 wt~ of the polymer is
within ~5.7% and -7.5Z ethylene about an average of
44.3%. Accordingly, the Inter-CD is 7.5~ when the given
wtZ of the polymer is 100%. The distribution may be
represented graphically as by curve 1 in Figure 3.
If the chains are broken into fragments, there
will be a new Inter-CD. For simplicity, consider first
breaking only molecule A into fragments show~ by the
slashes as follows:
EEEEP/EEEPE/EEPPE/EPPEP/PPEPP/PPPPP
Portions of 72.7~, 72.7%, 50%, 30.8%, 14.3Z and 02
ethvlene are obtained. If molecules B and C are simi-
larly broken and the weight ~ractions of similar compo-
sieion are grouped ~he new In~er-CD shown by curve 2 in
.
,
,
,
- - , . ~ .
-Z2-
FiOure 3 is obtained. The difference between the two
curves in the figure is due to Intra-CD.
Consideration of such data, especially near
the end point ranges, demonstrates that for this sample
at least 5~ of the chain contour represented by the cu-
mulative weight Z range (a) differs in ~omposition from
anoeher sec~ion by at least 152 ethylene shown as (b),
~he difference between the two curves. Th~ difference
is composition represented by (b) cannot be intermolecu-
lar. If it were, the separation process for the origi-
nal polymer would have revealed the higher ethylene con-
ten~s seen only for the degrad~d chain.
The compositional differences shown by (b~ and
(d) in the igure between original and frag~ented chain~
give minimum values for Intra-CD. The Intra-CD must be
at least that great, for chain sections have been
isolated which are the given difference in composi~ion
(b) or (d) from the highefit or lowest composition
polymer isolated from the original. We know in this ex-
ample that the original polymer represented at (b) had
sections of 72.7% ethylene and OZ ethylene in the same
chcin. It is highly likely that due to the inefficiency
of the ~ractionation process any real polymer with
In,ra-CD examined will have sections of lower or higher
ethylene connected along its contour than that shown by
the end points of the fractionation of the original
polymer. Thus, this procedure determines a lower bound
for Intra-CD. To enhance the detection, the original
whole polymer can be fractionated (e.g., separate mole-
cu'e A from molecule B from molecule C in the hypothet-
ic21 example) with these fractions refractionated until
~hey show no (or less) Inter-CD. Subsequent fragmen~a~
tion of this intermolecularly homogeneous fraction no~
re:eals the total Intra-CD. In principle, for the exam-
ple, if ~olecule A were isolated, fragmented, fraction-
ated and analyzed, he Intra-CD for the chain sections
wc-~ld be 72.7-0% = 72.7% rather ~han 72.7-50% = 22.7%
.
'.................. . ~ ,
'~L7~
-23-
seen by fractionating the whole mixture of molecules A,
B and C.
In order to determine the fraction of a
polymer which is intramolecularly heterogeneous in a
mixture of polymers combined from several sources or as
several modes in the case described here, the mixture
must be separated into fractions which show no further
he~erogenity upon subsequent fractionation. These
~ractions are subsequently frac~ured and fractionated to
reveal which are heterogeneous.
The fragments into which the original polymer
is broken should be large enough to avoid end effects
and to give a reasonable opportunity for the normal sta-
tistical distribution of segments to form over a gives
monomer conversion range in the polymerization. In-
tervals of ca 5 wt~ of the polymer are convenient. For
example, at an average polymer molecular weight of about
105, fragments of ca 5000 molecular weight are appropri-
ate. A detailed mathematical analysis of plug flow or
ba~ch polymerization indicates that the rate of change
of composition along the polymer chain ~ontour will be
most severe at high ethylene conversions near the end of
the polymeri~ation. The shortest fragments are needed
here to show the low propylene content sections.
The best available technique for determination
of compositional dispersity for non-polar polymers is
solvent/non-solvent fractionation which is based on the
thermodynamics of phase separation. This technique is
described in "Polymer Fractionation," M. Cantow editor9
Academic 1967, p. 341 ff and in H. Inagaki, T. Tanaku,
DeYelopments in Pol~mer Characterization, 3, 1 (1982).
.
For non-crystalline copolymers o ethylenP and
propylene, molecular weight governs insolubility more
than does compo~i~ion in a solvent/non solvent solution.
High molecular weight polymer is less soluble in a given
solven~ mix. A~so, ehere is a systema-ic correlation of
,
: ~ ,
': .
.
-24-
molecular weight with ethylene eonten~ for the poly~ers
described herein. Since ethylene polymerizes much more
rapidly than propylene, high ethylene polymer also tends
to be high in molecular weight. Additionally, chains
rich in ethylene tend to be less soluble in hydrocar-
bon/polar non-solvent mixtures than propylene-rich
chains. Thus the high molecular ~eight, high ethylene
chains are easily separated on the basis of thermodynam-
ics .
A fractionation procedure is as follows: Un-
fragmented polymer is dissolved in n-hexane at 23C to
form ca a lZ solution (1 g polymer/100 cc hexane).
Isopropyl alcohol is titrated into the solution until
turbidity appears at which time the precipitate is al-
lowed to settle. The supernatant liquid is removed and
the precipitate is dried by pressing between Mylar
(polyethylene terphthalate) film at 150C. Ethylene
content is determined by ASI~I method D-3900. Titration
is resumed and subsequent fractions are recovered an an-
alyzed until lOOZ of the polymer is collected. The
titrations are ideally controlled to produce fractions
of S-lOZ by weight of the original polymer especially at
the extremes of composition.
To demonstrate the bread-h of the distribu-
tion, the data are plotted as % ethylene versus the cu-
mulative weight of polymer as defined by the sum of half
the weight Z of the frac~ion of ~hat composition plus
the total weight % of the previously collected frac-
tions.
Another portion of the original polymer is
broken into fragments. A suitable ~ethod for doing this
is by thermal degradation-according to the following
procedure: I~ a sealed container in a nitrogen-purged
oven, a 2 mm thiek layer of the poly3er is heated for 60
minutes at 330C. This should be adequate to reduce a
105 molecular weight polymer to frag~ents of ca 5000 mo-
lecular weight. Such degradation does not change the
. -
.
: -
-25-
average ethylene content o the pslymer. This polymer
is fractionated by the same procedure as the high molec-
ular weight precursor. Ethylene content is measured~ as
well as molecular weight on selected ~ractions.
Ethylene content is measured by ASTM-D3900 for
ethylene-propylene-copolymers between 35 and 85 wt%
ethylene. Above 85Z ASTM-D2238 can be used to obtain
methyl group concentrations which are related to percent
ethylene in an unambiguous manner for ethylene-propylene
copolymers. When comonomers other than propylene are
employed no ASTM tests covering a wide range of ethylene
contents are available, however, proton and carbon 13
nuclear magnetic resonance czn be employed to determine
the composition of such polymers. These are absolut~
techniques requiring no calibra~ion when operated such
~hat all nucleii contribute equally to the spectra. For
ranges not covered by the AS~ tests for ethylene-propy-
lene copolymers, these nuclear magnetic resonance meth-
ods can also be used.
Molecular weight and molecular weight distri-
bution are measured using a Waters 150 gel permeation
chromatograph equipped with a Chromatix ~MX-6 on-line
li~ht scattering photometer. The system is used at
135C with 1,2,4, trichlorobenzene as mobile phase.
Showdex (Showa-Denko America, Inc.) polystyrene gel col-
umns 802, 803, 804 and 805 are used. This technique is
discussed in "Liquid Chromatography of Polymers and
Related ~aterials III," J. Cazes editor. Marcel Dekker,
1981, p. 207. No
corrections for column spreading are employed; however,
data on generally accepted standards, e.g., National Bu-
reau of Standards Polyethene 1484 and anionically
produced -hydrogenated polyisoprenes (an alternating
ethylene-propylene copolymer) demonstrate tha~ such cor-
rections on ~ /Mn or Mz/ ~ are less than .05 unit.
n is calcula~ed from an elu~ion time-molecular
~eight relationship wh~reas Mz/ ~ is evaluated using the
,
.
.
- . -
~- :
.
-26-
li~ht scâttering photometer~ ~he numerical analyses can
be performed using the co~eroially available co~puter
softwear GPC2, ~OL~7T2 availzble form LDC/2~ilton Roy-
R-viera Beach, ~lorida.
Since the tubular reactor is the preferred re-
ac~or system for carrying out processes in accordance
~-ith the present in~ention, the follo~ing illustrative
descrip~ions and exaDples are drcwn to that system, but
~ill apply to other reactor s~stems 2S will readily oc-
c~r to those skilled in the art having ~he benefit of
t~.e present disclosure.
In practicing processes in accordance wit~ the
p-~sent invention, use is preferably made of at le2st
c~e tubular reactor. Thus, in its simplest for~, such-~a
p~ocess would make use of ~.ut a single reactor. Howev-
er, Dore thaR one re2ctor ccn be used, either in paral-
l~l,or in series with multiple monomer feeds.
For example, various structures can be pre-
pared by adding additional ~onomer(s) during the course
of the poly~eriz2tion, 2S shown in Fig. 4, ~-herein com-
pcsltion is pl~tted versus position along the contour
l~gth of a polym~r chain. The structure show~ in curve
1 is obtair.ed by ree~ing all of the ~ono2~ers to the tu-
b~!cr reactor inlet or at the start of a b~tch reaction.
Ir. comparison, the structure depicted in curve 2 can be
~e by adding additional ethylene at a point along the
tL~e or at a time in a batch reactor, where the cha;ns
hcve reached about half their len~t~. Curve 3 requires
iple. feed additions. The structure depicted by
ct:rve 4 caTl be forhed if additional comonomer rather
t~.en ethylene is added. This structure permits a whole
et.ylen~ composition range to be omitted from the chain.
In each c2se, a third or more comonomers ~ay be added.
The co~position of the catalyst used to pro-
duce alp~2-olefin copoly~ers has a pro'ound effec~ on
cc?olymer product pro~er~ies such as co~positional
" ' ~ .. ' ,
.
,
~ ~ 7
d
-27-
dispersity and M~. The catalyst utilized in practicing
processes in accordance wi~h the present invention
should be such as to yield a controlled number of active
species, each of which must be capable of simultaneous
initiation of chains and must minimize chain transfer.
Each active catalyst species generated either by multi-
ple catalyst feeds or by a single catalyst feed that
generates multiple active species must prod~ce copolymer
product in accordance with the present invention, e.g.,
a copolymer of narrow MWD. - The extent to which a cata-
lyst species contributes to the polymeriza~ion can be
readily determined using the below described techniques
f~r characterizing catalyst according to the number of
active catalyst species.
Techniques for characterizing catalyst accord-
ing to the number of active catalyst species are within
the skill of the art, as evidenced by an article enti-
tled "Ethylene-Propylene Copolymers. Reactivity Ratio,
E~aluation and Significance," C. Cozewith and G. Ver
Str~te, Macromole~ules, 4, 482 (1971).
It is disclosed by the authors that copolymers
made in a continuous flow stirred reactor (CFSTR) should
have an ~D characterized by MW/Mn=2 and a narrow inter-
molecular compositional distribution when one active
catalyst species is present. By a combination of frac-
tionation and gel permeation chromatography (GPC) it is
shown that for single active species catalysts the com-
positions of the fractions Yary no more than ~3Z about
the average and the MWD (weight to number average ratio)
or these-samples approaches two (2). It is this latter
cbaracteristic (MW/~In OL about 2) that is deemed the
more important in identifying a single active catalyst
species. On the other hand, other catalysts gave
copolymer wi~h an compositional variation greater than
~10% about the average and multi-modal ~ often wit~
.
: .
- . . ',
:, . ~ ' .
-28-
M~/Mn greater than 10. These other catalysts are deemed
to have more than one active species.
Catalyst syste~s to be used in carrying out
processes in accordance with the present invention may
be Ziegler catalysts, which ~ay typically include compo-
Re~.tS selected from:
(a) a compound of a transition ~etal, i;e., a
~metal of ~roups I-B, III-B, IVB, ~'B, VI8, VIIB and VIII
of the Periodic Table, and (b) an organo~etal compound
o~ a metal o~ Groups I-A, II-A, II-B cnd III-A of the
P~riodic Table.
Tne preLerred catalyst syste~ in practicing
pr~cesses in accordance with the present invention com-
pr~'ses hydrocarbon-soluble ~anadium compound in which
t~e vanadium valence is 3 to 5 and organo-alumin~w~ com-
pc:nd, with the provision that the catalyst system
y_elds one active catalyst species which has the ca-
p~ility to produce ~arrow ~'~'D copolymers as described
2~ve. At least one of the vanadium co~pound/organo-
~luminum pair selected must also contain a ~alence-
bc~ded halogen.
In terms of for~ulas, vcncdi~m compounds use-
f~l in practicing processes in accordance with the pre-
`s~t invention could be:
(I) voclx(OR)3 x
where x = 0-3 and R = a hydrocarbon radical,
(II) VC14;
(III) VO(AcAc)2.
where AcAc = acetyl acetonate;
(IV) V(AcAc)3;
(V) V~)clx~AcAc)3-x~
where x ~ 1 or ~; Gnd
. . . .
- . . , ,: .
,
~ . ,
. .
.
~7~
-29 -
(VI 3 VC13 . nB,
Where n - 2-3 and B ~ Lewis base capable of
making hydrocarbon-soluble cQa:plexes with VC13, such as
te~rahydrofuran, 2-methyl-tetrahydrofuran and dimethyl
pyridine.
In formula I abQve, R preferably represents a
Cl to C10 aliphatic, alicyclic or aro~atic hydrocarbon
radical such as ethyl (Et), phenyl, isopropyl, butyl,
propyl, n-butyl, i-butyl, t-butyl, hexyl, cyclohexyl,
octyl, naphthyl, etc. Non-limiting, illustrative exam-
ples o~ formula (1) and (2) compounds are vanadyl tri-
halides, alkoxy halides and alkoxides such as VOC13,
VOC12(0Bu) where Bu = butyl7 and VO(OC2H5~3. The most
preferred vanadium compounds are VC14, VOC13, and
VGC12(OR).
As already noted, the co-catalyst is pref-
erably organo-aluminum compound. In terms of chemical
formulas, these compounds could be as follows:
AlR3. AltOR')R~.
AlR2Cl, R2Al-O-AlR'2,
AlR'RCl AlR2I;
A12R3C13
AlRC12 .
and mixtures thereof where R and R' represent hydro-
~arbon r~dicals, the same or different, as described
above with respect to the vanadium compound formula.
The most preferred organo-aluminum compound is an alumi
nu~ alkyl sesquichloride such as A12Et3C13 or A12(iBu~3-
C13. In one embodiment of the invention the aluminum
coapound can be described by the formula AlRnX3 n ~here
R is as ~reviously defined, X is halogen, preferably
chlorine and n can vary from 1 to 2.
When catalysts are desired that produce a sin-
gle 2ctive species, catalysts comprised of VOCl~ or VC14
with A12R2C13~ preferably where R is ethyl, have beeR
shcwn to be particularly effective. For best catalys~
performance, the molar amounts of catal~st components
., ~
..
- , . , ~
` ~` -` ' ' ,' . '-
,
.
`' - .: :
~30-
~ddPd to the reac~ion mixture should provide a molar ra-
tio of aluminum/vanadium (AltV) of at least about 2.
The preferred minimum Al/V is about 4. The maximum Al/V
is based`primarily on the considerations of catalyst ex-
pense and the desire to minimize the amount of chain
transfer that may be caused by the organo-aluminum com-
pound (as explained in detail below). Since, as is
known certain organo-aluminum compounds act as chain
transfer agents, if too much is present in the reaetion
mixture the ~ /kln of the copolymer may rise above 2~
Based o~ these considerations, the maximum Al/V can be
about 25, however, a maximum of about 17 is more pre-
ferred. The most preferred maximum is about 15.
Chain transfer agents for the Ziegler-catalyz-
ed poly~erization of alpha-olefins are well known and
are illustrated, by way of example, by hydrogen or
diethyl zinc for ~he production of EPM and EPDM. Such
agents are very commonly used to control the molecular
we~ght o~ EPM and EPDM produced in continuous flow
stirred reactors. For the essentially single active
species Ziegler catalyst systems used in accordance with
the present invention, addition of chain transfer agents
to a CFSTR reduces the polymer molecular weight but does
not a~ect the molecular weight distribution. On t~e
other hand~ chain transfer reactions during tubular re-
actor polymerization in accordance with the present in-
vention broaden polymer molecular weight distribution.
Thus ~he presence of chain transfer agents in - the re-
action mixture should be minimi2ed or omitted altogeth-
er. Although difficult to generalize for all possible
reactions,- the amount of chain transfer agent used
should be limited to those amounts that provide copoly-
mer produet in accordance with the desired limits as re-
gards ~D and composi~ional dispersity. It is believed
tha~ the ma~imum amount of chain transfer agent present
ir. the reaction mixture could be as high as about O . 2
mol~mol o~ transit;on metal , e . g., vanadium, again
.. . . . .
~ .
.
'
':
. ~ , .
7~
-31
pro~ided that the resul~ing copolyuer product is in ac
cordance with the desired limits as regards M~D and com-
positional dispersity. Even in the absence of added
chain transer agent, chain transfer reactions can occur
because propylene and organo-aluminum cocatalyst can al-
so act as chain transfer agents. In general, among the
organo-aluminum compounds that in combination with the
vanadium compound yield just one active species, the
organo-aluminum compound that gi~es the highest copoly-
mer molecular weight at acceptable catalyst activity
should be chosen. Furthermore, if ~he Al/V ratio has an
effect on the molecular weight of copolymer product,
that Al/V should b~ used which gives the highes~ molecu-
lar weight also at acceptable catalyst ac~ivity. Chain
transfer with propylene can best be limited by avoiding
excessive temperature during the polymerization as de-
scribed below.
~ Iolecular weight distribution is also
broadened by catalyst deactivation during the course of
the polymerization which leads to termination of growing
chains. It is ~ell known that the vanadium-based Zieg-
ler catalysts used in accordance with the present inven-
tion are subject to such deactivation reactions ~hich
depend to an extent upon the composition of the cata-
lyst. Although the relationship between active catalyst
lifetlme and catalyst syste~ composition is not known ~t
present, for any given catalyst, deactivatiQn can be
reduced by using the shortest residence ~im~ and lowest
temperature in the reactor that will produce the desired
monomer conversions.
~ olymerizations in accordance with the present
inYentiOn should be conducted in such a manner and under
condi~ions suf~icient to initiate propagation of essen-
tially all copolymer chains for each par~icular catalyst
species simultaneously. This can be accomplished by
utilizing the process steps and conditions described be-
low.
- : .
- - ~ ' . . .
7~
-32-
The ~a~aly~t com~onents are preferably pre-
.ed, that i~, reacted to form active catalyst outside
of the reactor, to ensure rapid chain initiation. Aging
o~ the premixed catalyst system, that is D the time spent
b~ the catalyst components (e.g.~ vanadium compound and
o-~anoaluminum) in cont~ct ~ith one another outside of
t~e reactor, must be kept within certain li~its. If not
a~ed for a sufficient period of time, the com~onent~
will not have reacted ~ith each other sufficiently to
yield an adequate quantity of active c2talyst species,
with the result of continued catalyst species formation
i~ the reactor, resulting i~ non-simultaneou.~ chain ini-
~lation. Also, it is ~no-~n that the activity of the
c~tGlyst speci~s will decrease with time so ~hat the ag-
ing ~ust be kept below 2 mGximum limit. The minimum ag-
ing period, depending on sueh ~actors 2S concentration
o catalyst components, te~perature and mixing equip-
m~nt, can be 2s low as abou~ 0.1 second~ The maximum
a~ing period is that period of aging af ter w~ich ~he
catalyst species has been de2ctivated to the point ~here
it canno~ effeeti~ely be used in the polymerization pro-
ce~s. In practice there is no appreciable advantage in
allowing the catalyst to age longer than a time sufficient
to fully react all of the available catalyst components
thereby generating all of the active catalyst species which
will be available for polymerization. Generally, the aging
time will ordinarily be about O~l seconds to about 200
seconds or even longer, usually about 0.5 seconds ~o lO0
seconds, preferably about l second to 50 seconds. The
premixing performed at low temperature such as 40C or
below. I~ is preferred that the mixing be performed at
25C or below, with 15C or below being most preferred.
Where more than one catalyst is com~ined into a
single catalyst feed st~eam, each catalyst and
- : : ,' ' ' ' :
.
-
-33~
cocatalyst can be premixed separately. The several pre-
mixed streams of catalysts species are then combined and
fed to the reactor. Alternately, the several pre-mixed
catalyst feed streams can be fed separately to different
points along the reactor.
The temperature of the reaction mixture should
also be kept with cer~ain limits. The temperature at
~he reactor inlet should be high enough to provide com-
plete, rapid chain initiation at the start of the poly-
merization reaction. The length of time the reaction
mix~ure spends at high temperature must be short enought
to minimize the amount of undesirable chain transfer and
catalyst deactivation reactions.
Temperature control of the reaction mixture is
co~plicated somewhat by the fact tha~ the polymerization
reaction generates large quantities of heat. This prob-
lem is, preferably, taken care of by using prechilled
eed to the reactor to absorb the heat of polymeriza-
tion. With this technique, the reactor is operated
adiabatically and the temperature is allowed to increase
duxing the course of polymerization. As an alternative
to feed prechill, heat can be removed from the reaction
~ixture, for example, by a heat exchanger surrounding at
least a portion of the reactor or by well-known autore-
frigeration techniques in the case o~ batch reactors or
multiple s~irred reactors in series.
h~ere an adiabatic reactor operation is used9
the inlet temperature of the reactor ~eed can be about
-80C to about 50C. The outlet temperature of the re-
action mixture can be as high as about 200C. The pre-
ferred ma~imum outlet temperature is about 70C. The
most preferred maximum is about 5~C. In the absence of
reactor cooling, such as by a cooling jacket, to remove
the heat of polymerization, the temperature of the re-
action mixture will increase from reactor inlet to out-
let by an amount dependent upon the heat of polymeriza-
tion~ reaction mixture specific heat and the percent of
-3~-
copolymer in the reaction mixture (weight of copolymer
per weight of solvent). For ethylene-propylene copoly-
meri2ation in hexane the temp,erature rise is about 13C
per weight percent of copolymer.
Having the benefit of the above disclosure,
those skilled in the art can determine the operating
te~perature conditions for making copolymer in accor-
dance with the present invention. For example, assume
an adiabatic reactor and an outlet temperature of 35C
are desired for an ethylene-propylene reaction mixture
in hexane containing 5% copolymer. The reac~ion mixture
will increase in temperature by about 13C for each
weight percent copolymer or 5 weight percent x 13C/wt%
= 65C. To maintain an outlet temperature of 35C, it
will thus require a feed that has been prechilled to
35C-65C = -30~C. In the instance that external cool-
ing is used to absorb the heat of polymerization, the
feed inlet temperature could be higher with the other
temperature constraints described above otherwise being
applicable.
Because of heat removal and reactor tempera-
ture limitat~ons, the preferred maximum copolymer con-
centration at ,he reactor outlet when this is the only
stream drawn from the reac~or is 25 wt/100 wt diluent.
The ~ost preferred maximum concentration is 15 wt/100
wt. When multiple streams o reaction mixture are wit~-
drawn from the reactor and each part of the reaction
mlxture withdrawn is blended with other parts of re-
action mixture withdrawn, the blend so formed has a pre-
ferred maximum copolymer concentration of about 25
ut./100 wt-. of diluent. The most preferred maximum is
15 wt./100 wt. diluent. In the case of either single or
multiple product stream withdrawal, there is no lower
li~it to concen~ration due to reactor operability, bu~
for economic reasons it,is preferred to have a copolymer
concentration o at least 2 wt/100 wt. ~ost preferred
is a concentration of at least 3 wt/100 ~t;
.
: .
'
z~
-3~-
The rate of flow of ~he reaction mixturethrough the reactor should be high enough to provide
good mixing of the reactants in the rad;al direction and
minimize mixing in the axial direction. Good radial
mixing is beneficial to minimize radial temperature gra-
dients due to the heat generated by the polymerization
reaction. Radial temperature gradients will tend to
broaden the molecular weight distribution of the copoly-
mer since the polymerization rate is faster in the high
temperature regions resulting from poor heat dissipa-
tion. The artisan will recognize that achievement of
~hese objectives is difficult in the case of highly vis-
cous solutions. This problem can be overcome to some
extent through the use of radial mixing devices such as
static mixers (e.g~, ~hese produced by the Kenics Corpo-
ration).
Residence time of the reac~ion mixture in the
mix-free reactor can vary over a wide range. The ~ini-
~um can be as low as about 1 second. A preferred mini-
~um is about 10 seconds. The most preferred minimum is
about 15 seconds. The maximum can be as high as about
3600 seconds. A preferred maximum is about 1~00 sec-
onds. The most preferred maximum is about 900 seconds.
~ ith reference to the accompanying drawings,
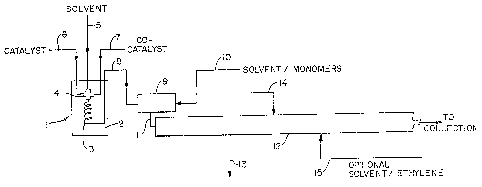
particularly Fig 1, reference numeral 1 refers to a pre-
mixing device for premixing the catalyst components.
For purposes of illustration, it is assumed that a
copolymer of ethylene and propylene (EPM~ is to be pro-
duced using as catalyst components vanadium tetrachlo-
ride and ethyl aluminum sesquichloride. The polymeriza-
tion is an adiabatic, solution polymerization process
using hexane solvent for both the catalyst system and
the reaction mixture.
The premixing device 1 comprises a temperature
control bath 2, a fluid flow conduit 3 and mixing de~ice
4 te.g., a mixing tee). To mixing device 4 are fed
hexane solvent, vanadium ~etr~chloride and ethyl
~J
. .
.
' ' ' '
. ' ' '' . ' ~. " '.
.
-3~
aluminum sesqui chloride through feed conduits 5 9 6 and
7, respectivelyO Upon being mixed in mixing device 4,
the resulting catalyst mixture is caused to flow within
conduit 3, optionally in the form of a coiled tubP, for
a time long enough to produce the active catalyst spe~
cies at the temperature set by the temperatur~ bath.
The temperature of the bath is set to give the desired
catalyst solution temperature in c~nduit 3 at the outlet
of the bath.
Upon leaving the premixing device 9 the cata-
lyst solution flows through conduit 8 into mixing zone 9
tc provide an intimate mixing with hPxane solvent and
reactants (ethylene and propylene) which are fed through
conduit 10. Any suitable mixing device can be used,
such as a mechanical mixer, orifice mixer or mixing tee.
For economic reasons, the mixing tee is preferred. The
residence time of the reaction mixture in mixing zone 9
is kept short enough to prevent significant polymer for-
~ation therein before being fed through conduit 11 to
~ubular reactor 12. Alternatively, streams 8 and 10 can
be fed directly to the inlet o reactor 12 if the flow
rates are high enough to accomplish the desired level of
intimate mixing. The hexane with dissolved monomers may
be cooled upstream of mixing zone 9 to provide the
desired feed temperature at the reactor inlet.
The tubular reactor is shown with optional
feed and eake off points. Where the catalyst comprises
only a single polymer species one or more take off
points, 13, are used to withdraw polymer fractions at
different points along the polymerization path. In or-
der to maintain constant flow, additional solvent may be
added to make up the volume of material withdrawn. Ad-
ditional catalyst and monomer can be introduced through
line, 14, or line, lS. Thc polymer withdrawn through
line, 13, is combined with all other fractions withdrawn
and collected with the reactor effluent for deashing and
finishing.
,
, - , ''
- '' '.' - ',
' ' '
-37~
h~ere ~ore than one catalyst species is used,
~ultiple premixing devlce~, 1, are used. The mixed cat-
21yst can be directed to mixing zone, 9, for mixing with
acditional catalyst species and monomer or ~he effluen~
from the premixing de~ices can be co~bined prior to the
~ixing zone .
~ .~ere a mixeure of VC14 and TOC13 is used as
the catalyst species in conjunction with,ethylaluminum
sesquichloride (EASC), the molar retio of VC14/VOC13 can
be about 0. 01 to about 100, more preferebly about 0.1 to
ab~ut lQ, most preferably ebout 0.5 to about 5. The
a~unt of ~he total polymer and the molecular weight of
ecsh componene will be determined by the ratio and ~he
feed locations and take off points 210ng the reactor.
The molar ratio of alkyl aluminum sesquihalide
~o vanadium components (VC14 plus VOC13) can be about 1
to about 40, preferably about 2 to about 40, more pref-
erably about 4 to about 20, ~ost preferably about 4 to.
~bout 10, e.g., about 5 to about 10. The alkyl group of
the sesquihalide is preferably a Cl-C6 alkyl group,
preferably ethyl. The halide can be bromine, chlorine
or iodine, preferably chlorine. The preferred aluminum
eo-catalyst is ethylaluminum sesquichloride (EASC). In
~h~s system the ~wo indepenGent, non-interacting, mu-
~- 211y compatible catalyst syste~s are VC14/EASC and
VOC13/EASC.
In a preferred embodi~ent~ a Lewis base moder-
~tor is incorporated into the catalyst system. The
~olar ratio of base to vanadium can be about 0 to about
5/1, preferably about 0 . 5/1 to about 2~1, more pref-
erably abeut 1/1 to about 1.5l1. Illustrative, non-
limiting examples of ~ewis bases suitable for use in the
pr2ctice of this invention are NH3 ~ phenol, cyclohexa~
no-.e . tetrahydrofuran, acetyl2cetone, ethyl silicate and
~r~-n-butyl-phosphate. The Le~is base suppresses some
long chain branching reactions when EPDM terpolymers are
prepared.
; " ~
.:
.
: : .
~ . ;
.- ' ' .
-38-
The polymer derî~ed from the process of this
invention is deashed aad finished using conventional
methods. Where t~e polymodal l~h~ is achieved by with-
drawing product and different points or times from the
reactor, the polymer streams are preferably blended and
a single deashing and finishing process used. The re-
sul~ is a thoroughly mi~ed, homogeneous polymer blend.
Alternately, each process stream can be finished inde-
pendently and combined by mechan-cal mixing.
Having ~hus described the above illus~ra~ive
reactor system, it will readily occur to the ar~isan
that many variations can be made within the scope of the
p.esent invention. For example, the placement and num-
ber of multiple feed sites, the choice or temperature
profile during polymerization and the concentrations or
reactants can be varied to suit the end-use application.
By practicing processes in aecordance with the
present învention, ethylene~alpha-olefin copolymers hav-
ing polymodal MWD with each molecular weight fraction
having very narrow MWD can be made by direct polymeri-
zation. Although narrow MWD copolymers can be made us-
ing other known techniques, such as by fractionation or
mechanical degradation, these techniques are considered
to be impractical to the extent of being unsuitable for
co3mercial-scale operation. With respect to ~PDM made
in accordance with the present invention, the products
have enhanced cure properties at a given Mooney Viscosi-
ty.
Where the polymodal molecular weight distribu-
tion is achieved by withdrawing polymer fractions from
the react~r, it will be evident from reference to this
disclosure that it is critical when or where polymer is
withdrawn from the reaction zone. This can be deter-
mi~ed without undue experimentation. For example, a pi-
lot plan~ scale tubula~ reactor can be ~quipped with a
~ultiplicity of t~ke off points. By running the reactor
and withdrawing polymer sa~ples from ~he system,
., ,
. . -
.
- -
- 39 -
molecular weight of the polymer at poin-ts along the re-
actor can be determined.
~ y converting the distance along the -tube to
time of reaction after introduction of catalyst, a plot
can be made of molecular weigh-t as a function of re-
action time for a given catalyst/monomer/solvent system~
The molecular weight/reaction time plot can be used to
position take off points. For flexibility in selecting
the product characteristics of a particular polymodal
MWD product, a multiplicity of take off points can be
installed, not all of which will be used in preparing a
particular product with predetermined specifications.
Similarly, inlet ports can be located at dif-
ferent locations for the in~roduction of additional
monomer or catalyst streams. By introducing fresh
catalyst and monomer downstream of the inlet, the MWD of
the polymer will be modified. So long as the polymer-
ization is carried~out in this manner the polymer will be
a polymodal MWD polymer of narrow MWD modes. Similar
results are achieved by introducing fresh premixed
catalyst with the additional monomer feed.
It will be evident from this disclosure to
those skilled in the art that the polymodal MWD polymers
of this invention can be prepared by blending the prod-
uct of runs prepared under different conditions or using
different catalyst. For example, one polymerization can
be conducted using VC14/E~SC as the catalyst and another
conducted using VOC13/EASC as the catalyst. The product
of the two runs can then be blended to form a bimodal
MWD polymer blend. Other variations can be used to gen-
erate polymer species of different Mw to prepare poly-
modal MWD compositions.
The advantages of the instant invention may be
more readily appreciated by reference to the following
examples.
. ' ' ' ' ' ` ~ .' :
- ,
''
:
.
~l~7~8
40-
Examp le
This example illustrates the method of this
in~ention for preparing an EPM wherein polymer product
is removed from the reactor at a point intermediate be-
tween the reactor inlet and outlet. The polymerization
was conducted in a 3/8 in. diameter tube and the resi-
dence time in the reactor was 30 seconds. A take off
port was located downstream of the inlet at a distance
equivalent to 1 second residence time.
Hexane was used as the solvent 9 VC14 as the
catalyst, and A12Et3C13 as the cocatalyst. Hexane is
purified prior to use by passing over 4A molecular
sieves (Union Carbide, Linde Div. 4A 1/16" pellets) and
silica gel (W. R. Grace Co., Davidson Chemical Div., PA-
400 20-4 mesh) to remove polar impurities whic~ act as
ca~alyst poisons. Gaseous, ethylene and propylene is
passed over hot (270C~ CuO (Harshaw Chemical Co.,
COl900 ~" spheres) to remove oxygen followed by molecu-
lar sieve treatment for water removal and then combined
wi~ hexane upstream of the reactor and passed through a
chiller which provided a low enough temperature to com-
pletely dissolve the monomers in the hexane.
A catalyst solution is prepared by dissolving
18.5 ~ of vanadium tetrachloride, VC14, in 5.0 1. of pu-
rified n-hexane. The cocatalyst consists of 142 g of
ethylaluminum sesquichloride, A12Et3C13, in 5.O 1. of
pùrified hexane. The two solutions are premixed at 10C
and aged for 8 seconds. Typical f ed rates and reacting
conditions are shown in Table I.
.
.
.
- ,
.
.
.
.~
7~
- 41 -
T~sLE I
Reactor Inlet Temperature ( C) -10
Reactor Outlet Temperature ( C) o
Reactor Eeed Ra-tes
Hexane (~g/hr) 60.3
Ethylene (kg/hr) 0.22
Propylene (kg/hr) 2.0
VC14 (g/hr) 2.22
A12Et3C13 (g/hr) 17
Catalyst Premixing Temperature (C) 10
Catalyst Premixing Time (sec) 8
Total Reactor Residence Time (sec) 30
A product stream is withdrawn from the take
off port at about 17 kg/hr and blended with effluent
from the reactor outlet. Approximately 20 wt% of the
polymer in the effluent came from the take off port.
The molec~Lar weight of the product from the take off
port is about one-half of that from the reactor outlet
with a similar MWD (MW/Mn) = 1.4, Mz/Mw = 1.3). The
product is deashed and stripped. The resulting polymer
is a bimodal MWD EPM with a theoretical ~ /Mn = 1.96 and
,IZ/MW = 1. 46 .
E~ample II
Example I is repeated except that no effluent
is taken from the take off port and two reactors in par-
allel are used. The feed rates listed in Table 1 are split so
that 17 kg./hr are passed through the reactor with one
second residence time, and the remaininy feed goes to the
other reactor. The residence times in these two reactors
are 1 and 30 seconds, respectively. Otherwise all
conditions are the same as in Example 1. The effluents from
the reactor outlets are blended. After steady state
is achieved, the blend is deashed, washed and stripped of
solvent. The resulting polymer is a bimodal MWD EPM with
a theoretical MWD as in Example I.
.
--
~. . ,
.
' ' ' ~ : '
' ' .
:
.
~.~7~ b
-b2-
Ex~mple III
Example I i~ repeated except that no effluent
is ~2ken from the take off port and VOC13/EASC is used
2S an additional catalyst~
The second cat~lyst sol~tion is prepared bg
dissolving 18.5g of VOC13 in 500 lo of purified hexane.
The cocatalyst consists of 142g of A12Et3C13 in 5. 0 1.
of purified hexane. The VC14 and VOC13 ar~blended with
coc~talysts in separate pre~ixing units and aged for ten
seconds. The two premixed catalyst streams are then
.mixed with the monomer/ hexzne stream and fed into the
recctor. Reactor residence time is 50 seconds. Other-
wise all conditions are the same as in Example I. After
s~eady state is achieved, the reactor effluent is de-
as~ed, washed and stripped of solvent. The resulting
?olymer is ea bimodal ~ EPM.
EYe--pleIV
Example I is repeated except that no effluent
is t2ken from the take off port. The catalyst system
used is vanadium oxytrichloride ~VOC13) and diethyl-
aluminum chloride (AlEt2Cl). O~herwise 211 conditions
~re ~he sa~e as in Ex2mple I; Ihis catalyst system pro-
duces at least two independent catalyst species, each of
which initiates a separate ~WD mode. After steady state
is achieved, the reactor effluent is deashed, washed and
stripped of solvent. The resulting polymer is a poly-
modal ~WD EPM.
Ex2nple V
`Example I is repe2ted except that no take offpo.t effluent is collected and the catalyst and feed
streams are splitO About 2/3 of the monomer/hexane
st~eam and 7~3 of the prem~'xed cztalyst are mixed an~
fed ~o the reactor inlet and the re~aining l/3 of t~e
mono~er/hexane feed is mixed with t~e remaining eatalyst
`` . ' ~, ' . ~."' ~ , .
'
~ 7X ~
~43-
Seream and fed into the re~ctor at a point midway be-
t~een the reactor inlet and outlet. The EPI~ product is
a poly~odal ~l~ polymer.
Ex2-ple VI
Exa~ple I is repected except that no effluent
is taken fron the take off port and a ca~zlyst reactiva-
t~r is used. The catalyst reactivator solution i~ pre-
pared by dissolving 30.5g of butyl perchlorocrotonate i.n
3.0 l of purified hexane. This solution is fed into the
reactor, at 3.6 g/hr along ~ith 50 g/hr of ethylene, at
a point ~idway between the reactor inlet and outlet.
Otherwise all conditions are the same as in Example I.
~fter steady state is achieved, the effluent is deashed,
washed and stripped of solvent. Ihe resulting product
is a polymodal I~ EPM.
It will be appreci2ted by those skilled in ~he.
art wbo have reference to this disclosure that where
rererence is made to t~e beginning of polymerization, in
a contînuous process, this is intended to mean the time
at ~-hich catalyst is introduced. Si~ilarly, the end of
the poly~eri~ation in a tubular reactor ~ecns at the
point where the polymerizationis terminated.
.~ere reference is ~ade to blends being made
by co~bining product or re~ction mi~tures withdra~ rom
the reactor at one or more times ~fter the start of
polyme?rization with product from the "reactor outlet" or
"co~pletion of poly~erization~' this langucge is intended
~o include the last product or reaction mixture with-
dre~n from the reactor for the purpose of forming the
blend whether or not the 12st product or reaction mix-
ture is obtained fro~ the physical reactor outlet or a~
the.2ctual co~pletio~ of poly&eri7ation9 notwithstanding
~he fact that product from the actual reactor ou~let or
ac;ual completion o~ poly~eri72tion is used for some
p~-pose other than blending with fra~tions o~ poly~er
i .harawn .
: .
' ~ ~, ',
- , . , , , ~
,