Note: Descriptions are shown in the official language in which they were submitted.
~ ~73~i3~
- 1 - 21489-6789G
This is a divisional application of applicatlon Serial
No. 490,301 filed September 10, 1985.
The parent application relates to a novel process for
the preparation of 4-phenylpyrrole derivatives of the formula I
r \ ~ Rl (I)
Rn N
R2
wherein Rl is CN, CHO or COO(Cl-C6)alkyl, R2 is hydrogen, CH2CH2CN
or CH2CH2COO(Cl-C6)alkyl, R is halogen, Cl~C6alkyl or Cl-C6halo-
alkyl, and n is 0, l or 2.
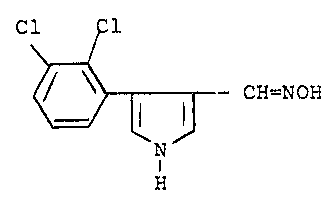
This application relates to 3-hydroxyiminomethyl-4-
(2,3-dichlorophenyl)pyrrole of the formula
Cl Cl
~CH=NOH
H
It should be noted that in this application the term
"invention" includes the subject matter of both the parent and
the divisional applications.
Depending on the indicated number of carbon atoms,
alkyl by itself or as moiety of another substituent, such as halo-
alkyl and the li~e, comprises e.g. the following straight chain
or branched groups: methyl, ethyl, propyl, butyl, pentyl, hexyl
etc., and the isomers thereof, e.g. isopropyl, isobutyl, tert-
36~
- la - 21489-6789G
butyl, isopentyl etc. Throughout this specification, a substituen-t
p:refixed by "halo" will be understood as meaning that said
substituent may be monohalogenated or perhalogenated. Halogen
and halo signify in particular fluorine, chlorine or bromine.
Hence haloalkyl denotes a monohalogenated to perhalogenated alkyl
radical, e.g. CHC12, CH2F, CC13, CH2Cl, CHF2, CH2CH2Br, C2C15,
CHBr, CHBrCl etc., with CF3 being preferred.
~ ;~73~
4~Phenylpyrrole derivstives of formula I, whereln n 1~ O, 1 or 2,
Rl i8 cyano and R2 i8 hydrogen or acetyl, are known n~ plant
funglcide~ from German Offenlegung~schrift 29 27 4801). As will be
shown below, compounds of formula I, wherein R1 is CHO or COO(C1-C6)-
al~yl or R2 i8 CHzCH2CN or CHzCHzCOO(Cl-C6)alkyl, an be converted
in simple manner into the known fungicldal 4-phenyl-3-cyanopyrroles
and thu~R have the character of in~ermediate~.
A proce~ for the pr~paration of 4-phenyl-~-cyanopyrrole derivatives
which i5 known fro~ Tetrahedron Letter~ No. 52, pp. 5337 S340,
19722), i~ dlsclosed in Ge~man Offenlegung~schrift 29 27 4801). In
this proceA~, known aY the TosMIC process, a cinnamic acid deri-
~ative of formula X
~X)
n _ CH3~ 02H ~ il_CN
(XX) CN3~ SO2CHzNC
i~ cyclised with to~yl methyl 10ocyanide (XX) ¦TosHIC], in the
presence of a strong base, e.g. sodium hydrid0, to give 4-phenyl-
3-cyanopyrrole derivative~ of formula tXXX). In the above formulae,
R is as defined for formula I and n i8 O, 1 or 2.
Although numerou~ pyrrole synthe~o3 are known (q.v. J.M. Patterson,
Synthesis 1976, pp. 281-3043)), only the TosMIC process outlined
abGve ha~ ~o far led direct to the fungicidally u3eful 4-phenyl-
3-cyanopyrrole derivatives. Howaver, reference 2) indicateR for the
preparstion of 4-phenyl 3-cyanopyrrole a yield of only 35 %, which
i8 low for indu~trial purposes. It ha~ been found that the reagent
TosMIC has grave di~advantages for industrial synt~eses. For
examplP, at elevated tempPrature~ above 90C (normal drying con-
dition~), TosMIC ha~ the propen~lty to decompo~e explo~ively. On the
~73~ 7
-- 3
other hand, resldual moisture consumes some of the base e~ployed
(danger of hydrolysl~/reduction in yleld). Further, TosMIC hAs
physiological ha~ards and causes severe irritation to the eyes and
3kin.
The shortcoming~ referred to above show that useful laboratory
methods are ~nsuitable for tha indu~trial production of 4-phenyl-
pyrrole derivative~. A novel, more economlc and envlronmentally more
acceptable process for the prepara~ion of thQse compounds in
surprlslngly high yleld haD now been found.
The novel proce3s of this invention for the preparation of the
4-phenylpyrrole derivatlves of the formula I as deflned at the
outYet comprises reactlng a phenacylamine of formula II
~ CH2-~H ~II)
R '~- 2
in the form of an acld add~tion salt, with a compound of ~ormula III
T-CH~CM-R1 (III)
to give an lntermediate of formula IV
~ CH2-~-CH-CH-R1 (IV)
R~ 2
and cyclislng thi~ compound of formula IV, ln the pre~ence of a
base, to a compound of formula I. In the formulae II, III and IV
above, the ~ub~tituents R1, R2 and R sre as defined for formula I,
T is a group selected ~rom -OZ, -N~R3)(R4~, -OCORa, -OS02~ , -SRc or
halogen, where Z is C1-C6alkyl, unsubstltu~ed or ~ubstituted phenyl,
an alkali metal atom or an alkaline earth ~etal atom, each of ~ and
independ~ntly of the other 19 C~-C~slkyl or unsubstituted or
sub~tituted phanyl, R~ i9 C~-C3alkyl, C1-C3haloalkyl or unsub-
stituted or substituted phenyl; and each of R3 and R4 independently
3~ 7 ~
of the other iB Cl-Csalkyl or, together with the ~mlne nitrogen
atom, form a saturated 5- or 6-member2d hetarocyclic rlng which
contains, as hetero atom, elther only the amlne nitrogan atom or a
further hetero atoM.
An unsubstltuted or ~ubstituted phenyl group 18 ln partlcular phenyl
or phenyl which i8 sub~tituted in the para-po~ition by halogen,
preferably chlorine or bromine, and by Cl-C3alkyl, prefersbly
methyl. Alkali metal atoms or alkaline earth metal atom~ may be Li,
Ns and R, preferably Na and K, or Mg, Ca, Sr and Ba, preferably Mg,
Ca and Ba. Where the -N(R3)(R4) group danotes a ~atursted S- or
6-membered heterocyclic ring containing N a~ hetero atom or a
further hetero atom, said ring ~ay be selected from the following
heterocyclic ring sy3tem3: pyrrolidine~ piperazine, perhydro-
thiazlne, morpholine, piperazine, oxazolldine, thia~olidlne,
lmlda~olldine, pyrazoline and the like. A further hetero atom i~
preferably N, O or S.
In the proce~s of this in~ention it i9 not necessary to i~olate tha
inter~ediate (IV) fir~t and then to cycli~e it to compound~ of
formula I. To the contrary, the reaction of (II) wlth (III) may also
be carried out direct in the presence of a base, utili~ing a ~ingls
reaction ves~el for both ~teps, to give the final product~. In thi~
procedure, the intermediate (IV) is further processed direct without
isolatlon. On the other hand, it may be convenient to prepare the
lntermediate ~IV) flrst in especially pure fo~m, e.g. by repeated
racryst~lliDation, and then to cycli0e it to a compound of formula
I. A preferred embodiment of the process of thi~ invention accord-
ingly coMpFlsQ~ reac~ing the phenacylamine II in the form oE an
acid sddition ~slt, in the pres2nce of a baae, direct with a
compound of formula III t~ glve the final product I.
The second preferrsd embodlment of the process comprises flrst
r reactlng the phenacylamlne II in the form of an $cid additlon ~alt,
in the absence of a base 9 to give the intermedlate (IV) and then
converting (IV3 to tI~ by cycli~ation in the presance of a baae.
~ ~73~
The react~nt~ (II), (III) and, where Approprlate, (IV) 9 are con-
venisntly employed in equimolar amounta. It i~ preferred to add an
equimolar amount or an excess of bsse.
Typical representatives of the compounds oE formula III, the li~t of
which 13 not exhaustlve, are the following compounds a) to t), of
which compounda a) to 1) are p~rticularly advantageous and therefore
preferred:
a) (CH3)2N-CH~CH-CN
b) (CzHs)2N-CH~CH-CN
c) ~ CH~CH-CN
d) ¦ ~-CH~CH-CN
e) O~ ~ -CH~CH-CN
f) NaO-CH~CH-CN
B) Ko-cH~cH-cN
h) (CH3)2N-CH~CH-COOCH3
1) (C2Hs)2N-CH~CH-COOCH3
k) (CH3)2N-CHnCH-CHO
i) (C2Hs)2 N-CN~CH-CHO
m) Cl-CH~CH-CN
n) Cl-CH~CH-COOCH3
o) CH30zSO-CH~CH-CN
p) [ C6H4CH3 ~ 4) ] -CH~CH-CN
q ) CH 3 0-CH~CH-CN
r) C2HsO-CH~CH-CN
~) C3N70-CH~CH-CN
t) [C6H4Cl(4~]0-CH'CH COOC~3
3~3~
The process of thls invention is conveniently carried out in an
inert solvent or mixture of solvents. Thus one or more lnert
solvents or diluents may be employed. Examples of suitable solvent~
and diluents are: allphatlc and aromatic hydrocarbons ~uch a~
benzene, tol~ene, xylene~, petroleum ether; halogenated hydrocarbons
such as chlorobenzene, methylene chloride, ethylene chloride,
chloroform, carbon tetrachloride, tetrachlorosthyleDe ethers and
ethereal compounds such a8 dialkyl ethers tdiethyl ether, dii~o
propyl ether, tert-butylme~hyl ether etc.), anisole, dio~cane,
tetrahydrofursn; nitril~s such as acetonitrile snd propionitrile;
N,N-dialkylated amides such as dimethylformamide; dimethylsulfoxide;
ketones such as acetone, diethyl kstone, methyl ethyl ketone;
alcohols, in particular methanol, sthanol) propanol~, butanols and
the llke; and water and aqueous two-p~sse mixtures and mixture~ of
the above solvent~.
The following solvents for example are suitable for the orgsnic
water-im~i~clble phase: ~liphatic and aromatic hydrocarbon~ such as
pentsne, xylenes etc.; halogenated hydrocarbons auch a~ dlchloro-
methane, chloroform, carbon tetrachloride, ethylene dichloride,
1,2 dichloroethane, tetrachloroethylene and the like, or allphatic
ethers such as diethyl ether, diiQopropyl ether, tert-butylmethyl
~ther and the like. The addition of a pha~e transfer c~tAlyst may be
Advsntageous. Examples of suitable phase tsansfer catalysts are:
tstraalkylsmmonium halides, hydrogen sulfatas or ~ydroxides, e.g.
tetrabutylammonlum chloride, tetrabutylammonium bromide, tetrabutyl-
ammonlum lodide, triethylbenzylammonlum chloride or triathylbenzyl-
ammonlum bromide, tetrapropyla~monlum chloride, tetrapropylammonium
bromlde or t~trapropylammonium iodlde etc. SuitablQ phase tran~fer
catalyst~ flre al~o phosphonium ~alts. The ammonlu~ salt of fos-
mula II it~elf scts as phas~ tranafer catalyst.
Part~cularly suitable solvent3 are nitriles and lowsr alkanol~,
prefersbly ~cetonitrlle and ethanol, aa well as mlxtures of
alkanolJwster ~ethanol/wster).
3~3~ _~
In all psrtlsl steps and in the single ve~sel reaction, the reaction
temperatur~s are generally in ~he range from 0 to tl20C, pre-
ferably froM ~30 to +80C.
Owing ~o the reduced thermfil stability o~ the stArting phenacyl-
amine, the compound of formula II i9 employed in the fDrm of it~
more ~table ammonium salt, which can be obtained by conventional
addition of an organic or i~organic acid to the fre~ amine.
~xamples of salt-forming acid~ are inorganic acid~, e.g. hydrohalic
acids such a~ hydrofluoric QCid, hydrochloric acid, hydrobromic acid
or hydriodic acid, as well &8 ~ulfuric acid, phosphoric acid,
pho~3phorous acid, nitric acid and the llke; and org~nic acid3 such
as acetic acid, trifluoroacetlc acid, trichloroacetic acid, pro-
pionic acid, glycollic acid, lactlc acid, s~cclnlc acid; benzoic
acid, cinnamic acid, oxalic acid, formic scid, benzene~ulfonic acid,
p-toluenesulfonic scid, methanesulfonic acid, sal~cyclic acid,
2-phenoxybenzoic acid or 2-acetoxybenzoic acid and the like.
PrefQrred sal~-forming acids are ~trong acidR such a~ the hydrohalic
acids, pho0phoric acid, nitric acld, and the sulfonic acids suoh a~
p-tolu~n~sulfonlc acld. Hydrochloric acid i9 especially preferred.
The reaction of (II) with (III) direct to give (I), or of (IV) to
giva (I~ conducted in ths presence of R base. Example~3 of
~uitable ba~e~ are inorganic ba3es such a3 the oxides, hydr~des,
hydroxides, carbonat~s, carboxylic acid ~alt~ and alcoholatas of
alkaline earth metal3, preferably o alkali metals, in particular of
~odium and potas~ium le.g. NaH, NHOH, KOH, Na2C03, K2CO3, CaC03,
CH3COONs1 C2HsCOOK, C2HsONa, CH30Na and ths like], pre~erably the
alkali metal alcoholates ~uch a~ ~odium ethylat~ or ~odium
m~thylate. Suitable organic baces are Q.g. triethylam~ne,
piperidine, pyridine, 4-dimethylaminopyridine and the like.
73~
In the proces~es of thls lnvention, intermedlates and final products
may be isolated from the reaction medium and, if de~ired, purifled
by one of the commonly employed methods, for example by extraction,
cry~tallis~tion, chromatography, distillation and the llke. However,
the preparation of the compound~ of ~ormula I can be carried out
generally in good yield and in excellent purity utilising a ~ingle
vea~el for bo~h reaction steps ~ithout i~olation of intermediates.
Preferred embodiments of the process of this invention are e.g.
tho~e which comprl~:
a) the u~e of starting material~ of formula II, wherein R is
hslogen, prefarably fluorine, chlorine or bro~ine, most pre-
ferably chlorine, n is l or preferably 2, with the proviso that,
if n i~ 2, the ortho- and meta~po~itions are particularly
prcferred, Rl i~ CN and R2 is hydrogen;
b) the us~ of reagents of formula III, wherein T i9 a group aelected
from -N(CH3)2, -N(C2Hs)2,
t ~ 0 ~ -OK or -ONa,
and R1 i~ CN, COOCH3 or CHO, preferably CN
c) the use of intermedintes of formula IY, wherein Rl, R2 and Rn are
as definad in a) and b) above;
d) the use of acid addition ~alts of formula II, which contaln, a~
acid component, a hydrohalic acid, preferably hydrochloric acid,
a sulfonic aoid, preferably benzenesulfonic or p-toluenesulfonlc
acid~ or ~ulfuric Acid;
e) carrying out the reaction of (II) with ~III) ~uch that th~
lnterm0diate IV 18 further proce~sed direot without ~olati~n;
37
,
_ 9 ~
f) csrrying out the process in the tempsrature ranga from ~30 to
+80C.
Accordingly, a particularly preferred embodlmeDt of the proces~ of
thc invention compri~es reacting 2,3-dlchlorophenacylamine in th0
form of an acld addition salt, preferably in the form of the
hydrochloride, with a co~pound of formula III, wherein Rl i~ CN and
T is a group ~slected from -N(CH3)z,
2 5 ) 2 ~ N~ \O , -OK or -ONa
._. . _ . "_ .
preferably -N(CH3)2, ~ ~ or ~ ~0
to give 3-(2,3-dichlorophenacylamino)acrylonitrile, and cyclising
thi~ intermediate, either as substanca or prefersbly in ~itu, in the
pre~ence of a base, preferably of 8 lower alkanolate, aodium
hydroxide, pota~sium hydroxide, sodium acetate, pota~lum acatate or
a tri-lower ~lkylsmlno, to give 4-(2,3-dichloroph~nyl)-3-cyano-
pyrrole.
Mo~t of the starting material~ of formula II are known or can be
p~epared in similar manner to the known representative~. However,
2,3~dichlorophenacylamine and the acld addition ~alts thereof are
novel. In view of its ~tructure, this compound iB deYtined for use
as lntermedlate for tha preparation of fungicidally active 4-(2,3-
dlchlorophenyl)-3-cyanopyrrole and thersfore constit~te~ an ob~ect
of this invention. It~ preparation wlll be described explicitly
below.
Compound~ of formula II, wherein R2 18 CH2CH2CN or CH2CH2COO(Cl-C6)-
~lkyl, c~n bs pr~psred e.g. a~ follows from the ~tarting phenacyl-
amines (II) (R2 - H): The acid addition ~alt (e.g. the HCl ~lt~of
an N-substituted phenacylamlne of formula II i~ reacted, in the
73~
, .
- lU -
presence of ~n equimolar amount of acrylonltrile or of a Cl-C6alkyl
~Rter of acrylic acid, preferably in th~ presence of one of the
basea specified above and under the cond~tions for the reactlon of
(II)~ with ~III) to give (I).
Within th~ scope of the present invsntion, typical rapre~entatives
of compounds of formula I ara for exampla tha compounds llsted in
Table 1.
Table l: Compounds of formula II
~ CHz-NH-R2 (II)
R Y-
Compound Rn R2
_ .
l.1 H
l.2 3-Cl H
1.3 2,4-C12 H
1.4 4-Cl H
1.5 4-F H
1.6 3-CH3 H
l.7 3-P H :
1.8 3-Br H
l.9 3-CF3 H .:
l.lO 2~Cl H
1.11 2,3-C12 H
1.12 2,5-C12 H
l.l3 2-Br H
l.14 2,6-Clz H
l.15 H CHzCHzCN
1.16 3-Cl CHzCHzCN
l.17 2-Cl C~2CH2CN
73~3~
Tabls 1 (continuation)
Compound _ _ _ .
l.18 2,3-C12 CHzCH2C~
l.l9 3-F CHzCH2CN
l.20 3-Cl CHzCH2COOCH3
l.21 2,3-C12 CH2CH2COOCH3
].22 2-Cl CH2CH2COOCH3
1.23 2,3-C12 CH2CH2COOC2Hs
l.24 2,3-C12 CHzCH2COOC3H7
l.25 2-Br-' CH2CHzCOOCH3
.
The compounds of formula III are in general commercially available
and thus known ~ub~tances or compounds which can be prepared in
similar manner to their known representatives.
The preparatlon of the intermediates of formula IV i~ an ob~ect of
the present invention and haa b~en described in detail abovs. These
intermediate~ IV can be converted by simple basic cycllsation into
the useful fungicid~ of formula I, have themselves fungicidal
activl~y, and accordingly constitute an es~entiQl ob~ect of ths
present invcntlon.
Withln the scope of thi~ invention, typical representat-lves o
in~srmediat~ of formula IV ara:
Table 2: Compounda of ths ~ormula
~ cHz-~-cH~cH-R
, , ~
Compound Rn RZ R,
2.1 H H CN
2.2 3-Cl _. _ CR
' '` '~ d '73~7
- 12 -
Table 2: (continuation)
Compound _ _ _~
2.3 2,4-Cl H CN
2.4 4-Cl H CN
2.5 4-F H CM
2.6 3-CH3 H CN
2.7 3-F H CN
2.8 3-Br H CN
2.9 3-CF 3 , H CN
. 2.10 2-Cl H CN
2.11 2,3-C12 M CN
2.12 2,5-Cl~ H CN
2.13 2-Br H CN
2.14 2,6-C12 H CN
2.15 2,3-C12 H COOCH3
2.16 H H CHO
2.17 3-Cl H COOCH3
2.18 3,4-C12 H COOCH3
2.19 2-Cl H COOCH3
2.20 2,3-C12 H COOC3H7
2.21 2,3-C12 CH2CH2COOCH3CN
2.22 2,3-C12 CH2CH2CN CN
2.23 H CH2CH2CN CN
2.24 3-Cl CHzCH2N CN
2.25 2-Cl CH2CH2CN CN
2.26 2,3-C12 CHzCH2COOC2Hs CN
2.27 3-F CH2CH2CN CN
2.28 3-Cl CM2CH2COOCH3CN
2.29 2-Cl CH2CH2COOCH3CN
2.30 2,3-C12 CHzCH2COOC3H7 CN
2.31 2-Rr CH2CH2COOCH3CN
2.32 2,3-C12 CH2CH2CN CHO
~! ~73~i3~
As mentloned at the outset, ~ome of the compound~ of formula I have
the character of interm0diates. These compounds sre the repre~en-
tatives of formula I hereinafter referred to a9 subgroup la, whereln
R i9 a~ defined for formula I; and in tho8e compounds in which R1
i9 CHO or CO0(C1-C6)alkyl, Rz lfl at the same tim~ hydrogen, C~IzCH2CN
or CHzCH2COO~C1-C6)alkyl, or in tho~e compounds in which Rl is CN,
Rz i9 at the same time CH2CH2CN or CH2CH2COO(C1-C6~alkyl. The~e
novel pyrrole derivatives, which also have fungicital propertie.q,
can be converted in simple manner into the fungicidal 4-phenyl 3-
cyanopyrrolss known from German 0ff~nlegung3schrift 29 27 480, a9
CHO and COO(C1-C5)alkyl can be converted into CN, and CH2CH2CN and
CH2CH2CO0(C1-C6)alkyl as ~ub~tituents at the pyrrole nitrogen atom
are easily removable group~. On account of the~e advantageous
properties, the co~pounds of ~ubgroup Ia constitute a further ob~ect
of the pre3ent invention.
Typical example~3 of compoundfl of ~ubgroup Ia are listed below.
Table 3: Compounds of formuls Ia
(Ia)
n
Compound R - r _ _ _ Rz
_ n _ _ _ _
3.1 H CHO H
3.2 3-Cl CHO H
3.3 2,4-Clz CHO H
3.4 4-Cl CHO H
3.5 4-F CH0 H
3.6 3-CH3 CHO H
3.7 3-F CHO H
3.8 3-CP3 CHO H
3.9 2,3-Clz CHO _
,
t'' ~ 73~j3~
Table 3 (continuatlon)
Co~po nd Rn . . _
3.10 2,6-Cl2 CHO H
. 3.12 3-C1 COOCH3 H
! 3.l3 2-Cl COOCH3 H
3.14 4-F COOCH3 H
3.15 2,3 C12 COOCH3 H
3.16 3-Cl CN CH2CH2CN
3.17 2-Cl CN CH2CHzCN
3.18 3-CH3 CN C}IzCH2CN
3.19 2,3-C12 CN CH2C~2CN
3.20 4-F CN CH2CH2COOCH3
3.21 2~Cl CN CH2CH2COOCH3
3.22 2,3-C12 CN CH 2 CH2COOCH3
3.23 2,3-C12 CHO CH2CH2COOCH 3
3.24 2,3-C12 COOCH3 CH2CH2CN
The conv~r~ion of CHO lnto CN can be effected in a msnn0r known per
se~ for example a~ follows: An aldehyde of formulA I (Rl ~ CHO) 19
converted st 0 to 100C, in sn lnQrt ~olvent (e.g. an alcohol, an
ether, pyrldine, triethylamine and the like) i~to the corra~ponding
oxime (syn/anti mixture), whlch i~ converted into the nitrlle by
treatment with a dehydrating agent (e.g. scetic anhydride, cyanuric
chlorlde/pyridlne, (PNCl2)3, dicyclohexyldlcarbodllmlde/CuCl2/trieth-
ylamlne, P2Os 9 tosyl chloride/pyridine, TiCl4/pyridine and the
llke).
If it is deaired to conver~ the es~er group COG(Cl-C6)alkyl lnto the
CN group, a start is be~t made from th0 free acid, which is prepared
ln a manner known per se by e~ter hydroly~i~ wlth an aqueous mlnars1
scid (a.g. HCl/h2O), in the pra~ence of a solubiliaer (e.g. alcohol9
dioxane, tetrahydrofuran and the like), most convenlently under
reflux tempera~ure. The free acld i~ then converted in~o the acid
amide either direct with ammonia at elevated tempersture or ~ia the
3~
- 15 - 21489-6789
acld chlorlde (-COOH + thionyl chloride ~ -COCl) wlth ammonla at
room temper~ture, snd the acid amide i9 converted to the nltrile
with one of the prevlou~ly mentloned dehydrating agents ln the
temperature sange from 80 to 220C.
I~ it i~ desired to ~orm the frce pyrrole ~y removal of the CHzCH2CN
or CH2CH2COO(CI-C6)slkyl radical, thi~ may be done e.g. by treaement
wlth a baa~ in the temperature range from -20 to +180C, in a
~uitable lnert solvent. Exemplary of sultabla reaction condition~
are:
a) sodium hydrlde in dimethylformamlde at 0C
b) ammonia/water/dioxane At 180C
c) potassium hydroxlde/~ater/alcohol at 100C.
Pr~paratory Rxample~
Example Pl: Prèpsrstion of
~f ~ CHz-~H2 HCl bsse \ ~ ll b
~CH3)2N-CH~CH-CN X
4-(2.3-Dlchlorphenyl)~3-cyanopyrrole
~) PrepsrAtlon of thc precur00r:
N-acetyl-2,3-dlchlorophenacylamine
lS0 g of 2,3-dichlorobenzoyl cyanide are hydrogrnated with element-
al hydrogen under normal pre~sure at 70C in 1.5 ~ of glacial scetlc
acld and 84.15 g of acetic anhydrlde over 5 g of PtO2. After
ab~orption of 112 % of th~ c~lcul~ted amount of hydrogan (time
ta~en: c. 5 hcurs), the hydrogenatlon 1~ dlscontinued, the raaction
mi:~tura 19 flltered 8nd the filtrate i~ concentrsted by evaporation.
The re~dual yellow oil is cryst~ ed by addition o hexane/
diethyl ether. The cry~tallln~ product 1~ i901ated by filtration and
J .r ~ ~
36~ ~
- 16 -
dried. M.p. 107-109C. IR (solid~KBr) in cm 1 3300 (NH); 1735 (C0)
1650 (C0). IH-~MR (CDCl3) in ppm: 2.08 (s,3H); 4.55 ~d,2H); 6.2-6.6
(broad 8, lH); 7.25 (m,3H).
b) Preparation of the precur~or:
2,3-dichlorophenacylamlne hydrochlorlde
50.0 g of the N-acetyl-2,3-dichlorophenacylamlne obtained in a) are
heated for 2 hours under reflux in 500 ml of hydrochloric acid. The
sllghtly turbld reaction ~olution i~ concentrated by evaporation and
the residuQ is digested wi~h ethyl acetate. The crystalline 2,3-di-
rhlorophenacylamine hydrochloride i9 i~olated by filtration and
dr1ed. Melting point: 217-218C. IR ~solid/RBr) in cm 1 1695 (C0).
(Another crystal modification ~how3 two carbonyl resonance band~ at
1690 and 1705 cm ~ H-NMR (DMS0, d6) in ppm: 4.54 (8, 2H~; 7.6 (t,
lH) 7.9 (~, 2H); 8.6 (8, 3H, replaceable with DzO).
c) Preparatlon of the fin~l product
4-(2,3-dichlorophenyl)-3-cyanopyrrole
20.0 g of 2,3-dichlorophenacylamine hydrochlorlde snd 10.0 g of
3-dimethyla~inoacrylonltrile are heated for 1 hour under reflux ln
300 ml of ethanol. Then an ethanolic ~olution of 30dium ethylate,
prepared from 2.1 g of Yodium and 30 ml of ethanol, i9 rapidly added
dropwise and thc reaction mixture i8 stirred for another 10 minute~
under reflux. The reaction mixture 19 cooled to room tcmperature and
thon poured into ice/hydrochloric scid snd the resultant mi~ture i~
stirred for 1 1/2 hours. Tha precipltate i9 i301ated by filtration,
washed with water and dried, affording 15.4 g (78 % of theory) of
title compo~nd with a meltlng polnt of 152-154C.
~7;3~
Example~ P2 to P~: Prep~ration of
P2: ~ CH~CH-CN C~ /Cl Cl
~._. ~ -CH2-NH2 HCl C~,
P3: j ~ -CH~CH-CN ba~e ~ ~ -C~
0_-
P4: 0/ ~ -CH~CH-CN
4 (2,3-Dlchlorophenyl)-3-cYanopyrrole
Pollowlng the proc~dure de~cribed in Exsmple Plc), but replacing
3-dlm~thyla~inoacrylonitrile by
N-plperidinylacrylonitrlle,
N-pyrrolidinylscrylonitrile, or
N-morpholinylacrylonltrile,
snd increa3ing the reactlon tlme from 1 hour to 3 to 4 hours, pure
4-(2,3-dichlorophenyl)-3-cyanopyrrole i8 obtsined in all three
Example~ in yield~ ranging from 76 to 85 % of theory. Melting point:
150-154C.
Example P5: Preparation of
~ NH-CH~CH-CN bas~
4-(2,3-di~hlorophenyl~-3-cyanopyrrole
a) Preparation of the intermediate:
3-(2,3-dichlorophenacYlamlno)acrylonitrile
20.0 g of 2,3-dichlorophenacylamine hydrochloride and 10.0 g of
3-dimethylamino6crylonitril~ ars heated for 1 hour under reflux in
300 ml of ethanol. After cooling lt to room temperature, the
reaction ~olutivn i~ poured into a mixtur~ of i~a/dilute hydrochlor-
~73~
- 18 -
ic acid. After e~tractlon with ethyl acetate, the combined extracts
are drled over Rodlum ~ulfate, filtered, and the filtrate i~
concentrated. The oily re~ldue i8 purifled by column chromatography
(~ilica gel: elution with a 4:1 mixture of toluene/ethyl acetate).
M.p. 125-127C. IR (~olid~KBr) ln cm : 3380 (NH)I 2200 (CN),
1715 (C0) 1625 (C~C). IH-NMR (DMSOd6) in ppm: 4.09 ~d, J ~ 15 Hz,
lH); 4.46 td, J ~ 7 Hz, 2H); 7.2 (q, lH); 7.4 ~broad s,lH);
7.45-7.85 (m, 3H). Ma~s ~pectrum: molecular pea~ at 254.
b) Preparation of the final product
4-2,3-dichlorophenyl)-3-cyanopyrrole
To 402 g of ths 3-(2,3-dichlorophenacylamino)acrylonitrilQ obtained
in a) i~ added 0.5 g of ~odium ethylate in 50 ~l of ethsnol. ThQ
resction mixtura i~ heated to ~eflux tempera~ure, cooled to room
temperaturP, poured into a mixture of dilute hydrochloric acid and
lce, and stirred for c. 1 hour. The precipitate i~ i~olated by
filtration, washed with water and dried, affording the title
compound in quantitative yield. Melting point: 149~-150C.
Example P6: (Formulaa, see Ex. P5)
a) Preparation of tha intermediata
3-(2l3-dichlorophenacylamino)acrylonitrlle
2 g of 2,3-tichlorophenacylamine hydrochloride, 1 g of 3-hydroxyacrylo-
nitrile, ~odium ~alt, and 20 ml of ~thanol are heated for 2 hour~
under reflux. The reactlon mixture i~ concentr~ted by evaporation
and the oily re~idue iB purlfied by column chromatography (~ ca
gel; elution with A 4:1 mixture of toluenet~thyl acetate), ~fford1ng
3~(2,3-dlchlorophenacylamino)acrylonitrlle in the cis/trans ratlo of
5:1. Melting point: 122-125C.
b) Preparation of the final product
4-(2~3 diehlorophenYl~-3-cy~nop~rrole
4.2 g of the 3-(2,3-dichlorophenacylamino)acrylonitril~ obtained in
a) are reacted ln 50 ml of ethanol with 0.5 g of sodium ethylate aR
de~cribsd ~n ~ample P5 b), ~f~ording the title compound ln quan~
titatlve yi01d. Meltlng point: 150-152C~
-- 19 --
Example P7: Preparstion of
C~ /Cl ~ /Cl
.\ , ~,_0 _ O_~ooC~ ~ -COO~ }1--o-C~
4-(2,3-dichlorophenyl)-3-cyanopyrrole
a~ Prepsration of 3-carbom~thoxY-4-~2,3-dichlorophenyl)pyrrole
10.7 g of 2,3-dichlorophenacylamine hydrochloride and 6 g of methyl
3-d~methylaminoacrylate ar~ heated for 2 hours under reflux ln
120 ml of ethanol. Then a solution of 4 g of sodium ethylate in
40 ml of ethanol is added dropwise and the reaction mixture 1~
heated for another hour under reflux. The reaction mlxture i8 ehen
concentrated by evaporation and the oily residu0 i8 purlfied by
column chromstography (~illca gel elution w~th a 3:1 mixturs of
toluene/ethyl acetate. Melting p~lnt: 205-206~C.
b) Preparation of the precursor
4-(2,3-dlchlorop~en~l)pyrrole-3-carboxyllc acid
3.2 g of the 3-carbomethoxy-4 (2,3-dichlorophonyl)pyrrole obtalned
in a) and 40 ml of a 1:1 mixture of methanol and 5N HCl nre stirred
for hour~ at 70C. Aft~r it has col~d to room te~perat~re, the
re~ction mlxture i9 poured onto iC8 and ~xtracted with e~hyl
acetate. The ester phase i8 in turn extractsd wit~ 10 % sodlum
hydroxide solution. The aqueou~ extract is washed twic~ with ethyl
acetate, acidifi~d with hydrochloric acid and extracted wlth ethyl
acetate. The organic phase i9 wa~h~d with water, dried G~er ~agne~-
ium sulfste and filtered. Tha flltrata i~ concentrat~d by e~apor~t
ion and the re~ultant 4-(2,3 dichlorph0nyl)pyrrol0-3-carboxylic acit
melt~ at 180-182C.
~3
- 20 -
c) Preparation of the final product:
4-(2,3-dlchlorophenyl)-3-cyanop~rrole
2.1 g of the free 4-(2,3-dichlorophenyl)pyrrole-3-oarboxylic acid
obtained in b) are dissolved in 30 ml of ethanol. Ths solut~on i~
made alkaline with concsntrated ammonia and then evaporated to
drynes~. The residue iB dls~olved in SO ml of ethanol. ~H3 gas iB
added ~0 atm) to this ~olution at room temperature in an autoclave
and the reaction mlxture iB kept for 15 hours at 22QC. The reaction
mixture, which has cooled to room temperature, is poured into
ice/HCl, the preclpitate is isolated by flltration and dried at
60C. The resultant powder i9 heated with 17 g of polypho~phoric
acid in an open ves~el at 180C, the hot mixture is dropped onto
ice, made alkaline with NaOH and extracted with ethyl acetate. The
combined extract~ are concentrated by 0vaporation and the re~ldue i8
purlfied by column chromatography (silica gel, elution with a 4:1
mixture of toluene/ethyl acetate), affording 4-(2,3-dichlorophenyl)-
3-cysnopyrrole of m.p. 148-150C.
Example P8: PreparatiQn of
C~ ~Cl C~ ~Cl
-CHO ~ -CH~NO~
a - -
4-(2,3-dichloroph~nyl)-3-cyanopyrrole
a) Preparation of 3--form~1-4~(2,3-dichlorophenyl)pyrrole
5.4 g of 3-dimethylaminoacrolein, 3.2 g of 2,3-dlchlorophenacylamine
hydrochlorlde and 60 ml of ethanol are heated for 1 1/2 hours under
reflux. Then a solution of ~odium ethylate in ethanol (prepared from
1 g of sodlum and 15 ml of ethanol) is sdded dropwis2 and the
reaction mixture is heated under reflux for another 30 mlnute~.
After it ha~ cooled to room temper~ture, the reaction mixture is
poured onto lce/water and neutrali~ed with hydrochloric acid. The
~73tCj3 ~
- 21 -
precipitate i8 washed with water, dried ~n vacuo, and tha dry
resldue 19 purified by column chromatography (sillca gel; elution
wlth a 4:1 mixture of toluene/ethyl acetate). M.p. 152-154C.
IR (aolid/KBr) in cm : 1655 (C0). IH-NMR (CDCL3) in ppm: 7.0 (broad
~, IH) 7.3 (m, 2H); 9.66 (8, lH), 11,9 (B, lH, H replaceable wlth
D20). Mass peak at 204. This ~ub~tance 1~ novelt has fungicldal
sctivlty, and falls within the ambit of the invention.
b) Preparatlon of hydroxylmlnomethyl-4-(2,3-dichlcrophenyl)pyrrole
5.0 g of the 3~formyl-4-(2,3-dichlorophenyl)pyrrole obtained in a),
1.7 g of hydroxylamine hydrochlorlds and 2.4 g of sodium acetate are
gtirred for 3 hours at 80C ln 80 ml of ethanol. After it has cooled
to room temperature, the reactlon mixture i9 poured onto lce and
stirred for 30 minute3. The precipitate iB isolated by filtration,
wa~hed with water and dried, affordlng 5.02 g of 3-hydroxyimino-
methyl-4-(2,3-dichlorophenyl)py~role a~ syn/antl mlxture of m.p.
158-160C. Thi~ substance i8 al~o novel, has funglcidal activity
and falls wlthin the amblt of the lnvention.
c) Preparation of the final product
4-(2,3-dichloropherlyl)-3-cyanopyrrole
3.2 g of the 3-hydroxyiminomethyl-4-(2,3-dichlorophenyl)pyrrole
obtain~d ln b) nre kept for S hours at c. 100C ln 50 ml of acetlc
anhydrlde, then cooled to room temperature, poured in~o ice/NaOH,
and the rasultant mlxture i8 stirred for 2 hours. The precipltate i8
dlssolved in ethyl acetate, wa~hed with water, and the e~ter pha~e
is dried over magnesium sulfate. The re~idue is purlf~d by column
chromatography (sillca gel; elution with a 4:1 mixture of toluene/
ethyl acetate). Melting point: 149 151C.
The compounds of formula I llsted in Table 4 are also prepared by
methods corre~ponding to tho~e descrlbed above.
~ 73~
- 22 ~
9 ~--ll 6-R1 tI)
R ' ~
Table 4
Compou~t R ~ - Rz I m.p; l~C~
_ n _ _
4.1 H CN H 120-123
4.2 3-Cl CN H 138-140
4.3 2,4-C12 CN H 1SO-152
4.4 4-Cl CN H 153-155
4.5 4-F CN H 137-139
4.6 3-CH3 CN H 109 111
4.7 3-F CN H 138-139
4.8 3-Br CN H 132-134
4.9 3-CF3 CN H 87-89
4.10 2~Cl CN H 136-138
4.11 2,3-Clz CN H 152 154
4.12 2,5-C12 CN H 13.7-142
4.13 2-Br CN H 135-138
4.14 2,3-C12 COOCH3 H 205 206
4.15 2,3-Cl2 CHO H 152-154
4.16 3-Cl COOCH3 H 187-189
4.17 3,~-C12 COOC~13 ~ 183 186
4.18 2-Cl COOCH3 H 198-200
4.19 2 9 3-C12 COOC3H7-i H 153-156
4.20 2,3-C12 COOC2~s ~ 149 151
4.21 2,3-C12 C~ CH2CH2CN
4.22 2,3-C12 CN CHzCH2COOCH3
__ _ . . ~ _ _ _ _ _
Th~ dc~crib~d proce~, including all partial ~3tep~, con~titu~c~ an
ob~ect of thi~ invention.