Note: Descriptions are shown in the official language in which they were submitted.
;~78~ 7
- 1 - 24205-716
- , This application is closely related to Canadian Application Serial No.~3i~y~ dated ~rch 5/ /~
(Agent's Docket No. 24205-715) filed on the same date.
This invention provides nucleoside analogs havinc
cyclopentane rins which can be used as substitutes fo-
purine nucleosides in the fields of biology, medicine
or gene manipulation ana as an antiviral agent.
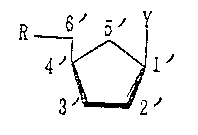
Derivatives of the compounds having the formula
_~' Y
3~ 2
wherein Y is guanin-9-yl or adenin-9-yl are the
e~amples of dideoxy analogs of purine nucleosides usec
in determination of base sequence in DNA
[Proc.Nat.Acad.Sci.USA,74,4563(1977)~. However,
2',3'-dideoxy analogs of purine nucleosides are so
susceptible to acids that cleavage occurs easily at
the glycosyl linkage, which is a great difficult~ in
their synthesis.
Recently it has been reported tllat 2',3'-dideoxy
analogs of purine nucleosides can act as inhihitors o
reverse transcriptase of virus origin, and hence these
analogs have attracted attention as therapeutics in
diseases due to RNA virus [Chemical and Engineering
News, January 27, number 28(1986~].
Although, as mentioned above, dideoxy nucleosides
and their carbocyclic analogs have been studied to
some extent, there are still many aspects to be
clarified. Therefore it is important to synthesize
and evaluate various analogs. This invention intends
to provide novel 2',3'-dideoxycarbocyclic nucleosides
,
' ;,s-, ., '
?., . .
. " :;.,~" .. . .. ..
78~i~L7
-- 2
which can be used as antiviral agents or for other
purposes.
The inventors completed this invention as the result
of their researches under the circumstances described
above to obtain novel and useful purine nucleoside
analogs. That is, this invention relates to
(lJ A compound of the formula (I~
~ 1' (I)
3 2'
wherein R is a hydroxyl group which may be protected
and Y is a purine base which may be protected, and the
salts thereof,
(2) a method of production of the compounds having
the formula (I) and the salts thereof, which comprises
subjecting a compound of the formula (II)
6~ 5~ Y
R ~ 1' (II)
~ ~ 2
3 l
R2 R,
wherein R is a hydroxyl group which may be protected,
either R1 or R2 is a hydroxyl group and the other
is a hydrogen, and Y is a purine base which may be
protected, to reduction reaction of 2' or 3'-hydroxyl
group, and
(3) an antiviral agent containing a compound of the
formula (I).
The protective groups of the hydroxyl group in
the compounds of the formula (I) or (II) are not
specifically limited as far as they are those used as
protective groups in nucleoside chemistry. In this
~78~
-- 3
invention the protective groups which are relatively
stable under alkaline conditions are favorably used,
such as alkylsilyl groups having 3-10 carbon atoms
(e.g. t-butyldimethylsilyl), alkyl or alkoxy cyclic
ethers having 4-10 carbon atoms (e.g. tetrahydrofuranyl and
its derivatives having 4-7 carbon atoms, tetra-
hydropyranyl and its derivatives having 5-8 carbon
atoms such as methoxytetrahyropyranyl), alkoxyalkyl
groups having 3-10 carbon atoms (e.g. ethoxyethyl,
methoxyethyl), and trityl and alkoxy substituted
trityl (e.g.monomethoxytrityi, dimethoxytrityl). When
the protective group is an acyl group, the hydroxyl
group can be protected in the form of aliphatic carboxylic
acid ester (e.g. straight chain or branched with 1-lO
carbon atoms), or arylcarboxylic acid ester (e.g.
with 5-30 carbon atoms).
Purine bases represented by Y include various
bases having purine ring skeleton which are used usua-
lly in the field of nucleic acid chemistry. Such bases
are exemplified by adenine, hypoxanthine, guanine,
isoguanine, xanthine, 3 deazaadenine, 7-deazaadenine,
8~azaadenine, and 2,6-diaminopurine, and bound via the
nitrogen atom at 9 position in the purine ring of the
compound having the formula (I) or (II).
~he protective group of purine base in a compouncl
of the formula (I) or (II), i.e. the amino protecti~Je
group at 2 or 6 position is any of those which can be
used usually in the field of nucleoside chemistry.
~or example an arylcarboxylic acid residue (with
5-30 carbon atoms) such as benzoyl for a protective
group of adenine and an aliphatic carboxylic acid
residue (straight chain or branched, with 2-lO
carbons) for a protective group of guanine are favora-
bly used.
~or production of a compound of the formula (I)
from a compound of the formula(II),preferably the hydroxyl
., ~ii .
. .
...........
1~7~35~7
4 --
group at 2' or 3' position in a compound of formula (II) is
thiocarbonylated at 0~80C or preferably at room temperature,
followed by reduction with a tri(lower alkyl) tin hydride, such
as tributyl tin hydride in the presence of an equivalent amount or
an excess of an azo compound radical initiator, such as a,'
azobisisobutylonitrile at 0-100C for 30 minutes to 2 hours, to
give a 2',3'-dideoxy derivative having the formula (I). The
thiocarbonylation can be favourably conducted by using thiocarbonyl
diimidazole for thiocarbonylation, phenyl chlorothiocarbonate for
phenoxythiocarbonylation. Further, S-methyldithiocarbonylation
may be carried also by using the mixture of carbon bisulfide and
methyl iodide. After this reduction, the 6'-hydroxyl protective
group, for example, the 4,4'-dimethoxytrityl group is easily
removed under acidic condition (e.g. treatment with acetic acid
or IN hydrochloric acid at room temperature), and moreover the
protective group of purine base can be removed under alkaline
condition (e.g. concentrated ammoniac water, IN-sodium hydroxide,
IM-sodium ethylate).
The compounds of formula (II) can be produced by, for
example/ the following procedure: the compounds of formula (II)
of which Y is an adenin-9-yl which may be protected can be
produced also by the method described in Japanese Patent
Application Laid-Open No. 62992/1975, Chemical & Pharmaceutical
Bulletin 24,2624(1976) or Nucleic Acids Symposium Series,
No. 16,141(1985). For example by the method described in
Japanese Patent Application Laid-Open No.62992/1975 or Chemical
& Pharmeceutical Bulletin 24,2624(1976), a compound in which in
~Z178~ L7
- 4a -
which in the formula (II) Y is adenin-9-yl, either Rl or R2 is
a hydroxyl group and the other is a hydrogen, and R is a hydroxyl
group is obtained by using aristeromycin as starting compound;
and a compound in which Y in the formula (II) is N6-benzoyl-
adenin-9-yl, R is a hydroxyl group protected with 4,4'-dimethoxy-
trityl, and Rl is a hydrogen and R2 is a
~785~
_ 5 _ 24205-716
hydro.~yl group is obtained by the method described in
Nucleic Acids Symposium Series described above. A
compound in which Y in the formula (II) is guanin-9-yl
or hypoxanthin- 9-yl which may be protected, R is a
hydro~yl group which may be protected, with a hydrogen
at 2' position and a hydroxyl group at 3' position is
obtained by the method described in Canadian Patent
Application Ser. No. 520,946 (see the Reference Examples
1~8).
On the Gther hand, the compound of formula ~II)
in which Y is 2,6-diaminopurine-9-yl, Rl is hydrogen,
R2 is hydroxylgroup can be synthesized as follows:
Hydroxyl group of the compound in which Y is adenin-
9-yl is protected, followed by Nl-oxidation with
hydrogen peroxide or metachloro perbenzoic acid, and
then the amino group at 6-pOsitioll is deaminated by
nitrous acid, and is heated with phosphorus oxychloride
(Japanese Patent Publication 4347/1967) to give
the corresponding 2,6-dichloropurine-
9-yl derivative. The chlorine at 6-position is
substitutecl by amino group, and deamination is con-
ducted by using sodium nitrite in aqueous acetic acid
to give the product of 2-chloro-6-hydroxyl-9-yl. Thus
obtained compound is subjected to amidation at 2-
position and after chlorination at 6-position, the
chlorine at this position is substituted by amino
group to give the desired compound.
The salts of the compounds having the formula (I)
in this invention include those formed with the amino
group in the purine base and a mineral acid
(e.g.hydrochloric acid, sulfuric acid, nitric acid),
anorganic carbo~:ylic acid (e.g. acetic acid, lactic
acid, tartaric acid, maleic acid, succinic acid) or an
organicsulfonic acid (e.g. methanesulfonic acid,
ethansulfonic acid, benzenesulfonic acid).
.
. ~ .
6 -
The campounds of th~ formula (I) in ~hls lnven-
tion provlde useful tools ~n gene clonin~. ~hat is,
the analogs derlved ~rom the compounds of this inven-
tion having cyclopentane ~ing are the ca~ocycllc
analogs of purine-2',3'-dldeoxynucleotid~, and are
ea~ ly aynthasized be~ause o~ ~b3Pnce of a glycosyl
lin~cage; and the tripho~phate ~erivatives thereo m~y
be used as age~a to 8 Op the DNA chs~n elongatlon in
determlnation o~ DNA ~e~u~nc~.
On ~he othex hand, the compounds of the ~ormulA
~I) have antiviral ~ctiv~ty againa~ DNA viruse~ or RNA
viruses. ~here may be mentioned as DNA viruse4
herpe~virus group (9.g. herpes simplex virus ~ype I or
II, cytomagalovlrus, Epstsln-9arr virus), adenovlru~
e (e.g. typ~ ), He~titis B vlrus or poxvirus7 ~s ~NA
viru3es hum~n immunodeficiency vlrus (HIV), which is a pathoger~
o~ ac~ired ~odeficiency syr~ome (A::DS), vesicular ~'camati-
tls vlrus, feline leukemia viru~, equine ln~ectious
anemic vlrus. Especially, the compound5 of the present
20 invention have patant antiviral activi~y aga~t RN~ viruses, in part~-
cular H~rLv-III(~u;nan T-cell Lyny?hotro~ic vin~ tYPe ~ V~TLV.IU)~ which
lr~ onc o~ H~l, as po33ible iniLibitor~ of reverse trælscriptase.
Accord.in~ly, .the c~mpound3 o~ the present lnven-
tion may be u3ed for txeatment agalnst var~ou~ vixal
ln~ectlons. A~ such viral in~ections, there may be
mentioned acquired immunode~iciency ~yndrome7
herpes gimp:Lex ~type I or II), varicella,
zostex, ker~titis, con~unctivitis or acute hepatitis
varlous opportunistic in~ection~, mallgnant tumor or
cen~ral nervous ~ymptoms which are brought out by
vlral in~ectlon and immuno~e~iciency.
The compound~ of the formula (I) and the ~h~eutlcally
acceptable ~alt~ thereof can be also u~0d as antiviral agent~ for
treatment o~ viru~-induced diseaq~ ln animals, par-
ticularly in mamalian animalY (laboratory an~als such
a3 rabbit, rat ~nd mouse~ pet animals such a8 aOg and
cat; human beingi livestock such as cattle, horse,
sheep and pig).
In general for the above pourpose, a suitable
effective dose of the compounds of the present
invention is in the range of 30-500 mg per kg
body weight per day, preferably in the range of
100-300 mg per kg body weight per day. The desired
dose is generally presented as two, three or four more
sub-doses administered at appropriate intervals
throughout the day. Administration may be any suitable
route including oral, rectal, nasal, topical (e.g.
buccal, sublingual), vaginal and parenteral (e.g.
subcutaneous, intramuscular, intravenous, intradermal).
The preferred route may vary with, for example, the
condition and age of the recipient. While the present
compounds can be administered alone, it is preferable
to present them as part of pharmaceutical formulation.
The formulations of the present invention comprise at
least one of the compounds of the formula (I), together
with one or more acceptable carrier thereof and
optionally other therapeutic ingredients.
The foxmulation may conveniently be presented in
unit dosage form. Formulations containing the com-
pound of the present invention for oral administration
include discrete units such as capsules or tablets;
powder or granules; solution or suspension; or an
oil-in-water liquid emulsion or a water-in-oil liquid
emulsion etc.
A tablet may be made by compression or molding,
30 optionally with one or more accessory ingredients.
Compressed tablets may be prepared by compressing in a
suitable machine the compound of the present invention in a form of a
powder or granules, optionally mixed with a binder
(e.g.hydroxypropyl-cellulose), lubricant (e.g. magne-
3r sium stearate), inert diluent (e.g. starch), pre-
servative, surface-active or dispersing agent.
~Z~ 7
Formulations for parenteral administration
include aqueous and non-a~ueous sterile injection
solutions which may contain anti-oxidants, buffers or,
bacteriostats; and aqueous and non-aqueous sterile
suspensions which may include suspending agents and
thickening agents. The formulations may be presented
in unit-dose or multi-dose containers, for example,
sealed ampules and vials, and may be stored in a
freeze-dried condition requiring only
the addition of the sterile liquid carrier, for
example water for injections, immediately prior to
use. Extemporaneous injection solutions and sus-
pensions may be prepared from sterile powders, gra-
nules and tablets.
Formulations for topical administration include
lozenges comprising the present compounds in a fla-
voured basis, usually sucrose and acacia or traga-
canth; pastilles comprising the present compound in
an inert basis such as gelatin and glycerin, or
sucrose and acacia; and monthwashes comprising the
ingredient to be administered in a suitable liquid
carrier.
Formulations for rectal administration may be
presented as a suppository with a suitable base such
as cacao butter.
Pormulations for vaginal administration may be
presented as pessaries, tampons, creams, gels, pastes,
foams or spray formulations containing in addition to
the present compound such carriers as are known in
the art to be appropriate.
Among the compounds of the formula (I),
particularly 2',3'-dideoxyaristeromycin (the compound
of Example 3) and 9-~(lS,4R)~4-hydroxymethyl-
cyclopentan-l-yl]guanine (the compound of Example 4)
inhibit potently growth of AIDS virus, and they are
more useful compounds accordingly.
8~L7
In the following, Rference Examples, Examples and
Test Example are described by which the invention is
explained in the concrete.
Reference Example 1
~ ynthesis of 9-[(lR,2S,3R,4R)-4-methyl-2-hydroxyl-
3,6-(tetraisopropyldisiloxanyl)dioxycyclopentan-1-yl]-
hypoxanthine
The C-analog to inosine (10 g, 37.5 mmol) was
dissolved in 200 ml of anhydrous DMF, 1,3-dichloro-
1,1,3,3-tetraisopropyldisiloxane t13 ml, 41 mmol) and
imidazole (11.3 g, 165 mmol) were added, and the mixture
was stirred at room temperature for 2.5 hours. The
reaction mixture was added dropwise to 2 liters of
water, and the precipitate was collected by filtration,
washed with water, further washed quickly with diethyl
ether and dried to give the title compound as a white
powder (17.2 g). A portion of the product was
recrystallized from dichloromethane to give crystals,
m.p. 135-138C.
Note: The C-analog to inosine is known by
Chemical & Pharmaceutical Bulletin 24,
2624(1976).
1~7~
-- 10 --
Reference Example 2
Synthesis of 9-[(lP~,2S,3~,4R)-4-methyl-2-phenoxy-
thiocarbonyloxy-3,6-(tetraisopropyldisiloxanyl)-
dioxycyclopentan-1-yl]hypoxantine
S The compound obtained in Reference E~le 1 (11.2 g, 22.3
mmol1 was dissolved in 300 ml of anhydrous acetonitrile,
dimethylaminopyridine (15.8 g, 53.5 mmol) and phenoxy-
thiocarbcnyl chloride (5 g, 29 mmol) were added and the
mixture was stirred at room temperature for 7 hours.
The solvent was removed under reduced pressure, and the
residue was dissolved in 250 ml of chloroform. The
solution was washed with 0.5 M potassium dihydrogen
phosphate solution (250 ml x 2) and then with water (200
ml), dried (anhydrous sodium sulfate) and concentrated
under reduced pressure to give a yellow syrup. This was
purified by silica gel chromatography (90 g; solvent:
CHC13 and CHCl3/MeOH = 60/1) to give the title
compound as a light-yellow grass-like substance (13.0
g) .
NMR(60MHz,CDCl3)~ ppm: 1.0-1.23 (28H, m),
2.13-2.43 (3H, m, H4', H5'1, 3.93-4.10 (2H, m,
H6'), 4.80-5~20 (2H, m, Hl', H3'), 6.00-6.20
(H, m, H2'), 7.03-7.50 (5H, m), 7.87 (lH, s),
8.13 (lH, s) s;singlet, m;multiplet
Reference Example 3
Synthesis of 9-[(lR,3S,4R)-4-methyl-3,6-
(tetraisopropyldisiloxanyl)dioxycyclopentan-1-yl]-
hypoxanthine
Anhydrous toluene ~30 ml) was added to the
compound obtained in Reference Example 2 (13.0 g, 20 mmDl),
followed by concentration under reduced pressure. The
residue was dissolved in 300 ml of anhydrous toluene,
and nitrogen gas was bubbled into the solution for 20
minutes. After addition of tributyltin hydride (11 ml,
40 mmol), the solution was heated at 80C and crystal-
-- 1 1 --
line ~ azobisisohutyronitrile ~AIBN) (820 mg) wasadced in 4 portlons at 15~minute intervals. A~ter 3
hours of heating with stirring, the solvent was removed
under reduced pressure. The oil obtained was purified
by silica gel chromatography (80 g; solvent: CHC13 and
CHC13/MeOH = 60/1 to 30/1) to give the title compound
as a colorless glass-like substance (10.4 g).
Recrystallization of a portion o~ the product from
ethanol gave colorless needles, m.p. 200-202C.
NMR(60MHz,CDC13)~ ppm: 0.93-1.20 (28H, s),
1.97-2.5~ (5H, m, H2', H4', H5'), 3-80-4-07
(2H, m, H6'), 4.4~-5.27 (2H, m, Hl', H3'),
7.87 (lH, s), 8.20 (lH, s)
Reference Example 4
Synthesis of 9-[(lR,3S,4R)-4-(monomethoxytrityloxy)-
methyl-3-hydroxylcyclopentan-1-yl]-(1-methoxymethyl-
hypoxantine)
The compound obtained in Reference E~mple 3 (9.8 g, 19.8
mmol) was dissolved in 240 ml of anhydrous dioxane,
sodium hydride (880 mg, 21.8 mmol) was quickly added to
the solution with ice cooling and stirring and, then,
the mixture was stirred at room temperature for 1.5
hours. Thereafter, methoxymethyl chloride (2 ml, 21.8
mmol) was quickly added to the mixture with ice cooling.
The whole mixture was stirred at room temperature for 3
hours.
The solvent was removed under reduced pressure and
the oily residue was dissolved in 200 ml of chloro~orm.
The solution was washed with 0.1 M triethylammonium
bicarbonate (TEAB) buffer (pH 7.5, 100 ml x 2) and
further with water (200 ml), dried (anhydrous sodium
sul~ate) and concentrated under reduced pressure to give
a syrup. Purification of the syrup by C18 silica gel
chromatography (~ 5.3 x 7.0 cm; solvent: acetone water,
55%-80%) gave a colorless glass-like compound (8.5 g).
1~78~ 7
- 12 -
This compound (8.0 g) was dissolved in 32 ml of
tetrahydrofuran (THF), tetrabutylammonium fluoride
trihydrate (TBAF~3H2O) (10 g) was added, and the
mixture was stirred at room temperature for 0.5 hour.
The solvent was removed under reduced pressure and the
remaining oil was dissolved in 100 ml of water. The
solution was washed with diethyl ether (100 ml x 2) and
was deprived of the tetrabutylammonium salt by treatment
on Dowex-50*resin (pyridine form 60 ml). The erfluent
and water washings (240 ml) were combined and concen-
trated and the concentrate was dehydrated azeotropically
with three portions of pyridine. The residue was
dlssolved in 100 ml of pyridine, monomethoxytrityl
chloride (MMTrCl~ (5.4 g) was added and the mixture was
stirred at 37C for 4 hours. The solvent was removed
under reduced pressure and the oily residue was
distributed between 0.1 M TEAB buffer (50 ml) and
CHC13 (100 ml), the organic layer was washed with
water (100 ml), dried (anhydrous sodium sulfate~ and
concentrated under reduced pressure and the concentrate
was subjected to dehydration by azeotropic distillation
with toluene to give a colorless syrup. 0.1 M-TEAB
buffer fraction and water washings were combined and
concentrated, whereby the unmonomethoxytritylated
compound was recovered. This compound was purified on
HP-20 resin (190 ml; solvent: water and 30~ ethanol-water)
and, after concentration and azeotropic distillation
with pyridine, monomethoxytritylated in the same manner
as mentioned above. Both the thus-obtained crops of the
0 title compound were combined and purified by silica gel
chromatography (80 g; solvent: CHC13/MeOH = 100/1,
60/1, 50/1) to give a colorless glass-like product ~6.1
g). A solution of a portion of this product in di~
chloromethane, when added dropwise to n-hexane, gave a
white powder-
N~R(60MHz,CDC13)~ ppm: 1.87~2.70 (5H, m, H2',
*Trade Mark
.~
. .
.
.
- 13 -
H4', H5'), 3.20-3.40 (2H, m, H6'), 3.4~ (3H,
s, CH30CH2), 3.80 (3H, s), 4.30-4.57 (lH, m,
H3'), 4.87-5.10 (lH, m, H1'), 5.47 (2H, s,
CH30CH2-N), 6.73-6.97 (2H, m), 7.17-7.53 (12H,
m3, 7.73 (lH, s), 7.98 (lH, s)
Reference Example 5
Synthesis of 1-L(lR,3S,4R)-4-(monomethoxytritylox~;)-
methyl-3-hydro~ycyclopentan-1-ylJ-(4-carbamoyl-5-amino-
imidazole)
The compound obtained in Reference E~ple 4 (6.1 g, 10.7mmol) was dissolved in 490 ml of ethanol and, with
heating under reflux, a warmed 5 M aqueous sodium
hydroxide solution (130 ml) was added quickly.
Refluxing was continued for additional 40 minutes. The
solvent was then removed under reduced pressure. The
oily residue was dissolved in 200 ml of chloroform and
washed with water (100 ml x 2), then with 0.1 M-TEAB
buffer (100 ml x 2) and further with saturated aqueous
sodium chloride solution (100 ml), dried (anhydrous
sodium sulfate) and concentrated under reduced pressure
to give a syrup. Purification of this syrup by silica
gel chromatography (90 g; solvent: CHC13/MeOH - 100/1
to 20/1) gave Zl colorless glass-like product (3.2 g).
solution of a portion of this product in chloroform,
when added dropwise to n-pentane with stirring, gave a
white powder.
Elemental analysis (%) for C30H32N404-0.5 H20,
molecular weight 5~1.616
Calcula~ed : C, 69.08; H, 6.38; N, 10.74
Found : C, 69.14; H, 6.09; N, 10.54
NMR(lOOMHz,CDC131~ ppm: 1.36-2.52 (5H, m),
3.00-3.40 (3H, m, H6', OH), 3.77 (3H, ~),
4.12-4.60 (2H, m, H1', H3'), 4.80-5.28 (2H,
bs, NH2), 5.64-6.44 (2H, bs, NH2), 6.76-6.94
(3T-T, m), 7.14-7.48 (12H, m) bs;broad singlet
1278~7
- 14 -
A _ t -
Synthesis cf 1-[(lR,3S,4R)-~(monomethoxytritylox-)-
methyl-3-hydroxycyclopentan-1-yl]-[4-carbamoyl-5-(N-
benzoyl-S-methylisothiocarbamoyl)aminoimidazole]
The compound obtained in Reference E~mple 5 (0.88 g, 1.-
mmol) was dissolved in 25 ml of anhydrous acetone and,
with heating under reflux, a solution of benzovl
isothiocyanate (260~1, 1.9 mmol) in acetone (8 ml) was
added dropwise over 10 minutes, followed by refluxing
for 50 minutes. The solvent was removed under reduced
pressure and the light-yellow glass-like substance
obtained was purified by silica gel chromatography (15
g; solvent: CHC13/~eOH = 50/1 to 30/1) to give a
light-yellow glass-like compound (0.87 g). A small
amount of acetone was added to this compound (0.84 g,
1.2 mmol) and the resultant syrup was converted to a
homogeneous solution by addition of 12.5 ml of 0.2 N
NaOH and sonication. Dimethyl sulfate (130 ~1, 1.4
mmol) was added with stirring and, then, vigorous
stirring was continued at room temperature for 1 hour.
The reaction mixture was mixed with CHCl~ (15 ml x 2)
for partition, the organic layer was washed with 0.1 M
TEAB buffer (15 ml x 3) and then with saturated aqueous
sodium chloride solution (20 ml), dried (anhydrous
sodium sulfate) and concentrated under reduced pressure,
and the residue was purified by silica gel chromatograp:ly
(15 g; solvent CHC13/MeOH = 100/1 to 60/lj. A small
amount of dichloromethane was added to the glass-like
substance obtained, the mixture was added dropwise to
hexane and the resultant precipitate was collected by
centrifugation and dried to give the title compound as a
powder (400 mg).
s~
- 15 -
lemental analysis (%) for C39H39N505S,
molecular weight 689.835
Calcula~ed : C, 67.90; H, 5.70; N, 10.15;
Found : C, 67.45; H, 5.45; N, 9.89;
NMR(lOOMHz,CDC13)~ ppm: 1.34-2.60 (5H, m), 2.52
t3H, s, SCH3), 3.04-3.44 (2H, m, H6'), 3.79
(3H, s, OCH3), 4.08-4.44 (lH, m, H3'),
4.60-5.00 (lH, m, Hl'), 5.64 (lH, bs, NH2),
6.72-6.94 (3H, m), 7.12-7.52 (15H, m), 7.BO-7.96
(2H, m), 11.35 (lH, bs, NH)
Reference Example 7
Synthesis of 9-[(lR,3S,4R)-4-monomethoxytrityloxy-
methyl-3-hydroxycyclopentan-1-yl~guanine
The compound obtained in Reference Example 6 (360 mg, 0.53
mmol) was added to a warmed 6 N sodium hyd~oxide (18 ml)
and the mixture was heated under reflux for 1 hour. The
product was extracted from the reaction mixture with
CHC13, and the extract was washed with 0.1 M TEAB
buffer (30 ml) and then with saturated aqueous sodium
chloride solution (30 ml), dried (anhydrous sodium
sulfate) and subjected to silica gel chromatography (8
g; solvent: CHC13/MeOH = 40/1 to 6/1). To the thus-
obtained glass-like substance was added a small amount
of acetone, the mixture was added dropwise to benzene
and the resultant precipitate was collected by cen-
trifugation and dried to give the title compound as a
powder (210 mg).
Elemental analysis (%) for C31H31N504-1.0H20,
molecular weight 555.633
Calculated : C, 67.01; H, 5.99; N, 12.60
Found : C, 6/.01; H, 5.69; N, 12.42
NMR(lOOMHz,DMSO-d6)~ ppm: 1.50-2.60 (5H, m~,
3.01 ~2H, bs), 3.98-4.20 (lH, m), 4.70-4.96 (2H,
m), 6.37 (2H, bs, NH~), 6.82-7.46 (14H~ m), 7.68
(lH, s, H8), 10.60 (lH, bs, NH)
1~785~7
- 16 -
Reference Example 8
. _
Synthesis of 9-I(lR,3S,4R)-4-hydroxymethyl-3 hydroxy-
cyclopentan-l-ylJguanine
The compound obtained in Reference Example 7 (180 mg, 0.33
mmol) was dissolved in 10 ml of 80~ acetic acid and the
solution was stirred at 40C for 4.5 hours. The solvent
was removed under reduced pressure and, further,
azeotropic distillation was conducted twice with water.
Water (10 ml) was added, the mixture was washed with
ether (10 ml x 2) and the water was removed under
reduced pressure. Thus was obtained the title compound
as colorless crystals (41 mg), m.p. ~46-248C.
[~]25=+/.7~ (C=0.5, DMF)
~max (nm): (H~O); 255, 278 (sh)
(H ) ; 257, 282
(OH ); 256 (sh), 273
Elemental analysis (~) for C11H15N5O3-0.5H2O-0.1C2H5OH,
molecular weight 278.886
- Calculated : C, 48.24; H, 6.00; N, 25.11
Found : C, 48.61; H, 6.41; N, 25.40
~IL2~78~
- 17 -
Example 1
N6-benzoyl-6'-O-(4,4'-dimethoxytrityl)-3'-O-
[(imidazol-1-yl)-thiocarbonyl]-2'-deoxyaristeromycin
N6-benzoyl-6'-O-(4,4'-dimethoxytrityl)-2'-
deoxyaristeromycin (2.5 g) was dissolved in 10 ml ofdry dichloromethane, to which thiocarbonyl diimidazole
(8.0 g) was added and stirred at room temperature for
20 hr. The reaction mixture was concentrated to
dryness, and subjected to purification with silica gel
chronatography (Kieselgel*60, Merck Co., 50 g,
solvent:ethyl acetate), to give a light yellow glassy
substance (yield 2~2 g).
NMR(90 MHz CDC13) ~ ppm: 3.80 (6H,S,2
CH30-), 8.35(1H,S,H8), 8.76(1H,S,H2).
Example 2
N6-benzoyl-6'-0-(4,4'-dimethoxytrityl)-2',3'-
dideoxyaristeromycin
The 3'-thiocarbonyl derivative obtained in
Example 1 (2.0 g) was dissolved in 20 ml of
dry dioxane, to which a solution of tributyl tin
hydride (4.5 g) in dry dioxane (10 ml) was added
dropwise with refluxing by heating. Meanwhile
crystals of ~,~'-azobisisobutylonitorile (500 mg) were
added little by little. The dropwise addition was
completed in 20 minutes and refluxing was continued
for further 2 hr. The solvent was evaporated off
under reduced pressure, and the oily residue was
purified with silica gel chromatography (40 g,
solvent:CHC13), to give a colorless powder (1.1 g).
NMR(9OMHz, CDC13) ~ ppm: 3.80 (6H,S,2
CH30-), 4.80-5.20(1H,m,H1,), 3.15(2H,d,2H6,),
8.76(1H,S,H2), 9.10 (lH,S,-NH-C-).
Example 3
2',3'-dideoxyaristeromycin
The compound obtained in Example 2 (1~0
*Trade Mark
;~ Y~.,.:.
.
.....
~i~785~7
- 18 -
g) was dissolved in a small amount of pyridine, to
which 50 ml of concentrated ammoniac water was added
and heated in a pressure-proof tube at 60C for 5 hr.
The reaction mixture was concentrated to dryness, to
which 80~ acetic acid (lO0 ml) was added and heated at
60C for 2 hr followed by concentration to dryness
un~er reduced pressure. The residue was dissolved in
water (100 ml) and washed twice with water. The
aqueous layer was concentrated to dryness, and the
residue was powderized in ether to give 2',3'-
dideoxyaristeromycin (0.23 g).
UVlmHa2O (n~);260
Elemental analysis (~) for CllH15N5O~H2O
with molecular weight 251.29
Calc.: C;52.57, H;6.82, Nj27.87
Found: C;52.83, H;6.95, N;27.54
The thus obtained 2',3'-dideoxyaristeromycin was
dissolved in the equivalent weight of lN-HCl. After
concentration, ethanol was added to the solution and
concentrated to dryness. This procedure was
repeated several times. The residue was
recrystralized from hot ethanol to give the crystals
of the hydrochloric acid salt. m.p. 173 - 175C.
Elemental analysis (%) for CllH15N5O~HCl~l/2H2O
with molecular weight 278.73
Calc.: C;47.40, H;6.15, N;25.12, Cl;12.72
Found: C;47.98, H;6.06, N;24.87, Cl;12.71
[a]D5=-6.79(C=0.61, H2O)
Example 4
The compound obtained in Reference Example 8 (2.5
g) was treated in a similar way to that in
Examples 1, 2, and 3, to give a crystalline powder of
9-[(lS,4R)-4-hydroxymethylcyclopentan-1-yl]guanine
(0.3 g). m.p. 269~C.
UV~max(nm):255,280(shoulder); UV~ma2x(nm):253,270
(shoulder); UVmax (nm):258 (shoulder), 270
-- 19 --
Elemental analysis (~) for C11H152N5 with
molecular weight 249.27
Calc.: C;53.00, H;6.07, N;28.10
Found: C;52.81, H;5. 86, N; 27. 83
[~]D =-4.74(C=0.57, DMF)
9-[(lS,4R)-4-hydroxymethylcyclopentan-1-yl]-
hypoxanthine is obtained when a hypoxanthine
derivative, instead of N6-benzoyl-6'~O-(4,4'-
dimethoxytrityl)-3'-O-[(imidazol-l-yl)-thiocarbonyl]-
2'-deoxyaristeromycin in Example 1 is treated in a
similar way to that in Examples 1-3.
Elementary analysis (%) for C11H14N4O2 with
molecular weight 234.25
Calc.: C;56.40, H;6.02, N;23.92
Found: C;56.81, H;6.33, N;24.25
Æxam~_e 5
9-[lS,4R)-4-hydroxymethylcyclopentan-1-yl]guanine
(1) Synthesis of 9-[llR,3S,4R)-4-hydroxymethyl-3-
hydroxylcyclopentan-1-yl]hypoxanthine
The compound (12.4g, 20 m mol) obtained in
Reference Example 3 was dissolved in 200 ml of toluene
and to the mixture was added tetrabutylammonium
fluoride (10.46 g, 40 m mol), followed by heating at
75C for 2 hr. The reaction solution was concentrated
to dryness and dissolved in water. The solution was
subjected to desaltation with 30 g of activated
charcoal, the crude product was recrystallized from a
mixture of methanol and ethylether to give colorless
crystals (4.6 g). m.p. 170C.
30 Elemental analysis (%) for C11H14N4O3-H2O
with molecular weight 268.27
Calc.: C;49.25, H;6.01, N;20.88
Found: C;49.08, H;5.86, N;20.81
(2) Synthesis of 9-[(lR,3S,4R)-4-monomethoxytrityl-
35 oxymethyl-3-hydroxylcyclopentan-1-yl]hypoxanthine
78~7
- 20 -
The crystaline compound (2.3 g, 9.2 m mol)
obtained by the method (1) described above was
dissolved in 100 ml of pyridine, followed by addition
of monomethoxytritylchloride (3.1 g, 10 m mol) and
then was stirred at room temperature for 5 hr. The
reaction solution was purified with silica gel
chromatography (80 g, solvent: CHC13/MeOH = 40/1 -
6/1) to give powdery desired product (4.3 g). A part
of the product was recrystalized from a mixture of
chloroform and ether. m.p. 244 - 246C.
Elementary analysis (%) for C31H30N4O4~H2O
with molecular weight 531.60
Calc.: C;70.04, H;5.~8, N;10.54
Found: C;70.39, H;5.77, N;10.38
(3) Synthesis of 9-[(lS,4R)-4-monomethoxytrityloxy-
methyl-cyclopentan-l-yl]hypoxanthine
The compound (4.32 g, 8.27 m mol) obtained by the
method (2) described above was dissolved in 70 ml of
toluene, followed by addition of thiocarbonyl-
diimidazol (2.2 g, 12.4 m mol) and stirred at room
temperature for 5 hr. The reaction solution was
concentrated to dryness, and the residue was purified
with silica gel chromatography (80 g, solvent:
CHC13/MeO~ = 100~1 - 60/1) to give pale yellow
powder (5 2 g). The thus obtained product was dis-
solved in 90 ml of toluene and reacted with tributyl
tin hydride (3.4 ml, 12.4 m mol) and~
azobisisobutylonitrile ~270 mg, 1.6 m mol) by the
similar method as described in Reference Example 3,
followed by purification with silica gel
chlomatography (100 g, solvent: ethylacetate/MeOH =
9/1) to give the desired product (1.63 g). A part of
the product was recrystalized from a mixture solution
of methanol and ethyl ether. m.p. 175 - 177C.
Elementary analysis (~) for C31H30~4O3-1/2H2O
with molecular weight 515.60
~78~
- 21 -
Calc.: C;72.21, H;6.06, N;10.87
Found: C;72.69, H;5.88, N;10.92
(4) Synthesis of 9-[(lS,4R)-4-hydroxymethyl-
cyclopentan-1-yl]guanine
The desired compound can be produced by similar
methods as described in Reference Examples 4 to 8
using the compound obtained in the step (3) described
above.
Exam~le 7
Tablets for oral administration of the antiviral
composition according to the present invention are
prepared as exemplified below:
Two hundred (200) mg of 2',3'-dideoxyaristromycin,
300 mg of lactose, 50 mg of starch and 2 mg of
magnesium stearate are mixed in methanol and after
removal of methanol with heating the mixture is molded
into tablets.
ExamDle 8
Injection for antiviral composition is prepared
as exemplified below:
~ive hundred (500) mg of 2',3'-dideoxyaristero-
mycin is dissolved in 10 ml of sterilized water and
adjusted to pH 6.0 with aqueous sodium hydroxide. The
solution is filtered with sterilized fi].ter and sealed
up in vial.
Text Examp~e 1
MATERIALS AND METHODS*
* Antimicrob. Agents Chemother,30,No.6,Dec.1986,933-937
Cells. An HTLV type I-carrying cell line, MT-4,and an
HIVHTLV III-producing cell line, Molt 4/HI~HTL~ III'
were used in this study. The cells were maintained in
RPMI 1640 medium supplemented with 10% fetal calf
serum, 100 IU of penicillin per ml, and 100 ~g of
streptomycin per ml at 37C in a CO2 incubator.
Virus and virus infection. HIVHTL~
,:
.-r.4, . .
- 22 -
from culture supernatants of Molt-4/HIVHTLv III as
previously described [Virolog~ 146, 272(1985)~. The
titer of this virus preparation was 6 x 10 PFU/ml.
Infection of MT-4 cells with HIvHTLv~IIIwas made at
multiplicity of infection of 0.002. Briefly, the
cells were mixed with virus solution and incubated for
1 h at 37C. After adsorption, infected cells were
washed and resuspended in fresh medium to a concentra-
tion of 3 x 10 cells per ml. This concentration
was cultured in both the presence and absence of
various concentrations of carbocyclic 2',3'-
dideoxynucleosides in a CO2 incubator at 37C for 6
days.
VHTLv_III~indUCed cytopathic effect
HIVHTLV III-induced cytopathic effect was analvzed by
mesuring the decrease in the number of viable cells.
The viable cells were counted by the trypan blue
exclusion staining method.
Y HTLV-III antigen expression.
HTLV-III-infected MT-4 cells with virus-specific
antigens were counted by an indirect [immuno-
fluorescence (IF)] method. Briefly, methanol-fixed
cells were incubated with diluted anti-HIVHTLv III
positive human serum for 30 min at 37~C. The pre-
parations were then washed for 15 min withphosphate-~buffered saline. The cells were then
incubated with fluorescein isothiocyanate-conjugated
rabbit anti-human immunoglobulin G (Dakoppatts A/S,
Copenhagen, Denmark) for 30 min at 37C and washed
again with phosphate-buffered saline. More than 500
cells were counted under a fluorescence microscope,
and the percentage of IF-positive cells was cal-
culated.
From the assay mentioned above, it was confirmed
that the compounds of the present invention have
obvious anti-HIVHTLV-III aCtivity- In the case of
,~;~
, . ~ ..... .
s~:
. ;.....
.... .... .
~ ~78~
- 23 -
2',3'-dideoxyaristeromycin, the effective con-
centration was 50-100 ~M and the cytotoxity was
observed at 5Q0-1,000 ~Mr respectively.