Note: Descriptions are shown in the official language in which they were submitted.
~284~99
4-lj913/-/CGC 1200
[2,5-Dihydro-2-oxo-5-phenyl-3-furanylJamines, methods of
manufacture, and use thereof in the production of angiotensin
converting enzyme inhibitors
The invention rslates to a novel process for the production of
angiotensin converting enzyme inhibitors which uses cheaper starting
materials, less toxic reagents, and allows for simple efficient and
inexpensive diastereoisomer separation over the conventional
synthetic means currently employed. Also within the ~cope of this
invention are the novel [2,5-dihydro-2-oxo-5-phenyl-3-furanyl]-
amine intermediates which make the process feasible and the method
of their manufacture.
Heretofore, the angiotensin converting enzyme inhibitors of
formula I
~ H - CH2 - CH2 - ~
2 '='
RH 2
wherein Rl is carboxy and R2 is carboxy or lower alkoxycarbonyl,
have been prepared by an expensive, time consuming, multistep
synthetic pathway requiring multiple purification steps which
culminated in a Borsch Reduction and an extremely difficult dia-
stereoisomeric separation. The details of such methods can be seen
~2~3~4~39
-- 2 --
in US Patents 4,410,520 and 4,473,575. A specific synthetic pathway
shown in the literature is given in Drugs of the Future, Vol. 9,
No. 5, 1984.
It is sn object of the invention to provide a synthetic pathway for
the manufacture of angiotensin converting enzyme inhibitors (ACEI)
of formula I, or salts thereof, which is simpler and less costly
than the known methods.
It is a further object of the invention to provide a synthetic
pathway for ACEIs which allows for the rapid resolution of the
product diastereoisomers.
It is another object of the invention to provide an ACEI synthetic
pathway which avoids the use of many of the highly toxic reagents
required in the known synthesis.
These and other important object~ are attained by the instant
invention which synthesizes the corresponding ACEI from 2-oxo-4-
phenyl-trans-3-butenoic acid by condensation with an appropriate
amine, R-NH2, to result in the novel intermediate R-N-12,5-dihydro-
2-oxo-5-phenyl-3-furanyllamine. This compound is then hydrogenated
whereby the furanyl ring becomes saturated, is opened, and the
resulting hydroxy function on the benzyl moiety hydrogenated to
result in a diastereomeric racemic mixture which can be separated
into an essentially pure diastereomer (S,S) by slurrying or re-
crystallizing the product from acetonitrile.
Hereinbefore and hereinafter the definitions used have meaning as
follows:
R is the residue ~1]-benzazepin-2-one-(3S)-3-yl which is substituted
by R1-CHz- in the l-position, that is on the ring-nitrogen.
~213~99
With the exception of the compounds of formula I where R1 denotes
carboxy, Rl and R2 are independently selected from carboxy and lower
alkoxycarbonyl.
The term "lower" defines groups having up to and including 7 carbon
atoms, preferably up to and including four carbon atoms and more
preferred one or two carbon atoms.
Lower alkoxycarbonyl is e.g. methoxycarbonyl, ethoxycarbonyl, propoxy-
carbonyl, or butoxycarbonyl, such as tert.-butoxycarbonyl.
Salts of the compounds of formula I may be formed with acids or bases,
and are preferably pharmaceutically acceptable salts, such as with
hydrohalic acids, e.g. hydrochloric acid.
Pharmacsutically acceptable salts are preferably metal or ammonium salts,
more particularly alkali or alkaline earth metal salts, e.g., the sodium,
potassium, magnesium or calcium salt; or advantageously easily crystal-
lizing ammonium salts derived from ammonia or organic amines, such as
mono-, di- or tri-lower (alkyl, cycloalkyl or hydroxyalkyl)amines, lower
alkylenediamines or lower hydroxyalkyl or aralkyl)alkylammonium bases,
e.g., methylamine, diethylamine, triethylamine, dicyclohexylamine,
triethanolamine, ethylenediamine, tris-(hydroxymethyl)aminomethane or
benzyltrimethylammonium hydroxide. The compounds of formula I form acid
addition salts, which are preferably such of therapeutically acceptable
inorganic or organic acids, such as strong mineral acids, for example
hydrohalic, e.g. hydrochloric or hydrobromic acid; sulfvdc, phosphoric,
nitric or perchloric acid; aliphatic or aromatic carboxylic or sulfonic
acids, e.g. formic, acetic, propionic, succinic, glycolic, lactic, malic,
tartaric, gluconic, citric, ascorbic, maleic, fumaric, hydroxymaleic,
pyruvic, phenylacetic, benzoic, 4-aminobenzoic, anthranilic, 4-hydroxy-
benzoic, salicylic, 4-aminosalicyclic, pamoic, nicotinic; methane-
sulfonic, ethanesulfonic, hydroxyethanesulfonic, benzenesulfonic,
p-toluenesulfonic, naphthalenesulfonic, sulfanilic or cyclohexylsulfamic
acid.
,~'
34499
- 3A -
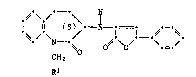
Essentially, the method entails reacting an amine of the formula II
,~-\ /'
NH2 (II)
¢H2
~1
with trans-2-oxo-4-phenyl-3-butenoic acid in the presence of an alcohol,
preferably a lower alkanol, more preferably ethanol, to obtain a novel
intermediate of the formula III
. ,. ~. ,.
~2~ 99
Thlese intermediates wherein R1 is carboxy or lower alkoxycarbonyl
are also within the scope of the invention. A reaction using
a~ilines instead of a compound of formula II is disclosed in J. Org.
Chem. 33, 3991-3993 (1968~.
The intermediate of formula III is then catalytically hydrogenated,
in the additional presence of an alcohol, preferably a lower alkanol
more preferably ethanol, to a compound of the formula IV
R ~ H - CH2 - CH2~ (IV).
~OH
The hydrogenation reaction saturates the double bond in the 2,5-di-
hydrofuran ring and causes the lactone bond to break, thereby
opening the ring. Since this results in a benzylic hydroxy function,
the hydrogenation continues so that the product of formula IV is
obtained. Although most any hydrogenation catalyst will suffice a
Pd/C cataly~t is especially prcferred.
surprising and important feature of the invention is that the
resolution of the S,SR racemic mixture into the desired S,S com-
pounds of formula I is simply and quickly accomplished by slurrying
the mixture with or recrystallizing it from acetonitrile. The S,S
diastereomer is generally obtained in greater than 98 % optical
purity.
When R2 in the desired compound of formula I is carboxy and Rl is
carboxy in the starting material, the synthesis is complete after
the recrystallization from or slurrying with acetonitrile. However,
if R1 was originally lower alkoxycarbonyl, reaction with e.g.
hydrogen chloride will yield the desired product. If the desired
product is to have R2 as a lower alkoxycarbonyl group, the product
of the crystallization or slurrying step can be reacted with the
desired lower alkanol, most preferably ethanol, in the presence of
thionylchloride. Further treatment of this product with e.g. about
4 N hydrochloric acid or about 1 N alkali metal hydroxide, such as
-` ~284~99
sodium hydroxide or potassium hydroxide at about 50C for at least
about 1 hour will selectively free the R1 carboxyl group without
affecting the RZ ester. Still further treatment with acid or base
will free the R~ carboxyl group as well. Even for this treatment
acids such as hydrochloric acid and bases such as sodium hydroxide
or potassium hydroxide are suitable.
In any case when a compound of formula I wherein R~ is free carboxyl
is desired, any alcohol which is otherwise non reactive with the
rest of the molecule and readily removable by known means is
suitable as the temporary esterification group for R~. As the free
Rl carboxyl group is desired in the compound of formula I, any
esterification group for the R~ carboxyl group is suitable in the
starting materials, provided it can be readily removed, although
such groups are not disclosed in detail .
The invention will be more specifically understood in terms of the
following example~, which are exemplary orlly and do not li~(lit the
scope of the invention. 'lemperatures are given in degrees Centi-
grade.
~xample 1:
a) Preparation of ethyl-3-l(2,5-dihYdro-2-oxo-5-phenyl-5S-3-furan-
yl)aminoJ-2,3,4,5-tetrah~dro-2-oxo-(3s)-l1J-benzazepine-1-acetate of
formula III
To a solution of 0.935 g (0.0055 moles) of 2-oxo-4-phenyl-trans-3-
butenoic acid in 8-10 ml of cold ethanol is added dropwise with
stirring at O~C an ethanolic solution of 1.3 g (0.005 moles) of
ethyl 3-amino-2,3,4,5-tetrahydro-2-oxo-(3S)-IlJ-benzazepine-1-
acetate. When the addition is complete the reaction mixture is
stirred at 20C for 1 hour, then allowed to warm to ambient
temperature. After 20-48 hours the product precipitates out as a
thick slurry and can be filtered to yield between 60-90 % of the
desired product depending on reaction time and temperature. The
product has a melting point of 144-146C (crude) or 146-148C after
recrystallisation.
~2~ 99
b) Pr~paration of the compound corresponding to formula I wherein R1 is
COOEt and_R2 is COOH
The unsaturated amino lactone of formula III (25 g, 0.06 mol) is
suspended in ethanol (1500 ml) and to this is added 4A molecular sieves
(50 g) and 5 g of Pd/C 5 %. The mixture is hydrogenated at ambient
temperature for approximately 20 hours until the theoretical amount of
hydrogen is consumed. The reaction mixture is filtered (to remove the
catalyst and molecular sieves) and the filter cake is washed with fresh
ethanol (about 1000 ml). The combined filtrates are refiltered through
Celite~ and concentrated to yield 25 g of a white solid (crude product).
The crude product is recrystallized from acetonitrile (about 200 ml,
80C), and cooled to yield 8.3 grams (1st crop) of the S,S diastereomer
compound of formula I wherein R1 = COOEt and R2 = COOH; m.p. 185-186C;
Rot. [~]Ds = -156.87 (1 % in ethanol~.
c) Preparation of the compound correspondin~ to formula I wherein R1 and
R2 are both COOEt
To a suspension of the compound described under b) [8.0 g (0.0188 mole)
in ethanol (~80 ml) cooled to 6C] is added dropwise thionyl chloride
3.2 ml (4.88 g, 0.41 mol) to give a clear solution. The reaction mixture
is refluxed for approximately 40 hours after which the reaction mixture
is monitored by thin layer chromatography, toluene/ethyl acetate (1:1)
and ethyl acetate, methanol, ammonium hydroxide solution (17:3:3) and
proved to be more than 90 % complete. The reaction mixture is evaporated
to dryness to yield the crude product which by HPLC analysis is more than
91 % pure S,S diastereomer of the compound of formula I wherein R1 and R2
are both COOEt.
d) Preparation of the compound of formula I wherein R~ is COOH and R2 is
COOEt (Alkaline conditions)
To a solution of the diester described under c) (1 g, 0.002 mol) in
ethanol (10 ml) is added a solution of sodium carbonate (212 mg,
0.002 mol) in 8 ml of water followed by a 1-molar solution of sodium
~,
~2~34~9
hydroxide (1.8 ml, 0.0018 mol). The reaction mixture is stirred at
ambient temperature and monitored by HPLC (C18 column, using a
water ~ methanol gradient over 20 minutes). ~en the reaction is
complete, the ethanol i5 removed under vacuum and the aqueous
residue is extracted twice with diethyl ether ~to remove any
unreacted starting material) and the aqueous layer is then adjusted
to a pH of 4.3 with 12N hydrochloric acid. The aqueous layer is
extracted exhaustively with methylene chloride. The extracts are
combined, dried ove~ sodium sulfate, and concentrated to yield
~400 mg (~46 %~ of the compound of formula I, wherein Rl is carboxy
and R2 is ethoxycarbonyl, being 1-carboxymethyl-3S-(lS-ethoxy-
carbonyl-3-phenylpropylamino)-2,3,4,5-tetrahydro-lH-[1]-benz-
azepin-2-one.
e~ Preparation of the compound of formula I wherein Rl is COOH and
RZ is COOEt (acid conditions)
A suspension of the diester described under c) (0.5 g, 0.01 mol) in
10 ml of 4N hydrochloric acid i~ heated to 5~"C for ab~ut 4 hours
after which time the reaction mixture become~ hornogenous and is
monitored by HPLC ~water/methanol 25:75) to yield >88.5 % of the
desired monoester of formula I. The reaction mixture is cooled and
the product crystallizes out of solution. The crvstals are filtered
and dried to yield as a crude product the compound of formula I
wherein Rl is carboxy and R2 is ethoxycarbonyl, being 1-carboxy-
methyl-3S-~lS-ethoxycarbonyl-3-phenylpropylamino)-2,3,4,5-tetra-
hydro-lH-Ll~-benzazepin-2-one which by HPLC is >96 % pure (impur-
ities are ~1 % diacid and 2 % diester).
f) Preparation of the compound of formula I wherein Rl and R2 are
COOH, as the hydrochloride salt.
To a suspension of the compound described unter d) (1 g, 0.002 mol)
in ethanol (about 10 ml) is added 4 ml of a 1.9 N solution of
potassium hydroxide. The reaction mixture is stirred at ambient
temperature for 1 hour, warmed to 50~C for 10 minutes, then cooled.
The ethanol is removed under vacuum and the pH of the remaining
aqueous solution is adjusted to pH 1 with 12 N hydrochloric acid
~28~9
and the desired product precipitates out of solution. The product is
filtered, washed with acetone, and dried to yield 600 mg of the
hydrochloride salt of the compound of formula I wherein Rl and R2
are carboxy, m.p. 278-280C.
Examples 2-4: Example 1 is followed except that Rl in the compound
of formula II i5 methoxycarbonyl, tert.-butoxycarbonyl, and carboxy
respectively. In the case where R~ is carboxy, hydrogenation of the
compound of formula III and recrystallizing from acetonitrile yields
the diacid directly.