Note: Descriptions are shown in the official language in which they were submitted.
1289972
BACKGROUND OF THE INVENTION
This invention relates to a process for producing
an acetyl-substituted aromatic compound which comprises
acetylating an aromatic compound such as an aromatic
hydrocarbon and a phenol with acetyl fluoride.
It is disclosed in Japanese Patent Application
Kokai (Laid-open) No. 135756/79 to produce a 2-alkyl-6-
acylnaphthalene by acylating a 2-alkylnaphthalene with
an acid fluoride in the presence of boron fluoride or
hydrogen fluoride and boron fluoride as a catalyst.
It is described in the above-mentioned inven-
tion that the presence of boron fluoride as a catalyst
component is essential and the absence of boron fluoride
results in low yield of the acylation product. However,
according to the experiment of the present inventors, it
has been found that when boron fluoride is used as a
catalyst component in the reaction of an aromatic compound
with acetyl fluoride, it is difficult to decompose the
resulting complex compound on account of the instability
of the aromatic ketone of the reaction product.
SUMMARY OF THE INVENTION
Accordingly, the present inventors have made
extensive studies to attain a process for producing an
acetyl-substituted aromatic compound with higher efficiency
~,; _ 1 _
~i`f;
~289972
1 including the steps of the decomposition of the complex
compound and the recovery of the catalyst component.
Consequently, it has been found that, in the reaction
of an aromatic compound with acetyl fluoride, even when
hydrogen fluoride alone is used as a catalyst the intended
acetyl-substituted aromatic compound can be obtained
in excellent yield,and further the complex compound formed
can be readily decomposed and the hydrogen fluoride
catalyst can be easily recovered. This invention has
been accomplished on the basis of the above findings.
DETAILED DESCRIPTION OF THE INVENTION
Thus, this invention provides a continuous process
for producing an acetyl-substituted aromatic compound which
5 comprises:
obtaining an acetyl fluoride by the reaction of
excess acetic anhydride with a substantially anhydrous hydrogen
fluoride wherein the ratio of excess acetic anhydride to
hydrogen fluoride is 0-5 mol~, and separating formed acetyl
0 fluoride by distillation,
making an aromatic compound selected from alkyl-
benzenes, alkylnaphthalenes, phenols and naphthols react with
the separated acetyl fluoride in the presence of substantiallv
anhydrous hydrogen fluoride as a catalyst, and
thermally decomposing the resulting complex compound
between the acetyl-substituted aromatic compound and hydrogen
fluoride.
-2-
~28997~
The aromatic compounds used as the starting
material in this invention include alkylbenzenes such as
toluene, xylene, trimethylbenzene, ethylbenzene, cumene,
and butylbenzene; naphthalene and alkylnaphthalenes
such as methylnaphthalene; phenols and naphthols; and
further aromatic ethers such as anisole and phenyl ether.
Particularly preferred are compounds in which the para
position to the substituent in the aromatic ring is
vacant,and naphthalenes having a substituent in the
iO 2-position.
-2a-
~ ~J
~899~7Z
1 Acetyl fluoride as the other starting material,
can be obtained by mixing acetic anhydride
with hydrogen fluoride to produce acetyl
fluoride according to the following equation (1)
and separating the free acid simultaneously formed.
(CH3CO)2O + HF . CH3COF + CH3COOH ............. (1)
It is essential here that the reaction (1)
mentioned above should be carried out using a slight excess
of acetic anhydride. Thus, when hydrogen fluoride is
in excess of the equivalen~, the maximum azeotropic mix-
ture combined hydrogen fluoride with the acid, whichleads to the loss of hydrogen fluoride, is formed to
become inseparable and further the acid formed is
contaminated with fluorine, so that special treatments
are required to remove it. When acetic anhydride is
in excess, such difficulties do not occur and hydrogen
fluoride can be quantitatively recovered as acetyl
fluoride. However, there is no need of a large excess
and the ratio of excess acetic anhydride to hydrogen
fluoride may be 5 mol% or below.
The apparatus used for generating acetyl
fluoride may be a conventional distillation column
having a number of plates necessary for fractionating
acetyl fluoride and the free acid. Acetic anhydride
,... .
~8997~
1 and hydrogen fluoride are fed to the appropriate plate
of the distillation column as a mixture or separately,
the column bottom is heated up to the boiling point of
the acid and the column top is given an appropriate
reflux. Such a conventional distillation operation
makes it possible to recover pure acetyl fluoride from
the column top and an acid containing no fluorine from
the column bottom.
Since the reaction proceeds at a high rate,
virtually no residence time is necessary. The reaction
can be conducted under an ordinary or an applied pressure,
for example 1 kg/cm2G, and either flow operation or
batchwise operation may be used. The difference of
boiling point between acetyl fluoride and the acid
formed is so large that the two can be easily separated.
The acetyl fluoride thus formed is used for
acetylation as the acetylating agent. The molar ratio
of acetyl fluoride to the starting aromatic compound
is 1 or below, 0.9 to 0.5 being particularly preferable.
The presence of excess acetyl fluoride decreases the
overall reaction rate (i.e. the space time yield of the
acetylation product).
As the catalyst, a substantially anhydrous
hydrogen fluoride is used. Its water content is
preferably 5% or below because the presence of water in
hydrogen fluoride causes a rapid decrease of catalytic
activity. In order to obtain a sufficient reaction rate,
the molar ratio of hydrogen fluoride to be used relative
; - 4 -
~289972
l to the acetylating agent is 5 or above, preferably in
the range of lO to 20. Hydrogen fluoride used in a
molar ratio of 20 or above gives little additional
effect and hence is not advantageous from the economical
viewpoint of the process.
The reaction temperature of acetylation is
0 to 70C, preferably lO to 50C. Since the reaction
rate increases as the temperature is increased but also
the side reaction increases, the optimum temperature is
selected from the above-mentioned range depending upon
the starting material used. When the starting compound
has a high melting point and further is insoluble in
hydrogen fluoride as in the case of aromatic hydrocarbons,
it is effective to use a suitable solvent in order to
make the reaction proceed smoothly. Preferable solvents
are those which can dissolve the starting compound well,
are chemically inert under reaction conditions, and have
a good compatibility also with the reaction liquid formed.
They include, for example, benzene or halogenated
hydrocarbons such as chlorobenzene, dichloromethane,
dichloroethane and ~Freon~ ~ Particularly, benzene is the
most suitable solvent in the present process because it
is not only substantially inert under the present
reaction conditions but also favorable as the solvent in
the step of recovering the catalyst from the reaction
mixture.
The amount of the solvent to be used in the
reaction is not specifically limited. The amount of
*Trademark for a line of fluorinated hydrocarbons.
12`89~7~
1 0.5 mole or below per mole of the starting compound is
usually sufficient.
Although the reaction pressure is varied
depending on the reaction temperature, usually it ranges
from atmospheric pressure to a slightly elevated
pressure of up to 2 kg/cm G.
The reaction proceeds in a homogeneous liquid
phase or, according to circumstances, in two-liquid
phases consisting of a starting aromatic compound phase
and a catalyst phase, so that there is no need of
vigorous stirring. Since the reaction is slightly
exothermic, a reactor provided with heat removal equip-
ment is used as required.
The acetylation reaction liquid thus obtained
is a solution of an aromatic ketone, and the acetylation
product in hydrogen fluoride. On heating the reaction
liquid, the affinity between the reaction product and
hydrogen fluoride is broken and hydrogen fluoride can
be easily vaporized and separated.
It is necessary to conduct the above-mentioned
catalyst recovery operation as rapidly as possible in
order to avoid the thermal degradation of the reaction
product. For this purpose, the catalyst recovery
operation is preferably conducted in a flow operation
using a multistage gas-liquid contact apparatus (i.e. a
distillation column). For catalyst recovery, heating
at a temperature of 40C or higher, particularly 40 to
100C, is necessary. The decomposition column is
6 --
q ~ .
. -:
1289g7Z
1 preferably fed with an amount of heat which is in excess
of that necessary for vaporizing hydrogen fluoride fed
to the column. It is advantageous in the process to
conduct the catalyst recovery operation under atmospheric
pressure or a slightly increased pressure of 2 kg/cm2G
or below. In order to make the thermal decomposition
of the complex compound between the acetyl-substituted
aromatic compound and hydrogen fluoride proceed smoothly,
the decomposition is preferably conducted by heating
the complex compound under reflux using as a diluent a
substance which has a boiling point such that it is
easily separable from hydrogen fluoride, has a good
compatibility with the reaction product, namely the
acetyl-substituted aromatic compound, and with hydrogen
fluoride, and is inert to hydrogen fluoride. Examples
of such diluent used include aromatic compounds such
as benzene and chlorobenzene. Particularly, benzene is
the most preferable diluent.
According to this invention, aromatic compounds
can be acetylated in a simple operation under low reaction
pressure, and hydrogen fluoride used as the catalyst
can be completely recovered and recycled. So this
invention is of great industrial advantage.
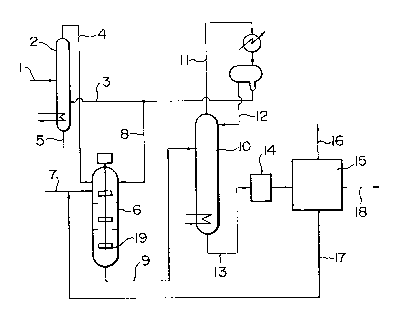
The accompanying drawing is a flow diagram
showing the acetylation process of this invention.
The process for acetylating an aromatic
compound according to this invention is illustrated below
with reference to Fig. 1.
- 7 -
~i! .
.
': ': ' ' ' '
- , : '
~,
~X8997Z
1 In Fig. 1, acetic anhydride is fed to the
middle plate of an acetyl fluoride generating apparatus
2 through pipe 1 and contacted there with heating with
hydrogen fluoride introduced through a pipe 3. The
acetyl fluoride formed is distilled out through an
outlet pipe 4 and acetic acid is withdrawn through a
pipe 5. The acetyl fluoride is fed to an acetylation
reactor 6 equipped with a stirrer 19 and is contacted
there with stirring with the starting aromatic compound
fed through a pipe 7 and with hydrogen fluoride fed
through a pipe 8. The reaction begins in two liquid
phases of the hydrogen fluoride phase and the starting
material oil phase, which then change into a homogeneous
liquid phase as the reaction proceeds. The reaction
liquid is drawn out through a pipe 9, led to a hydrogen
fluoride recovery column 10, and contacted there with
a diluent such as benzene which is being refluxed and
recycled. Hydrogen fluoride is separated by vaporization
and drawn out through a pipe 11. The column top vapor
is condensed by cooling and separated into layers. The
benzene phase is refluxed from a pipe 12 to the recovery
column 10; hydrogen fluoride is recycled to the acetyl
fluoride generating apparatus and the acetylation
; reactor (not shown in the Figure). From the bottom of
the hydrogen fluoride recovery column is withdrawn
through a pipe 13 the acetylation product, a crude product,
; which is then freed from trace amount of residual acid in
a neutralization and washing equipment 14 and distilled
-- 8 --
,
.
: .
: ` ' ' ~ ` `
~Z899~72
1 in a distillation apparatus 15, whereby the byproduct
is removed through a pipe 16, the unreacted raw material
is removed through a pipe 17 to be recycled to the
reaction step, and the final product is obtained through
a pipe 18.
A suitable solvent is used to make the reaction
proceed smoothly as re~uired. This is added to the
starting material in pipe 7. Fig. 1 shows a case
where the solvent and the diluent for decomposition are the
same. But, in the case where the solvent and diluent are
different, a process for recovering the solvent is included in
the overall process.
This invention will be further explained in detail
below with reference to Examples, but it is not limited thereto.
Example 1
Synthesis of acetyl fluoride
A stainless steel packed column having a
diameter of 50 mm and a height of 1000 mm provided with
a top reflux apparatus and a bottom reboiler was used
as the acetyl fluoride synthesizer. Acetic anhydride
(22.0 moles per hour) and hydrogen fluoride (21.0 moles
per hour) were mixed and fed continuously to the middle
plate of the packed column, and heat was supplied to the
reboiler with an electric heater at a rate of 260 Kcal
per hour. The synthesizer was operated under a pressure
of 1.0 kg/cm2G. While reflux was applied so as to keep
the temperature of the column top at about 35C, acetyl
g
- . , :
'-
:
.
128997Z
1 fluoride was distilled out of the column top at a rate
of 21 moles per hour, and a liquid mixture comprising
21 moles of acetic acid and 1 mole of acetic anhydride
was withdrawn every hour from the bottom. The yield of
acetyl fluoride relative to supplied hydrogen fluoride
was quantitative.
Acetylation of 2-methylnaphthalene
Two stainless steel vessels each equipped with
a stirrer, the first reactor having an inner liquid
volume of 6 Q and the second reactor having an inner
liquid volume of 4 Q, were connected in series to be
used as the acetylation reactor. A solution comprising
1.5 kg of 2-methylnaphthalene and 0.3 kg of benzene
was fed every hour to the first reactor. Simultaneously,
0.5 kg per hour of acetyl fluoride synthesi~ed above
and 2.5 kg per hour of hydrogen fluoride were also fed
to the first reactor.
The reaction temperature was adjusted to 25C
by passing cooling water through the jacket of the
reactor. The pressure in the reaction was 1 kg/cm2G.
The reaction mixture was continuously withdrawn from
the second reactor to be fed to the subsequent hydrogen
fluoride recovery column.
Recovery of hydrogen fluoride
The packed column used in acetyl fluoride
synthesis was employed as the hydrogen fluoride recovery
-- 10
. . .
' ~' '
.~ ' '
lZ8997Z
1 column. The hydrogen fluoride recovery column was
charged with benzene, and heat was supplied at a rate
of 300 Kcal per hour to the reboiler under a pressure
of 1 kg/cm G to keep the benzene refluxing.
Then the above-mentioned reaction mixture was
continuously fed to the upper part of the column at a
rate of 1 kg per hour, and the hydrogen fluoride recovery
column was continuously operated while being replenished
with benzene.
From the column top were distilled out
hydrogen fluoride and unreacted acetyl fluoride, while
from the column bottom were recovered every hour 243 g
of acetylated methylnaphthalene, 109 g of unreacted
methylnaphthalene, and 20 g of a high boiling point ~
product as the crude acetylation product. The acetylation -
product contained 75% of 2-acetyl-6-methylnaphthalene.
Example 2
Acetylation of toluene
Toluene was acetylated by using the same
apparatus and the same operation as in Example 1.
To the first reactor were fed every hour
0.9 kg of toluene, 0.4 kg of acetyl fluoride, and 2.0 kg
o hydrogen fluoride, and the reaction was conducted at
a reaction temperature of 40C and under a reaction
pressure of 1.5 kg/cm2G. The reaction mixture was
continuously withdrawn from the second reactor and
hydrogen fluoride was recovered in the same manner as in
1 1 _
12899'7Z
1 Example 1. The crude product obtained from the bottom
of the hydrogen fluoride recovery column had the
following composition: unreacted toluene 55%, methyl-
acetophenone 41%, high boiling point product 4%. The
methylacetophenone contained 97.5% of 4-methylacetophenone.
Example 3
Acetylation of m-xylene
A stainless steel autoclave of 500 ml volume
equipped with a jacket and a stirrer was used as the
acetylation reactor.
A solution of 56 g (0.9 mole) of acetyl
fluoride in 103.1 g (1 mole) of m-xylene was placed in
the autoclave, and then, with cooling, 300 g (15 moles)
of hydrogen fluoride was introduced thereinto. The
mixture was allowed to react at a reaction temperature
of 40C under a reaction pressure of 1.5 kg/cm2G for
1.5 hours. Thereafter, the reaction mixture was withdrawn
into ice water. The resulting oil layer was washed with
alkaline water and distilled to determine ~he amount of
20 high boiling point byproducts and analyzed by gas "
chromatograph to determine the yield of the acetylated
product.
Examples of acetylations conducted in the same
manner as mentioned above using various aromatic compounds
~- 25 as the starting material are summarized in Table 1.
.. :
' ' ~ ' -.`. ' '
.;
.
i289972
s~ ~
.~ O ~ O ~ 1
~ ~ ~ ~ ~ 1- co ~r co ~ r~ ~
~ o~ ~
o
,1
U aJ
o la~
~ ~ ~_
.,1 ~ ~ O tn O O O O
_i ~ ~ ~o ~
o
E~ .~ ~ ~ O' U~ O O O
,~ . ~ o
_
u~
~ ~ I`
~ ~ o' o o o o o o o
_
~ S aD
.~ ~_ ~ ~
~ ~ o ~ a~
:~ cn ~ o S~ 0
,~ ~ ~ a) ~ s ~ ~
æ H p, ,¢ p~
-- 13 --
: . ~
:-
1~89'37~
U~U700 ~
_ ~~ o o
la ~~1
'~n a~
~, ~ _, o
s ~ ~
o U ~ o s
~ s
o o~ ~ ~ ~ o o ~ ~ ~
.~ ~ ~ ~ s ~ ~ o o o
~ ~ ~ ~ o ~ a
~1 o ~ ~ o u s a) ~ Q~
lQ-~I ~ ~ Q. S S S
o ~ o s s a) _1 o ~4 Q~ Q,
C~ ~ ~ o o o
~ ~ o ~ s
o ~ ~
o I ~ o u u
~ S~ ~ S
u ~ ~ ~ ~ x x
O O O
S rl I Q S-l S ~::
a ~ o ~ ~ a~
~ ~ ~ S
I I ~ ~ I I I I
~ ~ ~I N ~ ~ t:~
U
U
~ ~ 3
o ~o o
U ~ ~
_l
~-~/ O~ co o r~
.,, ~-,1 ~ _,
_1 ~ ~ 3
.8 ~
O ~ -1
E- ~4 h
~1
U
'~:~
-- 14 --