Note: Descriptions are shown in the official language in which they were submitted.
32~3
- RAN 4093/77
This invention relates to methods for the prepacatisn of
novel tetrahydrocannabinol derivatives and to the use of
these derivatives as ceagents in improv2d immunoassays for
cannabinol metabolites in biological fluid samples.
Increases in the use of marijuana have led to the
development of assays for the detection of the primary
active constituent of the plant, L -tetrahydrocannabinol
(T~IC), and, more ~articularly, metabolites of THC in urine
and blood samples. These assays employ the use of labeled
cannabinol deri~ativea in conjunction with antibodies
against metabolites of the drug.
In practice, a blood or urine sample suspected to
con~ain cannabinol metabolites (including glucuronides and
other conjugation products) is contacted with the antibodies
in the presence of a labeled derivativeO To the extent that
cannabinol metabolites are present in the sam~le, there will
be competition for binding to the combining sites of the
an~ibodies, and the amount of the labeled derivative bound
26 w;ll be Leduced in 2ro~ortion to the degree of such
competi~ion.
; Descciptions of some representative immunoassays can be
found in O~Connor et al., J. Anal. Toxicol. 5:168 (1981),
Law et al., J~ ~nal. Toxicol. 8:14 (1984) and Childs et al.,
J. Anal. Toxicol. 8:220 (1984). In all of these references,
it is the displac~ment of some of the labeled cannabinol
derivative by metabolites in the assay samples that is t'he
basis of the assays desccibed. Superioc assay cesults will
be obtained when the labeled derivative is specifically
Klt/18011.87
~9;~ 3
. ,, `
-- 2 --
recognized by the antibodies and yet easily displaced by the
various products of cannabinol metabolism.
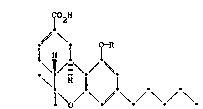
The present invention relates to novel derivatives of
cannabinol o the formula
T~
~\
i H i
I o
where R is an attachment chain selected ~rom the ~roup
consisting of a p-aminobenzyl group or a branched or linear
aminoalkyl group having ~rom 1 to 7 carbon atoms or an
organia or ~ineral acid addition sal ther~of, an isocyanate
or isothlocyanate derivative o~ the p-aminobenzyl or
aminoalkyl group, a aa~boxyl terminate~ derivative o~ the
aminoalkyl groue having from 1 to 7 additional carbon atoms
or a salt thereof, and an activated derivative o~ the
carboxyl terminated derivativeO
This invention further relates to the use of labeled
derivatives of the above compounds with antibodias against
cannabinol metabolites or derivatives for the detection of
aannabinol metabolites in blood or urine samplas. Detection
of the metabolites is facilitated by ~he use of the labeled
derivative compounds, which comprise the compounds of the
invention covalently soupled ~o a suitable raporter molecule.
.
,;. ~ .
~3~2~3
- 2a -
ln one aspect the invention provides a labeled
compound for use in the immunoassay of cannabinol
metabolites comprising ttrans-rac)~1-(3-aminopropoxy)-
6a,7,10,10a-tetrahydro-6,6-dimethyl-3-pentyl-6H-
dibenzo(b,d)pyran-9-carboxylic acid, ox an organic or
mineral acid addition salt thereof, covalently coupled to
a reporter molecule to facilitate detection.
In another embodiment the invention provides a
method for the immunoassay of cannabinol metabolites,
comprising mixing a sample suspected to contain cannabinol
metabolites with known amounts o~ a labeled compound
comprising a compound of the formula
To2H
, ~,j,, ,T~I
\o/ \.~ \./ \./ \
where R is an attachment chain selected from the group
consisting of a p-aminobenzyl group or a branched or
linear aminoalkyl group having ~rom 1 to 7 carbon atoms or
an organic or mineral acid addition salt thereof, an
isocyanate or isothiocyanate derivative o~ the p-
aminobenzyl or aminoalkyl group, a carboxyl terminated
derivative of the aminoalkyl group having from 1 to 7
carbon atoms or a salt thereof, and an activated
derivative of the carboxyl terminated derivative,
covalently coupled to a reporter molecule, and antibodies
against a cannabinol metabolite, which antibodies are also
capable of selectively binding the labeled compound,
separating the bound ~rom the free labeled compound,
measuring the amount of either the free or the bound
labeled compound and comparing the amount of the labeled
compound measured to values obtained ~rom samples
containing known amounts of a cannabinol metabolite.
The present invention may be more readily understood
by reference to the following figures, in which
2~
-- 3
Fig. 1 shows the formulae of the starting materials and
intermediates in~olved in the synthesis of (trans-rac)-l-
-(3 aminopropoxy)-6a,7,10,10a-tetrahydro-6,6-dimethyl-
-3-pentyl-6H-dibenzo(b,d)pyLan-g-carboxylic acid monohydro-
chloride;
Fig. 2 shows the formulae of the intermediates and
final p~oduct of the synthesis of (t~ans-rac)-6a,7,10,10a-
tetrahydro-~-t4 aminobenzyl)-6,6-dimethyl-3-pentyl-6H-
dibenzo(b,d)pyran-9-carbo,xylic acid; and
Fig. 3 is a graphical representation of a standard
curve for the immunoassay of THC, showing change in
absorbance at 49Z nm as a function of 9-carboxy-11-nor-
Q -tetrahydro-cannabinol concentration.
The immunoassays of the peesent invention are directed
to the assay of cannabinol metabolites in biological fluids
such as human blood and urine samples. As used herein, the
term "cannabinol metabolites" mean6 ~ or ~
tetrahydrocannabinol and the catabolic products of the~e
compounds, including conjugation products such as the
glucuronides. The term "cannabinol derivativell means a
chemically synthesized derivative of ~8 Ol a9
tetrahydrocannabinol.
Antibodies used in the invention can be prepared using
any one of a wide range of possible immunogens. These
immunogens comprise a cannabinol metabolite or derivative
conjugated to a suitable immunogenic carrier molecule.
Conjugation to a carrier is necessary because the cannabinol
metabolites or deriva~ives themselves are haptens (molecules
which are capable of specifically binding to an antibody but
incapable of eliciting antibody production, i.e., they are
36 not immunogenic).
3~Z13
-- 4
As used herein, the term "immunogenic carrier molecule~
means macromolecules which have the property of
independently eliciting an immunological response in a host
animal and which can be covalently coupled to the cannabinol
metabolite or derivative. Suitable carrier molecules
include, e.q., proteins; natural O synthetic polymeric
compounds such as polypeptides: polysaccharides etc.
Protein cacrier molecules are especially preferred. Once
coupled to a carrier molecule, the cannabinol metabolite or
derivative becomes immunogenic due to what is commonly known
as the "carrier effect".
Protein caLriers which may be used in this invention
include but are not limited to mammalian serum proteins such
as keyhole limpet hemocyanin human or bovine gammaglobulin,
human, bovine or rabbit serum albumin, or methylated or
other derivatives of such proteins. Other protein carriers
will be apparent to those skilled in the art. Frefeeably,
but not necessarily, the protein carcier will be foreign to
the host animal in which antibodies against the cannabinol
metabolite or derivative are to be elicited.
Covalen~ coupling to the carrier molecule can be carried
out by methods well known in the art, the exact choice of
which will be dictated by the nature of the functional
groups in the cannabinol metabolite or derivative and in the
carrier molecule available foL coupliny.
Once erepared, the immunogen can be used to induce the
formation of an~ibodies that are specific to cannabinol
metabolites in host animals by injecting the immunogen in a
host animal, preferably using a conventional adjuvant such
as E'reund's complete or incomplete adjuvant and the like.
Suitable host animals include rabbits, horses, goats, guinea
pigs, rats, cows, sheep, etc. The resulting antiserum must
be such that the antibodies contained therein, called
anti-THC antibodies, are capable of specifically binding to
-- 5
the cannabinol metabolites which are to be assayed and to
the labeled compounds of the invention bearing reporter
groups, as described below. The suitability of the
antiserum product can be rapidly ascertained by routine
experimentation.
In an illustrative embodiment, anti-THC antibodies
produced against three di~ferent con~ugates based upon three
THC derivative~ coupled at the 9 position were pooled for
use in the invention (see Example 14)o It again must be
stressed, however, that the exact nature of the conjugates
used to make the antibo~ies is not critical to ~he
invention, as long as the resulting antibodias have the
requisi~e broad specificity for THC metabolites.
Although whole antiserum can be used, the IgG fraction
is preferably isolated by salt fractionation, such as
ammonium sulphate precipitation, by DEAE chromatography or
hy other means known in ~he art.
Some o~ the novel cannabinol compounds of the present
invention are derivatives of a major metabolite of THC,
f 02H
i~o/l\o~
\l I ~] o\ /o\
ll-nor-~ (a )-THC-9-carboxylic acid,
in which the phenolic hydroxyl group i5 modified by
replacemen~ of ~he phenolic hydrogen by a ~-aminobenzyl
group or an amino-terminal alkyl group which can have from l
to 7 carbon atoms. Where an amino alkyl group is used, ~he
group may be branched or straight chain, such as
:~9;~ 3
-- 6
aminopropyl, aminoisopcopyl, aminobutyl, aminoisobutyl and
the like.
The amino compounds and intermediates of the invention
may be utilized as free bases or as acid addition salts of
organic o~ mineral acids. Etepresentative addition salts
that could be used include hydrochloride, hydrobromide,
sulfonate, methanesulfonate, nitrate, phosphate,
trifluoroace~ate, oxalate, maleate, succinate, acetate and
the like.
Other compounds of the invention are isocyanate or
isothiocyanate der~ivatives of the amino compounds, which can
be prepared by treating the p~aminobenzyl or amino-terminal
alkyl groups with phosgene and thiophosgene, respectively.
Still other compounds of the invention are carboxyl
terminated derivatives of the aminoalkyl compounds. Such
carbo~yl derivatives can be prepared by methods commonly
used in the art. For example, the amino-terminal alkyl
groups can be treated with chloro, bromo or iodo haloorganic
acids having from l to about 7 carbon atoms, to produce
carboxyl terminal deriva~ives. 5uch compounds can be
employed as such or as salts such as Na~, NH4~ and the
like. These carboxyl derivatives can be coupled to amino or
hydroxyl groups on suitable reporter molecules through the
use of coupling agents such as l-ethyl-3-(3-dimethyl-
aminopropyl) carbodiimide or, preferably, dicyclohexyl-
carbodiimide.
~ lternatively, the carboxyl com~ounds can be further
modified to produce activated derivatives such as
N-hydroxysuccinimide ester derivatives. Such derivatives
can be prepared by reac~ing the carboxyl derivatives with a
desired activating comeound such as N-hydroxysuccinimide in
the presence oE a coupling agent such as dicyclohexyl-
carbodiimide.
~3~ 3
-- 7
It mus~ be stressed that it is the availability of an
attachment chain at the phenolic hydroxyl group of THC that
imparts sueerior diagnostic pro~erties to the compounds of
~ this invention. The skilled artisan will immediately
recognize that through the use of the intermediates of the
invention, there are many other ways that similar at~achment
chains with similar functional groups suitable for coupling
to reporter groups can be prepared. For example, a carboxyl
teeminated derivative could be prepared directly without
proceeding by way of an amino derivative as described herein.
The available functional ~roups in the compounds of the
invention provide a convenient point for labeling by the
covalent coupling of an approeriate reporter group, to
facilitate detection in the immunoassays.
Suitable reporter groups for the labeled compounds
include, e.a., biotin (to be use in conjunction with
appropriately tagqed avidin): fluorescent, chemiluminescent
or bioluminescent groups; or radioisotopes such as H,
4C, 35S and lZ5l, which can readily be introduced
into the molecule in many forms well known in the art, due
to the availabil;ty of the amino and carboxyl groups in the
comeounds. Such groups can be detected and quantified by
liquid scintillation spectrometry, fluorescence
spectrosco~y, fluorescence polarization, etc. as appropriate.
Alternatively, conjugate compounds can be prepared in
which the compounds of the invention are covalently linked
to an enzyme by ~he free functional group, including but not
limited to various peroxidases, glucose oxidase,
~-galactosidase and alkaline phosehatase. Horseradish
peroxidase, which can be detected by spectrophotome~ric
analysis of i~s activity on a substrate such as pyrogallol
or o-phenylenediamine, is especially preferred. Where
-enzymes are used, the conjugate compounds can be used in
conjunction with conventional additives, buffers, diluents
lZ~32~3
-- 8
and enzyme stabilizers.
The cannabinol deLivatives of this invention make
superior reagents for use in immunoassay, in part because
they provide functional groups at which labeling can be
carried out. More, importantly, the presence of the
phenolic oxygen side chains creates compounds that can still
bind to anti-THC antibodies but which do so with reduced
affinity. As a result, they are more easily displaced from
the antibody combining site by assay sam~le cannabinol
metabolites. The immunoassays employing these labeled
compounds are thus extremely sensiti~e.
The anti-THC antibodies and labeled compounds of the
invention can be used in a variety of immunoassays for the
detec~ion of cannabinol metabolites. Such immunoassays
could take the form of a radiommunoassay, either in free
solution or solid state. Alternatively, enzyme immunoassays
could be carried out, again either in free solution or solid
state. SoLid state assays can be cacried out by the use of
solid particles onto which the antibodies have been
immobilized. Particles which could be coated with the
antibodies include, e.q., latex beads, li~osomes,
erythrocytes, polyacrylamide beads, polystyrene beads or
beads made of any of a number of other suitable polymers.
l~he immunoassays can be direct or indirect, with the
application of a second antibody directed against the
anti-THC antibodies.
In a preferred embodiment of the in~ention, a sample
suspected to contain cannabinol metabolites is mixed with
known amounts of a labeled compound of the invention and
known amounts of anti-THC antibodies adsorbed onto a
polystyrene bead. Following an incubation period, the bound
labeled compound is ~eparated from the free labeled
compound, and the amount of either ~he free or bound
compound is measured and compared to the values obtained by
. .
~3Z13
g
subjecting samples containing known amounts of a cannabinol
metabolite to the same analytical steps.
EXAMPLES
The followi.ng ace non-limiting examples which illustrate
tne synthesis o~ some of the novel cannabinol derivatives of
the invention and the use of one of these comeounds in an
enzyme immunoassay system. The chemical structures of the
intermediates and final product of the synthesis of
(trans-rac)-1-(3-aminopropoxy)-6a,7,LO,lOa-tetrahydro-6,6-
dimethyl-3-pentyl-6~-dibenzo-~b,d] pyran-9-carboxylic acid
monohydrochloride are shown in Fig. l. The chemical
structures of the intermediates and final product of the
synthesis of (trans-rac)-6a,7,lO,lOa-tetrahydro-l-
-(4-aminobenzyl)-6,6-dimethyl-3-pentyl-6H-dibenzo(b,d)~yran- -
-9-carboxylic acid are shown in F'ig. 2. The Roman numeral
designations of the compounds in the headings of Examples
1-13 refer to the structural formulae shown in Figs. l and 2.
EXAMPLE l
Preparation of
Ethyl 4-methYl-5-hydrox~-7-pentylcoumari-n-3-propionate (I)
A 5-liter flask equip~ed wi~h a mechanical stirrer,
thermo~eter and nitrogen bubbler was charged with 210.l g
(l.17 moles) of olivetol (5-pentyl-resorcine; ~ldrich),
300.0 g (L.30 moles) of diethyl-2-acetoglutarate (Aldrich)
and 180.0 g (L.117 moles) of phosphorus oxychloride. The
mixture was stirred at room temperature and slowly began to
thicken. After three days, the mixture had solidified, at
which time it was allowed to stand for an additional 7 days
without stirring.
'~he solid mixtuce (light green in color) was dissolved
in 2.0 l o~ methylene chlocide and transferred to a 6-liter
separatocy funnel. The organic layer was washed with lO l
~3;2~3
-- 10 --
of deionized water in five equal portions and dried over
anhydrous Na2S04, and the solvent was then stripped off
on a rotary evaporator (40C, SO mm Hg) to yield 672.7 g
(166%) of a pink solid. This solid was redissolved in 1.0 1
of ethyl acetate on a steam bath followed by the addition of
hexanes with continued heating. The solution was allowed to
cool to room temperature and then placed in a freezer
(-10C) overnight. Resulting crystals were filtered on a
~uchner funnel, washed with about 1.0 1 of a mixture of cold
hsxanes:ethyl acetate (2:1), and dried at 100C/10 mm ~Ig in
a vacuum oven for 20 hours to yield 243.5 g (60%) of light
pink crystals/ m.p. 118-120C.
EXAMPLE 2
Preparation of
7,LO Vihydro-l-hydroxy-3-pentyl-6H-dibenzo(b,d)pyran-6,9-
t8H)-dione (II)_
~ 5-liter flask equipped with a mechanical stirrer,
thermometer, addition funnel and nitrogen bubbler was
charged with 80.0 g (2.0 moles) of 60% NaH dispersion in
mineral oil ~Aldrich). The mineral oil wa6 removed by
washing the dispersion with L500 ml of hexanes (Fisher) in
three equal poetions. One hundred and sixty grams (0.462
mole) of compound I were added to the flask, and the two
solids were mixed mechanically. A 20C constant temperature
water bath was placed under the flask~ and 1.4 1 of
distilled dimethylsulfoxide (DMSO) was carefully added
dropwise so that the temperature of the mixture never rose
above 20C.
After 6 hours the addition was complete, and the bath
was removed. The mixture was stirred for an additional hour
and then placed in a freezer (-10C) overnight. The next
day the mixture was warmed to room temperature and poured
into an extractor filled with 12 1 of ice water and 250 ml
of concentrated HCl, when i~ was rapidly stirred for 2
hours. As the mixture was stirred, solids began to
~293213
precipitate. These solids were collected by filtration
through a Buchner funnel and washed with 200 ml of a
saturated NaHC03 solution followed by 250 ml of deionized
water. The solids were air dried and then dried in a vacuum
S oven (100C/L mm Hg) for 16 hours to yield 124.8 g (90~0) of
an off-white solid, m.~. 181-lB4C.
~5~
Prepara~ion of
7,8,9,10-Tetrahydro-l-hydroxy-3-pentylspiro-6-H-dibenzo
(d,b)-~Yran-9,2-(1'-3')-dioxolan-6-one (III~_
A 3-liter flask equipped with a thermometer, mechanical
stirrer, Dean-Stark trap, condenser and nitrogen bubbler was
charged with 20.0 g (0.32 mole) of ethylene glycol, 18.4 g
(0.06~ mole) of compound II, 1.0 g of p-toluenesulfonic acid
monohydrate and l.o 1 of toluene. The solution was refluxed
overnight, and 16.3 ml of azeotropic water were collected in
the tra~. The reaction was cooled to room temperature and
tran6ferred to a 2-liter separatory funnel, where it was
washed first with 300 ml of a saturated NaHC03 solution
and then with 300 ml of deionized water.
~ 'he toluene layer was dried over Na2S04 and then
strip~ed on a rotary evaporator (60C/50 mm ~lg) to a brown
oil that was redissolved in about 100 ml of methylene
chloride. Twenty ml of hexane were added to the solution,
and the mixture was placed in a free2er (-10C) overnight.
Crystals formed which were collec~ed by fll~ration on a
Buchner funnel, washed with 30 ml of hexane and dried
overnight in a vacuum oven (25~1 mm Hg) ~o give 14.7 g
(69%) of an off-white solid, m.p. 110-113C.
EXAMPLE 4
Preparation of
dl-1-Hydroxy-3-pentyl-6,6-dimethyl-6a,7-dihydro-6H-dibenzo
(d,b)pyran-9(BH)-one (IV)
A 3-liter flask equipped with a mechanical stirrer,
3~3
thermometer, heating mantle, addition funnel, condenser and
a nitrogen bubbler was charged with 12.5 g (0.5L5 mole) of
Mg turnings (Fisher) and 500 ml of anhydrous ether. Then,
73.1 g (0.515 mole) of iodomethane (Aldrich) was a~ded
dropwise to the suspension at such a rate as to maintain a
gentle reflux. Ater completion of the addition (about 30
minutes), the grey-black solution was refluxed for an
additional hour, at which time a solution of 16.9 g (0.040
mole) of compound III in 300 ml of anhydrous tetrahydrouran
(THF) was added dropwise to the refluxing solution over a
~eriod of 20 minutes. The mixture rapidly turned yellow,
and solids de~osited on the sides of the flask.
l'he resulting heterogeneous mixture was refluxed
overnight and then cooled to room tem~erature. The mixture
was quenched by the careful addition of S6 ml of lN HCl over
about 20 minutes to produce a gently refluxing solution. An
additional 210 ml of 6N HCl were added over 20 minutes, and
the dark green mixture was stirred for an additional hour
and transferred to a 2-liter separatory funnel, where the
aqueous layer was discarded.
The ethe~ layer was extracted in succession with 200 ml
of deionized water, 200 ml of a saturated NaHC03 solution,
and 200 ml of deion;zed water, after which the solution was
dried over Na2S04 and stripped on a rotary evaporator
t40C~50 mm Hg) to a dark green oil. Fifty ml of ether was
added, and the mixture was allowed to stand overnight at
room temperature. Crystals formed which were collected on a
Buchner funnel, washed twice with 25-50 ml of cold (-10C)
ether:hexane (1: L? and dried overnight in a vacuum oven
(25C/L mm Hg) to give 9.7 g (63%) of a light yellow
material, m.p~ 198-200C.
Concent~ation of the mother liquor on a rotary
evaporator (40OC/50 mm Hg) and the addition of 10 ml of
ether produced additional solids that Wee dried in a vacuum
~3132:~3
- 13 -
oven (Z5C/1 mm ~Ig) to give 1.3 g of a light green solid,
m.p. 193--19~C. Overall yield was 71~.
~XAMPLE 5
Preparation of
dl-6a~,7,10,10a-'retrahydro-1-hydLoxy-6,6-dimethyl-3-
p*ntYl-6H-dibenzo(b,d)pyran-9(8H)-one (V~ _
A 2-liter jacketed resin flask equipped with a
mechanical stirrer, dry-ice condenser, addition funnel and
gas inlet valve was charged with 1.5 1 of liquid MH3, and
0.15 g of Li wire (Alfa) was added, which turned the
solution blue. After 3 minutes, a solution of 32.0 g (0.102
mole) of compound IV in 500 ml of dry THF was added droewise
until the ~lue color aded. This process was repeated until
1~ a total of 2.7 g (0.39 g-atom) of Li wire had been added and
the addition of compound IV was complete (about 2 hr). The
blue solution was stirred for 15 minutes at -33C and then
quenched with 150 ml of saturated NH4Cl, added carefully
over a period of 10 minutes.
The NH3 was allowed to evaporate rapidly (over about
Z.5 hours), after which 500 ml of deionized water were
added. The pH of the solution was adjusted from 12 to 1
with the careful addition (over about 2 hr) of 800 ml of
concentrated HCl. The solution was extracted with 1.5 1 of
methylene chlo~ide in three equal portions, and the organic
phase was dried over Na2S04 and stripped on a rotary
evaporator (40C~50 mm Hg) to a yellow, oily solid.
The solid was redissolved in about 100 ml of chloroform
on a steam bath, and 500 ml of hexane were added. The
solution was concentrated in a rotary evaporator (35C/50 mm
Hg) until about 200 ml of distillate had been collected.
Crystals began to form and the mixture was allowed to stand
overnight at room temperature, after which the solids were
collected by filtration on a Buchner funnel and washed with
1.5 1 of warm (50C) hexane in three equal portions.
1~3;2~3
- 14 -
The washed solids were dried overnight in a vacuum oven
(25C/L mm ~Ig) to yield 27 g (84%) of an off-white solid,
m.p. 161-164C. Thin layer chromatographic comparison with
authentic standards in hexane:~tOAc (3:1) showed that the
solid material was primarily compound V, with some
contaminating cis isomer. Purification of the material was
cacried out by one of two methods.
In the first purification method, a high pressure liquid
chromatography (HPLC) Magnum 70 column packed with Partisil
40 silica and employing hexanes:EtOAc (4:L) as the mobile
phase was used. A 15-20 g sample was injected into the
column and 500 ml fractions were collected, with a total o~
8.5 1 of mobile ~hase applied. By this procedure, 72.1 g of
the cis/trans mixture was separated into the pure components
to yield 55.1 g (76%) of the trans compound V,
m.p. 163-165C and 10.3 g (14~) of the corresponding
c~s compound, m.p. 145-148C.
In an alternative procedure, isomerization was carried
out to isome~ize all o~ the mixture to the trans compound
V. A 500-ml flask equipped with a mechanical stirrer,
thermometer and nitrogen bubbler was charged with 11.9 g
tO.037 mole) of the above solid material, dissolved in 250
ml of methylene chloride and cooled to -10C (ice-salt
bath). The solution was charged wi~h 17,3 g ~0.129 mole) of
AlC13, which raised the internal reaction temperature to
-5C. The suspension was stirred at -5C ~ 5~C for 5
hours and then eoured in~o about 500 ml of ice water and
transferred to a separatory funnel, whsre the phases were
allowed to separate.
The methylene chloride layer was dried over anhydrous
Na~S04, concentrated on a rotary evaporator (40C/50 mm
Hg) to an off-white solid, suspended in 500 ml of hexane,
and heated to reflux on a steam ~ath for 15 minutes. The
warm suspension was filtered on a Buchner funnel, washed
13
with about 100 ml of boiling hexane, and dried in a vacuum
oven (2~C/10 mm ~Ig) overnight ~o yield 9.5 g (80%) of
white, solid compound V, m.p. 161-166.5C.
EXAMPLE 6
Preparation of
(trans-rac)-6a,7,8,9,10,10a-Hexahydro-l,9-dihydroxy-6,6-
dimethvl-3-~ntyl-6H-dibenzo(b,d2pyran-9-carbonitrile (VI)
A solution of 5 g (0.0158 mole) o~ compound V in 175 ml
Of methanol was added to a suspension of 5 g ~O.L02 mole) of
sodium cyanide in 20 ml of methanol, and the resulting
mixture was stirred at room temperature under nitrogen for 2
hours. A solution of 5.75 ml of glacial acetic acid in 50
ml of methanol was added to the mixture, and stirring was
continued for 30 ~inutes. The pH of the mixtuee was
adjusted to about 2 with anhydrous HCl, and the mixture was
stirred overnight under nitrogen.
The solvent was distilled off using a 40C water bath
~o and aspirator over a l-hour period, after which the residue
was partitioned between 75 ml of water and 100 ml of
methylene chloride. The aqueous layer was extracted with an
additional 100 m:L of methylene chloride, after which the
combi~ed organic layers were dried over anhydrous Na2S04
and evaporated in a rotary evaporator (40C/20 mm Hg) to
dryness. Fur~her drying was carried out for 0.5 hr at 0.5
mm Hg, to yield 5.5 g (100%) of compound VI as a light
yellow foam. This material was used without further
purification for the preparation of compound VII, although
an analytical sam~le was recrystallized from methylene
chloride/petroleum ether (L:10) to produce colorless
needles, m.p. 131-133C.
:~Z93~ 13
EX MPLE 7
Preparation of
(~rans-rac)-6a,7,8,9,10,10a-Hexahydro-l,9-dihydroxy-6,6-
dimethyl-3-pentyl-6H-dibenzo(b,d)pyran-9-carboxylic Acid
Methyl EsteL (VII) _
A stirred solution of 5.5 g (0.0160 mole) of compound vr
in 150 ml of methanol was treated by bubbling in anhydrous
HCl at 3C in an ice bath over a period of 1.25 hours to
satueation. The flask was capped with a septum and kept at
-20C for 48 hours, after which 75 ml of 6N aqueous HCl were
added. The mixture was evaporated in a rotary ~vaporator
t35C/20 mm Hg) and then at 0.5 mm Hg to produce an oil.
The oil was suspended in 150 ml of 50% aqueous methanol
and allowed to stand overnight at 25C. ~ copious white
precieitate formed which was collected by fi.ltration and
then dissolved in 250 ml of ethyl acetate. A small amount
of water was separated from the mixture, and the organic
layer was dried over anhydrous Na2S04 and then
evaporated in vacuo (30C/20 mm Hg) to dryness.
The residue was triturated with 50 ml of petroleum ether
(bp 30-60C). The solids were collected by filtration,
washed with 50 ml of petroleum ether in two equal portions,
and dried in vacuo (40C, 0.5 mm Hg) for 2 hours to yield
3.1 g (53%) of a colorless solid, m.~. 178-180C. The
mother liquors were evaporated to yield 1.3 g (22.5%) of a
yellow oil which NMR analysis showed to be the epimeric
hydroxy ester of compound VII. The total yield was thus 75%.
EXAMPLE 8
Peeparation of
(trans-rac)-6a,7,10,LOa-Tetrahydro-l~hydroxy-6,6-dimethyl-3-
pentyl-6~-dibenzo(b,d)pyran-9-carboxylic Acid Methyl Ester
(VIII)
A 50--ml eeaction flask equipped with a nitrogen bubbler
and a magnetic stirrer was charged ~;th 1.4 g (0.0037 mole)
~293Z~3
- 17 -
o~ compound VII, 10 ml of pyridine and 2.0 ml of thionyl
chloLide and then stir~ed at room temperature under nitrogen
for L hour. The mixture was quenched by pouring into 30 ml
of ice water and then extracted into 90 ml of ethyl acetate
in three equal portions.
The organic layer was dried over anhydrous Na2$04
and evaporated to dryness in vacuo (25C, 1 mm Hg) to yield
1.2 g of a solidified foam. This foam was triturated with
1~ 30 ml of petroleum ether (30-60C) to give 975 mg (73%) of a
light yeliow solid, m.p. 107-110C. An analytical sample
was recrystallized from ethel-hexanes (1:6) ~o give
colorless ~rystals, m.p. 139-141C.
1~ EXAMPLE 9
Preparation of
ttrans-rac)-l-(3-Aminopropoxy)-6a,7,10,10a-tetrahydro-6,6-
dimethyl-3-pentyl-6H-dibenzo(b,d)pyran-9-carboxylic Acid
Monoh~drochloride (IX)
Three hundred and fifty eight mg (0.001 mole) of
compound VIII were added to a suspension of 60 mg (0.0015
mole) of sodium hydride tAldrich, 60% in mineral oil,
prewashed with hexane) in 3.0 ml of-N,N-dimethylformamide
(DME'), and the mixture was s~irred at room temperature under
25 nitrogen for 30 minutes. Then, 560 mg (O.OOOZl mole) o
N-3-bromopropylphthalimide were added to produce a brown
solution which was stirred under nitrogen for 3 hours.
The mixture was poured into 20 ml of ice water and
extracted into 40 ml of ethyl ace~ate in two equal
portions. The organic layers were dried over anhydrous
NazSO4 and evaporated in vacuo (25C, 1 mm Hg) to yield
740 mg of a yellow oil. The oil was dissolved in 10 ml of
hexane:ethyl acetate (7:3) and puri~ied by column
chromatography in a 28 g silica gel column which had been
~repacked in hexane:ethyl aceta~e (7:3). 200 ml fractions
weee collected, and those containing the desired product as
~L2932~i3
shown by TLC on silica gel were evaporated in _acuo (Z5C, 1
mm ~Ig) to give 322 mg (57.9%) of a colorless foam.
All of the foam was dissolved in 10 ml of 15%
methylamine in methanol, stirred under nitrogen at room
temperature for l hour and then evaporated in vacuo
~25C, l mm Hg) to dryness. The residue was taken up in a
mixture of lO ml of 2N MaOH and lO ml of methanol and
stirred under nitrogen overnight. The mixture was then
evaporated in vacuo (25C, 50 mm Hg) to near dryness,
acidified with 6N HCl to pH 2, and extracted into 20 ml of
ethyl acetate in two equal portions. The organic layers
were dried over anhydrous NazSO4 and evaporated to
dryness in vacuo (25C, l mm Hg), and the residue was
triturated with ether to yield 90 mg (35%) of a colorless
solid, m.p. 255-257C.
EXAMPLE lO
Preparation of
(trans-rac)-9-HydLoxy-6a~7~8~9~lo~loa-hexahydro-l-(4-
nitrobenzyl)-6,6-dimethyl-3-pentyl-6~I-dibenzo(b,d)pyran-9-
carboxvlic Acid Meth 1 Ester ~X~
Y ~ , .
A solution of 3.80 g (0.01 mole) of compound VII (see
Example 8) in 150 ml of acetone was stirred under argon in a
500 ml three-neck flask, while 2.25 g (0.0~ mole) of
p-nitrobenzylbromide were added. Seven grams (0.05 mole) of
finely pulverized anhydrous K2C03 were added, and the
resulting suspension was stirred rapidly under argon for 18
hours at Z5C. Analysis by thin layer chromatography (TLC)
on silica gel plates in hexane-ethyl acetate (7:3) with lO~
CeS04/H2SO~, in which the resul~s were visualized by a
10% phosphomolybdic acid/ethanol spray followed by heating,
showed that ~he reaction was complete.
The mixture was filtered, and the residue was evaporated
in vacuo to afford a yellow oil. This oil was dissolved in
5 ml of ether and poured over a 30 g plug of silica gel
.
~L293Z13
-- 19 --
(70-230 mesh, E. Merck) in a sintered glass funnel. The
plug was then eluted with 400 ml of ether, and the solvent
was collected and evaporated to dryness on a rotary
evaporator at 30C.
Crystallization of the residue from ether-hexane
(1:5) afforded 4.7 g of a fluffy white solid, m.p. 120-123C.
~XAMPLE 11
Preparation of
(trans-rac)-6a,7,10,L0a-Tetrahydro-1-(4-nltrobutyl-6,6-
dimethyl-3-pentyl-6H-dibenzo(b,d)pyran-9-carboxylic Acid
Methyl Ester (XI)
A mixture containing 2.5 g t0.00488 mole) of compound X,
15 ml of pyridine and 7.75g (0.05075 mole) of POC13 was
stirred at room temperature argon for 2 hours. The mixture
was then poured into 100 ml of crushed ice and extracted
with two 100-ml volumes of CH2C12. The organic layer
was dried over Na2S04, filtered and evaporated to
dryness. The residue was taken up in 30 ml of CH2C12
and filtered through a pluy of 20 g of silica gel (70-230
mesh). The plug was eluted with 100 ml of 1:1 hexane-ether,
and the eluant was e~aporated to dryness on a rotary
evaeorator at 30C.
Z5
The residue, on trituration from 50 ml oE peteoleum
ether, afforded in two crops 1.65 g (68%) of colorless
prisms, m.p. 165-167C.
~XAMPLE 12
Preparation of
(trans-rac)-6a,7,10,10a-Tetrahydro~ 4-aminobenzyl)-6,6-
dimethyl-3-pentyl-6H-dibenzo(b,d)pyran-9-carboxylic Acid
Methyl Ester ~XII) _
A mixture containing 1.65 g (0.003 mole) of compound XI,
50 ml of C~zCl2, 100 ml of ethanol and 3.4 g of 85%
hydrazine hydrate (Aldrich) was Lapidly stirred as a
slurry. Five hundred milligrams of technical grade Raney
~2~3Z13
- 20 -
Nickel (pre-rinsed with three 5-ml volumes of ethanol) in
ethanol were then added in one portion. The mixture was
~apidly sticLed at room temeerature for 1 hour, after which
the catalys~ was removed by filtration over a pad of
Celite. The pad was rinsed with three 25-ml volumes each of
CH~C12 and ethanol, and the combined filtrates were
evapoLated on a rotary evaporator (40C, 5 mm Hg).
The residue was taken up in 25 ml of CHzCl2, passed
10 through a plug of 25 g of silica gel (70-230 mesh, ~. -
Merck), and eluted with 300 ml of hexane-ethyl acetate
(1:1). The combined eluant was evaporated on a rotary
evaporator (40C, Smm Hg) to afford 1.5 g of a yellow
semi-solid. This crude product was repurified as described
above and then crystallized from hexane-ether. A yield of
956 mg (62%) of a light yellow solid, m.p. 112-114C, was
obtained.
EXAMPLE 13
Pre~aration Of
(trans-rac)-6a~7,'10,10a-Tetrahydro-l-(q-aminobenzyl)-6,6-
dimethyl-3-pentyl-6H-dibenzo(b,d)-pyran-9-carboxylic Acid
A mixtuee containing 110 mg (0.22 mmole) of compound
XII, 10 ml of 2N NaO~I and 25 ml of ethanol was heated at
reflux under argon for 1 hour. This mixture was evaporated
on a rotary evaporator (30C, 0.5 mm Hg) to remove most of
the ethanol, bringing the volume to about 15 ml. The pH was
adjusted to 2 with 6N HCl, and the mixture was extracted
with two 30 ml volumes of CH2C12.
The organic layer was dried over Na2S04 and then
evaporated (30C, 10 mm Hg) to yield 105 mg o~ a brown
foam. ~'he amorphous amino acid could not be crystallized,
but analyses by 400 MH NMR, IR and mass spectroscopy were
consistent with the expected structure and showed that the
product was essentially homogeneous.
~293;~:~3
- 21 -
EXAMPLE 14
PeeParation of_~nti-THC Antiserum
one hundred mg of a cannabinol derivative having ~he
formula
~ C0
i
\/\~\,
\l i l! . -
15were dissolved in 20 ml of 30% methanol at pH 4.5. To this
solution, 8~ mg of 1-(3-dimethylaminoproQyl)-3--
ethylcarbodiimide hydrochloride were added and the p~ was
adjusted to 4.5. ~fter mixing at room temperature for 30
minutes, 100 mg of methylated bovine serum albumin were
added in 20 ml of water with mixing, and the ~H was again
adjusted to 4.5. 'rhe solution was mixed for 1 hour at room
temperature and then overnight at ~C, after which any
unconjugated cannabinol derivative was removed by dialysis
against 1000-ml volume changes of sodium phosphate buf~er at
4C, to produce a THC-methylated albumin conjugate.
one hundred mg of lipopolysaccharide (LPS; microbial
source) in 5 ml of water weEe combined with 1 ml of 1.0 M
WaI04 and treated for 2 hours a~ room temperature in the
dark. One ml of ethylene glycol was added, and the-mixture
was stirred foe an additional 30 minutes at room
temperature. The activated lipopolysaccharide was applied
to a Sephadex G-25 column equilibrated with water, and a
turbid effluen~ fraction was collected.
~Z93213
- 22 -
P~ive ml of tetLahydrofuran containing 38.~/5 mg of a
cannabinol derivati~e having the formula
/ \o/ \o/ \NH
. ,, ~ OH
~ wece added to the
acti~ated-LPS effluent, and the mixture was incubated for L8
hours at 22C in the dark. Two ml of a solution of 4.5
mg/ml NaBH4 in HzO were added, and the mixture was
incubated for 30 minutes at room temperature. Unconjugated
THC derivative was removed by dialysis for 2 days against
frequent changes of water and for 2 days against 0.l M
acetic acid, pH 3.5, to yield a l'HC-LYS conjugate.
One hundred mg of human IgG were dissolved in 7 ml of
0.2 M NaHC03, pH 9.6, and 108 mg of a cannabinol
derivative having the formula
~
O ~ in 2 ml of dioxane
were added. Nine additional ml of dioxane were added, and
the mixture was m~xed intermittently for 4 hours at 4OC,
after which l~0 ml of cold acetone were added. The mixture
was centrifuged for l0 minutes at 2,000 x g, and the pellet
~3Z13
- ~3
was washed once with 20 ml of cold acetone. Following
centrifugation for lO minutes at 2,000 x g, the pellet was
resuspended in lO ml of phosehate buffered saline (PBS).
Dialysis was caeried out for 3 days at 4~C against 2 changes
per day of lO00-ml volumes of PBS, to produce a THC-human
lg~ conjugate.
New Zealand white rabbits were injected intramuscularly
at the four axillary regions with 200 {g of T~IC in the
form of one of the above conjugates in l ml of ~reund's
complete adjuvant. Booster injections containing lO0 {g
of the same conjugates were given after 2 weeks in l ml of
Freund's incomplete ad~uvant, and antiserum was harvested
feom the ear veins after 4 weeks.
~cceptable anti-THC antisera from the three rabbits were
pooled for use as described below.
~XAMPL~ 15
Purification of Anti-THC IqG
Ten milliters of a saturated ammonium sulfate solution
were slowly added droewise to 20 ml of anti-THC antiserum,
with continual stirring at 23C. InitiaLly, a white
precipitate formed which was allowed to redissolve prior to
further ammonium sulfate addition. After about 6 ml of the
ammonium sulfate solution had been added, the precipitate
persisted, and from that poin~, the slow dropwise addition
has continued without pause. Stirring of the suspension was
continued for Z hours at room temperature, after the last of
the ammonium sulfate solution had been added, after which
the mixture was centrifuged at l,500 x g for 30 minutes at
room temperature. This procedure puts most of the IgG in
the pellet.
Aftec centeifugation, the supeLnatant fluid ~as
decanted, and the pellet was dissolved by the addition of
-~Z93;~i3
- 2~ -
T~IS/saline (composition) to a final volume of Z0 ml. The
LgG in the solution was reerecipitated by repetition of the
above ammonium sulfate addition and cent~ifugation steps,
after which the supernatant fluid was again decanted. The
pellet was dissolved in lO ml of 0.02 M Tris/saline, eH 7.5,
with 0.01~ thimerosal (sodium ethylmercurithiosalicylate).
To remove the ammonium sulfate from the IgG-containing
solution, dialysis was carried out overnight at 4C against
three 2~ er ~olumes oE 0.02 M Tris/saline. The material
in the dialysis bag was then brought to a final volume of 20
ml by the addition of 0.02 M Tris/saline. This solution was
centrifuged at 2,000 x g for 30 minutes at 4C, and a small
pellet which was formed was discarded. Protein concentration
in the final IgG-containing solution was then determined by
spectrophotometric analysis at 280 nm to be about 5.3 mg/ml.
EXAMPLE 16
PreParation of Anti-THC Beads
~ 4-litee vacuum 1ask was filled to the ~,500 ml mark
with l/4" frosted polystyrene beads (Clifton Plastics, Inc.,
Clifton Heights, PA), and 2,500 ml oE 95% ethanol were added
with s~irring. The ~lask was placed under vacuum
(l mm Hg) or 30 minutes a~ room temperature, and the
ethanol was decanted. The ethanol addition, vacuum
treatment and decanting steps were repeated twice more,
after which the beads were suspended in 3 l of deionized
water. The water was decanted, and the beads were twice
more washed in this fashion with 3-liter portions of
deionized water. Two liters of deionized water were then
added to the decanted beads, and the mixture was allowed to
stand overnight at Loom temperature. Throughout the washing
erocedure, it is important that the beads are never allowed
to dry out.
3;~i3
- 25 -
l~o coat the washed beads with antibodies against THC,
the anti-'~HC IgG solution described above was d-iluted to a
final protein concentration of Z5 ug/ml with 50 mM sodium
borate buffer, pH 8.0 (coating solution)~ The beads were
coated by adding 143 ml (enough to cover the beads) of the
coating solution to a measured volume of 250 ml of the
settled beads (about 1,000 beads) from which the water had
been decanted, and then letting the mixture stand for 18-24
hours at 18-26C.
Following the coating procedure, the coating solution
was decanted and discarded, and the beads were washed three
times at 4C with 500 ml volumes of a washing buffer
containing 0.01 M sodium phosphate in 0.15 M saline with
0.01~ thimerosal, pH 7.2. Each time, the beads were
thoroughly suspended in the washing solution by stirring,
and the solution was decanted after the beads had been
allowed to settle. Throughout this procedure, care was
taken to ensure that the beads did not dry out.
Next, the beads were treated to block unsaturated sites,
using one of two procedures. Throughout the procedures that
follow, the beads were handled only with plastic or glass
utensils.
~ 250 ml settled volume (about 1,000 beads) of the
washed anti-THC coated beads above was treated after
decanting with about 143 ml (enough to cover the beads) of a
45C blocking buffer containing 100 mM sodium phosphate with
1.0~ bovine serum albumin (BS~) and 0.05% thimerosal, pH
6.8, for 18-24 hours. Following the blocking buffer
~rea~men~, the beads were washed three times with 500 ml
portions of fresh blocking buffer at 4C. Each time, the
beads were suspended in the buffer with stirring and then
allowed to settle, and the huffer was decanted. For
storage, a quantity of the blocking buffer sufficient to
cover the beads was added, and the beads were kept at 4C
3;~:13
- 26 -
until needed.
Of course, the above procedures can be scaled ue as
desired, as lony as the relative proportions of beads and
solutions were maintained.
EXAMPLE 17
Preparation o THC Derivative-PQroxidase Coniuaates
Te~ milligrams of horseradish peroxidase rBoehringer
Mannheim or Seravac (Pel Freeze)] were activated for
conjugation by dissolving in 3 ml of deionized water,
stirring for l hour in the dark room at room temperature.
one hal~ ml of O.L M sodium metaperioda~e (sodium periodate)
was slowly added to the above solution over 20 minutes with
stirring in the dar~ room at room temperature. The sodium
periodate solution was prepared fresh immediately before
use, and the acti~ation reaction was stopped by adding 0.5
ml o 0.5 M ethylene glycol, chilled to 4-8C.
The mixture was immediately placed in dialysis tubing
(Spectrapor, molecular weight cutoff 12,000-14,000) and
d~alyzed overnight at 4C against ~hree l-liter changes of
0.01 M sodium acetate buffer, pEI 5.5. Following dialysis,
the material in the baq tvolume 4.0 ml) was placed in a lO
ml amber Wheaton vial.
For the conjuga~ion reaction, 4 ml of a 1:1 mixture of
ethanol:O.4 M sodium ace~ate bufferf pH 5.5 containing about
O.S6 mg of (trans-rac)~l-(3-aminopropoxy)-6a,7,10,LOa-
tetrahydro-6,6-dimethyl-3-pentyl-6H-dibenzo (~,d)
pyran-9-carboxylic acid monohydrochloride ~compound lX, see
E'ig. 1 and Example 9, su~ra~ were added dropwise with
stirring to the activated peroxidase solution and allowed to
Leact for 2 hours at room temperature in the dark.
32~
- 27 -
To stabilize the Schif~'s bases that had formed, l ml of
0.l M sodium cyanoborohydride was added to the peroxidase
conjugation mixture, and the mixture was stirred for 2 hours
at room temperature in the dark.
The stabiliæed conjugation reaction mixture was then
divided into four equal portions and ap~lied to four
Pharmacia PD-l0 columns which had previously been
equilibrated with buffer containing 0.2 M sodium phosphate,
pH 6.8 (chromatography buffer). The applied eortions were
run into the tops of the column beds and overlaid with 4 ml
of the chromatography buffer.
A volume o~ column eluate of about 12.4 ml was collected
from each column. Spectrophotometric analysis at 403 nm
showed that the recovery of peroxidase in conjugated form
was about 66%. taking the extinction coefficient to be 2.~.
Five millili.ters of the conjugate eluant solution were
diluted with 0.2 M sodium phosphate buf~er, pH 6~8 (diluted
THC-peeoxidase conjugate reagent), for use as described
below.
EXAMPLE l8
25THC-EnzYme ImmunoassaY Protocol
A series of ll x 75 mm polystryene test tubes was set up
containing duplica~e 50 ul aliquots of urine s~ecimens to be
analyzed or normal control or THC standard-containing urine
samples. The standard and noLmal control aliquots were
taken from normal urine samples containing 0, 50, l00, Z00
or S00 ng/ml added 9-carboxy-Ll-nor-~ -tetrahydro-
cannabinol (L00 ~g/ml in absolute ethanol, Research
Triangle Institute, Research Triangle Park, NC)o
Two-hundred-~ifty microliteLs of the diluted THC-~eroY~idase
conjugate reagent were added to each tube, and the tubes
were shaken. One anti-THC bead was then added to each tube,
;3LZS~3Z13
the contents of the tubes were mixed and the tubes were
incubated fo~ 30 minutes at room tempecature.
After the incubation, the beads were washed three times
by adding 3 ml aliquots of deionized water with shaking and
then aspirating the water. Two-hundred-fifty microliters of
a chromogen reagent containing 7.2 mg/ml o-phenylenediamine
dihydrochloride and 6 mM H202 in O.l M potassium citrate
buffer, pH 5.25, with O.l ml/liter of solution Kathon
CG (an isothiazolin solution from Rohm and Haas) added
as a preservative to each washed bead, and the tubes were
incubated for lO minutes at room temperature in the dark.
The reactions were then stopped by the addition of l ml of l
N sulfuric acid to each tube, and the absorbance of the
sample solutions was measured at 492 nm.
A plot of absorbance at 492 nm versus 9-carboxy~
nor-~ -tetrahydrocannabinol concentration for the
standards employed is shown in E'ig. 3.
EXAMPLE l9
C nical Sam~le AnalYsis
To establish the ~ensitivity and accuracy of the
THC-enzyme immunoassay of the invention, a series of 60
clinical urine samples was evaluated using the assay (EIA)
and ~he Roche THC radioimmunoassay ~IA) system, with the
reslllts shown in Table l.
3~3
- 29 -
Table 1
Comparison of EIA and RIA THC Analyses
THC ~na/ml) bY THC (nq/ml) by
Sample a b a b
No. EIA RIA _ Sample ~o.EI~ RIA
1 300 >2~0 31 >500 >2~0
2 ~500 >200 32 >500 >200
3 >500 >200 33 >500 >200
4 217 ~20Q 34 300 >200
10 5 3.5 0.0 35 >500 >200
6 >~00 >200 36 >500 >200
7 >500 >200 37 7.0 0.O
8 >500 >200 38 >500 >200
9 >500 >200 39 190 200
120 125 40 250 >200
11 5.0 0.0 41 10.0 0.0
1512 94 141 42 467 >200
L3 189 >200 43 194 106
14 47 50 44 465 >200
~500 >200 45 16.5 0.0
16 ~54 >200 46 >500 >200
17 217 190 47 >500 >200
18 110 86 48 >500 >200
19 180 >200 49 338 176
2020 500 ~200 50 >500 >200
21 ~500 >200 51 500 >200
~2 90 157 52 16.5 0.0
23 427 ~200 53 17.0 0.0
24 13 0.0 54 ~500 >20~
>500 >200 55 ~93 ~95
26 >500 >200 56 467 lg8
2527 >500 >200 57 340 158
28 ~66 ~200 58 >500 >200
2~ 335 >200 59 ~500 >200
8.0 0.0 60 13.5 0.0
_
EIA represents the THC-enæyme immunoassay of ~he
inventlon,
RIA represents the commercial THC radioimmunoassay
system of Roche Diagnostic Systems, a division of
Hoffmann-La Roche Inc., ~utley, New Jersey.
The threshold for a positive test in Table 1 was taken
to be 50 ng/ml 9-carboxy--11-nor-a -te~rahydro-
3;2~3
- 30 -
cannabinol in the EIA, and 100 ng/ml in the RIA. Based upon
these criteria for a positive test, only sample 18 showed a
discrepancy between the results obtained from the two
analytical methods. This samele was positive by EIA but
negati~e by RlA. Sample -18 was Letested by both methods and
found to be positive in the retests by bo~h RIA and EIA.
As a further check on the specificity of the THC-enzyme
immunoassay, a number of drugs not related to the
cannabinoids were added to pooled normal human urine samples
at a concentration of 10,000 ng/ml and tested by the
method. None of these compounds, which are listed in Table
2, were positive in the assay.
T~BL~ 2
Drugs TeYted ~or Cross-RPactivity
_
~cetaminophen Guaiacol glycerol ether
5 ~cetylsalicyclic acid Hydrochlorothiazide
~minopyrine Hydrocodone bitartrate
~mttryptyline Hydromorphone hydrochloride
~mobarbital - p-Hydroxyphenobarbital monohydrate
~mphetamine Imipramine
~mpicillin Lidocaine
~pomorphine Melanin
~scorbic acid Meperidine
10 ~tropine Methadone
Benzocaine d-Methamphetamine hydrochloride
Benzoylecgonine (cocaine metabolite)Methaqualone
Butabarbital Methyprylon
Caffelne Morphine
Calcium hypochlorite Naproxen
Chlordiazepoxide Niacinamide
Chloropheniramine Norethindrone
15 Chloroquine Oxazepam
Chlorpromazine Oxycodone hydrochloride
Cocaine hydrochloride Penicillin G
Codeine Pentobarbital
Cyclopentobarbital Phencyclidine
Dextromethorphan Phenobarbital
Dextropropoxyphene HCl Phenothi~zine
20 Diallylbarbituric acid Phenylbutazone
Diazepam Phenylpronanolamine
Dihydrocodeine bitartrate Procaine hydrochloride
5.6 Dihydroxyindole Promethazine
Diphenylhydantoin Propoxypherle
Ecgonine hydrochloride Quinine hydrochloride
Epinephrine Sodium secobarbital
25 Erythromycin Tetracycline
5striol Tetrahydrozoline
Gentisic acid Tr1fluoperazine
Glute~himide Zomepirac
Many modifications and variations of this invention may
be made without departing from its spirit and scope, as will
become apparent to those ~killed in ~he art. The specific
embodiments described herein are offered by way of example
only, and the invention is limited only by the terms of the
appended clalms.