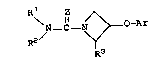Note: Descriptions are shown in the official language in which they were submitted.
~2~'~274 66l97-l77
MEI'HOD OF TREATING MUSCLE TENSION
MUSCLE SPASTICITY AND ANXIETY WITH
3-ARYL-OXYAZETIDINECARBOXAMIDES _
BACKGROUND OF THE INVENTION
1. Field of the Invention.
This invention rela-tes to a method of treating muscle
tension, muscle spasticity and anxiety in living animals,
including humans, with 3-aryloxyazetidinecarboxamides, certain of
which are novel.
2. Information Disclosure Statement
Certain o the compounds useful in the method of the
present invention are disclosed in U.S. Patent No. 4,226,861 as
having anticonvulsant activity and use in treating epilepsy having
the formula: O ~ R
R-NHC-N ~ O - ~ ?
wherein R is loweralkyl and Rl is hydrogen, aminocarbonyl or
trifluoromethyl.
Certain other of the compounds useful in the p.resent
invention are dlsclosed European Patent Application ~o.~102,194 A
publlshed March 7, 1984, as having anti-convulsant activity and
having the formula: 1
(R )n
2-C-~ 3 - ~
.
:'s'~
, ..
7~
- 2 - 66197-177
wherein Rl = H, F, loweralkyl, loweralkoxy, trifluoromethyl,
acetyl, or aminocarbonyl.
In European patent publication, it is stated that the
compounds of U.S. Patent 4,226,861, mentioned above, have muscle
relaxant property and based on another less reliable test states
that the compounds in the European patent publication do not have
muscle relaxant activity at an effective anticonvulsant dosage.
SUMMARY OF THE INVENTIO~
The 3-aryloxyazetidinecarboxamides useful in the method
of treating muscle tension, muscle spasticity and/or anxiety in
animals of this invention have the formula:
R z
N- C ~ Ar
2/ Y Formula I
R3
wherein Ar is selected from pyridyl in any of its positions
including pyridyl or halo substituted pyridyl, from phenyl or
phenyl substituted by 1 or 2 radicals selected from chloro, bromo,
iodo, fluoro, loweralkyl, loweralkoxy, nitro, aminocarbonyl,or
trifluoromethyl;
Z is oxygen or sulfur;
Rl and R2 are selected from hydrogen, loweralkyl, aryl,
allyl, substituted allyl, propargyl, cycloalkyl,
loweralkylcycloalkyl, cycloalkylloweralkyl, arylloweralkyl and
diloweralkylaminoloweralkyl, and Rl and R2 when taken together
with the adjacent nitrogen atom may form a heterocyclic amine
radical including azetidinyl, pyrrolidinyl~ piperidinyl,
.~.,~.
:~2~ 7~
homc~piperi~linyl; imidazolyl, piperazinyl, 4-substituted
pip~razinyl and mc>rpholir~yl;
R9 i~ ~elect~d from hydrogen, loweralkyl, aryl or
zlrylloweralkyl:
the geometrical i~omers including ci~, trans, (E)
~nd (Z) i~omer~ thereof, and the pharmaceutically acceptable
~alts thereof when Rl and/or R~ ~ave a ~alt-~orming basic
amino componen~ or when P~r is pyridyl.
The compounds of Formula I are novel except wherein Ar
is phenyl or phenyl ~ubsti'cut~d by trifluorome~hyl or
aminoc3rb~nyl ~t ~he ~;ame time Z i~ oxygen, R~ is hydrogen,
and Rl ænd R2 are a c:oTr~tination of hydrogen ~nd loweralkyl
and wher~in Ar is phenyl or phenyl sub~tituted by i~luoro,
loweralkyl, lowerallcoxy, tri~luoromethyl, acetyl or amino-
c~rbollyl ~t the same time 2 i5 oxygen ~nd Rl, R2 and R3
are hydrogen.
In the ~urther de~Einition of 8ynlbO18 ~n~ in the
~ormulas hereo~ and wher2 they appear elsewhere throughout
thi~ sp~ciication and in the claims, the terms havo the
follow~ng ~ignificance.
~h~ term "low~rallcyl" as u~ed herein, unla~s otherwise
sp~ci~i~d, includes straighlt and branched chain radicals
o~ up to eight carbons inclu6ive and i8 e~emplified by such
- grOUp8 rl8 me~thyl, ethyl, propyl, i~opropyl, butyl, ~ec-butyl,
25 t~rt-butyl, amyl, i~o~myl, hexyl, heptyl, and ~ctyl r~dis:al~
~nd ~ lik~. ThE term "loweralkoxy" has the ~ormula
-O-lower~lkyl .
The term "cycloalkyl" el~ u~d herein includes pri2narily
~:y~lic alkyl radical~ c~n~aining 3-9 car~on atoms inclu~ive
30 and ~ncludes ~uch group~ ac c:yclopropyl, cyclobu tyl, cyclo-
pentyl, cyclo~exyl, ~yclohep~yl ana th~ 1 ilce .
The term "loweralkylcycloalkyl" re~r~ to cyc:lc~alkyl
~ub~titute~d by alkyl ra~icals o~ 8 c:arbon length.
The tenn " cyclo~lkylloweralkyl" ref~rs tD il~ cyl::loalkyl
35 ra~ al lir~ced Vi21 1-8 caEbon ~lkyl ~h~ins, inc~luding
branl:he~ chains, ~o the ~mide ni~rogen.
~ he term "substituted ~llyl" mean~ 211yl ~u~itut~d
by ~lkyl radi~ in ~ny onle of it~ 3 po~i~ion~.
452
~ Z 9 ~
The term "aryl" under the definition o~ Rl, R2 and R3
re~rs ~o phenyl or phenyl ~ub~tituted by non-interferiny
r~dical~ such a~ halo, loweralkyl, loweralkoxy, nitroj cyano,
tri~luorome~hyl, carbomethoxy, carbo~thoxy, an~ the like.
The term l'arylloweralkyl" undex the d~inition ~f Rl,
R2 and R9 refers to ~n aryl group as defined ~bsve linked
via 1-8 carbon alkyl ~hainY, including brAnched ~hains, to
the amide nitrogen.
Th~ term '14-~ub titu~d-pipera~inyl" under the
de~inition o~ Rl and R2 refers t~ a piperazine radi~l
~ub~ituted by usual radical~ common in the art, including
aryl as de$ine~ above and loweralkyl as defin~d above.
As de~ined ~b~ve under Formula I, pharmaceu~ically
acceptable salt~q are included when Rl an~/or R2 have a salt-
15 $orming ~mino ~omponent or when Ar ~8 pyridyl. Salt~formingamino componQnt~ ~re prese~t, for axample, when Rl and/or
R2 are diloweralkylaminoloweralkyl, or when togather they
form piperazinyl or imidazololyl radicals. Pharmaceutically
accoptable aci~ addition ~alts are those salts which are
20 phy~iologically compatible in warm-bloode~l animal~. The
acid addition 9alt8 may be formed by either ~trong or weak
acid~. Repre~entative of ~trong acid~ are hydrochloric,
hydrobromic, ~uliEuric, and pho~phoric acid~. Repre~entativa
o:E U8efUl we~k acid~ nre ~umaric, mal~ic, succinic, oxalic,
25 citricl tartaric, h~xamic and the like.
A~ stat~d ~bove, the metho~ of ~his inv~n~ion employs
the Gompound~ of Foranula I to rel i~ve muscle tenq ion and
~pas~icity (i.e." to relax ~mscle~) ~nd/or ~nxi~ty in
an~mals, including human~ The ~roce~ure for tQ~ting
~0 compound~ for mu3~1e relaxant activity is ~he ~orphin~-
Indu~ed ~tr~ub-Tail-In-Mic~ ~st an~ the procedure for
indi~ating antianxiety re3pons~ i~ th~ Voyel Conflic~ T~t
ba~ed on ~hock ~uppr~2d ~rinking ~havi~r. ~th t~t~
~r~ giv~n in detail h~reinbelow und~r ~I~.h~rmacologic~
3~ ~&t Pr~c~dur~s."
~ he m~tho~ utilizing ~n~ or m~re o~ th~ ~mpoun~s o~
Formul~ I i8 a~apt~bl~ ~or tr~t~ng t~-ed ~nd ~pastàc
~2~4~7~
muscles and ~or treating anxiety in the same individual or
~r treating tensed and spa~tic muscles when anxiety i8 not
present or for treating anxiety when there are no muscle
pro~lems. Examples o~ effective muscle treatment would be
for muscular conditions arising from tetanus, whiplash,
stroXe, multiple schlerosis, cerebral palsy and the like.
Compounds of Formula I wherein Rl and R2 h~ve an
allyl, substi~uted allyl, propargyl or cycloalkyl value are
pre~erred ~or their potency as muscle r~laxants.
DETAILED DESCRIPTIO~ OF THE INVENTION
The methods used to prepare the muscle relaxant~ and
antianxiety agents of Fonmula I which are used in khe
method ~ this inv~ntion are cla~si~ied as ~ollows by
equation under Method Classes A, B, and C.
-- 6 --
Method Class A
Frt~m Phenoxyaze t id ine
El-N~ 0~ R2N=C=Z - R2 -NHC-N~ ~X) n
~~~~ + R P~ ~-C-Cl ~RlR2~-r-~ ~X)n
R3 R3
H-N~O~+ H2N-c-NHNo2 --> H2N C ~ (X)n
~ -C=Z
e~ ReNH2 ~ (~)-C=Z ~ H-N~O~ Xlr ( ~n
9 ~-NHC-~0~
R3
CH~N-(CH3) -~H3 + (~)-C~Z + H~ /7
x = o-8 : R3
~3 ~ ~ (CE~-(cH23-NH
R3
:: :
:
:
452
~Z~ 7
Method Class B
Via Aryloxy-l-ehlorocarbonylazetidine
(Ar=Phenyl, Substituted-phenyl, Pyridyl)
c~ N~O-Ar + R2NH2 ~ > R2HN-C-N~>_O-Ar
~9 ~ R9~
Cl-C-N ~ r + HN ~ CH2)n >(~ C-N ~ O-Ar
R~ R3_
Cl-C-N ~ O-~r ~ EN ~ R~ RB-~ N-C-~ ~ O-Ar
R9 R9
Cl_c_K?_O~r + a~ C--K?~O~r
20 Method Clas~ C
Via Methane (or Phenyl) suli~onyl azetidine
H-N~OSoæw R2-N=C=Z ~ R2NH-C-N~OSOz-W
2 5 R3 R3
W = CH3, phenyl, X
4-CH9-phen~ F Na~
: ~ X
~ z ~_
- : RS
: 35
: ~ :
: ~ ,
~52
Method Class A reactions comprise a process for
preparing phenoxy compounds of Formula I by reacting a
compound of the formula:
~-N ~ ~ ~X)n
with one of the following cl~sses of compou~ds:
a) R2N = C = Z
b) RlR2N_C_cl
c) H2N-C-NHN02
( ~ ~)2 N
or, e) and f) the product of ( ~ ~ -C=Z
and R2NH2 or (CH4 ~ N-(CH2)X-NH2
~ x 8 0 8
Method Class B reactions comprise a process for
preparing certain aryloxy compounds of Formula I by
reacting a compound of the formula:
Cl-C-~ ~ O-Ar
\~
: R3
with one of the ~ollowing classes o~ compoundso
a) R2NH2
: or3 b) a heterocyclic amino compound.
Method Class C reactions comprise a process ~or
preparing m-fluorophen~xy ~omp~unds of Formula I by
reacting a compound of the formula:
~5 , ~
H-~ ~ 0502W
~9 W=C~, C~H5, 4-CH3-C~H~
452
~ILZ9~Z~
g
with an isocyanate of the formula
R2 -~=C=Z
to give a compound of the formula
Z ~
R2NH-C_N ~ SO2W
and thereafter reacting with s~dium hydride and a meta-
fluoroph~ol of the f~rmula
X
oE~J~ F
to give a compound of the formula
R2NH-C-N ~ O
R3 F
Methods of preparing the starting phenoxyazetidines
used in Method class A are outlined by eauation in Chart I.
Methods of preparing the starting aryloxy-l-carbonylazetidines
used in Method Class B are outlined by equation in Chart II.
The ~tarting l-(diphenylmethyl)~3-hydroxyazetidines
used in Method class C route (See Chart III3 may be
prepared by the method of Anderson & Lok., J. ORG. CHEM.
~1972) ~7: 3953 from benzhydrylamine and an appropriate
epihaloh:ydrin. Cis and trans:isomers of the l-(diphenyl-
methyl~)-3-hydroxyazetidines when they exist may be separated
by chromatography. Starting ~-methylbenzyl-3-~zetidinol
i~ prepared by the method o~ Tetsuya Okutani, et al., CHEM.
PH~RM. BULL., Vol 22 (1974) p. 1490. Preparations 1-6
: ~5 illustrata the methods.
,
452
7 ~
- 10 -
Chart I
Preparation of Starting
l~Un3ub~tituted ph noxvaze~i~Lne~
R4* ~
~CH-N ~ OH (See Preparation~ 2 and 5)
CBH5 ~R3 l) CH3SO2Cl~Et3
\ 2) ~aOH, with or without
(X)n ~ X)n
¦DMF ati~n8 3 and 6)~ 4ee also U,S. Paten~
R~j ~ ~ X)n
C~H5 . 9 or H~ ~ Pa(0~)2/C ~ ~
(See Preparation 4) R3 ~****
*R4 = CH3: phenyl or subst. phenyl.
**X is restricted to substituents that are electron
withdrawings e.g.~ fluoro, chloro, bromo, iodo
or CF~.
~**When X = 4-cl, 4-Br, 4-I, 4-~ or 4-CF~, yields are
very low.
***~Exc~pt X cannot be chloro, bromo, or io~o in.the
produ~t a~ hydrogenolysis removes these radicals.
: : ~
452
4~7~
Chart II
Pxeparation of Starting
l-carbonyl-3-~rvlox~ azetidines
R4
CH-N ~ OH
C /Hs ~3 (See Preparation 1 for
R3 = CH~)
halo
\~halo
\ NaH
~DMF
C~s ~ ~( ) / ~-1;~
(See Preparation ~ ~
in Chart 1) . I c-c12
Z ~ ~ (Z=O or S)
Cl-C-N ~ OAr
\(
: R3
:
:
:
452
27
Chart I II
Preparat ion of Start ing
Methane~ ~or phenyl)- ~;ul:EonYlazetidine
R4-CH_N~oH WS02Cl ~ R~-CH-N~OSO2W
C~Hs - NEt3 Ce~Hs 3
toluene
R~ = CH~, Cs~s~ W = CH3, C~3Hs P~ .
-C3H4 -4CH3 Pd/~
~ ,
H-N/~_OS02 W
_
:
:
:
~ ~ ~ 4 2 7 ~ 452
- 13 -
Preparation 1
trans-1-(Diphenylmethyl~-2-m~thyl-3-azetidinol
oxalate r 1 11.
~ ~ .
A mixture of 126.4 g (0.72 mole) of diphenylmethylamin~
and 100 g (o.66 mole) of 3-bromo-1,2-ep~xybutane in 300 ml
o~ methanol was stirred while being protected from light
for 96 hr, then heated at reflux for ~0 hr as the color
changed from pale yellow to deep amber. A sample was assayed
by lH-nmr and showed 3 methyl doublets. A fine beige
precipitate was removed by filtration (diphenylmethylamine
hydrobromide) and the filtrate concentrated on a rotary
evap~rator to yield 174.6 g of crude oil. A 1.5 sample was
neutralized and placed on a 4 mm ~hick plate of a
Chromatotron~ and ~luted with 10~ ethyl acetate-toluene.
A total of sixteen fractions were collected which consisted
f 6 distinct spots by TLC The major component separated
was 700 mg and appeared to be the trans isomer. This
sample was converted to the oxalate salt. The main concen-
trate was converted to the free ba~e with ammonium
hydroxide and extracted into toluene which was dried over
magnesium ~ulfate a~d concentrated. The reaction residue
was dissolved in methanol and treated with 58 g o~ oxalic
acid, heated to give a homogenous solution and allowed to
cool after seeding with a sample of the ~rans oxalate
salt. Filtration yielded 62 g of white granular product,
m.p. 147-148.5C. A second ~rop of 26 g was also obtained.
The lH-nmr ~pectrum showed only a ~ingle CH9 doublet with j
(CH3-H) o~ 6.1 Hz which is consistent with the trans
compound.* Total yield o~ title compound was 88 g (38.80 .
*Robert H. Higgins and Norman H. Cromwell, J. Hetero. Chem.
8 ( 6 ) ~ 1059-62 ( 1971 ) .
Analysis: Calculated for Cl7Hl~NO-C2~204: C,66.46: H,6.16;
~,4 .o8
Found : C,66.38: HJ6~16;
NJ 4.07
.
452
1 2 ~
- 14 -
Preparation 2
l-(DiphenylmethYl)-~-azetidinol methanesulfonate (ester)
hYdrochloride Cl:l~
A mixture of 60.02 g (0.22 mole) of l-diphenylmethyl-
3-a~etidinol hydrochloride and 48.94 g (o.484 mole) of
triethylamine in 800 ml o~ toluene was stirred for 24 hr,
then cooled to 5C. in an ice bath and treated with 27.7 g
(0.24 mole) of methanesulfonyl chloride added at a rate
which maintained the temperature below 15C. The reaction
mixture was stirred for 3 hr and filtered to remove the
triethylamine hydro~hloride. The filtrate was treated with
40 g (0~242 mole) of 4-trifluoromethylphenol followed by
19.35 g (o.484 mole) of sodium hydroxide and 1.6 g (0.005
mole) o~ tetrabutylammonium bromide in 60 ml of water. .
The reac'ion mixture was stirred rapidly at reflux for 18 hrs,
then atirred for 72 hr while it cooled to an~bient temperature.
Th~ reaction mixture was transferred to a separatory ~unnel
and washed with 4 x 200 ml of water (emulsion). The
toluene phase was dried over magnesium sulfate and concen-
trated in vacuo to 82 g of oil. This residue was dissolved
in 200 ml of isopropyl alcohol and treated with 20 ml of
concentrated hydrochloric acid. Upon cooling, a solid
~eparated and was removed by filtration (5.1 g). The
$iltrate wa~ treated with isopropyl ether to give an oil
which W2S worked up later. The solid was identified by
spectral ~nalysis a~ the methylsul~onate of the starting
azetidinol. Recrystallization from isopropyl alc~hol gave
3.3 g o~ fine white crystalline material, m.p. 172-173C.,
(s~rinks 167C. 3 .
Analysis: Calculated for Cl~Hl~N03S~HCl: c,s7ro; H,5.70;
~,3-96
Found : C,57.80; ~,5.86:
N,3.92
"5~;
3~Z~ 4
- 15
Pre,pa ra t lon 3
]azetidine oxa~ate rl 11
A ~tirred 6}urry of :L.2 g (0.03 mol2) of sodium hydride
(60% dispersion in mineral oil) in 50 ml of dry DMF WZ18
treated with 3.45 g (0.01 mole) of trans-1-dip~enylmethyl-2-
m~thylazetidin-3-ol oxalate added in ~mall portion~. When
the a~dition was ~omplete and th~ ev~lution of hydrogen
c~as~d, the reaction wa~ heated to 80C. ~or 2 hr then 1.~4 g
(0.01 mole) of 3-fluoro-tri~luorornethylbenzene was added
13 dropwise. The reaction mixture w~ sti~red at Boc. for an
~dditional 18 hr. The reaction mixture was diluted with ice
water and extract~d with 3 x 25 ml of ~oluene. The extra~t~
were combined, dried o~rer magnesium aulfate, ~iltered, and
the filtrate treated with 1 g of oxalic acid~ . The resulting
15 ~olid wa~ collected by ~iltration. R~crystallization ~rom
acetone-isopropyl ether yielded 2.2 g (4~5O Of fine white
crystal~, m.p. 146-147C. Proton NMR ~hows it to be the
trans compound.
Analysis: Calculat~d ~or C2~H~2F9NO~C2H404: c,64.o6; H,4.96:
N,2.87
ao Found : C,64.26: H,4.~9:
Nl2.~9
Preparation 4
trans-2-Methyl-~-r3~(txil~1uoromethvl)phenox~ a.zetidine
oxalate ~
A methanol-warm water ~olution o 33 g (o.o68 mole) of
trans-l~diph~nylmethyl-2-methyl~ -(trifluor~methyl)
ph~noxy3azetidine oxalate was treated with amm~nium hy~roxide
unt~l ba~ic, then extracted with 4 x 150 ml of methylen~
chloride. The ~ombined m~thylene chl~ride extracts were
~0 waRh~ with wat~r, dri~d over m~gn~sium ~ul~ate, and
con~entrat~d i~ acuo to a pale yellow oil. ~his oil was
dissolved in 200 ml of 190 ~hanol plus 5 ml of ~ri~hyl-
amin~ ænd hy~rogenated on a Parr ~pparatus with 3.3 g of
5% pall~dium-on-cha~coal cataly~t with a 40 p8i atmosphere
o f hydrog~n at 70QC. ~or 12 hr. Ater the e~l~ulat~d ~moun~c
of hydrogen had b~en ~hsorb~d, ~he cat~ly t wa~ rem~ved by
452
~ 74
_ 16 -
~iltration and the ~iltrate concentrated in vacuo to yield
26.73 g o~ crude product. An 8 g portion was converted
to the oxalate ~;alt in isopropyl alcohGl yielding 6 . 1 g
o~ fine white powder; m.p. 155-156C. Total yield
extrapolated from the aliquot converted to the oxalate
salt was 84% of theory.
Analysis: Calculated for C~ 2F3NO CzH204: C,48.61; H,4.39;
N,4.36
Found : CJ48.67; H,4.38;
- N,4.34
Preparation ~
trans-l-(DiPhenylmethyl)-2-methyl-~-azetidinol methane-
Eulfonate (ester) hydrochloride ~
A ~olution of 6 g (0.025 mole) of trans-l-diphenyl-
methyl-2-methylazetidin-3-ol tobtained from the hydrochloride
salt by partitioning in organic solvent and aqueous base,
separating and avaporati.ng the organic phase) in 40 ml of
dry benzene was treated with 10 ml of triethylamine and
cooled to 5C. While stirring, the reaction mixture was
treated with 3.54 g (0.0~ mole) of methanesulfonyl chloride
at a rate to control the temperature below 10C. After
stirriny for 3 hx, TLC (20~ ethyl acetate/methylene chloride
on silica gel) showed the reaction to ~e incomplete. An
additional 1.14 g (0.01 mole) of methanesulfonyl chloride
was added and stirring continued for 1 hr. The reaction
25 mixture was dilute~ with 100 ml of water and the benzene
layer separated, washed with 300 ml of water, dried over
magnesium sulfate and concentrated to an oil. The oil was
di~solved in isopropyl ether and treated with ethereal
hydrogen chloride. The solid ~alt was- removed and recrystal-
lized from 190 ethanol to give 3.4 g (37%) of fluffy white
crystals, m.p. 152-153C~
Analysis: Caleulated for Cl~H21N03S HCl: C~58.77; H,6.o3;
~.81
Fou~d : C,58.68; ~,6.o8,
~,3.80
lZ94Z~ ~52
-- 17 --
PreParation 6
l-(Diphenvlmeth,yl)-7-r3-(trifluoromethyl~phen
azetidine.
~-Diphenylmethy~ hydroxyazetidine hydrochloride (I)
was prepared from benzhydrylamine and epichlorohydrin
according to Anders~n and Lok, J. O~g. Chem., ~7:3953 (1972).
I ~41.~3 g, 0.15 mole) and triethylamine (42 ml, 0.30 mole)
were ~tirred in toluene (250 ml) while methane sulfonyl-
chlori~e (12 ml, 0.15 m) was ad~ed dropwise over 10 minute3
with stirrin~ and the temperature was maintained between
4 and 12C. TLC (silica gel, 10~ ethyl acetate in methylene
chloride)at one hour ~howed all ~tartin~ materials had
r~ac~ed. The mixture wa~ filtered to remove the ~riethyl-
amine hydxo~hloride which was rinsed ~wice with toluene.
~he ~iltrate and washings ~ombined to about 450 ml of
solution. To this ~olution wa~ added m-tri~luoromethyl-
phenol (27.5 g, 0.17 mole),tetrabutyl ~mmonium bromide
(2.4 g), 50% 30dium hydroxide (24 g, 0.3 mole) and water
(24 ml) and the mixture was stirred vigorously and heated
to reflux under nitrogen for 2.5 hr. The toluene layer of
the mixtu~e wa~ s~parated and wa3h~d pnce with wat~r,
dried over sodium sulate ~nd evaporated to an oil. This
oil wa~ seeded and pumped on an oil pump overnight. A
solid caXe weighing 49.7 was obtained. Some of this solid
w~s dissolved in isopropanol with brief heating. Water
wa~ then added to cau~e ~light cloudines~. Th~ ~ixture was
~eeded ~nd c0012d to cause ~rystallization. ~he white soli~ wæ~
~ollected by filtration, wa~hed with 50% a~uous iæopropanol,
: and dried under va~uum overnight. Proton ~M~ ~howed slight
: cQntamination by sili~one oil, m.p. 82.5~C.
30 An~lysi~: Calculated for C29~4~F3No:C,72.05; ~,5.26; ~,3.65
Found :C,71.62; ~,5.29; N,3.61
3L294;27~ 45
- 18 -
The following examples illustrate preparation of
compounds encompassed by Formuila 1 and useful in the method
o~ treatment of the invention. The scope of the invention
i8 not limited by the exampl~s, howe~er.
Example 1
carbothioamide.
Crude 3-t3-(tri~luoromethyl)p~enoxy~azetidine from
catalytic debenzylation of 26.0 g (0.078 mole) ~ l-benzhydryl-
3_E3-(tri~luoromethyl)phen~xy~azetidine was dissolved in 100 ml
of methyl~ne chloride and treated dropwise under a ni~ro~en
~tmospher~ with a solu~ion o~ 5.0 g (o.o678 mole) of
methylisothiocyana~e in 15 ml o~ methylene chloride. The
reaction mixture was stirred for 16 hr at ambient temperature
and let stand over th~ weekend. The solution was filtered~ ~ . through a celitq~filter pad to remove a fine crystalline
precipitate and the filtrate w~s evaporated under xeduced
pressurQ. The xesidual oil was cryst~lliæed rom isopropyl
~ther to give 12.6 g of product, m.p. 79-86C. A 5.0 g
~ample was recrystallized ~rom i80propyl ether (charco~13 to
give 3.2 g, m.p. 8~-93C., which was shown by TLC on ~ilica
gel (10~76 methanol-toluene) to be contaminated by a lower
Rf material. -The ~iltrate was ~vaporated under reduced
pre~sure,combined with ~he 3.2 g of solid snd diss~lved in
100 ml of methylene chloride. The solution was ~tirr~d
with 25 g o~ silica gel for 0.5 hr and ~iltered through a
sintered glass filter. The ~ilica gel wa~ wa~hed with a
small volume ~f methylene chloride and the iltrate
evaporat~d under reduced pres~ure. The reYidual ~olid w~s
recry~tallized ~rom isopropyl ether to give 1.3 9 o~ pure
product, m.p. 96-98C.
An~lysis: Calculated ~or Cl2Hl9~N20S: C,49.65; H,4.51:
Found : C,49.58. ~,4 4~;
~,9-58
QC~e1nd~1~
lZ~27~ 452
-- 19 --
Example 2
-(2,6-Dimethylphenyl)-3{ ~(triflu~romethyl)p~enoxy
azetidinecarbothioamide.
Crude 3-~3-(trifluoromethyl)phenoxy~azetidine from
catalytic debenzylation of 30.0 g (0.078 mole) of 1-
benzhydryl-3-r3-(tri~luoromethyl)phenoxy~azetidine was
dissolved in 100 ml of methylene chloride and treated drop-
wise under a ni~rogen atmocphere with a solution of 12.7 g
~0.078 mole) of 2,6-dimethylphenylisothiocyanate in 25 ml
of methylene chloride. The pxoduct began to precipitate
during the addition and an additional 50 ml of methylene
chloride was added to faeilitate stirring. After stirring
overnight at ambient temperature, the product was collected
by filtration (13.5 g, m.p. 196-199C). A 6.o g sample
was recrystallized from isopropanol to give 5.~ g of
15product, m.p. 197-199C.
Analysis: Calculated for Cl~Hl~F3~20S: C,59.99; H,5.03;
N,7.~6
Found : C,60.01l; H,5.o4;
~,7-35
Example
20N-(Phenylmethyl~trifluoromethyl~hen
azetidinecarboxamide.
To a stirred and chilled (10-20C.) solution of 0.04
m4le of 3-t3-(trifluoromethyl)phenoxy3azetidine in 100 ml
of methylene chloride was added dropwise 6.12 g (o.o46 mole)
of benzyl isocyanate. The reaction mixture was stirxed at room
temperature for 2 hr and was filter~d. The filter cake was
washed with petroleum ether (2 x 50 ml), dilute a~ueous
so~ium bicarbonate (2 x 50 ml), and water (2 x 50 ml)~
yielding 12 g (86%)o Recrystallization twice from ethyl
acetate gave 9.0 g of clear white flakes~ m.p. 173.5-175C.
Analysis: calculated for CleHl7F9N202: C,61.71; H,4.89;
~, ~ .00
Found : C,61.57; H,4.87:
~,7-99
lZ94~7~ 452
20 --
ExamPl~ 4
-~? .6~DichlorophenYl)-3-r3-(trifluoromethyl)phen
l-azetidinecarbothioamide.
A solution of 0.04 mole of ~-~3-(trifluoromethyl)
ph~noxy~azetidine in 100 ml of absolute ethanol was stirred
in a tap water bath while 8.16 g (0.04 mole) of 2,6-di~hloro-
phenyl i~3othiocyanate was added all ~t on~:e. T~e reaction
was ~;lightly ext~th~nic and as the isothiocyanate began to
~i~solve, product began to precipitate. After ~tirring
~or 45 minutes the reaction mixture was heated on a steam
bath to assure that all the isothiocyanate dissolved, and
upon cooling, filtration yielded 15.2 g of white ~rystallin~
product. A portion of this material (7.9 g) was r~crystal-
lized fxom absolute ethanol to give 4.3 g of p~re cry~talline
powder, m.p. 195-197C.
Analysis: calculated or Cl7Hl9F9Cl2N20S: C,48.47; H,3 11:
Found : C,48,40: ~,3.07:
N,6.~4
Example ~
-L~-(Diethylamino)prop~l~ 3-(trifluorometh~l)
phenoxy~-l-azet inecarbothioamide oxal~t~L~Ll.
A solution of (0.0584 mole) of 3-~(trifluoromethyl)
p~enoxy~azetidine was stirred at 10C. while 10.66 g (0.0584
mole) of 3-(diethylamino)propyl iqothio~yanate was added
all at on~e. A~ter ~tirxing overnight a~ ambient temperature~
25 the reaction mixture was concentr~t~d at 50C. on a
rotary evaporator to a thicX ~yrup xesidue. The reRidue
was dissolved in i~opropanol and treated with 5.3 g of
oxalic ~id, warmed or~ a steam bath ~o di~olve ths acid,
and upon cooling, a ~oli~ ~alt precipitated. An ~ual
volume of isopropyl eth~r was added to en~ure complete
: pre~ipitation. ~iltration gave 26 g of crude product.
~:portion (13 g) was recrystalliz~d from isoprop~nol/
methanol/i~opropyl e~her (100/50/50) (cooled in ~ r~rig-
2rator) to yield upon ~iltration 7.5 g of whit~ p~oduc~,
m.p. 155-157C~ Proton NMR con~irmed that thi~ wa3 the
~xpected product.
452
lZ~4~7
-- 21 --
Analysis: Calculated for Cl8H~F3N30-CzH204: C,50.10; ~,~.89
N, .76
Found : C,50.02; H,5.97;
N,8.89
5Example 6
3-(Dimethylamino)propyl~-3-r~-(trifluoromethvl)
~henoxyl-l-azetidinecarbothioamide.
A stirred solution of o.o584 mole of 3-[3-(trifluor
methyl)phenoxy~azetidine at 10C. was treated with 8.42 g
lt) (0.0584 mole) of 3-~dimethylamino)propyl isothiocyana'ce
all at once and allowed to ~tir at an~ient temperature
overnight. The reaction mixture was tr~ated with 5.3 g
(0.0584 mole) of oxalic acid and diluted with 200 ml of
isopropyl ether which yielded only 3.8 g of product. The
15~ume was reduced to 100 ml at 50C. ln vacuo and diluted
wi~h 500 ml of isopropyl ether to yield an additional 15.~ g
of product. ~h~ combined solid m~terial Wa3 di~solved in
i~opropyl alcohol and upon cooling, a ~in~ precipitate
formed (~ dimethyl-1,3-propanediamine oxalate) which
was removed by ~iltration. The product fail~d to ~rystallize:
addition o~ isopropyl ether gave only an ~morphous gel.
After trying to obtain a more satis~actory product ~or
3 weeks, the reaction material was conver~d to the free
~ase and taXen up in isopropyl eth~r. ~he ether ~olution
was stirred with 300 ml of water overni~ht to remove the
diamine. The product crystallized ~s~the free base ~rom
the hetexogenous mixture and was ~iltered to give 11.~ g of
fine beige crystal~. Xework of the filtrate gave an
additional 2.3 g of product. A portion (8 g) was re~ry~tal- -
30 ~ lized from benzene/ligroin to yi~ld 5.~ g of very :Eineb~ige cry~tals which were dried at 82C. und~r vacuum,
m.p. 107-108C~ ~
Analysi : cal~ulated for C-~Hæ2F3~30S: CsS3.17 ~ J, 6.14;
~,11.63
~5Found : C,53.2~; X~6.15;
N,11.60
7 ~ 4~2
~xample 7
-(2-Propenyl~ 3-(trifluoromethyl~phenoxyl-1
azetidinecarboxamide.
A ~olution of 18.9 g (0.05 mole) of crude 3-~3-
(tri~luoromethyl)phenoxy~azetidine (contain~ an equhl
~olar amount of diphenylmethane) in 100 ml of i30propyl
ether was stirred under nitrogen while 4.16 q ~0.05 mole)
o~ 2-propenyl is~cyanate was slowly added. The reaction
mixture which was somewhat turbid, ~leared and ~fter 1 hr
a fine crystalline precipitate began to form. After
stirrin~ for 18 hrs, the product was removed by filtxation,
washed with fresh isopropyl ether and air dried to yield
9.5 g of white crystals, m.p. 75-76C.
Analysis: Calculated for Cl4HlsF3~202: C,56.00; ~,5.04;
~ 3
Found : C,55.98; H,5.05:
~,9-31
~æm~
N-Cycloprop~1~ 3-~trifluoromethyl)Phenoxy~
azetidinecarboxamide.
A mixture o~ 1.9 ~ (0.0~3 mole) of cyclopr~pylamine
and 4.9 g of l~1'-carbonyldiimidazole in 60 ml of tetra-
hydrofuran wa3 stirred at ambient temperature or 1 hr.
The clear solution which fonmed wa~ tre~ted with a solution
of (0.03 mole) o 3-C3-(trifluoromethyl)phenoxy~zetidi~e
in 20 ml of tetrahydrofuran. After ~tirring overnight, the
501id precipitate was removed by filtration to give 4.4 g
o~ gray-white powder. The CI mass spectrum showed a p~l
at 381 m/e which wa~ consist~nt with the expected product.
Recrystallization from benzene/ligroin gave 2.~ g of a
light gray powder, m.p. 152-15~C.
Analysis: Calculated ~or Cl~HlsFgN20: C,56.00 ~,5.04;
N99-3~
Found : C,55-97; ~.5~7;
N,9.28
.
~2
- 23 -
Exampl~_~
N-~3-~ViethYlaminc~?propvl~-3-t3-çtrifluorometh
ph no~ zetidinecarboxamide_oxalate (.2:3).
A mixture of 4.~ g (0.03~ mole) of 3-diethylamino-
propylamine and 4.9 g (0.0~ mol~) of l,l'-carbonyldiimidazole
in 60 ml ~ methylene ehlQride was ~tirred at ambient
tèmperature ~or 1 hr. The resulting ~olution wa~ treated
with ~-t~-(trifluoromethyl)ph~noxy~azetidine (~btained from
9.21 g (0.0~ mole) of the oxalate salt) in 30 ml of
methylene chloride. After stirring ~or 18 hr, the r~action
mixture was transferred to a ~eparatory ~unnel and washed with
3 x 20 ml of water, dried over magnesium sulfate and concen-
trated in vacuo to a dark oil. The.resi~ue (8 g) was
chromatosarphed on a 150 ~ neutral alumina column by eluting
with chloroform. Concentration of the initial fra~tion gave
15 the product as an amber oil which was dissolved in methyl-
isobutyl ketone and treated with 2 g of oxalic acid.
Dilution w$th isopropyl ether gave an oil which solidified
and was recrystallized ~rom ~cetone/isopropyl ~ther to
give 6.25 g (410 of beige crystals, m.p. 91-~3C.
Analysis: Calculated ~or Cl0H2~F3N302-1.5 C2H4O4
C,49.61: H,5.75, M,8.26
~ound C,49.56, H,5.73; N,8.24
Example 10
N 2 ( ~ luoromethYl)Phenoxy~
azetidinecarboxamide.
A solution of 8.7 g (0.04 mole~ of crude 3-~4-(tri-
fluoromethyl)phenoxy]az~tidine in 75 ml of i50pr~pyl ether
was stirred under ~ blanket o~ nitrogen while 4.2 g (0.05
mole) of 2-propenyl i~ocyanate wa~ added dropwi3e. After
tirring for 3 day~, no ~rystalline pr~duct precipitated.
T~e reaction mixture wa~ con~entr~t~ to ~ d~rX rs~ddi3h
oil. TLC (20% ethyl a~etate/me~hylene chloride on ~ilica
gel) showed at lea~t 6 ~p~tG, all well ~eparate~. The
residu~ was di~solved in chloro~orm, chromatograp~d on 8
350 g ~ilica gel column and eluted with ~hloro~orm until
the reddi~h forerun wa~ remo~ed. Th~ ~olumn was then ~luted
~2~94
24 -
with an ethyl acetate/chloroform gradient to 4% ethyl
~cetate. All the fractions were combined and concentrated
to give 4.8 of orange oil, which crystallized on standing.
Recrystallization from acetone/cyclohexane gave 3.~ g
(27.5%~ of beige crystals, m.p. 91-92.5~C.
AnalyQis: calculated ~or C14Hls~3N2o2: C,56 00 R,5.04:
~,g.33
Found : C,55.98: ~,5.17;
~.9.36
Exa~le 11
N-(Cyclc)propylmethyl? -;~-r3 (trifluoromethyl ~phenox~-
l-azetidinecarboxamide.
A solution of 2.6 g (0.024 mole~ o~ (aminomethyl)
cyclopropane hydrochloride in 50 ml of pyridine was stirred
under a blanket o~ nitrogen while 3.9 g (0.024 mole) of
l,l'-carbonyldiimidazol~ was added. A~ter stirring for
45 minutes, the TLC (5% methanol/methylene chloride on
~ilica gel) showed no reaction; therefore, 2 ml o~ tri-
ethylamine was added. The reaction mixture after 10
~inutes became cloudy and the TLC showed a new product.
~he reaction was treated with 6.2 g (0.02 mole) of 3-(~-
~trifluoromethyl)phenoxy~azetidine oxalate. After stirring
~or 1 hr, a sample was removed, and upon dilution with
waterJ a ~olid precipitated. The CI mas~ spectrum indicated it
was product. After 2 daysl the reaction w~s diluted wi~h
5 volumes of water and the resulting precipitate ~ollected
by filtra~ion to yield 6.5 g of pale yellow cry~talline
product. Recrystallization from ethanol/water produced
white plate-lik~ crystal~ which were dried at ~C. for
3 hr in a drying pistol under vacuum; weight of the product
was 5.8 g (92%), m.p. 1~2-1~3C.
Analysis: Calculated for l5Hl7F3N20z: C,57-32; H,~-45;
N,~.91
Found : C,57.22; H,5.44;
~,8.86
~ Z ~ 7 ~ 452
Example 12
!N-Diethyl-3-~3-(trifluoromethYl)phenoxy~ azetidine-
carboxamide.
~ . _
A stirred slurry of 5 g (0.0163 mole) of 3-~3-(trifluoro-
methyl~phen~xy azetidine oxalate in 50 ml of tetrahydrofuran
was treated with 5 ml of triethylamine and after 1 hr, 2.5 g
(0.018 mole) of die~hylcarbamoyl chloride wa~ added. Aft~r
stirring an additional 15 hr, the reaction wa treated with
10 ml of water and saturated with calcium chloride. The
tetrahydrofuran was decanted from the solid residue and
concentrated ln vacuo to an oil. The crude oil was chroma-
tographed on a Water's Prep-LC using 50% ethyl acetate/
toluene as the eluent. After concentration of the main
fractions 3.1 g (60.1%) of pale yellow oil was obtained.
Analysis: C~lculated for Cl5Hl9F3N202: C,56.96; H,6.o5;
~8.86
Found : C,56.69: H,6.01;
N,8.77
ExamPle l;~
N,N-DimethYl-3-~trifluoromethyl~n
azetidinecarboxamide.
A ~tirred slurry of 5 g (0.016~ mole) Of ~-~3-(trifluoro-
methyl)phenoxy]azetidine oxalate in 50 ml oP tetrahydrofuran
was treated with 5 ml of triethylamine and after 1 hr, 1.95 g
(0.018 mole) of dimethylcarbamoyl chloride was added. After
stirring an additional 15 hrJ the reaction mixture was
treated with 20 ml of w~ter and 10 g of calcium chloride.
The tetrahydrofuran layer was decanted and the residue
triturated with 20 ml of ~thyl acetate,then decanted. The
combined tetrahydrofuran and ethyl acetate ~olution was
concentrated in vacuo. The crude residue was chro~atographed
0 on a Waters Prep ~ sing 50% ethyl acetate/toluene as the
eluent. After concentration of the main fractions, 3.6 g
(76.6%) of pale yellow oil was obtained.
Analysis: Calculated for Cl9Hl5F3N202: C,54.17; ~,5.25,
~,~.72
Found : C,53.73; ~,5.20;
N,9.60
~ nqr k
l~52
4Z7
- 26 -
xample 14
-(2-Propynyl)-3-~3-(trifluoromethyl ph noxy~
azetidinecarboxamide.
A mixture of ~.9 g (0.02~ mole) o~ l,l'-carbonyl-
diimidazole and 1.32 g (0.024 mole) of 2-propynylamine in
50 ml of tetrahydrofuran was 6tirred at ambient temperature
for 1 hr, then treated with 6.2 g of ~-t~-(trifluorom2thyl3
phenoxy]azetidine. The reaction mixture was ~reated with
3 ml of triethylamine and stirred ~or 18 hr. ~he reaction
mixture was diluted with an equal volume of water and
~iltered to yield 8 g of wet product. ~ecrystallization
$rom isopropyl ether gave 3.8 g o~ gray solid, a ~xture ~f
product and the ~ymmetrical urea o~ starting 2-propynylamine.
A second recrystallization ~rom ethanol water yielded 2.6 g
(43.6%) of pur~ product, m.p. 105-106C.
Analy8is: Calculated for Cl~Hl3F~02: C,56.38: H,4.39:
.9-39
~ound : C, 56 . 32; H,4.34;.
N, 9 . 44
ExamPle 15
N-Cyclohex~1-3-~3-(trifluoromethyl)phenoxy~
azetidinecarboxamide.
- --
A stirred mixture of 5 g (0.016~ mole) of 3-~-(tri-
fluoromethyl)ph~noxyJazetidine oxalate an~ 2.04 g (0.018
mol~) o~ cycloh~xyl isscyanate in 50 ml of tatrahydrofuran
was treated with 2 ml uf triethylamine then stirre~ for
25 18 hr~ Dilution of the mixture with w~ter gave a solid
precipitate which w~s collected by filtration to yield 12 9
of crude produot. Recrystallization from ace~one/water
- gave 5 g ~f ~ine white ~rys~als, m.p. 148 150C. TLC (ethyl
aceta~e on 5il ica gel) ~howed a ~race of symmetrical cyclo-
~0 hexyl urea ~s well ~s` the produ~t. A s~cond re~ry~al-
lization fr4m isopropanol yielded 1.65 9 ~29.6%) o~ white
powdex; dri~d under 0.5 ~n/Hg vacuum" m.p. 153-154Q~.
Analysis: Calculated ~or Cl7H2aF~202: C~59.64: ~,6.18:
: ~,8.18
Found : C,59.52; ~,6-20;
~,~.17
:
27
-- 27 --
Exam~le 16
~-Cyclopropyl-3-~4-(trifluoromethyl)phenc)xy
azetidinecarboxamide.
A stirred slurry of 4.4 g (0.027 mole) of l,1'-carbonyl-
diimidazole in 50 ml of methylene chloride under nitrogen
was treated with 1.54 g (0.027 mole) of cyclopropylamine.
After a sh~rt (2 min) induction period, the lear ~olution
became suddenly exothermic, bringing ~he reaction to a
gentle r~flux. After 1 hr when the reaction mixture had
cooled to ambient temperature, 9.6 g (0.025 mole) of
3-~4-(trifluoromethyl)phenoxy~azetidine, 56.66% purity
(contains diphenylmethane) wa~ a~ded all at ~nce and stirring
continued ~or 18 hr. The reaction mixture was concentrated
on a rotary evaporator to give a partially crystalline
residue. The residue was partitioned between 30/60 petroleum
ether and water and the resulting waxy solid removed by
filtration. Recrystallization from isopropyl ether yielded
5.7 g (75.9~) of silver plate-like crystals, m.p. 145-147C.
After drying at 80C. under 0.5 mm/Hg vacuumJ the weight
was not diminished, m.p. 152-153C.
Analysis: Calculated for C~ 5F3N202: C,56.00, HJ5.04;
N~9.33
Found : CJ55.77; HJ4~98
NJ9.44
~xample 17
~-(CyclopropvlmethYl)~ 4-(trifluoromethyl)phenox~l-
l-azet idinecarboxamide
.... .. ... ........... ... ..
A stirred mixture of 4.4 q (0.027 mole) of 1J1~-
carbonyldiimidazole and ~.9 g ~0.027 mole) of (aminoethyl)
cyclopropane hydrochloride in 50 ml of methylene chloride was
treated with the dropwise addition of 2.73 g (0.027 mole)
of triethylamine. The reaction was exothermic. The mixture
was cooled while ~tirring ~or 1 hrJ then 9.6 g (0.025 mole)
of 3-~4-(trifluoromethyl)phenoxy]azetidine 56.66% (contains
diphenylmethane) was added all at once and stirri~g continued
for 18 hr. The reaction mixture was co~centr~ted ~n a
rotary evap~rator to give an amber residue. Trituration of
this xe~idue with ~o/60 petroleum ~ther gave only an
274 452
~ 28 --
insoluble oil. The trituration step was repeated with
2 x 20 ml of 30/60 petroleum ether and the residue treated
with water to yield a white solid. The solid was recrystal-
lized from isopropyl ether to yield 4.8 g (61.B%) of white
platelike crystals, after during at 80C. under 0.5 mm ~g
vacuum, m.p. 132-1~3C.
Analysis: Calculated for Cl5H117F3N202: C,57.~2; H,5.45;
N,~.91
Found : C,57.26; H,5.46;
N,8-93
Ex~m~ 18
-~3-(Dlethylamino~pro~yl~ S-L4-~tri:1uoromethYl)
phenoxy~ azetldinecarbothioamide.
A stirred solution of 1~92 g (0.005 mole) of 3-[4-
(tri~louromethyl)phenoxy]azetidineJ 56.66% (contains
diph~nylmethane) in 20 ml of isopropyl ether was treated
with o.88 g (0.005 mole) o~ 3-(diethylamino)propyl
isothiocyanate and ~tirred for ~.5 hr. The reaction mixture
was treated with 0.5 g of oxalic acid dissolved in 2 ml of
methanol. After stirring for 18 hr, the solid was collected
by filtration, yielding 1.9 g of fine tan powder, m.p.
147-150C. The solid was di~solved in water and treated
with dilute sodium hydroxide. An oil separated which
solidified and was collected by filtration. Recrystal-
lization from cyclohexane yielded 1.1 g (56.5%) of ~ine
25 tan crystals, m.p. 109-110C.
Analysis: Calculated for Cl8H26F3N30S: C,55.51; H,6.73;
~,10.79
Found C,55.68; H,6.67
N,10.73
- ~xample l9
N-c3-(Diethylamino)propyll-3-C4- ~ rifluoromethyl)
phenoxy~ azetidinecarboxamide oxalate ~1-21.
A ~tirred solution of 4.4 g (0.027 mole) of 1,1'-
carbonyldiimidazole in 50 ml of methylene chloride under
nitrogen was treated with the dropwise addition of ~52 g
~5 (0.027 mole) of 3-(diethylamino)propylamine. The reaction
mixture was stirred for 1 hr as the ~omewhat ~xothermic
reaction ~ooled to ambient temperature then tr~ated with
7~ 452
- 29 -
9.6 g (0.025 mole) of ~-t4-(trifluoromethyl)phenoxy~azetidine,
56.66% (contains diphenylmethane) all ~t once. A~ter
stirring for 18 hr, the reaction mLx~ure was concentrated on
a xotary evaporator and the residue dissolved in toluene.
The toluene solution was washed with ~ x 20 ml of water,
then treated with 2.5 g of oxalic acid in 10 ml o~ i~opropanol
The resulting solid was collected by filtration and
triturated with boiling acetone. After filtration, 1.8 g of
unidentified fine white precipitate formed which was
~eparated by filtration. The acetone solution was concen-
trated to a solid which was recrystallized from isopropyl
al~ohol/isopropyl ether to yield 9.2 g of crude product
t4 spots on TLC; 10~ methanol/methylene chloride on silica
gel). Recrystallization from methyl ethyl ketone yielded
6.8 g (49.1%) of fine white powder, m.p. 129-1~0C.
Analysis: Calculated for Cl~H~F3N30~-2C2H404:
C,47.74; H,5.46: N,7.59
Found C,47.82: H,5.68; N,7.76
ExamPle 2Q
N-(2-prop~ynyl L-~
~zetidinecarboxamide.
. ~
A solution of 4.4 g (0.027 mole) l,l'-carbonyl-
diimidazole in 50 ml of tetrahydrofuran was stirred under
nitrogen while 1.49 g (0.027 mole) of 2-propynylamine was
add~d with a syringe and needle through a ~eptum installed
in one neck o~ the reaction ~lask. After stirring for 2 hr,
9.6 g (0.025 mole) of 3-~4-(trifluoromethyl)ph~noxy~azetidine
(56.66~ purity; contains diphenylmethane) was added all at
once and stirring continued for an additional 18 hr. The
30 reaction mixture was diluted with ice-water and extracted
witb 30/60 petroleum ether to rem~ve the diphenylmethane.
The oily a~ueous portion was extracted with 4 x 50 ml o$
methylene chloride. These extracts were combinedJ dried
over sodium 6ulfate and concentrated to an amber oil on a
rotary evaporator. The oil solidified when triturat~d with
a small amsunt of i~opropyl ether (50 ml). Filtration
452
- 30 -
yielded 6.1 g o~ rose-tinted ~olid product. TLC (10~
methanol/methylene chloride on silica gel) showed a mixture
of 3 products and ~ome starting material. Recrystallization
iFrom ethanol/water gave the product in several small
fxactions. These were combined and recrystallized ~rom
isopr~pyl ether to yield 4.1 g of pale beige p~wder, m.p.
135-137C. TLC ~till ~howed 60me symmetrical 2-propynyl
urea. The solid was recrystallized again ~rom ethanol/
water to yield 3.5 g (46.9%) of pale yellow crystalline
product, m.p. 140-141C.
Analysis: Calculated for Cl4Hl3F3N202: C,56.38; H,4.39;
~,9-39
Found : C,56.34; H~4.36;
~,9.32
~xample 21
N-(?-Methyl-2-propenyl)-~-r4-(txifluoromethyl)phenoxy~-
l-azetidinecarboxamide.
A stirred solution of 3.6 g (0.022 mole) o~ 1,1'-
carbonyldiimidazole in 75 ml of methylene chloride under
nitrogen was treated with 1.6 g (0.022 mole) of 2-methyl-
2-propenylamine (added via a syringe and needle through a
~eptum placed in one neck of the reaction flask). After
stirxing for 1 hrJ 6.2 g (0.02 mole) of 3-~4-(trifluoro-
methyl)phenoxy~azetidine oxalate was added all at once
followed in 30 min with 5 ml of triethylamine and stirring
was continued for 3 hr. The reaction mixture was washed
with water (2 x 25 ml), dried over magnesium sulfate and
concentrated in vacuo. The oily residue solidified on
tanding and was recrystallized from isopropyl ether to
yield 3.7 g (58.9%) of fine white crystals, m.p. 101-102C.
Analysis: Calculated for C15Hl7F3~202: C,57-32 ; H,5-45;
N,8.91
Found : C,57.45; H,5.51:
N,9.23
2~ ~ ~ 7~ 452
- 31 -
Example 22
N-(?-Methyl-2-}~openyl)~ (trifluoxomethyl)phenoxv
l-azetidinecarboxamide.
A solution of 3.6 g (0.022 mole) of l,l'-carbonyl-
diimidazole in 75 ml of methylene chloride was stirred under
nitrogen while 1.6 g (0.022 mole) of methallylamine w~s
added with a syringe and needle through a ~eptum installed
in one neck of the reaction flask. The reaction was
slightly exothermic. The reaction mixture was stirred for
1 hr~ then treated with 6.2 g (0.02 mole) of 3-~3-(tri-
fluoromethyl)phenoxy~azetidine oxalatP followed in 0.5 hr
with 5 ml of triethylamine and stirring continued for 16 hr.
The reaction mixture was washed with 2 x ~0 ml of water,
dried over magnesium sulfate, and ~oncentrated on a rotary
evaporatvr to yield 6.7 g of oily residue which solidified.
1$ The residue was recrystallized fxom isopropyl ether to
yield 5.4 g (85.90 of fine white crystals, m.p. 90-91C.
Analysis: calculated for C15Hl7F3N202: C,57-~2; H,8-45:
N, .91
Found : C,57.20; H,5.50;
N,~-95
Example 2~
N-(3-MethYl-2-butenYl)-3-C4-(trifluoromethyl ~henoxy]-
l-azetidinecarboxamide.
A solution of ~.6 g. (0.022 mole) of l,l'-carbonyl-
diimidazole in 100 ml of methylene chloride was cooled in a
tap water bath and while 3tirring under nitrogen, 1.87 g
(0.022 mole) of 3-methyl-2-butenylamine was added dropwise.
After stirring for 1 hrJ 6.2 g (0.02 mole) of ~-~4-(tri-
fluoxomethy})phenoxy~azetidine oxalate was added all at
once followed in 0.5 hr with 5 ml o triethylamine and
~tirring continued for an additionaI 16 hr. The xeaction
mixtur~ was washed with 7 x:50 ml o~ water, dried over
magnesium sulfate and concentrated on a rotary svaporator
to yield a semi--~olid~residue. Trituration with isopropyl
ether and filtration yielded 7 g of crude produ~t whi~h
was recrystallized from ethanol-water to give 5.5 g ~87.8%)
of white ~rystals 9 m.p. 156.5-158 C.
~ ~ 4 ~ 452
Analysis: calculated for CloHleF3~202: C,58.53; B,~-8~;
Fou~d : C,58.81; ~ 5 ~9:
~,~.58
Example 24
-(3-Methyl-2-butenyl)-~-~3-(trifluoromethyl)phenoxy
A solution of 3.6 g (0.022 mole) of l,l'-carbonyl~
diimidazole in 100 ml of tetrahydrofuran was csolea with
a tap water bath and stirred under nitrogen while 1.6 g
(0.022 mol~) of 3-methyl-2-butenylamine was added with a
syringe and needle. The reaction mixture was stirred for 1 hr,
then treated with 6.2 g (0.02 mole) of 3-~3-~tri~luoro-
methyl)phenoxy~azetidine oxalate followed in 0.5 hr with
5 ml of triethylamine and stirring continued for 72 hr.
The reaction mixture was diluted with 500 ml of ice water
and extxacted with 6 x 50 ml o~ methylene chloride. The
combined extracts were washed with water, dried over
magnesium ~ulfate and concentrated to a solid residue on a
rotary evaporator. Recrystallization ~rom ethanol-water
yielded 6 g of white crystals, m.p. 143-144C.
Analysis: Calculated for Cl~Hl~F9N202: C,58.53; H,5.83;
N,8-5~
Found : C,58.46: H,5.86:
~,8.69
(E)-N-(2-~utenyl)-3-L4-(trifluoromethyl)phenoxy~
azetidinecarboxamide.
A mixture of 3.6 g (0.022 mole) of l,l'-carbonyl-
dii~idazole and 1.6 g (0.022 mole~ of trans-crotylamine
was stirred for 1 hr, then treated with 6.2 g ~0.02 mole)
~0 of 3-~4-(trifluorom~thyl)phenoxy]azetidine oxal~te and
followed in 0.5 ~ with 5 ml o~ triethylamine with stirring
continued for 16 hr. The partially crystalline mixture
was washed with 2 x 50 ml o~ wat~r, dried over magnesiu~
~ulfate and ~oncentrat~d on a rotary ~vaporator to a solid
35 residue, 14.2 g. Recrystallization fxom methanol-water
yielded 5.35 g (85.1%) of ~ine white crystals, m.p.
157 158C.
.
~ 4 452
- 33 -
Analysis: Calculated for C15Hl7F3N202: C,57-32; H~-45;
N, .91
Found: C,57.47, H,5.~9:
N,9.00
Example 26
(E)-
azetidinecarboxamide.
A ~olution of 3.6 g (0.022 mole) of l,l'-c~rbonyl-
diimidazole in 60 ml of methylen~ chlori~e was ~ooled in
an ice ba~h while ~tirring under nitrcg~n while 1.6 g
(0.022 mole) of tran~-crotylamine wa~ add~d dropwi~e.
After warming t~ ambient temperature, 602 g (0.02 mol~) o~
3-~3-(tri~luoromethyl)phenoxy~azetidine oxalate was added
all at once followed in 0.25 hr by 5 ml of triethyla~ine
with stixring continued for 72 hr. The reaction solution
15 was wa~hed with 2 x 50 ml of water, dried over magnesium
sulfate and concentrated on a rotary evaporator to a
soli~ residue, 7 g. Recrystallization from methanol-
wa~er ~ave 5.5 g o~ ~lightly yellow product. A second .
recrystallization with charcoal treatment from i~opropyl
ethar yielded 3.75 g (59.7%) of fine white cry~tals,
m .p . 12 7-12 8C .
Analysis: Calculated for Cl5Hl7F3N202: C,57.32; H,~.45;
~, .91
Found : C,57.35, H,5.47:
N,8.94
Example A..?~
N-Phenyl-3-~3-( ~r~ azet~dino-
carboxamide.
,
A stirred ~1urry o~6.2 g (0.02 ~ole) Of 3~r3-(tri-
fluorom~thyl)phenoxy~az~tidine oxalate in 60 ml of tetra-
hydrofuran wa~ treat~d with 5 ml of triethylamine followed
:by :2.62 g (0.022 mole) of phenyl i ocyanate and ~tirring
con~inued ~or 16 hr. ~he reaction mixture was diluted
with w~ter until an oil ~eparat~d which quickly ~olidified.
The aqueous tetrahydro~uran was:dec~nted and the ~olid
r~idue recrystalliYed from ~hanol-water to yield 5.3 g
(80.1f) o~ whit~ cry~t~l~, ~.p. 137-138c.
4 ~7 ~ 452
- 34 -
Analysis: calculated ~or Cl7Hl5F3N202: C,60.71; H,4.50
N, 8.33
Found : C,~io.Bl; H,4.47,
~,8-35
carb~xamide .
A tirred slurry of 6.2 g (0.02 mole) of 3-~4-(tri-
fluoromethyl)phenoxy~azetidine oxalate and 2.62 ~ (0.022
mole) of pher~yl isc>cyanate in 60 ml of tetrahydrofuran was
treated wi~h 5 ml of ~riethylamine and tirring continued
~or 16 hr. The reaction mixture was diluted with water
until an oil 3epar~ted. The tetrahydrofuran-water portion
was decanted ~nd the residue solidified on standing.
Recrystallization from ethanol-water yielded 3.5 g t~3.4
o~ fine white crystal~J m.p. 174.5-176C.
Analysis: calculated for Cl7HlS~3~202:C,60.71: H,4.50;
N,8.33
Found :C,60.91; H,4.53;
~,8.35
Exam~le 2~
trans-~,2-Dimet~Yl~ -(tri~luoromethyl)~henoxy~-l-
azetidinecarboxamide.
A stirred solution of 6 g (0.015 mole) o~ crude trans-
2-methyl-3-~3-(tri~luoromethyl)phenoxy]azetidine in 50 ml
of tetrahydrofuran was treated with 0.94 g (0.0165 mole) of
methyl isocyanate ~dded dropwlse and ~tir~d for 16 hr
under a blanXet o~ nitrogen. Dilution of the reaction
_ mixture with water produced~an oil whi~h solidified. After
decanting the a~ueous tetr~hydrofuran pha3e, the solid
residue was recrys~allized from:ethanol-water to yield ~.95 g
: (91.4%) of:fine white cry~tals, m.p. 104.5-106C.
~Analysis: Calculate~ ~or Cl3Rl5F3N202: C,54.17: H,5.25;
~,g.72
Found : C,54.50: ~,5.29;
71
;27~ 2
Example 30
txans-? -Methyl-3-r3-( trifluoromethyl)P}ienoxv~ -l-azetidine-
_arboxamide .
A mixture of 6 ~ (0.015 mole3 of crude trans-2-methyl-
~-~3-(trifluoromethyl)phenoxy]azetidine (purity 56.6%
contains diphenylmethane~and 2.4 g (0.0225 mole) ~f
nitrourea in 40 ml of acetone was treated with 4 ml of
water, then heated until a clear homogenous solution was
obtained. The reaction mixture was ctirred ov~rnight as
it cooled to ambient temperature and diluted with water
until an oil separated. The oil solidified and was
xecrystallized from ethanol/water, yielding 4.3 g of white
plate-like crystals: m.p. 117-118C. The product was
recrystallized from benzene, yielding 3.35 g (96.8%) of
crystals, m.p. 118-119C.
Analysis: Calculated for Cl2Hl3F3N202: C,52.56; H,4.78;
~,10.22
Found : C,52.54; H,4.74;
N,10.17
Example 31
trans-2 Methyl-N-(2-propenYl)-3-r~-ttrifluoromethyl)
phenoxY~ azetidinecarboxamide.
A solution o~ 6 g (0.015 mole) of crude trans-2-methyl-
~-~3-(trifluoromethyl)phenoxy~azetidine t~6.6%) in 50 ml
of tetrahydrofuran was treated with 1.54 g (0.0165 mole) of
2-propenyl isocyanate all at once and stirred under a
blanket of nitr~gen ~or 16 hr. The rea~tion mixtur~ was
diluted with water until an oil separa~ed. The oil failed
_ to crystallize and after 7 weeks it was triturated with
i~opropyl ether (3 x 25 ml). The combin~d triturates
g~ve 400 mg of white gr~nular ~ryst~ls (8.5 0 , m.p. 55-57 C.
~ Analysis: Calculated ~or Cl5Hl7F9N202 C,57 32; H,5.45;
~,8.gl
Found : C,57.~6; H,5.50;
N,8.97
27~ 452
- 36 -
Example ~2
3-(3-Chloro~henoxY)-N-methyl-l-azetidinecarboxamids.
A solution of l-chlor4carbonyl-3-(~-chlorophenoxy)
azetidine (0.01275 mole) in 20 ml of tetrahydrofuran was
treated with 4 ml (0.05 mole) of 40% a~u~ous methylamine
and fitirred for 16 hr. The reaction mixture was diluted
with water until asl oil began to separate, then extracted
with 3 x 50 ml of benzene. The combined extracts were
dried over magnesium sulfate and concentrated to a ~olid
which was recrystalliz~d from benzene/liyoin to-yield
1.2 g (40.0%) of fine white crystals, m.p. 140 141C.
Analysis: Calculated for CllHl3ClN202:C,54.89; H,5.44;
~ ,11.64
Found :CJ55OO5; H,5.58;
N,11.52
E amRle 33
~ 3-Chlorophenox~ ~N-~2-propenyl)-1-azetidine-
carboxamide._
~ solution of 5.4 g (0.017 mole) of l-chlorocarbonyl-
3-(3-chlorophenoxy)azetidine in 20 ml of tetrahydro~uran
was treated with 2.3 g (0.04 mole) oP 2-propenylamine and
~tirred for 2 hr. The reaction solution was concentrated
ln vacuo to a rose beige solid. Trituration of the solid
with water gave, a~ter filtering, 4.4 g o~ crude product.
A~ter drying, the ~olid was recrystallized with charcoal
treatment ~rom 2% acetone/isopropyl ether to yield 1.7 g
25 (37 5%) of paIe beige crystal~, m.p~ 87-89C.
Analysis: Calculated for Cl3Hl5Cl~202: C,58.54; H,5.67;
N,10.50
Found : C,58.4~: H,5.72;
~ ~ N,10.49
452
~2~ ~ ~ 7
- 37 -
Exam~le 34
N-Methyl-3- L2 -pyridinylox ~ -l-azetidinscarboxamide.
A 2M benzene &olution of p~o~gene (40 ml, o.o8 mole)
was added to a ~u~pension of 10 g of finely ground potassium
~arbonate in 40 ml of methylene chloride. The mixture was
~tirred for 15 min. at room temperature and 10 g (0.056 mole
o~ l-(l-phenylethyl)-3-(2-pyridyloxy)azetidine in 50 ml of
methylene chloride was added with mild coolin~. Ihe mixture
was stirred at room ~emperature for 1 hr ~nd concentrated
on a rotary evaporator (25~C./30 min). Th~ residue was
treated with 100 ml of tetrahydrofuran and cooled with an
ice bath. To the cooled, stirr~d mixture was added 20 ml
of 40~ aqueous methylamine. ~he mixtur~ was ~tirred for
20 min and partitioned between methylene chloride and
water. Th~ methylene chloride was dried over sodium sulfate
and concentrated. The residue was crystallized from
benzene-ethanol and recrystallized from ~thyl acetate-
i~opropyl alcohol. ~ield o~ title compound was 2.3 g
( 14 %), m .p . 165-168C.
Analysis: Calculat~d for CloHl3N~02: C,57.96; H,6.32:
M,20.28
Found : C,57.93: H,6.34:
N,20.12
Examlple ~5
N- ~ Propenyl)-3-(2-pvridinYloxv ~ l-azetidinecarboxamide.
To a stirred suspension of 10 g (0.072 mole) of finely
ground pota~sium carbonate in 90 ml of methylene ~hloride
was added 32 ml (0.062 mole) o~ 2M phosgene in ben~ene.
The mixture was fitirred $or 15 min and 8 g (0.031 mole) of
l-(l-phenylethyl)-3-(2-pyridyloxy)azetidine in 50 ml o~
methylene chloride was 2dded. The mixture was stirred at
~0 25C. for 2 hr and eoncentrated on a rot~ry evap3ra~0r at
25C. ~0 min and the residue wa~ ~reated with 100 ml of
tetrahydro~uran. The ~irred mixture was cooled with an ice
bath and ~reated dropwi~e with 4 g (0.07 mole) of allyl
amine. After ~tirring ~0 min at 2~C., the ma~eriæl wa~
partitione~ b~ween w~ter ~nd methylene ~hloride. The
: methylen~ ehloride was dried ~nd coneentrated. Th~ r~sidue
1~4~74 4
- ~8 -
was chromatographed on a Waters@ PREP-500 HPLC using a silica
column and eluting with 50% ethylacetate-hexane. The
produ~t was crystallized twice rom isopropyl ether. Yield
of title compound was 1.5 g (21%), m.p. 72-76 C.
Analysis: calculat~d for Cl2Hls~302: C,61.79; ~,6.48; N,18.01
Found : C,61.5~; HJ 6.50; ~,17.96
ExamPle 36
A 2M ~enzene solution of phosgene (~2 ml, 0.062 mole)
was added to a stirred suspension of 10 g of finely ground
potassium carbonate in 80 ml of methylene chloride. The
mixture was stirred for 15 min and 8 g (0.031 mole) of
~ phenylethyl)-3-(2-pyridyloxy)azetidine in 50 ml of
methylene chloride added. The mixture was stirred for 35
min and concentrated on a rotary evaporator (25C./30 min).
The residue was treated with 100 ml of tetrahydrofuran,
cooled with an ice bath and 20 ml of concentrated ammonium
hydroxid~ added slowly while stirring vigorously. The
mixture was stirred 1 hr at room temperature and partitioned
between m~thylene chloride and water. The water layer was
extracted 2 times with methylene chloride and the combined
organic layers were concentrated. The residue was
crystallized from benzene and recrystallized ~rom isopropyl
ether. Yield of title compound was 1.4 g, m.p. 133-137C.
Analysis: Calculated for C~HllN902: C,55.95, H,5.74; N,21.75
~ound : CJ55.73: HJ5~71; N,21.10
Example ~7
2-r ~ PhenYlethvl~ azetidinyloxy~pyridine.
The maleate salt of l-(l-phenylethyl)-3-azetidinol
(65 g,.2~ mole) was partitioned be~ween toluene and dilute
sodium hydroxide and was extracted once with toluene. The
organi~ layer was dried (~odium sul~ate) and co~centrated.
The residue was dissolved in 125 ml o~ dlmethylformamid~ and
~dded dropwi e ~ ~ stirred suspension o~ 9.~ g (.24 mole)
of sodium hydride (washed three times with isooctane) in
400 ml of dimethylformamide It 25-35C. ~he ~olution was
.
~ 4~2
- 39 -
heated to 65C. and 35 g (.22 mole) of 2-br~mopyridine was
added dropwise while heating to 75C. The solution was
stirred and heated at goc. for 1 hr followed by heating at
120C. for 2.75 hr. The mixture was stirred at room
temperature overnight and concentrated. The residue was
partitioned between water and isopropyl ether. The organi
layer was washed twice with water9 dried (sodium sulfate),
filtered and concentrated. Crude yield was 30 g. The residue
was distilled to give 8 g, bp 128-134 C./.01 ~ of product.
Analysis: calculated for Cl~Hl8N20: C,75.57; H,7.13; N,ll.01
Found : C,75.20; H,7.18; ~,10.90
Example 38
1-~3-~4-(Trifluoromethyl)phenoxy ~l-azetidinylcar
lH-imidazole.
A mixture of 1.7 g (0.01 mole) l,l'-carbonyldiimidazole
in 50 ml of tetrahydrofuran and 3 g (0.015 mole~ of
3-~4-(trifluoromethyl)phenoxy~azetidine was stirred for
6 hr. The reaction mixture was diluted with water and :
extracted with 3 x 50 ml o~ methylene chloride. The extracts
upon concentrating ln vacuo gave an ambex residue which was
dissolved in 20 ml of benzene and washed with dilute hydro-
chloric acid, then washed with waker. The benzene portion
was concentrated to give a semi-solid residue which when
- triturated with isopropyl ether gave 1.4 g of gray material.
Recrystallization from acetonitrile gave 1.3 g (41.8O of
fine gray crystals, m.p. 139-140C.
Analysis: calculated for Cl~H12F3N902: C,54.02; H,~.89;
N,13.50
Found : C,54.33; H,3.96;
N,1~.89
~
~
The ~ompound was prepared from the methane sulfonate
of 3-phenoxyazetidine and methylisocyanate as described in
Example 1 of U. S. Patent 4,226,851, m.p. 139-141C.
452
2 7
- 40 -
Exam~le 40
N-Methyl~ rifluoromethyl)phenoxyl-l- zetidine-
carboxamide.
The compound was prepared from 3-~4-(trifluoromethyl-
phenoxy~azetidine and methylisocyanate as described in
Example 3 of U. S. Patent 4,226,861, m.p. 154-157 C.
Example 41
N-MethYl-~-~( trifluoromethYl)phenoxvl ~
~arboxamide.
The compound was prepared from 3-~3~irifluoromethyl~-
phenoxy)azetidine and methylisoeyanate as described inExample ~ of U. S. Patent 4,226,861, m.p. 145-147C.
Example 42
N-Methyl-3-~ trifluoromethyl)phenox~-l-azetidine-
carboxamide.
The compound was prepared from 3-~2-(tri~luoromethyl)-
phenoxy)azetidine and methylisocyanate as des~ribed in
Example 5 of U. S. Patent 4~226,851, m.p. 134-1~6C.
Example 43
N-Meth~1-3-t2 ~mlnocarbonyl)phenoxY~-l-azetidine-
carboxamide.
The compound was prepared from 2-(3-azetidinyloxy)
benzamide and-methylisocyanate as described in Example 2
of U. S. Patent 4,226,861, m.p. 236-240C.
Example 44
-MethYl-7-C3-(aminocarbonvl~phenoxy~ azetidine=
carboxamide.
The compound was prepared from 3-(3-azetidinyloxy)
benzamide and methylisocyanate as described in Example 6
of U. S. Patent 4,226,861, m.p. 238 240C.
3 Example 4
N-Methyl-3-t4-(aminocarbonyl)phenoxy~ azetiaine
carbox~mide.
The compound was prepared from 4-(3-azetidinyloxy)
benzamide and methylisocyanate as described in Example 7
o~ U. S. Patent 4,226,861, mOp. 208-210C.
452
~ 2 ~4
- 41 -
Example 46
fTrifluorometh 1~ henox l-l-azetidinPcarboxamide.
~ ` Y ~ P _ X.~ _ .
A mixture o~ 30.6 9 (0.141 mole) of ~ (trifluoro-
methyl)phenoxy]aze~idine and 42 g (0.321 mole) of nitrourea
(800 in 500 ml of acetone was ~tirred for 5 days (5 days
not requir~d, but convenient~ at room temperature. The
mixture was iltered an~ the filtrate concentrated in vacuo.
The residue was partitioned between 150 ml of water and
100 ml of ethyl ~cetate and the layers ~eparated. ~he
a~ueous layer was washed with 100 ml of ethyl acetate. The
ethyl acetate layers were washed with 75 ml of 5~ aqueous
sodium hydroxide solution followed by 75 ml of water9
dried over sodium ~ulfate and concentrated in vacuo. The
residual oil was crystallized from ethyl alcohol-ethyl
acetate to give 22 g (60%) substantially the title compound.
15 Recrystallization twice from ethyl alcohol gave 9.9 g of
white crystalline solid, m.p. 151-152.5C.
Analysis: Calculated for CllHllF3N20z: C,50.77; H,4.26;
N,10 .76
~ound : CJ50~90; El,4.29;
NJ 10 .71
ExamE~e 47
N-EthYl-3-t3--(trifluoromethyl)ehenoxy~-l-azetidine
carboxamide.
To a stirred and c~illed (15-20C.~ solution of 00024
mole of ~-t3-(trifluoromethyl)phenoxy]azetidine in 50 ml
o dry benzene was added dropwise 1.99 g (0.028 mole) of
ethyl i~ocyanate. The reaction mixture was ~tirred at
room temperature overnight and was diluted with 50 ml of
methylene chloride. The solution was washed with 5% sodium
hydroxide ~2 x 50 ml), water (50 ml), aturated sodium
chloride (25 ml), dried over ~odium hydroxide, and concen-
trated in acuo. The sesidue (9.6 g) was twice recrystal-
lized from ethyl acetate-isopropyl ether t~ give 5.4 9 o~
a white ~olidJ m.p. 125-126C.
Analysis: Cal~ulated for C~Hl5F3N202: C,54.16: H,5.24:
~,9.72
~5 Found : C,54.247 H,~.23,
N,9.74
452
~ Z~ ~ 2 7
- ~2 -
Example 48
N~ MethylethYl)-3-r~-(trifluoromethyl)~henoxY~
azetidinecarboxamide.
To a stirred and chilled (10-20C.~ solution of 9.0 g
(0.042 mole) of 3-C3-(trifluoromethyl)phenoxy]azetidine
in 100 ml of dry methylene chloride was added dropwise 4.1 g
(o.o48 mole) of isopropyl isocyanate. The reaction mixture
was stirred at room temperature for 2 hr and was diluted
with 100 ml of methylene chloride. The solution was washed
with 5% sodium hydroxide t2 x 40 ml), water (50 ml),
saturated sodium chloride (50 ml), dried(sodium sulfate)
and concentrated in vacuo. The residue was crystallized
from ethyl acetate, affording 7.6 g (60.6%). Recrystal-
lization from ethyl acetate gave 5.0 g of clear white
needles, m.p. 150-151.5C.5 Analysis: calculated ~or Cl4Hl7F9N202: C,55.62; H,5.68;
~,9.27
Fou~d : CJ55.77~ H,5.68;
N,9.22
Example 49
-Propyl-3-~3 (trifluoromethyl)phenoxy~ azetidine-0 carboxamide.
.
To a stirred and chilled (10-15C.) solution of 0.027
mole of 3-~3-(trifluoromethyl)phenoxy~azetidine in 100 ml
~ of dry b~nzene was added dropwise 4.0 g (0.047 mole~ of
n-propyl isocyanate. The reaction mixture was stirred at
25 room temperature for 30 minutes. ~he benzene was washed
with dilute sodium bicarbonate (50 ml), water (25 ml),
saturated sodium chloride (25 ml), and dried(sodium
sulfate). The solution volume was reduced to 50 ml, and
30 ml of petroleum ether was add~d, yielding 6-5 g (81O
of product. Recrystallization from isopropyl ether--
isopropyl alcohol gave 6.o g of small white needles, m.p.
115-117~.
Analysis: Calculated for C14Hl7F3N202: C~55-63; H~5-67;
~,9.27
Found : C,55.65; H,5.68;
N,9-25
4 45
- 43 -
Exam~le ~0
N-Butyl-3-~3-(trifluoromethyl)phenoxyl-1-azetidine-
carboxamide.
.
A solution of 18.9 to.o5 mole) of crude 3-t3-(tri-
fluoromethyl)-phenoxy~azetidi'nc (contains an ~ual molar
amount o diphenylmethane) in 100 ml of isopropyl ether
was stirred under nitrogen while 4.96 g (0.05 mole) of
N-butyl isocyanate was slowly added. The clear reaction
solution became warm to the touch, and after 20 minutes
a white crystalline solid began to precipitateO After
stirring for 16 hr, the solid was removed by filtration,
washed with fresh isopropyl ether and air dried to yield
8 g of pale beige crystals, m.p. 108-109C.
Analysis: Calculated for Cl5Hl~F3N202: C,56.96; H,6.o5;
N,8.86
Found : C 9 56.78; H,6.o5;
N,8.83
_ample ~
N-Ethyl-3-C4-(trifluoromethyl)phenoxy~ azetldine-
carboxamide.
_
A solution of 6.5 g (0.03 mole) of crude ~-[4-ttri-
~luoromethyl)phenoxy]azetidine in 50 ml of isopropyl ether
was stirred under a blanket of nitrogen while 2.85 g (0.04
mole) of ethyl isocyanate was added dropwise. After
stirring for 2 hr at amhient temperature, a solid began
to precipitate, and after 4 hr, the solid was collected
by filtration to yield 3.7 g of beige product, m.p. 94-95C.
Rework of the filtrate gave only a trace of additional
~ product. The product was recrystallized from isopropyl
ether/hexane (treated with charcoal) to yield 2.61 9
(30%) of product, m.p. 109-110C.
Analysis: Calculated for Cl3HlsF~N2O2: C,54.17; H,5.25;
3 N,9.72
Found : C,54.40; H,5.33;
N,9.89
~4 ' 452
- 44 _
Example 52
-Butyl-3-L4-(trifluoromethyl)phenoxy-1-azetidine-
carboxamide.
A solution of 6.5 g (0.03 mole) of crude 3-~4-(tri-
~luoromethyl)phenoxy~azetidine in 50 ml of isopropyl ether
5 was tirred under a blanket of nitrogen while 4 9 (0.04
mole) of n-butyl isocyanate was added dropwise. The
reaction was slightly.-exothermic and after 30 min, a solid
~eparated. The ~olid wa~; collected by filtration after
3 hr to give ~.85 g of crystalline product, m.p. 135-136C.
After 24 hr, a second batch of crystals was obtained;
1.5 g; m.p. 132-134C. The two fractions were combined
and recrystallized from cyclohexane to yield 3.6 g of
product (38%), m.p. 136-137C.
Analysis: Calculated for Cl5Hl3F3N202: C,56.96; H,6.o5;
. ~,8.86
Found : C,57.12; H,6.1~;
~,8.93
Example 5~
M-Propyl-3-C, ~ trifluoromethyl)phenoxyl-l-azetidine-
carboxamide.
A solution of 8.7 g (0.0~ mole) of crude ~-~4-(tri-
~luoromethyl)phenoxy~azetidine in 75 ml of isopropyl ether
was ~tirred under a blanket of nitrogen while 4.3 g (0.05
mole) of n-propyl isocyanate was adde~ dropwise. After
~tirring for 2 hr, only a trace of crystalline precipitate
formed in the reaction mixture, a~d after stirring for
18 hr, filtration yielded only 1.1 g of product, m.p.
_ 112-I14C. The filtrate was concentrated in vacuo to give
: a dark amber residue. After 3 days, only 1.3 g of
additional crude product could be obtained from isopropyl
ether ~ exane. All of the reaction products were dissolved
in chloroform and chromatographed on a 200 g silica gel
column. Elution wi h chloroform gave a reddi~h forerun,
which wa~ discarded. The elution was changed to 2% ~thyl
acetate/chloroform, then to 4% ethyl acetate, and ~inally
to 2% methanol/chloroform. All the fractions wer~ combined
7~ 452
~ 45 ~
an~ ¢oncentrated to yield 4.1 g o~ white solid. Recrystal-
lization ~rom cyclohexane yielded 2.86 (23.6%) of ~ine
white crystalline pr~duct, m.p~ 119-120C.
Analy~is: calculat~d for C~H-7F3~z02: ~,55-63; ~,5.67;
~,9.27
Found : C,55.50; H,5.77;
~9-~9
,~
3-r4-(Trifluoromethy~
A solution of 9.6 g (0.025 mole) o~ 4-(trifluoro-
10 methyl)phenoxy~azet~dine 56.66% (contains ~iphenylm~thane~
in 50 ml of acetone was treated with 4~22 9 (0.045 mole) of
~itrourea and 5 ml o~ water. The mix~ure was h2ated on a
ho~ plate until a clear ~olution wa~ obtained then alIowed
to ~ool to ambient temperature ~uring the next 4 hr. The
reaction mixture wa~ diluted with 200 ml o~ ice water ~nd
an oil separat~d ~diphenylm~thane) which~wa~ dissolved in
30/60 petroleum et~r ~nd ~eparated. Upon ~t~nding, a fine
white prscipitate formed in the aqueous 801ution. Filtratio~
yielded 3.6 g of fine white crystal~, m.p. 176-178C.
After drying und~r 0.5 mm Hg vacuum at 80C., tha product
weight w~s reduced to 3.1 g (47.7%), m.p. 178-179C.
Analysi8s Calculated fox CllHl~F3N2~ ~,50.74; H,4.26,
N,10.77
Fou~a ~l: J50 .72; HJ4 .24;
~,10.72
azetidine~arboxamide.
.
A stirred z~urry o~ 6.2 g (0.02 mol~) o~ 3-~4-(tri-
~luoromethyl)phenoxy~a2etidine oxalate in 60 ml of ~e~ra-
hydro~uran was tr~ated with 1.8 y (0.022 mol~) o l-methyl-
~thyl i~o~yan~e ~nd a~r 0.5 hr, 5 ml o~ gri~hyl min~
was ~dded. A cl~ar yellow solution wa~ obtain~d ~nd WD8
~tirr~ for 18 hrJ then tr~t~ with 10 ml of wat~r,
y~olding 5.~ g t87070 o~ pal~ yellow ~ry~tals, ~.p.
151~15~
Analy~i30i c~lculat~a ~or C~4~l7F~N20~ :C,55.63; ~,5..67: ~,9.27
Fou~d sC,55.80; ~,5~71: ~,9.24
2~ 452
- 1~6 -
Exam~le ~6
D imethylethvl ) -3- ~ 4 - ( t r i~luorome~rl ~ phen~xv ] -
l~azetidinecarboxamide.
. .
A stirred slurry of 6.2 g (0.02 mole) ~f 3-~4-(tri-
~luoromethyl)p~enoxy]azetidinej oxalate in 60 ml of tetra-
hydrofuran was tr~ated with 2 g (0.022 mole) of 1,1-
dimethylethyl isocyanate and after 0.5 hr 5 ml of tri2thyl-
amine was added. The reacti~n slurry quickly turned to a
pale yellow solution which was stirred for 18 hr, then
treated with 10 ml of water. After 20 min~ th~ tetra-
hydrofuran portion was separated, dxied over magnesium
sulfate and concentrated on a rotary evaporator. ~he solid
re~idue was recrystallized ~rom isopropyl ether to yield
5 g (79.0%) o~ fine white çrystal~, m.p. 145-146C.
Analysis: Calculated for ClsHl~3N20~: C,56.96J H,6.o5;
~,8.86
Found : C,56.97, H,6.15:
N,8.86
~xample ~7
N-(2-Methy~propyl)-3 1 3- ~ rifluoromethyl)phenoxy
azetidinecarboxamide.
_ _ . . . . .
A stirred solution of 3.6 g (0.022 mole) of 1,1'-
carbonyldiimidazole in 100 ml of methylene chloride under
nitrogen wa~ treated with 1.6 g (0.022 mole) of 2-methyl-
propylamine (added via a ~yringe and needle through a septum
placed in one ne~k of the reaction flask). After stirring
for 1 hr, 6,2 g (0.02 mole~of 3-~3-(txifluoromethyl)phenoxy~
azetidine oxalate was ~dded all at once f~llowed in 30 min
with 5 ml of triethylamine and stirring continued for 18 hrO
The reaction mixture was washed with water (2 x 25 ml),
dried over magnesium sulfate and coneentrated 1n vacuo,
~Q yielding 13 g of crude ~olid residue. Recrystallizati~n
from ethanol/water yield~d 5.9 g of product having a pink
~as~. A ~econd recry~allization from cyclohexane yielded
4.8 g of fine white cry~tal~ m.p. 124-125C. Rewoxk of
the ~iltrates yielded 1.2 g a~ditional beige ~rystal~.
~5 Total yield of product was 75~6% of theory.
Analysis: Cal~ulated ~or Cl5~ F~202: C,56.96: ~,6.05; N,8.86
Found : C~57.01; HJ6.1~: ~,8.88
452
~4274
- 47 -
N-(l,l-Dimethyleth _ -3- ~-(trifluoromethyl)phenoxy~-
l-azetidinecarboxamide.
A ~tirred slurry of 6.2 g (0.02 mole) of 3-t3-(tri-
fluoromethyl)phenoxylazetidine oxalate in 60 ml of tetra-
hydrofuran was treated with 2 g (0.02 mole) of 101-dimethyl-
ethyl isocyanate followed in 30 min with 5 ml of triethyl-
amine. The clear solu~ion which developed was stirred fsr
18 hr. The reaction mixture was treated with 10 ml of
water and after 20 minJ the tetrahydrofuran porti~n was
separa ted, dried over magnes ium ~ul~ate, and concentrated
on a rotary evaporator. The solid residue was recrystal~
lized from ethanol/water, yielding 5.8 g (91-7 0 Of fine
white cry~tals, m.p. 105-107~C.
Analysiæ: calculated ~or Cl5Hl3F3N202: C556-96; H,8 8~:
Found : C,56.95: H,6,14:
~,8.92
Example 59
~-(2-Methylpropyl)-3- ~ tri~luoromethyl)Phen
azetidinecarboxamide.
A ~tirred solution of 3.6 g (0~022 mole) of 1,1-
carbonyldiimidazole in 100 ml of methylene chloride under
nitrogen was treated with 1.6 g (0.022 mole) of 2-methyl-.
propylamine (added via a syxinge ~nd needle through a
septum placed in one neck of the reaction flask). After
stirring ~or 1 hr, 6.2 g (0.02 mole) of 3-~4 (trifluoro-
methyl)phenoxy]azetidine oxalate was added all at once
followed in 30 min with 5 ml of triethylamine and stirring
was continued for 18 hr. The reaction mixture was washed
with water (2 x 25 ml), dried over magnesium sulfate, and
concentrated in vacuo. The solid resi~ue was r2crystalli~ed
_
from ethanol/water, yielding 5.4 g (85.4%~ of pale yellow
crystals.
~alysis: Calculated for Cl~Hl~F~202: C,56.g6; B,6.o5;
N,8.86
Found : C,57.02; H,6. o8 ,
1~52
4t274
~ 48 -
ExamPl~ 60
~- -~ r~henoxy~-l-azet idinecarboxamide .
A solution of 5.4 g (0.017 mole) of l-chlorocarbonyl-
3-(3 chlorophenoxy)azetidine in 20 ml of tetrahydrofuran
was treated with 3 ml of ammonium hydroxide and stirred for
1 hr. ~he reaction mixture was concentrated in vacuo to
a wet ~olid, 4 g, which was recry tallized after drying,
~rom benzene to yield 1.5 g o~ white ~rystalline powder,
m.p. 16~-164.5C. Yield calculated from the 1-(2-phenyl-
ethyl)azetidine compound was ~8.9%.
10Analy~is: cal~ulated ifor Cl~HllClN202: C,52099; H,4.89,
N,12.36
Found : C~52.99; H,4.91
~,12.32
~ FluorophenoxY)-N-methyl-l-azetidinecarboxamide.
15A stirred mixture of 5.4 g (0.02 mole) of 3-(~-fluoro-
phenoxy)-azetidine oxalate and 1.7 g (0.022 mole) of m~thyl
isocyanate in 20 ml o tetrahydrofuran was treated with
5 ml of triethylamine then ~tirring was continued for 3 hr.
~he reaction was diluted with water and the fine crystalline
precipitate obtained was collected by filtration and dried
at 60C. under vacuum to yield 3 g (66.9%) of product,
m.p. 155-156C.
Analysis: calculated for CllHl3FN202: C,58.92; H,5.84;
~,12.49
- Found : CJ58.93; H,5.91:
25N,12.26
ExamPle ~6?
3-~ Fluorophenoxv)-N-methyl-l-azetidinecarboxamide.
A stirred slurry of o.38 g (0.02 mole~ o~ 60% sodium
hydride as a mineral oil su~pension in 10 ml of dry
30 dimethylformamide was treated under nitrogen with the
dropwise addition of 1.12 g (0.01 mole) of ~-fluorophenol
in 20 ml of dimethylfonnamide. After 1 hr, the mixture was
heated at 90C. fox 20 min then 2.1 g (0.01 mole) of
~-methyl-~-r(methanesulfonyljoxy~ a~etidinecar~oxamide
~5 wa added as a ~olid. The reaction mixture was then
stirred at 90C. for 8 hr. The reaction mixture wa~
cooled by adding iFe water then further dilut~d ~o 200 ml
--- 12~27~ 452
-- 49 -
with water and extracted with 3 x 50 ml of methylen~
chloride. Less than 1 g of oil was obtained upon concen-
trations of the extracts. ~xtraction with 4 x 50 ml sf
benzene gave only a trace of product. The mass ~pectrum
(CI) showed the expected p+l at 225 m/e. The produ~t
~olidified on standing and was recrystallized from methanol/
water to yield 650 mg (29.0%) of flake-like sil~er
crystals3 m.p. 156-158~C.
~nalysis: Calculated for CllHl FN202: -
C,5~.92; H,5.84; N,12.49
Found C,58.93; H,5.91; ~,12.42
452
274
-- 50 --
Pha~TIacological Test Procedures
Muscle Relaxant Test
~ rhe test prt~cedure relied on to indicate positivemuscle relaxant activity i5 the morphine-Induc~d Str~ub Tail
in Mice Test de~cribed by G. D. ~ovak in DRUG DEVELOPMENT
RESEA~CH (19~2) 2: 383-~86, ~xcept 8 animals per group were
used per te~t rather than 10. The test i~ $umm~rized as
: ~ollow~: The test drug, ref rence drug, and control
articles to be admi~istered are prepared in saline, 0.5%
aqueous methylcellulose suspe~sion or other, depending on
solubility, in ~uch ~oncentration that the volum~ adminis-
tered i~ 10 ml/~g. The initial screening dose of the test
drug is u-~ually 100 mg~ky. Group~ of 8 mice are given an
IP dose of a compound or vehicle prepared as de~cribed
~bove. Aftex 15 min, mice are admin i8 ter~d morphine
~ulfate, 60 mg/kg, subcutaneously. Fîfteen minutes after
admini~tration of morphine ~i.e., 30 min after test compound
administration), mice were scored for presence of Straub
Tail defined as an elevation of the tail ~t least 90 degrees
from the horizontal. An ED50 value may be determined from
at least three logarithmically ~paced doses by the method
o~ Litchfield and Wilcoxon (1949), J. PH~RM~COL. EXP. THER.
~ : 99-113. Compared to a reference compound, methocarbamol~
which exhibited ~n ED50 of 75-110 in the above S~raub Tail
Test, the more active compounds of Formula I were 5-10 times
more active.
~ 2~ A35~ 3~
~ The test screening procedure relied on to indicate
: positive antianxiety resp~nse i8 a modification of the Vogel
Conflict Test whiah i~ ba~ed on ~hock-~uppressed drinXing
behavior in rat~ outlined by J. R. Vogel, et al in
PSYCHOP~RMACOLOGY 21: 1-7 (1971~. T~e procedure u~ed i~
: as follow~: The t:est re~erence and control articles to be
administered intraperitoneally in phy~iological salinQ,
0.5% aoueous methylcellu}ose or other ~epending on ~
solubility in such ~oncentration that the volume ~dminister~d
35 i~ 5 n~ sg. The initial ~creening dose of ~he t~st article
452
lZ~ 74
-- 51 --
is usually 100.0 mg/kg initially, and the reference drug
(diazepam) is 5 mg/kg.
Prior to dosing, rats are housed 2 per cage and deprived
of water for 48 hr and thereafter randomized into treatment
groups of five. Feed is available ad libitum. Thirty
minutes af~er dosing, each rat is placed individually in a
plexiglass cage measuring 18 cm in width, 13 ~m in height
and 29.5 cm in length and equipped with a stainl~ss steel
grid floor. The cage is ~overed with a plastic lid containing
holes to facilitate introduction of a water bottle t30 ml
plastic ~entrifuge tube) with a rubber stopper and metal
drinking tube. A Drinkometer circuit (Omniteck Electronics,
IncO, 3000 Cortona Road, Columbus, Ohio 43204j is conn~cted
between the drinking tube and the grid floor of the apparatus
80 that the rat ~ompletes the circuit whenever it licks the
tube. The procedure is to allow the rat to ind the drinking
tube and complete 20 licks (as displayed on the Drinkometer
digital readout) prior to the start of the experimental
~ession. Rats not xeaching this criterion are discarded.
A three minute experimental session is initiated by a 0.25
mA shock at the 20th lick. Rats that continue drinking
will experience a shock at each successive 20th lick. The
total number of shocks during the experimental session are
recorded as follows:
total licks + 1 = total shocks
Stati~tical analysis i8 performed by the Dunn's Multiple
Comparison Test described by O. J. Dunn (1964) TECH~OMETRICS
6(3):241-52. The mean number of sho~ks experienced by the
control group is compared with those of each drug-treated
group. Signi~icance is considered at pc 0.1. The higher
~he ~otal ~hocks compared to con~rol, the more active is
the compound. Active compounds may th~n be similarly tested
at reduced d~sage IllustrativelyJ one preferred compound,
35 the compound of Example 16J namely, ~-cyclopropyl-3-t4-(tri-
81uoromethyl)phenoxy~ l-azetidinecarboxamide wa~ active at as
low a dosage as 10 m~ per kg, where total number o~ shocks
taken as calculated above over ~ont~ol was 14/4.6 (P=0.03)
~ompared to ~b~ut 12-15/4 for diazepam.
452
4Z7
- 52 -
Pharmaceutical comp~sitions
The methods of treating anxiety, muscle tension, and
spasticity in mammals is best carried out by administering
as active ingredients in a pharmaceutical composition at
least one of the comp~unds of Formula I in associ2tion with
a p~armaceutical carrier or excipient. Ihe compounds are
thu6 presented in a therapeutic composition ~uitable for
oral, rectal, parenteral, subcutaneous, intramuscular,
intraperitoneal, or intravenous admini~tration. Thus, for
example, the composition for oral administration can take
the form of elixirs, capsules, tablets, or coated tablets
containing carriers conveniently used in the pharmaceutical
axt. Suitable tableting excipients include lactose, potato,
and maize starches, talc, gelatin, stearic and ~ilicic
acids, magnesium stearate and polyvinyl pyrrolidone.
For parenteral administration, the carrier can be
comprised o~ a sterile parenterally acceptable li~uid;
e.g., water or arachis oil contained in ampoules.
In compositions for rectal administration, the carrier
can be comprised of a suppo~itory ba~e, e.g., cocoa butter
or glyceride.
Advantageously, the compositions are formulated as
dosage units, each unit being adapted to supply a fixed
do~e of active ingredients. Tablets, coated tablets,
capsules, ampoules and suppositories are examples of
preferred dosage forms according to the invention. It is
only necessary that the active ingredient constitute an
effective amount; i.e., such that a suitable effective
dosage will be consistent with the dosage form employed.
The exact individual dc~ages as well as daily dosages will,
of course, be determined according to standard medical
principles under the direction of a physician or veterinarian.
Testing ~uggests that rompound~ of Formula I will be
efective in man or muscle relaxant effect~ ~t ~bout 4-40
mg~kg body weight per day. Illustratively, one compound,
nam~ly, ~-methyl-3-~3-(trifluorome$hyl3phencxy~ azetidine
carb3xamide ~Example 41), w~ found to be ~out 5 tïmes a~
3~Z~274
~ 53 ~ 66197-177
p~ent a~ Methocarbamol as a muscle relaxant.
The suggested regimen, ~or example, for the compound of
Example4l s a muscle relaxant appears to be about lO mg~kg/
day or for a 75 kg individual ~bout a unit do~age of 250 mg,
t.i.d.
E~fective daily dosages of compounds of Formula I as
antianxiety agents are projected to be about l-lO mg ~g/day
body weight based on animal data.
The pharmaceutical compositions according to khe
present invention in the ready-to-use dosage unit form, in
practical use, may be put into containers which bear
instructions that the composition should be used for treating
muscle tension, muscle spasticity and anxiety of animals
including humans.