Note: Descriptions are shown in the official language in which they were submitted.
7~25~
IMPROVED PROCESS FOR THE PREPARATION
OF N-ACETYL-P-AMINOPHENOL
Background of the Invention
This invention relates to the preparation of
N-acetyl-p-aminophenol by acetylation of p-aminophenol
in an aqueous medium, and more particularly, to an im-
proved process for the recovery of N-acetyl-p-aminophe-
nol from -the aqueous medium.
The analgesic N-acetyl-p-aminophenol, commonly
known in the art as acetaminophen or "APAP", is commer-
cially prepared by reaction of p-aminophenol ("PAP") with
acetic anhydride in an acidic aqueous medium. p-Amino-
phenol is conventionally prepared by catalytic hydrogen-
ation of nitrobenzene in a mineral acid system. After
purification of the reaction product for removal of res-
idual impurities such as 4,4'-diaminodiphenyl ether, the
p-aminophenol is precipi-tated as its alkali metal salt by
addition of a caustic solution or an alkali metal carbo-
nate or bicarbonate. The precipitated product is redis-
solved in acetic acid solution and acetic anhydride addedfor acetylation of the PAP to APAP.
In the co-assigned IJ.S. Patent No. 4 t 440,954 of
William R. Clingan et al, issued April 3, 1984, a process
is described for extractive removal of 4,4'-diaminodiphe-
nyl ether and related impurities from the aqueous reac-
tion product obtained upon catalytic hydrogenation of
nitrobenzene to PAP. Use of this process reduces the
level of impurities to be dealt with in the acetylation
of PAP to APAP. Irrespective of what process is used for
- ~z~
preparation and refining of the intermediate PAP,
APAP is produced by acetylation of PAP in acidic
aqueous solution.
APAP is recovered from the reaction solu-
tion by crystallization and filtration or centrifu-
gation, yielding a mother liquor that remains satur-
ated in APAP and contains a substantial proportion of
acetic acid. Unless this crude mother liquor is sub-
jected to further processing for recovery of at least
a portion of the APAP and acetic acid remaining in the
solution, a substantial yield loss may be suffered.
Commercially acetic acid has been recovered by dis-
tillation of the crude mother liquor, leaving a res-
idue which may be crystallized for recovery of a sec-
ond crop of APAP crystals. However, in this commer-
cial practice, the acetic acid fraction recovered typ-
ically has a strength of only about 20~ by weight to
25% by weight which limits the uses to which it can be
put, and its value. Color bodies in the mother liquor
are concentrated in the distillation residue and tend
to impart an undesirably strong color to the second
crop of APAP crystals. Additionally, corrosion pro-
blems may be encountered in distillation of the mother
liquor due to the presence of sulfur dioxide released
from the bisulfate salts conventionally added to an
APAP crystallization system to protect the APAP pro-
duct against air oxidation.
Statement of the Invention
__
The invention as claimed herein is in a
process for the preparation of N-acetyl-p-aminophenol
which comprises acetylation of p-aminophenol in an
aqueous medium to produce a crude aqueous reaction
---` ` lZ~2~
mixture, recovery of the N-acetyl-p-aminophenol pro-
duct by crystallization from an aqueous system com-
prising the reaction mixture, and separation of the
crystals from the crude mother liquor, said mother
liquor containing residual N-acetyl~p-aminophenol and
unreacted acetic acid, the improvement comprising re-
covering residual N-acetyl-p-aminophenol and acetic
acid from the crude mother liquor by liquid/liquid
extraction with a water immiscible organic solvent,
thereby producing an extract containing N-acetyl-p-
aminophenol and acetic acid.
The extract thus obtained may be distilled
to strip off the solvent and acetic acid.
The invention is further directed to such
a process in which the residue from the distillation
is mixed with water to reduce solubility of APAP in
the residue, and the resultant mixture is cooled to
crystallize APAP therefrom and produce a secondary
mother liquor. Crystallized APAP is separated from
the secondary mother liquor.
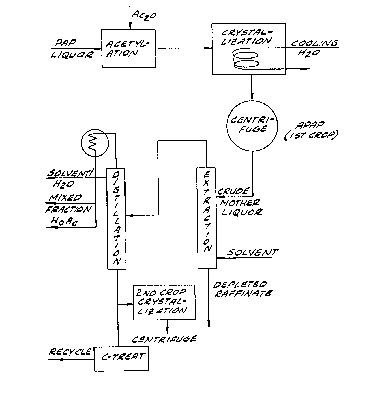
Brief Description of the Drawing
The single figure of the drawing is a sche-
matic flow sheet for the improved process of the in-
vention.
~``;?,`i~
~2~7~
Description of the Preferred EmbodimentS
In accordance with the present invention, an
improved process has been discovered for recovering acetic
acid from the crude mother liquor obtained upon the
crystallization of APAP from the reaction mixture that is
produced in the acetylation of PAP. In the improved
process, both acetic acid and residual APAP are removed
from the crude mother liquor by liquid-liquid extraction
and the acetic acid thereafter recovered from the extract
by distillation, leaving a residue containing ~PAP that may
be either recycled or subjected to further crystallization
for recovery of a second crop of APAP crystals.
Illustrated in the single figure of the drawing
is a schematic flow sheet of the improved process of the
invention. In this process, the slurry obtained by crystal-
lization of APAP from the acetylation reaction mixture is
centrifuged to produce the primary APAP product and a fil-
trate which is subjected to liquid-liquid extraction. Any
of a variety of conventional water-immiscible organic
solvents can be used for the extraction. However, to allow
recovery of substantially pure acetic acid the solvent
should have a volatility which differs significantly from
the volatility of acetic acid. It is further preferred
that the solvent not form an azeotrope with acetic acid.
Preferably the solvent should have a boiling point which
differs by at least about 20C from 118C, the boiling
point of acetic acid. Those solvents having a greater
volatility than acetic acid are more preferred. Solvents
which may be used include halogenated solvents such as
methylene dichloride, ethylene dichloride, and chloroform,
J~
ketones such as methyl ethyl ketone, methyl isopropyl
ketone, and methyl isoamyl ketone, and substantially
water-immiscible alcohols such as hexanol and amyl
alcohol. Preferably, however, the solvent is an ester of a
lower alcohol and a lowmolecular weight alkanoic acid such
as ethyl acetate, ethyl proprionate, methyl proprionate,
methyl acetate, an~ butyl proprionate, propyl formate, and
propyl acetate. Ethyl acetate is particularly preferred.
Extraction may be carried out by contacting the
crude mother liquor with thè solvent in conventional equip-
ment such as, for example, a stirred mixer/settler.
Preferably, extraction is conducted in a continuous
countercurrent extraction system having a plurality ofequilibrium stages and comprising means for promoting mass
lS transfer between the solvent phase and the aqueous phase~
Thus, the extraction may be carried out in a vertical-
packed column or in a centrifugal extractor such as that
sold under the trade designation ~Podbielniak" by
Baker-Perkins, or ~DeLaval" by TransAmerican DeLaval, Inc.
Preferably, however, the extraction is carried out in a
reciprocating plate column containing between about five
and about ten equilibrium stages.
While the temperature of extraction is not
critical, it should be below the temperature at which sig-
nificant mutual solubility of water and solvent is
incurred. This temperature varies with the identity of the
solvent. Generally, however, satisfactory results are
achieved where the extraction is carried out at a tempera-
ture in a range of about between 0C and about 30C.
By countercurrent extraction using a plurality of
stages, over ~0% of the APAP contained in the mother liquor
can be transferred to the extract. Recovery of acetic acid
* Trade Mark
.. .
?~
is even more efficient and can exceed 99%. Thus, aqueous
raffinate from the extraction, which contains ammonium
salts, sulfur dioxide, and color bodies can be discarded.
The extract is subjected to distillation for
recovery of the solvent and acetic acid, leaving a residue
containing APAP which can be recycled or subjected to
crystallization for recovery of a second crop of APAP.
In the distillation step, at least two separate
overhead fractions are preferably recovered, one comprising
principally solvent and water, and the other comprising
principally acetic acid and water. Where, for example, a
relatively volatile solvent such as ethyl acetate is used
for the extraction, the most volatile fraction recovered in
the distillation comprises solvent and water, while a
distinctly higher boiling fraction is substantially free of
solvent and comprises a high concentration of acetic acid
in water. In order to obtain a clean separation an inter-
mediate fraction, which comprises a mixture of all of three
of the vaporized components, may be collected. The vola-
tile fraction comprising solvent and water may be allowedto separate into a solvent layer and an aqueous layer, and
the solvent decanted for recycle to the extraction step.
The fraction comprising a high concentration of acetic acid
in water is removed from the process and used as a source
of acetic acid in other operations. Where an intermediate
fraction is obtained comprising solvent, water, and acetic
acid, the solvent may also be decanted and recycled and the
aqu~ous phase, comprising a relatively dilute solution of
acetic acid and water, may be used in other processing
operations or discarded.
Distillation is conveniently carried out batch-
wise to facilitate recovery of fractions of the desired
-
~Z~7~
composition. ~owever, in a high volume operation a con-
tinuous distillation column may be used with recovery of
sidestreams of the desired fractions. Whether batch or
continuous, distillation may be conveniently carried out at
atmospheric pressure, for example, in a packed column con-
taining at least one transfer unit. Distillation condi-
tions are readily controlled to provide an aqueous acetic
acid fraction having an acid strength of 65% by weight or
greater. If a column comprising five or more transfer
units is utilized, glacial acetic acid may be recovered.
The bottom fraction obtained in the distillation
contains APAP and typically about 25% by weight acetic acid
and may be subjected to crystallization for recovery of a
second crop of APAP or, alternatively, recycled to the
primary crystallization step. Where a second crop of
crystals is desired, the bottom fraction is mixed with
water to depress APAP solubility and then cooled to ambient
temperature or somewhat below in order to crystallize
APAP. Although the amount of water mixed with the bottom
fraction is not highly critical, it is preferably between
about 0.7 parts by volume and about 1.3 parts by volume per
part by volume of the bottom fraction. Roughly equal volu-
metric proportions are most preferred. If desired, the
mixture of water and bottom fraction may be refrigerated
~or crystallization, but the marginal increase in yield
thereby obtained may not justify refrigeration in many
cases.
` After crystallization is complete, the resultant
slurry of APAP in secondary mother liquor is fed to a cen-
trifuge or a filter for recovery of the second crop ofcrystals. The crystals are preferably washed with cold
water and dried under vacuum. In an alternative embodiment
of the invention, as noted above~ the bottom fraction
obtained by the ~istillation step may be recycled to the
primary crystallization step for recovery of further
amounts of APAP. Where the bottom fraction is recycled, it
is preferably treated by adsorption for removal of residual
amounts of impurities. Adsorption of impurities from the
bottom fraction is carried out by contacting the fraction
with an adsorbent such as activated carbon. Alternatively,
other adsorbents known to the art, such as clay, may be
used for treatment of the bottom fraction prior to~
recycle. As a further alternative, the bottom fraction may
be passed through an ion exchange column for separation of
impurities.
The following example illustrates the invention.
EXAMPLE
APAP was prepared by acetylation of PAP in
aqueous acidic solution. This solution was cooled for
crystallization of APAP and the crude mother liquor
separated by centrifugation.
An aliquot of the APAP mother liquor (500 mL) was
contacted under agitation with ethyl acetate (200 mL) and
the phases allowed to separate. The extract phase was
removed and the aqueous phase extracted with two additional
portions of ethyl acetate (200 mL each). The three
extracts were combined and subjected to fractional
distillation at atmospheric pres- sure. The following
fractions were collected:
~2~
Temperature ~olume
_(C) (mL) ~ sition
70 - 81 590 Ethyl Acetate + Water
81 - 94 37 Ethyl Acetate ~ Water +
Acetic Acid
94 - 106 62 Acetic Acid + Water
Approximately 30mL to 40mL of distillation heel remained in
the distillation pot. This heel was allowed to cool
slightly, after which deionized water (35 mL) was added
slowly to the pot. APAP started to crystallize as water
was added. The slurry was cooled to 20C and the crystal-
line APAP obtained was collected on a filter. These
crystal~ were washed with cold water and thereafter dried
at 70C under vacuum. Yield of APAP was 9.1 g, 57% based
lS on the APAP content of the crude mother liquor.
In view of the above, it will be seen that the
several objects of the invention are achieved and other
advantageous results attained.
As various changes could be made in the above
methods without departing from the scope of the invention,
it is intended that all matter contained in the above
description or shown in the accompanying drawings shall be
interpreted as illustrative and not in a limiting sense.