Note: Descriptions are shown in the official language in which they were submitted.
1%97~L3~
C-1~L 751
-- 1 --
TECHNICAL FIELD
The present invention relates to a continuous process
for the production of mononitrobenzene. In particular, the
invention relates to an improved, continuous adiabatic
process for the production of nitrobenzene.
BACRGROUND OF THE INVENTIOM
The process of nitrating benzene is old and well known
and has been commerically practiced ~or many years to yield
mononitrobenzene used, in turn, in the production of aniline.
10 Conventionally, the manufacture of nitrobenzene comprises the
batchwise, stepwise or continuous addition of mixed nitric
acid and sulphuric acid to benzene. This nitration reaction
may be conducted at temperatures controlled in the range of
60 - 70C with the removal of the heat of reaction or it may
15 be conducted under adiabatic conditions described by Castner
in U.S. Patent No. 2,256,999, where little or no heat of
reaction is remo~ed and in which the heat of reaction is
utilized in later stages of spent acid reconcentration. The
mixed nitric/sulphuric acid employed by Castner in his
20 adiabatic process comprises a mixture of 75% strength
sulphuric acid together with sufficient 63% strength nitric
acid such that the mixture has a content of 3~ nitric acid.
In an improved adiabatic process described by Alexanderson et
al in U.S. Patent No. 4,091,042, wherein the reaction is
25 carried out under superatmospheric pressure, the mixed acid
contains 3 - 8.5~ nitric acid, from 58.5 - 70.0% sulphuric
acid and not less than about 25% of water. In both the
Castner and Alexanderson et al processes, the mixed acid and
a stoichiometric excess of benzene are admixed and reacted
30 together under vigorous agitation at temperatures o~ 100C or
greater.
Since the acid phase and the benzene phase are not
miscible, the reaction rate and the reaction efficiency
between the phases are largely limited by mass transfer; that
35 is, by the ability to expose large interfacial areas of each
C-I-L 751
-- 2 --
of the phases to each other. As the interfacial areas are
increased, the reaction rate between the phases is enhanced.
In conventional nitrobenzene production facilities, these
interfacial areas are normally created by reacting the two
phases in one or more agitated vessels where high shear
forces are applied to the liquids. Alexanderson et al
described the use of "vigorous agitation~ to disperse the
benzene throughout the reaction mixture. In other similar
nitration processes, various means have been proposed to
10 bring together the immiscible phases. In the process of
Toischer et al (U.S. Patent No. 3,431,312), a cascade of
stirred reaction chambers is used in the nitration of
toluene. In the process of Terao et al (U.S. Patent
No. 3,160,669), a compartmentalized, elongated, baffled
15 reaction zone containing a series of agitating blades fixed
to a stirrer shaft are provided. In the process of Nilsson
(U.S. Patent No. 2,737,522), glycerine is nitrated with mixed
acid by injecting a pressurized jet of acid into a
venturi-shaped reaction zone to contact a similar jet of
20 ~lycerine within the zone where intimate mixing is caused to
take place. McKinney in U.S. Patent No. 2,951,866 describes
the use of a tubular reaction zone wherein separate streams
of polyhydric alcohol and nitrating acid are impinged upon
each other to form a turbulent reaction mixture. A similar
25 tubular reactor is described by Stow in U.S. Patent No.
3,111,538. Gebauer, in U.S. Patent No. 4,251,455, makes
reference to the process of German Patent No. 1,135,876
wherein the nitration of polyhydric alcohols is achieved by
impinging the two reactants upon each other. In the Chemical
30 Engineering Handbook (Perry), 6th Edition, a number of
methods are proposed to achieve intimate mixing or contact
between liquids including, for example, in-line motionless
mixers, mechanical agitation, gas agitation, jet mixers,
injectors, orifice mixers and nozzle mixers.
None of the aforesaid methods for achieving large
~979L3Z
C-I-L 751
-- 3 --
interfacial areas of contact between immiscible liquid phases
is completely satisfactory nor has any method, other than
mechanical agitation, been used commerically to an~ degree in
the manufacture of mononitrobenzene. These methods either
suffer from high capital and maintenance costs and high power
requirements, as in the case of agitated vesse~s, or they are
difficult to control in terms of optimum reaction efficiency
as in the case of impinging streams or jets. There,
therefore, remains a need for a benzene nitration process
10 which is economic to construct and operate, which is safe and
simple to control and which leads to optimum output of
reaction product at least possible cost.
DISCLOSURE OF T~E INVENTION
It is an object of this invention to provide a process
15 for the reliable manufacture of mononitrobenzene which
obviates or mitigates the known deficiencies of the prior art
processes.
It is a further object of this invention to provide a
process for the safe and energv-efficient manufacture of
20 mononitrobenzene on a continuous basis.
Therefore, according to this invention there is provided
a process for the continuous production of mononitrobenzene
or other organic nitro compound which process comprises
simultaneously and continuously introducing into a reaction
25 chamber separate liquid streams of a mixed nitrating acid
component and an immiscible organic component, one of the
said components, for example, the mixed acid component being
introduced into the said organic component through turbulence
inducing means which constricts the flow of said acid
30 component such as to cause its disruption to form fine
droplets of a desired size upon its emergence into the
reaction chamber, said turbulence inducing means further
causing said mixed acid to emerge in a flow pattern and at a
flow rate sufficient to cause the droplets so formed to come
35 into contact with a sufficient quantity of the organic
~L~97~3~
C-I-L 751
-- 4 --
component to provide for reaction between the said acid and
the said organic component to form an organic nitro compound.
Alternatively, the organic component may be introduced
through the turbulence-inducing means into the mixed acid
component within the reaction chamber to form the organic
nitro compound.
The means for causing disruption of one or the other of
the reactants may be any form of pressure atomiser i.e. a
device wherein the liquid is forced under pressure through an
10 orifice to discharge in the form of droplets of a size
acceptable for the purpose defined herein.
Thus, it will be appreciated that this process has the
advantage that the desired organic nitro compound product
can, in most instances, be produced in only one step without
15 reliance on liquid/liquid shear and so the use of the
expensive and energy inefficient shear mixing devices
typically required is avoided. In the event that less than
complete conversion of the organic component is achieved in a
single reaction chamber, a second chamber may be employed
20 wherein the product of the first chamber is subjected to
further reaction by exposure to additional amounts of one or
the other component in droplet form. Alternatively, a single
reaction chamber may be coupled with, for example, an in-line
mixer or orifice plate wherein a final or "polishing"
25 nitration is accomplished through intermixing of the phases..
Preferably, the flow of the organic component, for
example, benzene is constricted and atomized by means of an
orifice in said turbulence-inducing means wherein the path
length (Ln) through said orifice is sufficient so as to
30 provide for the greatest pressure gradient with minimum
losses in energy. The diameter of the orifice Do (m) should
be selected in accordance with the intended volume flow rate
Q (m3.s 1) and the desired droplet size. It can be shown
that maximum possible droplet size
D ~
Dmax Q3/4
~29~7~3Z
C-I-L 751
(assuming that no mechanism for coalescence exists) so that
~or constant drop size, if flow rate is increased, e.g.
7-fold, the nozzle diameter should be increased approximately
2-fold. Suitable orifice sizes for the purposes set out
herein may be in the range of about 0.001 m to about 0.02 m,
preferably from 0.005 m to about 0.015 m.
Preferably, the means for causing disruption of the
organic (benzene) component is a nozzle which discharges into
the reaction chamber, advantageously in a readily replaceable
lO manner for the purposes of nozzle exchange which nozzle is
adapted to constrict flow sufficiently to cause turbulence in
the stream of the benzene phase to provide for discharge of
dispersed single droplets of a size comparable to the eddies
in the flow created within the nozzle in use under operating
15 conditions. The advantage of this arrangement is that it
provides for localized break up of the benzene component
directly into the mixed acid component which provides for
localized energy dissipation and very efficient energy
transfer. Thus, preferred arrangements provide for local
20 energy dissipation rate (~) in the range of from 104 to 108
W/kg with most preferred rates being in excess of ]o6 W/kg.
Energy dissipation rate is routinely calculated (assuming
Newtonian liquid behaviour) from knowledge of the path length
Ln (m) through the orifice of the nozzle, the pressure drop
25 VPn (N.m 2) across the nozzle, the density ~F (kg.m 3) of
the mixed acid phase and the mean fluid velocity U (m.s.
all of which can be readily measured. The pressure drop
across the nozzle for a sharp edged orifice is shown by the
following equation:-
~;~g7~3Z
C-I-L 751
Pn ~ /2 ~ F U (1)
dt unlt tlme m
th~n the speciflc power dissipation ~ may be written as
~ F ~2)
5 w~lere ~Pn = ~ Pn and f rom ~1 )
Ln
we have ~ = /2 U3/Ln
By virtue of this invention, selected droplet sizes are
obtainable such that the average droplet size lies in a
narrow range so that high populations of droplets of less
than 8~um, preferably of about 4~um or less, down to about
0.5~um are consistently achievable. Ordinarily, it will be
found that for a given set of process conditions, droplet
sizes will lie within a relatively narrow range (save for a
small proportion of droplets which arise from coalescence of
formed droplets). Thus, for example, taking a flow rate of
say 20 l.m 1 for the benzene stream through a 4.6 mm diameter
orifiCe~ max Y
ax ~ (c8p ) /5 ~ /5
average
Daverage
C~I-L 7~1
-- 7
where ~ = interfacial tension (N.m
CD = drag coefficient of droplet
~C = density of the benzene phase (kg.m
~ = specific energy dissipation rate (W.kg 1)
U = dynamic benzene phase velocity ~m~.s 1)
Thus the droplet size, and hence the exposed interfacial
area, is controllable by flow rate and orifice dimensions.
Flow of the benzene component is isotropic, turbulent flow.
The velocities of flow and, hence, bulk Reynolds numbers (Re)
associated with these conditions are in the range of from
30,000 to 500,000, depending on plant throughput, and,
preferably, upwards of 50,000. The rate of flow of each
stream is, preferably, controlled to provide for ratios of
mixed acid component to benzene component by which a slight
excess tl - 10%) of benzene over the nitric acid content of
the mixed acid is achieved.
More preferably, the nozzle is one capable of
discharging a turbulent stream as a transient divergent sheet
producing a divergent pattern ~"solid cone") of droplets and
20 may or may not impart a rotational motion element to said
droplets. Such flow patterns may be obtained by use of
nozzles known from the spray-drying art.
The nozzle, preferably, includes internal baffles or
other means defining one or more tangential or helical
~5 passages to provide for a radial (helical) emergent flow
superimposed on a linear divergent flow to produce a
resultant helical flow which serves to enhance dispersion of
the droplets rapidly formed on discharge. The advantage of
this arrangement is that the helical flow creates a pressure
30 gradient along the notional jet boundary which facilitates
entrainment of the mixed acid component and mixing of
droplets with the continuously formed mononitrobenzene
reaction product.
The nozzle, preferably, has an exit cone angle of 70 or
C-I-L 751
-- 8 --
less. At 0 or very low exit nozzle cone angles, there is a
pronounced tendency to produce a collimated narrow stream of
the benzene component at higher stream velocities which is
unsatisfactory for efficient reaction rates.
Operating pressures (back pressure in no2zle) are
suitably in the range of from 10 psi to 200 psi, preferably,
30 psi to 135 psi and upwards, bearing in mind that the
higher the pressure used the greater the energy available for
droplet creation, the more efficient the chemical reaction
10 becomes. It is likely that pressure exceeding 160 psi would
be unnecessary for normal purposes.
The linear fluid velocity through the nozzle is
typically from 5 to 40 ms 1 and average droplet sizes of from
7 to 10 down to 1 or less,um are achieved.
As mentioned above, preferrsd nozzles are characterized
by short and narrow constrictions so that the stream of the
atomized phase passes rapidly through the nozzle constriction
under a high pressure gradient. Nozzles which will be
suitable for the purposes of this invention are commerically
20 available (Spraying Systems Co., Wheaton, Illinois, U.S.~.).
Preferably, the dimensions of the reaction chamber are
such as to minimize impingement of droplets on the walls of `-
the chamber so as to mitigate the problem of coalescence of
the droplets prior to complete chemical reactionO In other
25 words, the zone of droplet formation and initial dispersion
should desirably be remote from boundary surfaces.
Conveniently, the reaction chamber is a cylindrical vessel
having removable end closures, one of which has means
providing for removal of continuously formed reaction product
30 and waste acid. The removal of product is desirably
continuous but it is possible to provide for continual
removal of batches of product at selected intervals depending
upon the capacity of the reaction chamber and rate of
production of the organic nitro compound.
Preferably, also the mixed acid component is fed through
~æ97~3z
C-I-L 751
g _
a pipe passing directly into the reaction chamber in the
region of the organic (benzene) droplet discharge from the
noæzle and which is located adjacent to, but spaced
sufficiently from the nozzle to minimize coalescence of
droplets whilst enabling entrainment of the mixed acid stream
in said droplet discharge. A suitable arrangement is to
provide the nozzle centrally in an end wall of a cylindrical
vessel defining the reaction chamber and to have the pipe for
discharge of the mixed acid component passing through the
10 cylindrical wall to emerge at a position close to the nozzle
allowing said mixed acid stream to contact the benzene
droplets discharged by said nozzle and pass into the
continuously ~ormed reaction product.
The point or points of discharge of the mixed acid
lS component into the reaction chamber are capable of
substantial adjustment both laterally (i.e. at right angles
to the length dimension of the chamber), although probably
there will be a longitudinal position beyond which
insufficient entrainment (back mixing) of mixed acid will
20 occur and efficient chemical reaction will be defeated.
The invention in one preferred aspect provides a process
for producing mononitrobenzene comprising forming a turbulent
jet of benzene, preferably having a Reynolds number of
greater than about 50,000, subject to plant throughput, to
25 produce droplets of a selected size within the range of from
about less than 1 um to 10 um diameter and contacting said
jet continuously in the region of droplet formation with
mixed acid in an amount which is sufficient to provide
essentially complete conversion of nitric acid and produce
~o a mononitrobenzene containing substantially no
di- nitrobenzene.
Most preferably, the predominant droplet size is from
less than l~um to 2 ~m. "Size" means the number average
droplet diameter.
Employing prior art reaction apparatus wherein one
~97~32 C-I-L 751
-- 10 --
component is injected into a second component, use is made of
a velocity gradient between the components which provides a
shearing force which creates a series of small droplets.
Such shearing action is generally incapable of producing very
fine droplets except under extreme condition. In the process
of the present invention, no reliance is made on a velocity
gradient between the phases and consequent liquid/liquid
shear. Instead, fine droplets are produced from the organic
material which droplets are thereafter distributed throughout
10 the mixed acid. The degree of atomization and, consequently,
the droplet siæe of the organic component can be adjusted by
selecting the appropriate atomizing nozzle. The particle or
droplet size distribution of the acid component is narrow.
Although not specifically tested, it is envisioned that
15 the process of the present invention will also include the
step of introducing both the mixed acid and the nitratable
organic compound into the reaction chamber in the form of
atomized jets.
DESCRIPTION OF THE DRAWINGS
The invention will now be further described by way of
the following Examples and with reference to the accompanying
drawings in which:
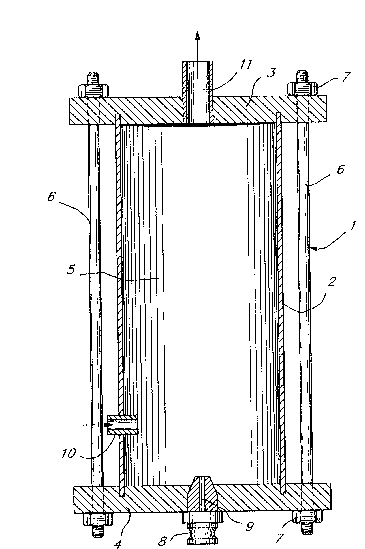
Figure 1 is a cross-sectional view of an embodiment of
a suitable reaction chamber apparatus used in the process of
25 the invention;
Figure 2 is a flow diagram of a typical adiabatic
mononitrobenzene continuous preparation process employing one
method of the invention; and
Figure 3 is a section through a nozzle suitable for the
30 purpose of this invention.
DESCRIPTION_OF THE BEST MODE
The invention is depicted in Figures 1 and 2 in terms of
the introduction of a reaction stream of mixed nitrating acid
containing from about 3 - 8.5% nitric acid, from 58.5 - 70.0%
35 sulphuric acid and not less than about 25~ of water into a
. .
~97~ C-I-L 751
stream of benzene or benzene-rich mononitrobenzene in the
form of a turbulent jet of discrete acid droplets within the
size range of about 1 to 10 um within a reaction chamber. It
will be understood that the process of the invention may be
reversed, that is, a turbulent jet of benzene may be
delivered into a stream or body of mixed acid as described in
the Example hereinbelow. It will also be understood that the
process will be applicable to other nitrations or other
chemical reactions which are mass transfer limited.
In Figure 1, a reaction chamber apparatus, generally
designated 1, is shown which consists of a cylindrical tube
2, upper end closure 3 and lower end closure 4. When
assembled as shown, tube 2 and closures 3 and 4 define a
chamber 5. The assembly can be held together, for example,
15 by bolts 6 secured by threaded nuts 7. Centrally located in
lower end closure 4 is an atomizing nozzle 8 having a narrow
passage 9 therein. Mounted in the side wall of chamber 5 and
passing through tube 2 is an inlet tube 10. This inlet tube
is adjustable both laterally ti.e. at right angles to the
20 longitudinal axis of the tube 2) and longitudinally (i.e.
along the length of the tube 2). Located in upper end
closure 3 is an exit or outlet port 11.
Reaction chamber apparatus 1 is adapted to receive a
turbulent spray of droplets of a mixed nitrating acid
25 component into a body of benzene with sufficient velocity to
effect contact at a micron particle size level. The benzene
component is continuously introduced into chamber 5 through
inlet tube 10 where it is entrained by a high velocity
atomized stream or spray of the acid component introduced
30 continuously into chamber 5 through passage 9 in nozzle 8.
The intermixing of the two phases permits rapid chemical
reaction between particles of a size as small as 2 microns or
less.
To achieve optimum reaction conditions between the two
35 components, several variable factors may be adjusted by trial
,. ~.... .. .
, . . .
~ 7~3Z
C-I-L 751
- 12 -
and error to produce the desired end product. The diameter
of chamber 5, the velocity of the atomized stream passing
into chamber 5 through nozzle passage 9, the type or angle of
spray achieved by nozzle 8, and the location of inlet tube 10
may all be manipulated to produce a desired end product in
the most effective manner.
The material of construction of the apparatus is,
essentially, of a corrosion resistant material, such as,
stainless steel or glass-lined steel. While the end closures
10 3 and 4 may be permanently fixed to the cylindrical tube 2,
it is preferred that closures 3 and 4 be removable for
cleaning and inspection of the inner chamber 5. Nozzle 8 is
conveniently adapted for easy replacement e.g. having a
threaded barrel for insertion in a corresponding tapped bore
15 in the end closure 4 and having an opposite end portion
adapted to receive a driving tool e.g. hexagonal flats
arranged to receive a spanner or socket.
The method of preparation of mononitrobenzene utilizing
the process of the invention will now be described with
20 reference to Figure 2. A reaction stream of mixed acid from
acid feed vessel 20 is pumped by means of metering pump 21
through acid preheater 22 and into reaction chamber 5 through
spray nozzle ~.
Simultaneously, a stream of benzene from benzene feed
25 vessel 23 is pumped by means of metering pump 24 into
reaction chamber 5 through orifice 10. The rate of flow of
each of the benzene and mixed acid components is controlled
by adjusting the operating rates of metering pumps 21 and 24
so that the reactants are delivered into the reaction chamber
30 in slight (1 - 10~) stoichiometric access of benzene and the
reaction temperature is maintained below 145C. Within
reaction chamber 5, the fine particles or spray oE mixed acid
reacts with the benzene to produce a mixture of substantially
homogeneous mononitrobenzene and spent acid which mixture is
35 continuously removed from reaction chamber 5 via line 25 to
C-I-L 751
- 13 -
continuous separator 26. As disclosed by Alexanderson et al
in U.S. Patent No. 4,09l,042, advantages can be gained by
maintaining conditions such that the nitric acid
concentration of the mixed acid is between 3 - 8.5% and the
water concentration is not less than about 25%. The spent
acid concentration should provide a sulphuric acid content of
from fi2-68% to maintain reaction rates and avoid denitration.
It may, in some instances, be desirable to subiect the
nitroben~ene/spent acid mixture exiting through line 25 to a
further refining step prior to delivery to separator 26 in
order to fully nitrate any residual, unreacted benzene which
may remain in the product. Such a refining step may take the
form (not shown) of, for example, a static mixer or the use
of an orifice plate, installed between the exit of chamber 5
and separator 26. Alternatively, a second reaction chamber
similar to chamber 5 may be employed in which the product
from line 25 is subjected to further nitration. At separator
26, the spent acid and crude nitrobenzene are separately
recovered. The crude nitrobenzene is directed to a washing
and purification step (not shown) and the hot spent acid is
directed to a concentrator (not shown) where it is restored
to its initial concentration by the removal of water by means
of external heat. The external heat requirement is reduced
since no cooling was applied at the reaction chamber.
E~AMPLE
In a pilot plant trial, an apparatus was prepared
consisting of a vertical stainless steel tubular reaction
chamber 43 cm in length and 7.5 cm in diameter. An atomixing
orifice, 0.5 mm in diameter and l.2 mm long, for the
introduction of benzene, was located centrally in a base
plate. An inlet for the introduction of mixed acid was
located in the side wall of the tubular chamber about lS cm
above the base plate and orifice. Mixed acid at about lO0 C
comprising a mixture of 5.08% by weight of nitric acid and
61.89% by weight of sulphuric acid was delivered into the
~297~L3Z
C-I-L 751
- 14 -
chamber at a rate of 573.2 ml/min. When steady acid flow was
achieved, benzene at ambient temperature was injected through
the atomizing orifice into the mixed acid at a rate of 75.2
ml/min. The reaction was continued for 30 minutes during
which time samples were taken for analysis. From the
analysis, the rate of convention, based on nitric acid, was
55.3% and on the organic phase, was 52.5%. Optimization of
these conditons in a full-scale plant can be expected to
produce close to 100~ conversion.
While the invention herein disclosed has been described
in terms of the particular process for the nitration of
benzene in the production of nitrobenzene, it will be
appreciated and understood by those skilled in the art that
other nitratable organic compounds may be reacted with mixed
15 acid employing the disclosed process. Amongst the nitratable
~rganic compounds, in addition to benzene, which may be
reacted employing process are, for example, toluene,
dimethylbenzene, halobenzene, naphthalene, methylnaphthalene,
halonaphthalene, halotoluene and halomethylnaphthalene.