Note: Descriptions are shown in the official language in which they were submitted.
5~
O.Z. l~050/383~3
Preparation of phenoxyalkanoltriazc,le compounds
and intermediat~s therefor
The present invention relates to a process for
preparing phenoxyzlkanoltriazole compounds in the form of
S their substantially pure diastereomers~ to intermediates
therefor and to processes for Preparing the intermediates
in the form of their substantially pure isomers.
It is known to use phenoxyalkanoltriazole compounds,
for example 8-(Z-fluorophenoxy)-4-~1,2,4-triazol-1-yl)-
3-hydroxy-2,2-dimethyloctane, as fungicides (EP 40 350).
These compounds, having two chiral centers in the mole-
cule, exist in the form of optical isomers and as di-
astereomers. The fungicidal action of the diastereomeric
pairs of enantiomers is not always the same, so that there
is interest in preparing the more active diastereomer.
It is known that this can be done by selective reduction
of the corresponding triazolyl ketones (DE 33 21 023~3).
However, the chemical yields and the isomeric purity of
the diastereomers obtained in the reduction is not al~Jays
satisfactory.
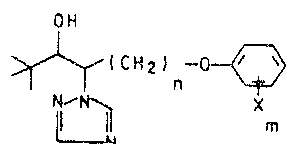
We have found a process for preparing a compound
of the formula I
OH
~CH~ -o
~N
where
X is hydrogen~ fluorine, chlorine, bromine, C1-C3-alkyl,
C1-C3-alkoxy, trifluoromethyl, phenyl or phenoxy,
m is 1, 2 or 3, the individual atoms or groups being
identical or different when m is greater than 1, and
n is 2, 3, 4, 5 or 6,
in the form of its substantially pure diastereomers by
reacting an alkene oxide of the formula II
--C--C--C--I CH2 ) --~
:L3~ s~
- 2 - o.Z~ Q050/38303
where X, n and m have the abovementioned meanings, in the
form of its substan~ially Pure E- or Z-isomer with tri-
a~ole in the presence of an acid amide and of an alkaline
compound at 130 - 230C.
S A substantially pure diastereomer contains 95
(by weight) or more of the diastereomer, based on the
total diastereomer content.
C1-C3-alkyl is for example methyl, ethyl, propyl
or isopropyl. C1-C3-alkoxy is for example methoxy, ethoxy,
propoxy ar isopropoxy. The reaction is carried out in the
presence of an alkali metal hydroxide, or of an alkaline
earth metal hydroxide or of a comparable alkali compound,
for example sodium hydroxide, potassium hydroxide, sodium
carbonate or potassium carbonate, in an acid amide, in
particular N-methylpyrrolidone or cdimethylformamide as a
diluent, and at from 130 to Z30C. This reaction surpris--
ingly preserves the spatial orientation of the various
substituents on the chiral centers. The spatial structure
of the end product is thus the same as that of the alkene
oxide used for the reaction. In this way it is possible
to obtain end products of defined spatial structure in a
simple reaction. With a high yield based on the chemical
conversion, a diastereomer which is not less than 95% pure
relative to the isomer content is obtained without problems.
The alkene oxide required for the reaction is ob-
tained by oxidizing an alkene of the formula III
+ C~=CH-(CH21 ~0
m
where X, n and m have the abovementioned meanings, in the
form of its substantially pure E- or Z-isomer with an
3û organic peracid in the presence of an organic liquid at
from -10 to ~30C. An organic peracid is for example
m-chloroperbenzoic acid or peracetic acid. This oxidation
is carried out for example in a chlorinated hydrocarbon,
~3~
- 3 - O.Z. 0050/38303
in particular in methylene chloride if the oxidizing agent
is m-chloroperbenzoic acid, or in a protic solvent, in
particular in aqueous acetic acid in the case of peracetic
acid as the oxidizing agent. If the oxidation is carried
S out with peracetic acid, this oxidizing agent can be formed
in situ from ~22 ancl acetic acid. The oxidation is car-
ried out at from -10 to -i30C, in particular at from ~15 to
~2SC~
The two process steps of oxiclation and reaction
with triazole give very high chemical yields with selec-
tivities, based on the isomers, of in each ~ase not less
than 95%. In this way the E-alkene of the formula III is
smoothly converted to the pair of RS/SR enantiomers of the
formula I and the Z-alkene of the formula III to the pair
of RRtSS-enantiomers of the formula I.
The problem of the diastereoselective synthesis
of I has consequently been reduced to the synthesis of
a stereochemically uniform ~- or Z-alkene of the formula
III.
The alkene of the formula III
+ CH--CH--~CH2~
where
X is hydrogen, fluorine, chlorine, bromine, C1-C3-alkyl,
C1-C3-alkoxy, trifluoromethyl, phenyl or phenoxy,
m is 1, 2 or 3, the individual atoms or groups being
identical or different when m is greater than 1, and
n is Z, 3, 4, 5 or 6,
is obtained in the form of its substantially pure E-iso-
mer by heating a compound of the formula IV
OR
f CH-CH2-~cH2) ~~
m
~3~
- 4 - o.Z. 0050/3~303
where
X is hydrogen, fluorine, chlorine, bromine, C1-C3-alkyl,
C1-C3-alkoxy, trifluoromethyl, phenyl or phenoxy,
m is 1, 2 or 3, the individual atoms or groups being
S identical or different when m is greater than 1, and
n is Z, 3, 4, 5 or 6, and
R is -COCH3 or -CSSCH3,
to 200 - 400C or 100 - 300C.
In the case of the acetate, the substance is heated
as such or in a high-boiling inert diluent to 200 - 400C.
If the reaction is carried out in substance, the range
from 250 to 350C is preferred. In the case of the
xanthogenate, it is sufficient to heat the compound to
100 - 300C, either in the presence or absence of a high-
boiling diluent. If the reaction is carried out in sub-
stance, ie. in the absence of a high-boiling diluent, the
preferred temperature range extends from 150 to 250C.
The resulting alkene of the formula III is distilled
under atmospheric pressure or reduced pressure. The
chemical yielcl is between 90 and 100X; the selectivity
based on isomer is not less than 95%. The preparation of
the acetate or xanthogenate can be effected in a conven-
tional manner from the alcohols described in DE 34 00 829.
Another way of preparing the alkene of the formula
2S III in the form of its substantially pure E-isomer com-
prises treating an alkene of the formula III which is pre-
sent in the form of its Z-isomer with short-~ave light in
the presence of diphenyl disulfide and of a hydrocarbon.
A hydrocarbon is for example an alkane or cycloaLkane
of 5 to 8 carbon atoms, for examPle pentane, hexane, cyclo-
hexane, heptane or octane. Short-wave light is for example
light which has the spectral distribution of a mercury
vapor high pressure lamp. The treatment can be effected
at customary temperatures, for example room temperature
35 (20C), or at any other temperature.
The alkene of the formuLa III in the form of its
substantially pure Z-isomer is obtained by reacting
~3~S~
- 5 - O.Z. 0050/33303
pivalaldehyde of the formula (CH3)3C-CHO ~ith a compound
of the formula V
(C~H5)3P--CH--lCH2) --
X
m
where
X is hydrogen, fluorine, chlorine, bromine, C1-C3-alkyl,
C1-C3-alkoxy, trifluoromethyl, phenyl or phenoxy,
m is 1, Z or 3, the individual atoms or groups being
identical or different when m is greater than l, and
n is 2, 3, 4, 5 or 6.
The reaction is carried out under salt-free con-
ditions, ie. in polar aprotic solvents in the absence of
dissolved metal cations (cf. HØ House, Modern Synthetic
Reactions, 2nd ed. 1972, pp. 7û7).
Suitable solvents for this reaction are linear or
cyclic ethers, in particular tetrahydrofuran. The reaction
takes place at 20 - 100C, in particular if tetrahydro-
furan is used as solvent at 50 - 60C (reflux temperature).
Removal of triphenylphosphine oxide after the reaction has
; ended gives the ~-alkene of the formula III in a high
chemical yield with a seleçtivity of not less than 90% in
respect of the Z-isomer. The intermediates required for
preparing the phosphorus compound are known (EP 40 350).
EXAMPLES
The example of preparing 8-(2-fLuorophenoxy)-4-
(1,2,4-triazol-1-yl)hydroxy-2,2-dimethyloctane as a pair
of 3R,4S/3S,4R-enantiomers is used to ilLustrate the in-
dividual pracess steps. The process described in the
examples can be used to synthesize the compounds listed
in the tables.
Preparation of acetate
OH OCOCH3
acetic anhydride ¦ ¦
-f-C ~o~ ~ - I -c ~o~3
~ F F
~3~5~
- 6 - O.Z. 00~0,'3830i
58 9 t~.23 mol) of the octanol derivative are refluxed
for 2 h in 200 ml of acetic anhydride in the presence of
1 ml of concentrated hydrochloric acid. Distillation
gives the acetate after an acetic acid/acetic anhydride
cut. 30iling point/0.05 mm Hg: 135C (65 g)
H-NMR (CDCl3): 0.9 s 9H, 1.25-1.65 m 6 H, 1.8 m 2 H,
7.05 s 3H, 4.0 t 2 H, 4.75 dd lH,
6.8 - 7.1 m 4H.
TA3LE 1
OCOCH3
CH2--(CH21--o~ 3
m
10 No. Xm n Physical data
IV/1 2-F 4 Boiling point
135C/0.05 mm Hg
IV/2 2-Cl 4
IV/3 2-CH3 4
15 IV/4 4-f 4
IV/5 4-CL 4
IV/6 4-CH3 4
IV/7 2,4-Cl2 4
IV/8 2,4,6-Cl3 4
Preparation of xanthogenate
OH 1. NaH O--C--S--CH3
-c-c ~ o~3 2. CS2 C-l ~O~>
3 . C H 3 I F
To 53 9 (0.23 mol) of the octanol derivative in a
solution in toluene is added in total 1 equivalent of sodium
hydride a little at a time. To obtain complete alcoholate
formation, the mixture is refluxed for 3 h. The tempera-
ture is then reduced to 20C, and CS~ is added dropwise in
~3(~5f~
- 7 - O.Z. 0050/333~3
the form of a solution in 100 ml of diethyl ether. About
1 h after the CS2 has been added, about 1.5 equivalents
of methyl iodide are added dropwise, and the mi~ture is
refluxed for 2 h. Filtration and evaporation af all the
readily volatile constituents under reduced pressure leaves
77 9 of the ~anthogenate as an almost colorless oil.
H-NMR (CDCl3): 0.95 s 9H, 1.3-1.9 m 8H, 2 55 s 3H, 4.3 t 2H,
5.72 dd 1H, 6.8-7.3 m 4H.
IR (coating): 2951, 15û7, 1456, 1Z81, 1223, 1206, 1110,
1051 cm 1.
TA~LE 2
S
O--C--S--CH3
--I--C ~I CH2 ) --~
X
m
No. X~ n Physical data
IV/ 9 2-F 4 NMR/IR see example
15 IU/10 2-Cl 4
IV/11 2-CH3 4
IV/12 4-F 4
IV/13 4-Cl 4
IV/14 4-CH3 4
20 IV/15 2,4-Cl2 4
IV/16 2,4,6-Cl3 4
Preparation of alkene from acetate
OCOCH3 H
--c--c ~,o~ -- --c--c~o~3
IIIa/l
81 g (0.26 mol) of acetate IV/1 are heated to 350C
in a metal bàth,with f;rst acetic acid and then alkane IIIa/1
distilling off through an attached column. 64 9 are obtained
~3~ 1S6
- 8 - O.Z. ~050/38303
of the alkene of boiling point 295C/760 mm Hg. rhe E-
configuration of the double bond can be unmistakably
identified through the POsition of carbons 3 and 4 in
13C_NMR (in CDCl3)
S C-3 142.1 ppm
C-4 124.2 ppm
Preparation of alkene from xanthogenate
O--C--S--C H 3 H
_l_1 ~o~ ~ -I~ ~0~
F H F
IIIi~11
20 9 of xanthogenate IV/9 (0.057 mol) are heated to
- 10 about 200C in a slow stream of N2. In the course of 2 h,
10.3 9 of alkene IIIa/1 are driven by the stream of N2 into
the receiver. The configuration of the double bond is un-
mistakably shown in 13C-NMR as being E:
C-3 142.1 ppm
C-4 124.2 ppm
Preparation of alkene IIIa/1 by isomerization
I short-wave
-C-- ~ ~0~ ~ C--C~ ~
H f F diphenyl H F
disulfide
II~b/1 IIIa/1
33 9 (0.132 mol) of Z-8-(2-fluorophenoxy)-2,2-dim-
ethyloct-3-ene IIIb/1 are dissolved in 4ûO ml of cyclohex-
ane. 2.83 9 (0.013 mol) of diphenyl disulfide are added, andthe solution is flushed with nitrogen for 30 min. The solu-
tion is subsequently irradiated with a mercury high pres-
sure lamp (125 W Philips HPK) through a Duran filter at an
internal temperature of 35C. After 3 h of exposure to
irradiation, the conversion is 95~. The reaction solution
is filtered, and the filtrate is distilled through a col-
umn. 25.8 9 of E-8-(2-fluorophenoxy)-2,2-dimethyloct-
3-ene IIIa/1 having a boiling point of û.2 mbar/90C
~3Q~516
- 9 - O.Z. 0~53/383~3
are obtained.
rAaLE 3a
_I-C ~ ~CH2~ - ~ E-configuration
H X
m
No. X~ n Physical data
. _ _
: 5 IIIa/1 2-f 4 aoiling point 295Ci760 mm ~9
IIIa/2 2-Cl 4
IIIa/3 2-CH3 4
IIIa/4 4-F 4
IIla/5 4-Cl 4
10 IIIa/6 4-CH3 4
IIIa/7 2,4-Cl2 4
IIIa/8 2,4,6-Cl3 4
Preparation of alkene IIIb/1
~ e I ~ -C- ~~~
( C 6 H5 ~ 3 P--CH I CH~ ~ 4--o~ c--c , ~c F
F H H
I I I b / 1
261.5 9 (0.5 mol) of tr;phenyl-~5-(3-fluorophenoxy)-
pentyl]-phosphonium bromide are prepared from equimolar
amoun~s of triphenylphosphine and 5-(2-fluorophenoxy)bromo-
pentane at 130 - 140C. After cooling down, the solidified
melt is comminuted and suspended in 1,000 ml of tetrahydro-
furan. 370 ml (O.SS mol) of a 1.5-molar butyl lithium
solution in n hexane are added dropwise with cooling. The
internal temperature is maintained at 20 - 30C while the
mixture is subsequently stirred for 1 h. To the resulting
brown solution is then added dropwise, in the course of
about 30 min~ a solution of 43 9 (O.S mol) of pivalaldehyde
56
- 10 - O.Z. ~~05~/333~3
in 150 ml of tetrahydrofuran. rhis is followed by stirring
at 20C for 20 h and refluxing at 60C for Z4 h until a
white, granular precipitate is formed.
The mixture is hydrolyzed with 2 l of water, the
organic phase is separated off, and the aqueous ~hase is
extracted a further three times with diethyl ether. The
combined organic phases are washed t~ice with water, dried
over Na2S04 and evaporated. To the residue are added
250 ml of ether to precipitate triphenylphosphine oxide.
After filtration with suction the filtrate is distilled
to give 33~5 9 of Z-8-(2-fluorophenoxy)-2,2-dimethyloct-
3-ene IIIb/1 as a colorless liquid having a boil ing point
of 127C/2 mbar.
TA8L8 3b
-C~ (CH2) -0 ~ Z-configuration
H H X
III b
No. Xm n Physical data
. ~
IIIbl1 2-F 4 80iling point 127C/2 mbar
IIIb/Z 2-Cl 4
I I Ib/3 2-CH3 4
20 I I Ib/4 4-F 4
IIIb/S 4-Cl 4
IIIb/6 4-CH3 4
IIIbl7 2,4-Cl2 4
IIIb/8 2,4,6-Cl3 4
Preparation of alkene oxicle
H F I \C/ \ ~ C ) ~3
IIIa/t IVa/l
~3~5~
- 11 - O.Z. 0050/38303
50 9 (0.2 mol) of E-8-(2-fluoropheno~y)-2,Z-dimethyloct-3-
ene lIIa/1 are dissolved in 300 ~l of methylene chloride.
While stirring, a solution of 39.7 g (O.Z3 mol) cf 3-chloro-
perbenzoic acid in 550 ml of methylene chloride is added
S drop~ise in the course of 20 min and the temperature is
maintained at 20C by cooling. Gradually a white pre-
cipitate (3-chloroben~oic acid~ is formed. Stirring is
continued at Z0C for 2 days. Thereafter all the start^
ing material has been converted (analysis by gas chroma-
tography). As workup, this is follo~ed by stirring ~Jith1,000 ml of 10% strength aqueous sodium sulfite solution
until peroxide is no longer detectable. The mixture is
brought to pH 7 with sodium hydroxide solution, the
methylene chloride phase is separated off, and the aqueous
phase is extracted once more with methylene chloride~ The
combined organic phases are washed twice with sod;um car-
bonate solution and twice with distilled water, dried over
sodium sulfate and evaporated at 40C in a rotary eva-
porator under reduced pressure to give 55.6 9 of a crude
product containing (gas chromatography) 90% of 2-~4-(2-
fluorophenoxy)butyl]-3-tert.butyloxirane IIa/1. According
to H-NMR, the ratio of E~ compound is 99:1.
H-NMR (CDCl3~: 0.9 s 9H, 1.5-1~7 m 4H, 1.8-1.9 ~ 2H,
2.49 m 1H, 2.82 m 1H, 4.05 t 2Hr
6.8-7.1 m 4H.
TAEILE 4a
I ,O~ ,H
f I tCH2) - ~ E-configuration
~v a
'
5~5g~
12 O.Z. 0~5~ 333.3
No. Xm n Physical data
IIa/1 2-F 4 1H NMR see example
IIa/2 2-Cl 4
IIa/3 Z-CH3 4
5 Ila/4 4-F 4
IIa/S 4-Cl 4
IIa/6 4-CH3 4
lIa/7 2,4-Cl2 4
IIa/8 2,4,6-Cl3 4
TALLE 4b
H H
(CH2~ - ~ Z-configuration
IVb m
No. Xm n Phys ical data
IIb/1 2-F 4
IIbf2 2-Cl 4
15 IIb/3 3-CH3 4
lIb/4 4-F 4
; IIb/S 4-Cl 4
Ilb/6 4-CH3 4
IIb/7 Z,4-Cl2 4
ZO IIb/8 2,4,6-Cl3 4
Preparation ûf triazole co~pound
H o H OH
-I-C -~IC-(cH2)4-O ~ N~N Na_~ -C-C ~ ~CH2)4 - ~
H F N~N~ F
pair of Ia/1 3R/4S-3S/4R-
enantiomers
~ 3C~5~
- 13 - O.Z. OOS0/38303
9.5 9 (0.036 mol) of E-2-(2-fluorophenoxybutyl)-3-t-outyl-
oxiran IIa/1 tabout 90% strength according to gas chroma-
tography), S0 ml of N-methylpyrrolidone, 1.5 9 (0.036 mol)
of sodiu~ hydroxide and 2.5 9 (0.036 mol) of 1,2,4-triazole
are added together and stirred at 180C. The reaction is
monitored by gas chromatography. After 4 h the conversion
is greater than 98~. N-methylpyrrolidone is distilled
off in a high vacuum, and the residue is dissolved after
cooling down in water/methylene chloride. The aqueous
phase is extracted once more ~ith methylene chloride The
combined methy~ene chloricle phases are washed with water
until acid-free, and dried over sodium sulfate and con-
centrated. The remaining brown oil (10.9 9) is dissolved
in 150 ml of cyclohexane by heating, and the solution is
stirred with 0.5 9 of active carbon for 30 min and filtered
off hot. The filtrate gives, on evaporation, 8.6 9 of 8-
(2-fluorophenoxy)-4-(1,2,4-triazol-1-yl)-3-hydroxy-2,2-
dimethyloctane as a colorless oil which crystallizes on
standing. The diastereomeric ratio according to NMR is0 (RR+SS):~RS+SR) = 1:99. Melting point: 87C.
TAPLE 6a
I 1~
--f--C~( CH2 ~ --o~3
N~N~ x
l m
pair of 3R,4S/3S,4R-enantiomers
No. Xm n Physical data
2S la/1 2-F 4 see examPle
Ia/2 2-Cl 4
Ia/3 2-CH3 4
Ia/4 4-F 4
Ia/5 4-Cl 4
30 la/6 4-CH3 4
Ia/7 2,4-Clz 4
Ia~8 2,4,6-Cl3 4
~l3~5~Z~5~
- 14 - O.Z. OOSO/38303
TA8LE 6b
OH
c l~ CH2 ) ~0~)
N~N~ X
~N m
pa i r of 3R,4R/3S,45-endnt iorners
No. Xm n Physical data
5 Ib/ 1 2-F
Ib/2 2-Cl 4
Ib/3 2-CH3 4
Ib/4 4-F 4
Ib/5 4-Cl 4
10 Ib/6 4-CH3 4
Ib/7 2,4-Cl2 4
Ib/8 2,4,6-Cl3 4
.