Note: Descriptions are shown in the official language in which they were submitted.
AHP-91 13
PATENT
~3 ~
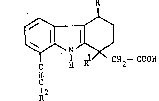
SUBSTlTUTED 2,3,4,9-TETRAHYD3~lH~ARBAZOLE-l-AC~TIC ACID
BACKGROUND OE THE INVÆNTION
a. Field of Invention
This invention relates to tricyclic acetic acid derivatives, to their
preparation and use.
More specifically, this invention relates to tricyclic acetic acid derivatives
in which the tricyclic portion thereof is characterized by having an indole
portion fused to a cyclohexane ring. Still more specificaliy, the compounds of
this invention are characterized as derivatives of the following tricyclic acetic
acid system:
7~2
CH2--COOH
2,3,4,9-tetrahydro-lH-carbazole-l-acetic acid in which the carbons at the l-, 4-end 8- positions are further substituted.
The tricyclic acetic acid compounds of this invention possess useful
pharmacologic properties; for instance, they exhibit analgesic and anti-
inflammatory activity at dose levels which do not elicit undesirable side effects.
The foregoing combination of attributes renders the compounds of this inventior
useful for the treatment of inflammatory or painful conditions in a mammal.
b. Prior Art
The closest prior art to the present invention is
,
- -2- ~ 3 ~ 6
Mobilio et al, United States Patent No. 4,616,028 issued October 7,
1986, United S-tates Patent No. 4,578,398 and United States Patent rlO.
4,584,312, and Asselin et al. United States Patent No. 4,057,559.
Mobilio et al, and Asselin et al, disclose analgesic and anti-inflamnatory
agents having the same heterocyclic ring system as the present invention
but without the l-, 4- and 8-substitutents of the present invention.
.
Demerson et al, United States Patent No. 3,939,178 discloses 1,3,4,9-
tetrahydropyrano[3,4-b3 indoles and 1,3,4,9-tetrahydrothiopyrano[3,4-b] indoles
having analgesic and anti-inflammatory activity. Related United States Patents
are Nos. 3,974,179 and 3,843,681.
Boehringer Mannheim EurQpean Patent 42593 generically discloses starting
materials useful for producing~ cardiotonic and beta-blocking agents. The
starting materiaIs include 1,2,3,4-tetrahydrocarbazoles with substituents selected
from the broad group including hydrogen, carboxy, lower alkyl and lower alkenyl.The starting materials are in each case also substituted with a reactive group
which distinguishes them from the compounds of the present invention.
Further removed, related patents that include tetrahydrocarbazoleacetic
acid derivatives useful as analgesic and anti-inflammatory agents are ~nited
States Patents 4,234,487; 4,264,500; 4,193,923; 4,158,007; 4,146,542, 39896,145 and
3,824,314; Japanese Patent J51032556; Netherland Patent NL 7,100,213 and Great
Britain Patent GB 1385620.
SUMMARY O~ THE INYENTION
The compounds of this invention are represented bX formula (I)
R
~ ~I)
C ~ R CH2- COOH
R2 ' : ;
:: :
n~ !
~ ', :
'' , '
~ ~ .
AHP-9113 mz
-3
wherein R and R2 are hydrogen or lower alkyl containing 1 to 4 carbon atoms and
Rl is lower alkyl containing l to 4 carbon atoms and the pharmaceutically
acceptable salts thereof.
A preferred aspect of the present invention is the group of compounds
represented by formula (I) wherein R is hydrogen, Rl is ethyl, R2 is as defined
above and the pharmaceutically acceptable salts thereof.
A still further preferred aspect of the present invention is the group of
compounds represented by formula (I) wherein R is hydrogen and Rl is ethyl and
R2 is hydrogen or methyl and the pharmaceutically acceptable s~lts thereof.
The most preferred compounds of the present invention are designated 8-
ethynyl-l-ethyl-2,3,4,9-tetrahydro-lH-carbazole-l-acetic acid and l-ethyl-2,3,4,9-
tetrahydro-8-(1-propynyl)-lH-carbazole-l-acetic acid.
The compounds of the present invention are prepared by a process in which
the ketone of structure (Il)
o C02CH3
1 (Il)
~l J R
R ~
wherein R and Rl are as defined above is reacted with ortho-iodophenylhydrazine
to form a phenylhydrazone. The hydrazone is further reacted in the presence of
acetic acid and borontriflouride-etherate to obtain compounds of structure (III~
~ (10
H R C2 CH3
: ~ :
:: : :
AHP-9 113 mz
--4--
wherein R and Rl are as defined ~bove.
Compounds of structure (III) or their carboxylic acids are reacted with
acetylenes of structure (V)
R2- C----C - R3 (V)
wherein R2 is hydrogen, lower alkyl containing 1 to 4 carbon atoms or trimethyl-silyl and R3 is hydrogen, lithium or copper in the presence or absence of
palladium catalyst such as tetrakis(triphenylphosphine)palladium(C)) or palladium
dichloride with added triphenylphosphine and in the presence or absence of a
copper cat~lyst such as copper iodide to obtain compounds of structure (VI) or
their acids
R
(~I)
I Rl C02CH3
C~ 2
wherein R, Rl and R2 are as defined above.
Compounds of structure (I), (III) and (VI) are obtained from their esters by
hydrolysis.
~ or basic hydrolysis a preferred embodiment involves subjecting the
tricy~lic ester to the action of a base, for example, sodium or potassium
carbonate, in the presence of sufficient water to effect hydrolysis of the ester.
The hydrolysis is performed using a suitable solvent, for example, methanol or
ethanol under a nitrogen atmosphere.
~ 3 ~ AHP-9113 mz
The reaction mixture is maintained at a temperature of from 25C to the
reflux temperature until hydrolysis occurs. Usually from 10 minutes to 48 hours
is sufficient for this hydrolysis. The reaction mixture is then rendered acidic
with an acid, for example, acetic acid, hydrochloric acid, s~furic acid and the
like, to release the free acid as a solid.
Alternatively, the tricyclic ester is hydrolyzed by subjecting the ester to
the action of a hydrolyæing agent which is a strong organic or inorganic acid, for
example, trifluoroacetic acid, p-toluenesulfonic acid, hydrochloric acid,
hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid and the like in a
suitable solvent at a temperature of at least 60C and preferably from 90C to
the boiling point of the mixture until the hydrolysis occurs. Usually from 5 to 48
hours are required for this hydrolysis. Suitable solvents include water, acetic
acid, aqueous alcohols and the like. If acid hydrolysis is used, the free acid is
formed directly. If necessary, the reaction mixture can be diluted with water toprecipitate the product.
DETAILED DESCl~IPTION O THE INYENTIC3N
The term '~ower alkyl" as used herein represents straight chain alkyl
radicals containing 1 to 4 carbon atoms and branched chain alkyl radicals
containing 3 to 4 carbon atoms and includes methyl, ethyl, propyl, isopropyl,
butylJ isobutyl, and the like.
The term "halogen" as used herein includes fluorine, chlorine, bromine and
iodine.
The compounds of formula (I) form salts with suitable pharmaceutically
acceptable inorganic and organic bases. These derived salts possess the same
activity as the parent acid and are included within the scope of this invention.The acid OI formula (I) is transformed in excellent yield into the correspondingpharmaceutically acceptable salts by neutrali~ation of said acid with the
appropriate inorganic or organic base. The salts are administered in the same
'
:
; :: ~ :
AHP-9 113 mz
--6 ~
manner as the parent acid compounds. ,Suitable inorganic bases to form these
salts include, for example, the hydro~ides, carbonates, bicarbonates or allcoxides
of the alkali metals or alkaline earth metals, for example, sodium, potassium,
magnesium, calcium and the like. Suitable organic bases include the following
amines; lower mono-, di- and tri-alkylamines, the alkyl radicals of which contain
up to three carbon atoms, such as methylamine, dimethylamine, trimethylamine,
ethylamine, di- and triethylamine, methylethylamine, and the like; mono, di- andtrialkanolamines, the alkanol radicals of which contain up to three carbon atoms,
such as mono-, di- and triethanolamine; alkylenediamines which contain up to sixcarbon atoms, such as hexamethylenediamine; cyclic saturated or unsaturated
bases containing up to six carbon atoms, such as pyrrolidine, piperidine,
morpholine, piperazine and their N-alkyl and N-hydroxyalkyl derivatives, such asN-methylmorpholine and N-(2-hydroxyethyl)piperidine, as well as pyridine.
Furthermore, there may be mentioned the corresponding quaternary salts, such
as the tetraalkyl (for example tetramethyl), alkyl-alkanol (for example
methyltrimethanol and trimethyl-monoethanol) and cyclic ammonium salts, for
example the N-methyl-pyridinium, N-methyl--N-(2-hydroxyethyl)-morpholinium,
N,N-dimethyl-morpholinium, N-methyl-N-(2-hydroxyethyl)-morpholinium, N,N-
dirnethylpiperidinium salts, which are characterized by good water-solubility. In
principle, however, $here can be used all the ammonium salts which are
physiologically compatible. ;
The transformations to the salts can be carried out by a variety of methods
known in the art. For example, in the case of salts of inorganic bases, it is
preferred to dissolve the acid of formula (I) in water containing at least one
equivalent amount of a hydroxide, carbonate, or bicarbonate . Advantageously,
the reaction is performed in a water-miscible organic solvent inert to the
reaction conditions, for example, methanol, ethanol, dioxane, and the like in the
presence of water. For example, such use of sodium hydroxide, sodium
carbonate or sodium bicarbonate gives a solution of the sodium salt~ Evaporationof the solution or addition of a water-miscible solvent of a more moderate
polarity, for example, a lower alkanol, for instance, butanol, or a lower alkanone,
for instance, ethyl methyl ketone, gives the solid salt if that form is desired.
: ~,
~ 3~ 6 ~l~IP-~113 m~
--7--
To produce an amine salt, the acid of formula (I) is dissolved in a suitable
solvent of either moderate or low polarity, for example, ethanol, acetone, ethylacetate, diethyl ether and benzene. At least an equivalent amount of the amine
corresponding to the desired cation is then added to that solution. If the
result;ng salt does not precipitate~ it can usually be obtained in solid form byaddition of a miscible diluent of low polarity, for example, benzene or petroleum
ether, or by evaporation. If the amine is relatively volatile, any excess can
easily be removed by evaporation. It is preferred to use equivalent amounts of
the less volatile amines.
Salts wherein the cation is quaternary ammonium are produced by mixing
the acid of formula (I) with an equivalent amount of the corresponding
quaternary ammonium hydroxide in water solution, followed by evaporation of
the water.
Included in the present invention are the diastereoisomers wherein the 4-
substituent other than hydrogen is either cis or trans to the acetic acid chain at
position one.
Also included in this invention are the optical isomers of the compounds of
formula (I) which result frorn asymmetric centers, contained therein. Such
isomers can be obtained in subs~tantially pure form by classical separation
techniques and by sterically controlled synthesis.
ANTI-INFLAMMATORY ACTIVITY
The useful anti-inflammatory activities of the compounds of formula (I) are
demonstrated in standard pharmaeologic tests, for example, the test designated:
Preventative Adjuvant Edema
The objective OI this test is to determine the ability of test drugs to
exhibit an acute anti-inflammatory effect in rats. This test is a primary screenfor anti-inflammatory drugs.
.
.
- . ,
,: . . '
'. . :
--`` 13~1~8~
Species:
Male Sprague Dawley rats (180-200g) are used. The animals have free
access to water but food is withdrawn 18 hours before testing.
Dru~ Preparations and Administration:
Freund's Complete Adjuvant is prepared by suspending 5 mg killed and
dried Mycobacterium butyricum (Difco) in 1 mL mineral oil. The test
compounds are dissolved, or suspended in 0.5~6 Tween 80 in distilled water
according to their solubility. For primary screening sll drugs are
administered by gastric lavage at the arbitrary dosage of 25 mg/kg, p.o. in
a volume of 0.5 mL/100 g body weight to groups of 10 animals.
Methodological Details:
.
The method is essentially that described by ~Ya:~ et al., J. Pharmacol. Exp.
Ther., 192, 166_171 (1975). Groups of rats are injected intradermally in the
left hind paw with 0.1 mL of Freund's Complete Adjuvant. The test
compound or vehicle is administered immediately before the adjuvant, 24
hours and ~8 hours after the adjuvant (day 0, 1 and 2). The injected hind
paw volume is measured before the injection of adjuvant and 24 hours after
the last drug administration (day 3) by means of a plethysmometer (Buxco
Electronics Inc). The difference between the hind paw volume on day 0 and
day 3 represents the edema volume. Etodolac (25 mg/kg, p.o.) is included
as a positive control.
Presentation of Results:
The mean edema volume (expressed as mL + SEM) is calculated for each
group and the percentage protection conferred by the drug Is calculated:
% protection = (c-t) 100
c
where c is the mean edema volume for the vehicle-treated (0.5% Tween 80
- in distilled water) controls and t is the mean edema volume for the drug
treated group.
* Trade-mark
:
;~ :
,,,,~'~ ~ ~
,
: . . . ~ .. . - : .
. . . : , . : .
. . . . . : : . -. - ::
3 3~ AHP-9113 mz
_g _
ANALGESIC ACTIVITY
A further test used to determine the utility of the c~mpounds of the
present invention is designated: Drug Effects on Phenylbenzoquinone-induced
Writhing in Mice
The objective of this test is to determine the ability of test drugs to inhibit
the nociceptive (pain) response of mice injected with a chemical irritant. This
test is a primary screen for both peripheral and centrally acting analgesic drugs.
Species:
Male Swiss albino mice ~15-2S g). The animals are fasted for 18 hours prior
to use but have free access to water.
Drug Preparation and Administration:
Drugs are dissolved or suspended according to their solubility in 0.5%
Tween 80 in distilled water. They are administered by gastric gavage in a
volume of 5 mL/kg. ~or primary screening all drugs are adrninistered at
the arbitary dosage of 25 mg/kg, p.o. to a group of lO mice.
Methodological Details:
A modification of the method of Siegmund et al, Proc. Soc. Exp. Biol.
Med., 95, 729-731 (1957) is used. Groups of 5 mice are dosed with the test
compound or vehicle control. Sixty minutes later the animals are injected
i.p. with 0.3 mL/20 g body weight of a 0.02% solution of
phenylbenzoquinone (PBQ; 2-phenyl-194-benzoquinone) and placed in
individual observation boxes. The number of writhing or abdominal
squirming movements made by each mouse during the folIowing 15 minutes
period is counted. The experiment is repeated with another group of 5
mice and the mean number of writhes per mouse for a group of lO mice is
calculated.
Presentation of ~esults:
Drug treated and vehicle-treated control groups are compared and the
percentage protection conferred by the drug is calculated:
- ~ .
,
, ' : ' ` ~ ~
AHP-9113 mz
Percentage protection = ~
where c = mean number of writhes in the control group
where t = mean number of writhes in the test drug group
Typieal results obtained for the compounds of the present invention in the
aforementioned tests are as follows:
Table I
Substituted 1,3,4,9-Tetrahydropyrano[3,4-b~ indole-l-acetic Acids
~CH2--COOH (I)
C
R2 ~:
Preventative Phenylquinone
Example Adjuvant Edema * Writhing in Mice *
42 0
2 2 0
* The numbers quoted are percent inhibition at 25 mg/kg.
The lack of side effects for the compounds of this invention are
demonstrated by standard acute toxicity tests described by R.A. Turner in
"Screening Methods in Pharmacology," Academic Press, New York and London9
1965, pp. 152-163 and by prolonged administration of the compound to warm-
blooded animals.
, .
.
~ 3 ~ HP-9113 mz
When the compounds of this invention are employed as anti-inflammatory
and analgesic agents in warm-blooded animals, they are administered orally,
alone or in dosage forms, i.e., capsules or tablets, combined with
pharmacologically acceptable excipients, such as starch, milk sugar and so forth,
or they are administered orally in the form of solutions in suitable vehicles such
as vegetable oils or water. The compounds of this invention may be administered
orally in sustained release dosage form or transdermally in ointments or patches.
The compounds of this invention may also be administered in the form of
suppositories.
The dosage of the compounds of formula I of this invention will vary with
the particular compound chosen and form of administration. ~urthermore, it will
vary with the particular host under treatment. Generally, the compounds of this
invention are administered at a concentration level that affords efficacy without
any deleterious side effects. These effective anti-inflammatorily and analgesic
concentration levels are usually obtained within a therapeutic range of 1.0 ~Ig to
500 mg/kg per day, with a preferred range of 1.0 llg to 100 mg/kg per day~ The
preferred antiinflammatory dose range is 1 mg to 2~ mg/kg b.i.d. The preferred
analgesic dose range is 1 ug to 4 mg/kg b.i.d.
The compounds of this invention may be administered in conjunction with
nonsterold anti-inflammatory drugs such as acetaminophen, ibuprofen and aspirin
and/or with opiate analgesics such as codeine, oxycodone and morphine together
with the usual doses of caffeine. When used in combination with other drugs, thedosage of the compounds of the present invention is adjusted accordingly.
The compounds of this present invention also possess antipyretic activity.
3 ~ j AIIP-9113 m~
-12-
The following examples further illustrate this invention.
EXAMPLE 1
8-Ethynyl-l-ethyl-2,3,4,9-tetrahydro-lH-carbazole-1-acetic Acid
(I, Rl= -C2Hs5 R2= H, R3=-C--CH)
a) Preparation of 2-Iodophenylhydrazine Hydrochloride
A mixture o~ concentrated hydrochloric acid (115.2 mL) and distilled water
(51.2 mL) was cooled in an ice water bath while 2-iodoaniline (100 g, 0.457 mol)was added slowly with vigorous stirring resulting in a thick tan suspension. Themixture was cooled to -5C and a solution ~f sodium nitrite ~35 g, 0.492 mol) inwater (51.2 mL) was added dropwise. The resulting yellow mixture was
maintained at 0C while a solution of tin (II)chloride dihydrate ~232 g, 0.9 mol) in
6N HCl (312 mL) was added dropwise over 2 1/2 hours, to form a pale yellow
slurry. After stirring at room temperature for 24 hours, the mixture was treatedwith 50% NaOH, added dropwise with vigorous stirring, until the aqueous layer
was basic. The free hydrazine was extracted with ether (3 2~1 L). The ether
layers were combined, washed with water (1 L), saturated NaCl solution (1 L),
concentrated to 500 mL and dried tMgSo4). With cooling and vigorous stirring9
HCl (gas) was introduced into the solution to yield an off white precipitate. This
was filtered, washed with ether and dried under vacuum at room temperature to
give an off-white solid (112 g, 91% yield) m.p. 149-150C
b) Preparation of 2-Ethylcyclohexanone
2-Ethylcyclohexanol (1.6 mol, 204 g, 226 mL) was stirred in 3.2 L of
acetone at 0C and treated with Jones reagent (prepared from 106~8 g of CrO3
suspended in 92 mL of concentrated sulfuric acid and diluted to 400 mL with
water) until the orange color persisted ( ~ 430 mL). Isopropanol was then added
to turn the solution green after which it was poured into 2 L of ether. The
product was washed with 6 x 500 mL of brine, dried over MgS04 and
~ ';
. ., : . . : ,
~ , .. . . . ..
~L3~14~ AIIP~9113 mz
--13--
stripped of solvent. Short path distillation (b.p. 80-85C at 25 mm) a~forded 184
g (1.46 moles, 91%) of 2-ethylcyclohexanone as a colorless oil.
c) Preparation of 2-Carbomethoxymethyl-2~ethylcyclohexanone
According to the procedure of E. Negishi and S. Chatterjee, Tet. Lett., 24,
1341 (1983), potassium hydride (417 mmol, 70 mL, ~ 6M in mineral oil) was placedunder nitrogen in a three-necked flask equipped with a rnechQnical stirrer and
was washed three times with petroleum ether (this washing can be omitted).
Tetrahydrofuran (200 mL, distilled from sodium/Ph2CO) was then added followed
by a solution of 2-ethylcyclohexanone, prepared in Step b) (50 g, 396 mmol) in
200 mI, of tetrahydrofuran added as a slow stream over ~ 15 minutes. The
addition was followed one minute later by 495 mL of lM Et3B in tetrahydrofuran
followed 1 hour later by 594 mmol (91 g, 56 mL) of methyl bromoacetate. The
yellow suspension was stirred for 2.5 hours, poured into 800 mL OI water (being
careful to decant away from excess KHI) and extracted with 4 x 300 mL of
petroleum ether. The combined organic phases were dried over sodium s~fate
and concentrated in vacuo. The product was distilled through a 6 inch Vigreux
column collecting the material boiling at 107-118 C at 0.8 mm (the two
regioisomers from the alkylation). This material was then purified by flash
chromatography (4 inches diameter column, 7.5% ethyl acetate in petroleum
ether eluent, 5 1/2 inches of silica gel) to afford 35.33 g (178.2 mmol, 45%) ofcolorless oil. The desired product is the lower Rf material of the two
overlapping spots on thin layer. Bf=D.23 in 10% ethyl acetate/petroleum ether~
About 5~10% oi the 2,6 regioisomer can be isolated as the top spot.
.
d) Preparation of l-Ethyl-8-iodo 2,394,9-tetrahydr~lH-carbazole-l-acetic
Acid Methyl Ester
Solid 2-iodophenylhydrazine hydrochloride (2S.6 g, 94.6 mmol), prepared in
Step a), was partitioned between lN NaOH (150 mL) and ether (200 mL). The
aqueous layer was washed with more ether (200 mL)~ The combined ether Iayers
were washed with saturated NaCl (50 mL), dried (MgSO4) and concentrated to
~ ' ' ~', ,
- .
~ 3~ 8~ AHP-~113 mz
_ ,
-14-
give 21.3 g (91 mmol) of a reddish oil. The oil was dissolved in benzene (250 mL)
and the ketoester, 2-carbomethoxymethyl-2-ethylcyclohexanone, prepared in
Step (c), (18 g, 91 mmol) was added. The mixture was refluxed using a Dean~tark
trap until starting materials were no longer present (usually around 20 hours) to
form the intermediate hydrazone ~TLC on silica gel in 10% ethyl acetate in
hexanes gave hydrazine Rf=0.09; keto-ester Rf=0.18; hydrazone Rf=0.37; indole
Rf=0.43). The reaction mixture was then cooled, concentrated to an oil ana
redissolved in glacial acetic acid (115 mL). According to the procedure of Snyder,
H.R. and C.W. Smi~h, JACS, 46, 2452 (1943), boron trifluoride etherate (20 g, 141
mmol) was added and the reaction mixture was heated at reIlu~E for 30 minutes.
The mixture was cooled and poured ints~ ice water (350 mL~ which was extracted
with ether (3 x 350 mL). The combined ether layers were washed with saturated
sodium bicarbonate solution, dried (MgSOD~) and concentrated to give a brown oil.
The product was then purified by ~lash chromatography (silica gel, 15 cm I.D. X
15 cm ht., 5% ethyl acetate in petroleum ether) to give 18.6 g (51%) of a light
reddish oil which slowly solidified on standing in the free2er.
NMR (CDC13) ~ 0.87 (3H, t, J=7.7 Hz), 1.55-2.2 (6H, m), 2.55-2.85 (4H, m), 3.75
(3H, s), 6.91 (lH, t)J 7.32-7.65 (2H, 2d), 9.35 (lH, br.s).
IR (neat) 3360, 3060, 3000-2840,1730 cm~l.
e) Preparation of 8-Ethynyl-l-ethyl-2,3,4,9-tetrahydr~lH-carbazole-l-
acetic Acid.
According to the procedure of S. Takahashi, et al, Synthesis, 627,1980,1-
ethyl-8-iodo-2,3,4,9-tetrahydro-lH-carbazole-l-acetic acid methyl ester,
prepared in Step d), (10 g, 25.2 mmol) was dissolved in diethylamine (100 mL) and
benzene (100 mL), then treated with Pd(PPh3)2C12 (350 mg, O.S mmol) and Cul
(150 mg, 0.78 mmol). The mixture was bubbled with nitrogen gas for ~ive
minutes, treated with trimethylsilylacetylene (17 mL, 11.8 g, 120 mmol), sealed
and stirred for sixteen hours at room temperature. TLC analysis (silica gel, 5%
ethyl acetate in petroleum ether) indicated clean formation of a new product
- ' , . ~ : ' .' ' . . ' '.
AHP-91 13 mz
-15-
(R~=0.5 vs 0.42 for starting material). The reaction mixture was concentrated,
dissolved in ether (200 mL), filtered, then concentrated again. The residue was
purified by flash chromatography (silica gel, 9.5 cm, I.D. x 15 cm ht, 5% ethyl
acetate in petroleum ether) to give the intermediate 8-TMS acetylene ester.
The ester was dissolved in methanol (150 mL) and treated with KOH (1.62 gm of
86.9% KOH, 25.2 mmol) dissolved in water (10 mL). The mixture was heated to
reflux for sixteen hours. The reaction mixture was concentrated to removc
methanol then partitioned between lN HCl (30 mL) and ether (150 mL). The
ether layer was dried ~MgSO4), concentrated and partially purified by flash
chromatography (silica gel, 9.5 cm, I.D. x 15 cm ht, 40% ethyl acetate in
petroleum ether). The fractions containing the product were concentrated then
chromatographed by reverse phase (Clgsilica gel from a WATERS PREP-500
cartridge, 7.5 cm, I.D. x 15 cm ht, 60% acetonitrile in water) to give the pure
product. The product was crystallized from acetonitrile-water by concentration
of the active fractions from the reverse phase column. The crystals were
collected and air-dried to give 3.8 g (54%) of 8-ethynyl-1-ethyl-2,3,4,9-
tetrahydro-lH-carbazole-l-acetic acid as a yellowish cryst~lline solid, m.p.
147 C.
IR (CHC12) 3470, 3400, 3315, 2105,1710 cm~l.
NMR (CDC13, 60 MHz) ~ 0.85 (3H, t, J=6.5 Hz), 1.7-2.1 (6H, m), 2.6-2.9 (4H, m),
3.33 (lH, s), 6.87-7.65 (3H, m), 9.15 (lH, br.s), 9.7 (lH, v.br.s).
MS (EI) m/z 281 (M+- C2Hs), 222 (M ~ - CH2CO2H).
Calculated C 76.84; H 6.81; N 4.98
Found C 77.08; H 6.92; M 4.98
C 77.15; H 6.98; N 5.00
' '',
AHP-9113 rnz
-
--16-
EXAMPLE 2
l-Ethyl-2,3,4,9-tetrahydro-8-~1-propynyl)-lH-carbazole-l-acetic Acid
(I, Rl=-C2Hs, R2= -H, R3=-C~--C-CH3)
a) Preparation of l-Ethyl-8-iodo-2,3,499-tetrahydro-lH-carbazole-l-acetic
Acid
l-Ethyl-8-iodo-2,3,4,9-tetrahydro-lH-carbazole-lacetic acid methyl ester,
prepared in Example 1, Step d), (45 g9 113 mmol) was dissolved in methanol (665
mL). Potassium carbonate monohydrate (37.5 g, 226 mmol), dissolved in water ~;(70 mL), was added and the mixture refluxed for 16 hours. The reaction mixture
was then cooled and concentrated to remove methanol. The residue was
partitioned between ether (1700 mL) and lN HCl (700 mL). The ether layer was
dried (MgSO4), then concentrated to give 41.6 g (96%) of crude acid. The acid
was purified by flash chromatography (silica gel, 15 cm, I.D. x 15 cm ht, 40%
ethyl acetate in petroleum ether) and recrystallized from ether-petroleum ether
to give 30.6 g (71%) of a tan solid, m~p. 173-175C.
b) Preparation of Copper (I) propyne
Propynyl lithium (3.96 g, 86.1 mmol) was added to THF (300 mL) and stirred
to form a suspension. Copper (I~ iodide (16.4 g, 86.1 mmol) was added and the
mixture heated at reflux for one hour. The mixture was then cooled and a bright
yellow precipitate was collected by filtration. The filtered product was washed
with plenty of water, ethanol and finally ether, then dried under vacuum to give5.84 g (86%) of the propynyl copper as a greenish yellow solid.
c) Preparation of l-Ethyl-2,3,4,9-tetrahydro-8-(1-propynyl)-lH-carbazole-l-
acetic Acid
l-Ethyl-8-iodo-2,3,4,9-tetrahydro-lH-carbazole-l-acetic acid, prepared in
Step a) (15.0 g, ~39.2 mmol), and cuprous propyne, prepared in Step b)~ (8.03 g9
:
- , -
~3~ AHP-9113 mz
-17-
78.4 mmol) were slurried in dry pyridine (190 mL) and bubbled with nitrogen gas
for ten minutes. According to the procedure of R.D. Stephens and CoE~ Castro,
Org. Chem., 28, 3313 (1963), the mixture was then refluxed under nitrogen for 16hours. The reaction mixture was cooled and poured into lN HCl (3 L), then
extracted with ether (2 x 750 mL). The combined ether extracts were washed
with saturated sodium chloride solution (750 mL), dried (MgSO4) and
concentrated to an oil. The product was purified by flash chromatography (silicagel, 15 cm I.D. x 15 cm ht, 40% ethyl acetate in hexanes) to give 9.9 g (86%) of a
brown viscous oil. The product was dissolved in ethyl acetate, treated with
decolorizing carbon, filtered, then recrystallized from ethyl ace~ate-petroleum
ether to give 5.9 g (45%) of an off-white powder, m.p. 174-177C.
IR (KBr) 3380, 3100-2860,1695,1610,1470,1260cm~l.
NMR (CDC13, 60 MHz) ~ 0.88 (3H, t, J=7.5 Hz), 1.3-2.15 (6H, m), 2.15 (3H, s), 2.5-
2.8 (4H, m), 6.85-7.55 (3H, m), 8.4 (lH, br.s), 9.7 (lH, v.br.s3.
MS ~EI) m/z 295 (M~), 266 (M+-C2Hs), 236 (M+-CH2CO2H).
Calculated C 77.26; H 7.17; N 4.74
Found C 77.15; H 7.13; N 4.71
, ~ :
-
: '