Note: Descriptions are shown in the official language in which they were submitted.
~L311~
HALOACETAL CO~POUND
BACKGROUND OF THE INV~3NTION
Field of the Invention
The present invention relates to a haloacetal compound
obtained in a process for the preparation of 2-(l0,ll-
dihydro-l0-oxodibenzo[b,f]thiepin-2-yl)propionic acid which
is of value as a pharmaceutically active compound.
Description of prior art
It is known that 2-(l0,ll-dihydro-l0-
oxodibenzo[b,f]thiepin-2-yl)propionic acid having the
following formula (I):
CH3jH
COOH O
(hereinafter refered to as dibenzothiepin deri~ative) shows
a high anti-inflamma~ory action as well as a high analgetic
action. T~e dibenzothiepin derivative is further known as
a practically valuable anti-inflammatory agent with little:
side effect. Por example, the dibenzothiepin derivative
and its pharmacological actions are described in Japanese
Pakent Provisional Publication No. 55(1980)-53282.
The above-mentioned~Patent Provisional Publ1cation
discloses a process for the prepara~ion of the dibenzo-
thiepin derivative wherein 3~ cyanoethy1)-6-pheny1thio-
:: :
:: :
-
- ' ` , ' ~` , ~
~ 31~ ~2
phenylacetlc acid is cycllzed to gi~e a dlbenzothiepin-
propionamide derivative and this derlvatlve is then
- hydrolyzed.
Another process for preparing the dibenzothiepin
5 derivatlve is disclosed in Japanese Patent Provisional
Publication No. 57(1982)-106678. Thi~ process comprlses
hydrolysls of a phenylacetate ester having a nitrile
group to give a dicarboxylic ~cid derivatlve and subse-
quent ring closure of the dicarboxylic acid derivative ln
10 the presence of a condensing agent such as sulfuric acid
or polyphosphoric acid,
Another process for preparing the dicarboxylic acid
derivative is disclosed in Japanese Patent Provisional
Publication No. 58(1983)-113168 which comprises a propio-
lS phenone derivative is once converted into a hydroxyacetal
compound. This process can be illustrated by the ~ollow-
ing equation:
O ~ 02R 7 CH3-C -C ~ CO
0502R" OR~C02R CC~R~cOzH
H OR'
wherein Y is chlorine or bromine, R is an alkyl group
ha~ing 1-5 carbon atoms or hydrogen, R' ~s an alkyl group
20 having 1-5 carbon atoms, and R" i~ methyl or p-tolyl.
~ '
~ 3 ~ 2
-- 3 --
While these known processes are employable for the
preparation of the dibenzothiepin derivative, these
processes have drawbacks in that the processes involve
complicated and multiple steps or the use of a toxic
reagent such as KCN. Accordingly, these known processes .
are not favourable as industrially employable processes.
SUMNARY OF THE INYENTION
According to the invention there is provided a
holoacetal compound having the formula:
7 (I)
C - .
2 O
wherein R1 i5 hydrogen or a lower alkyl group having 1-6
carbon atoms, R2 is a lower alkyl group having 1-6 carbon
atoms, and X is a halogen a~om.
The haloacetal compound i5 obtained in a process fox
the preparation of 2-(10,11-dihydro-10-
oxodibenzotb,f]thiepin 2-yl)propionic acid of the formula
(I): :
:; :
which comprises the steps:
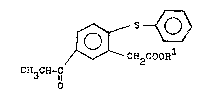
reacting a propiophenone derivative having the for~ula
(II~.
~ '
, .. ~ . , . , ~
(II)
CH2COOR
CH3CH2C
0
wherein Rl is hydrogen or a lower alkyl group,
with a halogenating agent to form a haloketone compound
having the ~ormula ( I II ):
~ ~ (III)
CH2COOR
CH3CH~
O
wherein Rl has the same meaning as defined above;
reacting the haloketone compound with a primary
alcohol having the formula (IV):
: 2
R OH (IV)
wherein R2 is a lower alkyl group,
and an orthoformate havlng th~ formula ~
HC(OR2)3~ (V)
wherein R has the same meaning as defined above,
to form a hal~aeetal compound having the formula (VI):
.:
: . . . :
~ 3 1 ~ ~ 9 ~
x o~2
CH3-CH C-~-S~> (VI)
CH2COOR
5 where~ n Rl and R2 both have the same meanings as deflned
above, an~ Rl is the szme as R2 where Rl o~ the ~ormula
(II) is hydrogen, and X is a halogen atom;
converting the haloacetal compound in the presence
of a zinc halide into a dicarboxylic acid ester havlng
10 the formula (VII ):
CH3
CH-~)--S~) ~VII)
COOR
2 : :
15 wherein Rl and R2 both have the same meanlngs as derined
above; and
hydrolyzing the dicarboxylic acld e3ter to give a
dicarboxyllc acid having th~ formula (VIII): ;
CH
/CH~ 5~ ~ (VIII)
COOH
:~ 3 ~
-- 6 --
and
converting the dicarboxylic acid ln the presence of
a condensing agent into the diben~othiepin derivative o~
the formula (I).
Further, 2-(10,11 dihydro-10-oxodibenzotb,f}thiepin
2-yl)proplonic acid can be also obtained by the same
proces~ as above except that the above-mentioned dicar-
boxyllc acid ester of the ~ormula (VII) is first cyclized
in the presence of a condensing agent to give a dibenzo-
10 thlepin ester derivatlve which is then converted lnto-the
desirsd dibenzothlepin derivative by hydrolysisO In more
detail, the diben~othlepin derivative of the rormula (I)
can be obtalned by the process comprising the steps of:
.converting the dicarboxylic acid ester having the
15 formula (YII):
CH3
/ CH~ 5 -<~ ( VI I )
COOR
CH2COOR
20 wherein Rl and R2 both have the same meanings as defined
above;
in the presence Or a condensing agent into a dibenzothie-
pin ester derivati~e having the rormula (IX):
:
~. 3 ~
.-
- 7 -
CH3CH ~ ~ (~X)
COOR2 1~
5 wherein R has the same meaning as defined above,
and
hydrolyzing the dibenzothlepin ester derivative to
give the dibenzothiepin derivative of the ~ormula (I).
Furthermore, 2-(10,11-dihydro-10-oxodibenzo~b,~]
10 thiepin-2-yl)propionic acid can be also obtained by the
same process as above except that the above-mentioned
haloacetal compound of the formula tVI~ ls directly con-
verted into the dicarboxylic acld o~ the formula (VIII).
In more detail, the dlbenzothlepin derivative o~ the for-
: 15 mula (I) ean be obtained by the process comprising the
steps Or: ~
converting the haloacetal compound having the
formula (VI):
X oR2
CH3-CH-C ~ -5- ~ (VI~ ~ ~
,,
CH COOR
wherein Rl and R2 both have the :same meanings as defined
: ~ above, and Rl ls the same as R2 where Rl of the ~ormula
25 (II) is~hydrogen,~nd X~is a halogen~ atom;~
in a proton-donating medium in thé~presence of a basic
~ compound into a~dicarboxylic acid having the formula
: (VIII)~
::
,
~ 3 ~
/CH~ s~ (VIII)
COOH
CH2COOH
5and
converting the dicarboxylic acid in the presence of
a condensing agent into the dibenzothiepin derivative o~
the formula ( I ) .
PREFERRED EMBODIMENT OF THE INVENTI~
In the formulae o~ the compounds employed ~or the
preparation of the dibenzothiepln derivative aocording to
the present lnvention, each of Rl and R2 ls the sæ~e as
or dif~erent ~rom each oth~r and is an alkyl group having
1-6 carbon atoms, preferably methyl~or ethyl. R may be
15 hydrogen ln the propiophenone derivati~e of the ~ormula
(II). In this oase, Rl in the~ormulae (VI3 and (~II)
gener ~ly is the same as R2 O~ the sam0 formula.
The f~rst step o~ the process~of the invention i3
for convertlng a propiophenone derivatiYe of the formula
20 (II) to a haloketone compound o~ the formula (III) u~ing
a halogenatlng agent.
As the halogenating~agent, a b~rominating agen~t~or a
chlorlnating agent~;is~generally employed. Pre~erre~ is
the b~rominatlng~agent, particularly,~bromine.
The halo~enating agent is emplo~ed ln an amount of
at least one mole~per one;mole o~ the propiophenone deri-
vative.
The reaction ls~pre~e~rably carried out at a tempera-
ture in the range of~room'temperature~,~,to 60C ~or a pe- ;
30 riod o~ 0.5 to 48 hours. ~The reactlon~can be carrled out
-
,.,. , , : , , . .:
:
.
..
~ 3 ~
g
in a solvent such as methanol, ethanol, ethylene tetra-
chloride, carbon tetrachloride, benzene, and toluene
which does not participate in the reaction.
The second step ls for converting the haloketone
5 compound of the formula (III) into the haloket ~ compound
of the formula (VI ) uslng a primary alcohol of the for-
mula (IV) and an orthoformate of the formula (V),
Preferred examples of the prlmary alcohols lnclude
methyl alcoho~ and ethyl alcohol. Preferred examples of
10 the ortho~ormates ~nclude methyl orthoformate ~nd ethyl
orthoformate.
The orthoformate ester and primary alcohol ~re used
in amounts of at least two moles and at least one mole,
respectively, per one mole of the haloketone derivatlve.
15 A great amount of the primary alcohol or orthoformate can
be employed ~or further serving as a reaction solvent.
The reactlon i9 preferably carried out at a tempera-
ture in the range of 80 to 130C ~or a period of 0.5 to
48 hours. The reaction can be carried out ln a 801vent
20 such as ethylene tetrachlorlde, carbon tetrachloride,
benzene, and toluene which does not participate in the
reaction.
As noted herelnbefore, i~ the Rl of the formula
(III) (proplophenone derivative) is hydrogen, the
25 hydrogen generally is replaced with R2 of the primary
~cohol in the reaction.
If desired, the first step and the second step can
be continuously per~ormed wlthout isolatin~ the product
of the first step, namely, the haloketone compound of the
30 formula (III). Ih this process, the continuous reaction
can~be carrled out by reacting the propiophenone deriva-
tive with the halogenating agent 9 primary alcohol and
ortho~ormate~ ~
The third step is for converting the hal oacetal com-
35 pound of the formula (VI ) in the presence o~ a zinc
3 ~ 2
-- 10 --
h ~ ide into a dicarboxyl~c acid ester of the ~ormula(VII).
Thls rearrangement reaction i8 generally carried out
in a solvent such as toluene 9 methyl alcohol, methyl
5 orthofomate, dichloroethane or trichloroethane which does
not partlcipate ln the reaction at a temperature of from
room temperature to r~fluxing temperature ~or a perlod of
0.5 to 24 hours.
An preferred example of the zlnc halide is zinc
10 bromide. The zinc h~ ide can be prepared in situ in the
reaction solution from zinc metal. Hydrogen hallde pro-
duced in the reaction reacts with the zinc metal to pre-
pare a zinc halideO
The zinc halide is preferably employed at least in a
15 catalystic amount.
If desired, the second step and the third step can
be continuously performed without i801ating the product
of the second step, namely, the haloketal compound of the
formula (VI)~ In this process, the continuous reactlon
20 can be carried out by reacting the haloketone compound
with the primary alcohol and ortho~ormate, and then fur-
ther heating the reaction mixture after addition of the
zinc h ~ide.
Altem ati~ely, the first step, the second step and
25 the third step can be comb~ned for directly converting
the propiophenone derivative into the dicarboxylio acid
ester without i801ating the haloketone compound and halo-
acetal compound. In thls process, the continuous reac-
tion can be carried out by first reacting the proplo
30 phenone derivative, halogenating agent, primary alcohol
and orthoformate and then further heating the reaction
mlxture after addition o~ the z$nc halide.
The fourth step is for converting the dicarboxylic
acid ester of the ~ormula (VII) into a dicarboxylic acid
35 of the formula (VIII~ through hydrolysis.
: ,' , .
.
~1 3 ~
1~ --
The hydrolysis can be carried out ln a co~ventional
manner, for lnstance, by heating the dicarboxylic acld
ester in an aqueous alkaline solutlon or in an aqueous
ac idic solution ~
The fifth step is for converting the dicarboxylic
acid of the formula t~III) into the dibenzothiepin derl-
vative of the formula (I) in the presence of a condensing
agent, Accordingly, this reaction is a rlng closure
reac tion .
The condesing agent is preferably employed ln a
weight amount of 1-30 times as much as the weight amount
of the dicarboxylic acid. Examples o~ the condensing
agents include sulfuric acid, polyphosphoric acid (pre-
ferably, 105%, 116%, or their mixture) and polyphosphoric
15 acld ester. The reaction is generally carried out at a
temperature of from room temperature to 150C ~or a
period of 10 min. to 15 hrs. After the reaction is com-
plete, the reaction liquid is lntroduced into water or a
mixture oi lce and water. Alternatively, water or a mix-
20 ture of ice and water can be introduced into the react~onliquid. To the aqueous mixture is th~n added an organic
solvent to extract the reaction product with the organlc
solvent. The organic solvent i9 then distilled off to
r~ over the reaction product. The reaction product can
25 be purified, for lnstance, by recrystallization,
Further details o~ the ring closure reaction are
described in the aforementioned Japanese Patent Provl-
sional Publication No. 57(1982)-10667B.
As described hereinbefore, the 3equence of the
30 hydrolysis and ring closure can be reversed. In more
detail, the dicarboxylic~a~id ester o~ the *ormula tVII)
i8 first con~erted into a dibenzothiepin ester derivative
of the formula (IX) in the presence of a condensing agent
and the dibenzothiepin es~er derivative is then converted
35 into the dibenzothiepin derivatlve of~the formula (I)
.
,
- . ~.
.
- 12 -
through hydrolysis. The reaction ~or rlng closure of the
dlcarboxylic ac 1 d ester and the reaction for hydrolysls
of the dlbenzothiepin ester derivative~ can be carried
out almost ln the s ame manner as described above,
The h~loacetal compound o~ the ~ormula (VI) can be
directly converted into the dicarboxyllc acld of the
formula (VIII)
This reaction can be performed ln a pro ton-donating
medi~:m in the presence of a basic compound. Examples of
10 the proton-donatlng medla include water, methyl alcohol,
ethyl alcohol, n-propyl alcohol, isopropyl alcohol and
e thylene glycol. The proten don~tlng medium can be us~d
in comblnation wlth e~ch other or in combination w.ith
other inert solvent. Other alcohol can be employed.
15 Examples Or the basic compound9 include sodlum hydroxice,
potassium hydroxide, sodium carbonate, potassium carbon-
at e, sodium hydrogen carbonate and pota3sium hydrogen
carbonate. Other basic compounds can be employed.
An aqueous sodlum hydroxide solution or aqueous
20 potassium hydroxide solution is preferably employed as a
combination of the proton-donating medlum and the ba~ic
compound.
The basic compound ls preferably employed ln an
amount corresponding to at leas~ two equivalent~ per ons
25 equivalent`of the haloacetal compound.
~ he reaction is generally carried out at a tempera-
ture ~rom 50C to refluxing temperature for a period of 1
hour to several tens hours.
The a~orementioned reaction for converting the pro-
30 piophenone derlvative into the haloacetal compound viathe haloketone compound and the reaction for dir~tly
converting the haloacet al compound into the dicarboxylic
acid can be combined to perform these reactions continu-
ously without isolating the intermediate compounds such
35 as the haloketone compound and haloacetal compound.
.
,
- ,
.
~ ~ ~ 3 ~
- 13 -
The ~ollowing examples ~urther deQcrlbe the present
invention.
Example 1
(1) Synthesls of methyl 5-(2-bromoproplonyl)-2-phenyl-
thiophenylacetate
In 300 ml o~ methylene chloride was dissolved 85.0 g
o~ methyl 5-propionyl-2-phenylthiophenyl acetate. To the
solution was dropwise added 40 g of bromine at room tem-
perature. A~ter the addition was complete, the mixture
10 was stirred for 30 minutes. To the mixture was then
added 160 ml o~ water. The mixture and water were well
mixed by stirring, and the organic layer was Qeparated.
The organic layer was washed with water, and the organic
sol~ent was removed under reduced pre~ure. To the re~i-
15 due was added 130 mI o~ methyl alcohol, and the mixturewas heated to glve a solutlon. The ~olution wa~ le~t
ovemight at a temperature below 15~C~ and the precipi- ~
tated crystals were collected. The crystals were recry-
stalllzed from acetone-he~cane to give 84 g of the desired
20 compo~d, m.p. 67. 5-68.0C.
(2) Synthe31~ of methyl 5-(2-bromo~ dlmethoxypropyl)-
2-p~enylthiophenyl acetate
A mixture o~ 15.72 g of methy} S-(2-bromopropionyl)
2-phenylthiophenylacetate obtain~d~in (1) above, 12.7 g
25 o~ methyl ortho~ormate, 0.38 g o~ methanesulfonIc acid
and 40 ml of methanol wa3 re~luxed ror 24 hours~and then
wa~ concentrated under reduced pressure. To the resldue ~ :
was added~ 100 ml Or: diethyl ether, and the mixture was
washed successively with 20 ml o~ saturated aqueou~ :
30 sodlum hydrogen carbonate solution~ 20 m~ of water~ and
: :,
:
- . .
:
. : , ,
~31~
- 14 -
20 ml o~ saturated brine solutlon. The mlxture was then
dried over anhydrous sodium sulfate. The solvent wa~
distilled to glve a colorless oil as a re~ldue. The oil
was purified to obtain 16.85 g of methyl 5-(2-bromo-1,1-
5 dimethoxypropyl)-2-phenylthiophenylacetate (purity go %)
as a colorless oil.
MMR (CDC13) :
1.52 (3H, d, J=8Hz), 3.21 (3H, s), 3.35 (3H, s),
3.61 (3H9 s), 3.87 (2H, S)3 4.45 (lH, q, J=8Hz),
7.1-~.5 (8H, m)
(3) Synthesis o~ methyl 5-(1-methoxycarbonylethyl)-2-
phenylthlophenylacetate
To methyl 5-(2-bromo-1,1-dimethoxypropyl)-2-phenyl-
thlophenylacetate ~haloacetal compound] obtained ln (2)
15 above were added 38 ml of toluene and 0.86 g of zlnc
bromide, and the resulting mixture was heated under
re~lux for 1 hour~ The m~xture was~cooled and then 100
ml o~ ether wa~ added. The re~ulting mixture was washed
successively wlth 30 ml of water and 30 ml o~ saturated
20 brine solution, and drled over anhydrou~ sodlum sulfate.
The solvent wa~ distilled of~, and the residue was dis-
tilled under reduced pressure to obtain 10.61 g of the
deslred compound (dicarboxylic acid ester) as a yellow
oil (yield 77 %, b.p. 212 -215C/2 mmHg).
25 NMR (CDCl3) S
1.49 (3H, d, J=7Hz), 3.61 (3H, ~)~ 3.67 t3H, s~,
3.82 (2H, ~), 3.5-3.9 (lH, m), 7.0-7.4 (8H, m)
(4) Syntehsis o~ 5~ carboxyethyl)-2-phenylthlophenyl~
acetle acid
. : - ~ . .. .
. .
- :, :
~L 3 ~
To 17.2 g of methyl 5~(1-methoxycarbonylethyl)-2-
phenylthiophenylacetate ~dlcarboxyllc acid ester3 ob-
t ~ ned ln (3) above wa added 125 ml of 2N aqueous sodium
hydroxide ~olution, and the resulting mlxture was heated
5 under reflux and stirring for 4 hour~. After the reac-
tion mlxture was oooled, it was ~dJust~d to pH 1 with 10
~ s~lfuric acid, and extracked with two portions of 150
ml of methylene chloride. The organic layer was washed
with 80 ml of saturated brine solutlon9 and then wa~3
îO dried over anhydrous sodlum sulfate. The dried layer wa~
concentrated to dryne~ under reduced pre3sure to obtain
pale brownish crude crystals. The crude cryst~s were
recrystallized from 30 ml o~ 1, 2-dichloroethane to o~tain
14.0 g of 5-tl-carboxyethyl)-2~phenylthiophenylacetic
15 ac~d as pale yellowish crystals (yleld 89 %), m.p. 145-
146~C.
(5) Synthesis of 2~(10,11-dihydro-10-oxodibenzotb,f]-
thiepin-2-yl)propionlc acid
To a solutlon o~ 63 g of polyphosphoric acid (11~ %)
20 in 63 ml Or methylene chloride was added 15, ~ g to.os
mol.) of 5-(1-carboxyethyl)-2 phenylthiophenylacetic aci~
bbtained in (4) above. The resulting mlxture was ~tirred
for 3.5 hrs. at 40C. To the reaction ~olution was add~d
a mixture of lce and water, and the resulting aqueou~ :
~5 mlxture was extracted with ethyl aoetate. The extract
was washed wikh saturated brine solution, and then drled
over anhydrous sodium sulfate. The solvent was distllled
off ~rom the dried extract under reduced pre~sure at a
temperature oP lower than 40C, and then re~idue was
30 recrystallized twice from a mixture of methylene chloride
and hexane to obtain 10.9 g of the 2-(10911-dihydro-10-
oxodibenzo~b,fJthiepin-2-yl)propionic ~cid as pale yellow
cry~tals (yield 73 ~),
3 ~
-- 16 --
(1 ) Synthe~is o~ methyl 2-(10,11-dihydro-10-oxodibenzo-
~b,f]thlepln-2-yl )proplonate
A mixture o~ 0.5 g of methyl S-(l-methoxycarbonyl-
5 ethyl )-2-~phenylthiophenylacetate ~dlcarboxyllc acid
ester] obtained in Example 1-(3) and 5.3 g o~ polyphos-
phorie acld (116 %) was stirred at a temperature o~ 60-
80C ~or 6 hours. The mixture wa~ cooled, and to thl~
was added a-mixture o~ ice and water to decompose exces-
10 slve polyphosphoric acid~ The resulting mixture wasextracted with ethyl acetate. The organic layer was
separated and washed successively with saturated brine
solution, aqueous saturated sodium hydrogen carbonate
solutlon and saturated brine solution. The wa~hed
15 extract was dried over anhydrou~ sodium sul~ate~ The
solvent was distilled o~f ~rom the dr~ed extract under
reduced pressure9 and then the reqidue was recrystallized
from a mixture of benzene and hexane to obtain 0.4 g o~
methyl 2-(10~ dihydro-10-oxodibenzotb,f~thiepin-2-
20 yl)proplonate (yield 89 %! m.p. 81.0 - 82.0C.
neat
IR cm : 1730, 1670
max
NMR ~CDcl3! S'
1.44 (3H, d, J=8Hz, ~ CH3~,
3 . 60 ( 3H , , -C0 2CH3 ~,
3.66 (lH~ q, J=8Hz, -CH) I :
6.96-7.60 ~5H7 m~ aromatic proton),
7.96-8.20 (lHI m9 aromatic proton)
30 (2) Synthesis o~ 2-(lO,ll~dihydro-10-oxodibenzo~bj~]-
thiepin-2-yl )proplonic acid
.
;
.
...... . .. . . .
~L 3 ~
_ 17 -
A mixture o~ 0.36 g of methyl 2-(10,11-dihydro-10-
oxod~benzQ~b,f]thiepln-2-yl)propionate, 4 rnl of methanol
and 3~7 ml Or an aqueous ~olution containing 0032 g of
sodium hydrogen carbonate was heated under re~lux and
5 stlrrlng for about 6 hours. After the reflux was com-
plete, the mixture was cooled and shaken wlth 29 ml o~ 8
% aqueous sodium hydro~en carbonate solution and 10 ml o~
methylene chlorlde. The aqueous layer was ssparated,
made acidic by addltion o~ conc. hydrochlorlc acld, and
10 extracted with ethyl acetate. The ethyl acetate layer
- was shaken and washed with saturated brine solution, and
then dried over anhydrous sodium sul~ate. Ethyl acetate
was distilled off from the dried extract under reduced
pressure to obtaln 0.34 g o~ a residue. The residue was
15 recrystallized from a mixture o~ methylene chloride and
hexane to obtaln 0.31 g of 2-(10,11-dihydro-lO~oxodi-
benzotb,f~thiepln-2-yl)propionic acid (yield 90 %).
(1) Synthe~is of methyl 5-(1 methoxycarbonylethyl)-2-
phenylthlophenylacetate
To a stirred mixture of 15.72 g (50 mmol.) of methyl
5-propionyl-2-phenylthlophenylacetate, 13.32 g ~125.5
mmol.) of methyl ortho~ormate, 20 ml of methanol and 20
ml ethylene tetrachlorlde was dropwise ad~ed ~der stir-
25 ring 8.39 g (52.5 mmol.) of brom`ine for a period of 30minutes. The resulting mlxture was further stirred at
room temperature for 30 minutes ~ and then wa~ heated
slowly to 110C for 1 hour, under distilllng o~f mate-
rials ha~ing a low boiling point. To the reaction mix-
30 ture was added 0.90 g (4.0 mmol.) of zinc bromide, andthe mixture was heated to ~10 C under re~lux for 3 hours.
me reaction mixture was cooled~ and 75 ml o~ water and
.
- ~ :
. ~, .
~ 3 ~ 3
- 18 -
40 ml of methylene chlorlde were added. The org~nic
layer was separated and the solvent was distilled off
under reduced pressure. The re~idue was di~tilled under
reduced pre~sure to obtaln 13.78 g o~ the desired product
5 as a yellow oll (yield 80 %, b.p. 212-215C/2 mmHg).
neat
IR cm :1740 (C=0)
max
NMR (CDC13) ~ :
1.49 (3H, ds J=7Hz) 3.61 (3H, s) 3.67 (3H, ~),
3.82 (2H, s), 3~5-3.9 ~lH, m), 7.0-7.4 (8H, m)
(2) The above-obtained methyl 5-(1 methoxycarbonyl-
ethyl)-2-phenylthiophenylacetate was treated in the same
manner as ln Example 1-(4) to -(5) to obtain 2-(10~11-
15 dihydro-10-oxodlbenzo~b,f]thiepin-2-yl~propionlc acid.
Example 4
(1) Synthesls of methyl 5-(1-methoxycarbonylethyl~-2-
phenylthiophenylacetate
To a stlrred mixture of 15~0 g (50 mmol. ) o~ 5-pro-
20 plonyl-2-phenylthiophenylacetic acid, 13~32 g (125~5
mmol. ) o~ methyl ortho*ormate, 20 ml o~ methanol and 20
ml ethylene tetrachloride was dropwise added under ~t~r-
rin~ 8.39 g (52.5 mmolO) of bromine ~or a period of 30
minut~s. The resulting mixture was further stirred at
25 room temperature for 30 minutes, and then was heated
slowly to 110C ~or 1 hour, under dl~tilllng off mate-
rials having a low boiling point. To the reaction mix-
ture wa~ added 0.90 g (4.0 mmol.) o~ zinc bromide, and
the mlxture was heated to 110C under re~lux for 3 hours.
30 The reaction mixture was cooled, and 75 ml of water and
40 ml of methylene chlorlde were added. The organic
~,
.
- ` ~ 3 ~ 2
-- 19 --
layer was separated and the solvent was distilled of~
under reduced pressure, The residue was dlstllled under
reduced pressure to obtaln 12.1 g of the de~ired product
as a yellow oil (yleld 70 %, b~po 212-215C/2 mmHg),
neat
IR cm 1 :1740 (C-0)
max
NMR ~CDC13~ ~ :
1.49 t3H, d, J=7Hz) 3.61 (3H, s) 3.67 (3H, ~),
3~82 (2H, ~), 3.5-3.9 (lH, m), 7.0-7.4 (8H, m)
(2) The above-obtaine~ me~hyl 5-(l~methoxycarbonyl_
ethyl)-2-phenylthiophenylacetate wa~ treated in the same
manner as in Example 1-(4) to -(5) to obtain 2-(10,11-
dihydro-10-oxodibenzotb,~thlepln-2~yl)proplonlc acid.
.
Example 5
(1) Synthesls of methyl 5~ methoxycarbonylethyl)-2~
phenylthiophenylacet~te
To a mixture o~ 15701 g (0.50 mol.) of methyl 2-
phenylthio-5-proplonylphenylacetate, 133.2 g(lo255 mol ?
20 of methyl orthoformate, 200 ml of methanol and 100 ml o~
ethylene tetr~chlorlde was added 2.62 g (0.040 gram atom)
o~ zinc powder, and the mixture was heated under stirring
to a temperature of 40 to 45C. To the solution was
dropped S3.6 g (O.S25 mol.) o~ bromine for 1 hour under
25 stirring, and then the temperature was maintained at a
temperature withln 42C and 45C for 30 minutes. The
reactlon mixture was gradually heated to raise the exter-
n~ temperature to 120C, under:dis~illing o~ materials
having a low bo~iling point (b.p. 32 to 65C). When
30 ~lmost all materials having a :low boiling~point were
distilled of~ and the lnternal temperature almost lowered
.
. .
--`~
`` ~ 3 ~
- 20 -
?
to approx. 805, to the reactlon mlxture was added 100 ml
ot ethylene tetrachlorlde, Then, the mixture was again
heated to raise the internal temperature to a temperature
of hlgher than 100C, under dlstilllng of~ remaining
5 msterials ha~ing a low boiling point. After the lnternal
temperature wa~ kept at a ~emperature o~ 100 to ~10~ for
1 hour, the reaction mixture wa~ cooled to ~pprox. 50C.
To the oooled reaction mixture was added 500 ml of water,
and the mixture wa~ stlrred ~or a whlle, Insoluble mate-
10 rials were ff ltered o~f over a Celite and then was washedwith three portions o~ 50 ml of 1,2-dichloroethane, The
organlc layer was separated from the ~iltrate and the
washings. The aqueous layer was extract~d with 150 ml o~
1,2-dichloroethane. The extract wa9 comblned with the
15 above-obtained organic layer and dried over anhydrous
sodium sulfate. ~he solvent was dlstllled ofr under
reduced pressure, and then the resldue was dlstilled
under reduced pressure ~o obtain 141 g o~ the desired
product as a yellow oil ~yleld 82 %3.
20 (2) me above-obtained methyl S~ methoxycarbonyl-
: ethyl)-2-phenylthiophenylacetate was treated in the same
manner a~ in Example 1-(4) to -(5) to obtain 2-(10,11-
dihydro-10-oxodlbenzoCb,f]thlepln-2-yl?proplonic acid.
,
:
25 (1 ) Synthesis of 5~ carboxyethyl S-2-phenylthiophenyl-
acetic acid :
:
A mlxture o~ 4.39 ~ ~10 mmol~) o~ methyl 5-(2-bromo-
1,1-dimethoxypropyl)-2-phenylth~ophenyl aeetate obtalned :
in Example 1-(2~ and 25 ml of 2N aqueous sodium Aydroxide
30 was heated under re~lux and stirrlng ~or~6 h~s. The:
reaction mixture was cooled~ ad~usted to pH 6.0 with 10 %
denotes trade mark
:
~.,., .. , ~ .. .. . . . .
,
~!~
` ~3~9~
-- 21 --
sul~uric acid, and washed with two portions o~ 16 ml of
methylene chloride. The mlxture then was adJusted to pH
1 with 10 % sul~urlc acid and extracted with two portions
o~ 16 ml of methylene chloride~ The organic layers were
S combined and washed with 16 ml o~ water, and dried over
anhydrous sodlum sulfate. The solv~nt wa~ then distllled
off, The residual crude crystals were recrystallized
from 1,2~dlchloroethane to obtain 2~49 g of th~ desired
product as whlte crystals ~yield 79 %), m.p~ 145-146C~
10 (2) The above-obtalned 5 (1-carboxyethyl)-2-phenylthio-
phenylacetic acid was treated in the same manner as in
Example 1-(5) to obtain 2-(10,ll-dihydro-10-oxodibenzo-
tb,f]thiepln-2-yl)pro pionic ac ld .
Example 7
15 (1) Synthesls o~ 5-(1-carboxyethyl)-2-phenylthiophenyl-
acetlc acld
A mixture o~ 4.39 g (10 mmol.) o~ methyl 5-(2 bromo-
1,1-dimethoxypropyl)-2-phenylthlophsnylacetate obtained
in Example 1-(2~, 3.45 g (lO mmol.) of anhydrou~ potas-
20 sium carbonate, ~6 ml Or methanol and 13 ml o~ water washeated ~er reflwc and stirring ~or 2 hours. The reac-
~on mixture was distllled to remove dlstillates havlng
bolllng points of below 100C. To the residue was added
10 ml o~ wa~er, and the mixture was heated under re~lux
25 and stirring for 12 hrs. ThB reaction:mixture was
treated in the sam~ manner as ln Example 6 ~1) to obtain
2.46 g o~ the desired product as white crystals (y$eld 78
%), m.p. 145-146C.
(2~ The above-obtalned 5-(1-carboxyethyl)-2-phenylthio-
30 phenylacetic acid was treated ln:the same manner as in
.~,.
. ~
~L 3 ~
- 22 -
.
Example 1-~5) to obtain 2-~10,11-dihydro-10-oxodibenzo-
~b,~Jthlepln-2-yl)propionic acid.
tl) Synthe9i~ of 5-(l-carboxyeghyl)-2-phenylthiopheny
ace tic ac id
A mlxture of 4.39 g (10 mmol.) of methyl 5-(2-bromo
1,1-dlmethoxypropyl)-2-phenylthiophenylaeetate obtained
in Example 1-(2), 3.45 g (25 mmol.) of a~hydrou~ potas-
sium carbonate, 13 ml of methanol and 13 ml o~ water wa~
10 heated under reflux and stirring ~or 40 hours. The reac-
tion mixture was treated in the same manner as in Example
6-(1) to obtain 2.41 g of the de~ired product as white
cr3rstals (y~eld 76 X), m.p. 145-146C.
(2) The aboYe-obtalned 5~ carboxyethyl)-2-phenylthio-
15 phenylacetic acid was treated in the same manner a~ ln
Ex amp le 1- ( 5 ) to obtain 2-(10,11-dihydro~ oxodibenzo-
~b, f 3thiepin-2-yl )proplonic ac ~d .
;'
Exarnp l e 9
(1) Synthe~i~ Or 5-(1-carboxyethyl)-2~phenylthiophenyl-
acetic acld
A mixtur~ of` 4.39 g (10 mmol. ~ o~ 5-(2-bromo~
dimethoxypropyl3-2-phenylthiophenylacetate ob~alned in
Example 1-(2), 4.20 g (50 mmol.:) o~ sod~lum hyd.rogen car-
bonate9 ~0 ml of: ethanol~ and 35 ml of water:was heated
25 under re~lux and stirring for 5 hours. The reaction mix~
ture was distilled to r~move:dist~llates having a boiling
point of below 100C. The resldue~ was then hea~ed under
reflux for 4 hours~ The mixture was~treated in the same
:
:~ 3 ~ 2
- 23 -
manner a~ in Example 6-(1) to obtain 2 .15 g of the
de3ired product as white crystal~ (yield 68 %~, m.p.
145-146C.
(2) The above-obtalned 5 (1-carboxyethyl)-2-phenylthio~
5 phenylacetic acid wa~ treated ~n the ~ame manner ~s in
Example 1-~5) to obtain 2-(10,11-dlhydro-10-oxodibenzo-
~b,f~thiepin-2-yl)propionlc acid.
Example lQ
(1) Synthesls of 5-(1-carboxyethyl)-2-phenylthiophenyl-
ace~ic acid
To a stirred mlxture of 78.6 g of methyl 5-propionyl
-2-phenylthiophenylacetate, 66.3 g of methyl orthoform~
ate, 100 ml of methanol and 50 ml ethylene tetrachloride
heated to approx. 45C was dropwise addsd under stirring
15 40.0 g o~ bromine heat~d to the ~ame temperature~ ~or a
period of one hour. The re~ulting mixture was iurther
stirred at the same temperature for 30 mlnutes9 and then
was heated slowly to approx. 100C (inner temperature3,
under distilling o~ almost all material~ havlng a low
20 bolllng point. To the reaction mixture was added 625 ml
of 2N aqueous sodium hydroxide solutlon. The mixture was
then heated under reilux and stirring for 7 hours~ under
dis tllllng o~f ethylene tetrachloride tog0ther wlth water
as an azeotropic mixture. The reactlon mixture was
25 cooled~ and methylene chloride was added,. The mixture
was made acldic to pH 1 by addition of 10h sulfuric acid
under stirr~ng ~or extracting the reaction product with
the methylene chloride. The methylene chloride layer was
separaked, washed with saturated brine solutlon and dried
30 over anhydrous sodium suliate. The dried methylene
chl oride layer was evaporated to dryness ~der redueed
,,
.
3 ~ 2
-- 24 --
pressure to glYe 69 . 9 g of the reaction product as pale
yellowish crude crystal~. ~he crude cry~tal~ were
recrystallized from 140 ml of 1,2-dichloroethane to
obtain 63. 2 g o~ the desired product ~yleld 80 %) .
5 ( 2 ) Synthe sls o~ 2- ( 10 ,11-dihydro- 1 0-oxodib enzo-
~b, f ~thiepin-2-yl )propionic acld
In 120 ml o~ dichlc~roethane was diYsolved 45.0 g of
5~ carboxyethyl)-2-phenylthiophenylacetic acid obtalned
ln (1) above under heating. To the re~ulting solution
10 was added 315 g of polyphosphorlc acid (105 %)0 The was
heated under an atmo~pheric pressur~ to approx. 100C to
distilled off dichloroethane. After dichloroethane was
distilled off, the mixture was cooled to approx~ 80C,
and stirred at the temperature for 3.5 hours. The mix-
15 ture was then cooled, and to the m~xture was added 180 mlof dichloroethane. To the resulting mlxture wa9 portion-
wise added ~der stirr~ng 180 ml ~ water, under keeplng
the mixture at 50C (inner tem~erature). The organic
layer was separated, and the aqueous layer wa3 repeatedly
20 extracted with dichloroethane. The dlchloroethane ~olu-
tions were combined and dried over anhydrou-~ sodium sul-
fate. The solvent was then disti lled off ~der reduced
. pressure. The residue was recrystallized from a mixture
of methylene chloride and n-hexane to obtain 36 . 3 g of
25 the desired product (yleld 86 %).