Note: Descriptions are shown in the official language in which they were submitted.
1312077
FP-1679
Process for preparinq 1,5-benzothiazepine derivatives
SUMMARY OF THE INVENTION
This invention relates to a process for preparing 1,5- :
benzothiazepine derivatives and a pharmaceutically accept- - ~ : able salt thereof which is useful as a pharmaceutical
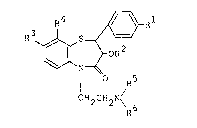
compound and represented by the formula:
R4 ~ Rl
~ ~ -OR2 (I)
N
¦ R5
CH2CH2N\ 6
wherein R is a lower alkyl group or a lower alkoxy
group, R is hydrogen or a lower alkanoyl group, one -
of R3 and R4 is a lower alkyl group or halogen atom,
and the other is hydrogen, and each of R5 and R6 is a .
lower alkyl group. .
- ~:
The 1,5-benzothiazepine derivatives (I) and a pharmaceuti- -~
cally acceptable acid addition salt thereof are useful
25 pharmaceutical compounds having an excellent hypotensive~- -
~ ~~ , . ~ ,
~. r~
- 1312~77
-- 2
activity, cerebral or coronary vasodilating activity
and/or platelet aggregation-inhibiting activity, and among
the compound (I), a compound wherein R2 is hydrogen atom
is also useful of as an intermediate for synthesis of
pharmaceuticals.
DESCRIPTION OF TH~ PREFERRED EMBODIMENTS
Examples of the compound (I) of the present invention may
include compounds in which R is a lower alkyl group, or a
lower alkoxy group, R2 is hydrogen or a lower alkanoyl ;
group, either one of R3 or R is a lower alkyl group or
halogen atom, and the other is hydrogen atom, and each of
R5 and R6 is a lower alkyl group.
-
In the above-mentioned examples of the 1,5-benzothiazepine
derivative (I), the lower alkoxy group, the lower alkyl
group and the lower alkanoyl group include an alkoxy group --~
of one to six carbon atoms, an alkyl group of one to six
carbon atoms and an alkanoyl group of two to six carbon
atoms, respectively. Preferred examples of these group ~ -
are an alkyl group of one to four carbon atoms and an
alkanoyl group of two to five carbon atoms. ; -~
According to the present invention, the compound (I) or a
pharmaceutically acceptable salt thereof can be prepared ; -
by reacting a compound represented by the formula~
.:: .. . :. :
H~
wherein Rl, R2, R3 and R4 have the same meanings as ~ : -
defined above,
' '.-.' ':
1312~77
- 3 -
with an aminoethanol represented by the formula: -
R \
N-CH -CH -OZ (III)
R
wherein R5 and R6 have the same meanings as defined
above, and Z is hydrogen, a lower alkylsulfonyl
group, an arylsulfonyl group or sulfo group,
to condensation reaction and, if necessary, convert it to a
salt thereof according to a conventional method.
The above condensation reaction, when the starting compound
(III) wherein Z is hydrogen is employed, can be practiced - ~
15 suitably in the presence of a dehydrating agent. As the -
dehydrating agent, for example, a mixture of triphenylphos-
phine and diethylazodicarboxylate, a mixture of triphenyl-
phosphine and dimethylazodicarboxylate, a mixture of
sulfuryldiimidazole and sodium hydride can be suitably
20 used. This condensation reaction may be preferably prac- -
ticed in a suitable solvent (e.g., chloroform, dichloro-
ethane, dichloromethane, acetone, diethyl ketone, methyl -
ethyl ketone, ethyl acetate, methyl acetate, ethyl propion- -
ate, methyl propionate, dimethylformamide, diethylformate,
25 dimethylacetamide, N-formylmorpholine, N-acetylmorpholine,
dioxane, tetrahydrofuran, ether, dimethoxyethane, diglyme, -
toluene, benzene, xylene, etc.) at 0 C to 150 C.
On the other hand, when the compound (III) wherein Z is a
30 lower alkylsulfonyl group (e.g., methylsulfonyl group,
ethylsulfonyl group), an arylsulfonyl group (e.g., benzene-
sulfonyl group, tosyl group) or sulfo group, the condensa-
tion reaction of said compound and the compound (II) can
be practiced preferably in the presence of an alkali y
35 reagent. Examples of the alkali reagent may include
alkali metal hydroxides (e.g., potassium hydroxide, etc.), :
, , ,
~312~77
alkali metal carbonates (e.g., potassium carbonate, etc.),
alkali metal hydrogen carbonates (e.g., potassium hydrogen -
carbonate, etc.), alkali metal hydrides (e.g., sodium ~-
hydride, etc.). This condensation reaction should prefer-
ably be practiced in a suitable solvent (e.g., methyl -
ethyl ketone, diethyl ketone, acetone, dimethylsulfoxide,
dimethylformamide, ethyl acetate, methyl propionate, ethyl -~ -
propionate, tetrahydrofuran, dioxane, ether, dimethoxy-
ethane, diglyme, toluene, benzene, xylene, acetonitrile, -
10 dimethylacetamide, diethylforma~lide, N-formylmorpholine, ~ -
N-acetylmorpholine, etc.) at 0 C to 150 C. ~ ~
The compound (I) thus obtained can be easily converted - - -
into a pharmaceutically acceptable acid addition salt, for -~
15 example, by treating with an acid, if necessary. Examples -
of such pharmaceutically acceptable acid addition salts ;~
may include inorganic acid addition salts such as hydro-
chloride, hydrobromide, hydroiodide, perchlorate, sulfate,
phosphate, etc.; and organic acid addition salts such as : -~
20 oxalate, maleate, fumarate, tartrate, methanesulfonate, etc. -~
Since the reaction of the present invention as mentioned :
above can proceed without accompanying any racemization,
by using an optically active compound (II) as the starting ~
25 material, the compound (I) can be obtained as an optically i-~ :
active compound. ';
The 1,5-benzothiazapine derivatives (I) or pharmaceuti- ~-
cally acceptable acid addition salts thereof of the pre- ;
sent invention have excellent hypotensive activity, cere-
bral or coronary vasodilating activity and/or platelet
aggregation-inhibiting activity as mentioned above, and -
can be used for therapy and prevention of brain diseases -~
such as cerebrovascular contraction, cerebral ischemia, : :
35 cerebral infarction, etc., or cardiac diseases such as ~ - -
stenocardia, cardiac infarction, etc. Also, among the
- . ' .
- .: .
` 1312~77
compound (I), the compound wherein R2 is hydrogen is also
useful as a synthetic intermediate, since this compound can
be converted by acylation to a compound (I) wherein R2 is a
lower alkanoyl group.
The starting compound (II) to be used in the present
invention can be prepared according to the methods as
described in U.S. Patents No. 4,567,175, No. 4,590,188 and
No. 4,665,068, respectively.
Also, the compound (I) of the present invention and the
compound (II) include either two kinds of stereoisomers (that -~
is, cis and trans-isomers) or four kinds of optical isomers
(that is, (+)-cis, (-)-cis, (+)-trans and (-)-trans isomers)
and mixtures thereof based on asymmetric carbon atoms (two)
in the molecule.
Example 1
To 80 ml dichloromethane solution of 3 g of (+)-cis-2-(4- -
methoxyphenyl)-3-acetoxy-8-chloro-2,3-dihydro-1,5-benzo-
thiazepin-4(5H)-one and 2.3 g of triphenylphosphine were
added 15 ml dichloromethane solution of 779 mg of 2-(di-
methylamino) ethanol over 20 minutes, then 15 ml dichloro-
methane solution of 1.52 g of diethylazodicarboxylate at room
temperature over 20 minutes. The mixture was stirred for 20
.
1312~77
- 5a -
hours at room temperature, followed by condensation under
reduced pressure. The residue was dissolved in ethyl acetate ~
and insolubles were removed by filtration. The filtrate was --
extracted with a 10 % hydrochloric acid, and the aqueous
layer was made alkaline with potassium carbonate and
extracted with chloroform. The extract was washed with ~-
water, dried and evaporated under reduced pressure. The
residue was converted to maleate and re- ~
''':
1312~77
crystallization from ethanol gave 3.55 g of (+)-cis-2-(4-
methoxyphenyl)-3-acetoxy-5-[2-(dimethylamino)ethyl]-8-
chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-one.maleate.
Yield: 79.2 %.
mp. 158 to 160 C.
Example 2 - -
After stirring a mixture of 354 mg of 2-(dimethylamino)-
ethanol and 159 mg of a 60 ~ sodium hydride in 14 ml of ~ -
dimethylformamide at room temperature for 20 minutes, 787
mg of sulfuryldiimidazole was added at - 40 ~C to the
mixture and the mixture was stirred at the same tempera-
ture for one hour. Subsequently, 5 ml dimethyl sulfoxide
solution of 1.0 g of (+)-cis-2-(4-methoxyphenyl)-3-acet-
oxy-8-chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-one was
added at - 40 C and after gradual warming, the reaction
mixture was stirred at room temperature for 20 hours.
20 After completion of the reaction, methanol and chloroform -
were added. The mixture was washed with water and dried,
followed by removal of the solvent, and the residue was
separated by column chromatography. After removal of the -
starting lactam by elution with chloroform-ethanol (20 :
1), the oily product subsequently eluted was converted to
maleate and recrystallized from ethanol to give 777 mg of
(+)-cis-2-(4-methoxyphenyl)-3-acetoxy-5-[2-(dimethylamino)-
ethyl]-8-chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-one.-
maleate.
mp. 158 to 160 C.
Example 3
To 30 ml acetone solution of 1.86 9 of (+)-cis-2-(4-
methoxyphenyl)-3-acetoxy-8-chloro-2,3-dihydro-1,5-benzo-
" ~
, ' -
1312077
- 7 -
thiazepin-4(5H)-one were added 2.4 g of powdery potassium
carbonate and 1.1 g of 2-(dimethylamino)ethyl methane-
sulfonate-hydrochloride, and the mixture was refluxed
under stirring for 10 hours. After completion of the
reaction, inorganic materials were removed by filtration,
to the residue were added a 10 % hydrochloric acid and -
ethyl acetate, and extracted with a 10 % hydrochloric -~ -
acid. The extract was washed with water, dried and then - :-
condensed under reduced pressure. The residue was con-
verted into hydrochloride and recrystallized from a mix-
ture of acetone-ethanol to give 2.10 g of (+)-cis-2-(4- -~
methoxyphenyl)-3-acetoxy-5-[2-(dimethylamino)ethyl]-8- -
chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-one.hydro-
chloride. Yield: 86.5 %. -
:
mp. 127 to 131 C (decomposed). --
Example 4 :~
. ~,
To 50 ml dimethylsulfoxide solution of 3.36 g of (+)-cis-
2-(4-methoxyphenyl)-3-hydroxy-8-chloro-2,3-dihydro-1,5-
benzothiazepin-4(5H)-one was added 1.35 g of potassium
hydroxide under ice-cooling and, after stirring at room
temperature, 244 mg of 2-(dimethylamino)ethyl methanesul-
fonate hydrochloride was added, followed by stirring at
room temperature for 16 hours. After completion of the -
reaction, the mixture was poured into ice-water and the ;
mixture was extracted with a 1 : 1 mixture of chloroform
and ethanol. The extract was washed with water, dried and
then evaporated under reduced pressure. Recrystallization
of 3.48 g of the residue from a mixed solvent of ethyl
acetate and hexane gave 3.06 g of (+)-cis-2-(4-methoxy- -
phenyl)-3-hydroxy-5-[2-(dimethylamino)ethyl]-8-chloro-2,3-
dihydro-1,5-benzothiazepin-4(5H)-one. Yield: 75.2 %. --
mp. 122 to 124 C.
'' ~, ' .,-
..~
: ~' '
1312~77
Example 5
To 30 ml acetone-0.5 ml water solution of 1.68 9 of (+)-
cis-2-(4-methoxyphenyl)-3-hydroxy-8-chloro-2,3-dihydro-
1,5-benzothiazepin-4(5H)-one were added 2.40 9 of potas-
sium carbonate and 1.09 9 of 2-(dimethylamino)ethyl
methanesulfonate-hydrochloride, followed by refluxing for
20 hours. After completion of the reaction, inorganic - --
materials were removed by filtration, to the residue were
added a 10 ~ hydrochloric acid and ethyl acetate and the
organic layer was extracted with a 10 % hydrochloric acid.
The hydrochloric acid layers were combined and made alka-
line with potassium carbonate and then extracted with
ethyl acetate. The extract was washed with water, dried,
and evaporated under reduced pressure. Recrystallization
of the residue from a mixture of ethyl acetate-n-hexane
gave 1.40 9 of (+)-cis-2-(4-methoxyphenyl)-3-hydroxy-5-[2-
(dimethylamino)ethyl]-8-chloro-2,3-dihydro-1,5-benzothiaze- ~
pin-4(5H)-one. Yield: 68.8 ~i. -
mp. 123 to 125 C.
Example 6
In Example 3, by use of 2-(dimethylamino)ethyl benzene-
sulfonate.hydrochloride in place of 2-(dimethylamino)ethyl
methanesulfonate, (+)-cis-2-(4-methoxyphenyl)-3-acetoxy-5-
[2-(dimethylamino)ethyl]-8-chloro-2,3-dihydro-1,5-benzo-
thiazepin-4(5H)-one.hydrochloride was obtained.
This product had the same physical data as the product in
Example 3. -
~-
Example 7 - -
In Example 3, by use of 2-(dimethylamino)ethyl toluene-
1312~77 ~
g
sulfonate.hydrochloride in place of 2-(dimethylamino)ethyl
methanesulfonate, (+)-cis-2-(4-methoxyphenyl)-3-acetoxy-5-
l2-(dimethylamino)ethyl]-8-chloro-2,3-dihydro-1,5-benzo-
thiazepin-4(5H)-one.hydrochloride was obtained.
This product had the same physical data as the product in
Example 3.
:. . .
Example 8
': ':
In Example 3, by use of 2-(dimethylamino)ethyl sulfate in
place of 2-(dimethylamino)ethyl methanesulfonate.hydro- ; -
chloride, (+)-cis-2-(4-methoxyphenyl)-3-acetoxy-5-[2-(di-
methylamino)ethyl]-8-chloro-2,3-dihydro-1,5-benzothiazepin-
4(5H)-one.hydrochloride was obtained.
This product had the same physical data as the product in :
Example 3.
20 Examples 9 to 14 -
"'. ' .
By treating the corresponding starting materials in the
same manner as in Examples 1 to 5, the following compounds - -
were obtained.
(9) (+)-cis-2-(4-methoxyphenyl)-3-hydroxy-5-12-(dimethyl- ~- -
amino)ethyl]-9-chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-
one.perchlorate ~hydrate
~ "'' .- '
mp. 190 to 192 C. ~
: .,:' ~,
(10) (~)-cis-2-(4-methoxyphenyl)-3-acetoxy-5-[2-(dimethyl-
amino)ethyl]-9-chloro-2,3-dihydro-1,5-benzothiazepin-4(5~1)-
one.hydrochloride monohydrate
-
mp. 140 to 143 C.
1312~77
-- 10 --
(11) (+)-cis-2-(4-methylphenyl)-3-hydroxy-5-~2-(dimethyl-
amino)ethyl]-8-methyl-2,3-dihydro-1,5-benzothiazepin-4(5H)-
one
mp. 142 to 143 C (recrystalli7ed from ethyl acetate).
(12) (+)-cis-2-(4-methylphenyl)-3-acetoxy-5-[2-(dimethyl-
amino)ethyl]-8-methyl-2,3-dihydro-1,5-benzothiazepin-4(5H)-
one.hydrochloride
mp. 184 to 186 C (recrystallized from a mixed solvent
of isopropanol and ether).
This product, when recrystallized from a mixed solvent of
acetone and isopropanol exhibits a melting point of 190 to
192 C.
Fumarate of this product:
mp. 196.5 to 198.5 C (recrystallized from isopropanol).
Maleate of this product: ~ -
mp. 173.5 to 175.5 oc (recrystallized from ethanol).
'~-:
This product, when recrystallized from methanol exhibits a
melting point of 172.5 to 174 oc and, when recrystallized
from water, gives crystals exhibiting a melting point of -
191.9 C, thus having the properties of crystals polymor-
phism.
.,
Methanesulfonate of this product:
mp. 124 to 128 C (recrystalllzed from isopropanol).
(13) (+)-cis-2-(4-methylphenyl)-3-acetoxy-5-[2-(dimethyl-
amino)ethyl~-8-methyl-2,3-dihydro-1,5-benzothiazepin-4(5H)-
one.maleate
. .
1312~77
-- 11 --
mp. 194 to 197 C (decomposed) (recrystallized from
ethanol).
¦~]20 + 83.7 (c=0.362, methanol).
Oxalate of this product:
mp. 179 to 180 C (recrystallized from ethanol).
[~]20 + 88.2O (c=0.288, methanol).
(14) (-)-cis-2-(4-methylphenyl)-3-acetoxy-5-[2-(dimethyl-
amino)ethyl]-8-methyl-2,3-dihydro-1,5-benzothiazepin-4(5H)-
one.oxalate
mp. 179.5 to 181 C (decomposed) (recrystallized from
ethanol).
[~]D ~ 83.8 (c=0.333, methanol).
Maleate of this product: -
mp. 195 to 197.5 C (decomposed) (recrystallized from
ethanol).
[~]20 _ 83.6 (c=0.50, methanol). - :
Fumarate of this product:
mp. 210.5 to 212.5 C (decomposed) (recrystallized from
ethanol).
[~20 _ 91.3 (c=0.323, methanol).
L-(+)-tartrate of this product:
mp. 140 to 143 C (recrystallized from a mixed solvent
of ethanol and ether).
. .,, . , " ~ , " . , ~. , , - . .