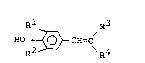Note: Descriptions are shown in the official language in which they were submitted.
3 ~
HYDROXYSTYRENE DERIV~TIVE
BACKGROUND OF THE INVENTION
The present invention relates to a novel
hydroxystyrene derivative or a salt thereof, which has
antiallergic activity, 5-lipoxygenase inhibiting
activity, antibacterial activity, tyrosine kinase
inhibiting activity, ultraviolet (hereinafter referred to
as "W") absorbing activity and reverse transcriptase
inhibiting activity and is useful as an intermediate for
preparing various organic compounds, and relates to an
antiallergic agent, a 5-lipoxygenase inhibiting agent, an
antibacterial agent, a tyrosine kinase inhibiting agent,
an W ab~orber and a reverse transcriptase inhibiting
agent containing the same as an active ingredient.
The compound of the present invention is a
novel compound which has not yet been reported in a
literature and is first synthesized by the present
inventors.
~ he objects of the present invention will
become apparent from the description hereinafter.
SUMMARY OF THE INVENTION
. . _ . . .
It has now been found that a novel
hydroxystyrene derivative of the present invention is a
useful intermediate for preparing various organic
compounds and has itself antiallergic activity, 5-
lipoxygenase inhibiting activity, antibacterial activity,
tyrosine kinase inhibiting activity, W absorbing
activity and reverse transcriptase inhibiting activity.
In accordance with the present invention, there
is provided a hydroxystyrene derivative represented the
formula (I~:
Rl R3
HO ~ CH=C (I)
R2-~-' \R4
,~,
- 2 - ~.3 ~
wherein when Rl and R2 are the same or different and each
is phenyl group, benzyl group or phenethyl group, or
is a group having the formula: R50- in which R5 is
hydrogen atom, an alkyl group having 1 to 5 carbon atoms
or benzyl group and R2 is benzyl group or a group of
PhSCH2 in which Ph is phenyl group, hereinafter the same,
R3 and R4 are taken together to represent a group having
the formula: -CONH-CS-S-, a group having the formula:
-CONH ~ , a group having the formula: -CONH ~ S02-
or a group having the formula: -CO-N=C-S- in which
NH(CH2)nlR6
R6 is a group having the formula: ~ (Xl)ml [in which
xl is hydrogen atom, a halogen atom, methyl group, ethyl
group, an alkoxyl group having the formula: R70~ (in
which R7 is methyl group or ethyl group), nitro group,
aminosulfonyl group or amino group, and ml is 1 or 2],
pyridyl group, furyl group or thienyl group, and nl is O
or an integer of 1 to 3; when Rl and R2 are the same or
different and each is phenyl group, ben~yl group or
phenethyl groupr or Rl is a group having the formula:
R50- in which R5 is as defined above, and R2 is benzyl
group, R3 is cyano group and R4 is carbamoyl group, or R3
and R4 are taken together to represent a group having the
formula: -CO-Y-CH2CH2- in which Y is oxygen atom or -NH-,
or a group having the formula: -CO-N-NH-CO-; and
when Rl and R2 are the same or different and each is an
alkyl group having 1 to 3 carbon atoms, R3 and R4 are
taken together to represent a group having the formula:
-CO-N=C-S- in which nl and R6 are as defined above,
NH(CH2)nlR6
or a salt thereof.
Also, in accordance with the present invention~
there is provided an antiallergic agent, a 5-lipoxygenase
inhibiting agent, an antibacterial agent, a tyrosine
_ 3 ~ 3 ~ ~ 7 ~ ~
kinase inhibiting agent, an W absorber or a reverse
transcriptase inhibiting agent containing the
hydroxystyrene derivative (I) or a pharmaceutically
acceptable salt thereof as an active ingredientO
DETAILED DE _RIPTION
The compound having the formula (I) of the
present invention can form a salt with a base or an
acid. The salt of the present invention may be any which
can be formed from the compound of the present invention
and the base or the acid.
~ xamples of the salt with the base are, for
instance, (1) a salt with metal, especially an alkali
metal salt, an alkaline earth metal salt and a salt with
aluminum; (2) an ammonium salt; and (3) an amine salt,
especially a salt with methylamine, ethylamine,
diethylamine, triethylamine, pyrrolidine, piperidine,
morpholine, hexamethyleneimine, aniline or pyridine, and
the like.
Examples of the salt with the acid are, for
instance, (1) a salt with an inorganic acid, especially a
salt with hydrochloric acid, sulfuric acid, phosphoric
acid, nitric acid or carbonic acid; (2) a salt with an
organic acid, especially a salt with a carbo~ylic acid
such as formic acid, acetic acid, propionic acid,
succinic acid, oxalic acid, tartaric acid, maleic acid,
lactic acid, benzoic acid, anthranilic acid or salicylic
acid; a salt with a sulfonic acid such as p-
toluenesulfonic acid or methanesulfonic acid; a salt with
an amino acid such as glycine, methionine or lysine; and
the like.
When the salts are employed for the
antiallergic agent, the 5~1ipoxygenase inhibiting agent,
the antibacterial agent, the tyrosine kinase inhibiting
agent, the W absorber or the reverse transcriptase
inhibiting agent, the pharmaceutically acceptable salts
should be employed.
As typical examples of the compounds of the
:~ 3 ~
-- 4
invention, the compounds (1) to (45) are shown in Table 1
by showing the groups Rl, R2, R3 and R4 in the formula
(I), and further, exemplifying the group R6 and nl in
case that R3 and R4 are taken together to represent a
group having the formula: -CO-N=C-S- . Also, the
NH(CH2)nlR6
molecular formula, molecular weight, melting point, and
data of elementary analysis of each compound of (1) to
(45) are shown in Table 1. The results of lH-NMR
spectrum analysis and IX spectrum analysis of the
compounds (1) to (45) are shown in Table 2.
~3~ 3
~ ,
~.~ ~ ~ ~n ~
~ ~U) ^ U~ _ ~ _ ~ _ ~ _ s
0 3~ o n2 Ln o ~ O ~ O ~ ,~
o In o u~ z ~r Z ~r Z Ln JJ
Z ~ ~:
n a~ na r~ O
~ ~ m~ CO m~ m
U
U
~1 0
~o ~:
I I . I
~,
~! ~ 1~ ~ ~o~
~ o o o o o
, C~ ,u Y C'
N ~ Uc S
~`J
s m ~ ~uq
~ ~ r n
O O
U Z
- 6 - ~ J
_ o In o
~_ ~ .
Z E4d-~
.~n . l
~q ~ co a~
t~ _ C~
.
c m _ ~ r
~ ~: - a~ ~ o ~o 1`
~_
~ 0~
c~ O~
~ ~ ~ r~l oo O
~ V _ ~ ~ ~~1 ~
o o o oo
X ~ o
O ~ o ~1
~ Z
- 7
_~ l
0V ~
~ .,, u~ cn ~ u~ cn :~
a~
0 3 o ~r O a~ O ~1 0 O ~) .,
Z In Z ~ Z ~o Z ~D Z ~ -L~
r~ 1 O
~: o ~ r~ ~ o
~ ~ ,~
~: ~
~ l l l l l
~D~
1:~ I U~
U C~
~; O Z Z O
D~
~; Ln ~
~a ~
~1 ra
cO~ ~ ~ o
o o
c~ z
- 8 - ~ 3
.
~, ~p t~
~-
Z ~_ oo ~ o
~_
.U~ .
u~ ~ u~ 1 o o
o
~
:C
, ~ o
E o _ ~ ~ ~ ul m
1~
.
In C
_ U~
.
-- ~o ~ ~ CO
C~ In U~
C~
r~ ~ oo oo l`
~ _ ~ ~ o o
O _ ~
~ Lr~
,1 ~ ~ oa~
.,, ~ _
~: C) O O O O O
O -- ~ .1~
~: ~ o a~ o
~ a~
a~
.~ ~U~
C~
O O
C) Z
~ 9
~JJ l
0 3 ~ 0~1` otY) ~ 0~ a~ ~-- ~
~1 Z ~ Z U') Z ~1 Z ~ Z ~ ~
o O
~ ~ m ~ m Ln ;~ r ~
a~
O N -- N -- N -- N
~ l
~D~ l l l l l
~r ~ O
~ o o o o Z
t`l S .C p~ P ~ .
~ ~r S
~:
a ~
O ~ Z ~
- 10 - :~3~ b~ ~
~ ~''1 .-1 N ~1 ~)
~) _ I` o a~ Il) ~1
.
~_ ~ CO ~
.,1
z ~a v
~: ~ ~ l N ~--1 O
O ~ ~ O a~ In C~
~ ~ l
.Ul .
o~ ~ r~
~ C) ~p u~
ra C~ ~
m
~ ~ 0
V ~ _
O _
E~
.
~a ~ ~ O 1`
_ ~ a~
o~ ~
-- o
c~ r~ l` ~ I` t`
~o o ~ ,~
_ ~ 0
E4- o ~r r oo r~
~ 0
a~ ~ ~ co a~
~: ~ ~ N
-~ V _
V S:: ~) O O O O O
O V V .1_~ V ~1
O O --
X R~ o IJ7 0 1~
~ 11') ~ 0 r-
'1:1 ~1 ~I N r~l r l
.~
~:
O O ,~ ~ ~ ~ n
~1 ,
O O
Z
e ~ N ~ ~
~ a~ O -- ~-- O ^ O ^ O -- ~::
0 3 ~ N O In Z ~ N 00 N ~ ,1
r ~ r~ r Z Lr~ V
s~ ~ ~ O ~ ~~ ~ ~ O O
~ ,1 tq ~ m ~ D v
:~ V ~r r~ N t~ ~ r~ --
O
~ l ll l l l
l~ I I t
m O
Z O Zo Z;_s Z_~
o o
~;
~: ~
~: ~
V E~ ,1 ,1 ,1 ~I N
l ~o~ z;
- 1 2
~ o a~ l
~ _ ~ r o.
C~- ~ ~
Z ~ 0 o ~J ~ ~::
~_ a~ w
~_ ~ I
,~n .
ul ~ I~ a~ co ~o In
dP
u~ Ln ~ ~r Lr
m
_
~ ~0- In Ln
E~
CJ ~ ~ O
~ ~ C~
~ I~
c)
~ ~ o a~
~0- ~ co ~
co
~o n ~ o
o o o o o
o --
~: ~ o 1~
Ln U~ ~ o
'JJ
O O ~ I~ o
U ~ ~
O O
U Z
- 13 ~ P) ~ `'i
_ I
r~) r~l r~
~ Q~O -- O -~ O ~ O -- ~ ^ r
0 3r~ In r~ ~ ~ O ~ --I O 1` .
q~Z~ ~r) Z~D r`') Z'~' ~ Z~ . Z ' ~
fl~ _I N ~I r~ ,1 ~ ,~ ~ r~ ~ O
tC ~ tC o ~ rl~ r~7 ~o U
:~ rl~ r~) co r~ ~ r~7 r~
U ,~
~ ~01 U ~ l
O ~
~; I I I I S
O O 0 5, ~ C~
r~ Z Z Z Z ~ Z
C~ C~ C) y O
r~l ~ ~ ~
~ m P:~ :r w ~
o o
~ Lt~ r~) O ~0 p,
.~ ~
O O ,1 ~ r~
U O O r~ r~l r~l r~ r~
~l 3 ~
-- 14
~ o~ ~
dP ~ U~ ~ O
~- CO
::
Z ~ o ~ co
~ - ~ o
~o- o~
,tQ .
~ ~ ~ ~ o o o
~1 U _ ~D N 0~ 1~ CO
.
~a ~ ~ Ln u~
~ ~I
~a ~ 1
~ ~ dP
a~ ~o - u~
W
.
O ~ 0~ ~ ,1
C~ dP 01) ~
~ -- o o a~
O
~ _ Ln O W
O _ O O ~
~ I~
~ r~ o ~ u~ n
.,~ ~ _ ~1 ~'d ~ N ~I
O O O O O
a) o -- J~
~ Q~ ~
a)
.,, ~a
O O ~
c~ O O N N N N N
O Z
- 1 5 - ~ 3 ~
_ I
~ ~ ~ ~ ~ .~s
~ ~ ~ U~ ~
o 3 O ~t) o ~ o ~I o ~ o ,_1 ._~
~D ~ ~ ~ ~D ~ U~ ~ U~ .
~ ~ Z Z ~ Z~ Zco ~r oD N O
11~ ~1t~ C)~ ~ ~~ I` t`J O N t` t)
,, :: m~
~ a
a~ ~o1 U C~
~ P, ~0 s ~ s
~r
~ ~`J ~ N N
~ m
U~
--æ c3_t¢
o C~ C~ Uo
s '~ m
tl ~ S C
,~ m m ~~
~o ~ U
O O ~ I` OD cr~ o
r~
l C~ Z
:~ 3 ~
-- 16
~:i ~
V _
~ Lr) ~o In In U-
:~
Z r
S~ _ ~ O U~ D ~O~
~_ Lr
.U~ .
_
~1 ~ ~ .
~
0
h trl
~ ~_ ~ o 0
~P .
~ ~ _ Ln
.
~_ o ~
~ -- In C~
r~
,~
O _ U~ O U~ ~ ~
IL~ ~
~ ~P ~ a~ I` ~r
.~ ~ ~ ~
~ ~ C~ O O O O O
,1.. -1 0 ~ ~ ~ J~
a) o --
u~ ~ ~ In ~Y
~r ~ ~ I` ~
~ . f`~
.
O O w 1~ 0 a~ o
C~ O O ~
~ Z
- 1 7 ~ 3
1~1~ l
~I C U~ ~
0 3O r-l O OO IY) O 1-'1 0 N C
LIN 1~ ~ 1N 11'1 ~i In N Lt7 C
~ la ~ ~ ~ o ~ tl ~ O
~ ~(~ ~ t~ I o N r-l U
~ ~m ~ m ~
~ ~a~
O a) N N N N N
~I) o c,~ V l~
~0 ~
~1 0
~D~ ~ S
\~
U~
C C ~ C C
, m e~
u~
-)~Z u--~ Z C~_r
Z Z Z Z iZ
~ O ~ O o
I
m m~
O
mN
C~; y '' y Y
::~; ~C
C :1
O O
o ~
o o
~ Z
~ 3 ~
-- 18
~ ~ l
C) ~ ,~
U~
. ~
.
Z ~ u~ c
c ,_ a~ o a~ ~
~0,-
,u~ ~
~n
:~ t~
a c~
0
m
~a r~ o oo ~ ~P
V ~ Ul ~ U~ O
~0 -
O ~ ~ ~ o In C~
0 _ ~ a~ o o ~D
C~
ro ~
o ~_ ~r ~ o o ~D
r~
C ~ ~1 OD ~r r_
.~ .,. _
V C U o o o o o
.~.
~
U) ~1 00 ~ 1
~a
V :~
O O ~ ~ ~ ~ In
u e
u zo
1 9 ~
~'3 ~ O~Ln O~ oP~l 3
~J O a~ o~ z~ u) zOJ U~ z ~
O U UN U UN N O
~ ,1 ~1 ~t 'I 'I
~ ~ ~ U ~ o ~
~ U~ U~ U~ U~ U~
t ~Y U~
~ ~ ~ ,1 ,1
o e~ O ~ O
- 2 0 _~ e~ ~ ~ 7 ~,
o
~ _ ~ o .t. ~o ~ l
~ ~ o
U- ~
.
Z ~_ ~ o
~0
,u~ .
~? ~a t~ ~ u~
a
0 ~0~ - U~ 9 Ul
~ t':
~ ~ ~0 ~ ~ C;~,~
.u ~ _a~
~ O-' In L~
.
~_~ O ~ O ~O
~1 ~P .
0 _ ~ Cl~
C~ ~
~a ~ o
~: _ 1~
d~ . . . . .
o _ ~ a~ I~ o ~
~ ~ o~
-~ JJ _ U
,~ ) - ~ ,U U
aJ O -- ~_
1 ~1
~ co
O O ~ r~ o
c~ o o ~
~ z
~J- l
e .~ ~ n ~
h 11~ ~ G
0 3 O ~ O U~ O t~O O Lt~ O ~ ,~
~J Z ~ Z Z Z Z
u t~ ~ o 1~) It O
(lS ~1 ~ 1~ ~ O ~ O~
~ ~ ~ ~ :q ~ ~'
~ O ~ ~ ~
~
Z~ I
~o~$ ~ ~O~C,Q<~Z
~ O
~r ~
m~ ' qN I ~ I m~
-z ~ x ~_~
Z Z Z Z; Z
~) C) O o
P~
~ .Y, .Y, .' .Y, .Y,
_1~ pr~ m~
Y Y ~' Y Y
.r~
c~
l ~ Z
22 - ~ 3
.
~ ~ CO
U- ' '` ~
Z ~ CO
~ _ ~
E4-
~n .
_ un
.
U -
~:
r~
_
~ ~ _ Ln
.
I~D ~ a~
c~ ~ ~
~ _ ~
U Ln
U
~ ~ I~
o _ co r~
~ ~O ~ OD 00 ~1
.~ ~ _ C~
~ ~ o ~ o o o
,,-,, O ~ ~a ~ ~ ~
~ Ln a~ o o
~O o t~ a~
~a
.
O O ,~
~ O O
~ Z
~ 23
Table 2
Compound lH-~MR spectrum IR spectrum
No. ~ (ppm) (cm 1)
1 CDCQ3/DMSO-d6 = 1/1;7.3-7.7(13H,m), KBr; 3540, 3150l
9.01(1H,br~, 13.4(1H,br) 3050, 1700, 1590
2 CDCQ3/DMSO-d6 = 1/1;4.03(4H,s), 7.0- KBr; 3330, 3300,
7.4(13H,m), 9.27(1H,br), 13.55(1H,br) 1680, 1570
3 CDCQ3/DMSO-d6 = 1/1;6.7-7.8(16Hrm), KBr; 3550, 3180,
8.33(1H,s), 8.6(1H,br), 10.4(1H,br) 3050, 1695, 1620,
1590
4 CDCQ3/DMSO-d6 = 1/1;4.05(4H,s), 6.5- KBr; 3380, 3200,
7.3(16H,m), 7.45(1H,s), 9.0(1H,br), 1685, 1585
10.2(1H,br)
CDCQ3/DMSO-d6 = 1/1;3.97(4H,s), 7.1- KBr; 3450, 3200,
7.8(16H,m), 7.75(1H,s), 9.5(1H,br) 3060, 1680, 1600
6 CDCQ3/DMSO-d6 = 2/1;1.40(3H,t), 4.10(2H,
q), 4.16(2H,s), 4.70(2H,d), 7.0-7.7(15H,
m), 9.1-9.6(1H,br), 9.7-lO.O(lH,br)
7 CDCQ3/DMSO-d6 = 1/1;4.15(2H,s), 6.9 KBr; 3440,3260,
(2H,s), 7.0-8.6(8H,m), 10.0(2H,br~ 1670,1575
8 CDCQ3/DMSO-d6 = 1/1;4.18(2H~s), 5.18 KBr; 3520, 3120,
(2H,s), 6.8-7.6(13H,m), 9.7(lH,br) 3050, 2850, 1675,
1570
9 CDC~3/DMSO-d6 = 1/1;0.98(3H,t), 1.2- KBr; 3480, 3130,
1.9(4H,m), 4.05(2H,t), 4.17(2H,s), 3050, 2850, 1675,
6.97(2H,s), 7.0-7~3(5H,m), 7.42(1H,s), 1570
9.45(1H,br), 13.4(1H,br)
CDCQ3/DMSO-d6 = 1/1;0.95(3H,t), 1.3- KBr; 3480, 3130,
2.0~4H,m), 3.93(2H,s), 4.02(2H,t), 3020, 2950, 2850,
6.8-7.4(7H,m), 7.45(1H,s), 9.28(1H,br) 1685, 1570
11 CDCQ3/DMSO-d6 = 1/1;4.19(2H,s), 6.7- KBr; 3420, 3180,
7.8(12H,m), 9.3(2H,br), 1003(1H,br) 1705, lS90
.
- continued
3 ~
- 24
- continued
.
Compound lH-NMR spectrum IR spectrum
No. ~ (ppm) (cm 1)
12 CDCQ3/DMSO-d6 = 1/1;4.22(2H,s), 5.25 KBr; 3505, 3150,
(2H,s), 6.7-7.7(16H,m), 8.87(1H,d), 3080, 3050, 3020,
9.3(1H,br), 10.3(1~,br) 1670, 1615, 1580
13 CDCQ3/DMSO-d6 = 1/1;3.97(3H,s), 4.00 KBr; 3400, 3170,
(2H,s), 6.7-7.6(11H,m), 8.77(1H,d), 3060, 1690, 1620,
9.2(1H,br), 10.4(1H,br) 1610, 1580
14 CDCQ3/DMSO-d6 = 1/1;0.94(3~,t), 1.3- KBr; 3160l 3130,
1.9(4H,m), 3.94(2H,s), 4.00(2H,t), 6.5- 3060, 3020, 2950,
7.5(12~,m), 8.9(1H,br), 10.4(1H,br) 1685, 1610
CDCQ3/DMSO-d6 = 1/1;7.3-7.8(12H,m), KBr; 3500, 3475,
7.85(2H,s), 8.15(1H,s), 9.25(1H,s) 3300, 3200, 2205,
1710, 15~0
16 CDC~3/DMSO-d6 = 1/1;4.00(4H,s), 7.1- KBr; 3400, 3320,
7.3(10H,m), 7.4(2H,br), 7.57(2H,s), 2205, 1660, 1565
7.90(1H,s), 9.5(1H,br)
17 CDCQ3/DMSO-d6 = 1/1;2.93(2H,t-d), 4.00 KBr; 3360, 1720
(4H,s), 4.30(2H,t), 7.0-7.3(13H,m), 1645, 1590
9.0(1~,br)
18 CDCQ3/DMSO-d6 = 1/1;2.77(~H,m), 3.30 KBr; 3400, 3200,
(2H,m), 3.97(4H,s), 6.8-7.5(13H,m), 2900, 1685, 1640,
7.8(1H,br), 8.8(1H,br) 1600, 1580
19 CDC~3/DMSO-d6 = 1/1;7.0-8.0(16H,m), KBr; 3530, 3220,
8.48(1H,s), 8.53(1H,s), 9.3(1H,br) 3080, 1720, 1660,
1620, 1570
CDCQ3/DMSO-d6 = 1/1;4.00(4H,s), 7.0- KBr; 3150, 3060,
7.9(16H,m), 8.3(1H,s), 8.35(1H,s), 3020, 1700, 1655,
9.8(1H,br) 1620, 1570
21 CDCQ3/DMSO-d6 = 1/1;1.43(3H,t), 3.97 KBr; 3520, 3380,
(2H,s), 4.12(2H,q), 7.1-7.3(6H,m), 3170, 2205, 1685,
7.43(2H,br), 7.60(1H,d), 8.00(1H,s), 1575
9.30(lH,br)
- continu~d
~ 3 ~ J ~
- 25
- continued
CompoundlH-NMR spectrum IR spectrum
No. ~ ~ppm) (cm 1)
22 CDCQ3/DMSO-d6 = 1/1;3.87(3H,s), 3.93 KBr; 3500, 3370,
(2H,s), 7.1-7.3(6H,m), 7.40(2H,br), 3170, 2200, 1665,
7.60(1H,d), 7.98(1H,s), 9.5(1H,br) 1570
23 CDCQ3/DMSO~d6 = 1/1;3.92(2H,s), 7.06 KBr; 3440, 3310,
(lH,d), 7.1-7.3(5H,m), 7.4(2HIbr), 3250, 2210, 1660,
7.53(1H,d), 7.87(1H,s), 9.4(2H,br) 1590, 1570
24 CDCQ3/DMSO-d6 = 1/1;3.90(2H,s), 7.1- KBr; 3480, 31~0,
7.8(12H,m), 8.38(1H,dd), 9.9(2H,br) 1710, 1650, 1600,
1570
25 CDCQ3/DMSO-d6 = 1/1;4.75(2H,d), 7.3- KBr; 3570, 3200,
7.7(18H,m), 8.8(1H,br), 9.84(1H,t) 2850, 1690, 1635,
1610, 1570
26 CDCQ3/DMSO-d6 = 1/1;4.00(4H,s), 4.82 KBr; 3300r 3200,
(2H,d), 7.1-7.3(18H,m), 9.0(1H,br), 3010, 2880, 1660,
9.78(1~,t) 1610, 1590, 157
27 CDCQ3/DMSO-d6 = 1/1;4.13(2H,s), 4.72 KBr; 3550, 3180,
(2H,s), 6.37(2H,d), 6.90(2H,s), 7.2- 2800, 1660, 1620,
7.5(6H,m), 7.57(1H,d), 9.8(3H,br) 1580
28 CDCQ3/DMSO-d6 = 2/1;1.40(3H,t), 4.10(2H,
q), 4.16(2H,s), 4.70(2H,d), 7.03-7.73
(15H,m), 9.10-9.60(1H,br), 9.7-lO.O(lH,br)
29 CDCQ3/DMSO-d6 = 1/1;0.97(3H,t), 1.3- KBr; 3520, 3200,
2.0(4H,m), 4.03(2H,t), 4.13(2H,s), 3050, 2950, 2880,
4.72(2H,s), 6.9-7.5(13H,m) 1680, 1615, 1595
30 CDCQ3/DMSO-d6 = 1/1;1.02(3H,t), 1.3- KBr; 3520, 3200,
1.9(4H,m), 4.03(2H,s), 4.08(2H,t), 3020, 2900, 2870,
4.59(2H,s), 6.88(2H,s), 7.1-7.7(11H, 1670, 1590
m), 8.0(1H,br)
31 CDCQ3/DMSO-d6 = 1/1;4.17(2H,s), 4.87 KBr; 3500, 3200,
(2H,s), 5.17(2H,s), 6.9-7.6(16H,m), 3060, 2770, 1680,
9.8(2H,br) 1630, 1610, 1590
. . . _ . . .
- continued
- 26
P' rd~
- continued
_ _ . . .. . _ . _ . _
Compound lH-NMR spectrum IR sp~ctrum
No. ~ (ppm) (cm 1)
_ _ _
32 CDcQ3~l.3o(l2H,d)f 3.12(2H,m), 7.10
(2H,d), 7.41(2H,s), 7.52(1H,br), 7.90
(lH,s), 10.21(1H,br)
33 CDCQ3/DMSO-d6 = 10/1;1.20(12H,d), 3.30
(2H,m), 4.70(2H,s), 7.13(2H,s), 7.30
(5H,m), 7.56(1H,s), 9.30-9.80(1H,br)
34 CDCQ3/DMSO-d6 = 10/1;1.23(12H,d),
2.96(2H,t), 3.40(2H,m), 3.80(2H,q),
7020-7.40(7H,m), 7.53(1H,s), 8.40-
8.70(1H,br), 9.46(1H,t)
CDCQ3;1.23(12H,d), 3.36(2H,m), 4.76
(2H,d), 6.86-7.50(6H,m), 7.67(1H,s),
7.90-8.40(1H,br), 9.23-9.66(1H,br)
36 CDCQ3/DMSO-d6 = 10/1;1.26(12H,d), 3.36(2H,m),
4.70(2H,s), 7.20(2H,s), 7.33(4H,s), 7.07
(lH,s), 8.00-8.40(1H,br), 9.10-9.70(1H,br)
37 CDCQ3/DMSO-d6 = 10/1;1.23(12H,d), 3.33
(2H,m), 4.70(2H,d), 7.20 7.47(5H,m), 7.67
(l~,s), 7.80-8.20(1H,br), 9.20-9.60(1H,br)
38 CDCQ3/DMSO-d6 = 10/1;1.16(12H,d), 3.33
12H,m), 3.73(3H,s), 4.70(1H,s), 6.80~2H,
d), 7.16(2H,s), 7.30(2H,d), 7.60(1H,s),
7.85-8.20(1H,br), 9.00-9.60(1H,br)
39 CDCQ3/DMSO-d6 = 10/1;1.23(12H,d), 2.33(3H,s),
3.36(2H,m), 4.70(2H,d), 7.06-7.26(6H,m),
7.66(1H,s), 8.0-8.3(1H,br), 9.30(1H,t)
40 CDCQ3/DMSO-d6 = 10/1;1.23(12H,d), 3.36
(2~,m), 4.87(2H,d), 7.16(2H,s), 7.50
(lH,s), 7.60(2H,d), 8.20(2H,d), 8.2-8.6
(lH,br), 9.67(1H,br)
- continued
- 27 - ~ 3 1.~ 3
- continued
_
CompoundlH-NMR spectrum IR spectrum
No. ~ (ppm) !cm~l)
. _ . . . _ . . .
41 CDCQ3/DMSO-d6 = 10/1;1.23(12H,d),
3.16(2H,s), 3.33(2H,m), 4.80(2H,s),
6.96-7.90(7H,m), 8.0-8.4(1H,br),
9.63-9.76(1H,m)
42 CDCQ3/DMSO-d6 = 10/1;1.26(12H,d)~ 3.30
(2H,s), 3.36(2H,m), 4.66(2H,d), 6.63
(2H,d), 7.06(2H,d), 7.20(2H,s), 7.56
(lH,s), 8.4-8.8(1H,br), 9.5-9.7(1H,br)
43 CDCQ3/DMSO-d6 = 10/1jl.27(12H,d), 3.36
(2H,m), 4.80(2H,d), 6.36(2H,s), 7.26
(2H,s), 7.43(1H,s), 7.73(1H,s), 7.8-803
(l~,br), 9.1-9.5(1H,br)
44 CDcQ3/DMso-d6 = 10/1;1.26(12H,d), 3.36
(2H,m), 4.96(2H,d), 6.9-7.3(5H,m), 7O73
(lH,s)~ 7.8-8.4(1H,br), 9.40(1~,t)
45 CDC~3;1.23(12H,d), 3.2312E,m), 4.86
(2H,d), 7.06-7.46(5H,m), 7.66(1H,d),
7.76(1H,s), 8.50(1H,d), 8.7-9.1(lH,br)
. , . _ . . _ . _ _ ~
- 28
The compound having the formula (I) of the
present invention can be prepared by any processes as far
as the compound can be obtainedl and there are
exemplified the following processes (a), (b) and (c) as
the preparation processes.
(a) The compound having the formula (I) can be
prepared by a condensation reaction of a benzaldehyde
having the formula tII):
R8
Rl OO--~CHO ( I I )
R
wherein R8 and R9 are the same or different and each is
an alkyl group having 1 to 3 carbon atoms, phenyl group,
benzyl group or phenethyl group , or R8 is a group having
the formula: RllO- in which Rll is hydrogen atom, an
alkyl group having 1 to 5 carbon atoms or benzyl group,
and R9 is benzyl group or a group: PhSCH2, and R10 is
hydrogen atom, an alkyl group having 1 to 3 carbon atoms,
an alkyl group substituted with ethers, e.g.
methoxymethyl group or methoxyethoxymethyl group, benzyl
group, an acyl group having the formula: COR12 in which
R12 is hydrogen atom or an alkyl group having 1 to 3
carbon atoms, or a trialkylsilyl group such as
trimethylsilyl group or tert-butyldimethylsilyl group;
and a compound having the formula (III):
~ R13
C~2 (III)
~Rl 4
wherein R13 is cyano group and R14 is carbamoyl group, or
R13 and R14 are taken together to represent a group:
-CO-Y-CH2CH2- in which Y is oxygen atom or a group:
N(CoR15)- in which R15 is hydrogen atom or an alkyl
group having 1 to 3 carbon atoms, a group:
~ 3 ~ 3
- 29
-CO-N~N~CO-, a group: -CON~-CS-S-, a group: -CO~
or a group: -CON~ ~ SO2-;
or a compound having the formula: (IV):
N ~ NU(C~2~n2R16 ~IV)
wherein R16 is a group having the formula: ~ (X2)m2
[in which x2 is hydrogen atom, a halogen atom, methyl
group, ethyl group, an alkoxyl group having the formula:
R17O- (in which R17 is methyl group or ethyl group),
nitro group, aminosulfonyl group or amino yroup/ and ml
is 1 or 2]l pyridyl group, furyl group or thienyl group,
and n2 is 0 or an integer of 1 to 3;
in the absence or presence of an acid or a base as a
catalyst.
Examples of the acid used as the catalyst in
the above-mentioned reaction are, for instance, a proton
acid such as sulfuric acid, benzenesulfonic acid or p-
toluenesulfonic acid, a Lewis acid such as boron
trifluoride, and the like.
Examples of the base used as the catalyst are,
for instance, ammonium or its salt, an or~anic base such
as piperidine, pyrrolidine, monoethanolamine, p~ridine,
morpholine or 1,8-azabicyclo [5.4.0] undeca-7-ene or a
salt thereof~ an alkali metal salt of or~anic acid such
as sodium acetate or potassium acetate, an alkali metal
hydroxide such as sodium hydroxide or potassium
hydroxide, an alkali metal amide such as lithium
diisopropylamide, an alkali metal alcoholate such as
sodium methylate or potassium butylate, an alkali metal
hydride such as sodium hydride or potassium hydride, and
the like.
When R10 in the starting material is remained
- 30
in the obtained product as an alkvl, an alkyl group
substituted with ethers, ben~yl, an acyi, or
trialkylsilyl group due to noncatalytic reaction or the
kind of catalyst employed, the desired compound can be
obtained by eliminating R10~ For eliminating R10~ when
R10 is an alkyl group or an alkyl grou~ substituted with
_~~e_~ cleavage re-c-ion which is carried out by using a
Lewis acid such as aluminum chloride~ boron trifluoride or bo-
ron tribromide or a proton acid such as hydrogen bromide or
trichloroacetic acid, other ether bond cleavage reaction,
or the like can be adopted. When R10 is benzyl group,
catalytic reduction reaction can be employed which is
carried out by using a noble metal catalyst such as
palladium carbon, as well as the above-mentioned ether
bond cleavage reaction. When R10 is an acyl group, R10
can be eliminated by hydrolysis reaction which is carried
out by using a base such as an alkali metal hydroxide
such as sodium hydroxide or an alkaline earth metal
hydroxide such as barium hydroxide. ~hen R10 is
trialkvlsily~ grou~, R10 c2n be eliminated with water,
me_hanol, an acld, fluorine ion, or the like.
When the reaction is carried out by employing
an N-acyllactam and an acyl group is remained in the
obtained product, the acyl group can be eliminated by
hydrolysis reaction using a base such as alkali metal
hydroxide such as sodium hydroxide to give the desired
compound.
(b) The compound having the formula (I) can be
pre?ared, according to O. Ister et al. [~elvetica Chimica
Acta (~elv. Chim. Acta), 40, 1242(1957)], G. A. Howie et
21. ~Journal of Medicinal Chemistry (J. Med. Chem.), 17,
840~1974)], ~. Wamhoff et al. [Synthesis, 331(1976)], and
the like, by reacting a benzaldehyde having the formula
(V):
R18
~0 ~C~O (V)
Rl9
s~
- 31 ~
wherein R18 and R19 are the same or different and each is
an alkyl group having 1 to 3 carbon atoms, phenyl group,
benzyl group or phenethyl group, or R18 is a group: R200-
in which R20 is hydrogen atom, an alkyl group having 1 to
5 carbon atoms or ben2yl group, and R19 is benzyl group
or the group: PhSC~2;
with an ylide having the formula (VI~:
R21
(Ar)3P ~ (VI)
R22
wherein Ar is an aryl group, R21 is a cyano group, and
R22 is carbamoyl group, or R21 and R22 are talcen together
to represent a group having the formula:
-CO-Z-C~2C~2- in which Z is oxygen atom or -NH-, a group
having the formula: -COI-N~-CO-, a group having the
Ph
formula: -CON~-CS-S-, a group having the formula:
-CONH ~ or a group having the formula: -CONH ~ S02-;
or an ylide having the formula (VII):
(Ar)3P1 S
O~`N ~ NH(CH2)n3R23 (VII)
wherein R23 is a group having the formula: ~ (X )m3
[in which X3 is hydrogen atom, a halogen atom, methyl
group, ethyl group, an alkoxyl group having the formula:
R240- (in which R24 is methyl group or ethyl group),
nitro group, aminosulfonyl group or amino group, and m3
is 1 or 2], pyridyl group, furyl group or thienyl group,
and n3 is O or an integer of 1 to 3.
The above-mentioned reaction (b) is carried out
according to the so-called Wittig reaction. For the
ylide in the reaction (b), a ylide derived from a
trialkyl phosphine such as tributyl phosphine or a
triaryl arsine such as triphenyl arsine can also be used
- 32
as well as the above-mentioned ylide (VI) or (VII~.
(c) The compound, which is one of the
embodiments of the present invention, having the formula
(VIII):
R25
HO ~ CH S (VIII)
R26 O~N~N:E~(CH2)n4R27
wherein R25 and R26 are the same or different and each is
an alkyl group having 1 to 3 carbon atoms, phenyl group,
benzyl group or phenethyl group, or R25 is a group having
the formula: R23O- in which R28 is hydrogen atom, an
alkyl group having 1 to 5 carbon atorns or benzyl group,
R26 is benzyl group or the group: PhSCH2, R27 is a group
having the formula:
~ (X )m [in which X4 is hydrogen atom, a halogen
atom, methyl group, ethyl group, an alkoxyl group having
the formula: R29O- (in which R29 is methyl group or ethyl
group), nitro group, aminosulfonyl group or amino group,
and m4 is 1 or 2], pyridyl group, furyl group or thienyl
group, and n4 is 0 or an intenger of 1 to 3, can be
prepared, according to M. T. Omar et al. [Acta Chimica
Academiae Scientiorum ~ungaricae (Acta Chim. Budapest)],
83, 359(1974); Indian Journal of Chemistry (Ind. J.
Chem.) 20B, 849(1981)], by reacting a compound having the
formula (IX):
R30
HO ~ CH ~ S (IX)
R31 O N ~ S
H
wherein R30 and R31 are the same or different and each is
an alkyl group having 1 to 3 carbon atoms~ phenyl group,
benzyl group or phenethyl group, or R30 is a group having
- 33 - ~ 3 ~
the formula: R32O- in which ~3~ is hydrogen atom, an
alkyl group having 1 to 5 carbon atoms or benzyl group,
and R31 is benzyl group or a group: PhSC~2;
or a compound having the formula (X):
R33
HO~C~-r -- S ( X )
R34 ~ N ~ SR35
wherein R33 and R3~ are the same or different and each is
an alkyl group having 1 to 3 carbon atoms, phenyl group,
benzyl group or phenethyl group, or R33 is a group having
the formula: R36O- in which R36 is hydrogen atom, an
alkyl group having 1 to 5 carbon atoms or benzyl group,
R34 is benzyl group or a group: PhSCH2, and R35 is an
alkyl group having 1 to 3 carbon atoms;
with an amine having the formula (XI~:
H2N(C~2)n5R37 (XI)
wherein R37 is a group having the formula~ )m5
[in which X5 is hydrogen atom, a halogen atom, methyl
group, ethyl group, an alkoxyl group having the Cormula:
R38O- (in which R38 is methyl group or ethyl group),
nitro group, aminosulfonyl group or amino group, and m5
is 1 or 2], pyridyl group, furyl group or thienyl group,
and n5 is 0 or an integer of 1 to 3.
The novel hydroxystyrene derivative (I) of the
present invention or a salt thereof is useful as an
intermediate for preparing various organic compounds, and
also useful as an antiallergic agent/ 5-lipoxygenase
inhibiting agent, an antibacterial agent, a tyrosine
kinase inhibiting agent, an W absorber or a reverse
transcriptase inhibiting agent.
That is, the hydroxystyrene derivative can be
expected to be used as an antiallergic agent and the
like, by its antiallergic activity. By its 5-
lipoxygenase inhibiting activity, it can be expected to
- 3~ - ~3~
be used as an antiasthmatic agent, an antiinflammatory
agent, agents for the treatments of psoriasis, nephritis
and myocardial infarction, an agent for preventing
myocardial infarction and the like. By its antibacterial
activity, it can be expected to be used as an
antibacterial agent. By its tyrosine kinase inhibiting
activity, it can be used as an antiasthmatic agent, an
antiinflammatory agent, an anti~cancer agent, a
carcinogenesis preventing agent, a metastasis-preventing
agent, an agent used for the treatment of mental disease
and the like. By its W absorbing activity, it can be
expected to be used for the prevention of erythema
solare, used for preventing the deterioration of
materials of organic high molecular weight compounds due
to ultraviolet rays, and the like. Also, by its reverse
transcriptase inhibiting activity, it can be expected to
be used as an agent for preventing virus infections.
The above-mentioned activities of the compound
of the present invention are specifically described by
the following tests. In Tables 3 to 9, each compound No.
corresponds to the compound No. in Tables 1 and 2.
The antiallergic activity of the compound of
the invention is proved by the tests of inhibitory
activity against passive cutaneous anaphylaxis
(hereinafter referred to as "PCA") reaction, inhibitory
activity against antigen-induced anaphylactic shock and
inhibitory activity against antigen~induced airway
constriction.
(1) Inhibitory activity against homologous PCA reaction
in rats
Antiserum was prepared according to I.
Mota [Immunology, 7, 681(1964)] and the PCA reaction was
conducted according to Maruyama et al. [Folia
Pharmacologica Japonica, 74, 179(1978)].
Preparation of antiserum
An ovalbumin solution dissolved in
physiological saline (2 mg/mQ) was injected
intramuscularly into both thighs of male Wistar rats
~ 35 ~ ~ 3 ~
weighing 200 to 260 g in a volume of 0.5 mQ/lOO g body
weight, and pertussis vaccine tsordetella pertussis, 2 x
10l/mQ, Chiba Serum Institute) was intraperitoneally
administered at 1 mQ/rat. Twelve days after
sensitization, blood was taken from posterior aorta under
ether anesthesia and antiserum was obtained and stored at
-80C until use.
PCA reaction
Groups of 4 male Wistar rats each, weighing 180
to 210 g, were used. Back of the rats was shaved and
each 0.05 mQ of antiserum diluted 32 times with
physiological saline was injected intradermally at four
sites on the back. After 48 hours, 1 mQ of a mixture of
ovalbumin (2 mg/m~) as an antigen and Evans blue (10
mg/mQ) in the volume ratio of 1 : 1, which was dissolved
in physiological saline was injected intravenously into
the tail. Thirty minutes later, the rats were bled to
death under ether anesthesia and the back skin of the
rats was removed. The blue-dyed area formed by pigment
exudation was measured and an inhibition rate (%) was
calculated as compared with control according to the
following equation.
A - B
Inhibition rate (~ x 100
A
A: Blue-dyed area in the control group
B: Blue-dyed area in the test compound group
A test compound suspended in a 2.5 ~ aqueous
solution of gum arabic containing 0.2 % Tween 80 was
administered orally in a volume of 0.5 mQ/lO0 g body
weight l hour before the injection of antigen. To the
control group, only the vehicle was administered.
Tranilast which was a positive control compound was
administered orally 30 minutes before the injection of
antigen. The result shown in Table 3 proves that the
compound of the present invention shows an excellent PCA
- 36
reaction inhibitory activity.
~able 3
_ _
5Compound No. Dose (mg/kg)Inhibition rate ~)
23 100 29
32 100 21
33 100 50
10 34 100 48
100 43
37 100 21
39 100 65
41 100 25
15tranilast 300 40
(2) Inhibitory activity against antigen-induced
anaphylactic shock in actively sensitized guinea pigs
Antigen-induced anaphylactic shock death was
observed according to John P. Devlin [Pulmonary and
Antiallergic Drugs~ John Wiley & Sons, 155(1985~]
employing actlvely sensitized guinea pigs.
Each 100 mg/kg of body weight ovalbumin
dissolved in physiological saline was injected into
gluteus and into peritoneal cavity of male guinea pigs
weighing 250 to 350 g. Three days later, the animals
were further injected intraperitoneally with ovalbumin
(100 mg/kg body weight) to conduct booster. Those
animals were employed for testing 3 to 4 weeks after the
sensitization.
Groups of 4 or more actively sensitized guinea
pigs each were pretreated by subcutaneously injecting
pyrilamine (1 mg/kg body weight) 30 minutes before
antigen inhalation to suppress histamine-dependent
response and propranolol (1 mg/kg body weight) to enhance
the response induced by other than histamine 10 minutes
before the antigen inhalation.
r~
The animal was placed in a desiccator with a
capacity of about 5 Q and 0.5 % aqueous solution of
ovalbumin in the state of aerosol was inhaled with
ultrasonic type nebulizer for five minutes. ~naphylactic
shock death was observed and the animals survived for 90
minutes or more after antigen inhalation were estimated
to be protected. All the animals of the control group
died due to anaphylactic shock. The results are shown in
Table 4. The compounds of the present invention and
therapeutic antiasthmatic agent (tranilast, theophylline)
were administered orally 30 minutes before the antigen
inhalation. The result shown in Table 4 proves that the
compounds of the present invention shows an excellent
inhibitory activity against anaphylactic shock.
Table 4
. _ _
Compound No.Dose (mg/kg) Protecting effect*
. . _
20 35 10 2/4
36 100 1/4
37 100 1/4
38 100 1/~
39 100 1~4
25 41 10 1/4
tranilast 100 0/4
theophylline 30 2/4
control - 0/20
... . . _ . _ _ _ .. . .
(note) * Number of survivors/Number of animals used
(3) Inhibitory activity against antigen-induced airway
constriction in actively sensitized guinea pigs
According to Orange and Moore [Journal of
Immunology (J. Immunol.), 116, 392(1976)], an emulsion of
a solution of ovalbumin dissolved in phvsiological saline
(2 mg/mQ) and Freund's complete adjuvant (Difco
Laboratories), mixed in the equal volume was injected
~^3~cr ~;~
- 38
into peritoneal cavity of guinea pigs in the volume of 1
mQ/guinea pig to sensitize them. Three or four weeks
later, airway contraction caused by antigen-antibody
reaction was measured in accordance with Konzett Rossler
[Archiv f~r Experimental Pathologie und Pharmakologie
(Arch. Exp. Path. Pharmak.), 195, 71(19~0)]. That is,
the sensitized guinea pigs (5 guinea pigs/group) were
provided with artificial respiration by inserting a
tracheal cannula under urethane anesthesia (1.5 g/kg body
weight, intraperitoneal administration), and then,
gallamine at 1 mg/kg body weight was injected
intravenously) to stop spontaneous respiration of the
guinea pigs. Inhalation of 0.5 % aqueous solution of
ovalbumin was conducted using a nebulizer for 1 minute to
increase antigen-induced airway constriction, at the same
time, airway pressure was recorded through a
transducer. Test compound was administered into jugular
vein (i.v.) of the guinea pig 3 minutes before the
antigen inhalation or administered orally (p.o.) 2 hours
before the antigen inhalation. The control group
received the vehicle. As a positive control compound,
theophylline which was a drug for anti-asthma was used.
The effect of the compound was estimated bY calculating
the maximum value of airway constriction (%) in
comparison with the control group, according to the
following equation.
A - B
Inhibition rate (%) = ~-- x 100
A
A: Maximum value of airway constriction iII the
con-trol group
B: Maximum value of airway constriction iII the
test compound group
The result shown in Table 5 proves that the
compounds of the present invention shows excellent
- 39 - ~ t~
inhibitory activity against antigen-induced airway
constriction.
Table 5
. _ . . .
Compound ~oute of Dose Inhibition rate
No. administration (mg/kg) (~)
. . _ _ . . _
7 i.v. 1 25
8 i.v. 1 43
9 i.v. 1 20
11 i.v. 1 52
11 p.o. 30 26
12 i.v. 1 32
26 i.v. 1 68
15 3~ i.v. 2 23
33 i.v. 2 26
37 i.v. 1 21
39 i.v. 1 59
42 i.v. 5 33
20 ~3 i.v. 5 21
i.v. 1 42
theophylline i.v. 1 31
_ ... .. . _
5-Lipoxygenase inhibiting activity of the
compound of the present invention was measured referring
to the method for measuring 5-lipoxygenase activity by K.
Ochi et al. [Journal of Biological Chemistry (J. Biol.
Chem.), 258, 5754(1983)].
Sterilized 2 % solution of casein (pH 7) was
injected intraperitoneally into Hartley guinea ~igs in a
volume of 5 mQ/100 g body weight. Fifteen hours later,
the guinea pigs were killed and peritoneal exudate cells
thereof were collected. After the exudate cells were
washed with 17 mM Tris-HCQ buffer (pH 7.4) containing
O.74 ~ ammonium chloride to remove contaminating
erythrocytes, the residual cells (leukocytes) were washed
with buffer A (130 mM NaCQ, 1 mM EDTA, 25 mM sodium
- 40
phosphate, pH 7.4). The washed cells were suspended in
bufer B (50 mM sodium phosphater 1 mM EDTA, Ool
gelatin, pH 7.~) at 108 cells/mQ, sonicated and
centrifuged a~ 10,000 x g for 20 minutes under the cold
atmosphere. The obtained supernatant was further
centrifuged at 105,000 x g for 60 minutes under the cold
atmopshere. The cytosol fraction was obtained and used
as an enzyme solution of 5-lipoxygenase.
The enzyme solution was preincubated with the
test compound in the presence of lmM CaCQ2, 1 mM reduced
glutathione (GSH) and 2n~ ATP at 30C for 5 minutes in
0.2 m~ of a reaction mixture and the mixture was further
incubated at 30C for 5 minutes by adding 20 ~M [1-14C]
arachidonic acid t0.1 yci) thereto. The test compounds
were dissolved in ethanol to give the reaction mixture
containing 2 % ethanol as a final concentration. Only
ethanol was added to the reaction mixture as a control
group.
To the reaction mixture were added 2.~ m~ of a
mixture of chloroform and methanol (2/1 by volu~e) and
0.3 m~ of 40 mM citrate to stop the reaction. The
mixture was vortexed and an organic solvent layer was
evaporated to dryness under nitrogen gas. After
dissolving the dried organic layer in a fixed amount of
the mixture of chloroform and methanol 12/1 by volume~,
it was spotted on a silica gel plate (Kiesel gel 60F254,
E. Merck) and 5-lipoxygenase products were separated
using the solvent system of an organic solvent layer of
ethyl acetate/water/2,2,4-trimethylpentane/acetic acid =
30 11/10/5/2 by volume. After the radioactive position of
the product was determined by means of a radioautography,
an area equivalent to that of 5-hydroxyeicosatetraenoic
acid (hereinafter referred to as ''5-HETE'I) was scraped
off, and then its radioactivity was measured with a
liquid scintillation counter. With regarding the amount
of the generated 5-HETE as the 5-lipoxygenase inhibiting
activity, the inhibition rate (%) in comparison with the
control group was calculated according to the following
~ 3 ~
- 41
equation.
A - B
Inhibition rate (~ ~~~ x 100
A
A: Value of radioactivity in the control group
B: Value of radioactivity in the test compound
group
The 5-lipoxygenase inhibiting activity of the
compounds of the present invention is shown in Table 6.
The result shown in Table 6 proves that the compounds of
the present invention sufficiently inhibits 5-
lipoxygenase activity.
Table_6
. . . _ . . _
Compound Concentration* Inhibition rate
20No. (lIM) (~6)
_ _ _ . . ... _
2 10 78
4 10 85
7 1 88
8 1 61
- 9 1 91
1 ~3
11 1 87
12 1 84
13 1 86
14 1 27
16 10 28
83
22 10 63
_ _ _ _ _
- continued
- ~2 - ~ 3 ~ ~y .~
- continued
.. _ _ . ... _ _ ..
Compound Concentration* Inhibition rate
No. (~M)
-- . . . _ _.
23 10 ~5
24 1 48
1 23
26 1 84
29 1 86
1 87
31 1 49
33 10 82
~9
36 10 84
37 10 89
38 10 88
39 10 87
87
41 10 89
42 10 88
43 ~ 53
1 3
_ _ . . . _ _ _ _ ., _5 (note) * Concentration of the test compound in the
reaction mi~ture
The antibacterial activity against Gram-
positive bacteria of the compound of the present
30 invention was measured according to a standard method of
Nippon Kagaku Ryoho Gakkai [Nippon Kagaku Ryoho Gakkaishi
tJournal of the Chemical therapy of Japan), 29,
76(1981) ] .
As for gram-positive bacteria, after
35 cultivation in Mueller Hinton broth medium (made by Difco
Co., Ltd.), there was prepared a bacterial suspension for
inoculation containing about 106 of the bacteria per 1 mQ
of the medium. On the other hand, the test compound was
~ 3 ~
- ~3
added to Mueller Hinton ager medium (made by Difco Co.,
Ltd.) so as to give a serial dilution ager medium. Then,
the above-mentioned bacterial suspension for inoculation
was streaked to each agar medium for about 2 cm with a
nichrome wire (inner diameter: about 1 mm).
After that the each agar medium was cultured at
37C for lB to 20 hours~ the growth of the test bacteria
was determined. The minimum concentration of the test
compound, which completely inhibited the growth of the
test bacteria, was decided as a minimal inhibitory
concentration (hereinaf-ter referred to as "MIC").
As for acid-fast bacteria, after cultured in
glycerol broth medium, there was prepared a bacterial
suspension for inoculation containing about 106 of the
bacteria per 1 mQ of the medium. On the other hand,
there were prepared some glycerol Czapek agar plating
media with adding the test compounds, and thereto the
bacterial suspension for inoculation was streakedO
After the each agar plating medium, to which
the acid-fast bacteria was streaked, was cultured at 37C
for 40 to 42 hours, MIC was determined as defined
above.
As the result, each MIC of the test compounds
(1)l (2), (4), (11), (15), (16), (19) and (20) against
25 Micrococcus luteus IFO 13867 was not more than 6 ~g/mQ,
not more than 6 ~g/mQ, not more than 6 ~g/mQ, 12 ~g/mQ~
60 ~g/mQ, not more than 15 ~g/mQ, 50 ~g/mQ and not more
than 50 ~g/mQ respectively; each MIC of the test
compounds (1), (2), (11), (15), (16), (19), (20) and (41)
30 against Bacillus subtilis IFO 3134 was not more than 6
~g/mQ, not more than 6 ~g/mQ~ not more than 6 ug/mQ, 100
~g/mQ, not more than 15 ~g/mQ, 100 ~g/mQ, 50 ~g/mQ and 25
~g/mQ respectively; each MIC of the test compounds (1),
(2), (11), (15), (16), (19), (20) and (41) against
35 Staphylococcus aureus IFO 12732 was 12 ~g/mQ, 12 ~g/mQ,
25 ~g/mQ, 60 ~g/mQ, not more than 15 ~g/mQ, 100 ~g/m~,
100 ~g/mQ and 50 ~g/mQ respectively; and each MIC of the
test compounds (1), (15), (16), (19), (28), (30), (31),
- 44 ~ i3
(32), (33), (34), ~35), (41), (42), (43), (44) and (45)
against Mycobacterium sme~matis ATCC 607 was 6 ~g/mQ, not
more than 15 ~g/mQ, not more than 15 ~g/mQ, not more than
5 ~g/mQ, not more than 15 ~g/mQ, not more than 6 ~g/mQ~
not more than 6 ~g/mQ, not more than 6 ~g/mQ, not more
than 15 ~g/mQ, not more than 6 ~g/m~, 15 ~g/mQ, 25 ~g/mQ,
not more than 15 ~g/m~, not more than 6 ~g/m~, 2 ~/mQ
and not more than 6 ~g/mQ respectively.
Consequently, it was found that the compounds
of the present invention were effective on both gram-
positive and acid-fast bacteria.
Tyrosine kinase inhibiting activity of the
compound of the present invention was measured referring
to a method for measuring tyrosine kinase activity by G.
Carpenter or by S. Cohen et al. [J. Biol. Chem.~ 254,
4884(1979); J. Biol. Chem., 257, 1528(1982)].
Cell line A-431 derived from human epidermoid
carcinoma (ATCC CRL1555) was cultured at 37C under the
condition of 5 % CO2 in Dulbecco's modified Eagle's
medium (made by NISSUI PHARMACEUTIC~L CO., LTD.)
containing 10 ~ by volume fetal bovine serum, 50 ~g/mQ of
streptomycin, 50 IU/mQ of penicillin G and 50 ~g/mQ of
kanamycin. The obtained cel]s were treated according to
the above-mentioned method of Cohen or Carpenter et al.
to give membrane preparation containing epidermal growth
factor receptor-tyrosine kinase complex (hereinafter
referred to as "membrane preparation"). The membrane
preparation was employed in the following measurement
without solubilization.
A test compound dissolved in dimethylsulfoxide
(hereinafter referred to as "DMSO") was added to a
mixture of 20 mM of N-2-hydroxyethylpiperazine-N'-2-
ethanesulfonic acid buffer (pH 7.4), 1 mM of MnC~2, 7.5
~g of bovine serum albumin and the membrane preparation
(10 ~g as protein). After incubation at 0C for 5
minu-tes, 100 ng of epidermal ~rowth factor (hereinafter
referred to as "EGF") was added thereto and the mixture
was further incubated at 0C for 15 minutes. [r-32P]RTP
(3000 Ci/mmol, 0.1 ~Ci~ was added thereto to make final
volume of 70 ~Q. After incubation at 0C for 15 minutes,
50 ~Q of the reaction mixture was soaked into Whatman 3
MM filter paper (made by Whatman Ltd.) and immediately
S the reaction was stopped by an aqueous solution of 10
by weight trichloroacetic acid containing 10 mM sodium
pyrophosphate. The filter paper was sufficiently washed
with the same solution and then washed with ethanol, and
dried. Radioactivity present in the filter paper was
measured by liquid scintillation counter (A). Also,
radioacti~ity was measured in case of the reaction
without EGF (B), the reaction without the test compound
(C), and the reaction without both EGF and the test
compound (D) as a control.
Tyrosine kinase inhibition rate (%) was
calculated by the following equation.
(C - D) - ~A - B)
Innhibition rate (~ - x 100
C - D
The result proves that the compounds of the
present invention shows excellent tyrosine kinase
inhibitory activity.
There is shown each tyrosine kinase inhibition
rate of the compounds of the present invention in Table
7.
*Trade-mark
~ 3 ~
- 46
Table 7
.
Compound Concentration* Inhibition rate
No. t~M3 _ (%)
_ . . . _ _ . __ . _ . _ _ _
1 l 23
2 1 20
3 1 45
4 1 74
1 42
7 lO S9
8 lO 69
9 lO 50
1 - 40
11 lO 52
12 10 30
14 1 ~3
l 100
16 l lO0
17 1 25
18 1 87
19 1 74
l 46
21 l 98
22 1 84
23 10 60
24 1 37
72
26 lO 62
27 1 58
28 1 66
29 1 63
1 70
31 lO ~1
32 1 74
33 10 69
- continued
~ 3 ~ ~ t)~~
- 47
- continued
Compound Concentration* Inhibition rate
No. (llM) (%)
_
34 1 63
1 70
36 10 59
37 10 83
1 38 10 85
39 10 43
~0 10 21
41 10 70
42 1 85
~3 10 95
44 1 80
.. .. _ .
(note) * Concentration of the test compound in the
reaction mixture
Additionally, the compounds of the precent
invention have W absorbing activity and thus there are
expected to use the compounds as the W absorber in order
to prevent a living body from dermatitis solaris
(generally called as sunburn), prevent organic high
molecular materials (e.g. plastics, gum, paints and the
like) from declining by W -ray or prevent photographs and
pictures from discoloring by UV-ray.
Each W absorption spectrum of the compounds of
the present invention was measured according to the
conventional method, in which methanol was used as a
solvent, and thereby molar extinction coefficient thereof
was calculated. The results were shown in Table 8. It
is found that, as shown in Table 8, the compounds of the
present invention rather strongly absorb W -ray~
- ~8
Table 8
. _ . . . _ ~
Compound ~ max molar extinction
No. (nm) coefficient
4 257 1.87 x 104
361 1.80 x 104
. _ . . _ _ . _
271 2.04 x 104
348 2.11 x 10
. . ~ . _ . ._
249 1.51 x 104
347 2.40 x 104
.. . . _
18 304 1~87 x 104
_ _ . _
There was found the following point by using
reverse transcriptase derived from Moloney-Murine
Leukemia Virus (hereinafter referred to as "M-MLV").
The compound of the present invention was
dissolved in DMSO to give a 100 mM solution thereof.
Then, the solution was diluted with water containing DMSO
to give a solution of the test compound having a defined
concentration. A mixed ratio of DMSO and distilled water
was adjusted so that the concentration of DMSO at this
time is 10 % by volume and a final concentration of DMSO
at the beginning of a reaction is 1 % by volume.
The thus prepared solution of the test compound
0 was added to a solution containin~ 50 mM of Tris-~CQ
buffer (pH 8.3), 8 mM of MgCQ2, 30 mM of NaCQ, 50 mM of
dithio threitol (made by Wako Pure Chemical Industries
Ltd.), 0.2 mM of thymidine-5'-triphosphate (m~de by
Pharmacia K. K.) and 6 U/mQ of reverse transcriptase
derived from M-MLV (made by Pharmacia K. K.) at 37C for
30 minutes. After there was added thereto 10 ~g/mQ of
polyadenylic acid, 0.01 U/m~ of oligodeoxy thymidylic
acid (made by Pharmacia K. K.) and 10 ~Ci/mQ of [methyl-
' i ."
- 49
3~] thymidine-5'-triphosphate (made by ~mersharn ~apan
Co., Ltd., 47 Ci/mmol) to give a reaction mixture, the
mixture was further incubated at 37C for 30 minutes,
followed by cooling with ice to stop the reaction.
The radioactivity incorporated into
deoxyribonucleic acids was measured according to the
method of Linteril et al (Science, 170, ~47 to 449
(1967)) After 50 ~Q of the reaction mixture was soaked
into DE-81 filter paper (made by Whatman Ltd.), the
filter paper was washed with 5 % by weight of Na2HP04
solution for three times, distilled water and ethanol
successively by one and then dried. Radioactivity
contained in the filter paper was measured by liquid
scintillation counter to give the each radioactivity of
the test solution groups.
On the other hand, the same procedure as above
was carried out using DMSO-distilled water without the
test compound instead of using the test solution, to give
the value of radioactivity of a control group.
Reverse transcriptase derived from M-MLV
inhibition rate (%) was calculated by the following
equation.
A - B
Inhibition rate (%) = x 100
A: radioactivity of the control group
B: radioactivity of the test solution group
The typical examples of reverse transcriptase
derived from M-MLV inhibiting aclivity of the compounds
of the present invention are shown in Table 9.
The results proves that the compounds shown in
Table 1 have strong inhibiting activity against reverse
transcriptase derived from M-MLV and thus it can be
expected that the compounds show sufficient growth
inhibiting effect on retrovirus having reverse
~ ~ 6
transcriptase.
Table 9
5CompoundConcentration*Inhibition rate
No. (~M) (%)
. _ _
1 1 96
2 1 95
10 5 10 87
6 10 98
7 1 98
8 1 98
9 1 73
1510 10 61
11 1 94
59
19 1 75
97
2024 1 91
26 10 76
27 1 73
31 10 61
42 10 50
_ _ _ _ _
(Note) * Concentration of the test compound in the
reaction mixture
Acute toxicity test
Groups of 6 female ICR mice each, each mouse
weighing 23 to 26 g, were employed. The compounds (1) to
(45) suspended in an aqueous solution of 2.5 % gum arabic
containing 0.2 % Tween 80 were administered orally to
each mouse in a volume of 0.1 m~/10 g body weight. The
general symptoms of the mice were observed for two weeks
after the administration. The LD50 (mg/kg) values were
estimated from the ratio of the number of dead mice to
the number of mice used. ~s a result, there were
51
observed no dead mice at a dose of 500 mg/lcg. The LD50
of the compounds (1) to (45) of the present invention was
estimated to be not less than 500 mg/kg, which proved a
low toxicity of the compounds of the present invention.
Preparations and Dosage
The antiallergic agents, 5-lipoxygenase
inhibiting agents, antibacterial agents~ tyrosine kinase
inhibiting agents, W absorber or reverse transcriptase
inhibiting agents of the present invention can be
administered orally, rectally, or parenterally in
pharmaceutical dosage form, for example, tablets,
capsules, fine subtilaes, syrups, suppositories,
ointments, injections and the like.
For an excipient of the antiallergic agents, 5-
lipoxygenase inhibiting agents, antibacterial agent,tyrosine kinase inhibiting agents, W absorber or reverse
transcriptase inhibiting agents o~ the present invention,
organic or inorganic phar~aceutically acceptable
excipient material is employed in a solid or liquid
state, which is usually inactive and suited for oral,
rectal or parenteral administration~ Examples of such
excipient are, for instance, crystalline cellulose,
gelatin, lactose, starch, magnesium stearate, talc,
vegetable or animal fat and oil, gum, polyalkyleneglycol,
and the like~ The ratio of the compound of -the present
invention having the formula (I), contained in the
antiallergic agents, 5-lipoxygenase inhibiting agents,
antibacterial agents, tyrosine kinase inhibiting agents,
W absorber or reverse transcriptase inhibiting agents as
an active ingredient in the formulation may vary in the
range from 0.2 to 100 %.
The antiallergic agents, 5-lipoxygenase
inhibiting agents, antibacterial agents, the tyrosine
kinase inhibiting agents, W absorber or reverse
transcriptase inhibiting agents of the present invention
may contain other antiallergic agents, 5-lipoxygenase
inhibiting agents, antibacterial agents, tyrosine kinase
inhibiting agents, W absorber, reverse transcriptase
- 52
inhibiting agents or any other drugs, which are
compatible with the agents of the present invention. In
this case, it is needless to say that the antiallergic
agents, 5-lipoxygenase inhibiting agents, antibacterial
agents, tyrosine kinase inhibiting agents, W absorber or
reverse transcriptase inhibiting agents of .he present
invention may not be the principal ingredients in the
formulation.
The antiallergic agents, 5-lipoxygenase
inhibiting agents, antibacterial agent, the tyrosine
~inase inhibiting agents, W absorber or reverse
transcriptase inhibiting agents of the present invention
are administered at a dose where the desired activity is
generally achieved without any side effects.
Though a practical dose should be determined by
a physician, the compound of the present invention having
the formula (I), which is an active ingredient of the
agents of the present invention, is generally
administered at a dose from 10 mg of 10 9, preferably
from 20 mg to 5 g, for an adult a day. The antiallergic
agents, 5-lipoxygenase inhibiting agents, antibacterial
agents, tyrosine kinase inhibiting agents, W absorber or
reverse transcriptase inhibiting agents of the present
invention can be administered as a pharmaceutical
formulation which contains 1 mg to 5 gl preferably 3 mg
to 1 g of the compound having the formula (I) as an
active ingredient.
The present invention is more specifically
described and explained by means of the following
Examples. It is to be understood that the present
invention is not limited to Examples, and various changes
and modifications may be made in the invention without
departing from the spirit and scope thereof.
Example 1
[Preparation of the compound (1)]
In 100 mQ of benzene were dissolved 1.37 g of
3,5-diphenyl-4-hydroxybenzaldehyde and 0.82 g of
~ 3 ~
- 53
rhodanine, and thereto 0.1 m~ of piperidine and 0.5 m~ of
acetic acid were added. The mixture was heated under
reflux for 5 hours in Dean-Stark apparatus while removing
water produced. After cooling, the deposited crystals
were filtered and subjected to crystallization from a
mixed solvent of benzene and acetone to give 1.2 g
(yield: 62 ~) of the compound (1).
The melting point and data of elementary
analysis of the obtained compound (1) are shown in Table
1. And results of lH-NMR and IR of the obtaine~ compound
(1~ are shown in Table 2.
Examp~
[Preparation of the compound (4)]
In 70 mQ of benzene were dissolved 1.51 g of
3,5-dibenzyl-4-hydroxybenzaldehyde and 0.67 g of
oxyindol, and thereto 0.1 m~ of piperidine and 0.5 mQ of
acetic acid were added. The mixture was heated under
reflux for 5 hours in Dean-Stark apparatus while removing
water produced. After cooling, the solvent was distilled
away under reduced pressure. The obtained residue was
dissolved in 200 m~ of chloroform, washed with water and
dried with sodium sulfate. Chloroform was distille~ away
under reduced pressure, the residue was subjected to
crystallization from ethanol to give 600 mg (yield: 29 ~)
of the compound (4).
The melting point and data of elementary
analysis of the obtained compound (4) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(4) are shown in Table 2.
Example 3
[Preparation of the compound (5)]
In 70 mQ of benzene were dissolved 0.61 g of
3,5-dibenzyl-4-hydroxybenzaldehyde and 0.39 g of 2~-1,4-
benzothiazine-3(4H)-one-1,1-dioxide, and thereto 0.1 mQ
of piperidine and 0.5 m~ of acetic acid were added. The
mixture was heated under reflux for 5 hours in Dean-Stark
- 54
apparatus while removing water produced. After cooling,
the solvent was dlstilled away under reduced pressure.
~he obtained residue was subjected to a column-
chromatography (carrier: silica-gel) and eluted with
mixed solvent of chloroform/methanol (98/2: v/v).
fraction containing the desired compound was concentrated
and the obtained residue was subjected to crystallization
from benzene to give 180 m~ (yield: l9 %) of the compound
(5).
The melting point and data of elementary
analysis of the obtained compound (5) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(5) are shown in Table 2.
Example 4
[Preparation of the compound (7)]
To lO0 mQ of benzene were added 2.6 9 of 5-
phenylthiomethylprotocatechuic aldehyde, l.33 g of
rhodanine, O.l mQ of piperidine and 0.5 m~ of acetic
acid. The mixture was heated under reflux for 5 hours in
Dean-Stark apparatus while removing water produced.
After cooling, the deposited crystals were filtered off
from the reaction mixture and the obtained crystals were
recrystallized from ethanol to give 2.78 g (yield: 74 %)
of the compound (7).
The melting point and data of elementary
analysis of the obtained compound (7) are shown in Table
1. And results of l~-NMR and IR of the obtained compound
(7) are shown in Table 2.
Example 5
[Preparation of the compound (ll)]
In 70 mQ of benzene were dissolved 0.78 g of 5-
phenylthiomethylprotocatechuic aldehyde and 0.4 g of
oxyindol, and thereto O.l m~ of piperidine and 0.5 m~ of
acetic acid were added. The mixture was heated under
reflux for 5 hours in Dean-Stark apparatus while removing
water produced. Af ter cooling, the deposited crystals
o~
- 55
were filtered off from the reaction mixture and washed
with benzene. And the obtained crystals were
recrystalliæed from a mixed solvent of benzene and
acetone to give 1.0 g (yield: 90 ~) of the compound (11).
The melting point and data of elementary
analysis of the obtained compound (11) are shown in Table
1. And results of l~-NMR and IR of the obtained compound
(11) are shown in Table 2.
Example 6
[Preparation of the compound (12)]
A condensation of 0.7 9 of 3-benzyloxy~4
hydroxy-5-phenylthiomethylbenzaldehyde and 0.27 g of
oxyindol was carried out in the same manner as in the
above Example 1. And the obtained residue was subjected
to a column-chromatography (carrier: silica~gel) and
eluted with mixed solvent of chloroform/methanol (98/2:
v/v). After a fraction containing the desired compound
was concentrated under reduced pressure, the fraction was
subjected to crystallization from ethanol to give 0.62 g
(yield: 66 ~) of the compound (12).
The melting poin~ and data of elementary
analysis of the obtained compound (12) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(12) are shown in Table 2.
Example 7
[Preparation of the compound (15)]
In 200 mQ of benzene were dissolved 2.90 g Gf
3,5-diphenyl-4-hydroxybenzaldehyde and ~40 mg of -
cyanoacetoamide, and thereto 0.1 m~ of piperidine and 0.5
mQ of acetic acid were added. The mixture was heated
under reflux for 5 hours in Dean-Stark apparatus while
removing water produced. After the solvent was distilled
away under reduced pressure, the obtained residue was
subjected to a column-chromatography (carrier: silica-
gel) and eluted with a mixed solvent of
chloroform/methanol (98/2: v/v). A fraction containing
~ 56
the desired compound was concentrated and the obtained
residue was subjected to crystallization from a mixed
solvent of benzene and acetone to give 11.15 g (yield: 32
%) of the compound (15).
The melting point and data of elementary
analysis of the obtained compound (15) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(15) are shown in Table 2.
Example 8
[Preparation of the compound (17)~
To 50 mQ of acetonitrile were added 760 mg of
3,5-dibenzyl-4-hydroxybenzaldehyde and 1.04 g of ~-
triphenylphosphoranylidene-y-butyrolactone. The mixture
was heated and stirred overnight at 80C. After cooling,
the deposited crystals were filtered and subjected to
crystallization from ethanol to give 450 mg (yield: 48 %)
of the compound (17).
The meltin~ point and data of elementary
analysis of the obtained compound (17) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(17) are shown in Table 2.
Example 9
[Preparation of the compound (18)]
In 50 mQ of dried benzene was suspended 0.6 g
of sodium hydride under nitrogen atomosphere, to which a
solution of 1.73 g of 3,5-dibenzyl-4-methoxymethoxy-
benzaldehyde and 1.27 g of N-acetylpyrrolidone dissolved
in 20 mQ of benzene was added dropwise r subsequently
heated and stirred overnight at 50C. After cooling, the
reaction solution was added to an ice water and extracted
with chloroform. The solvent was distilled away from the
obtained extract under reduced pressure. The obtained
residue was dissolved in 50 mQ of dried methylene
chloride, which was added with ~ mQ of trifluoroacetic
acid and stirred for 3 hours at room temperature. The
solvent was distilled away from the solution under
- 57
reduced pressure, the obtained residue was subjected to a
column-chromatography (carrier: silica-gel) and eluted
with mixed solvent of chloroform/methanol (98/2: v/v). A
fraction containing the desired compound was concentrated
and the obtained residue was sub~ected to crystallization
from ethanol to give 450 mg (yield: 21 %) of the compound
(18).
The melting point and date of elementary
analysis of the obtained compound (18) are shown in Table
1. And results of l~-NMR and IR of the obtained compound
(18) are shown in Table 2.
Example lO
[Preparation of the compound (l9)]
In lO0 mQ of benzene were dissolved 1.37 g of
3,5-diphenyl-4-hydroxybenzaldehyde and 0.88 g of l-
phenyl-3,5-pyrazolidinedion, and thereto 0.1 mQ of
piperidine and 0.5 mQ of acetic acid were added. The
mixture was heated under reflux for 5 hours in Dean-Stark
apparatus while removing water produced. After cooling,
the deposited crystals were filtered and subjected to
crystallization from ethanol to give 600 mg (yield: 28 %)
of the compound (l9).
The melting point and data of elementary
analysis of the obtained compound (l9) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(l9) are shown in Table 2.
Example 11
[Preparation of the compound (25)]
To lO0 mQ of benzene were added 1.37 g of 3,5-
diphenyl-4-hydroxybenzaldehyde, 0.82 g of rhodanine, 0.1
mQ of piperidine and 0.5 m~ of acetic acid. The mixture
was heated under reflux for 5 hours in Dean-Stark
apparatus while removing water produced. The deposited
crystals was filtered off from the reaction mixture.
After drying, the deposited crystals were heated under
reflux for 5 hours with l.l mQ of benzylamine in 50 mQ of
58 ~ 3~
ethanol. After cooling, the solvent was distilled away
under reduced pressure. The residue was subjected to a
column-chromatography (carrier: silica-gel) and eluted
with mixed solvent of chloroform/methanol t100/2: v/v).
After a fraction containing the desired compound was
concentrated under reduced pressure, the fraction was
subjected to crystallization from ethanol to give 0.60 g
(yield: 26 ~) of the compound (25).
The melting point and data of elementary
analysis of the obtained compound (25) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(25) are shown in Table 2.
Example 12
[Preparation of the compound (26)]
To 100 mQ of benzene were added 3.02 g of 3,5-
dibenzyl-4-hydroxybenzaldehyde, 1.33 g of rhodanine, 0.1
mQ of piperidine and 0.5 mQ of acetic acid. The mixture
was heated under reflux for 5 hours in Dean-Stark
apparatus while removing water produced. The deposited
crystals were filtered off from the reaction mixture.
After drying, the deposited crystals were heated under
reflux for 5 hours with 2.2 mQ of benzylamine in 100 mQ
of ethanol. After cooling, the solvent was distilled
away under reduced pressure. The obtained residue was
subjected to a column-chromatography ~carrier: silica-
gel) and eluted with mixed solvent of chloroform/methanol
(100/2: v/v). ~fter a fraction containing the desired
compound was concentrated under reduced pressure, the
fraction was subjected to crystallization from ethanol to
give 2.0 g (yield: 41 %) of the compound (26~.
The melting point and data of elementary
analysis of the obtained compound (26) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(26) are shown in Table 2.
Example 13
[Preparation of the compound (28)]
59 - ~ 3 ~ ~ d~~
To lO0 mQ of ethanol were added ~.04 g of 5-(3-
ethoxy-4-hydroxy-5-phenylthiomethylbenzylidene)-rhodanine
obtained by the condensation reaction of 5-phenylthio-
methylethylvanillin and rhodanine in the same manner as
S above and 2.2 m~ of benzylamine. The mixture was heated
under reflux for 5 hours. After cooling, the solvent was
distilled away under reduced pressure. The obtained
residue was subjected to a column-chromatography
(carrier: silica-gel) and eluted with chloroform. AEter
a fraction containing the desired compound was
concentrated under reduced pressure, the fraction was
subjected to crystallization from ethanol to give 1.36 g
(yield: 38 %) of the compound (28).
The melting point and data of elementary
analysis of the obtained compound (28) are shown in Table
1. ~nd results of lH-NMR and IR of the obtained compound
(28) are shown in Table 2.
Example 14
[Preparation of the compound (30)]
To 50 mQ of ethanol were added 0.80 g of 5-(3-
n-butyloxy-4-hydroxy-5-benzylbenzylidene)-rhodanine
obtained by the condensation reaction of 3-n-butyloxy-4-
hydroxy-5-benzylbenzaldehyde and rhodanine in the same
manner as above and 0.44 m~ of benzylamine. The mixture
was heated under reflux for 5 hours. After cooling, the
solvent was distilled away under reduced pressure. The
obtained residue was subjected to a column-chromatography
(carrier: silica-gel) and eluted with a mixed solvent of
chloroform/methanol (lO/l: v/v). After a fraction
containing the desired compound was concentrated under
reduced pressure, the fraction was subjected to
crystallization from ethanol to give 0.72 g (yield: 76 %)
of the compound (30~.
The melting point and data of elementary
analysis of the obtained compound (30) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
~30) are shown in Table 2.
- 60 - 1 .3 ~
Example 15
[Preparation of -the compound (33)]
In 30 mQ of ethanol was dissolved 966 mg of 5-
(3,5-diisopropyl-4-hydroxybenzylidene~-rhodanine, and
thereto 624 mg of benzylamine was added. The mixture was
heated under reflux for 5 hours. Ethanol was distilled
away under reduced pressure, and the obtained residue was
dissolved in chloroform. After washing with water, the
solution was concentrated to dryness. The obtained
concentrate was subjected to a column-chromatography
(carrier: silica-gel) and eluted with chloroform. A
fraction containing the desired compound was collected,
concentrated and dried to give 660 mg (yield: 56 %~ of
the compound (33).
The melting point and data of elementary
analysis of the obtained compound (33) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(33) are shown in Table 2.
Example 16
[Preparation of the compound (34)]
In 30 mQ of ethanol was dissolved 966 mg o~ 5-
(3,5-diisopropyl-4-hydroxyben~ylidene)-rhodanine, and
thereto 726 mg of phenethylamine was added. The mixture
was heated under reflux for 12 hours. Ethanol was
distilled away under reduced pressure, and the obtained
residue was dissolved in chloroform. After washing with
water, the solution was subjected to a column-
chromatography (carrier: silica-gel) and eluted with
chloroform. A fraction containing the desired compound
was collected, concentrated, dried and subjected to
crystallization to give 600 mg (yield: 68 %) of the
compound (34).
The melting point and data of elementary
analysis of the obtained compound ~3~) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(34) are shown in Table 2.
~ J3
- 61
E~ample 17
[Preparation of the compound (35)]
In 30 m~ of ethanol was dissolved 966 mg of S-
(3,S-diisopropyl-4-hydroxybenzylidene)-rhodanine, and
thereto 773 mg of p-fluorobenzylamine. The mixture was
heated under reflux for 7 hours. Ethanol was distilled
away under reduced pressure, and the obtained residue was
dissolved in chloroform. After washing with wa-ter, the
solution was subjected to a column-chromatography
(carrier: silica-gel) and eluted with chloroform. A
fraction containing the desired cornpound was collected,
concentrated and dried to give 660 mg (yield: 52 %) of
the compound (35).
The melting point and data of elementary
analysis of the obtained compound (35) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
~35) are shown in Table 2.
Example 18
~Preparation of the compound (39)]
In 30 mQ of ethanol was dissolved 966 mg of 5-
(3,5-diisopropyl-4-hydroxybenzylidene)-rhodanine~ and
thereto 726 mg of p-methylbenzylamine was added. The
mixture was heated under reflux for 12 hours. Ethanol
was distilled away under reduced pressure, and the
obtained residue was dissolved in chloroform. After
washing with water, the solution was subjected to a
column-chromatography (carrier: silica-gel) and eluted
with chloroform. A frackion containing the desired
compound was collected, concentrated, dried and subjected
to crystallization to give 900 mg (yield: 30 %) of the
compound (39).
The melting point and data of elementary
analysis of the obtained compound (39) are shown in Table
1. And results of l~-NMR and IR of the obtained compound
(39) are shown in Table 2.
Example 19
- 62
[Preparation of the compound (41)]
In 30 mQ of ethanol was dissolved 966 mg of 5-
(3,5-diisopropyl-4-hydroxybenzylidene)-rhodanine, and
thereto 681 mg of p-aminosulfonylbenzylamine
hydrochloride and 606 mg of triethylamine. The mixture
was heated under reflux for 6 hours. Ethanol was
distilled away under reduced pressure, and the obtained
residue was dissolved in chloroform. After washing with
water, the solution was subjected to a column-
chromatography Icarrier: silica-gel) and eluted with a
mixed solvent of chloroform/ethanol (9/1: v/v). A
fraction containing the desired compound was collected,
concentrated and dried to give 400 mg (yield: 27 %) of
the compound (41).
The melting point and data of elementary
analysis of the obtained compound (41) are shown in Table
1. And results of l~-NMR and IR of the obtained compound
(41) are shown in Table 2.
Example 20
[Preparation of the compound (42)]
In 30 mQ of ethanol was dissolved 1.61 g of 5-
(3,5-diisopropyl-4-hydroxybenzylidene)-rhodanine, and
thereto 1.30 g of p-aminobenzylamine was added. The
mixture was heated under reflu~ for 5 hours. Ethanol was
distilled away under reduced pressure, and the obtained
residue was subjected to a crystallization f rom
chloroform to give 570 mg (yield: 56 %) of the compound
(~2).
The melting point and data of elementary
analysis of the obtained compound (~2) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(42) are shown in Table 2.
Example 21
[Preparation of the compound (44)]
In 30 mQ of ethanol was dissolved 966 mg of 5-
(3,5-diisopropyl-4-hydroxybenzylidene)-rhodanine, and
~ ~Q~ ~)~
- 63
thereto 707 mg of 2-aminomethylthiophene w~s added. The
mixture was heated under reflux for 3 hours. Ethanol was
distilled away under reduced pressure, and the obtained
residue was dissolved in chloroform. After washin~ with
water, the solution was subjected to a column-
chromatography ~carrier: silica-gel) and eluted with
chloroform. A fraction containing the desired compound
was collected, concentrated and dried to give 300 mg
(yield: 24 %) of the compound (44).
The melting point and data of elementary
analysis of the obtained compound (44) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
~44~ are shown in Table 2.
Example 22
[Preparation of the compound (45)]
In 30 mQ of ethanol was dissolved 966 mg of 5-
(3,5-diisopropyl-4-hydroxybenzylidene)-rhodanine, and
thereto 648 mg of 2-aminomethylpyridine was added. The
mixture was heated under reflux for 4 hours. Ethanol was
distilled away under reduced pressure, and the obtained
residue was dissolved in chloroform. After washing with
water, the solution was subjected to a column-
chromatography (carrier: silica-gel) and eluted with a
mixed solvent of chloroform/ethanol (20/1: v/v). A
fraction containing the desired compound was collected,
concentrated and dried to give 200 mg (yield: 17 ~) of
the compound (45).
The melting point and data of elementary
analysis of the obtained compound (45) are shown in Table
1. And results of lH-NMR and IR of the obtained compound
(45) are shown in Table 2.
In addition to the ingredients used in the
Examples, other ingredients can be used in the Examples
as set forth in the specification to obtain substantially
the same results.