Note: Descriptions are shown in the official language in which they were submitted.
1 3 ~ 6~693-4103
STARBURST CONJUGATES
The present invention concerns the use oE dense star
polymers as carriers for selected materials (the "carried"
material). In recent years polymers referred to as dense star
polymers or Starburst polymers have been developed. It has
been found that the size, shape and properties of these dense
star polymers or starburst can be molecularly tailored to meet
specialized end uses. Starburst polymers have significant
advantages which can provide a means for the delivery of high
concentrations of carried material per unit of polymer, controlled
delivery, targeted delivery and/or multiple species delivery or
use.
This application is one of three closely related patent
applications serial Nos. 544,734, 544,735 and 544,736 all filed
on August 18, 1987. Application serial No. 544,734 deals with
all cases in which the carried material is a pharmaceutical
material, application serial No. 544,736 deals with all cases in
which the carried material is an agricultural material and the
present application deals with all the remaining cases in which
the carried material is neither pharmaceutical nor agricultural.
In its broadest aspect, the present invention is
directed to polymer conjugate materials comprising dense star
polymers or starburst polymers associated with carried materials
which carried materials are other than agricultural or pharma-
ceutical carried materials (hereinafter these polymer conjugates
will frequently be referred to as "starburst conjugates" or
Trade-mark
f'~
:. .
-la-
64693-4103
1 3 1 ~ 5, 2 ~
"conjugates"), processes for preparing these conjugates,
compositions containing the
~3~2~
conjugates, and methods of using the conjugates and
compositions.
The conjugates of the present invention are
suitable for use in a variety of applications where
specific deliver~ is desired. In a preferred
embodiment o~ the presen~t invention, the starburst
conjugates are comprised of one or more starburst
polymers associated with one or more agents.
The starburst conjugates offer significant
benefits over other carriers known in the art due to
the advantageous properties of the starburst polymers.
Starburst polymers exhibit molecular architecture
~- 15 characterized by regular dendritic branching with
radial symmetry. These radially symmetrical molecules
are referred to as possessing "starburst topology".
These polymers are made in a manner which can provide
concentric dendritic tiers around an initiator core.
The starburst topology is achieved by the ordered
assembly of uniform (within each tier) organic
repeating units in concentric, dendritic tiers around
an initiator core; this is accomplished by introducing
multiplicity in a geometrically progressive fashion
through a number of molecular generations. The
resulting highly functionalized molecules generations
have been termed "dendrimers" in deference to their
branched (tree-like) structure as well as their
oligomeric nature. Thus, the terms starburst oligomer.
and starburst dendrimer are encompassed within the term
~tarburst polymer.
Covalent bridging of the starburst dendrimers
through their reactive terminal groups produces a class
of topological polymers, with size and shape controlled
35,444-F"B" -2-
.
..,, ,... ., ~, . .. ,. ,.. ~ ~
. . .
~ 3 ~
domains, which are referred to as "starburst bridged
dendrimers", whic~ term is also encompassed within the
term starburst polymer.
The following description of the figures aid in
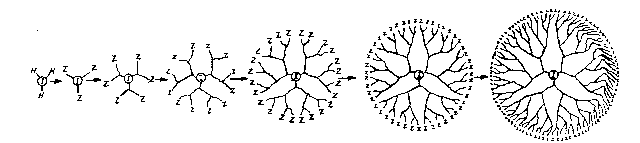
understanding the present invention7
Fi~ure 1 depicts various generations of starburst
dendrimers.
Fi~ure 2A depicts a dendrimer having un~-ymmetrical
tunequal) branch junctures.
Fi~ure 2B depicts a dendrimer having symmetrical
(equal) branch junctures.
The starburst polymers are illustrated by
Figure 1 wherein ~ represents an initiator core (in
thi~ Pigur~ a tri-functional initiator core, shown by
the ~ar left drawing); Z represents a terminal group;
shown in the first instance by the second drawing from
the left, referred to as a starbranched oligomer; A, B,
C, D, and E represent particular molecular generations
of 3tarburst oligomers, called dendrimers; and (A)n~
(B)n~ (C)n, (D)n~ and (E)n represent starburst bridged
dendrimers.
The starburst dendrimers are unimolecular
assemblage~ that possess three distinguishing
architectural features, namely, (a) an initiator core,
~b) interior layers (generations, G) composed of
repeating units, radially attached to the initiator
core, and (c) an exterior surface of terminal
functionality (i.e., terminal functional groups)
attached to the outermost generation. The size and
shape of the starburst dendrimer and the functional
35,444-F"B" _3_
13~2~
64693-4103
groups present in the ~endrlmer can be controlled by the cholce of
the lnitiator core, the number of generatlons ~i.e., tiers)
employed ln creatlng the dendrimer, ~nd the cholce o~ ~he repeat-
ing units employed at each generation. Since ~he dendrimers can
be isolated at any particular generatlon, a means ls provlded for
obtaining dendrlmers havlng desired propertles.
The cholce o~ the starburst dendrlmer components a~fects
the propertles of the dendrlmers. The lnltlator core type can
affect the dendrlmer shape, produclng (dependlng on the cholce of
initiator core)~ for example, spheroid-shaped dendrimers, cylin-
drical or rod-shaped dendrimers, ellipsoid-shaped dendrimers, or
musllroom-shaped dendrlmers. Sequential bulldlng of generatlons
(l.e., generatlon number and the sl~e and nature of the repeatlng
unlts) determlnes the dimenslons of the dendrlmers and the nature
of their lnterlor.
Because starburst dendrlmers are branched polymers con-
talnlng dendrltlc branches having functlonal groups dlstributed on
the periphery of the branches, they can be prepared wlth a varlety
of propertles. For example, starburst dendrlmers, such as those
deplcted ln Flgure 2A and Flgure 2B can have dlstinct propertles
due to branch length. The dendrlmer type shown ln Flgure 2A (such
as Denkwalter U.S. Patent 4,289,872) possesses unsymmetrlcal (un-
equal segment) branch ~unctures, exterlor (l.e., surface) groups
(represented by Z'), lnterior moletles (represented by Z) but much
less lnternal void space. The dendrlmer type shown ln Figure 2B
possesses symmetrlcal (equal segment) br~nch ~unctures wlth sur-
face groups (represented by Z',), two
, '
~5~ ~ 3 1 ~ ~211
different interior moieties (represented respectively
by X and Z) with interior void space ~Jhich vaires as a
function of the géneration (G). The dendrimers such as
those depicted i-n Figure 2B can be advanced through
enough generations to totally enclose and contain void
space, to give an entity with a predominantly hollow
interior and a highly congested surface. Also,
starburst dendrimers, when advanced through sufficient
generations exhibit "starburst dense packing" where the
surface of the dendrimer contains sufficient terminal
moietie~ such that the dendrimer surface becomes
congested and encloses void spaces within the interior
of the dendrimer. This congestion can provide a
f 15 molecular level barrier which can be used to control
diffusion of mater-ials into or out of the interior of
the dendrimer.
Surface chemistry can be controlled in a
predetermined fa~hion by selecting a repeating unit
which contains the desired chemical functionality or by
chemicall~ modifying all or a portion of the surface
functionalities to create new surface functionalities.
In an advantageous use of the dendrimers, the
dendrimers can themselves be linked together to create
polydendric moieties ("starburst bridged dendrimers")
which are also suitable as carriers.
In addition, the dendrimers can be prepared so
as to have deviations from uniform branching in
particular generations, thus providing a means of
adding discontinuities (i.e., deviations from uniform
branching at particular locations within the dendrimer)
and different properties to the dendrimer.
35,444-F"B" -5-
-6- ~ 3 ~ 2ll
The starburst polymers employed in the
starburst conjugates of the present invention can be
prepared according to methods known in the art, for
example, U.S. Patent 4,587,329.
Dendrimers can be prepared having highly
uniform size and shape and most importantly allow for a
greater number of functional groups per unlt of surface
area of the dendrimer, and can have a greater number of
functional groups per unit of molecular volume as
compared to other polymers which have the same
molecular weight, same core and monomeric components
and same number of core branches as the starburst
polymers. The increased functional group density of
the starburst polymers may allow a greater quantity of
material to be carried per dendrimer. Since the number
of functional groups on the dendrimers can be
controlled on the surface and within the interior, it
also provides a means for controlling the amount of
agent carried per dendrimer.
I An analogy can be made between early generation
starburst dendrimers (i.e. generation -1-7) to
clas~ical spherical micelles. The dendrimer-micelles
analogy was derived by comparing features which they
had in common such as shape, size and surface.
35,444-F"B" -6- _
_7_ ~3~2~
Table I
Parameter Regular Classical Starburst Dendrimers
Shape Spherical Spherical
Size 20-60A 17~ 7fl
(diameter)
Sur~ace 4-202 Z=6-192
aggregation number (generation = 2-7)
area2/surface group 130-80A2 127-75~2
10 (~ ~
Z ~s the ~umb~r of surface groups; 1A= 10~1 nm;
1A = 10~ nm
f 15 In Table I, the shape was verified by scanning
transmission electron micrographs (STEM) microscopy and
intrinsic viscosity (rl) measurements. The size was
verified by intrinsic viscosity (~) and size exclusion
chromatography (SEC) measurements. The surface
aggregation numbers were verified by titrmetry and high
field NMR. The area/surface group was calculated from
SEC hydrodynamic measurements.
The first five generations of starbur~t
polyamidoamine (PAMAM) dendrimers are microdomains
which very closely mimic classical spherical micelles
in nearly every respect (i.e. shape, size, number of
surface groups, and area/surface group). A major
difference, however, is that they are covalently fixed
and robust compared to the dynamic equilibration of
nature of micelles. This difference is a significant
advantage when using these microdomains a~
encapsulation devices.
35,444-F"B'~ -7-
-8- 131~3~
As further concentric generations are added
beyond five, congestion of the surface occurs. This
congestion can lead to increased barrier
characteristics at the surface and manifests itsel~ as
a smaller surface area per head ~surface) groups as
shown in Table II.
35,444-F7'B" -8-
~ 3 ~
_9_
"~ ~ ~ ''C
N
N ~
r1 U~
~` ~ ¢ 1 ~3
C ~ S 0~¢ ' ¦ S
~ o a
U~ ~ O NO --
H 01 ~
~1 N~ S ~ X
.~
S...... ~ ~I N ~ r r~ g S~
1~ tr~ l _I N N '~ N ~
C O ~y~
. ~
~ . ~ .
o.~l N No~l~ N 0 31 S~ S
CO U~ '7 N 1
" ~
~ O
~ ~ O ~D ~ ~ '~ O
O ~ .
'
u~ U U ~ C
a ~ O
Ul U~ U ~ U~ ~ ~ g
t E 3 a ~ D. a o o~
35, 444-F "B" -9-
~ ~L 3 ~
64693-4103
For example, amine termlnate~ generations 5.0, 6.0, 7.0,
.0 and 9.0 have decreased surface areas of 104, 92, 73, 47 and
32A2 per Z group, respec~ively. Thls characterlstic corresponds
to a transltlon from a less congested mlcelle-llke surface to a
more congested bllayer/monolayer barrier llke surface normally
assoclated with vesicles (llposomes3 or Langmuir-Blodgett type
membranes.
If this surface congestlon is lndeed occurrlng, the
change ln physlcal characterlstlcs and morphology should be
observed as the generations increase ~rom the intermediate
generatlon (6-8) to the more advanced generatlons (9 or 10). The
scannlng transmlsslon electron mlcrographs (ST~M) for generations
o 7.0, 8.0 and 9.0 were obtalned after removlng the methanol
solvent from each of the samples to provide colorless, llght
yellow solld films and staining wlth osmium tetrao~ide. The
morphological change preclicted occurred at the generatlon G = 9.0
stage. The interlor microdomalns at generation, G = 9.0, measure
about 33A ln dlameter and are surrounded by a colorless rim whlch
ls about 25A thlck. Apparently the methanollc solvent has been
entrapped wlthln the 25A outer membrane-llke barrler to provlde
the dark stalned interlor. Thus, at generation = 9.0, the
starburst PAMAM ls behaving topologlcally like a vesicle
(llposome). However, thls starburst ls an order of magnltude
smaller and very monodlspersed compared to a llposome.
Consequently, the present dendrlmers can be used to molecularly
encapsulate solvent fllled vold spaces of as much dlameter as
about 33A ~volume about 18,000A3) or more.
Slnce the nurnber of functional groups on the dendrimers
can be~controlled on the surface and wlthln
. ~ ~
' ., '
2 ~
the interior, it also provides a means for controlling
the amount of carried material to be delivered per
dendrimer. In one embodiment of the presen~ invention,
the dendrimers are targeted carriers o~ agents capable
of delivering the carried agent (material) to a
particular locus.
Dendrimers suitable for use in the conjugates
of the present invention include the starburst polymers
described in U.S. Patents 4,507,466, 4,558,120,
49568,737 and 4,587,329.
In particular, the present invention concerns a
starburst conjugate which comprises at least one
f 15 starburst polymer associated with at least one carried
material. Starburst conjugates included within the
scope of the present invention include those
represented by the formula:
(P)x * (M)y (I)
wherein each P represents a dendrimer;
x represents an integer of 1 or greater;
each M represents a unit (for example, a molecule,
atom, ion, and/or other basic unit) of a carried
3 material, said carried material can be the same carried
material or a different carried material;
y represents an integer of 1 or greater; and
35,444-F"B" -11-
.
-12~ 2 ~
* indicate~ that the carried material i~ associated
with the dendrimer.
Preferred starburst conjugates of formula (I)
are those in which M is a signal generator such as
fluorescing entities9 signal reflector such as
paramagnetic entities, signal absorbers such as
electron beam opacifiers, fragrance, pheromones, or
dye; particularly pre~erred are those in which x-1, and
y=2 or more.
Also included are starburst conjugates of
formula (I) wherein the dense star dendrimers are
covalently linked together, optionally via linking
f 15 groups, so as to form polydendric assemblages (i.e.,
where x~1).
As u~ed herein, "associated with~' means that
the carried material(s) can be encapsulated or
entrapped within the core of the dendrimer, dispersed
partially or fully throughout the dendrimer, or
attached or linked to the dendrimer, or any combination
thereof. The association of the carried material(s)
and the dendrimers may optionally employ connectors
and/or spacers to facilitate the preparation or use of
the starburst conjugates. Suitable connecting groups
are grQups which link a targeting director (i.e., T) to
the dendrimer (i.e., P) without significantly impairing
the effectiveness of the director or the effectiveness
of any other carried material(s) (i.e. 7 M) present in
the starburst conjugate. These connecting groups may
be cleavable or non-cleavable and are typically used in
order to avoid steric hindrance between the target
director and the dendrimer, preferably the connecting
group3 are stable (i.e., non-cleavable). Since the
35,444-F"B" -12-
-13- ~ 3~2~
size9 shape and functional group density of the dense
star dendrimers can be rigorously controlled, there are
many ways in which the carried material can be
associated with the dendrimer. For example, (a) there
can be covalent, coulombic, hydrophobic, or chelation
type association between the carried material(s) and
entities, typically functional groups, located at or
near the surface of the dendrimer; (b) there can be
covalent, coulombic, hydrophobic, or chelation type
association between the carried material(s) and
moieties located within the interior of the dendrimer;
(c) the dendrimer can be prepared to have an interior
: which is predominantly hollow allowing for physical
r' 15 entrapment of the carried materials within the interior
~void space) wherein the release of the carried
material can optionally be controlled by congesting the
sur~ace of the dendrimer with diffusion controlling
moietie~; or (d) various combinations of the
2~ aforementioned phenomena can be employed.
Dendrimers, herein represented by "P", include
the dense star polymers described in UOS~ Patents
4,507,466, 4,558,120, 4,568,737 or 4,587,329.
Carried materials, herein represented by "M",
which are suitable for use in the starburst conjugates
include any materials, other than pharmaceutical or
agricultural materials, which can be associated with
the starburst dendrimer without appreciably disturbing
the physical integrity of the dendrimer, for example,
metal ions such as the alkali and alkaline-earth
metals; signal generators such as fluorescing
entities; ~ignal reflectors quch a~ paramagnetic
entities; signal absorbers such as an electron beam
opacifiers; pheromone moieties; fragrance moieties; dye
35,444-F"B" -13- _~
-14-
~1 3 ~
moieties; and the like. Carried materials include
scavengin~ agents such as chelants or any moieties
capable of selectively scavenging a variety of agents.
The starbursts conjugates of formula (I) are
prepared by reacting P with M, usually in a suitable
solvent, at a temperature which facilitates the
association of the carried material (M) with the
starburst dendrimer (P).
Suitable solvents are solvents in which P and m
are at least partially miscible and inert to the
formation of the conjugate. If P and M are at least
partially miscible with each other, no solvent may be
r` 15 re~uired. When desired, mixtures of suitable solvents
can be utilized. Examples of such suitable solvents
are water, methanol, ethanol, chloroform, acetonitrile,
toluene9 dimethylsulfoxide and dimethylformamide.
The reaction condition for the ~ormation of the
starburst conjugate of formula (I) depends upon the
particular dendrimer (P), the carried material (M), and
the nature of the bond (*) formed. Typically, the
temperature can range from room ternperature to reflux.
The selection of the particular solvent and temperature
will be apparent to one skilled in the art.
The ratio of M:P will depend on the si~e of the
dendrimer and the amount of carried material. For
3 example, the molar ratio (ratio of moles) any ionic M
to P is usually 0.1-1,000:1, preferably 1-50:1 and more
preferably 2-6:1. The weight ratio of any organic M to
P is usually 0.1-5:1, and preferably 0.5-3:1.
35,444-F"B" -14-
-15- ~3~2~
Other starburst conjugates are those conjugates
which contain a target director (herein designated as
"T") and which are represented by the formula:
(T)e * (P)x * (M)y (II)
wherein
each T represents a target director;
e represents an integer of 1 or greater; and
P, x, *~ M9 and y are as previously defined herein.
Preferred among the starburst conjugates of
formula (~) are those in which M is a signal generator,
signal reflector, or signal absorber. Also preferred
are those conjugate3 in which e-1; and those in which
x=1 and y=2 or more; and particularly preferred are
those in which x=1, e=~l, and y=2 or more. Most
preferred are those in which M and T are associated
with the polymer via the same or different connectors.
The starburst conjugates of Eormula (Il) are
prepared either by forming T*P and then adding M or by
forming P*M and then adding T. Either reaction scheme
is conducted at temperatures which are not detrimental
to the particular conjugate component and in the
presence of a suitable solvent when required. To
control pH, buffers or addition of suitable acid base
i~ used. The reaction conditions are dependent on the
type of association formed (*), the starburst dendrimer
u~ed (P), the carried material (M), ahd the target
director (T). Alternatively, P and M can be chelated,
35,444-F"B'i -15-
,. ............. .
~3~6~
usually in water, before conjugation to T. The
conjugation with T is carried out in a suitable buffer.
The ratio of T:P is preferably 1:1. The ratio
of M:P will be as before.
Target directors capable of targeting the
starburst conjugates are entitie~ which when used in
the starburst conjugates of the present invention
result in at least a portion-of the starburst
conjugates being delivered to a desired target,
chemical functionalities exhibiting target specificity,
and the like.
In the absence of a target director (or in the
presence of a target director if de~ired), due to the
number of functional groups which can be located at or
near the surface of the dendrimer, all or a sub3tantial
portion of such functional groups can be made anionic,
cationic, hydrophobic or hydrophilic to effecti~ely aid
delivery of the starburst conjugate to a desired target
of the opposite charge or to a hydrophobic or
hydrophilic compatible target.
Preparation of the conjugates of formula (II)
using a P with a protected handle (S) is also intended
as a process to prepare the conjugates of formula (II).
The reaction scheme is shown below:
S*P loadin~ ~ S*P*M deprotection D P*M
T*P*M linking ¦
where
35,444-F"B'1 -16- _
-17- ~ 3~ ~3 2~
S*P represents the protected dendrimer;
S*P*M represents the protected dendrimer
conjugated with m;
P*M represents the dendrimer conjugated
with M (starburst conjugate);
T*P*M represents the starburst conjugates
liked to the target director.
Suitable solvents can be employed which do not
effect P*M. For example when S is t- = , S
can be removed by aqueous acid.
The ~tarburst conjugates can be used for a
f 15 variety of in vitro applications such as radio-
immunoassay~, electron microscopy, enzyme linked
immunosorbent assay~, nuclear magnetic resonance
spectroscopy, and contrast imaging, and immuno-
scintography, in analytical applications; or used as
starting materials for making other useful agents.
The present invention is also directed to
starburst conjugate compositions in which the ~tarburst
conjugates are formulated with other suitable vehicles.
The starburst conjugate compositions may optionally
contain other active ingredients, additives and/or
diluents.
The preferred ~tarburst polymer for use in the
starburst conjugates of the present invention is a
polymer that can be described a~ a starburst polymer
having at least one branch (hereinafter called a core
branch), pre~erably two or more branches~ emanating
from a core, said branch having at least one terminal
group provided that (1) the ratio of terminal groups to
35,444-F"B" -17-
-18- ~ 3~2~
the core branches is more than one, preferably two or
greater, (2) the density oP terminal groups per unit
volume in the polymer is at least 1.5 times that of an
extended conventional star polymer having similar core
and monomeric moieties and a comparable molecular
weight and number of core branches~ each of such
brancheq of the extended conventional star poly~ler
bearing only one terminal group, and t3) a molecular
volume that is no more than about 80 percent of the
molecular volume of said extended conventional star
polymer as determined by dimensional studies using
scaled Corey-Pauling molecular models. As used herein,
the term "dense" as it modifies "star polymer" or
"dendrimer" means that it has a smaller molecular
volume than an extended conventional star polymer
having the same molecular weight. The extended
conventional star polymer which is used as the base for
comparison with the dense star polymer is one that has
the same molecular weight~ same core and monomeric
components and same number of core branches as the
dense star polymer. By "extended" it i~ meant that the
individual branche~ of the conventional star polymer
are extended or stretched to their maximum length,
e.g.; a~ such branches exist when the star polymer is
completely solvated in an ideal solvent for the star
polymer. In addition while the number of terminal
groups is greater for the dense star polymer molecule
than in the conventional star polymer molecule, the
chemical structure of the terminal groups is the same.
Dendrimer~ used in the conjugates of the
present invention can be prepared by processes known in
the art. The above dendrimers, the various coreactants
35,444-F"B" -18-
~ 1 9 - .
and core compound~, an~ proces~ for their preparation
can be a~ defined in U.S. Patent 4,587,329.
The ~tarburst dendrimers, for use in the
starburst conjugate~ of the present invention, can have
terminal groups which are sufficiently reactive to
undergo addition or substitution reactions. Examples
of such terminal groups include amino, hydroxy 9
mercapto, carboxy, alkenyl, allyl, vinyl, amido, halo,
urea, oxiranyl, aziridinyl7 oxazolinyl, imidazolinyl,
sulfonato, phosphonato, isocyanato and isothiocyanato.
The dendrimers differ from conventional star or 3tar-
branched polymers in that the dendrimer~ have a greater
concentration of terminal groups per unit of molecular
volume than do conventional extended star polymers
having an equivalent number of core branches and an
equivalent core branch length. Thus, the density of
terminal groups per unit volume in the dendrimer
usually i~ at least about 1.5 times the density of
terminal groups in the conventional extended star
polymer, preferably at least 5 times, more preferably
at least lQ times, most preferably from 15 to 50 times.
The ratio of terminal groups per core branch in the
starburst dendrimer is preferably at least 2, more
preferably at least 3, most preferably from 4 tc 1024.
Preferably, for a given polymer molecular weight, the
molecular volume of the starburst dendrimer is less
than 70 volume percent, more preferably from 16 to 60,
3 most preferably from about 7 to 50 volume percent of
the molecular volume of the conventional extended star
polymer.
Preferred starburst dendrimers for use in the
starburst conjugates of the present invention are
characterized as having a univalent or polyvalent core
35,444-F"B" -19- _
--20 1 3 ~ ~ ~ 2 ~
that i9 coYalently bonde~ to dendritic branches. Such
ordered branching can be illustrated by the following
sequence wherein G indicates the number of generation~:
G = 1 G = 2
N - - N -
~ ~
H
H H
G = 3
. N -
,~ ~
~ ~ ~
N- f N N N
3 H H H / H H H H H
35,444-F"B" -20-
-21- ~ 3 2 ~
Mathematically, the relationship between the
number (#) of terminal groups on a dendritic branch and
the number of generationq of the branch can be
represented as follows:
NrG
# of terminal groupq per dendritic branch =
wherein G is the number of generations and Nr is the
repeating unit multiplicity which is at least 2 as in
the case of amines. The total number of terminal
groups in the dendrimer iY determined by the followingo
f 15
NcNrG
# of terminal groups per dendrimer =
wherein G and Nr are as defined before and Nc
represents the valency (often called core
functionality) of the core compound. Accordingly, the
dendrimer~ of this invention can be represented in its
component parts as follows:
~ Terminal
(Core~ (Repeat Unit~N G l Moiety N G
I C
Nr-l /
35,444-F"B" -21-
-2~- ~ 3 ~
wherein the Core, Terminal Moiety, G and Nc are as
de~ined before and the Repeat Unit has a valency or
functionality of Nr ~ 1 wherein Nr i~ as defined
before.
A copolymeric dendrimer which is a preferred
dendrimer for the purposes of this invention i9 a
unique compound constructed of polyfunctional monomer
units in a highly branched (dendritic) array. The
dendrimer molecule is prepared from a polyfunctional
initiator unit (core compound), polyfunctional
repeating units and terminal units which may be the
same or different from the repeating units. The core
compound i~ represented by the formula ~ (ZC)Nc
wherein ~ represents the core, zc represents the
functional groups bonded to I and Nc represents the
core functionality which i~ preferably 2 or more, most
pre~erably 3 or more. Thus, the dendrimer molecule
comprise3 a polyfunctional core, ~, bonded to a number
(Nc) of functional gro~ps, zc, each of which is
connected to the monofunctional tail of a repeating
unit, X1Y1(Z1)N1~ of the first generation and each of
the Z groups of the repeating unit of one generation is
bonded to a monofunctional tail of a repeating unit of
the next generation until the terminal generation is
reached.
In the ~dendrimer molecule, the repeating units
are the same within a single generation, but may differ
from generation to generation. In the repeating unit,
X1Y1(Z1)N1, X1 represent~ the monofunctional tail of
the first generation repeating unit, y1 repre~ents the
moiety constituting the first generation, z1 represent~
the functional group of the polyfunctional head of the
repeating unit of the firYt generation and may be the
35,444-F"B" -22-
-23- ~3~rj~
same as or different from the functional groups of the
core compound, ~ (ZC)Nc~ or other generations; and N1
is a number of 2 or more, most preferably 2, 3 or 4,
which represents the multiplicity of the polyfunctional
head of the repeating unit in the first generation.
Generically, the repeating unit is represented by the
formula XiYi(Zi)Ni wherein "i" represents the
particular generation from the first to the t-1
generation. Thus, in the prePerred dendrimer molecule,
0 each zl of the first generation repeating unit is
connected to an x2 of a repeating unit of the second
generation and so on through the generations such that
each zi group ~or a repeating unit XiYi(Zi)Ni in
lS generation number "i" is connected to the tail (Xi~1)
of the repeating unit of the generation number "i~l".
The final or terminal of a preferred dendrimer molecule
comprises terminal units, XtYt(Zt)Nt wherein t
represents terminal generation and xt, ytJ zt and Nt
may be the same as or different from xi, yi~ zi and Ni
except that there is no succeeding generation connected
to the zt groups and Nt may be less than two, e.g.,
zero or one. Therefore the preferred dendrimer has a
molecular formula represented by
~5
35,444-F"B" -23- _~
-24- l 3 ~
~ n is ~ ~ n LS
1~ where i is 1 to t-1
wherein the symbols are as previously defined. The
function is the product of all the values between its
defined limits. Thus
f 15
n Nn - (N1)(N2)(N3)..~(Ni-2)(Ni-l)
n=1
which is the number of repeat units, XiYi(Zi)Ni,
comprising the ith generation of one dendritic branch
and when i is 1, then
IIO = 1
n=1
In copolymeric dendrimers, the repeat unit for one
generation differs from the repeat unit in at least one
other generation. The preferred dendrimers are very
symmetrical as il]ustrated in structural formulas
described hereinafter. Preferred dendrimers may be
converted to functionalized dendrimers by contact with
another reagent. For example, conversion of hydroxyl
in the terminal generation to ester by reaction with an
aoid chloride gives an ester terminally functionalized
dendrimer. This functionalization need not be carried
out to the theoretical maximum as defined by the number
35,444-F"B" -24- _~
-25- ~ 2 ~
of available functional groups and, thus, a
functionalized dendrimer may not have high symmetry or
a precisely defined molecular formula as is the case
with the preferred dendrimer.
In a homoplymer dendrimer, all of the repeat
units, XiYi~2i)Ni, are identical. Since the values of
all Nl are equal (defined as Nr)~ the product function
representing the number of repeat units reduces to a
simple exponential form. Therefore, the molecular
formula may be expressed in simpler form as
( ~ (Zc)N ) ~ i yi (zi)N~ n ~ (XtY (Z )N~ N Nr
where i = 1 to t-1
This form still shows the distinction between
the different generations i, which each consist of NcNr
(i-1) repeating units, XiYi(Zi)Ni. Combining the
generations into one term gives:
3o
35,444-F"B" -25-
-26- ~3~ ~2~
Z )N )~X y (zi) Nr(t ~(X Y (Z )Nt) NCNr(t-l)
or
1C core repeat unitremoval unit,
1' ((~ )N )~XrYr(~r)Nr)( y (Z )Nt)
Nr-1
wherein xryr(zr)Nr is the repeating unit which is used
in all generations i.
Consequently, if a polymer compound will fit
into these above formulae, then the polymer is a
starburst polymer. Conversely, if a polymer compound
will not fit into these above formulae, then the
polymer is not a starburst polymer. Also r to determine
whether à polymer is a starburst polymer, it is not
necessary to know the process by which it was prepared,
but only`whether it fits the formulae. The formulae
3 also demonstrate the generations (G) or tiering of
dendrimers.
~ learly, there are several ways to determine
the ratio of agent (M) to dendrimer (P) which depend
upon how and where the association of P*M occurs. When
there is interior encapsulation, the weight ratio of
35,444-F"B" -26-
- 27~
M:P usually is 10:1, preferably 8:1, more preferably
5:1, most preferably 3:1. The ratio can be as low as
0.5:1 to 0.1:1. When interior stoichiometry is used,
the weight ratio of M:P is the same as for interior
encapsulation. When exterior stoichiometry i3
determined, the mole/mole ratio of M:P given by the
following formulae:
M P
(A) 5 NCNtNr6~l 1
(B) 3 NCNtNr~ 1
,.
f 15 (C) 1 NCNtNr
where Nc means the core multiplicity, Nt means the
terminal group multiplicity, and Nr~m,eans branch
juncture multiplicity. The NCNtNr~ ~ term will result
in the number of Z groups. Thus, for example, (A)
above will result when proteins, enzymes or highly
charged molecules are on the surface; (B) above when it
is octanoic acid; (C) above when it is carboxylate ions
or groups.
Of course other structures of ~arious
dimensions can be readily prepared by one skilled in
the art by appropriately varying the dendrimer
components and number of generations employed. The
dimensions are 3ignificant in that they are small. A
linear polymer of comparable molecular weight would
have a radius of gyration, (in its fully extended
form), that would be much larger than the same
molecular weight dendrimer.
35,444-F"B" -27- _~
: , .
~3~$~
64693-4103
Llnking target directors to dendrlmers ls another aspect
of the present lnventlon. In preferred embodlments of the present
inventlon, a reactive functional group such as a carboxyl,
sulfhydryl, reactive aldehyde, reactlve ole~lnic deriva~ive,
l~othlocyanato, lsocyanato, amlno, reactlve aryl halide, or
reactive alkyl hallde can convenlently be employed on the
dendrlmer. The reactive functional groups can be lntroduced to
the dendrimer uslng known technlques, for example
(1) Use of a heterofunctlonal lnitiator ~as a star~lng
material for syntheslzlng the dendrlmer) which has lncorporated
into lt functional groups of different reactivity. In such
heterofunctional lnitiator at least one of the functlonal groups
will serve as an inltiation si~e for dendrlmer formation and at
least one of the other functional groups wlll be avallable for
linking to a target director but unable to inltiate dendrimer
synthesls. For example, use of protected anlllne allows further
modificatlon of NH2 groups within the molecule, without reactlng
the NH2 of the anlline.
The functlonal group whlch will be available for linking
to a target dlrector may be part of the lnitlator molecule ln any
one of three forms, namely:
(a) In the form in which lt will be used for linking
with the target dlrectors. Thls ls posslble when
none of the synthetic steps lnvolved in the
dendrlmer synthesls can result ln reactlon at this
center.
(b) When the functlonal group used for linking to the
targeting dlrector is reactive in
28
~T
'~
-29- ~3~ ~5~
the synthetic steps involved in the
dendrimer synthesis, it can be protected
by use of a protecting group, which
renders the group unreactive to the
synthetic procedures involved, but can
itself be readily removed in a manner
which does not alter the integrity of the
remainder of the macromolecule.
(c) In the event that no simple protecting
group can be found for the reactive
functionality to be used for linking with
the targeting director, a synthetic
precursor can be used which is unreactive
f 15 in all the synthetic proecedures uqed in
the dendrimer synthesis. On completion of
the synthesis, this functional group must
be readily convertible into the desired
linking group in a manner which does not
alter the integrity of the remainder of
the molecule.
(2) Coupling (covalently) the desired reactive
functional group onto a preformed
dendrimer. The reagent used must contain
a functionality which is readily reacted
with the terminal functional groups of the
dendrimer. The functional group to be
ultimately used to link with the targeting
agent can be in its final form, as a
protected functionality, or as a synthetic
precursor. The form in which this linking
~unctionality i~ used depends on itq
integrity during the synthetic procedure
to be utilized, and the ability of the
35,444-F"B" -29-
,
: ' ,
3 ~ 3 ~
~inal macromolecule to with~tand any
conditions necessary to make this group
available for linking. For example, the
preferred route for PEI uses
~ ~N02
Example~ of heterofunctional initiators ~or use
in (1) above, include the following illustrative
examples:
f 1~
H2N ~ CH2NH2
2C
CNHCH2CH2NH2
H2N~ CH2CH
CNHCH2CH2NH2
30 ' 8
35,444-F"B" -30-
-31~ 2~
/CNHCH2CH2NH2
(CH3)3 COCNH~ O~> CH2C~H
\ _ ` CNHCH2CH2NH2
~ ~CH2NH2
( CH3 ) 3 COCN~ CH2CH
CH2NH2
,, lC
H2N ~CH2CH
CH2NH2
3C H2N ~ CH2CH2NH2
35, 444-F"B" -31~
, .,
.
~3~~ :IL3:1 ~52l~
/CH2NH2
~2N ~,>--CH2CH
CH2NH2
02N--<~(~ CH2CH2NH2
t' 1 _
2C - CH2NH2
/ \ /
02N <~- CH2CH ; and
\
CH .NH2
2~
.
35, 444-F"B" -32- ~
-33-
~ 3 ~ 2 ~
f H2CH2NH2
CH2NCH2CH2NH2
02N C~2CH
CH2NCH2CH2NH2
CH2CH2NH2
There are several chemistrie of particular
f 15 importance:
1) Starburst Polyamidoamides ("PAMAM") Chemistry;
2) Starburst Polyethyleneimines ("PEI") Chemistry;
3) Starburst PEI compound with a surface of PAMAM;
4) Starbur~t Polyether ("PE") Chemistry.
ModiYications of the dendrimer surface
functionalities may provide other useful functional
~5 groups such as the following:
-OP03H2, -P03H2, -P03H(-~ po3(-2), _C02(-1), -S02H,
-S02( 1), -S03H 9 -S03(-1), -NR1R2~ -R5, -OH, -ORl,
-NH2, polyetherq, perfluorinated alkyl, -CNHR1, -COH,
1- ,.
O O
35`,444-F"B" -33~
.. ~3~~ ~3~
-(CH2)n ~ -N=C ~
R3 R4
-NHCH2 ~ -(CH2)n ~ ~ -(cH2)
1 R3 R4 N
wherein
R represents alkyl, aryl or hydrogen;
f 15 Rl represents alkyl, aryl, hydrogen, or
~ (CH2)n ~
-N X
(CHz)n
R2 represent~ alkyl, aryl, or
~ (CH2)n ~
-N X
~CH2)n
, ` 30
- 35,444-F"B" -34- _~
-35~ 2 ~
R3 represents -OH, -SH, -C02H, -S02~, or -S03H;
R4 represents alkyl, aryl, alkoxy, hydroxyl, mercapto,
carboxyl, nitro, hydrogen, bromo, chloro, iodo, or
fluoro;
R5 represents alkyl;
x represents NR, O or S; and
n represents the integer 1, 2 or 3.
The choice of functional group depends upon the
particular end use for which the dendrimer is designed.
The following examples further illustrate the
invention but are not to be construed as a limitation
on the scope of the invention. The lettered examples
concern the preparation of starting materials; the
numbered examples concern the preparation of product.
Example A: Preparation of 2-Carboxamido-3-(4'-nitro-
phenyl)-propanamide.
p-Nitrobenzyl malonate diethylester (2.4 grams
(g), 8.13 mmole) was dissolved in 35 ml of methanol.
The solution was heated to 50-55C with stirring and a
stream of anhydrous ammonia was bubbled through the
solution for 64 hours. The solution was cooled and the
white, flocculant product was filtered and
recrystallized from 225 milliliters (ml) of boiling
methanol to afford 1.85 g (7.80 mmole) of bis amide in
96~ yield (mp _ 235.6C(d)).
The structure was confirmed by MS,lH and 13C NMR
spectroscopy.
35,444-F"B" -3
-36- 13~
Anal: Calc. for c1oH1 104N3
C H N
Theo: 50.63 4~69 17.72
Found: 50.75 4.81 17.94
Example B: Preparation of l-Amino-2-(aminomethyl)-3-
(4'-nitrophenyl)propane.
2-Carboxamido-3-(4'nitrophenyl)propanamide (2.0
1 g, 8.43 mmole) was slurried in 35 ml of dry tetrahydro-
furan under a nitrogen atmosphere with ~tirring. To
this mixture was added borane/tetrahydrofuran complex
(106 ml, 106 mmole) via syringe. The reaction mixture
was then heated to reflux for 48 hours during which
time the suspended amide dissolved. The solution was
cooled and the tetrahydrofuran was removed in vacuo
using a rotary evaporator. The crude product and
borane residue was dissolved in 50 ml of ethanol and
this solution was purged with anhydrous hydrogen
chloride gas~ The solution was refluxed for 1 hour and
the solvent removed in vacuo. The crude hydrochloride
salt was dissolved in 15 ml of deionized water and
extracted with two 50 ml portions of methylene
chloride. The aqueous layer was cooled in an ice bath
under an argon blanket and 50~ sodium hydroxide was
slowly added until basic pH-11.7. The basic aqueous
layer was extracted with four 25 ml portions of
methylene chloride and these combined extracts were
evaporated (rotary) to give 1.45 g of amber colored
oil. This oil was triturated with diethyl ether (50
ml) and filtered under pressure through a short silica
gel (grade 62 Aldrich) column. The column was washed
with 100 ml of ether and the combined filtrates were
vacuum evaporated giving 1.05 g (5.02 mmole) o~ the
35,444-F"B" -36-
37 ~3~
titled diamine as a clear oil (mp = 275-278C(d) bis
HCl salt).
The structure was conflrmed by MS, 1H and 13C NMR
Spectroscopy.
Anal: Calc. for C1oHl7N3o2cl2
C H N
Theo~ 42.57 6.07 14.89
Found: 43.00 6.14 15.31
Example C: Preparation of 1-Amino-2-(aminomethyl)-3-
(4'-aminophenyl)propane.
Borane/tetrahydrofuran solution (70 ml, 70
mmole) was added under nitrogen via a cannula needle to
a flask containing 4-amino-benzyl malonamide (1.5 g,
7.24 mmole) with stirring. The solution was brought to
reflux for 40 hours. The colorle~s solution was cooled
and excess tetrahydrofuran was removed by rotary
evaporation leaving a clear gelatinous oil. Methanol
(50 ml) was cautiously added to the oil with notable
gas evolution. Dry hydrogen chloride was bubbled
through the suspension to effect dissolution and the
solution was then refluxed for l minute. The
methanol/HCl was rotary evaporated and the resulting
hydrochloride salt was carried through the same
dissolution/reflux procedure again. The hydrochloride
silt obtained was dissolved in lO ml of water and
cooled in an ice bath under argon. Concentrated sodium
hydroxide (50%) was added slowly with stirring to
pH=11. The aqueous portion was then extracted with 2 X
100 ml portions o~ chloro~orm which were combined and
filtered through a short ~ilica gel plug without
drying. The solvent was removed in vacuo (rotary)
35,44~-F':B" -37-
-38~ J~ 2'1
affording the title compound (0.90 g, 5.02 mmole) in
70% yield (Rf=0.65 - CHC13/MeOH/NH40H conc - 2/2/1).
The structure was confirmed by 1H and 13C NMR and used
without further purification.
Example D: Preparation of 6-(4-Aminobenzyl)-1,4~8,11-
tetraaza-5,7-dioxoundecane.
4-Aminobenzyl malonate dimethylester (2.03 g,
8.43 mmole) was dissolved in 10 ml of methanol. This
solution was added dropwise to a stirred solution of
freshly distilled ethylene diamine (6.00 g, 103.4
mmole) in 10 ml o~ methanol under nitrogen over a 2
hour period. The clear solution was stirred for 4 days
f 15 and thin layer chromotograph~ (TLC) analysis indicated
total conversion of diester (Rf = 0091) to the bis
amide (Rf = 0.42 - 20~ conc NH40H/80~ ethanol). This
material was strongly ninhydrin positive. The methanol
and excess diamine were removed on a rotary evaporator
and the resulting white solid was vacuum dried (19-1
mm, 50C) overnight to afford crude product (2.45g, 8.36
mmole) in 99% yield. An analytical sample was
recrystallized from chloroform/hexane, MP - 160-161C.
The mass spectral, 1H and 13C NMR data were consistent
with the proposed structure.
Exam~le E: Reaction of Mesyl Aziridine with 1-Amino-2-
(aminomethyl)-3-(4-nitrophenyl)propane.
3 l-Amino-2-(aminomethyl)-3-(4-nitrophenyl)-
propane (400 mg, 1.91 mmole, >95% pure) was dissolved
in 10.5 ml of absolute ethanol under nitrogen. Mesyl
aziridine (950 mg, 7.85 mmole) was added to the stirred
diamine solution as a solid. The reaction was stirred
at 25C for 14 hours using a magnetic stirrer and during
35,444-F"B" -38- _~
_39_ ~3~
this period a white, gummy re~idue formed on the sides
of the flask. The ethanol was decanted and the residue
was triturated with another 15 ml portion of ethanol to
remove any unreacted aziridine. The gummy product was
vacuum dried (101mm~ 25C) to afford the tetrakis methyl
sulfonamide ~1.0 g, 1044 mmole) in 75% yield (Rf = 0.74
- NH40H/ethanol - 20/80). The structure was confirmed
by 1H and 13C nuclear magnetic resonance (NMR)
spectro~copy.
Example F: Preparation of 2-(4-Nitrobenzyl)-1,3-(bis-
N,N-2-aminoethyl)diaminopropane.
The crude methylsulfonamide (650 mg, 0.94
f 15 mmole) wa~ dissolved in 5 ml of nitrogen purged,
concentrated sulfuric acid (98%). This solution was
maintained under nitrogen and heated to 143-146C for 27
minutes with vigorous stirring. A slight darkening was
noted and the cooled solution was poured into a stirred
solution of ether (60 ml). The precipitated white salt
cake wa~ filtered and immediately dissolved in lO ml of
deionized water. The pH of the solution was adjusted
to pH=11 with 50~ NaOH under argon with cooling. The
resulting solution was mixed with 90 ml of ethanol and
the precipitated inorganic salts were filtered. The
solvent was removed from the crude amine under reduced
pressure and to the resulting light brown oil was added
190 ml of toluene under nitrogen. The mixture was
stirred vigorously and water wa~ removed through
azeotropic distillation (Dean-Stark trap) until the
remaining toluene acquired a light yellow color (30~40
ml remaining in pot). The toluene was cooled and
decanted from the dark, intractable residues and salt.
This solution wa stripped of solvent in vacuo and the
resulting light yellow oil was vacuum dried (0.2 mm,
35,444-F"B" -39- _
`~` 13l6~
64593-4103
35C) overnlght affordlng ~10 mg of the product ~60~) whlch was
characterlzed by MS lH and 13C NMR.
E~ample G: Preparatlon of a starburst polymer
(containing an aniline derivative) o~ one half
generation represented by the ~ollowing scheme:
~ 11 H2c=cHcocH3
--~ O ,>--CH2CH (CNHCH2CH2HN2 )
CH OH
Compound #1 3
2 ~ C~l2cH(cNHcH2cH2N(cH2cocH3)2)2
Compound #2
Methyl acrylate ~2.09 g, 24 mmole) was dissolved in
methanol (15 ml). The compound 6-(4-aminobenzyl)-1,4,8,11-
tetraaza-5,7-dioxoundecane (1.1 g, 3.8 mmole) (i.e., Compound #1,
preparatlon descrlbed ln Example D) was dlssolved ln methanol (10
ml) and was added slowly over 2 hours wlth rlgorous stlrrlng to
the methyl acrylate solutlon. The reactlon mlxture was stlrred
for 48 hours at ambient temperatures. The solvent was removed on
the rotary evaporator malntalnlng the temperature below 40C. The
ester (Compound #2) was obtalned as a yellow oll (2.6 g). No
carboxyethylatlon of the anlllne functlon was observed.
,
-41- ~31~2~
Example H: Preparation of a starburst polymer
(containing an aniline moiety) of one generation;
represented by the following scheme:
Compound #2 + H2NCH2CH2NH2
CH30H
O O
H2N ~ -cH2cH(cNHcH2cH2N(cH2cH2cNHcH2cH2NH2)2)2
Compound #3
.
The ester (Compound #2) (2.6 g, 3.7 mmole) was
dissolved in methanol (100 mlS. This was carefully
added to a vigorously stirring solution of ethylene
diamine (250 g, 4.18 mole) and methanol (100 ml) at
such a rate that the temperature did not rise above
40C. After complete addition the reaction mixture was
stirred for 28 hours at 35-40C ~heating mantle). After
28 hours no ester groups were detectable by infrared
specroscopy. The solvent was removed on the rot;ary
evaporator at 60C. The excess ethylene diamine was
removed using a ternary azeotrope of toluene-methanol-
ethylene diamine. Finally all remaining toluene was
azeotroped with methanol, Removal of all the methanol
3 yielded 3.01 g of the product (Compound #3) as an
orange glassy solid.
35,444-F"B" -41-
- -~2- ~ 3~6~2~
Example I: Preparation of a ~tarburst polymer
(containing an aniline moiety) of one and one half
generations represented by the following scheme:
O
Compound #3 + ~2C=CHCOCH3
CH30H
O O
H2N~ CH2CH ( C'NHC~2C~2N ( Cl~2cl~2cN~lcH2c~l2N ( C}~2C~i2CoC113 ) 2 ) 2 ) 2
Compound #4
The amine (Compound #3) (2.7 g, 3.6 mmole) wa3
dissolved in methanol (7 ml) and was added slowly over
one hour to a stirred solution of methyl acrylate t3.8
g, 44 mmole) in methanol (15 ml) at ambient
temperatures. A slight warming of the solution was
observed during the addition. The solution was allowed
to stir at ambient temperatures for 16 hours. The
solvent was removed on the rotary evaporator at 40C.
After removal of all the solvent and exces methyl
acrylate the ester (Compound #4) was obtained in 4.7 g
yield as an orange oil.
35,444-F"B" -42-
~ _43_ ~3~
Example J: Preparation of a starburst polymer
(contain.ing an aniline moiety) of one half generation -
represented by the following scheme:
,.
H2~ ~ -cH2cH(cH2NH2)2 + ~2C-CHCOCH3
CH30H
Compound #5
H2N- ~ -CH2CH(CH2N(CH2cH2cOcH3)2)2
Compound #6
The triamine (Compound #5, the preparation of
this oompound is shown in Example C) (0.42 g, 2~3
mmole) was dissolved in methanol (10 ml) and was added
dropwise over one hour to methyl acrylate (1.98 g, 23
mmole) in methanol (10 ml). The mixture was allowed to
stir at ambient temperatures for 48 hours. The solvent
was removed on the rotary evaporator, maintaining the
temperature at.no higher than 40C. The exces~ methyl
acrylate waq removed by repeated azeotroping wif,h
methanol. The ester (Compound #6) was isolated as an
orange oil (1.24 g).
35,444-F"B" -43-
~ 3 ~
-~4
Example K: Preparation of a star~urst polymer
(containing an aniline moiety) o~ one generation;
represented by the following scheme:
Compound ~,~6 + H2NCH2CH2NH2
CH30H
H2N ~ CH2cH(cH2N(cH2cH2cNHcH2cH2NH2)2)2
Compound #7
r` 15 The ester (Compound #6) (1.24 g, 2.3 mmole) was
dissolved in methanol (50 ml) and was added dropwise
over two hours to ethylenediamine (73.4 g9 1~22 mole)
in methanol (100 ml). A small exotherm was noted,
vigorous stirring was maintained. The solution was
left to stir at ambient temperatures for 72 hours. The
solvent was removed on the rotary evaporator at 60C.
The excess ethylene diamine was removed using a ternary
azeotrope of toluene-methanol-ethylenediamine. Finally
all remai~ing toluene was removed with methanol and
then pumping down with a vacuum pump for 48 hours gave
the amine (Compound #7) (1.86 g) as a yellowtorange
oil.
3o
35,444-F"B" -44- _
~45- ~3~6~2~
Example L: Preparation of a starburst polymer
(containing an aniline moiety) of one and one half
generations; represent by the following scheme:
Compound ~7 + H2C-CHCOCH3
CH30H
.
O O
H2N- ~ -cH2cH(cH2N(cH2cH2cNHcH2cH2N(cH2cH2cocH3)2)2)~
Compound #8
, 15 The amine (Compound #7) (1.45 g, trace of
methanol remained) waq dissolved in methanol (100 ml)
and was added slowly over 1~ hours to a stirred
solution
of methyl acrylate (5.80 g) in methanol (20 ml). The
solution was allowed to stir for 24 hours at room
temperature. Removal of the solvent followed by
repeated azeotroping with methanol enabled the removal
of all the exces~ methyl acrylate. After pumping down
on a vacuum pump for 48 hours the ester (Compound #8)
was isolated as an orange oil (2.50 g, 1.8 mmole).
Example M: Hydrolysis of 4.5 generation dendrimer and
preparation of calcium salt.
3 4.5 Generation PAMAM (ester terminated,
initiated off NH3) (2.11 g, 10.92 meq) was dissolved in
25 ml o~ methanol and to it was added 10% NaOH t4.37
ml, 10.92 meq) (pH - 11.5-12). After 24 hours at room
temperature, the pH was about 9.5. After an additional
35,444-F"B" -45-
, .'
-46- ~3~2~
20 hours, the solution was rotovaped, 50 ml of toluene
added, and rotovaped again.
The resulting oil was dissolved in 25 ml of
methanol and precipitated as a white gum upon addition
of 75 ml of diethyl ether. The liquid was decanted,
and the gum was rotovaped to give a very fine off-white
powder which upon drying gives 2.16 g of product (98%
yield). No ester groups were found upon NMR and
infrared analysis.
The sodium salt of 4.5 Generation PAMAM (ester
terminated, initiated from NH3) was replaced by for the
t- 15 calcium via dialysis. The sodium salt (1.03 g) was
dissolved in 100 ml of water and passed through hollow
fiber dialysis tubing (cut off - 5000) at 3 ml/minute.
The exterior of the tubing was bathed in 5~ CaC12
solution. This procedure was then repeated.
The resulting solution was again dialyzed, this
time against water, then repeated two additional times.
Evaporation provided 0.6 g o~ wet solid, which
was taken up in methanol (not totally soluble) and is
dried to give 0.4-5 g of off-white crystals.
c369H592Q14lN91ca24 Calc. 10.10% Ca+~
M Wt. = 9526.3 Calc. - C-4432.1, H-601.8, 0-2255.9,
N-1274.6, Ca-961.9)
Theo: C-46.5, H-6.32, N-13~38, Ca-10.10
Found: C-47.34, H-7.00, N-13.55, Ca-8.83
35,444-F"B" -46-
_47_ ~ 2~
Example N: Preparation of dendrimers with terminal
carboxylate groups.
Half-generation starburst polyamidoamines were
hydrolyzed to convert their terminal methyl ester
groups to carboxylates. This generated spheroidal
molecules with negative charges dispersed on the
periphery. The dendrimers hydrolyzed ranged Prom 0.5
generation (three carboxylates) to 6.5 generation (192
carboxylates).
The products could be generated as Na+, K+, Cs+
or Rb+ salts.
Example 0: N-t-butoxycarbonyl-4-aminobenzyl malonate
dimethylester
4-Aminobenzyl malonate dimethylester (11.62 g,
49 mmol) was dissolved in 50 ml of 5-butanol:water
60:40 with stirring. Di-t-butoxydicarbonate (19.79g,
90 mmol) was added and the reaction mixture stirred
overnight. The butanol waq removed on the rotary
evaporator, resulting in a yellow suspension of the
product in water. ~xtraction into methylene chloride,
drying (MgS04~ and evaporation gave a yellow oil (21.05
g, contaminated by di-t-butoxydicarbonate).
recrystallization Prom 2-propanol:water (75:25) yield
pale yellow crystals (11.1 g, 33 mmol, 67%). The
structure was confirmed by 13c NM~ and purity checked
by hplc analysis (spherisorb ODS-1, 0.05M H3P04 pH 3:
CH3CN 55:45). The material was used without Purther
purification.
35,444-F"B" -47-
-48~
Example P N-t-butoxycarbonyl-6~(~-aminobenzyl)-
1,4,8,11-tetraa~a-5,7-dioxoundecane
N-t-butoxycarbonyl-4-aminobenzyl malonate
dimethylester ~8.82 g 26 mmol), prepared in Exarnple 0,
was dissolved in 50 ml of methanol, This solution was
added dropwise (2 hours) to a solution of freshly
distilled ethylenediamine (188 g 3.13 mole) and 20 ml
of methanol, under a nitrogen atmosphere. The solution
was allowed to stir for 24 hours. The ethylene
diamine/methanol solution was removed on the rotary
evaporator. The product was dissolved in methanol and
toluene added. Solvent removal on the rotary
evaporator gave the crude product as a white solid
- 15 (10.70 g contaminated with ethylenediamine). The
sample wa3 divided into two samples for purification.
Azeotropic removal of ethylenediamine with toluene,
using a soxhlet extractor with sulphonated ion exchange
beads in the thimble to trap the ethylenediamine,
resulted in partial decomposition of the product,
giving a brown oil. The remaining product was isolated
as a white solid from the toluene on cooling (2.3 g
approximately 50 percent). Analysis of a 10 percent
solution in methanol by gas chromatography (Column,
Tenax 60/80) showed no ethylenediamne detectable in the
sample (<0~1 percent). The second fraction was
dis~olved in methanol to give a 10 percent solution (by
weight) and purified from the ethylenediamine by
reverse osmosis, using methanol as the solvent. (The
membrane used was a Filmtec~ FT-30 , in an Amicon TClR
thin channel separator, the ethylenediamine crossing
the membrane.) The product waq isolated as a white
~olid (2.7 g), in which no detectable amounts of
ethylenediamine could be found by gas chromatography.
The 13C NMR data and hplc analysis (Spheri30rb ODS-1,
35,444-F"B" -48-
-49- ~31~2~
0.05M H3P04 pH 3:CH3CN 55:45) were consistent with the
proposed structure. The product was used with no
further puri~ication.
Example Q: Preparation of a starburst dendrimer of one
half generation from N~t-butoxycarbonyl-~-(4-
aminobenzyl)-1,4,8,11-tetraaza 5,7-dioxoundecane
N-t-butoxycarbonyl-6-(4-aminobenzyl)-1,4,8,11-
tetraaza-5,7-dioxoundecane (5.0 g 13 mmol), prepared in
Example P, was dissolved in 100 ml of methanol. Methyl
acrylate (6.12 g, 68 mmol) was added and the solution
stirred at ambient temperatures for 72 hours. The
reaction wa~ monitored by HPLC (Spherisorb ODSl,
Acetonitrile: 0.04M Ammonium acetate 40:60) to optimize
conversion to the desired product. The solution was
concentrated to 30 percent solids, and methyl acrylate
(3.0 g 32 mmol) was added. The reaction mixture was
stirred at ambient temperatures until no partially
alkylated products were detectable by HPLC (24 hours).
Removal of the solvent at 30C by rotary evaporation,
and pumping down at 1 mm Hg for Z4 hours gave the
product as yellow viscous oil, yield 7.81 g. The 13C
NMR data was consistent with the proposed structure.
The product was used without further purification.
Example R: Preparation of a starburst dendrimer of one
full generation from N-t-butoxycarbonyl-6-(4-
aminobenzyl)-1,4,8,11-tetraaza-5,7-dioxoundecane
3o
The half generation product (Example Q) (7.70
g, 10.45 mmol) was dissolved in 75 ml of methanol and
added dropwise over 2 hours to a 3tirred solution of
ethylenediamina (400 ml, 7.41 mol) and methanol (50
ml). The reaction mixture was stirred at ambient
temperatures for 48 hours. The ethylenediamine and
35,444-F"B" -49- _~
.. .
-50- ~ 3~ ~2~
methanol were removed by rotary evaporation to give a
yellow oil (11.8 g contaminated with ethylene diamine).
The product was dissolved in 90 ml of methanol, and
purified from the ethylenediamine by reverse osmosis
(Filmtec FT-30 membrane and Amicon TC1R thin channel
separator, methanol as solvent). After 48 hours, no
ethylenediamine could be detected by gas chromatography
(Column, Tena~ 60/80). Removal of the solvent on the
rotary evaporator, followed by pumping down on a vacuum
0 line for 24 hours gave the product as a yellow glassy
solid (6.72 g). Analysis by HPLC, PL~P-S column,
acetonitrile:O.015M NaOH, 10-20 percent gradient in 20
min.) and 13C NMR analysis was consistent with the
proposed structure.
Example S: Preparation of a starburst polymer of one
and one half generation from N-t-butoxycarbonyl-6-(4-
aminobenzyl)-1,4,8,11-tetraaza-5,7-dioxoundecane
The one generation product (Example R) (2.14 g,
25 mmol) was dissolved in 12.5 ml of methanol, and
methyl acrylate (3.5 g~ 39 mmol) in 5 ml of methanol
was added. The solution was stirred at ambient
temperatures for 48 hours, monitoring the progress of
the reaction by HPLC (Spherisorb ODS-1, acetonitrile:
0.04M ammonium acetate, 60:40). A second aliquot of
methyl aorylate was added (3.5 g 39 mmol) and the
reaction mixture stirred at ambient temperatures ~or a
further 72 hour~. Removal of the solvent on the rotary
evaporator gave the product as a yellow oil (3.9 g)
after pumping down overnight with a vacuum pump. The
product was used with no further purification.
i
35,444-F"B" -50- ~
-51-
Example T: Preparation of a starburst polymer of two
full generations from N-t-butoxycarbonyl-6-(4-
aminobenzyl)-1,4,8,11-tetraaza-5,7 dioxoundecane
The one and one half generation product
(Example S) (3.9 g, 2.5 mmol) was dissolved in 50 ml of
methanol, and was added dropwise over 2 hours to a
stirred solution of ethylenediamine (600 g, lO mol) and
methanol (50 ml). The solution was stirred at ambient
temperatures under an atmosphere of nitrogen for 96
hours. The ethylenediamine/methanol was removed on the
rotary evaporator to give a yellow glassy solid (4.4 g
contaminated with ethylenediamine). A 10 percent
solution of the product was made in methanol, and
purified from the ethylene diamine by reverse osmosis
f 15 (membrane used as a Filmtec FT-30, in an Amicon TClR
thin channel separator) until no ethylenediamine could
be detected by gas chromatography (Column, Tenax 60/80.
Removal of the solvent gave the product a~ a yellow
glassy solid (3.52 g). The 13C NMR data and HPLC
analysis (PLRP-S column, acetonitrile:O.015 M NaOH, 10
to 20 percent gradient in 20 minutes, were consistent
with the proposed structure.
Example U: Reaction of the two generation starburst
with Bromoacetic Acid to give a methylene carboxylate
terminated starburst dendrimer
The second generation product (Example T) (0.22
g, 0.13 mmol) was dissolved in 15 ml of.deionized water
3 and the temperature equilibrated at 40.5C. Bromoacetic
acid (0.48 g, 3.5 mmol) and lithium hydroxide (0.13 g,
3.3 mmol) were dissolved in 5 ml of deionized water,
and added to the reaction mixture. The reaction pH was
carefully maintained at 9, with the use of a pH stat
(titrating with O.lN NaOH), at 40.5C overnight.
35,444-F"B"' -51-
-52- ~3~
Monitoring by reverse phase HPLC, (~pherisorb ODS-1
column, eluent 0.25 M H3P04 pH 3 [NaOH]; acetonitrile
85:15) con~irmed the s~-nthesis of predominantly a
single component.
Example V: Preparation of Isothiocyanato functionalized
second generation methylene-carboxylate terminated
starburst dendrimer
Five ml of a 2.8 mM solution of the second
generation methylenecarboxylate terminated starburst
dendrimer (Example U) was diluted with 20 ml water and
the pH adjusted to 0.5 with concentrated hydrochloric
acid. After one hour at room temperature the mixture
, 15 wa9 analyzed by HPLC to verify the removal of the
butoxycarbonyl group and then treated with 50 percent
sodium hydroxide to ~ ~g the pH to 7. A pH stat
(titrating with 0.1 N NaOH) was used to maintain the pH
at 7 and 225 ~l thiophGsgene was added. After 15
minutes at room temperature the pH of the mixture was
adjusted to 5 with 1N HCl. The mixture washed with
chloroform (20 ml x 2) then concentrated on a rotary
evaporator at reduced pressure. The residue recovered
0.91 g is a mixture of the isothiocyanate and salts.
Example W: Preparation o~ second generation starburst
polyethyleneimine-methane sulfonamide
To a solution of 125 g N-methanesulfonyl-
aziridine in 50 ml ethanol was added 25.0 g tris(2-
aminoethyl)amine. The solution was stirred at room
temperature ~or 4 days. Water was added to the
reaction mixture as needed to maintain the homo~eneity
of the solution. The solvent was removed by
distillation in vacuo to give the 2nd generation
35,444-F"B" -52- _
-53- ~3~
starburst PEI-methane sulfonamide as a yellow glass
(161 g).
Example X: Cleavage of methane sulfonamides to form
second generat'on starburst polyethyleneimine
A solution of 5.0 g of second generation
starburst PEI-methane sulfonamide, from Example W in 20
ml of 38 percent HCL was sealed in a glass ampoule.
The ampoule was heated at 160C for 16 hours, then
cooled in an ice bath and opened. The solvent was
removed by distillation in vacuo and the residue
dissolved in water. After adjusting the pH of the
solution to greater than or equal to 10 with 50 percent
, 15 NaOH, the solvent was removed by distillation in vacuo.
Toluene (150 ml) was added to the residue and the
mixture heated at reflux under a Dean-Stark trap until
no more water could be removed. The solution was
filtered to remove salts and the filtrate concentrated
in vacuo to give 1.9 g second generation starburst PEI
as a yellow oil.
Example Y: Preparation of third generation starburst
polyethyleneimine-methane sulfonamide
To a solution of 10.1 g second generation
starburst PEI, from Example X, in 100 ml ethanol was
added 36.6 g N-methanesulfonylaziridine. The solution
was stirred at room temperature for 1 week. ~ater was
3 added as needed to maintain the homogeneity of the
solution. The solvent was removed by distillation in
vacuo to give third generation ~tarbur~qt PEI-methane
sulfonamide as a yellow glass (45.3 g).
35~444-F"B" -53- __
-5LI ~3~$2~
Example Z: Cleavage of methane sulfonamides to form 3rd
gen starburst polyethyleneimine
: The methane sulfonamide groups of third
generation starburst PEI-methane sulfonamide (~.0 g),
from Example Y, were removed by the same procedure as
described for the second generation material in Example
X to give 2.3 g third generation starburst PEI as a
yellow oil.
Example AA: Preparation of a methylenecarboxylate-
terminated second generation starburst polyamidoamine
(initiated from ammonia)
r 15 The second generation starburst polyamidoamine
(2.71 g, 2.6 mmmol) and bromoacetic acid (4.39 g, 31.6
mmol) were dissoLved in 30 ml of deionized water and
the pH adjusted to 9.7 with 5N NaOH using a pH stat.
The reaction was maintained at this pH for a half hour,
and the temperature was slowly raised to 60C and was
maintained at 60C for three hours at constant pH. The
pH was raised to 10.3, and the reaction mixture
remained under control of the pH stat at ambient
temperatures overnight. The reaction mixture was
refluxed for a further four hours prior to work up.
Removal of the solvent, and azeotropin~ the final
traces of water with methanol gave the product as a
pale yellow powder (8.7 g, contaminated with sodium
bromide). The 13C NMR spectrum wa~ consistent with the
proposed ~tructure (with some contamination due to a
small amount of defected material as a result of some
monoalkylation).
35,444-F"B" -54-
. '
.
:, . ..... - - ,
. .
-~55-
Example BB: Preparation o~ a methylenecarboxylate
terminated second generation starburst
polyethyleneimine (initiated from ammonia)
The second generation starburst
5 polyethyleneimine (2.73 g, 6.7 mmol), from Example AA,
and bromoacetic acid (11.29g, 81 mmol) were dissolved
in 30 ml of deionized water. The pH was slowly raised
to pH 9.5 maintaining the temperature below 30C. The
temperature was raised slowly to 55C, and the reaction
10 pH maintained at 9.5 for 6 hours with the aid of a pH
stat (titrating with 5N NaOH). The pH was raised to
10.2, and maintained at that pH overnight. Removal of
the solvent on the rotary evaporator, and azeotroping
f the final traces of water using methanol, gave the
5 product as a yellow powder (17.9 g, contaminated with
sodium bromide). The 13C NMR spectrum was consistent
with the proposed structure (with some COntamin~ltiOn
due to a small amount of defected material as a result
20 of some monoalkylation).
Example CC: Preparation of a 3.5, 4O5~ 5.5 and 6.5
generation starburst PAMAM
~tef~arJol/c
2 To a 10 wt%~solution of 2.46 g 3 generation
5 PAMAM starburst was added 2.32 g of methyl acrylate.
This mixture was allowed to sit at room temeprature for
64 hr. After solvent and excess methyl acrylate
removal, 4.82 g of product was recovered (105~ of
30 theoretical).
Preparation of higher 1/2 generation starburst PAMAM'S:
' , .
Generations 4.5, 5.5 and 6.5 were prepared as
described above with no significant
35,444-F"B" -55- _
-
-56- ~316~
differences in reactant concentrations, reactant ratios
or reaction times.
Example DD: Preparation of a ~, 5 and 6 generation
starburst PAMAM:
To 2000 g of predistilled ethylenediamine was
added 5.4 g of 4 1/2 generation starburst PAMAM as a 15
wt% solution in methanol. This was allowed to sit at
room temperature for 48 hrs. The methanol and most of
the excess ethylenediamine were removed by rotary
evaporation under water aspirator vacuum at temperature
less than 60C. The total wt o~ product recovered was
ô.07 g. Gas chromatography indicated that the product
still contained 34 wt% ethylenediamine at this point.
A 5.94 g portion of this product was dissolved in 100
ml methanol and ultrafiltered to remove the residual
ethylenediamine. The filtration was run using an Amicon
TClR thin channel recirculating separator equipped with
an Amicon YM2 membrane. An in-line pressure relief
valve was used to maintain 55 psig (380 kPa) pressure
across the membrane. The 100 ml was first concentrated
to 15 ml by forcing solvent flow exclusively through
the membrane. After this initial concentration, the
flow was converted to a constant volume retentate
recycle mode for 18 hrs. After this time~ 60 ml of
methanol was passed over the membrane to recover
product still in the module and associated tubing. The
product was stripped of solvent and 2.53 g of 5
generation starburst PAMAM was recovered. Analysis by
gas chromatography indicated 0.3% residual
ethylenediamine remained in the product.
35,444-F"B" -56- __
. . .
.
~57~ 13~2~
Preparation of generation 4 and 6 proceeded as
above with the only difference being the weight ratio
of ethylenediamine to starting material. To prepare
4th generation this ratio was 200:1 and for 6th
generation this ratio wa~ 730:1.
~xample 1: Preparation of a product containing more
than one rhodium atom per starburst polymer.
2.5 Gen PAMAM (ester termlnated, initiated off
NH3) (0.18 g, 0.087 mmole) and RhC13~3H20 (0.09 g, 0.3
mmole) were mixed in dimethylformamide (DMF) (15 ml)
and heated for 4 hours at 70C. The solution turned
crimson and almost all of the rhodium was taken up.
f 15 The unreacted rhodium was removed by filtration and the
solvent removed on the rotary evaporator. The oil
formed was chloroform soluble. This was washed with
water and dried (MgS04) before removal of solvent to
~ield a red oil (0.18 g). The NMR spectrum was
recorded in CDC13 only minor differences were noted
between the chelated and unchelated starburst.
Dilution o~ some of this CDC13 solution with ethanol
followed by NaBH4 addition resulted in rhodium
precipitation. RhC13~3H~0 is insoluble in chloroform
and in chloroform starburst solution thus confirming
chelation.
Example 2: Preparation of a product containing
chelated with a starburst polymer
3o
3.5 Generation PAMAM (ester terminated,
initiated o~f NH3) (1.1 g, 0.24 mmole) was dissolved
with stirring into acetonitrile (50 ml)O Palladium
chloride (0.24 g, 1.4 mmole) was added and the solution
was heated at 70-75C (water bath) overnight. All the
PdC12 was taken up into the starburst. Removal of the
35,444-F"B" -57- _
,
- _5~_ ~3~
solvent and recording the NMR in CDC13 confirmed that
chelation had occurred. Dilution of the CDCl~ solution
with ethanol and addition of NaBH~ resulted in
precipitation of the palladium. The chelated product
(1.23 g) was isolated as a brown oil.
Example 3: Preparation of a product containing
~ffi~seee~ with a starburst polymer
~J~ r~ScL~
~ A sample of 5-carboxyfluorescein (0.996 g) and
starburst polyethyleneimine (Gen-2.0; amine terminated,
initiated off NH3) (0.202 g) were mixed in 10 ml of
methylene chloride and 5 ml of methanol and allowed to
reflux for 10 minutes. Upon filtering, an insoluble
red powder (0.37 g) was obtained (mostly unreacted 5-
oarboxy fluorescein). From the filterate was isolated0.4 g o~ a brilliant-red solid which exhibited a
softening point of 98-103C and foamed to a brilliant
red melt at 175-180C; NMR spectra (D20~hof thi~ product
were consistent with dendrimer having ~ ~s~ bound
to the surface
Example 4: Preparation of a product containing
fluoro~oei-n with a starburst polymer
ol~ ~s~e/~7
In a procedure similar to that described in
Example 3, starburst polyethyleneimine (Gen-2.0; amine
terminated, initiated off NH3) was reacted with
fluorescein isothiocyanate to give a brilliant-red
iridescent solid which was suitable for use as a
fluorescent labelling reagent.
35,444-F"B" -58-
.
~ 3 ~
-59-
Example 5: Hydrolysi~ of 4.5 generation dendrimers and
preparation of calcium salt.
4.5 Generation PAMAM (ester terminated,
initiated off NH3~ (2.11 g, 10.92 meq) was dissolved in
25 ml of methanol and to it was added 10% NaOH (4.37
ml, 10.92 meq) (pH = 11.5-12). After 24 hours at room
temperature, the pH was about 9.5. A~ter an additional
20 hours, the solution was rotovaped, 50 ml of toluene
added, and rotovaped again.
The resulting oil was dissolved in 25 ml of
methanol and precipitated as a white gum upon addition
of 75 ml of diethyl ether. The liquid was decanted
off, and the gum was rotovaped extensively to give a
very fine off-white powder which upon further drying
gives 2.16 g of product (98% yield). No ester ~sroups
were found upon NM~ and infrared analysis.
The sodium salt of 4.5 Generation PAMAM (ester
terminated, initiated from NH3) was exchanged for the
calcium salt via dialysis. The sodium salt (1.03 g)
was dissolved in 100 ml of water and passed through
hollow fiber dialysis tubing (cut off = 5000) at 3
ml~minute. The exterior of the tubing was bathed in 5%
CaCl2 solution. This procedure was then repeated.
The resulting solution wa~ again dialyzed, this
time against water, then repeated two additional times.
3 Evaporation provided 0.6 g of wet solid, which
was taken up in methanol (not totally soluble) and is
dried to give 0.45 g of off~white crystals.
.
c369H592ol41N9lca24 Calc. - 10.10% Ca++
35,444-F"B" -59- _~
,
-60-
M Wt. - 9526.3 Calc. - C-4432.1, H-601.~, 0-2255.9,
N-1274.6, Ca-961.9)
Theo: C-46.5, H~6.32, N-13.38, Ca-10.l0
Found: C-47.34~ H-7.00, N-13.55, Ca~8.83
Example 6: Preparation of dendrimers with terminal
carboxylate groups.
Hal~-generation starburst polyamidoamines were
hydrolyzed to convert their terminal methyl ester
groups to carboxylates. Thi3 generated spheroidal
molecules with negative charges dispersed on the
periphery. The dendrimers hydrolyzed ranged ~rom 0.5
generation (three carboxylateq) to 6.5 generation (192
carboxylates).
The products could be generated as Na+, K+, Cs+
or Rb~ salts.
Example 7- Encapsulation of R(+) - Limonene in
Polyamidoamine Starburst Dendrimers
A 5-50 weight percent solids solution in
methanol of ~tarburst - PAM~M dendrimer (M.W. about
175,000; generation - 9.0) is added dropwise to (R(+)
limonene in methanol until saturated. The solu~ion is
stirred at room temperature (about 25C) for several
hours and then devolatized on a Buchi rotovap at room
temperature to give a solid product. Warming at
temperatures greater than 80C gives solvent insoluble
products which retain substantial amounts of (R~)-
limonene in an encapsulated form. Theqe product3 are
excellent prototypes for slow release of (R~)-limonene
as a fragrance and deodorizer product.
35,444-F"B" -60-
~3~24
64693-4103
Example ~: Encapsulation of Heavy Metal Salts in
Polyamindoamlne Starburst Dendrimers
A 5-50 weight percent solids solutlon in water of
starburst PAMAM dendrimer (M.W. about 350,000; gen~eration = 10.0)
ls stirred as a saturated solutlon of lead acetate ~Pb(C2~302)2]
is added dropwise. The solution ls stirred at room temperature
(about 25C) for several hours and then ~evolatilized on a B~chi
rotorap to give solld products. Scanning transmls~ion
electronmicrograph of these products showed that these heavy m0tal
l~ salts are encapsulated ln the lnterlor of the dendrlmers. These
fllms contalnlng heavy metal salts are useful as s~lelds for
absorblng electromagnetlc radlatlon.
ExamPle 9: Encapsulatlon of Fluoresceln (water
soluble) Dye ln Polyamldoamlne Starburst Dendrlmers
A 50-50 welght percent sollds solutlon (H20/CH30H) of
starburst-PAMAM dendrlmer (M.W. about 175,000; generation = 9.0)
is stirred as fluoresceln, dlsodlum salt (Acld Yellow 73, Cl.
45350; Uranine; ; avallable from Aldrlch Chemical Co. (Milwaukee,
WI) is added untll saturated. The solutlon ls stlrred at room
temperature (about 25C) for several hours and then devolatlllzed
at room temperature to give a colored solld product. These dye
encapsulated dendrlmers are excellent reference probes for
calibrating ultrafiltration membranes.
ExamPle lO: Preparation of dendrlmers wlth termlnal
fluorescent groups
A. Reactlon of Amine Terminated Dendrlmer wlth N-
Dansyl A~lrldlne
Trade-mark 61
~f~
~3~ 2l~
64693-4103
A sample (1.5 g, 1.6 x 10-3 mole) of starburst
polyethylenelmlne (LPEI), G = 3.0, terminal groups (Z) = 12, M.W.
= 920) ls dissolved ln 20 ml of methanol. The solutlon was
stlrred and 0.884 g (3.84 ~ 10-2 mole) of a solutlon of N-dansyl
azirldine (ICN Biomedlcals, Costa Mesa, CA) is added dropwise over
a period of 20 mlnu~es. The reactlon mlxture ls allowed to stlr
at room temperature overnlght. Removal of solvent under vacuum
glves a solld product. NMR and lnfrared analysls lndlcates that
the product ls covalently bonded dansyl groups ln the surface of
the dendrlmer.
B. Reactlon of Amlne Termlnated Dendrlmers wlth Dansyl
Chlorlde
A solutlon of starburst pol~amldoamine (?..0 g, 1.9 x
10-4 mole) (lnitiated from ammonla, G = 4.0, termlnal groups (Z) =
24, M.W. = 5,147) in 30 ml of water ls stlrred ln a 3-neck flask
wlth 80 ml of toluene whlle a solution of dansyl chlorlde (1.23 g,
4.5 x 10-3 mole) (5-dlmethyl-amino-1-naphthalenesulfonyl chloride,
from Aldrlch Chemlcal Co., Mllwaukee WI) ~n 40 ml of toluene is
added dropwise whlle coollng wlth lce. Concurrently, a solutlon
of 10~ NaOH (13.3 mole, 10% excess) is added to the reactlon
mixture to glve an olly ball. The product ls washed wlth water,
dlssolved ln methanol, and preclpltated wlth dlethyl ether to glve
a solld product. NMR and infrared analysls ls consistent with
covalently bonded dansyl groups in the dendrimer surface.
62
-63- 13~6~2ll
Example 11: Demonstration of multiple chelation of
iron by a sodium propionate tsrminated sixth generation
starburst polyamidoamine.
The sodium propionate terminated sixth
generation polyamidoamine (initiated from ammonia)
(97.1 mg, 2.45 mol.) was dissolved in 1.5 ml of
deionized water. Addition of 0.5 ml of 0.5N HC1
reduced the pH to 6.3. Ferric chloride was added (0.5
ml of 0.1.2M solution, 0.051 mmol) producing a light
brown gelatinous precipitate. On heating at 60C for
0.5 hours, the gelatinous precipitate became soluble,
resulting in a homogeneous orange solution. The
solution wa~ filtered through Biogel P2 acrylamide gel
tlO g, twice) isolating the orange band (free of halide
contamination). Removal of the solvent in vacuo gave
the product as an orange film (30 mg). Analysis was
consistent with chelation of approximately 20 moles of
ferric ions per mole of starburst dendrimer.
35,444-F"B" -63-
-64-
~31~2~
Table III
Theoretical
Found
Na4Fe20H 1 28SB NasFe20H 1 27SB Na6Fe20H 1 26sg
Na0.39,0.24 0.25 0.31 0.3
(0.31 0.1%)
Fe3.14,3.11 3.05 3.05 3.04
(3.12 0.02%)
C 47.11 49.87 49.84 49.81
H 7.33 7.31 7.30 7.29
N 14.81 14.49 14.48 14.47
o ~ 25.03 25.02 25.01
Mwt. 36632.23 36654.21 36375.18
SB = C1s21H2467N379573
These results confirm chelation of 20~2 moles of ferric
ions per mole of starburst dendrimer.
35,444-F"B" ~64- _~