Note: Descriptions are shown in the official language in which they were submitted.
132~848
The present invention relates to 5-substituted uridine
derivatives which are novel substances and 5'-trityl-5-
substituted uridine derivatives which are useful as
intermediates for preparing the S-substituted uridine
derivatives. The 5-substituted uridine derivatives of this
invention have an excellent anti-cancer effect and are useful
as anti-tumor agents.
5-Fluorouridine (hereinafter referred to as "FUR"),
which was synthesized in 1959, is known for its excellent
activity against malignant tumors (U.S. Patent No. 2885398).
~owever, FUR has a problem on clinical use because of its
high toxicity.
Many attempts have been made to resolve this problem by
converting FUR into various derivatives (Japanese Unexamined
Patent Publication Nos. 64280/1975; 52183/1976; 91997/1982;
246196/1986). However, such attempts failed to give useful
derivatives.
5-Trifluoromethyl-2'-deoxyuridine (hereinafter referred
to as "F3TdR") represented by the formula
-- 1 -- .
X
.
' ~ '' .
.. . ..
1326848
HN~ C F3
o
HO
~ .,.
OH
ha~ an anti-tumor activity [Cancer Research 24, 1979 (1964)~
and a strong antiviral activity ~Cancer Research 30, 1549,
l97a]. In view of these activities, various investigations
have been ~ade on the utility of F3TdR as pharmaceuticals,
but without developing a useful compound.
'
. ~, . .. .
132~848
3 --
According to the present invention, there are
provided:
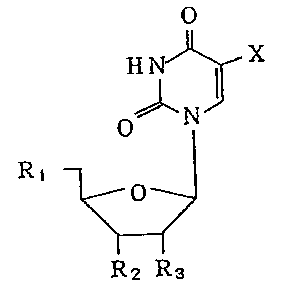
(1) a 5-substituted uridine derivative represented by the
formula
o
J~ X
H~
o N ( I )
R1 ~
y~ .
R2 R3
wherein X is fluorine atom or trifluoromethyl group, R
and R2 each represent (a) a group represented by the
formula -OSi-(R4)(R5)(R6) (wherein R4, R5 and R6 are the
same or different and each represent alkyl group having 1
to 10 carbon atoms, a group represented by the for~ula
-(CH2)nPh (wherein n is 0 to 2 and Ph is phenyl group) or
a group represented by the formula ~ R7)(R8)(0H)
(wherein R7 and R8 are the same or different and each
represent lower alkyl group)), (b) hydroxyl group, (c)
aminoacyloxy group in which the amino group may optionally
be substituted with lower alkyl group or (d)
carboxylalkylcarbonyloxy group, R3 is a group represented
by the formula -OSi-(R4)(R5)(R6) (wherein R4, R5 and R6
.
~ ' . "'
-, ,
,, - : : .
132~848
4 --
are as defined above), hydrogen atom, hydroxyl group,
aminoacyloxy group in which the amino group may optionally -
be substituted with lower alkyl group or
carboxylalkylcarbonyloxy group, provided that at least one
of Rl, R2 and R3 is a group represented by the formula
-OSi-(R4)(R5)(R6) (wherein R4, R5 and R6 are as defined
above) and that when X is fluorine atom, R3 is not
hydrogen, or a pharmaceutically acceptable salt thereof,
and
(2) a S-substituted-5'-trityluridine derivative
represented by the formula
O
R2' Ra
wherein X is fluorine atom or trifluoromethyl group, R2'
is hydroxyl group or a group represented by the formula
-OSi-(R4)(R5)(R6) (wherein R4, R5 and R6 are the same or
different and each represent alkyl group having 1 to 10
carbon atoms, a group represented by the formula -(CH2)nPh
~wherein n is O to 2 and Ph is phenyl group) or a group
. . .. . . . .
1326848
^osi -
represented by the formula -S~-(R7)(R8)(OHJ (wherein R7
and R8 are the same or different and each represent lower
~lkyl group)), R3' is hydrogen atom, hydroxyl group or a
'group represented by the formula -OSi-(R4)(R5)(R6)
(wherein R4, R5 and R6 are as defined above), provided
that at least one of R2' and R3' is a group represented by
the formula -OSi-(R4)(R5)(R6) (wherein R4, R5 and R6 are
as defined above) and that when X is fluorine atom, R3' is
not hydrogen atom.
According to the present invention, there i5
further provided an anti-tumor agent containing as an
effective component the compound of the formula (I) or a
pharmaceutically acceptable salt thereof.
According to the present invention, there is
further provided a method for treating tumors,
characterized by administering to a mammal an effective
amount of the compound of the formula (I) or a
pharmaceutically acceptable salt thereof.
The 5-substituted uridine derivatives of the
formula (I) according to this invention have a lower
toxicity and a more excellent anti-tumor effect than FUR
and F3TdR, hence useful as medicaments. The 5'-trityl-5-
substituted uridine derivatives of the formula ~II) are
useful as intermediates for preparing the compounds of the
formula (I).
. . .
, , :
~, .
1326848
-- 6 --
Examples of aminoacyloxy groups with the amino
group optionally substituted with lowe~ alkyl group which
are represented by Rl, R2 and R3 in the formula (I) are
acyloxy groups, particularly alkylcarbonyloxy groups,
having 2 to 6 carbon atoms, which may be substituted with
one or two amino groups wherein one or two hydrogen atoms
attached to the nitrogen atom may optionally be
substituted with lower alkyl group, particularly alkyl
group having 1 to 4 carbon atoms, such as methyl, ethyl,
propyl or the like. Examples of such acyloxy groups are
glycyloxy, N,N-dimethylglycyloxy, alanyloxy, -amino-
isobutyryloxy, -aminobutyryloxy, ~-N,N-dimethylamino-
butyryloxy, N,N-diethylalanyloxy, valyloxy, leucyloxy,
isoleucyloxy, ornithinyloxy, lysinyloxy, ~,3-
di~dimethylamino)-propionyloxy, etc. Examples of
carboxylalkylcarbonyloxy groups are those having 3 to 6
carbon atoms such as carboxylmethylcarbonyloxy, 2-
carboxylethylcarbonyloxy, 2-carboxylpropylcarbonyloxy, 3- .
carboxylpropylcarbonyloxy, 4-carboxylbutylcarbonyloxy,
etc.
Examples of alkyl groups having 1 to 10 carbon
atoms represented by R4, R5 and R6 are straight- or
branched-chain alkyl groups such as methyl, ethyl, propyl,
isopropyl, butyl, sec-butyl, tert-butyl, pentyl, neo-
pentyl, hexyl, 2,3-dimethyl-2-butyl, heptyl, octyl, nonyl,
.
,. . ~ ' , '. " ' . ' ~ , .
~''' ' ' ' .
`, ~ . " .' " .' ', :
- `
13268~8
7 --
decyl, etc. Examples of lower alkyl groups represented by
R7 and R8 are straight- or branched-chain alkyl groups
having 1 to 6 carbon atoms such as methyl, ethyl, propyl,
isopropyl, butyl, sec-butyl, tert-butyl, pentyl, neo-
pentyl, hexyl, etc.
Of the compounds of the formula ~I), preferred
compounds are those wherein X is fluorine atom, one or two
of Rl, R2 and R3 represent~s) a group of the formula -OSi-
(R4')~R5')(R6') (wherein R4', R5' and R6' are the same or
different and each represent alkyl group having 1 to 8
~carbon atoms, benzyl group, 2-pjhenylethyl group or a group
represented by the formula -6~-~R7)(R8)(0H) (wherein R7
and R8 are the same or different and each represent lower
alkyl)), the remaining one or two of Rl, R2 and R3
represent(s) hydroxyl group, aminoalkylcarbonyloxy group
with the amino group optionally substituted with lower
alkyl group or carboxylalkylcarbonyloxy group. Of these
preferred compounds, more preferred are the compounds
wherein Rl is the group represented by the formula -OSi-
(R4')(R5')(R6') (wherein R4', R5' and R6' are as defined
above), R2 and R3 are the same and each represent hydroxyl
group, aminoalkylcarbonyloxy group with the amino group
optionally substituted with lower alkyl group or
carboxylalkylcarbonyloxy group, and the compounds wherein
Rl is hydroxyl group, R2 and R3 are the same and each
13268~8
represent the group represented by the formula -OSi-
(R4')(R5')(R6') (wherein R4', R5' and R6' are as defined
above).
Also preferable are the compounds of the formula
(I) according to the invention wherein X is
trifluoromethyl group, R3 is hydrogen atom, one of Rl and
R2 is a group represented by the formula -OSi-
(R4')(R5')(R6') (wherein R4', R5' and R6' are the same or
different and each represent alkyl group having 1 to 8
carbon atoms, benzyl group, 2-phenylethyl group or a group
1 ;y represented by the formula -~-(R7)~R8)10H) ~wherein R7
and R8 are the same or different and each represent lower
alkyl group)), the other of Rl and R2 iq hydroxyl group,
aminoalkylcarbonyloxy group with the amino group
optionally substituted with lower alkyl group or
carboxyalkylcarbonyloxy group, or both of Rl and R2 are
the group represented by the formula -OSi-~R4')(R5')(R6')
~wherein R4', R5' and R6' are as defined above).
More preferred compounds of the formula (I) are
those described in items (i) and (ii) below:
(i) compounds of the formula (I) wherein X is fluorine
atom, one or two of Rl, R2 and R3 repreqent(s) tert-
butyldimethylsilyloxy group, dimethyloctylsilyloxy group
or benzyldimethylsilyloxy group, the remaining one or two
of Rl, R2 and R3 represent(s) hydroxyl group, glycyloxy
, ' , . : :
,
- 9 - 13268~8
group with the amino group optionally substituted with
lower alkyl group or carboxyethylcarbonyloxy group and
(ii) compounds of the formula (I) wherein X is
trifluoromethyl group, R3 is hydrogen atom, one of Rl and
R2 is tert-butyldimethylsilyloxy group or
benzyldimethylsilyloxy group, the other of Rl and R2 is
hydroxyl group, glycyloxy group with the amino group
optionally substituted with lower alkyl group or
carboxylethylcarbonyloxy, or both of Rl and R2 are tert-
butyldimethylsilyloxy group or benzyldimethylsilyloxy
group.
As the more preferred compounds of the type (i),
there may be mentioned the compound~ of the formula (I)
wherein X is fluorine atom, Rl is tert-butyldimethylsilyl-
oxy group, dimethyloctylsilyloxy group or benzyldimethyl-
silyloxy group, R2 and R3 are the same and each represent
hydroxyl group, glycyloxy group with the amino group
optionally substituted with lower alkyl group or carboxy-
ethylcarbonyloxy group; and compounds of the formula (I)
wherein Rl is hydroxyl group, and R2 and R3 are the same
and each represent tert-butyldimethylsilyloxy group,
dimethyloctylsilyloxy group or benzyldimethylsilyloxy
group.
Especially preferred examples of compounds of
the formula (I) are as follows: -
.
. ..
.
~ ~ .
- lo - 132~8~8
5'-0-tert-butyldimethylsilyl-5-fluorouridine,
2',3'-bis(0-tert-butyldimethylsilyl)-5-
fluorouridine,
5'-0-dimethyloctylsilyl-5-fluorouridine,
5'-0-benzyldimethylsilyl-5-fluorouridine,
5'-0-tert-butyldimethylsilyl-2',3'-bis(0-
dimethylglycyl)-5-fluorouridine,
5'-0-tert-butyldimethylsilyl-2'-deoxy-5-
trifluoromethyluridine,
5'-0-tert-butyldimethylsilyl-2',3'-bis(0-2-
carboxyethylcarbonyl)-5-fluorouridine.
Preferred examples of compounds of the formula
~II) which are the intermediates of the invention are as
follows:
2'-0-tert-butyldimethylsilyl-5'-0-
triphenylmethyl-5-fluorouridine,
3'-0-tert-butyldimethylsilyl-5'-0-
triphenylmethyl-5-fluorouridine,
2',3'-bis(0-tert-butyldimethylsilyl)-s'-o-
triphenylmethyl-5-fluorouridine,
2'-0-benzyldimethylsilyl-5'-0-triphenylmethyl-5-
fluorouridine,
3'-0-tert-butyldimethylsilyl-5'-0-
triphenylmethyl-2'-deoxy-5-trifluoromethyluridine.
Described below are processes for preparing the
' ~ . ,.
.~ .
32~848
compounds of the formula (I) according to invention. The
compound of the formula (I) can be prepared by any of the
following processes A, B and C.
Process A
The compound of the formula (I) according to the
invention can be prepared, as shown in a reaction scheme
below, by reacting the compound of the formula (III) with
the halogenosilyl compound of the formula (IV) in a
~olvent in the presence of a baqic catalyst.
. .
J~ X
~ R ,,
o N . I
HO I ~ .R5 --Si--X
lCoY R6
. ~f .
HO R~ .
(m) (IV)
O
. ' J~ X ' ' "'
H~
o N~
Rlo
~l~ \,
Rn R~2
(I' )
.~
.: ` :~ : .
,. ... ... .. ... .
1' . , ; . `." " ' ~ . .
` ..
. :
. , ....... ~:
- . . . .
- 12 - 1 32 6848
In the formulae, X, R4, R5 and R6 are as defined
above, Xl is halogen atom, Rg is hydrogen atom or hydroxyl
group, Rlo and Rll are hydroxyl group or a group
represented by the formula -OSi-(R4)(R5)(R6) (wherein R4,
R5 and R6 are as defined above), R12 is hydrogen atom,
hydroxyl group or a group represented by the formula -OSi-
(R4)~R5)(R6) (wherein R4, R5 and R6 are as defined above),
and at least one of Rlo, Rll and R12 is a group
represented by the formula -oSi-(R4)(R5)(R6) (wherein R4,
R5 and R6 are as defined above), with the proviso that
when X is fluorine atom, Rg and R12 are not hydrogen
atom. Specific examples of halogen atoms represented by
Xl are chlorine, bromine and iodine atoms.
Insofar as the solvent used does not adversely
affect the reaction, the solvent is not specifically
limited. A wide range of conventional solvents can be
used without specific limitation. Examples of useful
solvents are benzene, toluene, xylene and like aromatic
hydrocarbons, ether, tetrahydrofuran, dioxane and like
ethers, acetonitrile, pyridine, dimethylformamide,
dimethylsulfoxide and like aprotic solvents, etc. These
solvents are usable singly or at least two of them can be
used in mixture.
Suitable examples of the basic catalyst are
pyridine, dimethylaminopyridine, 2,6-lutidine, imidazole,
,
~ 13 ~
1326848
triethylamine and like organic bases, etc.
The amount of the basic catalyst to be used is
about 1 to about 10 moles, preferably about 1.5 to about 4
moles, per mole of the compound of the formula (III). The
amount of the halogenosilyl compound of the formula (IV)
to be used is about 0.5 to about 10 moles, preferably
about 0.8 to about 3 ~1 moles, per mole of the compound of
the formula (III).
The reaction temperature is 0 to about 80C,
preferably room temperature to about 50C. The reaction
time is variable depending on the kinds of the solvent and
the basic catalyst to be used, but is usually about 0.5 to
about 20 hours.
Process B
A 5'-trityl-5-substituted uridine derivative of
the following formula (II')
O
NJ~x
R,3 R,4
wherein X i9 fluorine atom or trifluoromethyl group, R13
, -
, ~
. .
13268~8
is hydroxyl group or a group represented by the formula-OSi-(R4)(R5)(R6), R14 is hydrogen atom, hydroxyl group or
a group represented by the formula -OSi-(R4)(R5)(R6), and
at least one of R13 and R14 is a group represented by the
formula -OSi-(R4)(R5)(R6), with the proviso that when X is
fluorine atom, R14 is not hydrogen atom; and R4, R5 and R6
herein are as defined above, is subjected to reaction for
removal of trityl in the presence of an acid catalyst,
giving a compound of the invention represented by the
formula ( r~ ) given below:
HNJ~x ,.
o~N (I' )
HO
~o~l
R~ ~14
wherein X, R13 and R14 are as defined above.
Solvents usable in this reaction include the
same solvents exemplified above with respect to process
A. Examples of suitable acid catalysts are formic acid,
acetic acid and like organic carboxylic acid, toluene-
sulfonic acid and like organic sulfonic acids, etc.
-
~ '
- 15 - 1 326848
The amount of the acid catalyst to be used is
about 0.01 to about 10 moles, preferably about O.OS to
about 10 moles, per mole of the S'-trityl-S-substituted
uridine derivative of the formula ~II').
The reaction temperature is 0 to about I30C,
preferably room temperature to about 80C. The reaction
time is about O.S to about 10 hours although variable
depending on the kinds of the solvent and the basic
catalyst to be used.
The intermediate of the invention, i.e., S'-
trityl-5-substituted uridine derivative of the formula
~II') can be prepared, as illustrated in a reaction scheme
below, by reacting a S'-trityl-5-substituted uridine
derivative of the formula ~V) which is a known compound
with the halogenosilyl compound of the formula ~IV) in the
presence of a basic catalyst.
O
H ~ ~ X R~
C - 0 ~ + Rs - S i - X~ t~)
H O R~
~: (V) (IV)
- 16 - 1326848
In the formulae, Rg, x and xl are as defined
above.
The reaction conditions such as solvent, basic
catalyst, reaction temperature, reaction time, the amounts
of reactants, etc. are the same as specified in respect of
process A.
Process C
The compound of the invention represented by the
formula
H~ ~
o N ( VI)
R,5 ~
Rl6 Rl7
wherein X is as defined above, Rl5 and R16 represent a
group of the formula -OSi-(R4)~R5)(R6), aminoacyloxy group
with the amino group optionally substituted with lower
alkyl or carboxylalkylcarbonyloxy group, Rl7 is a group
represented by the formula -OSi-(R4)(R5)(R6), aminoacyloxy
group with the amino group optionally substituted with
lower alkyl group, carboxylalkylcarbonyloxy group or
. .
", -
~` ~
132~848
hydrogen atom, at least one of Rl5, R16 and R17 is a group
represented by the formula -OSi-(R4)(R5)(R6) and at least one
of R15, R16 and R17 i8 aminoacyloxy group with the amino
group optionally 6ubstituted with lower alkyl group or
carboxylalkylcarbonyloxy group, with the proviso that when X
is fluorine atom, R17 i8 not hydrogen atom: and R4, Rs and R6
herein have the same meanings as above, can be prepared by
reacting the compound obtained by process A or B and
repre~ented by the formula
H NJ~_ x
o N ( Vl )
R~s '
R~8 Rl7
wherein X is as defined above, Rls' and Rl6' represent
hydroxyl group or a group represented by the formula -osi-
(R4)(Rs)(R6), Rl7' is hydrogen atom, hydroxyl group or a
group represented by the formula -OSi-(R4)(Rs)(R6)~ at least
one of Rls', R16' and Rl7' is a group represented by the
formula -OSi-(R4)(Rs)(R6), and at least one of R1s', R16' and
R17' is
- 17 -
.
:
- ~ . . . . .. . . .
- 18 - 1326~8
hydroxyl group, with the proviso that when X is fluorine
atom, R17' is not hydrogen atom; and R4, R5 and R6 herein
are as defined above, with the carboxylic acid of the
following formula (VIII) or a reactive derivative thereof,
or an anhydride of the dicarboxylic acid of the following
formula (IX) in the presence of a basic catalyst using or
without using a condensation agent. The compound of the
formula (VIII) or the formula (IX) or their reactive
derivative is caused to react with the hydroxyl group
repreRented by at least one of R15 ! R16 and R17
compound of the formula (VII).
R18COOH (VIII)
In the formula, R18 is aminoalkyl group, particularly Cl-
C5 aminoalkyl group, wherein the amino group may
optionally be substituted with lower alkyl group.
HOOC-Rlg-COOH (IX)
In the formula, Rlg is alkylene group, particularly Cl-C4
alkylene group.
Examples of the reactive derivative of
carboxylic acid of the formula (VIII) are acid halide,
acid anhydride, etc. The use of a condensation agent
which is not critical in the invention, enables smooth
progress of reaction. Examples of useful condensation
agent are N,N-dicyclohexylcarboxylimide, 2-chloro-1-
methylpyridinium tosylate, etc. The amount of the
, 1 '..~' ~ ' .
` ` `, ~
lg- 1326848
condensation agent to be used is about 2 to about 6 mole-s,
preferably about 2 to about 4 moles, per mole of the
compound of the formula (VII).
Solvents useful in this reaction include, for
example, methylene chloride, 1,2-dichloroethane,
chloroform and like halogenated hydrocarbons, ether,
tetrahydrofuran, dioxane and like ethers, etc. These
colvents are usable s~ngly or at least two of them can be
used in mixture. Examples of suitable basic catalysts are
pyridine, dimethylaminopyridine, 2,6-lutidine, imidazole,
triethylamine and like organic bases, etc. The amount of
the basic catalyst to be used is about 0.1 to about 20
moles, preferably about 5 to about 10 moles, per mole of
the compound of the formula ~VII).
A suitable amount of the compound of the formula
(VIII) or the compound (IX) to be used i9 about 1 to 6
moles, preferably about 2 to about 4 moles, per mole of
the compound of the formula (VII). The reaction
temperature is 0 to about 60C, preferably 0 to about
30-C. Although the reaction time is variable depending on
the kinds of the solvent and basic catalyst to be used,
the reaction is completed usually in about 0.1 to about 48
hours.
The 5-substituted uridine derivative of the
invention thus obtained can be easily separated and
- ~ .
. ~ ,
:
- ~326848
purified by conventional separation and purification means
such as recrystallization, reprecipitation, column
chromatography or the like.
While usable per se as a drug for treating a
malignant tumor, the compound of the invention can be made
into a pharmaceutically acceptable salt to facilitate its
dissolution in water and its absorption in the body. The
pharmaceutically acceptable salt contains an acid
component capable of forming a salt in combination with
the aminoacyloxy group wherein the amino group may
optionally be substituted with lower alkyl group of the
compound of the formula (I) according to the invention or
an alkali component capable of forming a salt in
combination with the carboxylalkylcarbonyloxy group of the
compound of the formula ~I), and is not particularly
limited insofar as the salt thus formed can exhibit the
desired efficacy and is nontoxic or of low toxicity in the
living body. Examples of the acid component are hydrogen
chloride, hydrogen bromide, sulfuric acid, nitric acid,
phosphoric acid and like inorganic acids, and p-
tolunenesulfonic acid, benzenesulfonic acid, formic acid,
oxalic acid, succinic acid, malic acid, citric acid,
tartaric acid and like organic acids. Examples of the
alkali component are sodium, potassium and like alkali
metals, calcium, magnesium and like alkaline earth metals,
-
: . . ' ' ' ~ ' ~
- 21 - 1326848
ammonia, methylamine, dimethylamine, piperidine,
cyclohexylamine, triethylamine and like primary, secondary
and tertiary amines, etc.
The salt can be prepared by a conventional
process for producing a salt, for example by reacting the
compound of the formula (I) with theoretical amount of the
acid or alkali component in a suitable solvent.
When the salt is soluble in a solvent, the
desired salt is produced by addition of a solvent
incapable of dissolving the salt or by lyophilization.
When the salt is fully insoluble in a solvent, the desired
salt is obtained by filtering the formed salt. ~he salt
obtained in this way can be purified with use of MCI gel
(product of Mitsubishi Chemical Industries Limited, Japan)
or the like.
The compound of the invention, when used as an
agent for treating malignant tumors of mammals including
humans, may take pharmaceutical dosage forms including
parenteral preparations such as injections, suppositories,
eye drops, aerosols and the like and oral preparations
such as tablets, coated tablets, powders, granules,
capsules, liquids and the like. Oral preparations are
generally preferred. The above preparations are
formulated in a manner known in the art. Por the
formulation of solid preparations for oral administration,
.
. ~ , . . .
: ~: - .
.
- 22 - 1326848
an excipient, and if desired, a binder, disintegrator,
lubricant, coloring agent, corrigent, flavor, etc. are
added to the compound of the invention, and then tablets,
coated tablets, granules, powders, capsules or the like
are prepared in a conventional manner. For the
formulation of injections, a pH adjusting agent, buffer,
stabilizer, isotonic agent, local anesthetic or the like
is added to the active ingredient of the invention, and
injections for subcutaneous, intramuscular or intravenous
administration can be prepared in a conventional manner.
For the formulation of suppositories, a base, and if
desired, a surfactant are added to the active ingredient
of the invention, and the suppositories are prepared in a
conventional manner. The excipients useful for the solid
preparations for oral administration are those generally
used in the art, and useful examples are excipients such
as lactose, sucrose, sodium chloride, starches, calcium
carbonate, kaolin, crystalline cellulose, methyl
cellulose, glycerin, sodium alginate, gum arabic and the
like, binders such as polyvinyl alcohol, polyvinyl ether,
polyvinyl pyrrolidone, ethyl cellulose, gum arabic,
schellac, sucrose, water, ethanol, propanol,
carboxymethylcellulose, potassium phosphate and the like,
lubricants such as magnesium stearate, talc and the like,
and further include additives such as usual known coloring
. . :
.
'
.
.. . .
.. - . . . .
- 23 - 1326848
agents, disintegrators and the like. Examples of bases
useful for the formulation of suppositories are, for
example, oleaginous bases such as cacao butter,
polyethylene glycol, lanolin, fatty acid triglycerides,
Witepsol (trademark, Dynamite Nobel Co., Ltd.) and the
like. Liquid preparations may be in the form of aqueous
or oleaginous suspension, solution, syrup, elixir and the
like, which can be prepared by a conventional way using
usual additives.
The amount of the compound (I) of the invention
to be incorporated into the pharmaceutical composition of
the invention varies with the dosage form, solubility and
chemical properties of the compound, administration route,
administration scheme and the like. Preferably the amount
is about 10 to about 15 w/w% in the case of oral
preparations, and about 0.1 to about 1 w/w~ in the case of
injections which are parenteral preparations.
The dosage of the compound (I) of the invention
is suitably determined depending on the individual cases
taking symptoms, age and sex of the subject and the like
into consideration. Usually, the dosage in the case of
oral administration is about 100 to about 800 mg per day
for an adult in 2 to 4 divided doses, and the dosage in
the case of injection, for example, by intravenous
administration is 2 ml (about 1 to about 10 mg) which is
- 24 - 1326848
administered once a day for an adult wherein the injection
may be diluted with physiological saline or glucose
injection liquid if so desired, and slowly administered
over at least S minutes. The dosage in the case of
suppositories is about 1 to about 300 mg which is
administered once or twice a day at an interval of 6 to 12
hours wherein the suppositories are administered by
insertion into the rectum.
Given below are Preparation Examples. In the
Preparation Examples that follow, the compound numbers
correspond to the compound numbers used in the Examples to
be described later.
Preparation Example 1 : Tablets
Compound 1 S0 g
Lactose 200 g
Corn starch 80 9
Hydrolyzed starch 20 g
Potassium stearate 10 9
360 9
Compound 1, lactose, corn starch and hydrolyzed
starch were mixed, and granulated by adding water to
prepare an active paste. After drying overnight at 45 C,
the granules were sieved. Potassium stearate was added
thereto and the tablets weighing 360 mg and having a
diameter of 10 mm were produced by means of tabletting
13268~8
- 25 -
machine.
Preparation Example 2 : Capsules -
Compound 4 25.0 9
Lactose 150.0 g
Corn starch 40.0 g
Talc 5.0 9
Per capsule 200 mg
Compound 4, lactose and corn starch were mixed
and pulverized. After addition of talc, the mixture was
placed into hard gelatin capsules.
Preparation Example 3 : Injection~
To Compound 40 (50 g) and 400 9 of glucose was
added distilled water for injection with stirring until
the total volume became 10 liters. The mixture was
filtered for sterilization and placed into 2-ml colorless
ampoules, and nitrogen gas was aerated therein followed by
sealing, thereby producing injection preparations each
having a volume of 10 ml per ampoule.
EXAMPLES
Given below are Examples of the present
invention.
Example 1
Preparation of 5'-O-tert-butyldimethylsilyl-5-fluoro-
uridine ~Compound 1)
A 1.50 9 quantity of 5-fluorouridine (5.72
, : ' : .
.: :
- - - - -: .-. . . .... . .
- ~'-: "' .
. .
~" `
- 26 - 1326848
mmoles) was dissolved in 5 m~ of N,N-dimethylformamide.
To the solution were added 520 mg (7.64 mmoles) of
imidazole and 633 mg (4.20 mmoles) of tert-butyldimethyl-
silyl chloride, and the reaction was conducted at room
temperature for 15 hours. The reaction mixture was ice-
cooled, and 40 mQ of water was added thereto. The reaction
mixture was extracted three times with 40 mQ of ethyl
acetate. The organic layers were combined, washed three
times with 50 m~ of water and washed three times with 50
m~ of a saturated aqueous sodium chloride solution and
dried over anhydrous magnesium sulfate. The solvent was
evaporated off under reduced pressure, and the residue
thus obtained was subjected to silica gel column
chromatography (chloroform : methanol = 20 : 1). The
eluate was concentrated and the residue was recrystallized
from ether, giving 1.3 9 of the title compound as white
crystals. Yield : 60%.
Example 2
~a) Preparation of 5'-O-trityl-2'-O-tert-butyldimethyl-
silyl-5-fluorouridine (Compound A), 5'-O-trityl-3'-O-tert-
butyldimethylsilyl-5-fluorouridine (Compound B) and 5'-O-
trityl-2',3'-bis (O-tert-butyldimethylsilyl)-5-
fluorouridine (Compound C).
- A 5.0 9 quantity (9.99 mmoles) of 5'-O-teityl-5-
fluorouridine was dissolved in 70 m~ of N,N-dimethyl-
' ',' ~:
- 27 - 132~848
formamide. To the solution were added 1.35 9 (19.8
mmoles) of imidazole and 1.78 g (11.8 mmoles) of tert-
butyldimethylsilyl chloride, and the reaction was effected
at room temperature for 12 hours. The reaction mixture
was ice-cooled, and extracted with lS0 mQ of ethyl acetate
after adding 30 m~ of water. The extract was dried over
anhydrous magnesium sulfate. The solvent was evaporated
off under reduced pressure, and the residue was subjected
to silica gel column chromatography ~chloroform : methanol
= 100 : 1~, giving 1.3 9 (yield 17.8%) of Compound C
having Rf value of 0.65 (chloroform : methanol = S0 : 1),
0.90 9 (yield 14.6%) of Compound B having Rf value of O.S0
(chloroform : methanol = S0 : 1) and 0.75 g (yield 12.2%)
of Comopund A having Rf value of 0.40 (chloroform :
methanol = 50 : 1).
~b) Preparation of 2'-O-tert-butyldimethylsilyl-5-
fluorouridine ~Compound 2), 3'-O-tert-butyldimethylsilyl-
S-fluorouridine ~Compound 3) and 2',3'-bis (O-tert-
butyldimethylsilyl)-5-fluorouridine (Comopund 4).
To 1.2 9 (1.64 mmoles) of 5'-O-trityl-2'-O-tert-
butyldimethylsilyl-5-fluorouridine (Comopund A) obtained
above was added 5 m~ of 80% aqueous solution of acetic
acid, and the mixture was stirred at 80C for 1 hour.
After the reaction, the reaction mixture was evaporated
under reduced pressure and the residue was subjected to
- :.
. ~, ,. - , .. . .
- 28 - 13268~8
silica gel column chromatography (chloroform : methanol =
20 : l). The eluate was concentrated and the residue
obtained was recrystallized from ether, giving 0.40 9 of
Compound 2 as white crystals. Yield : 64.9~.
Following the above procedure and using 0.80 g
(l.09 mmoles) of 5'-O-trityl-3'-O-tert-butyldimethylsilyl-
5-fluorouridine (comopund B), 0.35 9 of Compound 3 was
prepared as white crystals. Yield : 85.3%. Similarly,
from 1.1 9 (1.50 mmoles) of 5'-O-trityl-2', 3'-bis (O-
tert-butyldimethylsilyl)-5-fluorouridine (Compound C),
0.65 9 of Compound 4 was prepared as white crystals.
Yield : 88.3%.
Example 3
Preparation of 2', 3', 5'-tri(O-tert-butyldimethylsilyl)-
5-fluorouridine (Compound 5), 2',5'-bis(O-tert-
bùtyldimethylsilyl)-5-fluorouridine (Compound 6) and
3',5'-bis(O-tert-butyldimethylsilyl)-5-fluorouridine
(Compound 7)
A 2 9 ~uantity of 5-fluorouridine (7.63 mmoles)
was dissolved in 5 mQ of N,N-dimethylformamide. To the
solution were added 1.24 g (18.3 mmoles) of imidazole and
2.98 9 (l9.1 mmoles) of tert-butyldimethylsilyl chloride,
and the mixture was stirred at room temperature for 10
hours. The reaction mixture was ice-cooled, and 60 m~ of
water was added thereto. The reaction mixture was
. . , - .
. . . .
.
- 29 - 13268~8
extracted with 300 mQ of ethyl acetate. The extract was
washed three times with 50 mQ of a saturated aqueaus
sodium chloride solution and dried over anhydrous
magnesium sulfate. The solvent was evaporated off under
reduced pressure, and the residue thus obtained was
subjected to silica gel column chromatography (benzene :
ether = 17 : 1), giving 0.97 g ~yield 21%) of Compound 5
having Rf value of 0.40 (benzene : ether = 17 : 1), 0.99 9
(yield 26.4%) of Compound 7 having Rf value of 0.30
~benzene : ether = 17 : 1) and 0.1 g (yield 2.7%) of
Compound 6 having Rf value of 0.12 (benzene : ether
= 17 : 1).
Similarly, Compounds 8 to 34 shown in the
following table I were prepared.
Table I and Table II each show ~ values (ppm) of
lH-NMR (solvent DMSO, internal standard TMS) and melting
points (C) of Compounds 1 to 34 as well as Compounds A to
C which are the intermediates of the present invention.
In the following description, the values of coupling
constant J in lH-NMR spectrum data is expressed in terms
of Hz.
.
, , ~ .
., ~ . .. .
:
1326848
-- 30 --
C '
~, l ~'
~: 11
~o ~
Z a~, "_,,.,,,~_
'' ~ '' In :,
~=~ :~ OO-D~O~OO
0~ Z7~ c~ _ a~oo~r~o
~ ~C N ~
~ ~ .) ~q t~
N ¦ W ¦
'.'
.
.~
- 31 - 1 32 68~8
C ~ ~
~ a~ ~
~_, l . ~
~ a~ r-
. ~ e ~
I 11 ~ 11
~_
~O
C~
. . . P~ e~
c~ ~ 0
l ~ ~ ~
:: a~oo~oo~oo In~OO~C`l~OO
_ OOO~D~ . Ooomt~~
. . . . . . .~ . . . . ~ ~ .
oo~m~oo~ oo~moo~
o ~l o
~ p~ ~
~q ~ :~
~ o o o o o o
- - - -
: p~ ~: ~: ~:
-
--
- 32- 1326848
C
t-
~,
~ . ~
.
~ ~ U~
0 0 u~ 0 u~ 0 F ~ D
.~ ~
~ ~P P P ~ ~ ~-I ,
Z ~C~,,,.,_,'_
:~ ~rooo-nOo~O~OO,~
_ o o o ,
. . . . . . . . . . '~
oooooo~u~
o
~ P~ ~
L~ - ~
~C _ . ._
~` ~
.
. ~ . . . -, , :
- .
, ` . ~ ~, . . .
:. : , ` ~ .
- \ :
13268~8
-- 33 --
__ .
~^ l
r-
.
~ ~: 11
~o ~
cn ~
P: . ~ q
Z ~ ~ ~ ~ ~ '' '
l ~ r
_ 000~1~00000~0 .
........... ~
O O O O O O O O ~ U~ 00
''
:: -
C~ ~ t )--u~
1 ~ ~' I x
~ In .,
~ . . ._. '
" . ; ~, .. .. .
1326848
-- 34 --
O
~ ~ .
. t`
tq 11
Co ~
~ n O
P~
~ N
Z
I ~ ~
P Cl~ O _1 1~ 00 00
_ ooooo~t~cr,
. . . . . . .--I
~ ~ p~
:q C.~
O O O
_ ~:
-
- ` ~
1326848
-- 35 --
G .
~ l
~D ' D
00
~D
~o ~ ~ ~~ ~ ~ R
~ ~ . '
Z ~O-DO~,.I,,~ . ,,
I ~
~ ~OOO~C~
_ O ~ D O t- ~ ,
........ ~
OOOO~U~ l
--cn--~ ~ :~
O~
L~L I
,: , ,. . -
.
.
1326848 :
-- 36 --
o N :,
~ ~ c
_.~ O o
~ N q
~ _~ .
E E E~ ~ E E . E ~ ~ E
. ~ ~
11 ~ 11
~_ N ~ ~ C`l ~1 1_ ~ C`~ ~ ~1 1
_ ~ _ _ `~ ~ ,D
N O O C~ O rQ D E ~ . ~ O O O D D
~ a~ q ~ t- O U~
.` .... ¢~ l ~ ....
Z D O ~ ~~ ~ ~0 0. ~ _
I I I ------''d' -- I I 1' 1 ------~s~
~ ~~ NO00C`~O~N00 t`ON00~1~ ~00
_ ~ o~o~tn~_, . o~o~a~ .
.......... _1 ......... ~
. ~ ~i a~
~-t~: ~ ¢ ~ - ~:
,~ ~ ~ P P ~ C.)--V~
:C~ O O O O O O
~ '; ' ~ ,
: _ 1~ ~i _~$
~,~ ~a
~:~ oo . a~
.,
~ ~ .
.
t
.
.. ~ : : : ~ :
1326848
-- 37 --
,~
~ U~
E3 ~ E E E E o
11 ~1 0 11
~O ~ ,D ~
t,q C~l O O O .D ~ E~ ~ ~ u~ oo ,D ~ E C~ ~
O
~ ~ ~ ~ ~ /
z ~ o ~ ~ ~ ~ ~ ~ ~,
I ~
:r OOC~OONOO~U~OO ~c~1t~rtn~r t`OO
_ ~r~O~C`J~t-C~, DU~ ~,
......... -1 ...... .~'
o o o ~1 ~ In In 1~ C;O ~ O ~ 1~ ~ t' ~
~ :~ P: ~ -c.) ~ cn ~ :~: ~:
Cg O O . I O~- O O
`: ~ ~: __
~ ~_~
. . . . . ,: : ~, . . .. .
.. , ~
- 38 - 13268~8
C -
8. ~ N
~ ~ O .
t ` ~ N
.
E E ~ . E EE .
, :q ~o ' ~ ,
P~ In 11 ~ ~P: 11
~_ 1~ ' ~ ~ ~ N
~o ~ ~ ~ ''
u~ N D ~ ~ ~ N 'O
P: ~ m
~ . ~ ~ ~ P~
Z ~ ~ J
I I ''`~ D --' I '''--'' I I ''1
P:~ 00 00 ~D ~r 00 ~100 O N ~ 00 C`l 00 ~ O C~
_ ~ . ~ ~o ~/ ~ t~ C`l d' ~ .
... ,... ~ ........ '1
~ u~ m
-, . ': `.
, - ~. - ~ ,
; - : . .
- 1326848
- 39 --
.
C o .
.'
~ ~ ,~
~ l . ~
o~
o ~ ~
. ~ . e ~E .
11 ~ 11 ~ 11
~- ~ ~ ~ ~ ~
~o ~ -~ ~ ~ ~ ~ ~
,,~ ~ EOOD~ E~ ~ ~nOODD EOO~
~: ~ ~ ~ o ~ ~ ~o ~
I ~ I I ~ In . .
~ ~ ~ ~D ~ ~ O~ O C~l -D OO C~ r o ~ oO
_ ~ D o m t~ a~ . O ~ O
ooo,~ ~
~. ~ In .. ...
13268~8
-- 40 --
-- N 1 ~ :
~C`l ~ ~C~
~3 E E E ~
P~ :~ 11 ~ 1
U~
~o ~ ~ ~D ~ ~'' ~D
0 ~OODD e~ ~ 0 u~D~ E~~J ~
P: ~ ~O ~ ~ d~ P:
~ ~ c
Z ~ ~
I ~ ~ ~ I '' ~ `_ '_ I '' ~ ~ I '' O
P: oo ~ ~r o oo o o ~ oo ~ In O
_ OC`l~D~I~OOO . ~C~ro~ .,
........ '~ ........
O C`l ~ ln ~ ~ -D OO ~ o ~ ~
_ _ _
o
~ ~I~ C~I~
:~, P~
~ ~ ~ t~ :~ c.) ~ P~
~ ~ O . O O O O O
~ N p! ~Y N ~
. _ _
~:~
~ ~ ...
.
:: :
~ -.
.~:
13268~8
_ 41 --
C ~
8 0 n
ê E ê. ~ c~
~ r: ~11 P:~ 11
In ~ ~ I
'O
~ e o ~ ~ ~ e ~ ~
~ ~ ~ ~ O ~ ~ ~ ~
~P~ C ~ X~
Z ~ C~ ~ '
I ~ O
:~: ~ ~ ) ~ ~ ~ a~ .D ~r oo ~ c~ ~ a: a~
_ ,~ . ~ a~ ~ ~ o
..~ .~
o~m~u~oo~ oo~u~
X ~ X I X
~:¢ P: c~ c~ :r ~ .
C~ o o o o o o
_ N _ N _
., ~ ~~ ~
. . .: .
13268~8
-- 42 --
_ ~ o
~ o o
~ ~: 'C .
:~:
C`l ~ C`l
E . E .
.
:~ 11 ~ 11
I
~O ~ ~.D ~ '~ "D
. cro.Q .Q E ~ ~ ~ ~o.D ~ E
N ~ ~ ~
~ :~ m . P~ x ~ ~
Z ~ c~ ~ ~ a~ ,,,
I ~ ,
p: oo~o~ :>oo oc~o~roo
_ D a~ ~ ~ Ln r~ ~ .
. . . . ~ . . . . . .--
oo~u~ oo~u~ ~
-
C~ In ~ In C~ ~n
~_ N -- N N -- N
~ ~q ~) P ~ ~ ~
' ~ O O l O l O .~
_ N C'~ _ C~l C'~
P~ ~ ~ P$ P~ p!
~ O ~ ,
,.
.
t ~ ~ :
1326848
-- 43 --
C ~
o
o ~ ,
a
00
..
e ~e . ~e
P~ P~
~q 11 ~ :~ 11
U~ ~ ~ C`l U~
~o ~ ~ ~ ~ ~ D
O O D ~ e ~ ~ O 00 ~ D
C~
. . ~ ~ :q ~ ~ . .
~z;~ ~
II I `~
:~OOC7~0C`l~OO OOOOI~NI~OO
_oot~omt~ . OOdl~t`~
. . ~ . . . .
o~mu~ o~m~moO~
c~ N ~ N N o~ N
P$ C~ 1
C~ C~
~_ P'-P ~ P
C~ P ~ c~
C~ O O O O O O
_ _
_ N ~ _ N ~
C`l tn
j
. .
1326848
-- 44 --
: 00
~ o l
. ~ oo
11
C~ ~ -~
~o ~ D
0 00 ~ ~ ~ ~ ^
C~ ~O ~
. . P~ ~ 1
z ~ ~ ~ t~ ' ~'
`~ ~
~ O 00 N 00 d~ ~D 00 ~ ~ ~ 00
_ co ~r ~`1 ~ t` _1 . O Cl~ O .
. ~ . ~
0 ~ 0 0 ~ 1~ 00
~: : ~ r :~: c~ c~ ~ :r:
~ O O O O O O
':
_ C~l ~ _ ~ ~
: '' ~ ~Y ~ ~: ~:
~ ,
~ l N
.
.~
, - . . ~. ~
~ . ~ ~ , . . . . . . . .
13268~8
__ ._
CL C2. '.'
o o
~: ~
. ..
E ~ ~ c~
t 11 P~ 11
~_ I
~O ~ rO
~ODD F~ . ~ ~ O D D
m P~
Z ~ ~ ~ ~ r~ ~
l ~ ~ r ~ ~ I ~ ~ ~ ~ ~ .. .
p: u~ r~oo a~oo~r~u~
_ o oo ~ o~ ~ . o oo ~ ~ ~ .
....... -~ ...
oo~oo--l oo~
~ ~ N ~ ~ U
~ :r c~ ) P ~ ~ c~
cn O O O O O O
_ N ~O _ N t'~
~ ~Y p: p: ~ p!
; æ
.~ ~ ~_
C`l C`l
.
. ~ ..
,. ~ .
-: . -, ; , . ~
....
.... ... ~
- 46 - 1326848
.. . __
C
o o~ .~
~:
_
o ~ e~
e e . e
`
p:~ In 11 P:
~O ~ ~ .`
~qOO.,DD eo~ ~ ~o~ eo~
P ~o
~ ~ r ~ 1
Z ~ ~ ~ ~,
I
~ OOO~OONO~CO ~ rOl~C`lOt~O~
_ _,~ . ~ ~,
....... ~ .......
c~ ~ ~ m ~ ~ ~ ,l ~ ~ ~ u~
. ~ ~ N ~ (~ N
N I N N I N
'- ~ ~ '-~
c~ ) ~ c~
~ ~ ~: O O O O l O
. _ N ~ _ N I:rl
. P~ p: P~ p! P~ ~
.. _ _ _
C~l C`l
' ' . ' ~
- 47- 13268~8
~o~ o l
_ ~ ~
a:
.
. P~
~ ~ U~ 11 ~P~
Co
. ~oP~ ~o~ ~ U~OO~ ~ .
. ~ . ~ r , o ~ o , , ,
~ ~ ~ P~
Z
I ~ '' '' I '' O _~ I I I ~ ~ '' I ''
~ ~mo~o~a~
_ ~ ~ oo ~ . ,I r~ ~r ~ ~ t~ oo ,
c~i~lnlnln~D~ ooc~
N ~ [~ N ~3
. . ~ p '-- ~ ~ ' ~
~~C~ ~ c~ c~ ~ ~
C~ O N O O O
p:: p!: P: .. p:~ P
~ o ~
... . ~.
.
.. : . , . ~ : . ~ ~ .
:, .. : . . ~ :
-
- 48- 13268~8
3~ s
C ~ O O
~:
.
. ~
P: ~ 11 q ~ ;~ 11
~o N N In ~ ~ N N 1
~D
~n D 00 C`l ,D ,D E~ ~ ~ u~ ~D 00 N D D ~ ~ ^
P: ~O ~ N ~ q ~O t--N ~ ~ ~ ~
~ ~ . , , ~ ~ , , . P P~ P:~ P ~
Z ~ N d~ '' ~ N
I
q O 00 ',D 00 ~1~ 00 N ~ ~ 00 ~D 00 ~ ~rl 00 CO 01
_ ~ t/ O ~ ~ ~ ~~ r O ~ ~ N .
........ '1 ........ ~
O O N ~ It~l It~ In 00 _1 O O N ~ 1~ 00 ~
~ P ~ ~q ~ P
Cg O O O O O O
-- N ------ ~ ~
P~ ~ ~: P: ~: ~:
;, '
_ 49 _ i326848
C
~D .
...
o
.
P: ~ t`
oo~ 11
~_ C~ U~
~o
D O .D .D ~ .D
C~
~ . . ~ ~ ~
Z ~,,,,~_,',
I I I `~
:~ 0 0 ~ ~ ~ OO
oo ,~ .
. . . . . . .--
o~m~t-
~
N N
~ O
~ ~ ..
C~ C)~ ) = ~
_
_ N cr~
~: ~: P:
1326848
~ 50 -
o
~:
ô
~
n
r~ ~ 1
~o ~
P
q ~0
tq
z; ~o a~ ~ ~ ~,,, ~, ~, ~_
='~ Z /\ I `~
a~ Z~ / --~Y :~ ~o o o o In Irl oo
o o - P~: _ O 00 ~ O
cd \/ O O ~
E~
~3~ ~ ~o
~\ ~ p~ l ~
N
¢
O
- ` ~
- 51 - 13268~8
. . ..
C
o C~ o .
~ . ~ ~
_~ CO
E ~ E . E ~ .
.
11 ~ P: 11
C~ C~
^^^^^ ~e D
~ OO~ ~
P ~ ~ 1
~q m ~ m ~
Z ~ a~ ~ ~ ~ `~
I ''~ I I ~ D
P: c~l o ~ ~ o u~ r- ~ ~ D OO C`l ~ _l o c~
_ ~ c~ ~ . o o ~ oo ~ o cr~ r o ,
........ ~ ....... --I
OO~U~ ~ Ooooo~t_~
,
3 C~ ~ ~ I
P ~ ~ C ~ -'
C ~ P C~--cn--c~)
O O O O
I I I I
N ~ N C')
P: ~;
.
~ ~ C~
:
1~2~8~8
- 52 -
Example 4
Preparation of 5'-O-tert-butyldimethylsilyl-5-
trifluoromethyl-2'-deoxyuridine (Compound 35)
A 1.0 9 quantity of 5-trifluoromethyl-2'-
deoxyuridine (3.6 mmoles) was dissolved in 3 mQ of N,N-
dimethylformamide. To the solution were added 0.49 9 (7.2
mmoles) of imidazole and 0.64 g (4.3 mmoles) of tert-
butyldimethylsilyl chloride, and the reaction was
conducted at room temperature for 10 hours. The reaction
mixture was treated in the same manner as in Example 1
with the exception of using chloroform/methanol (30 : 1)
as an eluent for silica gel column chromatography, giving
0.6 9 of the title compound as white crystals having a
melting point of 199 to 200C. Yield : 41%.
lH-NMR (internal standard TMS, solvent d6-DMSO, ~ value,
ppm)
11.9 (lH, b, N-3H)
8.1 ~lH, s, 6-H)
6.03 (lH, t, J=6.8, 1'-H)
5.28 (lH, b, -OH)
4.00 to 4.28 (lH, b, 3'-H)
3.84 to 4.00 (lH, m, 4'-H)
3.68 to 3.84 (2H, m, 5'-CH2)
2.00 to 2.32 (2H, m, 2'-CH2)
0.85 (9H, s, t-Bu)
: -
. . ~ '
1326848
- 53 -
0.05 (6H, s, -Si(CH3)2)
Example 5
Preparation of 3', 5'-bis(O-tert-butyldimethylsilyl)-5-
trifluoromethyl-2'-deoxyuridine (Compound 36)
A 1.0 g quantity of 5-trifluoromethyl-2'-
deoxyuridine (3.6 mmoles) was dissolved in 5 mQ of N,N-
dimethylformamide. To the solution were added 1.02 g (15
mmoles) of imidazole and 1.14 g (7.60 mmoles) of tert-
butyldimethylsilyl chloride, and the reaction was
conducted at room temperature for 18 hours. The reaction
mixture was treated in the same manner as in Example 4 to
prepare 1.68 g of the title compound as white crystals
having a melting point of 93 to 94C.
Yield : 88.5~.
H-NMR ~internal standard TMS, solvent d6-DMSO,
value, ppm)
11.9 (lH, b, N-3H)
8.1 ~lH, s, 6-H)
6.03 (lH, t, J=6.1, l'-H)
4.20 to 4.44 (lH, b, 3'-H)
3.80 to 4.00 ~lH, b, 4'-H)
3.60 to 3.80 (2H, b, 5'-CH2)
2.04 to 2.36 (2H, m, 2'-CH2)
0.87, 0.86 (18H, s, t-Bu)
0.08, 0.05 (12H, s, -Si(CH3)2)
., :
.
.. ~
- ` `
13268~8
- 54 -
Example 6
Preparation of 5'-O-trityl-3'-O-tert-butyldimethylsilyl-5-
trifluoromethyl-2'-deoxyuridine (Compound D) and 3'-O-
tert-butyldimethylsilyl-5-trifluoromethyl-2'-deoxyuridine
(Compound 37)
A 3.2 g quantity of 5'-O-trityl-5-trifluoro-
methyl-2'-deoxyuridine (5.9 mmoles) was dissolved in
5 mQ of N,N-dimethylformamide. To the solution were added
0.82 g (12 mmoles) of imidazole and 1.26 g (8.3 mmoles) of
tert-butyldimethylsilyl chloride, and the reaction was
conducted at room temperature for 10 hours. The reaction
product was purified in the same manner as in Example 4,
giving 3.0 g of Compound D in an amorphous form. Yield :
94%.
H-NMR (internal standard TMS, solvent d6-DMSO,
value, ppm)
11.9 (lH, b, N-3H)
8.16 (lH, s, 6-H)
7.36 ~15H, m, Ph)
6.07 (lH, t, J=7.0, l'-H)
4.20 to 4.48 (lH, m, 3'-H)
3.80 to 4.04 (lH, m, 4'-H)
3.00 to 3.40 (2H, m, 5'-CH2)
2.08 to 2.44 (2H, m, 2'-CH2)
0.80 (9H, s, t-Bu)
. . .
' ,' :` ' ," "~' ,
; ,. ~ , .
. ~
. ,
_ 55 _ 1~2~848
0 03, -0.06 (6H, s, -Si(CH3)2)
A 5 mQ of 80~ aqueous solution of acetic acid
was added to 2.9 g (5.38 mmoles) of the 5'-O-trityl-3'-O-
tert-butyldimethylsilyl-5-trifluoromethyl-2'-deoxyuridine
obtained above, and the mixture wàs stirred at 60C for 1
hour. After the reaction, the reaction mixture was
extracted with a saturated aqueous sodium chloride
solution-ethyl acetate and the extract was dried over
anhydrous magnesium sulfate. Purification was conducted
in the same manner as in Example 4, giving 0.32 g of
Compound 37 in an amorphous form. Yield : 16%.
H-NMR (internal standard TMS, solvent d6-DMSO,
6 value, ppm)
11.8 ~lH, b, N-3H~
8.67 (lH, ~, 6-H)
6.05 (lH, t, J=6.1, l'-H)
5.26 (lH, b, -OH)
4.24 to 4.52 (lH, b, 3'-H)
3.72 to 3.88 (lH, m, 4'-H)
3.40 to 3.72 (2H, m, 5'-CH2)
2.00 to 2.40 (2H, m, 2'-CH2)
0.87 (9H, 5, t-Bu)
0.08 (6H, s, -Si(CH3)2)
Example 7
Compound 38 was prepared in the same manner as
.
::: .
1326848
- 56 -
above.
5'-O-triisopropylsilyl-5-trifluoromethyl-2'-
deoxyuridine (Compound 38)
Melting point 171 to 171.5C
~-NMR (internal standard TMS, solvent d6-DMSO,
6 value, ppm)
11.9 ~lH, b, N-3H)
8.06 (lH, s, 6-H)
6.02 (lH, t, J=6.8, l'-H)
5.32 (lH, d, J=4.4, -OH)
4.08 to 4.32 (lH, b, 3'-H)
3.64 to 4.00 (3H, m, 4'-H, 5'-CH2)
2.08 to 2.32 (2H, m, 2'-CH2)
0.60 to 1.32 (21H, m, -Si(iso-Pr)3)
ExamPle 8
Preparation of 5'-O-tert-butyldimethylsilyl-2', 3'-bis(O-
dimethylglycyl)-5-fluorouridine (Compound 39)
To 60 m~ of a solution containing 2.5 9 (6.65
mmoles) of Compound 1 in methylene chloride were added 2.0
9 (19.9 mmoles) of dimethylglycine, 5.3 9 (43.9 mmoles) of
N,N-dimethylaminopyridine and 6 g (20 mmoles) of 2-chloro-
l~methylpyridinium tosylate. Thereafter the mixture was
stirred at room temperature for 4 hours. After the
reaction, the reaction mixture was extracted with ethyl
acetate and water. The organic layer was washed with 0.1%
132g848
cooled and diluted hydrochloric acid, and dried over
magnesium sulfate. The organic layer was evaporated off,
giving 3 9 of Compound 39 in an amorphous form.
Yield : 82.6%
H-NMR (internal standard TMS, solvent d6-DMSO,
value, ppm)
0.12 (6H, s)
0.90 (9H, s)
2.21 (6H, s)
2.26 (6H, s)
3.17 to 3.41 (4H, m)
3.87 (2H, ~)
4.24 (lH, m)
5.37 (2H, m)
5.99 (lH, m)
7.98 (lH, d, J=6.8)
11.96 (lH, br)
- ~
Preparation of 5'-O-tert-buty]dimethylsilyl-2', 3'-bis(O-
dimethylglycyl)-5-fluorouridine malate (Compound 40) and
tosylate (Compound 41)
To 20 mQ of a solution containing 560 mg (1.0
mmole) of Compound 39 in ether was added 10 mQ of a
solution containing 287 mg (2.14 mmoles) of L-malic acid
in ether, and the mixture was stirred at room temperature
'. . : . :~ , ~
. . :: . ,
13268~8
- 58 -
for 1 hour. The crystals precipitated were filtered,
giving 800 mg of Compound 40 (yield : 98~). Similarly, 10
mQ of a solution containing 96 mg (0.7 mmole) of p-
toluenesulfonic acid in ether was added to 10 mQ of a
solution containing 200 mg (0.36 mmole) of Compound ~9 in
ether. Thereafter the mixture was stirred with ice-
cooling for 30 minutes. The crystals precipitated were
filtered, giving 245 mg of Compound 41. Yield : 95~.
Compound 40 Melting point 95 to 97C
H-NMR (internal standard ~MS, solvent d6-DMSO,
value, ppm)
0.12 (6H, s)
0.90 (9H, s)
2.26 t6H, s)
2.32 (6H, s)
2.40 to 2.60 (4H, m)
3.20 to 3.52 (4H, m)
3.88 (2H, m)
4.08 to 4.32 (3H, m)
5.41 (2H, m)
6.00 (lH, m)
6.00 to 7.60 (6H, br)
7.98 (lH, d, J=6.9)
11.90 (lH, br)
Compound 41
,
: '. - ~ .: ': . . .. , .... '
, . . .
- 59 - 1326~8
Amorphous
H-NMR (internal standard TMS, solvent d6-DMSO,
value, ppm)
0.18 (6H, s)
0.96 l9H, s)
2.33 (6H, s)
2.88 (6H, s)
2.93 (6H, s)
3.90 to 4.41 (7H, m)
5.51 (2H, m)
6.19 (lH, m)
7.11 to 7.21 (4H, m)
7.50 to 7.58 (4H, m)
7.99 (lH, d, J=6.6)
10.04 (2H, br)
12.07 ~lH, br)
Example 10
Preparation of 5'-0-(2,3-dimethyl-2-butyl)dimethylsilyl-
2',3'-bis(O-dimethylglycyl)-5-fluorouridine (Compound 42)
To 20 mQ of a solution containing 1 g (2.47
mmoles) of Compound 14 in methylene chloride were added
763 mg (7.41 mmoles) of dimethylglycine, 1.99 g (16.32
mmoles) of N,N-dimethylaminopyridine and 2.23 g (7.43
mmoles) of 2-chloro-1-methylpyridinium tosylate.
Thereafter the mixture was stirred at room temperature for
. ~ . . , . '
" '' ' , ~ '' .
: .
- 60 - 132~8~8
2 hours. After the reaction, the reaction mixture was
extracted with ethyl acetate and water. The organic layer
was washed with 0.1% cooled and diluted hydrochloric acid,
and dried over magnesium sulfate. The organic layer was
evaporated off, giving 1.24 9 of Compound 42 in an
amorphous form. Yield : 88%
lH-NMR (internal standard ~MS, solvent d6-DMSO,
6 value, ppm)
Compound 42
Amorphous
0.15 (6H, s)
0.85 (6H, s)
0.86 (6H, d, J=6.4)
1.40 to 1.80 (lH, m)
2.21 ~6H, 8)
2.26 (6H, s)
3.08 to 3.72 (4H, m)
3.86 (2H, m)
4.21 (lH, m)
5.36 (2H, m)
5.99 (lH, m)
7.94 (lH, d, J=6.6)
11.98 (lH, br)
ExamPle 11
Preparation of 5'-0-(2,3-dimethyl-2-butyl)dimethylsilyl-
- ~ `
1~26~48
- 61 -
2', 3'-bis(O-dimethylglycyl)-5-fluorouridine malate
(Compound 43)
To 20 mQ of a solution containing 0.5 9 (0.87
mmoles) of Compound 42 in ether was added 5 mQ of a
solution containing 233 mg (1.74 mmoles) of L-malic acid
in ether, and the mixture was stirred at room temperature
for 1 hour. ~he crystals precipitated were filtered,
giving 687 mg of Compound 43. Yield : 93.7%.
Compound 43
Amorphous
lH-NMR (internal standard TMS, solvent d6-DMSO,
: 6 value, ppm)
0.15 (6H, s)
0.85 (6H, B)
0.86 (6H, s)
1.40 to 1.80 (lH, m)
2.25 ~6H, s)
2.31 (6H, s)
2.40 to 2.60 ~4H, m)
~: 3.20 to 3.72 ~4H, m)
.
3.87 (2H, m)
4.04 to 4.32 (3H, m)
5.36 (2H, m)
5.98 (lH, m)
5.60 to 7.40 (7H, br)
1326848
- 62 -
7.96 ~lH, d, J=6.8)
Example 12
Preparation of 5'-0-tert-butyldimethylsilyl-2', 3'-bis(O-
2-carboxyethylcarbonyl)-5-fluorouridine (Compound 44)
To 40 mQ of a solution containing 2.0 9 (5.44
mmoles) of Compound 1 in methylene chloride were added
2.18 g (21.76 mmoles) of succinic anhydride and 5.32 g
~43.5 mmoles) of N,N-dimethylaminopyridine, and the
mixture was stirred at room temperature for 8 hours.
After the reaction, the reaction mlxture was subjected to
extraction after addition of 10~ aqueous solution of
citric acid and 500 m~ of ether. The organic layer was
washed with water and dried over magnesium sulfate. After
evaporating the organic layer, the residue was
recrystallized from n-pentane, giving 2.5 g of Compound 44
having a hygroscopic property.
Yeild : 76.6~.
Compound 44
H-NMR (internal standard TMS, solvent d6-DMSO,
value, ppm)
0.11 ~6H, s)
0.90 (9H, s)
3.33 to 3.49 ~8H, m)
3,86 (2H, m)
4.18 (lH, m)
,
.
1326848
- 63 -
5.28 to 5.32 (2H, m)
6.02 (lH, m)
7.97 (lH, d, J=6.8)
12.00 to 12.23 (3H, br)
Example 13
Preparation of 5'-O-tert-butyldimethylsilyl-2', 3'-bis(O-
2-carboxyethylcarbonyl)-5-fluorouridine dipotassium salt
(Compound 45)
To 100 m~ of a solution containing 2.4 g (4.17
mmoles) of Compound 44 in ethyl acetate was added 3.2 g
(17.38 mmoles) of potassium 2-ethylhexanoate, and stirred
at room temperature for 14 hours. The crystals
precipitated were filtered and purified with MCI gel (50
9, H2O _ H2O : CH3CN = 1 : 1, product of Mltsubishi
Chemical Industries Limited, Japan). The eluted fraction
was lyophilized, giving 560 mg of Compound 45. Yield :
20.6%.
Compound 45
Melting point 195 to 197C
H-NMR (internal standard TMS, solvent D20,
value, ppm)
0.18 (6H, s)
0.94 ~9H, s)
2.41 to 2.70 (8H, m)
3.g8 (2H, m)
.
,
1326848
- 64 -
4.40 (lH, m)
5.43 (2H, m)
6.17 (lH, m)
7.95 (lH, d, J=5.9)
Example 14
Preparation of 5'-O-tert-butyldimethylsilyl-3'-O-
dimethylglycyl-5-trifluoromethyl-2'-deoxyuridine (Compound
46) and 5'-O-tert-butyldimethylsilyl-3'-O-dimethylglycyl-
5-trifluoromethyl-2'-deoxyuridine malate (Compound 47)
Compounds 46 and 47 were prepared in the same
manner as in Examples 10 and 11.
Compound 46
Amorphous
H-NMR (internal standard TMS, solvent d6-DMSO,
value, ppm)
0.10 (6H, s)
0.89 (9H, s)
2.00 to 2.60 (2H, m)
2.29 (6H, s)
3.00 to 3.76 (3H, m)
3.76 to 4.00 (2H, m)
4.20 (lH, m)
5.21 (lH, m)
6.07 (lH, t, J=2.9)
8.17 (lH, s)
.
': : ' ~ :
.. : -
- . .
- 65 - 1~2~8~8
Compound 47
Amorphous
H-NMR (internal standard TMS, solvent D2O,
value, ppm)
0.06 (6H, s)
0.86 (9H, s)
2.20 to 2.60 (4H, m)
2.34 (6H, s)
3.37 (2H, s)
3.68 to 3.88 (2H, m)
4.00 to 4.28 ~2H, m)
4.32 to 5.08 (4H, b)
5.21 ~lH, m)
6.11 (lH, t, J=3.7)
8.14 ~lH, d, J=0.9)
Exam~le 15
Preparation of 2',3',5'-tri~O-tert-butyldimethylsilyl)-5-
trifluoromethyluridine ~Compound 48)
: To 6 m~ of a solution containing 596 mg ~2.1
; mmoles) of 5-trifluoromethyluridine in N,N-dimethyl-
formamide were added 1.14 9 ~16.7 mmoles) of imidazole and
then 1.27 9 (8.4 mmoles) of tert-butyldimethylchloro-
silane, and the mixture was stirred at room temperature
for 17 hours. After the reaction, the reaction mixture
was subjected to extraction after addition of 30 m~ of
; , ' ': ' , ' ' : ,
, . . .
1326848
- 66 -
water and ethyl acetate (30 mQ x 3). The extract was
washed with water (20 mQ x 3) and a saturated aqueous
solution of sodium chloride ~20 mQ x 1), and dried over
anhydrous magnesium sulfate. The solvent was evaporated
off under reduced pressure, and the residue thus obtained
was subjected to silica gel column chromatography by
elution with 3~ methanol/chloroform, giving 1.28 g of
Compound 48. Yield : 93%.
Compound 48
Amorphous
H-NMR (internal standard TMS, solvent CDCQ3,
value, ppm)
0.05, 0.09, 0.13, 0.16, 0.17 ~18H, each s)
0.93, 0.97, 1.00 ~27H, each s)
3.73 to 3.93 ~2H, m)
4.08 to 4.23 ~3H, m)
6.12 (lH, d, J=5.7)
8.20 ~lH, d, J=l.l)
8.83 (lH, br)
Example 16
Preparation of 2',3',5'-tri(O-tert-butyldimethylsilyl)-5-
fluorouridine (Compound 49)
Compound 49 was prepared in the same manner as
in Example 15.
Comopund 49
- :
- 67 - 132~8~8
Amorphous
lH-NMR ~internal standard TMS, solvent CDCQ3, ~ value,
ppm)
0.04, 0.05 (12H, each s)
0.88, 0.89 (18H, each s)
2.33 (3H, s)
2.89 (6H, s)
3.30 to 3.64 (2H, m)
4.05 to 4.38 (5H, m)
5.76 (lH, m)
7.16 ~2H, m)
7.57 (2H, m)
8.02 (lH, d, J=7.04)
10.02 (lH, br)
11.94 ~lH, m)
Pharmacological Test
Cells of mouse-transplantable tumor Sarcoma 180
~5 X 106 cells) were subcutaneously transplanted in the
back of male mice of ICR/JCL strain (weighing 27 to
30 g). A solution or suspension of a test compound in a
physiological saline solution containing 0.1~ Tween 80 was
administered intraperitoneally to mice (7 mice in each
group) at a dose of 0.1 ml/10 g mouse body weight three
times, namely on the 1st, 5th and 9th days, after the day
of the transplantation.
. . ,, .: ........ : :
~' '' ' ; .:
.: . , ' , .:
13268~8
- 68 -
A physiological saline solution of the same type
but free of the test comopund was given in the same way as
above to a control group.
On the 12th day after the transplantation, the
tumor was weighed to calculate the average weight of the
tumors for each dose in the group to which the test
compound was given, and the weight was compared with the
corresponding weight in the control group to determine the
tumor growth inhibition ratio for each dose.
Table III below shows the results.
.
13268~8
- 69 -
Table III
CompoundDoseTumor growth Number of
(mg/kg/day)inhibition death
ratio (%) (per 7 animals)
2 0 1 6 0
F U R 3 5 54 0
_ 7
2 0 1 6 0
F3 T d R 4 0 3 6 0
52 0
1 6 0 6 5 5
44 0
7 0 5 5 0
1 0 0 6 9 0
140 82 0
._
58 0
2 7 0 7 1 0
1 0 0 8 3 0
1 4 0 9 o 6
5 0 3 1 0
4 7 0 4 8 0
1 0 0 6 2 0
1 4 0 7 0 0
.
5 0 1 7 0
7 0 3 0 0
1 0 0 5 1 0
1 4 0 6 4 0
,
36 0
8 70 53 0
1 0 0 6 3 0
1 4 0 8 0 0
,
r . :.
:
:' '- ' - ~
1326848
- 70 -
Table III
CompoundDose Tumor growth Number of
(mg/kg/day) inhibition death
_ ratio (~) (per 7 animals)
53 0
1 3 7 0 6 5 0
1 0 0 8 1 0
1 4 0 8 8 5
_
66 0
1 6 7 0 7 8 0
1 0 0 8 0 0
_ _ 1 4 0 8 3 4
2 0 4 2 0
49 0
82 0
.. . . .
2 0 3 5 0
3 8 4 0 5 3 0
8 0 9 0 6
.
42 0
. . 70 55 0
. _ 1 0 0 8 2
59 0
4 3 7 0 7 5 0
1 0 0 _ 7
37 0
4 5 . 7 0 5 0 0
1 0 0 7 8 0
'' ' '
'' ~ ' ' '
' ' ' ',
'
- 71 - ~32~8~8
As seen from Table III, the compounds (I) of the
invention have higher anti-tumor activity and lower
toxicity compared with FUR and F3TdR.
, . . . , ~ . -