Note: Descriptions are shown in the official language in which they were submitted.
F9
0019/MW14 1328877
- 1 - 17700IA
''."
TITLE OF T~E INVENTION
3-Æ TO HMG-CoA REDUCTASE INHIBITORS -~
BACKGROUND OF T~E INVENTION
Hypercholesterolemia is known to be one of
the prime risk factors for ischemic cardiovascular
disease, such as arteriosclerosis. Bile acid
sequestrants have been used to treat this condition;
they seem to be moterately cffective but they must be
20 consumed in large quantities , i.e. several grams at -
a time and they are not very palatable.
;~ MEVACOR~ (lovastatin), now commercially
available, is one of a group of very active
antihypercholesterolemic agents that function by
limiting cholesterol biosynthesis by inhibiting the
enzyme, HMG-CoA reductase. In addition to the
natural fe~rmentation products, mevastatin and
-~ lovastatin, there are a variety of analogs thereof,
produced by microbial, enzymatic and synthetic
techniques.
''' .;
.. ..
1328877
F9
0019/MW14 - 2 - 17700IA
The naturally occurring compounds and their
analogs have the following general structural
formulae:
xo~ ~2 R
OR H
R R
~: 15
wherein: .
l is hydrogen. Cl_s alkyl or Cl-s alkyl
substituted with a member of the group
consisting of phenyl, dimethylamino, or
acetylamino; and
R* is
: '
~ 25
3~
~CH2 ~ ' '
H
Qa~,b ~ M :`-
~ ~ .
3~
~: ' ' '
: '
:
1 3288 77
F9 --.
0019/MW14 - 3 - 17700IA
wherein I
Q is R3-lC- or R3-CH; ~3 is H or OH or Q is -~HCH20H; :.
CH~
5 M is -~HR4t R4 is hydrogen or hydroxy; X is
CR5R6, O, S, or NH; R5 and R6 are H, OH, or
oR7 where R7 represents a phosphoryl or acyl
moiety; ~-.
R2 is hydrogen or methyl; and a, k. c, and d
represent single bonds, one of a, k. ~ or d ~ -
represents a double bond, or both ~ and or
both k and d represent double bonds provided .
that when a is a double bond, Q is -C= or
H3
~ ~
C= and when d is a double bond, M is =~ and ~.
~ H H - ::~ provided that when R5 or R6 is OH or oR7 or
X is 0, S, or NH, a, k. and c are single ~:
bonds.
U.S. Patent 4,517,373 discloses hydroxy :~
containing compounds represented by the abave general ~ .
formula wherein R* is
:- -
':
o I o I .- '. ' .
C1 1 0alkyl~o CH2 C1 1 0alkYlJ~o CH/
ND C~3~C~ ~OH
CH3 -
: , .
1328877
F9
0019/MW14 - 4 - 17700IA
U.S. Patent 4,537,859 and U.S. Patent
4,448,979 also disclose hydroxy-containing compounds
represented by the above general formula wherein R* is
o I o
1X~f~3
HO ANb H
These compounds are prepared by the action --
of certain microorganisms on the corresponding : :
non-hydroxylated substrates. One such organism :- -
~: described in ~.S. 4,537,859 is of the genus Noc~rdia.- U.K. Patent 2,075,013 discloses hydroxy -~
.- .
containing compound~ represented by the above general -:
formula wherein R* i8:
/CH2 -
. ~2_~o CH2
~ M~ ~
Rl ~ OH ~-
':: .
,
~ ~' '''
1328877
,......
- 5 - 17700IA
wl~ereill Rl is H or Me, and R2 is M or acyl.
Canadian Patent Application Serial No. 540,097,
Inamine et al, filed June 19, 1987, discloses 6-substituted
compounds of the above general formula wherein R* is: .
' ,
o I ~ ~ '
~C ~ ,CI-~2
R o C~ 2
~ ~3
R
. .
¦~ wherein R is CH20H, CH20~R4, Co2R7 or ~NR8R9; -.
and Rl, R4, R7, R8 and R~ are broadly defined organic
moieties.
U.S. Patents 4,604,472 and 4,733,003 disclose ~ :
compounds of the above formula wherein R* is: -~
2S
: .,
~ I '. '
~ cH ~ :
., I x Cl H2 ''~' .
, ~ \ ~CH3
~2 : :
R
~ , .
;~ "
' .
, ~ ~.;; . . , -
1~28877
F9
0019/MW14 - 6 - 17700IA
wherein X represents a hydrogen atom or a
2-methylbutyryl group, Y represents a hydrogen atom
or a methyl group and Rl and R2 are the same or
different and each represents an oxygen atom or a
group of formula =N-oR3 where R3 is a hydrogen or
alkyl moiety.
DETAILED DESCRIP~ION OF THE INVENTION
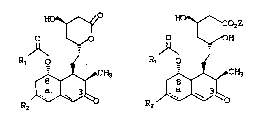
This invention relates to HMG-CoA reductase `~
inhibitors of structural formulae (I) and (II). -
~o ~ozZ ~`-`
~ ~ ~ ~ OH :
Rl o ~ R1 0 ~ ~:
R2 R2 ~ CH3 -
`~
~:
wherein-
RI is selected from: ~
(1) Cl 10 alkyl; :`~ ---
(2) substituted Cl_lo alkyl in which one or
more substituent(s) is selected from .
(a) halogen,
(b) hydroxy, ~
~:- (c) Cl_lo alkoxy, :~ .
- (d) Cl_5 alkoxyearbonyl,
.
(e) Cl_5 acyloxy,
: ~ . -' .
1328~77
F9
0019/MW14 - 7 - 17700IA
(f) C3-8 cycloalky
(g) phenyl,
(h) substituted phenyl in which
the substituents are X and Y, ~ .
(i) Cl-10 alkYlS(O)n in which n
is 0 to 2,
(i ) C3-8 cycloalkyls(o)n~
(k) phenylS(O)n, - .
lo (1) substituted phenylS(O)n in
which the substituents are X
and Y, and
(m) oxo;
(3) Cl_10 alkoxy; ~ -
(4) C2 l0 al~enyl;
(5) C3-8 cycloalkyl;
;~ (6) substituted C3_8 cycloalkyl in which - .
- one substituent is selected from
a) Cl 10 alkyl ~-
:~ 20 (b) substituted Cl_10 alkyl in
which the substituent is
~ selected from ~.
:~ (i) halogen,
(ii) hydroxy,
(iii) Cl_10 alkoxy,
(iv) Cl_5 alkoxycarbonyl,
(v) Cl_5 a
(vi) phenyl,
(vii) substituted phenyl in
which the substituents
are X and Y
(viii) Cl_lo alkYlS(O)n~
(ix) C3-8
~- cycloalkylS(O)n,
-~: (x) phenylS(O)n,
1 328877
F9
0019/MW14 - 8 - 17700IA
(xi) substituted phenylS(O)n ~ . .
in which the
substituents are X and
Y, and
(xii) oxo, ,
(C) Cl_lo alkYlS(O)n~
~d) C3_8 cycloalkylS(O)n,
(e) phenylS(O)n,
(f) substituted phenylS(O)n in
which the substituents are x
and Y,
(g) halogen,
(h) hydroxy, ~. :
(i) Cl_lo alkoxy,
; (j) Cl 5 alkoxycarbonyl,
~ (k) Cl_5 acyloxy~
:~ (1) phenyl, and
(m) substituted phenyl in which
the substituents are X an* Y;
(7) phenyl; .
~ (8) substituted phenyl in which the
:~ substituents are X and Y; . :
(9) amino; ~ --
(10) Cl_5 alkylamino;
(11) di(Cl_5 alkyl)amino;
(12) phenylamino;
(13) substituted:phenylamino in which the
substituents are X and Y;
(14) phenyl Cl_10 alkylamino;
~: (15) substituted~:phenyl Cl 10 alkylamino in
which the substituents are X and Y;
~ (16) a member selected from
,:.~ , : : '-' ~ -'"
.
.,
, ~ : ;
~- 1328~77 :
F9 :
0019/MW14 - 9 - 17700IA :
,
(a) piperidinyl,
(b) pyrrolidinyl,
(c) piperazinyl, .
(d) morpholinyl, and
(e) thiomorpholinyl; and
(17) R3S in which R3 is selected from
(a) Cl_10 alkyl,
(b) phenyl, and
(c) substituted phenyl in which the
substituents are X and Y;
R2 is H, CH3, or C~20H;
: 15 X and Y are independently selected from:
a) 0~,
~: b) halogen,
c) trifluoromethyl,
d) cl-3alko~y~ --
e) Cl_3alkylcarbonyloxy,
f) phenylcarbonyloxy,
g) Cl_3alkoxycarbonyl,
h~ phenyloxycarbonyl,
i) hydrogen; - .
j) Cl_5alkyl;
: Z is selected from
;~ (1) hydrogen;
~ (2) Cl~5alkyl;
;~ 30 (3) substituted Cl_5alkyl in which the
substituent is selected from
(a) phenyl, . ~-
,
`
1328~77
F9
0019/MW14 - 10 - 17700IA
(b) dimethylamino, and
~c) acetylamino, and
(4) 2,3-hydroxypropyl;
halogen is Cl or F;
_ is a single bond or a double bond;
and pharmaceutically acceptable salts of the compound
(II) in which Z is hydrogen.
1 0 , ,
Except where specifically defined to the
contrary, the terms ~alkyl~ alkenyl~ acyl~
~aryloxy~ and ~alkoxy~ include both the straight-chain : ~
and branched-chain species of the term. -
~ 15 One embodiment of this invention is the ~:
i~ class of compounds of formulae (I) and (II) wherein:
~ .
Rl is selected from:
`~ (1) Cl_l0 alkyl;
(2) substituted Cl_l0 alkyl in which one
or more substituent(s) is selected from
(a) halogen,
~ (b) hydroxy,
¦~ (c) Cl_lo alkoxy,
(d) Cl_5 alkoxycarbonyl,
(e) C1_5 acyloxy~
f) C3-8 cycloalkyI, ~ ~ -
(g) phenyl,
~ h) substituted phenyl in which the
l- 30 substituentæ are X and Y, and :
~: (i) oxo;
(3) C3-8 cycloalkyl;
(4) substituted C3_8 cycloalkyl in which one
substituent is selected from
~,, ~:~ . : -
-~
1~28g77
F9 ;:
0019/MW14 ~ 17700IA
(a~ Cl_l0 alkyl,
(b) substituted Cl_l0 alkyl in which the
substituent is selected from
s (i) halogen,
(ii) hydroxy,
(iii) Cl_l0 alkoxy
(iv) Cl_5 acyloxy,
(v) Cl_5 alkoxycarbonyl, ..
(vi) phenyl,
(vii) substituted phenyl in which
the substituents are X and Y, and
(viii) OxO, ,~
(c) halogen,
lS (d) hydroxy,
: (e) Cl_lo alkoxy,
(f) Cl_5 alkoxycarbonyl,
(g~ Cl_5 acyloxy,
~: (h) phenyl,
~` 20 (i) substituted phenyl in which the -
substituents are X and Y;
. (5) phenylamino;
;-: (6) substituted phenylamino in which the
substituents are X~ and Y;
(7) phenylCl_l0alkylamino; and
~ (8) substituted phenyl Cl_l0 alkylamino in which
:~ the substituents are X and Y; ::
:
X and Y are independently selected from~
(a) OH~
(b) F, ; -~
(c~ trifluoromethyl, :-
d) Cl_3~alkoxy,
~; (e) hydrogen,
( ) 1-5 Y
, ~ ' ' '
::
, ~ ,
1328~77
F9
0019/MW14 - 12 - 17700IA
In one subclass are the compounds of
formulae (I) and ~II) wherein Rl is Cl_10 alkyl.
Illustrating this subclass are those
compounds of formulae (I) and ~II) wherein:
Rl is 2-butyl or 2-methyl-2-butyl; and
R2 is H or CH3. . .
Exemplifying this subclass are the following
compounds:
(1) 6(R)-[2-[8(S)-(2-methylbutyryloxy)-2(S),6-
dimethyl-3-oxo-1,2,3,7,8,8a(R)-hexahydronaphthyl-
l(S)]ethyl]-4(R)-hydroxy-3,4,5,6-tetrahydro-2H-
pyran-2-one; - :
(2) 6(R)-t2-~8(S)-(2,2-dimethylbutyryloxy)-2(S),6- ~ .
dimethyl-3-oxo-1,2,3,7,8,8a(R)-hexahydronaphthyl- ~:
l(S)]ethyl]-4(R)-hydroxy-3,4,5,6-tetrahydro-2~- ~
pyran-2-one; -~:
(3) 6(R)-[2-[8(S)-(2-methylbutyryloxy)-2(S)-methyl-3- ~ -
oxo-1,2,3,7,8,8a(R)-hexahydronaphthyl-l(S)]ethyl]-
; 4(R)-hydroxy-3,4,5,6-tetrahydro-2H-pyran-2-one;
(4) 6(R)-[2-[8(S)-(2,2-dimethylbutyryloxy)-2(S)-
methyl-3-oxo-1,2,3,7,8,8a(R)-hexahydronaphthyl- :
l(S)]ethyl~-4(R)-hydroxy-3,4,5,6-tetrahydro-2H- ~ -
pyran-2-one;
(5) 6(R)-t2-[8(S)-(2,2-dimethylbutyryloxy)-2(S),6(R)- -~
dimethyl-3-oxo-1,2,3,5,6,7,8,8a(R)-octahydro- -~. .
naphthyl-l(S)]ethyl]-4(R)-hydroxy-3,4,5,6-tetra- -
hydro-2H-pyran-2-one;
; ~'.' ~
'' ~--'-' :'
1~2~77
F9
0019/MW14 - 13 - 17700IA
(6) 6(R)-[2-[8(S)-(2,2-dimethylbutyryloxy)-2(S)-
methyl-3-oxo-1,2,3,5,6,7,8,8a(R)-octahydronaphthyl
-l(S)]ethyl]-4(R)-hydroxy-3,4,5,6-tetrahydro-2H-
py~an-2-one.
(7) 6(R)-[2-[8(S)-(2-methylbutyryloxy)-2(S),6(R)-
dimethyl~3-oxo-1,2,3,5,6,7,8,8a(R)-octahydro-
naphthyl-l(S)]ethyl]-4(R)-hydroxy-3,4,5,6-tetra-
hydro-2H-pyran-2-one;
(8) 6(R)-[2-[8(S)-(2-methylbutyryloxy)-2(5)-
methyl-3-oxo-1,2,3,5,6,7,8,8a(R)-octahydronaphthyl
-l(S)]ethyl]-4(R)-hydroxy-3,4,5,6-tetrahydro-2H-
pyran-2-one.
-: The compounds of formulae (I) and (II)
~ wherein R2 is methyl and a is a double bond, may be
`~; prepared from lovastatin or simvastatin or its : ::
analogs having a 6-methyl group by one of the -:.:
2~ following microbiological procedures:
(a) adding the substrate to a growing culture of :-
Nocardia autot~ro~hica for a suitable incubation
period followed by isolation, and derivatization -~:
~: if desired; : ~ ;
(b) collec:ting a culture of the bioconverting ::
microorganism and contacting the collected cells
with thé substr~ate; or
(c) preparing a cell-free, enzyme-containing extract
from the cells of the bioconverting microorganism
~ 30 and contacting this extract with the substrate.
i - Cultivation of the bioconverting micro-
organism of the genus Nocardia can be carried out by
conventional means in:a conventional culture medium
.~ , .
" ., ~ , ", . , ,., . . , . , ... . - . . - ., . ~, . . . . . . ,. . . ~
1328377
F9
0019/MW14 - 14 - 17700IA
containing nutrients well known for use with such
microorganisms. Thus, as is well known, such culture
media contain sources of assimilable carbon and of
assimilable nitrogen and often inorganic salts.
Examples of sources of assimilable carbon include
glucose, sucrose, starch, glycerin, millet jelly,
molasses and soybean oil. Examples of sources of
assimilable nitrogen include soybean solids
lo (including soybean meal and soybean flour), wheat
germ, meat extracts, peptone, corn steep liquor,
dried yeast and ammonium salts, such as ammonium ~ -
sulphate. If required, inorganic salts, such as
sodium chloride, potassium chloride, calcium carbonate
or phosphates, may also be included. Also, if
desired, other additives capable of promoting the
production of hydroxylation enzymes may be employed
in appropriate combinations. The particular
cultivation technique i3 not critical to the process ~ -
of the invention and any techniques conventionally
used for the cultivation of microorganisms may equally
be employed with the present invention. In general, ~ ;
of course, the techniques employed will be chosen
;~ having regard to industrial efficiency. Thus, liquid
culture is generally preferred and the deep culture
method is most convenient from the industrial point -~-
of view.
Cultivation will normally be carried out
~- under aerobic conditions and at a temperature within
the range from 20 to 37C., more preferably from 26O - ~-
to 28C.
,, ~,'.,~ ~ '.
:''. .. :-
132g~77
F9
0019/MW14 - 15 - 17700IA
Method (a) is carried out by adding the
substrate to the culture medium in the course of
cultivation. The precise point during the cultivation
at which the starting compound is added will vary
depending upon the cultivation equipment, composition
of the medium, temperature of the culture medium and
other factors, but it is preferably at the time when
the hydroxylation capacity of the microorganism begins
lo to increase and this is usually 1 or 2 days after
beginning cultivation of the microorganism. The
amount of the substrate added is preferably from 0.01
to 5.0% by weight of the medium, more preferably from
0.05 to 0.5%, e.g., from 0.05 to 0.1% by weight.
After addition of the substrate, cultivation is
continued aerobically, normally at a temperature
within the ranges proposed above. Cultivation is
normally continued for a period of from 1 to 2 days ~ -
after addition of the substrate.
In method (b), cultivation of the micro-
-~ organism is first carried out under conditions suchas to achieve its maximum hydroxylation capacity;
this capacity usually reaches a ma~imum between 4 and
5 days after beginning the cultivation, although this
2s period is variable, depending upon the nature and
temperature of the medium, the species of micro-
organism and other factors. The hydroxylation -
capacity of the culture can be monitored by taking
samples of the culture at suitable intervals, deter-
mining the hydroxylation capacity of the samples by
contacting them with a substrate under standard
conditions and determining the quantity of product
obtained and plotting this capacity against time as a
.
132~877
F9
0019/MW14 - 16 - 17700IA
graph. When the hydroxylation capacity has reached
its maximum point, cultivation is stopped and the
microbial cells are collected. This may be achieved
by subjecting the culture to centrifugal separation,
filtration or similar known separation methods. The
whole cells of the cultivating microorganism thus
collected, preferably, are then washed with a
suitable washing liquid, such as physiological saline
or an appropriate buffer solution.
Contact of the collected cells of the micro-
organism of the genus Nocardia with the substrate is
generally effected in an aqueous medium, for example
in a phosphate buffer solution at a pH value of from -
5 to 9. The reaction temperature is preferably within
the range from 20 to 45C., more preferably from 25
to 30C. The concentration of the substrate in the
reaction medium is preferably within the range from
0.01 to 5.0% by weight. The time allowed for the
reaction is preferably from 1 to S days, although
this may vary depending upon the concentration of the
substrate in the reaction mixture, the reaction tem~
perature, the hydroxylation capacity of the micro-
organism (which may, of course, vary from species to
- 25 species and will also, as explained above, depend
upon the cultivation time) and other factors.
The cell-free, enzyme-containing extract s~
employed in method (c) may be obtained by breaking
-~ down the whole cells of the microorganism obtained as -
described in relation to method (b) by physical or -
chemical means, for example by grinding or ultrasonic
treatment to provide a disintegrated cellular mass or -
by treatment with a surface active agent or an enzyme
- ''`'~'' ;'
.
:, .
F9 1~28877
0019/MW14 - 17 - 17700IA
to produce a cellular solution. The resulting cell-
free extract is then contacted with the substrate
under the same conditions as are described above in
relation to method (b~.
The microorganism useful in the novel
process of this invention is of the genus Nocardia. -~
Of particular importance are the known strains of
microorganism, Nocardia autotrophica, subspecies
canberrica, ATCC 35203 of the culture MA-6181 and
subspecies amethystina ATCC 35204 of the culture
MA-6180 of the culture collection of Merck & Co.,
Inc., Rahway, New Jersey. A sample of the culture
designated ATCC 35203 and ATCC 35204 is available in
the permanent culture collection of the American Type
Culture Collection at 12301 Parklawn Drive,
Rockville, MD 20852.
~fter completion of the conversion reaction
by any of the above methods, the desired compound can
be directly isolated, separated or purified by
conventional means. For example, separation and
purification can be effected by filtering the
reaction mixture, extracting the reæulting filtrate
with a water-immiscible organic solvent (such as -
ethyl acetate), distllling the solvent from the -~
extract, subjecting~the resulting crude compound to
column chromatography, (for example on silica gel or
alumina) and eluting~`the column with an appropriate
eluent, especially in an HPLC apparatus. - :
~ Where the acyl~moiety of formulae (I) or
is~ other than 2-methylbutyryl or 2,2-dimethyl-
butyryl, the acyl moiety of lovastatin may be
hydrolyzed and the hydroxyl group r~esterified with ~ -~
- :
:
:.:
::
1328877
an apyropr~ate alkanoyl halide followillg the
procedure in U.S. Patent 4,444,784. The alkanoyl
halide can be formed by standard transformations such ~ -
as substitution with an alkyl halide or other
appropriate electrophile at an acidic C-H site on an
available starting material. See ~or example U.S.
Patents 4,766,145 and allowed pending applications
S.N. 536,266 and S.N. 536,225, both of Hoffman et al,
10 filed June 10, 1988.
Starting material (1) where R2 is CH20H
may be prepared following the procedures in copending
Application S.N. 540,097 Inamine et al, filed June -~.
19, 1987. ~ :.
The compounds of formulae (I) and (II) may :~
also be prepared following the synthetic methodology
in Scheme 1.
'',"
',, ' ' '''' ~ ~'
',-","'''''''
~' ~ , .
1328877
F9 : -
0019/MW14 - 19 - 17700IA
SCHEME_l
"'W TW
A 'f TCl A ~
R, R~
0 (1) (2)
~0 TO~P
1 ) PhS~X A ~ n-Bu3snH- A
;~ 15 2) H20~ }1 P~BN ~CH3
R 2~ ~ .
OH
~ (3~ (4)
:; ~ 2 0
TO~D TO~P
:~ Jl~ ~ CH,Cl,~e:t3N J~ .
PCC/~l 3 1 ~EI CF35Ol 1~3 ~C~3
: ~ 25 R~," Rl~` osll~3
6)
:~ ~0 H~W -~
~ . 30 o ~ O o ~ O
Pd(OAc~l J'~ u~NF J'~ -
C~{3CN, 20C ~H HC~AC
Rl~J~ Rl~J~O
($~ .
:~ T = A hydroxy protecting group such as trialkylsiiyl. ::
~ R12 = H, CH3 or CH20T.
1328877 - ~
F9
0019/MW14 - 20 - 17700IA
Starting material (1) is treated with a -~-
reagent suitable for protecting the alcohol group at ~
the lactone 4-position. Examples of suitable reagents -
are trialkylsilyl chlorides, dialkylarylsilyl
chlorides and dihydropyran.
The diene (2) is treated with a halogenating
agent such as phenylselenyl chloride or bromide or
phenylsulfinyl chloride, preferably phenylselenyl ; -~
10 chloride, in an approximately equimolar ratio in an - ~ -
inert æolvent at about -80~C, for approximately 20 -
minutes; illustrative of such inert solvents are -~
methylene chloride, ether and the like. After a
standard workup the product residue is dissolved in
15 an ethereal solvent, chilled to about 0C and oxidized :-
with an agent such as 30% hydrogen peroxide or a -~ -
peroxy acid such as peroxybenzoic acid to yield a - :
halohydrin analog (3).
Intermediate (3) i8 treated with a halide . ~.
20 reducing agent such as a trialkyltin hydride or a -
~; triaryltin hydride, preferably tri-n-butyltin hydride
~ and a radical initiator~ such as azobisisobutyro-
-~ nitrile (AIBN) in an inert solvent such as benzene at
a temperature between 70C~and 100C preferably about
90C for 0.5 to S~hours preferably 2 hours, to yield -
compound (4~
Compound (4) is treated with pyridinium `
chlorochromate (~PGC) on aluminum oxide in toluene to
yield the enone (5). Compound (5) is contacted with ~ ;
trimethylsilyl trifluoromethanesulfonate and an amine
to yield the trimethylsilyl ether diene (6). -
.,
: ~ . ,~.
:~ ... .
1328877
F9
0019/MW14 - 21 - 17700IA
Compound (6) is treated with palladium acetate in
acetonitrile to form dienone (7). Hydroxyl
protecting groups are removed by treatment with
tetrabutyl a~monium fluoride and acetic acid in
tetrahydrofuran to yield product (I).
Enone (5) can be converted to compounds of
formula (I) wherein a is a single bond by treatment
with tetrabutyl ammonium fluoride in acetic acid.
Alternatively the compounds of formulae ~I)
can be prepared following the synthetic outline of
Scheme 2.
~5
31 - .
~ 20
.3
' :
~',',
.f~ ,' ~' '
,` .
1328877 : ~:
F9
0019/MW14 - 22 - 17700IA
SCH~ 2
. ::
HO~;O HO~P HO~;O : ~ -
~CI~ ~.~ ~CP13A 1~ ~O Cl ~
10 R1 - H i E;tOAc. OC R1 Q H i ~ R1 ~ H
~CH3 ~ CH3 ~--
~8) ¦Cdba)3Pd2. THF
lS . ¦(iPrO)3P 55C .
HO~P o HO~
~: 20 R~ ~CH3
on R2 OH
10) ( 11 ) . ~
CH2Cl2, 3, 5-dinethyl-
pyrazole
PCC
~ ~. ....
. .....
~D HO~
~ ~ 30 ~ R1 ~fCH + ~CH3
R~ Rz
132887~
F9
0019/MW14 - 23 - 17700IA
Diene starting material (1) is converted to
epoxides (8) and ~9) by treatment with m-chloro-
peroxybenzoic acid at about 0C. The mixture of
epoxides is then contacted with tris
(dibenzylideneacetone)-dipalladium(0) and
triisopropoxy phosphine to yield the mixture of
hydroxy dienes (10) and (11). This mixture is then
oxidized with PCC attenuated with 3,5
dimethylpyrazole to yield 5-one compound (12) and
product (I).
Enone (5) of Scheme 1 can also be formed
from hydroxyl protected epoxide (9) or the mixture of
epoxides (8) and (9) as shown below:
~pO TO~pO
Il ~ 1l ~o
2 ~- O i R, 0 H
~ Ph2P PCl2 ~CH~
R~ Z~ Ph~3, 1 20C
(9) (5) : ~
- :
': -
Compound (5) can then be employed in Scheme 1
to form product (I).
~' ~''':',"
.' ,,
132~877 ~
F9
0019/MW14 - 24 - 17700IA
Where the reaction conditions of the above
noted chemical transformations would be deleterious ..
to the substituents in the 8-acyloxy moiety, the
acetoxy group can be employed as a protecting group
which after the elaboration elsewhere in the molecule
can be removed by hydrolysis to give the 8-hydroxy -
derivative which then can be acylated according to
the general procedures described in U.S. Patent
lo 4,661,4~3.
Where the product formed by the above
described synthetic pathways is not the desired form
of that compound, then that product may be subjected
to one or more further reactions such as hydrolysis,
disilylation, salification, esterification, acylation,
ammonolysis or lactonizaton by conventional methods. -
Preferred metal salts are salts with alkali
~ metals, such as sodium or potassium, salts with -:
- alkaline earth metals, such as calcium or magnesium,
~` 20 or salts with other metals such as aluminum, iron,
zinc, copper, nickel or cobalt, of which the alkali
metal, alkaline earth metal, and aluminum salts are -
preferred, the sodium, calcium and aluminum salts -
being most preferred. -
2s Preferred amino acids to form amino acid
sal~s are basic amino acids, such as arginine,
~ lysine, a,~-diaminobutyric acid or ornithine.
i Preferred amines to form amine saltR include
~; dibenzylamine, ethylenediamine, morpholine, and
~;~ 30 tris(hydroxymethyl)aminomethane. Also preferrred is
` ammonia to form the ammoniun~æalt.
Esters are preferably the alkyl esters, such
as the methyl, ethyl, propyl, isopropyl, butyl,
, ~ .
1~ ~r - :. .,
,
1328877
F9
0019/MW14 - 25 - 17700IA
isobutyl, or pentyl esters, of which the methyl ester
is preferred. However, other esters such as phenyl-
Cl_5alkyl, dimethylamino-Cl_5alkyl, or acetylamino-
Cl_5alkyl may be employed if deæired.
Metal salts of the carboxylic acids offormula (II> may be obtained by contacting a
hydroxide or carbonate with the carboxylic acid of
formula (II). The aqueous solvent employed is
preferably water, or it may be a mixture of water
with an organic solvent, preferably an alcohol (such
as methanol or ethanol), a ketone (such as acetone),
an aliphatic hydrocarbon (such as hexane) or an ester
(such as ethyl acetate). It is preferred to use a
mixture of a hydrophilic organic solvent with water.
Such reactions are normally conducted at ambient
temperature but they may, if desired, be conducted
with heating or cooling.
Amine salts of the carboxylic acids of
formula (II) may be obtained by contacting an amine
in an aqueous solvent with the carboxylic acid of
formula (II); Suitable aqueous solvents include
water and mixtures of water with alcohols ~such as
methanol or ethanol), ethers (such as diethyl ether
:~ 25 and tetrahydrofuran), nitriles (such as acetonitrile)
or ketones (such as acetone); it is preferred to use
aqueous acetone as the solvent for this reaction.
The reaction is preferably carried out at a
temperature of ambient or below, more preferably a
temperature of from 5 to 10C. The reaction
immediately goes to completion. Alternatively, a
metal salt of the carboxylic acid of formula (II)
(which may have been obtalned as described above) can
,'.
1328877
F9
0019/MW14 - 26 - 17700IA
be dissolved in an aqueous solvent, after which a
mineral acid salt (for example the hydrochloride) of
the desired amine is added, employing the same
reaction conditions as when the amine itself is
reacted with the carboxylic acid of formula (II) and
the desired product is then obtained by metathesis.
Amino acid salts of the carboxylic acids of
formula ~II) may be obtained by contacting an amino
acid in aqueous solution with the carboxylic acid of
formula (II). Suitable aqueous solvents include
water and mixtures of water with alcohols (such as :
methanol or ethanol) or ethers (such as tetrahydro-
furan).
Esters, preferably alkyl esters, of the
carboxylic acids of formula (II) may be obtained by
contacting the carboxylic acid of formula (II) with ~
an appropriate alcohol, preferably in the presence of
an acid catalyst, for example a mineral acid (such as ~
hydrochloric acid or sulphuric acid), a Lewis acid :
- (for example boron trifluoride) or an acidic ion
exchange resin. The solvent employed for this
reaction is not critical, provided that it does not
adversely affect the reaction; suitable solvents
include the alcohol itself, benzene, chloroform,
ethers and the like. Alternatively, the desired ~ I
product may be obtained by contacting the carboxylic
acid of formula (II) with a diazoalkane, in which the
, alkane moiety may be substituted or unsubstituted.
I 30 This reaction is usually effected by contacting the
acid with an ethereal æolution of the diazoalkane.
As a further alternative, the ester may be obtained
by contacting a metal salt of the carboxylic acid of
F9 1328877
0019/MW14 - 27 - 17700IA
formula (II) with a halide, preferably an alkyl
halide, in a suitable solvent; preferred solvents
include dimethylformamide, tetrahydrofuran,
dimethylsulfoxide and acetone. Finally, esters may
also be obtained from the lactone of formula (I) by
reaction with an appropriate alkoxide in an absolute
alkanol. All of the reactions for producing esters
are preferably effected at about ambient temperature,
lo but, if required by the nature of the reaction
system, the reactions may be conducted with heating
or cooling.
Lactones of the carboxylic acids of formula
(I) may be obtained by lactonizing the carboxylic
acids of formula (II) under ordinary conditions known
to one skilled in the art.
The intrinsic HMG-CoA reductase inhibition ~-
activity of the claimed compounds i9 measured in the
in vitro protocol published in J. Med. Chem., 28, p.
347-358 (1985).
Representative of the intrinsic HMG-CoA
reductase inhibitory activities of the claimed
compounds is the relative potency of 6(R)-[2-[8(S)- -
(2,2-dimethylbutyryloxy)-2(S),6-dimethyl-3-oxo-1,2,3,-
7,8,8a(R)-hexahydronaphthyl-l(S)]ethyl]-4(R)-hydroxy-
3,4,5,6-tetrahydro-2H-pyran-2-one which exhibited an
IC50 value of 2 ng/ml when compared to an IC50 value - -
of 4.2 ng/ml for simvastatin. Compound 6(R)-[2-[8(S)-
(2,2-dimethylbutyryloxy)-2(S), 6(R)-dimethyl-3-oxo-
1,2,3,5,6,7,8,8a(R)-octahydro-naphthyl-l(S)~ethyl]-
4(R)-hydroxy-3,4,5,6-tetrahydro-2H-pyran-2-one;_
exhibited an IC50 of 20ng/ml.
'...
~ . . .
. ~ . ..
F9 1328877
0019/MW14 - 28 - 17700IA
The compounds of this invention are useful
as antihypercholesterolemic agents for the treatment
of arteriosclerosis, hyperlipidemia, familial
hyperch~lesterolemia and the like diseases in humans. -
They may be administered orally or parenterally in
the form of a capsule, a tablet, an injectable
preparation or the like. It is usually desirable to
use the oral route. Doses may be varied, depending
on the age, severity, body weight and other conditions
of human patients but daily dosage for adults is -
within a range of from about 2 mg to 2000 mg
(preferably 10 to 100 mg) which may be given in two
to four divided doses. Higher doses may be favorably
employed as required.
The compounds of this invention may also be
coadministered with pharmaceutically acceptable
nontoxic cationic polymers capable of binding bile
acids in a non-reabsorbable form in the gastro-
intestinal tract. Examples of such polymers include
cholestyramine, colestipol and poly[methyl-~3-tri-
methylaminopropyl)imino-trimethylene dihalide]. The
relative amounts of the compounds of this invention
and these polymers is between 1:100 and 1:15,000.
Included within the scope of this invention
is the method of treating arteriosclerosis, familial
hypercholesterolemia or hyperlipidemia which comprises
administering to a subject in need of such treatment
;~ a nontoxic, therapeutically-effective amount of the
compounds of formulae (I) or ~II) or pharmaceutical
~ compositions thereof.
;
1328877
- 29 - .
The following examples illustrate the
preparation o~ the compounds of tlle formulae ~I) and
~II) and their incorporation into pharmaceutical
composi~ions and as such are not to be considered as
limiting the invention set forth in the claims
appended hereto.
EXAMPL~ 1
Preparation o~ 6(R)-~2-[8(S)-(2,2-dlmethyl-
butyryloxy)-2(S),6-dimethyl-3-oxo-1,2,3,7,8,8a(R)-
hexallydronaphthyl-l(S)]ethyl]-4(R)-hydro~y-3,4,5,6-
~e~Lhydro-2H-pvran-2-one
Utilizing the general procedure for the
bioconversion of sodium salt of 7-[1,2,6,7,8,8a(R)- :
hexahydro-2(S),6(R)-dimethyl-8(S)-(2,2-dimethyl-
butyryloxy)-l(S)-naphthyl]-3(R),5(R)-dihydroxy-
¦ heptanoic acid as described in co-pending patent
application Serial No. 5~0,097, Inamine et al,
filed June 19, 1987, the above titled compound was
~;~ isolated as a minor product.
~ .
~ 25
~ ' :
1~ :
::.
~' ~.' '. '
..
~ :',.
' ' " ' ~ ' ' ' ' . ' - ~ .
132~77 ~:
F9
0019/MW14 - 30 ~ 17700IA
The following media are utilized in the
bioconversion reactions described below:
Medium A &rams per liter
distilled water
Yeast extract 4.0
Malt extract 10.0
Nutrient broth 4.0
lO Dextrose 4
pH 7.4
Medium sterilized for 20 min. at 121C
~ Medium B Gramæ per liter
; lS distilled water
Dextrose 10.0
Polypeptone 2.0
Meat extract 1.0
Corn steep liquor 3.0
pH 7.0
Medium sterilized for 20 min. at 121C
I. Culture Conditions and Bioconversion
A lyophilized tube of Nocardia autotrophica
subsp. canberrica ATCC 35204 (MA-6180) was used to
inoculate 18 ~ 175 agar slants (Medium A) which were
incubated at 27C for 7 days. The slant culture was
s washed with 5 ml of sterile medium B and transferred
to a 250 ml flask containing 50 ml of sterile medium
~;~ 30 B. This first stage seed was grown at 27OC on a 220 -
rpm shaker and, after 24 hours, 2 ml was transferred -
to another flask of sterile medium B.
~ : -
~ . .
... , . ., . . , ; .. ", . .. " . . .. , , , , ,, ., ., ..... .. ,,. ,. , .. ~ . " " .. .... .. ~ . ~ .. . . -. -, -
1328~77
F9
0019/MW14 - 31 - 17700IA
Grown under the above conditions, the second
seed was used to start the bioconversion culture: 20
ml of the seed culture was placed in 400 ml of
sterile medium B in a 2L flask. After the culture
had grown for 24 hours, 80 mg of the sodium salt of
7-[1,2,6,7,8,8a~R)-hexahydro-2(S),6(R)-dimethyl-8(S)-
(2,2-dimethylbutyryloxy)-l(S)-naphthyl]-3(R),5(R)-di-
hydroxyheptanoic acid was added to each flask. The
incubation was continued for 28 hours or until no
7-[1,2,6,7,8,8a(R)-hexahydro-2(S),6(R)-dimethyl-8(S)-
(2,2-dimethylbutyryloxy)-l(S)-naphthyl]-3(R),5(R)-di-
hydroxyheptanoic acid could be detected by HPLC. The
whole broth was clarified by centrifugation followed
by filtration through Whatman No. 2 filter paper.
II. HPL~ Methods
Aliquots of whole broth could be analyzed
for 7-[1,2,6,7,8,8a(R)-hexahydro-2(S),6(R)-dimethyl-
8(S)-(2,2-timethylbutyryloxy)-l(S)-naphthyl]-3(R),
5(R)-dihydroxyheptanoic acid derivatives by HPLC. -
~-~ Filtered broth could be injected directly (10 to 20
~1) or after dilution with methanol. The compounds
were separated on reverse phase columns utilizing a
gradient from 35 to 45 percent aqueous acetonitrile
at flow rates ranging between 1 and 3 ml/min.
Addition of glacial acetic acid or H3PO4 (0.1 ml/L
mobile phase) was required for the separation of the
free acids. Derivatives of 7-[1,2,6,7,8,8a(R)-
hexahydro-2(S),6(R)-dimethyl-8(S)-(2,2-dimethyl-
butyryloxy)-l~S)-naphthyl]-3-(R),5(R)-dihydroxy~ ~-
heptanoic acid were detected by monitoring the
absorbance at 238 nm, as well as the absorbance ratio
of 238 nm/228 nm. The desired products, ~ `
6(R)-[2-[8(S)-(2-alkylacyloxy)-2(S),6-dimethyl-3- ~
.. ~ . ~ : . .
~328377
F9
0019/MW14 - 32 - 17700IA
oxo-1,2,3,7,8,8a(R)-hexahydronaphthyl-l(S)]ethyl]-
4(R)-hydroxy-3,4,5,6-tetrahydro-2H-pyran-2-one, were
detected by monitoring the absorbance at 293 nm.
III. 6(R)-[2-[8(S)-(2,2-dimethylbutyryloxy)-2(S),6-
dimethyl-3-oxo-1,2,3,7,8,8a(R)-hexahydronaphthyl-
l(S)]-ethyl~-4(R)-hydroxy-3,4,5,6-tetrahydro-2H-
pyran-2-one
19 Following the general procedure described
above, the pH of the whole broth from the bio-
conversion of twenty kilograms of the sodium salt of
7-[1,2,6,7,8,8a(R) hexahydro-2(S),6(R)-dimethyl-8(S)-
(2,2-dimethylbutyryloxy)-l(S)-naphthyl]-3(R),5(R)-di-
hydroxyheptanoic acid (12,700 liters) was adjusted to
~`~ 4.0 with 2N sulfuric acid and was then extracted with
ethyl acetate (2x4500 1.). The whole broth extraction
was followed by an extraction of the ethyl acetate
solution into lN sodium bicarbonate (20% by volume)
20 and the aqueous e2tract was then washed with ethyl ~ -
~3~ acetate. To the aqueous extract was then added
methylisobutylketone (MIBK, 570 I.) and the pH of the - ~
aqueous phase adjusted to 3.1 using 7.2N sulfuric -~-
acid. The MIBK extract of the acidified aqueous
~ 25 phase was then separated from the aqueous phase which
J~ was then extracted with a second time MIBK (570 1.).
' The MIBK extracts were combined, filtered through
,~ diatomaceous earth,nazeotropically dried and
3 concentrated in vacuo to 870 liters. The MIBK
solution was heated to 95C, and then treated with
trifluoroacetic acid (0.9 1.) in MIBK (23 l.).
,- :
~4
~: "' .
_ 33 _ ~32~77
A~ter about 15 minutes, the mixture was cooled to
2s~C and washed successively with lN sodium
bicar~onate (0.5 volumes3 an~ water (2x0.5 volumes).
The organic phase was concentrated ~a vacuo and the
residue dissolved in acetonitrile, which was then
diluted to 30% acetonitrile using 0.02M phosphate
buffer at pH=7. Aliguots (1/3) which contain
approximately 700 gm. of
. 6(R)-[2-[8(S)-(2,2-dimethylbutyryloxy)-6-hydroxy-
methyl-2(S)-methyl-1,2,6,7,8,8a(R)-hexahydro-
naphthyl-l(S)]ethyl]-4(R)-hydroxy-3,4,5,6-tetra-
hydro-2H-pyran-2-one were ehromato~raphed over an
SP-207 ~300 1, brominated copolylner o~ styrene and
divinylbenze~e, Mitsubishi Co.) column. Elution with
acetonitrile/buffer <30%, 37%, 47%, 57%,) and
acetonitrile/water (67%j gave the above titled .~ -~
product and the 6-hyd~oxymethyl compound as a
mixture. The desired product may be ~urther puri~ied -~-
by removing most of the 6-hydroxymethyl compound by
crystallization by dissolving the mixture in
isopropyl acetate (IPAC) or methyl-~-butyl ether
(MTBE> and then adding the solution to a non-polar
solvent (g-heptane, cyclohexane or petroleum ether).
IV. Isolation of 6(R)-[2-[8(S)-(2,2-dimethyl-
butyryloxy)-2(S),6-dimethyl-3-oxo-1,2,3,7,8,8a(R)- ~
hexahydronaphthyl-l(S)]ethyl]-4(R)-llydroxy-3,4,5,6- - --
tetrahydro-2H-pvran-2-one
The crystallization mother liquors from Step
III were concentrated to an oil and then dissolved in
toluene:methanol:acetonitrile (8:1:1, V:V:V) to a
final volume of 100 ml. This solution was charged to
* Trade Mark
, . , , i , , "
132~77
_ 34 -
a 10 liter column of Sepha~ex LH-20 (Pharmacia Inc.)
equilibrated with hexane:toluelle:lnethanol (3:1:1,
V:V:V) and eluted wi~h this solvell~ at a Elow rate of
100 ml/min.
The desired compound eluted between 11 and
14 column volumes and the rich cut eluant was
collcentrated to a solid. The pro~uct was Purther
puri~ied by preparative reverse pllase hplc on a C18
column (21.4 mm ID x 30 cm) eluted with a linear
gradient starting 10 minutes a~ter injection ~rom 25%
acetonitrile in water to 75% acetonitrile in water
over 40 minutes at a ~low rate of 10 ml/ min. The
fractions containing the desired product (eluting at
2~ minutes) were combined and concentrated to yield
about 400 mg. of the desired product in cry~talline
~orm.
13C N~IR Data (CD2C12, ~c=53.8 ppm)
~ pm
9.4 36.5 67.0 ~
10.6 . ~6.8 76.0 :
24.1 37.7 123.1 :
2524.3 39.0 124.5
24.4 39.6 144.3
~- 24.9 42.7 154.9
32.9 43.3 170.2
33.4 63.1 177.6
203.4
* Trade Mark
.
.
.
.
1~28877
19/MW14D - 35 - 17700IA
MS analysis showed a weak M+ ion at m/z 432 and
fragment ions at m/z 316 and 173 (base).
W spectrum exhibited a ~max = 290 nm, with ~=21,900.
In a similar fashion Nocardia autotrophica
subsp. canberrica ATCC 35203 (MA6181) was utilized in
the bioconversion reaction with the sodium salt of
7-[1,2,6,7,8,8a(R)-hexahydro-2(S),6(R)-dimethyl-8(S)-
(2,2-dimethylbutyryloxy)-l(S)-naphthyl~-3(R),5(R)-di-
hydroxyheptanoic acid to afford the desired products.
Additionally, the æodium salt of
7-[1,2,6,7,8,8a(R)-hexahydro-2(S),6(R)-dimethyl-8(S)-
(2-methylbutyryloxy)-l(S)-naphthyl]-3(R),5(R)-
dihydroxyheptanoic acid, the sodium salt of ringopened lovastatin, was subjected to analogous
bioconversion reactions utilizing both N. autotrophic
Qubsp. amethystina ATCC 35204 (MA6180~ and N. -
autotrophic subsp. canberrica ATCC 35203 (MA6181) to
afford 6(R)-[2-[8(S)-(2-methylbutyryloxy)-2(S),6-
dimethyl-3-oxo-1,2,3,7,8,8a(R)-hexahydronaphthyl-
l(S)]-ethyl]-4(R)-hydroxy-3,4,5,6-tetrahydro-2H-pyran- -
2-one.
EXAMPLE 2
Preparation of 6(R)-[2-[8(S)-(2,2-dimethylbutyryloxy)-
2(S),6-dimethyl-3-oxo-1,2,3,5,6,7,8,8a(R) Octahydro-
naphthyl-l(S)]-ethyl]-4(R)-hydroxy-3,4,5,6-tetrahydro-
2H-~yran-2-one
Thirty milligramæ of the dieneone product of
30 example 1 (Rl = 2 methyl-2-butyl, a = double bond), -
dissolved in 3 ml of ethyl acetate, was hydrogenated -~
(1 atm H2, room temperature) over 6 mg of 10%
palladium on carbon for 30 hours. Removal of the
~ :,- ,. .
: ' '. .:
-,, - -:. .
- '~' .
1328877
F9
0019/MW14 - 36 - 177001A
catalyst by filtration and evaporation of the solvent
afforded the title compound. IR(film): 1718 cm -1,
1665 cm -1. MS(EI): m/z 434 (M+).
EXAMPLE 3
Preparation of 6(R)-t2-t8(S)-(2,2-dimethylbutyryloxy~-
2(S),6-dimethyl-3-oxo-1,2,3,7,8,8a(R)-hexahydro-
naphthyl-l(S)~-ethyl~-4(R)-hydroxy-3,4,5,6-tetrahydro-
lo 2E-pyran-2-one
(a) 6(R)-[2-~8(S)-(2,2-Dimethylbutyryloxy)-2(S),6(R)-
dimethyl-1,2,6,7,8,8a(R)-hexahydronaphthyl-l(S)]-
ethyl]-4(R)-(t-butyldimethylsilyloxy)-3,4,5,6-
tetrahvdro-2H-pYran-2-one (2') ~ -
~E~-Butyldimethylsilyl chloride (8 g, 52
mmol) was added to a stirred solution of 6(R)-[2-
[8(S)-(2,2-Dimethylbutyryloxy)-2(S),6(R)-dimethyl-
1,2,6,7,8,8a(R)-hex:ahydronaphthyl-l(S?]ethyl]-4~R)-
hydroxy-3,4,5,6-tetrahydro-2H-pyran-2-one (20 g, 48
mmol~ and imidazole (6.8 g, 0.1 mol) in DMF (150 mL)
at 0C. The resulting mixture was stirred at 0C for
5 minutes, then warmed to room temperature and
stirred for~5 hours. TLC analysis of an aliquot
indicated that the reaction~was complete. The -~
2s reaction mixture was~ poured into cold water and
extracted with ether. The ether~eal extract was
washed with dilute hydrochloric acid, water and 5%
sodium~bicarbonate solution.~ After;drying over
MgS04,-the organic extract was filtered and the
filtrate was concentrated in vacuo to afford the
desired product as a eolorless, viscous oil: NMR
(CDC13) ~ 0.84 (3~, t, J = 7~z), 0.89 (3H, d, J =
~:
- ~ .
1328~77
F9
0019/7.~W'14 - 37 - 17700IA
:
7~z), 0.90 (9H,s), 1.09 (3H, d, J = 7Hz), 1.11 (3~,
s), 1.12 (3H, s), 4.30 (H, m), 4.60 (H, m), 5.33 ~E,
m), 5.51 (H, m), 5.77 (H, d of d, J = 10, 6Ez), 5.98
(H, d, J = lOHz).
(b) 6(R)-[2-~5(S)-Chloro-4a(S)-hydroxy-8(S)-(2,2- ~-
dimethylbutyryloxy)-2(S),6(R)-dimethyl-1,2,4a,5,
~,7,8,8a(S)-octahydronaphthyl-l(S)]ethyl]-4(R)-
lo (t-butyldimethylsilyloxy)-3,4,5,6-tetrahydro-2H-
pyran-2-one (3'~
A solution of phenylselenyl chloride (10 g,
52 mmol) in methylene chloride (50 mL) waæ added
dropwise to a stirred solution of compound 2' (25.2
g, 48 mmol) in methylene chloride (350 mL) cooled in
a dry ice/i-propanol bath (-780C). The resulting
mixture was stirred at -78C for 20 minutes, poured
;~ into cold water (300 mL) and extracted with ether
twice (400 mL, then 150 mL). The combined extracts ~ --
were dried (MgS04), filtered and concentrated to
l afford an oily residue which was dissolved in -
l~ tetrahydrofuran <300 mL). This solution was chilled ~
in an ice bath (0C), and 30% hydrogen peroxide (15 - -
-~ mL) was added. The resulti;ng mixture was stirred at
2s 0C for 5 minutes, then warmed to room temperature ~-
and stirring continued for 1 hour. The reaction
~ mixture was poured into cold water and extracted with - -
j~ chloroform three times (400 mL, then 2 x 100 mL). :-
The combined extracts were dried (MgS04), filtered
~; 30 and concentrated to yield a residue which was
purified by flash chromatography on a silica gel
column. Elution with hexane:ethyl acetate (5:1/v:v)
~ removed the impurities. Further elution with ~ -
1: . :,
,~
~ :
132~877
F9
0019/MW14 - 38 - 17700IA
hexane:ethyl acetate (4:1/v:v) provided the title
compound as a pale yellow gum which later solidified
on standing: mp 117-8C, NMR (CDC13) ~ 0.075 (3~, s),
0.08 (3H, s), 0.85 (3H, t, J = 7~z), 0.88 (9~, s),
0.89 (3H, d, J = 7Hz), 1.15 (3H, s), 1.16 (3H, s),
1.32 (3H, d, J = 7Ez), 1.58 (2H, q, J = 7Hz), 3.39
(H, ~), 4.05 (H, bs), 4.30 (~, m), 4.60 (H, m), 5.32
(H, m), 5.59 (~, d, J = llHz), 5.79 (H, d .of d, J =
lo 11, 6Hz).
Anal. Calcd. for C31H53C106Si: C, 63.61; H, 9.13.
Found: C, 63.80; H, 9.04.
(c) 6(R)-[2-[4a(S)-hydroxy-8(S)-(2,2-dimethylbutyryl-
oxy)-2(S),6(R)-dimethyl-1,2,4a,5,6,7,8,8a(S)-
octahydronaphthyl-l(S)]ethyl]-4(R)-~t-butyldi-
methylsilyloxy)-3,4,5,6-tetrahydro-2H-pyran-2-one
(4l~
Tributyltin hydride (7.06 ml, 26.25 mmol)
20 and azobisisobutyronitrile (AIBN) (0.82 g, 5.0 mmol)
were added to a magnetically stirred solution of
.
chlorohydrin 3l (8.78 g, 15 mmol) in benzene (100 :
ml). The resulting solution was refluxed for 2
hours, cooled and concentrated in vacuo to a viscous
yellow oil which was stirred with pet ether (200 ml)
at -15C (icelacetone bath) to provide 4~. as a
fluffy, colorless solid (6.9 g, mp 97-9C). The
iltrate was extracted with CH3CN (4 x 50 ml) to
~- remove all of the product contained in the pet
ether. The CH3CN extracts were combined and
concentrated to a colorless oil which was purified by
flash chromatography on a æilica gel column. Elution
with ethyl acetone/hexane (l:3/v:v) gave a colorless
, ' '', . '
~28~77
F9
0019/MW14 - 39 - 17700IA
solid ~1.0 g) which was stirred in pet ether (25 ml)
at 0C to remove some tin residues. The mixture was
filtered to provide the product 4' as a colorless
solid. M.P. 103-4C, nmr (CDC13) ~ 0.07 (3H, s),
0.08 (3H, s), 0.88 (9H, s), 1.15 (3E, s), 1.16 (3H,
s), 1.20 (3H, d, J = 7Hz), 2.78 (H, s), 4.28 (H, m),
4.58 (H, m), 5.30 ~H, m), 5.58 (H, d, J = lOHz), 5.67 ;
(H, dd, J = 10, 5Hz).
lo Calcd. for C31H5406Si: C, 67.59; H, 9 88
Found: C, 67.20; H, 9.99.
"
(d) 6R-[2-t3-oxo-8(S)-(2,2-dimethylbutyryloxy)-2(S)6-
(R)-dimethyl-1,2,3,5,6,7,8,8a-octahydronaphthyl :
~; 15 l(S)]-ethyl~-4(R)-(t-butyldimethylsilyloxy)-3,4,5,
6-tetrahydro-2H-~yran-2-one. (5
7.2 g (12 mmol) of compound (4~) was -
combined with 60 ml of toluene and 42 g of pyridinum :
chlorochromate/aluminum oxide. The mixture was
20 stirred and heated on a steam bath for 20 minutes --~
after which time tlc showed the reaction to be
~ complete.~The mixture was cooled, filtered and the
;~: solids washed with warm toluene (4 X 50 ml). The ~ -
solvent was evaporated to yield an amber gum. Nmr . .:
(CDC13) ~0.073 (3H;,~s), 0.079 (3H, s), 0.804 (3H, t,
J = 7 Hz), 0.881 (9H, s), 1.026 (2H, d, J = 6 Hz),
1.036 (3H, d, J = 6 Hz), 1.10 (6H, br s), 2.55 - 2.66 ~:
(3H, m), 4.276 (H, m), 4.588 (H, m) 5.42 (H, m),
5.910 (H, d, J = 1.5 Hz)
,~'' . .,',.~ '' .
~'~' -.`' . ,'
1328~77
F9
0019/MW14 - 40 - 17700IA
(e) 6(R)-t2-[8(S)-(2,2-dimethylbutyryloxy)-2(S),6(R)-
dimethyl-3-(trimethylsilyloxy)-1,2,6,7,8,8a(R)-
hexahydro-l(S)]ethyl]-4(R)-(t-butyldimethylsilyl-
oxy)~3.4~5~6-tetrahvdro-2H-pvran-2-one. (6'~
The amber gum product of Step 3d was
dissolved in methylene chloride and cooled to 0C
under argon. The solution was treated with
triethylamine (7.2 ml, 50 mmol) followed by slow
addition of trimethylsilyl trifluoromethanesulfonate
(5.4 ml, 28 mmol) while maintaining the temperature
, below 3C. After stirring at 0C for 15 minutes (tlc
! showed the reaction to be complete by 5 minutes) the
¦ dark solution was diluted with methylene chloride
(100 ml), washed with sat. NaHC03 (100 ml), dried and
the solvent evaporated. " -
(f) 6(R)-~2-[8(S)-(2,2-dimethylbutyryloxy)-2(S),6-
dimethyl-3-oxo-1,2,3,7,8,8a(R)-hexahydronaphthyl-
l(S)]ethyl]-4(R~-(t-butyldimethylsilyloxy)-
3.4.5 6-tetrahvdro-2~-~vran~ ne. (7')
~; The dark-amber residue of Step (3e) was
dissolved in acetonitrile/tetrahydrofuran. Palladium`I
(II) acet~ate (3.0g, 13.0 mmol) was added to the
mixture and the mixture stirred at room temperature
~ for 22 hours, at which time tlc showed the reaction
'~ to be complete. The mixture was filtered through a 3
3~ cm~pad, of silica gel and then washed with ethyl
acetate (150 ml), and the solvent evaporated. Nmr
(CDC13) ~ 0.076 (3H, s) 0.082 (3H, s) 0.752 (3H, t, J
= 7 Hz) 0.883 (9H, 9) 1.033 (3H, d, J = 7 Hz) 1.059
(3H, s) 1.065 ~3H, s) 1.804 (3H, s) 4.295 (H, m) ~
4.606 (~, m) 5.408 (H, m) 5.781 (H, br s), 6.136
(H, br s).
's ~ :
:
:',:; .'''` - -
1328~77
F9
0019/MW14 - 41 - 17700IA ~-
(g~ 6(R)-[2-[8(S)-(2,2-dimethylbutyryloxy)-2(S),6- ~-
dimethyl-3-oxo-1,2,3,7,8,8a(R)-hexahydronaphthyl-
l(S)]ethyl]-4(R)-hydroxy-3,4,5,6-tetrahydro-2H-
furan one. (I)
The dark brown gum of Step (3f) was -~
dissolved in tetrahydrofuran, and to this was added a ~ -
mixture of tetra-n-butyl ammonium fluoride (30 ml)
and acetic acid (5.6 ml). The combined mixture was
lo stirred at 50C for 4 hours, cooled, diluted with
ethyl either (400 ml) washed with water (5xlO0 ml),
dried and the solvent evaporated. The residue -~ -
solidified to a brown mass. The brown mass was -
chromatographed on a 50 mm LP column U8 ing
hexane-ethylacetate, 1:1 for the first 10 fractions
(25 ml fractions) then 1:2 for 11, then 1:4. The
titled product was found in fraction3 25-53, m.p. - -
160-174C. This chromatographed product was then
recrystallized from ethyl acetate (30 ml)-hexane (30
ml). After drying at 60OC for 2 hours under a vacuum
the titled product was obtained with M.P. 179-180C. -~
Nmr (CDC13) ~ 0.758 (3H, t, J = 7.4 Ez) 1.035 (3H, d,
~; J = 7.4 Hz) 1.063 (3H, s), 1.069 (3H, s), 1.867 (3H,
s), 2.63 (H, ddd, J = 1.47, 3.64, 12.6 Hz), 2.749 (H,
dd, 4.94, 12,6 Ez) 4.398 (H, m), 4.645 (H, m), 5.424
(H, m), 5.781 (H, br~), 6.138 (H, brs)
- Anal. Calcd- for C25H366 C, 69-42; H~
~ Found: C, 69.73; H, 8.54
.:
;~ 30 ~XAMPL$ 4
~; Preparation of 6(R)-t2-t3-oxo-8(S)-(2,2-dimethyl-
-~ butyryloxy)-2(5), 6(R)-dimethyl-1,2,3,5,6,7,8,8a- -
octahydronaphthyl-l(S)]ethyl]-4(R)-hydroxy-2,4,5,6-
tetrahvdro-2H-pyran-2-~ne. ~-
; '.", :'
~32g~77
F9
0019/MW14 - 42 - 17700IA
A solution of compound (5') (500 mg, 0.9
mmol) of example 3 in acetic acid (42 ml) and water
(15 ml) was heated at 70C for 3 hours. After
cooling, the reaction mixture was diluted with water
and extracted with ether. The ethanol extract was
washed with water five times, then washed with
aqueous sodium bicarbonate and brine. After drying
and filtration, the filtrate was evaporated to afford
a residue which was purified by flash chromatography
on silica gel column. Elution of the column with 30%
of acetone in methylene chloride gave the title
compound as a solid: mp 117-8C; nmr (CDC13) ~ 0~80
(3H, t, J = 7 Hz), 1.02 (3H, d, J = 7 Hz), 1.04 (3H,
d, J = 7 Hz), 1.10 (6H, s), 2.64 (H, m of d, J = 18
¦~ Hz), 2.72 (H, d of d, J = 18, 4 Hz), 4.3H (H, m),
4.65 (H, m), 5.44 (H, m), 5.92 (~, bs).
~ Anal. Calcd- for C25H386 C, 69.0 ;
¦~ Found: C, 68.85; H, 8.65
EXAMPLE 5-9
Following the procedure of Example 3 and
~ substituting an equivalent amount of reactant (A) for
¦ simvastatin in step (a), the product (B) is formed.
j ' '. .
'
~ -
,
.' ,
,~ :
13288 77 - ~
F9 `-
0019/MW14 - 43 - 17700IA --~
HO ~ HO ~ O
~ ~ CH3
R2 R
A B
: . :
Example
' :.
Rl = 2-butyl, R2 = CH3;
~: 6 Rl = 2-butyl, R2 = H;
7 Rl = 2-methyl-2-butyl, R2 = H;
8 Rl = 2-methyl-2-butyl, R2 - CH2OH;
9 Rl 5 2-butyl. R2 = CH2H
.~ - ' .
EXAMPLE 10
Preparation of Ammonium Salts of Compounds II ;.
The lactone (1.0 mmol) from Example 1 is
dissolved with stirring in 0.1N NaO~ (1.1 mmol) at
ambient temperature. The resulting solution is
cooled and:~acidified by the dropwise addition of lN
HCl. The resulting mixture is:extracted with diethyl
~; 30 ether and the:extract washed with brine and dried -~
(MgSO4). The MgSO4 is removed by filtration and the
. ~ . ,,
' ,~ ~ ~ '' .,
-:
.
1328377
F9
0019/MW14 - 44 - 17700IA
filtrate saturated with ammonia (gas) to give a gum
which solidified to provide the ammonium salt.
EXAMPLE 11
Preparation of Alkali and Alkaline Earth Salts of
Compounds II
To a solution of 42 mg of lactone from
Example 1 in 2 ml of ethanol is added 1 ml of aqueous
lo NaOH ~1 equivalent). After one hour at room
temperature, the mix~ure is taken to dryness in vacuo
to yield the desired sodium salt.
In like manner, the potassium salt is
prepared using one equivalent of potassium hydroxide,
and the calcium salt, using one equivalent of CaO.
EXAMPLE 12 -
Preparation of Ethyle~ediamine Salts of Compounds II
To a solution of 0.50 g of the ammonium salt -
20 from Example 10 in 10 ml of methanol is added 0.75 ml
of ethylenediamine. The methanol is stripped off
under vacuum to obtain the desired ethylenediamine
salt. ~
~ ~ -
EXAMPL~ 13
Preparation of Tris(hydroxymethyl)aminomethane Salts
of Compounds II
To a solution of 202 mg of the ammonium salt
from Example 10 in 5 ml of methanol is added a ~-
solution of 60.5 mg of tris(hydroxymethyl)
aminomethane in 5 ml of methanol. The solvent is
removed in vacuo to afford the desired tris(hydroxy-
methyl)aminomethane salt.
'
- ~ ' " ;; ' ~ ; ; '; , A .:. "~
.
1328877
F9
0019/MW14 - 45 - 17700IA
`
I ~XAMPL~ 14
Preparation of L-Lvsine Salts of Compounds II
A solution of 0.001 mole of L-lysine and
0.0011 mole of the ammonium salt from Example 10 in
15 ml of 85% ethanol is concentrated to dryness in
vacuo to give the desired L-lysine salt.
..
lo Similarly prepared are the L-arginine,
L-ornithine, and N-methylglucamine salts.
.,
EXAMPLE 15
Preparation of Tetramethylammonium Salts of CQmpounds II :
A mixture of 68 mg of ammonium salt from ~
Example 10 in 2 ml of methylene chloride and 0.08 ml ~: ;
~1; of 24% tetramethylammonium hydroxide in methanol is
~ diluted with ether to yield the desired tetramethyl-
3~ ammonium salt.
~`
XAMPLE 16
Preparation of Methvl Esters of Com~ounds II
To a solution of 400 mg of lactone from
Example 1 i~ 100 ml~of absolute methanol is added 10 ~ :
25 ml O.l M sodium methoxide in absolute methanol. This ~:
solution is allowed to stand at room temperature for
one hour, then is diluted with water and extracted
twice with ethyl acetate. The organic phase is
separated, dried (Na2S04), filtered and evaporated in
Y~9Q to yield the desired methyl ester.
:
~'
. ~
1328377
F9
0019/MW14 - 46 - 17700IA
In like manner, by the use of equivalent
amounts of propanol, butanol, isobutanol, t-butanol,
amylalcohol, isoamylalcohol, 2-dimethylaminoethanol,
benzylalcohol, 2-acetamidoethanol and the like, the
corresponding esters are obtained.
EXAMPLE 17
Preparation of Free Dihydroxy Acids
lo The sodium salt of the compound II from
Example 11 is dissolved in 2 ml of ethanol-water
(1:1; v:v) and added to 10 ml of lN hydrochloric acid
from which the dihydroxy acid is extracted with ethyl
acetate. The organic extract is washed once with :
water, dried (Na2S04), and evaporated in vacuo with a
bath temperature not eæceeding 30C. The dihydroxy :
acid derivative slowly reverts to the corresponding,
parent lactone on standing, but is stable at a pH
above 7. :
: 20
~XAMPLE 18
As a specific embodiment of a composition of
this invention, 20 mg of lactone from Example 1, is -:
formulated with sufficient finely divided lactose to
provide a total amount of 58Q to 590 mg to fill a :.
size 0, hard-gelatin capsule.
: :
~ .
.