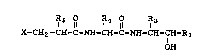Note: Descriptions are shown in the official language in which they were submitted.
: ~ 1329~8~
~ HA432
. ~ .
., - .
.,
':
. . .
,.- :
, - .
" r ' - ::
N-HETEROCYCLIC ALCOHOL DERIVATIVES
:j The present invention is directed to
.~ N-heterocyclic alcohol derivatives, and more
; ~ 5 particularly concerns such compounds useful as
~,, renin inhibitors.
i. !
:~ ' . i : : :'
i~¦ In accordance with the pre ent invention
,~¦ novel compounds which are inhibitors of renin, and ~ :
~I 10 therefore useful as cardiovascular agents, are ~ ~ -
disclosed. These compounds have the formula
. '',
.. , :
.~.
:. 15
. . ;.
~ ~ '
".,,
~ , ' . '
., .~,,"~
::~`'`3j 20
''~ 1
,, , :
.,i,
,,
i 25
! ~
': ''1 .
. ,~ , "' `
~, .
j . ~ '
~ 1329~80
HA432
-2-
I 5 I R4 1l 1 3
,' X-CH2-CH-C-NH-CH-C-NH-CH-CH-R
OH
~'.,i .
~ 5 including pharmaceutically acceptable salts
.. j o
1 thereof, wherein X is R6-~CH2)m-A-N-C-,
"i"31 0 I R1o
R6-(CH2)m-A-C- R6-(CH2)m-A-O-C- 1
" 10 R6 - ( CH2 )m-A-S-, R6 - ( CH2 3m-A-SO-, R6 ( CH2 ~m-A-SO2 t : - -
;- R6-(CH2)m-A-IN-SO2-, R6-~CH2)m~A~C~S~~
R
lo
O o :,
~1 15 R6-(CH2)m-A-(O)p-l-~ CN C; ~ ~;
1 P .... -
i A
C~2)
'
R1 is a fuIly saturated, partially saturated,
~ or unsaturated monocyclic N-heterocyclic ring of 5
i~ or 6 atoms containing at least one N atom or a
-¦ bicyclic ring in which such N-heterocyclic ring i5 fused to a benzene ring. The N-heterocyclic ring
can also include an O or S atom or up to three
~ additional N atoms. The N heterocyclic ring is
;~ attached to -CH- by way of an available carbon atom.
OH
An available N atom in the N-heterocyclic ring can
be substituted with an N-protecting group such as
~1 :
:, ~
~.
~ ~ 1 32~ ~8 0
,~, .
~-` ~ HA432
3-
~ ~- CH2-O-CH2~ SO2 ~ CH3, or 2,4-dlnitrophenyl,
;~ Q :
~ or loweralkyl, -(CH2)n- ~ , or -(CH2)n-cycloalkyl. :~
-j 5 Similarly, an available C atom ln the monocyclic
ring or an available C atom in the benzene portion
~i of the bicyclic ring can be substituted with lower . I
,1 alkyl, -(CH2)n- ~ , or -(CH2)n-cycloalkyl.
' 1 1 0 ,, ~ ,
Preferred N-heterocyclic rings are - :
R2 N O
Nr ~ N ~ ~ Nr t
'''D;~ R2 :~
S R9
:~ ~ ~ Rg, ~ ~ N-R2 , - ~ S , ~:
~ 20 - N---N N ~ N
.~ R2 Rg Rg
~ ~ ' ~ tt~- Rg, ~ ~ Rg, ~ ~ Rg
25 N ~ I I N
Rg R2
~ N~ 1~ Rg
:: N N - R2
~ .
`~!
'l
. ` ~
~ ~ 1329680
.
~ 4_ HA432
.
;
S O
R and ~ ~ Rg
N N -- ;~
R2 is -CH2-O-CH2~ SO2 _ ~ - CH3,
2,4-dinitrophenyl, hydrogen, lower alkyl,
, i. ~ ::
-(CH2)n ~ or (CH2)n-cycloalkyl;
:~ .10
CN represents a heterocyclic ring of the
, . i . .~:
:~ I formula
.~ (CH2)a :~
'~ 15
, Y N-
~ (CH2)b
:: :
.~ 20 wherein Y is -CH2, O, S, or N-Rg, a is an integer
from 1 to 4, and b is an integer from 1 to 4
provided that the sum of a plus b is an integer
from 2 to 5 and such heterocyclic rings wherein
one carbon atom has a lower alkyl substituent;
:1 25 R3 and R5 are independently selected fxom
hydrogen, lower alkyl, halo substi~uted lower
alkyl, -(CH2)n-aryl, -(CH2)n-heterocyclo,
-(CH2)n-OH~ -(CH2)n-o-lower alkyl, -(CH2)n-NH2,
( HZ)n SH, -(CH2~r-S-lower alkyl,
~, `' , ~
I'
''~ .
~ 13296~
, ~ . . . :
~ 5-
~ ,
: . : :
, -, . ,
~ ( 2)n (CH2)g~OH, -(CH2)n-O-(CH2)g~NH2,
. ~ .,. Il .
.-~f~Z -(CH2)n~s-(cH2)g~OH~ ~(C~2)n~c~H'
~:`'-. NH
i S -(cH2)n-S-(CHz)g-NH2~ -(CH2)n-NH-c~ , l
-:i NH2 ~: .
~ O
, ~
n n~J ? n~
~ N N
~ ;1 10 R8
,~' and -(CH2)n-cycloalkyl;
. R4 is selected from hydrogen, lower alkyl,
~ halo substituted lower alkyl, -(CH2)n-aryl, 1
''I -(CH2)n-heterocyclo, -(cH2)n~OH~ -(CH2~n-O-lower
1 15 ~ 2)n NH2, -(CH2)n-SH, -(CH2) -S-lower
:l alkyl, -(CH2)n-O-(CH2)g-O~, -(CH2)n-O-(CH2)g-NH2,
,j i O
'~Z -~CH2)n-S-(CH2)g-OH, -(C~2)n-C-OH, ~ :
`~ NH :
Z 20 2)n S (CH2)g-NH2, -(CH2) -NH-C//
i N~2
,iZ
i ( 2)n C N~2~ ~(CH2)n ~ ~N-R7, -(CH2) ~ N,
N NJ
.Z ~ I
::, 25 1 7 R8
-(CH2)n-cycloalkyl, -(CH2)n ~
i l 30 -(CH2)n - ~ ~ , -(CH2~n - ~\ ¦ I ::
N N NH
l -(CH~n~ (CH2)n- <\ 3 and,
:~, 35 : :
~ Z
~' ~ ' '~' ' "'
1 3 2 9 S 8 0
,-i.
~ 6- HA432
~,~' O :
~ (CHz)
`:~, R6, R6' and R6" are independently selected from
hydrogen, alkyl, aryl, pyridyl and cycloalkyl, or R6
~, and R6' taken together with the atom to which they ::~are bonded may form a ring of 3 to 5 carbons;
m, m' and m" are zero or an integer from 1 to 5; -~::
I n is an integer from 1 to 5;
p and p' are zero or 1;
g is an integer from 2 to 5;
~- 15 R7 is -CH2-O-CH2- ~ or -CH
8 iS 2,4-dinitrophenyl, -C-O-CH2 ~ , :~
-SO2- ~ CH3, or -C~2-O-CH2 ~ ;
Rg is hydrogen, :Lower alkyl, -(CH2)n , ::
.¦ or -(CH2)n-cycloalkyl;
Rlo is -(CH2)m,-R6';
A and A' are a single bond or -(CH)-(CH2)~"-R6".
,
:
: :
, . ~
:: ~
: :i
1 3 2 ~ ~ 8 ~
; ... , ;~ ~ ` ... -
- HA432
7-
' ~1' :'
s- ~ Thls lnventlon ln its broadest aspects
~-, relates to the compounds of formula I above, to
i~, compositions and the method of using such compounds
5 as antihypertensive agents. -
'~,/J The term lower alkyl used in defining various
~'i symbols refers to straight or branched chain
!.,..,,,, radicals having up to seven carbons. Similarly,
~`lthe terms lower alkoxy and lower alkylthio refer to
such lower alkyl groups attached to an oxygen or
sulfur. The preferred lower alkyl groups are
straight or branched chain of 1 to 5 carbons.
,The term cycloalkyl refers to saturated rings
iof 4 to 7 carbon atoms with cyclopentyl and
-15 cyclohexyl being most preferred.
~iiThe term halogen refers to chloro, bromo and
fluoro.
, ,~
;~The term halo substituted lower alkyl refers
~-~to such lower alkyl groups described above in which
~'20 one or more hydrogens have baen replaced by chloro,
bromo or fluoro groups such as trifluoromethyl,
which is preferred, pentafluoroethyl,
2,2,2-trichloro~thyl, chloromethyl, bromomethyl,
etc
, ::
,~ 25 The term aryl refers to phenyl, 1-naphthyl,
, .. ~ . . .
2-naphthyl, mono substituted phenyl, 1-naphthyl, or
,~i 2-naphthyl wherein said substituent is lower alkyl
of 1 to 4 carbons, lower alkythio of 1 to 4
carbons, lower alkoxy of 1 to 4 carbons, halogen,
30 hydroxy, amino, -NH-alkyl wherein alkyl is of 1 to
i~ 4 carbons, or -N(alkyl)2 wherein alkyl is of 1 to 4
,~ ~, ,,~ . .
;.:.
s 3 ~:
~ `~
1 3 2 ~ ~ 8 0
-8- HA432
carbons, di or tri substituted phenyl, 1-naphthyl
or 2-naphthyl wherein said substituents are
~`, selected from methyl, methoxy, methylthio, halogen,
- and hydroxy.
The term heterocyclo refers to fully
saturated or unsaturated rings of 5 or 6 atoms
containing one or two O and S atoms and/or one to
four N atoms provided that the total number of
¦ hetero atoms in the ring is 4 or less. The hetero
ring is attached by way of an avaiiable carbon
atom. Preferred hetero groups include 2-thiazolyl,
2- and 4~imidazolyl, 2- and 3-thienyl, 2- and
3-furyl, 2-, 3- and 4-pyridyl. The term hetero
also includes bicyclic rings wherein the five or
six membered ring containing 0, S and N atoms as
~ defined above is fused to a benzene ring. The
-! preferred bicyclic ring is benzimidazolyl.
, ~
~ . :
,
, '
'.
',, , ,"
:'1
.:. :
';} ~, :
'`''`' 1 ' ,:
.1 ,,
. . ",
`', 'G'' ~ 3 2 ~ ~ Q n
U O U
HA432
The compounds of formula I are
-~,; prepared by coupling an amine of the formula
~-f II R4 O R3
H2N-CH-C-NH-CH-CH-
OH
, .,
~I with the compound of the formula
~, ,. " I
l 10 R5 O
- III X-CH2-CH-C-OH
.,-, - .
;1 in a solvent, e.g. dimethylformamide, and in the
l presence of a coupling agent, e.g. dicyclohexyl-
¦ 15 carbodiimide.
To make the amine of formula II, a starting
material, H-R1, is treated with n-butyl lithium to
obtain a compound of the formula
'- ~1 , -
IV Li-R1.
` ~J Compound IV is thereafter re
acted with an
aldehyde of the formula
,,,~'. ,
VR3 O
- Prot-NH-~H-CH ;
(wherein Prot is an amino protecting group, ~-
e.g. t-butoxycarbonyl)
~,
.' '. .'~
:; ~ i` , ~ ,
.
. .'~ . . ' ., ~ ~
'.~ ~ '.' ',''`' '
2 ~ 6 g O
~ HA432
-: ~
-;; to provide the protected amine of the formula
~ VI R3
3 Prot-NH-cH-cH-Rl
OH
~!
Compound VI, in a solvent such as ethyl
acetate, can be deprotected by means known in the
art, e.g. by treatment with hydrogen chloride, to
provide an amine of the formula
R3
~, VIIH2N-CH-CH-
OH
The amine of formula II can then be prepared by
.~ I
reacting the deprotected amine of formula VII with
an N-protected amino ac:id of the formula
'
VIII R4 1l
Prot-~-CH-C-OH
in the presence of a coupling agent, such as
dicyclohexylcarbodiimide, and thereafter removing
the protecting group by known means, e.g. treatment
with hydrogen chloride in the case of a
t-butoxycarbonyl protecting group.
To make th~ compounds of formula I wherein X
is R6-(CHz)m-~-C-, a compound of the formula
t
:
3 2 ~ ~ ~ 0
~ HA432
. .;: ~,: :: :
IX o
. ~ ',, R6-(CH2 )m-A-C-CH2Br
(the preparation of which has been described, for
example, in Tetrahedron Letters, 26, 5611-5615,
~i 1970) is coupled with a diethyl malonate derivative
s having the formula
,
, .. .
R5 O
X HC~C-OCH2CH3
=0
OCH2CH3
.
in a solvent, e.g. tetrahydrofuran, and in the
j 15 presence of a base, e.g. sodium hydride, to provide
, a compound of the formula
:,~
~3 XI 0 R5 0 --
(cH2)m-A-~-cH2-c-c-ocH
, 20 C=O
OCH2CH3 ;
~l Compound XI in a solvent, e.g. aqueous ethanol,
is treated in a strong base, such as sodium hydroxide,
and thereafter with hydrochloric acid and heat
to provide the compounds of formula III where
~`~, O
~¦ X is R6-(C~2)m-A-C-. Reaction with compound II, as
above, provides the corresponding compounds of
30 formula I.
. . i ~ .
:: ,
,.
;.
~:
:; - 1 3 2 ~ 6 8 0 :~
~ HA432
..,, .,,--, ~
i ~ To make the compounds of formula I where X
~,-,,.. ,, o o
- ~ is R6-(CH2)m-A-N-C- or CN_C_ and
Rl o
::' 5 R5 is -(CH2)n-aryl and n = 1, a compound of the
~,l formula
:1,
XII Ar
CH O
11 11
- 10 C - C-OCH2CH3
, I
-~:; ' CH2-C-OH ~ ~ ~
(the preparation of which has been described in J.
~ 15 Amer. Chem. soc., 90, 3495, (1968)), is
.,: hydrogenated in the presence of a palladium on
carbon catalyst to provide a compound having the
formula
- 20 XIII Rs l -
CH-C-OCH2CH3
:~1 CH2-C-OH
,.,., O . ~ ~
r"i,
,: ~ 25 Compound XIII is reacted with a compound of :.
the formula
XIV R6-(CH2)m-A-NH ~::
Rlo
30.
.
'~
:: ~ ',
~ : ' : :~,
'`''~1 : ~ ~.
3 2 ~ 6 8 0
HA432
13-
, ~ r~
'''':: XV ~NH
- in the presence of a catalyst, such as hydroxybenzo-
triazole, and dicyclohexylcarbodiimide to provide
the ethyl ester of the formula
..,
,.,
- ,
XVI I R5 O
R6-(CH2 ~m-A-N-C-CH2 CH C OCH2C~3
. Rlo
., 10
~ or
. XVI I CN_C_CH2 CH C OC~2 CH3
Compound XVI or XVII, in a solvent such as :
~i aqueous ethanol, is treated with a strong base, e.g.
. . sodium hydroxide to provide the compounds of formula
~- :4 20 o - o
; III wherein X is R6-(CH2)m-A-I-C-, or CN_C_ ;
o
and R5 is -(CH2)n-aryl and n = 1. Reaction with
compound II, as above, provides the corresponding ~ :
. 25 compounds of formula I.
-~ Alternatively, to make the compounds of
,,:,.. 1 O ,.
~, formula I where X is R6-(CH2)m-A-N-C-, CN C or :~
. .1 0~ 1 o
~ 30 R6-(CH2)m-A-O-C-, and R5 is -(CH2)n-aryl and n = 1
.;;: to 5, a dialkylmalonate of the formula
.~. . I - i,.,~:,
~ !
~; 3~
32~68~
-14- HA432
. ~.
-~ : XVIII o
CH2-C-Oalkyl
,. C=O
~ . I
~. Oalkyl
-, 5
. in a solvent, such as tetrahydrofuran, is treated
~:. with sodium hydride and thereafter reacted with a ~ :
. compound of the formula
~ 10 XIX R5-Cl or R5-Br
:
to provide a compound having the formula
XX Rs
; 15 CH-C-Oalkyl
C=O
Oalkyl . :
~` Compound XX, in a solvent such as aqueous
~I 20 ethanol, is treated with a strong base, e.g. sodium : ~:
hydroxide, and thereafter with hydrochloric acid to
~ provide
'~ ~ XXI Rs
~1 25 CH2-C-OH.
: .~ "
~j Compound XXI is treated with benzyl alcohol
and 4-dimethylamino pyridine in a solvent, e.g.
methylene chloride, in the presence of dicyclohexyl-
~¦ 30 carbodiimide to provide the ester of the formula
', ' ;~;;
~j ,
:.:, ~:
,.,': ~........................ -
...~ j
1 3 2 ~ ~ 8 Q
:' f ~
: - HA432
-15-
XXII R5 O
', ~ CH2-C-OCH2--.~
which is treated with diisopropylamine and n-butyl
lithium in a solvent such as tetrahydrofuran, and
-~, thereafter reacted with t-butyl bromoacetate to
. provide
'. `,
.~: XXIII O R5 O
i~. 10 t-BuO-C CH2-CH-c-O
s
i~ Compound XXIII, in a solvent, such as methylene
~ 3 chloride, is treated with a strong acid, e.g.
;~ ~ trifluoroacetic acid, to provide a compound of the
.. l 15 formula
, ,: , 1 .
XXIV HO-C-CH2-CH-C-OCH2 ~
~;. 20 Compound XXIV, in a solvent, such as tetra- ~:
hydrofuran, is coupled with R6-(CH2)m-A-
~,. R
,!", ~-~ or R6-(CH2)m-A-OH in the presence of a
.:. cata~yist, such as hydroxybenzotriazole or dimethyl- :;:
.: : .
~ 25 aminopyridine, and dicyclohexylcarbodiimide to
~ l provide the compounds of formula III where X is . .
O O o :~
~ 6 (CH2)m A N C , N-C-, or R6-(CH2)m-A-O-C- ~ .
' , R l o
30 and Rs is -(CH2)n-aryl and n = 1 to 5. Reac~ion ;
~ with compound II, as above, provides the corresponding :
s. compounds of formula I.
s~ ,. ..
;ii.,,,, '~
~ ,
.,~,..~., .
i" ,., I
."
,
~ ~ 3 2 9 ~ ~ 0
-16- HA432
To make the compounds of formula I where X
: is R6-lCH2)m-A-S-, a compound of the formula
~,'
XXV 11 ~5 11
5HO-C-CH-C-OH
is reacted with dimethylamine in the presence of
formaldehyde to provide a compound of the formula
10 XXVI I Rs
~ HO-C - C ~ C-OH
'' ,CH2
~N : ~i :
' CH3. CH3
:.l 15
Compound XXVI is heated to provide the
:~ acrylic acid of the formula ~:~
XXVII R5 O
20H2C=C--C-OH.
,~ :
i; ~ Compound XXVII, in a solvent such as
piperidine, is reacted with a compound of the
. formula
~ ~ ~5
r ~ XXVIII R6-(CH2)m-A-SH :; :
~¦ to provide ~j~
"
30 XXIX R5 l
- R6-(CH2)m-A-S-CH2-CH-C-OH,
'!: I
', l :,
i', j ~ ~
:.1 1 .
~" " .
.,. .~,' .
132~6~0
17 HA432
, ::
. ::
that is, the compounds of formula III wherein X is
R6-(CH2)m-A-S-. Reaction with compound II, as
above, provides the corresponding compounds of
formula I.
, .
Alternatively, a compound of the formula
XXVII may be esterified by reaction with ethanol
" in the presence of dicyclohexylcarbodiimide and a
j catalyst such as dimethylaminopyridine to give a
:,
~ compound of the formula
,, 1 0
R5 O
XXVIIa H2C=C ~ C-OCH2CH3
Compound XXVIIa, in a solvent such as
ethanol is then reacted with a compound of the
formula XXVIII in the presence of a base such as
sodium ethoxide to give a compound of the formula
.,
~":'
XXIXa R6-(CH2)m-A-S-CH2-CH-c-ocH2cH3
~ Compound XXIXa is treated with sodium
i~ hydroxide to give compound XXIX.
When X is R6-(CH2)m-A-SO-, compound XXIX in
~j 25 a solvent, e.g. methanol, is treated with hydrogen
~3 peroxide. When X is R6-(CH2)m-A-SO2-, compound
XXIX, in a solvent such as methanol, is treated
with potassium monopersulfate. The resulting
~`l species of formula III can be reacted with compound -
l 30 II, as above, to provide the compounds of formula I
i;¦ wherein X is R6-(CH2)m-A-SO- and R6-(CH2)m-A-S02-,
: 7 respectivel~.
''i'''1 .
. , , ' .
.. ~ ~ . .
: ~
l3296~
HA432
l8-
,~ ~
To make the compounds of formula I where X
O .
11
is R6-(CH2)m A-(O)p P and p and p'are l, a
(O)p,
A'
(IcH2)
. R6 '
: compound of the formula
., : .
0
11
XXX R6-(CH2)m~A~(O)p-P-H :
(b)p, -:
A'
( ~CH2 )m,
R6'
1 is reacted wi~h the acrylic acid of formula XXVII
-.1 in dichloromethane and in the presence of bis-
~1 (trimethylsilyl)acetamide to provide the compound
O
of formula III where X is R6-(CH2)m-A-(O)p-1-
(1)P' : :
~ A'
'~1 (CH2i~m, :
~6'
Reaction with compound II, as above, provides the
I corresponding compounds of formula I.
~I To make compouds of the formula I where X is
':~' O
~30 R6-(CH2)m-A-(O)p-P- and p and/or p' are 0, a
.
: A'
~ 2 )m~
: ~ .:
"
~ ~ 329~0
-19- HA432
" .
. "
compound of the formula XXX wherein p and p' are 1
is reacted with a compound of the formula
XXXI R6-(cH2)m-A-MgBr
:~
The resulting species is then reacted with the -:
acrylic acid of the formula XXVII to provide the -~
compound of the formula III where X is
! ..
! 10 R6-(CH2)m-A-(O)p-P- and elther p or p' or
( O~
A'
(R~2)m~
1 15 both are 0. Reaction with compound II, as above,
¦ . provides the corresponding compounds of formula I.
To make compounds of the formula I where X
O :'-.' ',' ~' ~. ', , -
is R6-(CH2)m-A-C-S-, a compound of the formula
O
XXXII R6-(CH2~m-A-c~-sH
¦ is reacted with an acrylic acid of formula XXVII
25 to give a compound of ~Ihe formula i~
Rs ~:
XXXIII R6-(CH2~m-A-C-S-CH2-CH-C02H
.~ .
¦ 30 Reaction of compound ~XXIII with compound II, as
above, provides the corresponding compounds of ~ .
I formula I.
:`
:: ~
l32~6~a
: HA432
-20-
To make compounds of the formula I where X =
HS, a compound of the formula I where X =
R6-(CH2)m-A-C-S- is treated with ammonium
hydroxide solution.
To make compounds of the formula I where X =
R6-(CH2)m-A-N-SO2-, a compound of formula XXXIII is
,I Rl o
:;, treated with ammonium hydroxide solution to give a
compound of the formula
~1 R
:~
. XXXIV HS-CH2-CH-CO2H
1 15 The compound of formula XXXIV is esterified, for
;i example, by treatment with ethanol and dicyclo-
,< hexylcarbodiimide in the presence of a catalyst, -
;~ such as dimethylaminopyridine, to give a compound
of formula
, 20 : - :
:-; :.'- :.
~ XXXV HS-CH2-CH-COCH2CH3
:¦ The compound XXXV is treated with chlorine gas in
25 a solvent such as aqueous acetic acid, to give the ~ :
compound
O R5 1l
Cl-S-CH2 -CH-C-OCH2 CE3~3
1 . ~.
''i ' ' " ':
'
,','`
:
:
3~9~
, . .
i,. .
-~1- HA432
?::
" .
which is reacted with an amine
.:
XXXVII R6-(CH2)m-A~
.' - Rlo
~;
to give a compound of the formula
. Rs
XXXVIII R6-(CH2)m-A-N S-CH2-CH-C-OCH2CH3 . :-
1 10 Rlo O :~
Compound XXXVIII is saponified with a strong
base, such as sodium hydroxide, to give a compound
of the formula
R , ```;;
XXXIX R6-(CH2)m-A-I - I-CH2CH-CO2H . . --
R1o
,
Reaction of compound XXXIX with compound I I, as
above, provides the corresponding compounds of
formula I.
In the above reactions, if any of R3, R4 and
l Rs are -(CH2)n-aryl wherein aryl is phenyl,
1-naphthyl, 2-naphthyl substituted with one or more
:~ hydroxy or amino groups, -(CH2)n-heterocyclo
wherein heterocyclo is an imidazolyl, -(CH2)n-NH2,
' j ~NH
~ -(CH2)n-SH, -(CH2)n~OH/ -(CH2)n~NH~C\ or
i 30
.
'i
:' , :
:~ .
.
. .
~329~8~
-22- HA432
~:
:
o
-(CH2)n-C-OH, then the hydroxyl, amino, imidazolyl,
- mercaptan, carboxyl, or guanidinyl function should
be protected during the reaction. Suitable
protecting groups include benzyloxycarbonyl,
t-butoxycarbonyl, benzyl, benzhydryl, trityl,
tosyl, etc., and nitro in the case of guanidinyl.
The protecting group is removed by hydrogenation,
, treatment with acid, or by other known means
^' 10 following completion of the reaction.
The various peptide intermediates employed in
, above procedures are known in the literature or can
-~ be readily prepared by known methods. See for
example, the Peptides, Volume 1, "Major Methods of
Peptide Bond Formation", Academic Press (1979).
Preferred compounds of this invention are
those of formula I wherein 1- 1
O EIsCzO O O
H5C2O ~
is - < ~ , ~ ; or~
i N N
, R3 is straight or branched chain lower alkyl
~-~ 25 of 3 to 5 carbons, -(CH2)n-cyclopentyl,
-(CH2)n-cyclohexyl~ or -(CH2)n ~ , wherein n
is an integer from 1 to 3;
R4 is hydrogen, straight or branched chain
lower alkyl of up to 5 carbons, -(CH2)4-NH2,
. . :l :,
.
' :
., ~
:,
: : ~
2 9 ~ 8 0
HA432
23-
:i -
, is,i, ~ .
-CH2 ~ JNH -CH2 rJN C
~` 5 -C~2 ~ ~ , -C~2 ~ , -C~ ~ O~
~ .
'~ 10 -CH ~ , -CH2 - @ N , -CHz ~ N
-(CH2) ~ , -~CH2)n
S
(CH2)n ~ ~ , (C~2)n ~
.~ NH
.I 20
,; ~; 2r~ ;
~ N
.3 Q~o2 ~ ~.
:;' ~ "'' :
~, 25 NO2
, R5 is straight or branched chain lower alkyl
of up to 5 carbons, -CH2 ~ , -(CH2)2~
I' ;
~,i .:
~.~
. . ~
.,~1, ' ~
'
"`
~: :
329~8a
; ~: HA432
~ --24-
.,. , ~,
-CH2- ~ OCH3, -CH2-(oZ-naphthyl),
-CH~ 4-naphthyl), -CH ~ OH, -CH2-cyclopentyl,
-CH2-cycloheXYl, -CH2 ~ , -CH2 ~ N, -CH2~
-CH2 ~JNH, or CH2~,~
! H
I Most preferred are those compounds of formula
Z I wherein
¦ R8 is cycloalkyl, morpholinyl, ethyl or
I 15 ethoxy;
I N , NH S
R1 is - ~ ¦ <N ~ ~N 3
:
i 20 R3 i5 -CH2- ~ or -CH2-CH ; :~
CH3
Z /CH3
R~ is -CH2-CH or -C~2 ~ NH; and,
~1 CH3 N
R5 is -CH2 ~ or
1~ -CH2~
Z The compounds of formula I form salts with a ~ :
i variety of inorganic and organic acids. The
30 nontoxic pharmaceutically acceptable salts are
I preferred, although other salts are also useful in
: . '~
: ~ :
:: ~
~ : 1 3 ~
HA432
.;- ~ 25
,.- .
isolating or purifying the product. Such
- pharmaceutically acceptable salts include those
~` formed with hydrochloric acid, methanesulfonic
acid, sulfuric acid, acetic acid, maleic acid, etc.
The salts are obtained by reacting the product with
an equivalent amount of the acid in a medium in
l which the salt precipitates. `~
;~ The compounds of formula I contain asymmetric
j centers when any or all of R3, R4 and Rs are other
~l 10 than hydrogen and at the carbon to which the -OH
group is attached. Thus, the compounds of formula
I can exist in diastereoisomeric forms or in
l mixtures thereof. The above-described processes ;
¦ can utilize racemates, enantiomers or diastereomers
as starting materials. When diastereomeric
products are prepared, they can be separated by
conventional chromatographic or fractional
crystallization methods.
The compounds of formula I, and the
pharmaceutically acceptable salts thereof, are
antihypertensive agents. They inhibit the
~ conversion of angiotensinogen to angiotensin I and
', therefore, are useful in reducing or relieving
angiotensin related hypertension. The action of
the enzyme renin on angiotensinogen, a
pseudoglobulin in blood plasma, produces
angiotensin I. Angiotensin I is converted by
i angiotensin converting enzyme (ACE) to angiotensin
, II. The latter is an active pressor substance
which has been implicated as the causative agent in
several forms of hypertension in various mammalian
t329680
~-~ HA43
26-
species, e.g., humans. The compounds of this
invention intervene in the angiotensinogen ~
~renin) ~ angiotensin I (ACE) ~ angiotensin II
sequence by inhibiting renin and reducing or `
1 5 eliminating the formation of the pressor substance
-i angiotensin II. Thus by the administration of a
I composition containing one (or a combination) of
, the compounds of this invention, angiotensin
l dependent hypertension in a species of mammal
! 10 ~e.g., humans) suffering therefrom is alleviated.
A single dose, or preferably two to four divided
daily doses, provided on a basis of about 100 to -
1000 mg, preferably about 250 to 500 mg per kg of
body weight per day is appropriate to reduce blood - `
pressure. The substance is preferably administered
orally, but parenteral routes such as the
subcutaneous, intramuscular, intravenous or
l intraperitoneal routes can also be employed.
The compounds of this invention can also be
formulated in combination with a diuretic for the
treatment of hypertension.
A combination product comprising a compound
~-l of this invention and a diuretic can be
administered in an effective amount which comprises
a total daily dosage of about 1000 to 6000 mg,
~' preferably about 3000 to 4000 mg of a compound of
i this invention, and about 15 to 300 mg, preferably
about 15 to 200 mg of the diuretic, to a mammalian
species in need thereof. Exemplary of the
~':! 30 diuretics contemplated for use in combination with
a compound of this invention are the thiazide
., .
, .
8 ~
-c~ HA432
~ 27-
..
.
: ~.:
diuretics, e.g., chlorothiazide,
l hydrochlorothiazide, flumethiazide,
¦ hydroflumethiazide, bendroflumethiazide,
methylclothiazide, trichloromethiazide,
polythiazide or benzthiazide as well as ethacrynic
acid, tricrynafen, chlorthalidone, furosemide,
musolimine, bumetanide, triamterene, amiloride and
'A, spironolactone and salts of such compounds.
' The compounds of formula I can be formulated
s 10 for use in the reduction of blood pressure in ;~
s compositions such as tablets, capsules or elixirs
for oral administration or in sterile solutions or
suspensions for parenteral administration. About
100 to 500 mg of a compound of formula I is
15 compounded with physiologically acceptable vehicle,
carrier, excipient, binder, preserv tive,
stabilizer, flavor, etc., in a unit dosage form as
called for by accepted pharmaceutical practice.
The amount of active substance in these
20 compositions or preparati.ons is such that a
suitable dosage in the rcmge indicated is obtained.
The present invention will now be described
by the following examples, however, the invention
; should not be limited to the details therein.
:3 - ~
3 2 9 6 8 0
,,~- .
HA432
~28~
....
..,
Exam~le 1
N-[(S~-2-Cyclohexyl-l-[(R)-hydroxv(lH-
imidazol-2-yl)methYllethyll-N2-[4-(4- -~
morpholinyl~-1,4-dioxo-2-(phenylmeth~
butyll-L-histidinamide, isomer B,
¦ dihvdrochloride
I A. (Phenylmethyl)butanedoic acid, 1-ethyl ester
A mixture of ~-ethyl-~-benzal succinate (16.3
- g, 70 mmols; prepared as described by Cohen and
Milovanovic, J. Ame~. Chem. Soc., 90, 3495, (1968))
~, and 10~ palladium on carbon (1.2 g) in absolute
ethanol (250 mL) was hydrogenated at 30 psi on a
Parr apparatus for 24 hours, after which it was
filtered and concentrated. The residue was
I dissolved in ether, dicyclohexylamine (70 mmols)
i was added, and the mixture was filtered. Hexane -
, was added to the filtrate and the crystals which
-~ 20 formed on cooling to -30C were collected. The
solid product was recrystallized from ethyl acetate
¦ to give the title A dicy~clohexylammonium salt (10.7
g). The salt was dissolved in ethyl acetate and
washed with aqueous lN hydrochloric acid. The
ethyl acetate layer was dried and concentrated.
The residue was dissolved in ether and was
extracted with sodium bicarbonate 601ution. The
combined aqueous layers were acidified by addition -
of concentrated hydrochloric acid and extracted
with ethylene acetate. The extract was dried over
anhydrous magnesium sulfate and concentrated to
give the title A compound as a pale yellow oil
which solidified on standing overnight (6.2 g, 38%
¦ yield).
::
~" ~
`
~32~ HA432
, 9_
.,:.. - ~ , .
B. ~-(Phenylmethyl~-y-oxo-4-morpholinebutanoic
~i- acid, ethyl ester
l~ To a mixture of the title A compound (3.05 g,
15 mmol) and morpholine (1.3 mL, lS mmol) in
tetrahydrofuran (50 mL) and dimethylform`amide (5
mL) were added l-hydroxybenzotriazole hydrate (2.1
g, 15 mmol) and dicyclohexylcarbodiimide (3.1 g, 15
mmol). The mixture was stirred at 25C for 16
hours, after which it was filtered and
concentrated. The residue was dissolved in ethyl
acetate, washed sequentially with lN hydrochloric
~, acid and saturated aqueous sodium bicarbonate
solution, dried over anhydrous magnesium sulfate,
and concentrated. The residue (3.6 g) was flash
chromatographed on Merck silica gel (180 g),
eluting with 1:1 hexane:ethyl acetate to give the
title B compound as a clear, colorless oil (3.4 g,
75%), Rf 0.25.
C. -(Phenylmethvl)-y-oxo-4-morpholinebutanoic
acid
A mixture of the title B compound (3.3 g,
10.8 mmol) and lN aqueous sodium hydroxide solution
11 mL, 11 mmol) in absolute ethanol (11 mL) was
, 25 stirred for 22 hours at 25C, after which it was
concentrated in vacuo. The residue was dissolved
in water, washed with ether, acidified by addition
of lN hydrochloric acid (50 mL), and extracted
three times with ethyl acetate. The combined
30 extract was dried over anhydrous magnesium sulfate
and concentrated and the residue was crystallized
from ethyl acetate/hexane to give the title C
compound as a white solid (2 g, 67%), m.p.
133-134C.
] 35
* Trade-mark
" ~ ~329sg~
HA432
,,-., ~
,,, . :
D. (S)-2-[[(1,1-Dimethylethoxy)carbonyl]-
~, amino]-2-phenylmethyl-1-ethanol
To a solution containing N-[(l,l-dimethyl-
, ethoxy)carbonyl]-L-phenylalanine (10 g, 37.7 mmole~
; 5 in dimethylformamide (40 ml) is added solid sodium
bicarbonate (4.75 g, 56.6 mmole) and iodomethane
;, (16 g, 113 mmole). The mixture is heated at 40
under argon for 12 hours, the cooled and the
reaction mixture partitioned between water (150 ml)
and ether (250 ml). The organic layer is rinsed
with 2% aqueous sodium bicarbonate (2 x 100 ml),
2% aqueous sodium bisulfite (100 ml), water (2
x 100 ml), and brine, dried over magnesium sulfate,
and concentrated in vacuo to give 10.5 g of
N-[(l,l-dimethylethoxy)carbonyl]-L-phenylalanine,
' methyl ester as an oil.
i To a solution containing N-[(l,l-dimethyl-
^', ethoxy)carbonyl]-L-phenylalanine, methyl ester (10
'l g, 35.8 mmole) dissolved in a mixture of
! 20 tetrahydrofuran (190 ml) and absolute ethanol (190
~I ml) is added lithium chloride (6.09 g, 143.2
j mmole~. The resulting homogeneous solution
1 is treated with sodium borohydride (5.42 g, 143.2
;, mmole) and the reaction is stirred at room
temperature under argon for 24 hours. The reaction
mixture is next filtered using ether (~700 ml) to
rinse the filter cake. The resulting filtrate is
rinsed with water (3 x 200 ml) and brine (200 ml),
dried over magnesium sulfate, and concentrated in -
~ 30 vacuo to give 9 g of crude product. Recrystal~
¦ lization from ether/hexane gives 7.59 g of the
~¦ title D compound,; m.p. 94-96.
Analysis calc'd for Cl4H2lNO
C, 66.90; H, 8.42; N, 5.57;
Found: C, 66.80; H, 8.57; N, 5.38.
,
HA432
~ E. [(S)-2-Cyclohexyl-l-(hydroxvmethyl)ethyl
;~ carbamic acid, l,l-dimethYlethyl ester
A solution of the title D compound (7 g, 27.8
mmole) in methanol (70 ml) is hydrogenated at 55
psi on a Parr Shaker using 5% rhodium on alumina
' (500 mg) as catalyst. After 17 hours, the reaction
mixture is filtered and concentrated in vacuo to
yield 7.36 g of the title E compound as an oil.
Analysis calc'd for Cl4H27NO3:
C, 65.33; H, 10.57; N, 5.44
Found: C, 64.94; H, 10.55; N, 5.23.
F. (S)-(2-Cyclohexyl-l-formyleth~l)c~rbamic
acid, l,l-dimethylethyl ester
A solution of the title E compound (4.6 g,
17.9 mmole) in methylene chloride (40 ml) is added
~ to a mixture of Dess-Martin periodinane reagent (8
-~ g, 19 mmole) [prepared according to Dess et al., J.
Org. Chem., Vol. 48, p. 4155 (1983)] and t-butanol
, 20 (1.5 g, 19 mmole) in methylene chloride (70 ml)
-~ which had been stirred at room temperature before
`I the addition. A slight exotherm (to 32) results.
i After 30 minutes, the reaction mixture is quenched
in ether (800 ml), resulting in the separation of a
-I 25 white solid. A mixture of sodium thiosulfate
~j pentahydrate (31.3 g, 126 mmole) in saturated ~ `
I sodium bicarbonate solution (200 ml) is added, with
~ stirring. The resulting two-phase mixture is
;~ separated and the organic phase is washed with
water, saturated sodium bicarbonate (2 x 100 ml),
water, and brine, dried over magnesium sulfate, and
il concentrated in vacuo to give 3.8 g of the title F
~I compound as a colorless oil.
,
3 2 9 ~ ~ 0
... ~ .
HA432
32-
~,, -: ~-
'
- G. [(lS)-l-(Cyclohexylmethyl)-2-hydroxy-2-[1-
[(phenylmethoxy)methyl]-lH imidazol-2-yll-
ethyl]carbamic acid, 1,1-dimethylethyl ester
2.5 M n-Butyllithium solution in hexane (12
ml, 31 mmole) is added to a solution of 1-[(phenyl-
, methoxy)methyl]-lH-imidazole (5.3 g, 28 mmole) in
tetrahydrofuran (90 ml) ~t -70 under argon. After
stirring for 15 minutes, the title F compound (3.6
g, 14 mmole) in tetrahydrofuran ~36 ml) is added
dropwise over a period of 5 minutes at a reaction
temperature of -65 to -70. After 2 hours at -70,
the bath is warmed to 0 and saturated ammonium
chloride (25 ml) is added followed by ether ~300
ml) and water ~2 x 50 ml) and brine, dried over
magnesium sulfate, and concentrated in vacuo. The
, resulting crude product (8.4 g) is flash
chromatographed eluting with acetsne-petroleum
ether (1:4) to give 580 mg of the title G (fast
moving isomer), 370 mg of a mixed fraction and 2 g
of a slow moving isomer.
' H. [R-(R*,S*]l-a-(1-Amino-2-cyclohexylethyl)~
'~ [(phenylmethoxy2methyl]-lH-imidazole-2-methanol ' ~
A solution of the title G compound (3.92 g, ~ ~;
8.83 ~mols) in ethyl acetate (200 mL) was cooled to
0C and hydrochloric acid gas was bubbled through
the solution for 30 minutes. The mixture was then
stirred for 3.5 hours as it warmed to room
i temperature~ after which it was concentrated in
~'1 30 vacuo to give the title H compound as a white power
' (3-56 g 97%).
:
~ .
:
' ~::
,.: ~': . ,~' : .
6 8 0
.. '. . . ~ .
~ 33-
... ~ ~ -
~. .. ~ - .
-~:
'~ I. L-Histidine, methyl ester, dihydrochloride
~;~ To a stirred solution (ice-bath) of
i L-histidine ~38.75 g, 240 mmol) in methanol (500
; ml), thionyl chloride (27.2 ml,.375 mmol) was added
in drops. After fifteen minutes the ice bath was
removed and the reaction mixture was stirred at
room temperature for one hour. Then after
refluxing for 48 hours, it was concentrated in
vacuo. The separated crystals were filtered using
methanol for washing (48.93 g). The methanolic
solution on dilution with ether afforded additional
10 g of the title I compound, m.p. 208-209.
. .
J. N,l-Bis[(l,l-dimethvlethoxy)carbonyl]-L-
! 15 histidine, methvl ester
~ To a suspension of the title I compound (24.2
-i g, 100 mmol~ in methanol (80 ml) were added
triethyl amine (28 ml, 200 mmol) and di-tert-butyl
dicarbonate (48 g, 220 mmol). After 3.5 hours, it
~ 20 was filtered and the met:hanolic solution
I concentrated i~ vacuo. The residue was taken into
1 chloroform and washed with 10% citric acid. The
i crude product on crystallization from iospropyl
ether afforded 23.1 g of the title J compound, m.p.
; 25 88-95C. After evaporation and redissolution of ~ -~
the mother liquor (15.75 g) in methanol (50 ml)
di-tert-butyl dicarbonate (10 g, 45.9 mmol) was
added. After stirring the reaction mixture
overnight it was evaporated, taken into chloroform
~3 30 and washed with 10% citric acid. The residue after
chromatography over silica gel yielded 6.4 g of
~I homogeneous title J compound.
~ ,." ":
~ : :
.3296~0
. ~ .
~- HA432
34-
. ~ -
. ~
K. N-[(l,1-Dimethylethoxy)carbonyl]-3-[(phenyl-
methoxy)methyl]-L-histidine, methyl ester,
monohydrochloride
To a solution of the title J compound (24.7
g, 66.9 mmol) in dry methylene chloride (156 ml),
benzylchloromethyl ether (11.6 ml, 88.6 mmol) was
added and the reaction mixture stirred at room
temperature for 5 hours. After concentration in
: vacuo and on dissolution in ethyl acetate (100 ml),
the title K compound crystallized out (17.85 g,
65%), m.p. 152-153C.
" .
1,. N-[(1,l-DimethylethoxY~arbonYl] 3-[(phenyl-
methoxy)methyl]-L-histidine
~ 15 The title K compound (18.66 g, 43.8 mmol) was
;; dissolved in methanol (50 ml). Aqueous sodium
hydroxide (lN, 92 ml) was added followed by water
- 83 ml). After keeping the reaction mixture at room
temperature for 90 minutes it was further diluted
by the addition of water (650 ml) and acidified to
~;I pH 4.5 using aqueous hydrochloric acid. The
J aqueous solution was extracted with chloroform.
The chloroform solution was evaporated and the
residue was crystallized from ethyl acetate (15.13
g, 92%), m.p. 155-157C.
~ M. [(1,1-Dimethylethoxy)carbonyl]-N-[(lS,2R)~
`~, (cyclohexYlmethyl)-2-hydroxv-2-[1-[(~henYl-
~! methoxy)methyl]-lH-imidazol-2-yl]ethYl~-3-
[(~henylmethoxy)methyl]-L-histidinamide
~¦ To a solution of the title H compound (3.06
g, 7.35 mmols), 1-hydroxybenzotriazole hydrate
(1.13 g, 7.35 mmols), and the title L compound
(2.76 g, 7.35 mmols) in tetrahydrofuran (20 mL)
~ 35 were added triethylamine (2.06 mL, 14.7 mmol) and
.'.i ' . '
,
3 2 ~ ~ 8 0
~.. . . ~. `
HA432
35-
,- . ~ ., .
; dicyclohexylcarbodiimide (1.52 g, 7.35 mmol). The
mixture was stirred for 18 hours at 25C, after
which it was filtered. The filtrate was diluted
with ethyl acetate, washed with saturated sodium
bicarbonate solution, dried over anhydrous
magnesium sulfate and concentrated. The residue
(4.92 g) was chromatographed on Merck silica gel,
eluting with ethyl acetate:pyridine:acetic
acid:water (80:20:6:11) to give the title M
compound as the major product (3.98 g, 77%).
N. N-[(lS,2R)-1-(Cyclohexylmethyl~-2-hydroxy-2-
Ll-[(phenylmethoxy)methYll-lH-imidazol-2-yl]-
ethyl]-3-l(phenylmethoxy~methyl]-L-histidinamide
A solution of the title M compound (3.88 g,
- 5.53 mmol) in ethyl acetate (200 mL) was cooled to
0C in an ice bath and hydrochloric acid gas was
bubbled through the solution for 30 minutes. The ~ ;
I resulting mixture was then stixred for 2.5 hours as
i 20 it warmed to 25C, after which it was concentrated "~
~ to small volumie. The resulting white precipitate
;~ was collected on a PTFE filter to give the title N
compound as a white powder (3.33 g, 85%), m.p.
143-157C.
O. ~4-(4-Morpholiny1)-1,4-dioxo-2-(phenYlmethyl)
butyl]-N-[(lS,2R~ (cyclohexylmethyl)-2-
hydroxY-2 Ll-[(phenylmethoxy)methyl]-lH-imidazol- .
2-yl~ethyi]-3-[(phenYlmethoxY)methyl]-L-
¦ 30 histidinamide, isomer B `-
To a mixture of the title C compound (610 mg, ` ~
~¦ 2.2 mmols), l-hydroxybenzotriazole hydrate (337 mg, ~ `
2.2 mmols) and the title N compound (1.47 g, 2
mmols~ in tetrahydrofuran (a mL) at 0C were added
triethylamine ~0.84 mL, 6 mmols) followed by
:
, :
1 3 2 9 ~ g O
-~ ~A432
-~ --- - -36-
- ~..,. - ~ .
, ~: . .::: .~
dicyclohexylcarbodiimide (453 mg, 2.2 mmols). The
resulting mixture was stirred for 18 hours as it
warmed to 25C, after which it was fitered. The
filtrate was diluted with ethyl acetate, washed
with saturated sodium bicarbonate solution, dried
over anhydrous magnesium sulfate and concentrated.
ThP mixture (1.64 g) was chromatographed on Merck
silica gel (165 g), eluting with ethyl
acetate:pyridine:acetic acid:water (80:20:6:11) to
give isomer A of the title 0 compound (550 mg, 32%)
and isomer B of the title 0 compound. The B isomer
of the title 0 compound was further purified by
I preparative HPLC to give 580 mg of the title 0
- compound.
P. -N-[(S~I-2-Cyclohexyl-l-[(R)-hYdroxy(lH~
¦ imidazol-2 -Yl ~methyl]ethvl]-N2- L4- ( 4-
i morpholinyl)-l,4-dioxo-2-(~henylmethyl
¦ butyll-L-histidinam de, isomer B,
dihydrochloride ~-~
A mixture of the t:itle 0 compound (isomer B)
~, (580 mg, 0.53 mmols) 20% palladium hydroxide on
carbon (120 mg), and l N aqueous hydrochloric acid
~j (1.17 mL, 1.17 mmols) in methanol (15 mL) was
1 25 hydrogenated under a slow stream of hydrogen for l9
:! hours. The mixture was then filtered and -
;i concentrated to dryness. The residue t350 mg) was
dissolved in lN aqueous hydrochloric acid (10 mL)
, and concentrated to dryness in vacuo. The residue
'I 30 was redissolved in lN hydrochloric acid and
concentrated to dryness again, then was dissolved
in water, millipore filtered and lyophilized to
give the title compound as a fluffy white solid
(330 mg, 90%).
:'
~ 3 ~
~ 37-
.
.,
E~sample 2
: ~
~.,'
[4-(4-Morpholinyl)-1,4-dioxo-2-(1-naph-
thalenylmethyl)butyl]-N-[(lS,2R~ (cyclo-
hexylmeth~ 2-hydroxy-2-(lH-imidazol-2
Yl)ethyll-L-histidinamide, isomer B,
dihvdrochloride
A. (1-Naphthalenylmethyl)propanedioic acid
diethYl ester
To a suspension of sodium hydride (8 g of 60%
dispersion in mineral oil, 200 mmols) in
tetrahydrofuran (200 mL) was added diethyl malonate
, (30.5 mL, 200 mmols) dropwise over 15 minutes; gas -
j 15 evolution was observed. When the addition was
complete, the mixture was stirred for 10 minutes at
25C. A solution of 1-chloromethyl naphthalene
(35.5 g, 200 mmols) in tetrahydrofuran (20 mL) was
added dropwise over 15 minutes, after which the
mixture was heated at reflux under argon for 18
; hours. Excess lN hydrochloric acid was then added
and the mixture was extracted with ether. The
~extract was washed with saturated agueous sodium
bicarbonate solution, dried over anhydrous
magnesium sulfate, and concentrated i~ vacuo. The
residue was transferred to a flask fitted with a -
vacuum distillation head. Unreacted diethyl
malonate (bp 45-55C/0.2 torr) and l-chloromethyl
j naphthalene (bp 120-130C) were distilled off and
~ 30 the residue was recooled to 25C. The distillation
¦ residue (43.5 g) was crystallized from petroleum
ether at -30C to give the title A compound as a
l low melting (mp about 30C) solid (27.3 g, 46%).
.: '.'~
:
1 3 2 9 6 8 ~
~- - HA432
~ 38-
'' .~ - ~ -:
,
B. 1 NaphthaleneproPanoic acid
A solution of the title A compound (10 g,
33.3 mmols) in ethanol (70 mL) and 1 N aqueous
sodium hydroxide solution (70 mL, 70 mmols) was
stirred for 2 hours at 25C then for two hours at
reflux. The ethanol was removed in vacuo and the
remaining aqueous mixture was diluted wlth water
and washed with ether. The aqueous layer was
acidified by addition of lN hydrochloric acid (100
mL). Dioxane (100 mL) was then added (for
solubility) and the mixture was heated at reflux
for 18 hours, after which it was concentrated in
vacuo. Water and ethyl acetate were added to the
~ residue and the mixture was extracted three times
'! 15 with ethyl acetate. The extract was washed with lN
- hydrochloric acid, dried over anhydrous magnesium
sulfate, and concentrated. The residue was
triturated with ether to give the title B compound
as a white solid (4.6 g, 60%), m.p. 151-153C.
¦ ~ C. 2-Na~hthalene~roPanoic acid, ~henylmethvl ester
l1 To a solution of the title B compound (7 g,
,~1 35 mmol), benzyl alcohol (3.8 mL, 37 mmol), and
~, 4-dimethylamino pyridine (420 mg, 3.5 mmol) in
1 25 methylene chloride (175 mL) at 0C was added
dicyclohexylcarbodiimide (7.6 g, 37 mmol). The
, resulting mixture was stirred for 2 hours at 0C
`I then for 18 hours at 25C, after which it was
~il filtered. The filtrate was washed with water and
brine, dried over anhydrous magnesium sulfate, and
concentrated. The residue (10.9 g) was dissolved
in benzene and filtered through a pad of coarse
silica gel (180 g). The filtrate was concentrated
~, to give the title C compound as a clear, colorless
-i 35 oil (9.66 g, 95%).
i~` 1329~80
HA432
-39-
- ,,
..-
D. (2-NaPhthalenylmethyl)butanediolc acid,
4~ -dimethy~ethyl) l-(phenylmethYl ester
' To a solution of diisopropyl amlne (4.4 mL)
ln tetrahydrofuran (58 mL) at -10C under argon was
added n-butylllthlum (11.3 mL) of 2.6 M solution in
hexane). The mixture was stirred at that
temperature for 15 minutes, then was cooled to
-78C. A solution of the title C compound (8.5 g,
29 mmols) in tetrahydrofuran (10 mL) was added
dropwise to the cold solution, after which the
solution was stirred for 15 minutes at -78C.
Tert-butyl bromoacetate (5.2 mL, 32 mmol) was then
added and the mixture was stirred for 15 minutes at
-78C, then for 2 hours as it slowly warmed to
25C. The mixture was then poured into excess lN
hydrochloric acid and extracted with ethyl acetate.
The extract was washed with saturated sodium
bicarbonate solution and brine, dried over
anhydrous magnesium sul:Eate, and concentrated. The ;~
~l 20 residue (8.06 g) was flash chromatographed on
silica gel (500 g), eluting first with ~i
benzene:hexanes (2:1) to bring off a yellow colored
band then with benzene and finally with
. benzene:ethyl acetate (4:1), to give the title D
~, 25 compound as a colorless oil which crystallized on
standing (6.62 g, 56%), m.p. 64-66C.
, ~
'.'!, E. (2-NaphthaIenYlmethYl)butanedioic acid, 1
(phenylmeth~l) ester -
30To a solution of the title D compound (9.5 g,
23.5 mmols) in methylene chloride (50 mL) at 0C
was added trifluoroacetic acid (50 mL). The
mixture was stirred for 2 hours at 0C, after which -
. ' ,
'`i :: :
':
:: :
2 ~ ~ ~ 0
HA432
-40-
:
it was concentrated to dryness. The residue was
crystallized from acetonitrile to give the title E
~' compound (6.89 g, 84%), m.p. 124-126C.
F. (2-Naphthalenylmeth~l)butanedioic acid, 1-
(~henylmethYl) ester, (+)-isomer
The title E compound ~5.8 g, 16.6 mmol) was
dissolved in ether (100 mL) and (-)-ephedrine (2.75 ~
g, 16.6 mmol) was added. The mixture was stored at -
-30C for three days resulting in a waxy solid
which was collected. The solid was recrystallized
twice from 35 mL of ethyl acetate and 165 mL of
~, ether, then twice more from 50 mL of ethyl acetate
and 150 mL of ether, resulting in a solid of
i 15 constant melting point and specific rotation (3.0
g, m.p. 114.5-115.5C, [a]D=+31.7). The crystalline
solid was then partitioned between ethyl acetate
and lN hydrochloric acid and was extracted with ;~
ethyl acetate. The extract was dried and conc~ntrated
~, 20 to give the title F compound, [a]D=~54.8 (methanol), ;~
~1.89 g).
G. a-(2-Naph~halenYlmethyl)-y-oxo-4-morpholine-
butanoic acid, phenYlmethYl ester, ~+)-isomer
, 25 To a mixture of the title F compound (1.89 g,
5.4 mmol, [a]D=+54.8)~ morpholine (0.525 mL, 6
mmols), and l-hydroxybenzotriazole (730 mg, 5.4
mmol)-in tetrahydrofuran (15 mL) was added
dicyclohexylcarbodiimide (1.1 g, 5.4 mmol). The
resulting mixture was stirred at 25C for 20 hours,
after which it was filtered. The filtrate was
diluted with ethyl acetate, washed with lN
'
.
1329680
... . .
-41- HA432
hydrochloric acid and saturated sodium bicarbonate
solution, dried over anhydrous magnesium sulfate,
~: ~nd concentrated to give the title G compound (2.1
g, 93%). [~]D=+32.1 (c 4, methanol).
H . a - ( 2-~aphthalenylmethYl L-y-oxo-4-morpholine- ~:
, butanoic acid, (-)-isomer
A mixture of the title ~ compound (2.1 g, 5 :~
. mmol) and 1 N sodium hydroxide solution (5 mL) in ::~
~ 10 dioxane (10 mL) was stirred at 25C for 16 hours, ;~
after which it was diluted with water and washed
with ether. The aqueous layer was acidified (HCl)
and extracted three times with ethyl acetate. The
extract was dried over anhydrous magnesium sulfate
. 15 and concentrated to give the title H compound as a
colorless glass (1.58 g, 97%). :~
I. [R-(R*,S*I]-N-[l-(Cyclohexylmethyl2-2-hydroxy~
1 2-(lH-imidazol-2-yl)ethyll-L-histidinamide
j 20 A mixture of the title M.compound from
9 Example 1 (1.46 g, 2 mmols), 20% palladium hydroxide
on carbon (350 mg, Aldrich), and 1 N hydrochloric
, acid (4 mL, 4 mmols) in methanol (15 mL) was
. hydrogenated under a slow :~
,' 25 stream of hydrogen for 20 hours at 25C, after
which it was filtered (PTFE filter) and :
! concentrated to dryness. The residue (1.1 g) was
;, dissolved in acetic acid (30 mL) and dry
hydrochloric acid gas was bubbled through the
solution for 30 minutes at ~5C. The mixture was
then stirred at 25C for two hours, after which it :
was concentrated in vacuo. The residue was
1329~80 HA432
. -42-
.
~ triturated with acetonitrile to give a white solid
. ,
(833 mg). The solid was dissolved in excess lN
' hydrochloric acid and was concentrated to dryness;
the process was repeated a total of three times to
give the title I compound as a white solid.
J. [4-(4-Morpholinyl)-1,4-dioxo-2~(1-naph-
thalenylmethyl)butyll-N-[(lS,2R)-1-(cYclo-
he~methyl)-2-hydroxy-2-~lH-imidazol-2
~l)ethyll-L-histidinamide, isomer B,
~ dihydrochloride
;~l To a solution of the title H compound (164 ~;~
mg, 0.5 mmol), 1-hydroxybenzotriazole hydrate (84
mg, 0.55 mmol) and the title I compound (276 mg,
~t 0.55 mmol) in dimethylformamide (2.5 mL) at 0C
~3 were added triethylamine (0.24 mL, 1.65 mmol) and
dicyclohexylcarbodiimide (113 mg, 0.55 mmol). The
resulting mixture was stirred for 18 hours as it ;~
warmed to 25C, after which the mixture was
concentrated to dryness in vacuo. The residue was
dissolved in a mixture o~ methanol (3 mL) and lN
hydrochloric acid (2 mL~, filtered, and
~' concentrated. The residue (814 mg) was
chromatographed on Merck silica gel, eluting with
ethyl acetate:pyridine:acetic acid:water
(50:20:6:11). Fractions containing the major
;, product (Rf 0.25) were combined and concentrated.
~' The residue was dissolved in excess lN hydrochloric
acid and concentrated to dryness three times. The
reddish residue (295 mg, 74%) was chromatographed
on HP-20, eluting with a gradient from 0.OlN
hydrochloric acid to methanolic hydrochloric acid
(O.OlN). Fractions containing the major product
i . :
:
::
:
132~
HA432
43-
. -
. ::.
were combined and partially concentrated, then
lyophilized to dryness. The residue was
relyophilized from water to give the title compound
as a fluffy white solid (1~0 mg, 29%).
Exam~l~ 3
[4-CvclohexY~ 4-dioxo-2-~phenylmethy
butyl]-N-~ ~S,3 ~ 1-(cyclohexylmethyl~-2
hydroxY-2-(lH imidazol-2-yl)ethyl]-L~
histidinamide, isomer B, dihydrochloride
A. (2-Cyclohexyl 2~oxoethyl)(~henyl~ethyl~
ProDanedioic acid, diethYl e ter ~-
lS To a suspension of sodium hydride (8 g, 0.2 -
mole) in tetrahydrofuran (200 mL) under argon was
added diethylbenzyl malonate (50 g, 0.2 mole).
3 The mixture was stirred for 30 minutes
~ 1 .
at 25C after the addition was complete, resulting
in a homogeneous solution. Bromomethyl cyclohexyl
ketone (20.5 g, 0.1 mole; the preparation of which
I has been described in Tetrahedron Letters, 26,
5611-5615, (1970)) was then added over 10 minutes,
. after which the mixture was warmed to 50C and
stirred at that temperature for 16 hours. The
mixture was then poured into excess lN hydrochloric
acid and the resulting mixture was extracted with
ether. The extract was washed once in lN
hydrochloric acid and twice in sodium hydrogen
carbonate, dried over anhydrous magensium sulfate
and concentrated to a yellow oil. The oil was
distilled and the residue passed through a short
::
:::
:
~329~80
~ 44-
. . ~ - - -.
column of silica gel, using stepwise gradient
elution. Elution with 1:1 hexane:ethyl acetate
jgave the title B compound as a yellowish oil (28 g,
75%).
d 5
~ B. ~-(Phenylmethyl~-y-oxocYclohexanebutanoic acid
1, A mixture of the title A compound (28 g,
0.075 mmol) and 1 N sodium hydroxide solution (150
mL, 0.15 mole) in ethanol (150 mL) was stirred at
10 75C for 15 hours, after which it was diluted with --~
water (500 mL) and washed with ether. The aqueous
solution was then acidified in concentrated
hydrochloric acid and extracted twice with ethyl
acetate. The extract was dried and concentrated to
15 a yellow oil (18 g). The residue was dissolved in -
dioxane (180 mL), concentrated hydrochloric acid (1
~ mL) was added, and the mixture was stirred at 100C
`~I for 22 hours. The mixture was then concentrated to
j an orange oil. The oil was dissolved in lN sodium
hydroxide solution (125 mL) and the mixture was
' stirred for 18 hours at 75C, after which it was
- diluted with water and washed with ether. The
agueous solution was made acidic with hydrochloric
acid and extracted twice with ether. The extract
was dried over anhydrous magnesium sulfate and
; concentrated to give a yellow oil (12 g~. After
standing at 25C for one week, the compound
crystallized. The crystals were triturated with
petroleum ether to give the title B compound as a
30white powder (8.4 g, 41%), m.p. 71-73C. ~ ~
1 :
:
:
: :
~:~
1329680
A HA432
;; -45-
, :.
,
~ C. [4-Cyclohexyl-1,4-dioxo-2 (phenylmethyl)-
. ::.i.
butYl~-N-[(2S,3R)-l-(cyclohexylmethyl)-2-
~f hydroxY-2-(lEI-imida7.ol-2-yl)ethYl]-L-
- histidinamide, isomer B, dihydrochloride
. .
To a solution of the title I compound from
Example 2 t754 mg, 1.5 mmols) l-hydroxybenzo- ~ -~
triazole hydrate (230 mg, 1.5 mmols), and
the title B compound (230 mg, 1.5 mmols) in
dimethylformamide (7 mL) at 0C were added
triethylamine (0.68 mL, 4.9 mmols) and
dicyclohexylcarbodiimide (309 mg, 1.50 mmols). The
resulting mixture was stirred for 18 houxs at 25C,
after which it was concentrated to dryness.
i Methanol (9 mL) and 1 N hydrochloric acid (6 mL)
t 15 were added to the residue. The resulting solution
j - was filtered and the filtrate was concentrated to
-~, dryness. The residue (2.56 g) was chromatographed
on Merck silica gel; fractions containing the major
product (Rf 0.2) were collected and concentrated.
~' 20 The residue (640 mg) was further purified by
i preparative HPLC. Two major fractions (Isomer A
and Isomer B) were separately collected and
l concentrated. The resulting trifluoroacetate salts
-! were not water soluble, thus the residues were
25 separately dissolved in excess 1 N hydrochloric
acid and reconcentrated three times to convert to
the soluble hydrochloride salts. The residues were
then dissolved in water, charcoal filtered, and
lyophilized to give Isomer A (162 mg, 16%) and
Isomer B (134 mg, 13%) of the title compound.
.
,, , ~ ~,
'` ! ~ :
,: i
: .
~ 3 2 ~ 6 8 ~ HA432
-~6-
Example 4
:~
[3-[(Cyclohex~lthio)methyl]-1-oxo-3-~henyl-
propyl] N-[(lS,2R)-1-(cyclohexylmethYl)-2-
hYdroxy-2-(lH-imidazol-2-yl)ethyl]-L-
histidinamide, dihydrcchloride
., .
A. ~-Methylenebenzenepropanolc acl_
; Benzyl malonic acid (13 g, 0.067 mole) was
mixed with aqueous dimethylamine (7.6 g, 0.068
mole, 40%) and formalin (5.4 g, 0.068 mole, 37%) in
water (150 ml). The voluminous solid which formed
in 15 minutes was filtered after 2 hours, washed
with water and dried partially in air to give 20.8
g. The solid was melted in a 170 oil bath and
heated for 10 minutes, until amine evolution had
stopped and bubbling had virtually ceased. The
cooled product, a mobile liquid, was acidified with
, 10% potassium hydrogen sulfate, extracted with
hexane, dried over sodium sulfate and evaporated to
give 6.3 g of solid. The aqueous filtrates from
the Mannich reaction were allowed to stand
overnight and were then heated at 100C on a steam
cone until bubbling ceased. Cooling, acidification
and extraction as above gave another 1.2 g of solid
for a total of 7.5 g of benzyl acrylic acid.
B. -[(Cyclohexylthio)methYl]benzenepropanoic
I acid
To a solution of the title A compound (8.1 g,
50 mmol) in piperidine (16 mL) under argon was
added cyclohexyl mercaptan (6 mL, 50 mmol).
The mixture was stirred under argon at
100C for 24 hours, after which it was cooled to
25C and acidified by addition of concentrated
8 ~
~ 47_
- ~ .. , - ~
",., , ,,: .
hydrochloric acid. The mixture was then saturated
with sodium chloride and was extracted twice with
ether. The ether extracts were combined and
extracted with 1 N sodium hydroxide solu~ion. The
basic aqueous extract was made acidic by addition
,~ of concentrated hydrochloric acid and was extracted
j .
with ethyl acetate. The extract was dried over
anhydrous magnesium sulfate and concentrated. The
residue (4 g) was purified by flash chromatography
; 10 on silica gel, eluting with 1:1 ethyl
acetate:hexanes, to give the title B compound as a
colorless oil.
:,
C. [3-[LCyclohexylthio)methyl]-l-oxo-3-phenyl-
~ropyl]-n-[(lS,2R)-l-(cYclohexylmethYl)-2-
.' hydroxY-2-~lH-imidazol-2-yl)ethyl]-L
histidinamide, dihydrochloride
To a solution of the title B compound (557
mg, 2 mmol), l-hydroxybenzotriazole hydrate (337
~ 20 mg, 2.2 mmol) and the title I compound
I from Example 2 tl.l g, 2.2 mmol) in dimethyl-
formamide (8 mL) at 0C were added triethylamine
(0.92 mL, 2.2 mmol) and dicyclohexylcarbodiimide
(453 mg, 2.2 mmol). The resulting mixture was
25 stirred for 18 hours at 25C,
after which it was concentrated to dryness. The
residue was dissolved in a mixture of methanol (9
mL~ and 1 N hydrochloric acid ~6 mL), filtered, and
the filtrate was concentrated to dryness. The
30 residue (3.68 g) was flash chromatographed on -~
silica gel, eluting with ethyl acetate:pyridine:
acetic acid:water (80:20:6:11) to yield a major
fraction having Rf = 0.31 (940 mg). This pinkish m
.l material was further purified by chromatography on
HP-20 (400 mL), eluting with a gradient from 0.01 N
-
~'
~ ~ - 13296~0
-. HA432
-48-
hydrochloric acid to o.ol N hydrochloric acid in
~- ~ methanol. Fractions were monitored by HPLC
analysis; those containing the desired products
~- (7.1 min and 8.3 min. YMC S-ODS column, 4.6 x 150
~`ce. S mm, 1.0 mL/min of 66% aqueous methanol containing
--j,, 0.01% phosphoric acid, A = 220 nm) were combined
and partially concentrated in vacuo to remove
methanol, then frozen and lyophilized. The residue
was relyophilized from water to give the title
compound as a fluffy, off-white (pinkish) solid
(386 mg, 28%) .
Example 5
[2-[(Cyclohexylsulfinyl)methyl]-1-oxo-3-
phenylpropyll-N-[(lS,2R~ (cyclohexvl-
~, methyl)-2-hydroxy 2-(lH-imidazol-2-yl)-
-~ ethyl]-L-histidinamide, isomer 2B, tri-
fluoroacetate (1:2) salt
A. a-[(Cyclohexylsulfinyl)methyl]benzene-
ro~anoic acid, isomers 1 and 2
A solution of the title B compound from
~ , Example 4 (1.95 g, 7 mmol) and hydrogen peroxide
1 25 (4.76 mL, 42 mmol) in methanol (35 mL) was stirred
~¦ at 25C for two hours, after which additional
hydrogen peroxide was added (0.45 mL). The mixture
was stirred for 1.5 additional hours (3.5 hours
I total), then made acidic by addition of sulfuric
;~ 30 acid (35 mL). The mixture was checked for presence
of peroxides using starch-iodine test paper
(positive test). Saturated aqueous sodium
thiosulfate solution was added until a positive
peroxide test was no longer obtained. The mixture
¦ 35 was then diluted with an equal volume of ethyl
$
3 2 9 ~ 8 0
~ 49_ HA432
. -
- .
. . - ~ -
acetate and stirred for 10 minutes, after which it
was filtered and extracted with ethyl acetate. The
-,
~ ~ extracts were combined, dried over anhydrous
.''r" ' ~ magnesium sulfate, and concentrated. The residue
- ;~ 5 (2.3 g) was crystallized from 95% ethanol to give
isomer 1 o~ the title A compound (670 mg, 32%, m.p.
153-154C). The mother liquor was diluted with
ether and cooled to give a white solid (m.p.
118-120C) which was recrystallized from ethanol to
give isomer 2 of the title A compound (365 mg, 18%,
m.p. 123-124C).
B. [2-[(CYclohexylsulfinyl)methyl]-l-oxo-3
phenylpropyll-N-[(lS,2R)-l-(cyclohexyl-
methYl?-2-hydro_y-2-(lH-imidazol-2-yl)-
ethyll-L-hist dinamide, isomer 2B, tri-
fluoroacetate (1:2L salt
.
To a solution of the isomer 2 of the title A
compound (539 mg, 1.83 mmol), l-hydroxybenzo-
triazole hydrate (306 mg, 2 mmol), and the
title I compound from Example 2 (998 mg, 2 mmol) in
dimethylformamide (7 mL) at 0C were added
triethylamine (O.85 mL, 6 mmol) and dicyclo-
hexylcarbodiimide (412 mg, 2 mmol). The
resulting mixture was stirred for 19 hours at 25C,
after which it was concentrated to dryness. The
, residue was dissolved in a mixture of methanol (10
! mL) and 1 N hydrochloric acid (7 mL), filtered and
;~¦ 30 concentrated. The residue (3.6 g) was flash
chromatographed on silica gel, eluting with ethyl
I acetate:(pyridine:acetic acid:water) 2:1(20:6:11).
I Fractions containing the major "product" (Rf 0.30)
were combined and concentrated to give an isomeric -~
J
`1 ~ ~ . .
~: :
3 2 9 ~ 8 ~
HA432
50-
~',,-'"' ~ '
mixture (720 mg). The isomers were separated by
preparative HPCL to give isomer 2A of the title B
; compound (280 mg, 31%) and isomer 2B of the title B
compound (150 mg, 17%).
t Example 6
[2-[(Cyclohexylsulfonyl)methyl~ oxo-3-
phenylProPYl] N-[(lS,2R)-1-(cyclohexyl-
methyl ? -2-hydroxY-2-(lH-imidazol-2-yl)-
ethyl]-L-histidinamide, isomer B, tri-
fluoroacetate (1:2) salt
A. a-[(CyclohexYlsulfonyl)methyl]benzene
proPanoic acid
To a solution of the title B compound from
Example 4 (2.78 g, 10 mmol3 in methanol (40 mL) at
0C was added a solution of potassium monoper- -~
' - sulfate (9.21 g, 30 mmol) in water (40 mL). The
1 20 resulting mixture was stirred for 2 hours as it
,I warmed to 25C, after which it was extracted with
chloroform. The extract was dried over anhydrous
magnesium sulfate and concentrated. The residue
(3.02 g) was crystallized from ethyl
acetate/hexanes to give the title A compound (2.90
g, 94%), m.p. 109-111C.
.:t B. [2-[(Cyclohexylsulfonyl~methyl]-1-oxo-3-
,l phenylpropYll-N-[(lS,2R)-1-(cyclohexyl-
! 30 methyl)-2-hYdroxY-2-(lH-imidazol-2-yl)-
ethyll-L-histidinamide, isomer B, tri-
,l fluoroacetate (1:2~ salt
To a solution of the title A compound (1.24
g, 4 mmol), 1-hydxoxybenzotriazole hydrate (612 mg,
~i~ 35 4 mmol) and the titl~ I compound from
. .~. .:
: ~ I I . ' / ' ;
`~: 1329~gO
- HA43Z
51-
Example 2 (2 g, 4 mmol) in dimethylformamide (15
mL) at 0C were added triethylamine (1.76 mL, 12.6
,.
mmol) and dicyclohexylcarbodiimide (824 mg, 4 mmol).
-~ After being stirred for 18 hours at
; 5 25c, the mixture was concentrated to dryness. To
the residue were added methanol (20 mL) and 1 N
hydrochloric acid (14 mL). The mixture was
filtered and the filtrate was concentrated to
dryness. The ~esidue was chromatographed on silica
gel to give an isomeric mixture Rf 0.28 (1.54 g).
The isomers were separated by preparative HPLC.
Fractions containing the individual isomers were
partially concentrated and lyophilized. The
residues were relyophilized from water to give
isomer A of the title B compound (730 mg, 21%) and
isomer B of the title B compound (520 mg, 15%).
Example 7
[2-[(DiethoxYPhosphinyl)methvl]-l-oxo-3-
phenylpro~vll-N-[(2S,3R)-l-(cyclohexvl-
m_~hyl)-2-hydroxy-2-(lH-imidazol-2-yl)ethyl-
L-histidinamide, isomer B, trifluoxoacetate
2) salt
A. a [~DiethoxyphosPhinyl~meth-yl]benzene- -
propanoic acid
Bis(trimethylsilyl~acetamide (5 g, 0.024 i~
mole, 6 ml) was added, at room temperature, to a
, 30 solution of benzyl acrylic acid (2 g, 0.012 mole)
j and diethyl phosphite (3.3 g, 0.024 moles) in
dichloromethane (30 ml). The mixture was stirred
at ambient temperature for 30 minutes, concentrated
¦ in vacuo at room temperature, and the residue was
~ 35 heated at a bath temperature of 100-110 for 16
:' ~:.' ;'.
3 ~ ~ 6 ~ 0
-52- HA432
hours. The colorless oil reaction mixture was
dissolved in ethyl acetate and extracted with 5%
sodium bicarbonate (pH 9-10). The aqueous alkaline
~ solution was washed with ether and acidified to a
- 5 pH of 1-2 with concentrated hydrochloric acid. The
- colorless oil that separated was extracted into
ethyl acetate, washed with brine, dried over
anhydrous magnesium sulfate and concentrated in
vac~o to give a colorless oil ~3.6 g).
B. [2-[(DiethoxYphosphinyl)methy~ oxo-3-
~enylpropyl]-N-[(lS,2R)-l-(cyclohexylmethyl)-
2-hydroxY-2-[1-[(phenylmethoxY)methyl]-lH-
imidazol-2-yllethyll-3-[(phenylmethoxy)methyl]-
L-histidinamide, isomers A and B
To a solution of the title J compound from
Example 1 (1.46 g, 2.0 mmols), l-hydroxybenzo-
;, triazole hydrate (337 my, 2.20 mmols), and
the title A compound (6G0 mg, 2.20 mmols) ln
~3 20 tetrahydrofuran (8 mL) at 0C were added
triethylamine (O.92 mL, ~.6 mmols) and
dicyclohexylcarbodiimide (453 mg, 2.20 mmols).
The resulting mixture was stirred for 18 ~ ;~
.l hours at 25C and filtered. Ethyl acetate was ~i
¦ 25 added to the filtrate and the resulting solution
was washed with saturated sodium bicarbonate
solution, dried over anhydrous magnesium sulfate
;¦ and concentrated. The residue (1.75 g) was flash
¦ chromatographed on Merck silica gel, eluting with
1 30 ethyl acetate:pyridine:acetic acid:water (100:20
¦ 6:11), to give isomer A of the title B compound
(680 mg, 38%) and isomer B of the title B compound
(620 mg, 35%).
: "~
~. ;~
: j; : . . ,: : .
.. ' " ~ .,'~',.. `".~,
~ 3 2 9 6 8 0 HA432
53-
C. [2-[(DiethoxYPhos~hinYl)methYl~ oxo 3-
phenylpropy_~-N-~(2S,3R~ (cyclohexyl-
:~ . methyl)-2-hydroxy-2-(lH-imidazol-2-yl)ethyl-
L-histidinamide, isomer B, trifluoroacetate
~: 5 (1:2) salt
~ ~-
A mixture of the isomer B of the title B
compound (600 mg, 0.68 mmols), 20% palladium
hydroxide on carbon (150 mg), and 1 N
hydrochloric acid (1.5 mL, 1.5 mmols) in methanol
(15 mL) was hydrogenated under a slow stream of
hydrogen for 18 hours at 25C, after which it was
filtered and concentrated to dryness. The residue
(470 mg) was flash chromatographed on Merck silica
gel, eluting with chloroform:methanol:ammonium
hydroxide (100:25:1.25). Fractions containing the
major component were combined and concentrated.
`~ The residue (300 mg) was dissolved in excess 1%
aqueous trifluoroacetic acid solution and ~:~
reconcentrated. The residue was then dissolved in
20 water ~50 mL), treated with activated charcoal, ~:
l millipore filtered, and lyophilized to give the ¦~:
;1 title compound as a fluffy white solid (320 mg,
.1 66%).
! ~:
'`l .
il `
~:~;; ' ; ;'
':,`1~ ' "'`'` ~
~32~68~
- HA432
-54-
,~
Example 8
. . .
~ [(S)-2-[(Benzoylthio)methyl~ oxo-3-phenyl-
-~ propyll-N-[(lS,2R)-l-(cyclohexylmethyl-2-
hydroxy-2-(lH-imidazol-2-Yl)ethyl]-L-
histidinamide, trihydrochloride
A. a-[(Benzoylthio)methyl]benzenepropanoic acid
A mixture of the title A compound from
Example 4 (13.7 g, 85 mmol) and thiobenzoic acid
(15 mL, 127 mmol) in methylene chloride (170 mL)
was stirred under argon at reflux temperature for
3 days, after which it was concentrated in vacuo.
The residue was crystallized from ether/hexane to
l 15 g:ive 13.7 g of the title A compound as a colorless
¦ solid, m.p. 99-100C.
B. (S)-a-[(BenzoylthioLmethyl]benzene~ropanoic
! acid
A solution of the 1:itle A compound (14.5 g,
48.0 mmol) and (R)-(+)-a--methylbenzyl amine (6.25
mL, 48 mmol) in ether (250 mL) was stored at -4C
for two days, resulting in a gummy solid. The
i solvent was decanted and the solid was
recrystallized 3 times from hexane/methylene
chloride (1:1) then twice from isopropanol to give
I a 1:1 salt, m.p. 131-132C, ~a]D = -25.0 (c = 1,
CHCl3). The salt was dissolved in ethyl acetate
and washed with 1.0 N hydrochloric acid. The
organic layer was dried over anhydrous magnesium
sulfate and concentrated to give 1.57 g of the -;
title B compound as a white solid, m.p. 65~66C,
[~]D = ~53 3 (c = 1, CHC13).
' '~'~''`''
'~ ~.: '.~:
1 .
!
` ~ 3 2 ~ 6 ~ 0
- HA432
~ -55-
:.: : .
~- :
C. [(S)-2-[(Benzoylthio)methyl]-l-oxo-3-phenYl-
prop~l]-N-[(lS,2R)-l-(cyclohexylmethyl-2-
hydroxy~2-(lH-imidazol-2-yl)ethyll-L-
.~ .
histidinamdie, trihydrochloride
To a solution of the title B compound (1.42
g, 4.73 mmol), l-hydroxybenzotriazole hydrate
(724 mg, 4.73 mmol) and the title I compound from
Example 2 (2.22 g) in dimethylformamide (20 mL) at
0C were added triethylamine (2.2 mL, 16 mmol) and
dicyclohexylcarbodiimide (975 mg, 4.73 mmol). The
resulting mixture was stirred at 25C for 18
hours, after which it was filtered and
concentrated. The residue was chromatographed on
silica gel, eluting with e~hyl acetate:(pyridine:-
acetic acid:water) 2:1(20:6:11). Fractions
containing the major product were combined and
-~, concentrated. The residue was dissolved in excess -
.,
! 1 N hydrochloric acid and concentrated in vacuo.
I The residue was then lyophilized from water to
give the title compound as a fluffy white solid.
~! Example 9
~1 :
I . [(S)-2-(Mercaptomethyl)-l-oxo-3-~henYl-
-I 25 ~ro~yl]-N-[(lS,2R)-l-(cyclohexylmethYl)-
2-hydroxy-2-(lH-imidazol-2-yl2eth~1]-L-
I histidinamide, trifluoroacetate (1:2~ salt
-~ A mixture of [(S)-2-[(benzoylthio)methyl]-1-
;~' 30 oxo-3-phenylpropyl]-N-[(lS,2R)-l-(cyclohexylmethyl-
~ 2-hydroxy-2~(1H-imidazol-2~yl)ethyl]-L-histidinamide,
, :
:, . '~
'I :
,
.
~ ~329~8~
- HA432
56-
. ~
:,.. ..
~- trihydrochloride (270 mg, 0.35 mmol) and
- concentrated ammonium hydroxide solution (1 mL) in
methanol (5 mL) was stirred at 50C for 7 days
(additional ammonium hydroxide (2 mL) was added
- 5 after 2 and 6 days). After 7 days, the mixture
was concentrated in vacuo and the residue was
purified by preparative HPI,C. Fractions containing
the major product were concentrated in vacuo. The
residue was dissolved in water and lyophilized to
give 45 mg of the title compound as a fluffy white
powder.
Examples 10-30
Following the procedures of Examples 1-9,
i 15 additional compounds within the scope of this
invention can be prepared having the formula ~ ~
R5 IR4 1l R3 - ~ :
. X-CH2 -CH-C-NH-CH-C-NH-CH-CH-R
OH
"
~ wherein the substituents are as defined below.
,
, ~'':','~,:-:
:, : ',:.
`~1
,~ :
....
1 3 2 9 6 8 0
5 7 - EIA4 3 2
:,. :~ , - .
'~ ; . ~: /= $
~r: ~-Z~ Z _ ~ :
r~
~ ~ m
I I ~ ~
P~i N
u ~ I N
.,
1~ 20 ~: [~
. g~ Z~ ~
' 25 I , : ~
,` O=U O=U ' :': ~:.
X ' O u o=~la_o
1 31) ~ ¦ U
'I . :~ ~:
.
. ¦ Z ~I N ~"
, . . ,~ ::: ;
35 1~
. .
: ~ :
: : :
l::
: ~ 11 32~o
5 8 - HA4 3 2
.
1~: ~ 1 ~Z
. . ~ 5 N _ N
., ~ _ ~ U ~ ~ :
`' ~ ~ $
N U N N
~, 15 X ~ ~ N
.3
,.,~ '"`:-:
~ 20 a: (~
'! X ~ U '.. .
, ,N~ N .. ~
~. U U
,i:i ~25
i', O=u~- O ~1 ~ '- '' ''''' ~'
,-
~ X I ~ N
. 1 ~Z~ N o= pl =o o~
: ~ J ~ O ' :, "
3 0 O u
~ . U U '
1-1
:
1 .
.~ ~
:
3 2 ~
HA4 3 2
5 9-
~ I N
Q N
~. 5 Z~ v~ ~z-m u~
\I=Z
~ \rz-;c
Zl~ Q m ~Z
Z~
, J I y ~ N
Y
~
U ~ ~ $
j . Z~ I N
~ .,
2 0 u U U
ZZ , Z 5 N
,Z
Z;~Z ~,
O~
, ~ . X ~ ~o= ~--o ~n o= u~ _o : :
`I 30 ~oJ
l! O ;~$ ~ ,
~1 z a~ a~ o ~1
35 ~
:
:`` :
132~680 HA432
: . --60--
, ~
U
5 ~ Z
. N ~ N :'J
' ~ ~ ~ 5~ 5 '''"''''
'',1 10 ~
., U ~ N
.~ l l l
15 ~ z
u ~ u .~
~ I , . "-~
~ ~ ~ N
., ~ N ' N ~ N ;
. ~ 25 ; ; :
( P:
U
.~ Iq O=U~=O ~ O=U~=O
"I x o_~_o u o=~=o
$''' ~ b
`,, .
o
~ ' ~; ~ ) ~ N
~, 35 lil
:'1
~`'' .
:
1~2~0
- 61- HA4 3 2
:~ :
m ~ m
',,
~ q ~ ~? ~
, ~ _ ~I N
V
,, ~ I ~ I I ::
0 m
: .:
1S ~ L
I .
,,, .: ~
,",,,, t~
C~
::,
:~1 30 o=l-U o~
~ ~1 æ
~ :; X ~ N ~J ~ : :
; . 3 5 ~1 . :
~ ,
, ~
: '~
,~: ~
~:,
1329680
,. .
~ - 6 2 - HA4 3 2
':',
" :'
~ ! ~ P (~ z
~i 5 I ~.
~i ~
,,
P;
,~ ~c 1~ ... ,, .. ~.
N
,
'~
u~
~:
25 1 1 =
. ~ .
Z I o `'
3 5 ~3
.1 .
~` ,