Note: Descriptions are shown in the official language in which they were submitted.
5010P/1286A 1 3 3 o ~ q o
- 1 - 17358
TITLE OF TH~ INVENTION
TETRAHYDROCARBAZOLE ESTERS
BACKGROUND OF T~E INVENTION
This invention relates to prostaglandin
antagonists useful in treating a variety of
conditions, such as allergic asthma where excessive
contractile activity of prostaglandins and
prostaglandin biosynthetic intermediates occur.
These compounds antagonize the actions of
contractile prostaglandins, such as PGF2a,
PGG2, PGH2, PGD2 and TXA2. The use of agents
which act as prostaglandin antagonists oEfers new
approaches to therapy in a number of disease states.
For esample, certain prostaglandins, such as
PGF2X, PGD2, PGG2, and PGH2, are potent
bronchospastic agents. Indeed human asthmatics have
been shown to be especially sensitive to the
bronchial constricting action of PGF2a.
,
1 3300~0
5010P/1286A - 2 - 17358IA
The compounds of the present invention are
also antithrombotic agents. Thus, they are useful in
the treatment and/or prevention of thromboembolic
diseases such as arterial thrombosis and those
involving platelet deposition, e.g. prothesis.
In addition to the involvement of
contractile prostaglandins in asthma, prostaglandins
are known to play a role in other allergic conditions,
as well as, diarrhea, hypertension, angina, platelet
aggregation, cerebral spasm, cerebral ischemia,
arrythmia, circulatory shock, sudden death, athero-
sclerosis, myocardial ischemia, premature labor,
spontaneous abortion, dysmenorrhea, glomerular
nephritis, and systemic lupus erythematosis.
15 Consequently, the compounds of this invention will -
alleviate the above mentioned diseases.
In addition to the prostaglandin antagonist
actions, the compounds of this invention are
inhibitors of the biosynthesi~ of 5-lipo-ygenase
metabolites of arachidonic acid, such as 5-HPETE,
5-HETE and the leukotrienes. Leukotrienes B4,
C4, D4 and E4 are known to contribute to variaus
disease conditions such as asthma, psoriasis, pain,
ulcers and systemic anaphyla~is. Thus inhibition of
the synthesis of such compounds will alleviate these
and other leukotriene-related disease states.
The compounds of the present invention may
be used to treat or prevent mammalian (especially,
human) disease states such as erosive gastritis;
erosive esophagitis; ethanol-induced hemorrhagic
erosions; hepatic ischemia; no~ious agent induced
. ~
~ damage or necrosis of hepatic, pancreatic, renal, or
".. ~,._.. ~ .
- : -
.
.
~: ' ~ '` ` ' ' ' ' , . `
1 3300q3
5010P/1286A - 3 - 17358IA
myocardial tissue; liver parenchymal damage caused by
hepatoxic agents such as CC14 and D-galactosamine;
ischemic renal failure; dissase-induced hepatic
damage; bile salt induced pancreatic or gastric
damage; trauma- or stress-induced cell damage; and
glycerol-induced renal failure.
Certain 9-benzyl-1,2,3,4-tetrahydro-
carbazole acetic acids or esters thereof are shown as
chemical intermediates in the preparation of
carbazoles that are known in the art as anti-
inflammatory, analgesic and anti-rheumatic agents
(see U.S. Patent 3,896,145 and British Patent
1,385,620). Certain 9-benzyl-1,2,3,4-tetrahydro-
carbazole carboxylic acids are known in the art as
anti-inflammatory, analgesic and anti-rheumatic
agents (see U.S. Patents 3,868,387; 4,009,181;
3,905,998 and 3,758,496), and 9-benzylcarbazole
carboxylic acids (U.S. Patents 3,956,295 and
4,057,640) and 9-benzylcarbazole acetic acids and
esters thereof ~U.S. Patent 3,896,145 and ~ritish
Patent 1,385,620) are known as anti-inflammatory,
analgesic and anti-rheumatic agents. None of these
compounds, however, are shown to be prostaglandin or
thromboxane antagonists or inhibitors of leukotriene
biosynthesis.
,, .
1 330090
5010P/1286A - 4 - 17358IA
DESCRIPTION OF THE INVENTION
One embodiment of the present invention is a
pharmaceutical composition containing a compound of
Formula I:
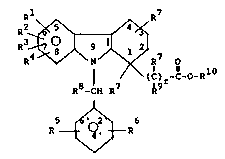
s
Rl R7
R2~\~
R3
¦ / (C) r-C-O-R
R3-CH R7 ~9
~ 6
R ~ R
I
wherein:
R , R2, R3, R4, R5 and R6 are
each independently selected from:
(1) hydrogen;
(2) alkyl having 1 to 6 carbons;
(3) alkenyl having 2 to 6 carbons;
(4) -(CH2~nM
wherein n is 0 to 3 and M is
CJRll
b) halogen;
c) CF3;
d) sRll;
e) phenyl or substituted phenyl;
f) COOR12;
1 3300qO
5010P/1286A - 5 - 17358IA
g) -C-R13;
h) -NH-C-R14
i) _NR12R12;
j) -NHSo2R15;
k) -C-CH2OH;
1) -SORll;
m) coNR12R12;
n) _so2NRl2Rl2;
S02R
p) N02;
q
r) -O-C-NR12R12;
~ 14
s) -O-C-OR
t) CN;
u) N3;
R7 is H or alkyl of 1 to 6 carbons;
R8 is H or alkyl of 1 to 6 carbons;
each R9 is independently H, OH, Cl to
C4-O-alkyl or alkyl of 1 to 4 carbons;
R10 is lower alkyl, substituted or
unsubstituted 2-phenethyl, substituted or
unsubstituted benzyl, or substituted or unsubstituted
phenyl
~.~
1 3300qO
5010P~1286A - 6 - 17358IA
each R~ independently H; Cl to C6
alkyl; benzyl; phenyl or substituted phenyl;
each R12 i~ independently H, phenyl,
benzyl or Cl to C6 alkyl;
each R i~ independently H,
(CH2)mCOOR12 wherein m i~ 0 to 4, Cl to C6
alkyl, CF3, phenyl, or substituted phenyl;
each R14 is independently Cl to C6
alkyl, benzyl or phenyl;
each R15 i~ independently Cl to C6
alkyl, 4-methylphenyl, phenyl, or CF3;
r is 1 to 6
or a pharmaceutically acceptable salt thereof.
Preferred compositions of the present
invention contain a compound of formula I
wherein:
~p
- "
1 3300qO
5010P/1286A - 7 - 17358IA
R , R , R3, R4, R5 and R6 are
each independently selected from:
(1~ hydrogen;
(2) alkyl having 1 to 6 carbons;
(3) alkenyl having 2 to 6 carbons;
(4) -(CH2)nM
wherein n is 0 or 1 and M is as defined
previously for Formula I;
and the remaining substituents are as
defined previously for Formula I.
More preferred compositions of the present
invention contain a compound of Formula I.
wherein:
R , R , R3, R4, R5 and R6 are
each independently selected from:
(1) hydrogen;
(2) alkyl having 1 to 6 carbons;
(3) alkenyl having 2 to 6 carbons;
(4) M, wherein M is as defined initially
for Formula I;
r is 1 or 2; and the remaining substituents
are as defined initially for Formula I.
Most preferred compositions of the present
invention contain a compound of Formula I.
wherein:
R , R2, R3, R4, R5 and R6 are
each independently selected from:
(1) hydrogen:
(2) alkyl having 1 to 6 carbons;
(3) M wherein M is
a) ORll;
b) halogen;
1 3300~0
5010P~1286~ - 8 - 17358IA
C) C~;
A) sR~l;
~) COOR12;
f) -C-R13;
g) -SORll;
h) _coNRl2Rl2;
~ ) -so2NRl2Rl2;
j ) -S2R
k) -o-C-R13;
1) C~;
m) N3;
each R9 i8 ~ndependently H, or alkyl of
to 4 carbon~;
R10 i~ lower alkyl;
r ~8 1 and the remaining substituents are as
defined initially for Formula ~.
Another embodiment of the present invention
relate~ to no~el compound~ of Formula I:
~,.",,~.~'
1 330090
5010P/1286A - 9 - 17358IA
~70
R8-CH R7 ~)r-~-O-R10
5~R6
wherein:
R , R2, R3, R4, RS and R6 are
each independently selected from:
(1) hydrogen;
(2) alkyl having 1 to 6 carbons;
(3) alkenyl having 2 to 6 carbons;
(4) -(CH2)nM
wherein n is 0 to 3 and M is
a) oRll;
b) halogen;
c) CF3;
d) sRll;
e) phenyl or substituted phenyl;
f) COOR
g) -C-R13;
h) ~NH~C~Rl4;
,A~
1 330090
5010P/1286A - 10 - 17358IA
i) NR12R12;
j) -NHSo2Rl5;
k) -C-CH2OH;
1) -SORll;
m) _coNRl2Rl2;
n) _so2NRl2Rl2;
o) _so2Rll;
p) NO2;
q) -o-C-R13;
r) -O-C-NR12R12;
O
s) -o-C-oR14;
t) CN;
u) N3;
provided that when R1, R2, R3 or R4
is alkyl havlng 1 to 6 carbons, it is not
located at position 6;
R7 i8 H or alkyl of 1 to 6 carbons;
R is H or alkyl of 1 to 6 carbons;
each R i5 independently H, OH, Cl to5 C4-O-alkyl or alkyl of 1 to 4 carbons;
R10 is lower alkyl, substituted or
unsubstituted 2-phenethyl, substituted or
unsubstituted benzyl,or substituted or unsubstituted
phenyl:
. .
1 330090
5010P/1286A ~ 17358IA
each Rll independently i5 H; Cl to C6
alkyl; benzyl; phenyl or ubstituted phenyl;
each R12 is independently H, phenyl,
benzyl or Cl to C6 alkyl; and,
each R13 is independently H,
(CH2)mCOORl2 wherein m i3 0 to 4, Cl to C6
alkyl, CF3, phenyl, or sub~tituted phenyl;
each R14 is Cl to C6 alkyl, benzyl or
phenyl;
each R15 is Cl to C6 alkyl,
4-methylphenyl, phenyl, or CF3;
r is 1 to 6
or a pharmaceutically acceptablP salt thereof.
As used herein, the terms ~each
independently~ or the equivalents thereof are employed
to describe a number of possible position isomers
and/or structural variations. For e~ample, as
described above, the following unit is attached to
position 1 of the tetrahydrocarbazole ring:
1 3300qG
5010P/1286A - 12 - 17358IA
R7 q
-(C)r-C-O-R
R9
The letter r represents possible alkane
chains of from 1 to 6 carbon atoms, each having the
R7 and R9 substituent groups. On each carbon
atom of the alkane chain, the R7 and/or R9
substituent may be different. The above description
therefore contemplates structures such as the
following for the segment -(CR7R9)r-:
CH H H
1 3 1
(- ~C - C - C) , (- CH2 -)
OH H H
H H H HH H H H
I
(--C -- C -- C--) , (--C -- C -- C -- C -- C--) ,
OH CH3 OH H OH CH3 H H
H Hl Hl
(-C - C - C-) , and the like.
H CH3 H
Substituted phenyl, substituted benzyl, and
substituted phenethyl signifies the presence of 1 or
2 substituents on the benzene ring selected from Cl
to C3 alkyl, halogen, CN, CF3, COOR12, CH2COOR12, or
Cl to C3 alkosy.
The alkyl groups referred to above may be
straight chain or branched or may include cycloalkyl
qroups. As used herein, the term ~lower~ as applied
~ 1 330093
5010P/1286A - 13 - 17358IA
to alkyl, acyl, alko~y and the like, unless stated
otherwise refers to groups having 1 to 6 carbon
atoms. Halogen or halo means fluoro, chloro, bromo
and~or ~odo.
Pharmaceutically acceptable salts of the
compounds described herein are included within the
scope of the present in~ention. Su~h salts may be
prepared from pharmaceutically acceptable non-to~ic
bases including inorganic bases and organic bases.
Salts derived from inorganic bases include sodium,
potassium, lithium, ammonium, calcium, magnesium,
ferrous, zinc, copper, manganous, aluminum, ferric,
manganic salts and the like. Particularly preferred
are the potassium, æodium, calcium and magnesium
salts. Salts derived from pharmaceutically acceptable
organic non-to~ic bases include salts of primary,
secondary, and tertiary amines, substituted amines
including naturally occurring substituted amines,
cyclic amines and basic ion e~change resins, such as
isopropylamine,tri-methylamine, diethanolamine,
diethylamine, triethylamine, tripropylamine,
ethanolamine, 2-dimethylaminoethanol, 2-diethylamino
-ethanol, tomethamine, lysine, arginine, histidine,
caffeine, procaine, hydrabamine, choline, imidazole,
betaine, ethylenediamine, glucosamine, methyl-
glucamine, theobromine, purines piperazine,
1
1 3300qO
5010P/1286A ~ 14 - 17358IA
N,N-dibenzyl- ethylenediamine, piperidine,
N-ethyl-piperidine, morpholins, N-ethylmorpholine,
polyamine resins and the like.
Preferred novel compounds of the present
invention comprise the compound of Formula I
wherein:
R , R2, R3, R4, R5 and R6 ar
each independently selected from:
(1) hydrogen;
(2) alkyl having 1 to 6 carbons;
~ 3) alkenyl having 2 to 6 carbons;
(4) -(CH2)nM
wherein n is 0 or 1 and M is as defined
previously for the compounds of Formula
I;
and the remaining substituents are as
defined previously for the compounds of Formula I.
More preferred novel compounds of the
present invention comprise a compound of Formula I
wherein:
Rl R2 R3 R4 R5 nd R6 are
a
each independently selected from:
(1) hydrogen;
(2) alkyl having 1 to 6 carbons;
(3) alkenyl haYing 2 to 6 carbons;
(4) M, wherein M is as defined initially
for the compounds of Formula I;
r is 1 or 2; and the remaining substituents
are as defined initially for the compounds of Formula
I.
Most preferred novel compounds of the
present invention comprise the compound of Formula I
wherein:
. . .
. ~ ~
1 330093
5010P/1286A - 15 - 17358IA
R , R , R , R , and R are each
independently selected from:
~1) hydrogen;
~2) al~yl having 1 to 6 carbons;
(3) M wherein M is
a) ORll;
b) halogen;
c) CF ;
d) SR~l;
e) COOR12;
o
Il 13
f) -C-R
g) -SORll;
h) _coNRl2Rl2;
i) _so2NRl2Rl2;
; ) -S02R
k) - o-C-R13;
1) CN;
m) N3;
R6 is located at position 3' or 4' and is
selected from:
(1) alkyl having 1 to 6 carbons;
(2) M wherein M is
' ' 1 1
b) halogen;
c) CF ;
d) sR~l;
e) COOR12;
~ ol 13
f) -C-R
~.~
1 330090
5010P~1286A - 16 - 17358IA
g) -SORll;
h) ~CNRl2Rl2;
S ~ ) -S02R
k) -o-8-R13;
1) CN:
m) N3;
pro~ided that when Rl, R2, R3 or R4
is alkyl having 1 to 6 carbons, it is not located at
position 6;
each R9 is independently H, or alkyl of 1
to 4 carbons;
~10 is lower alkyl;
r is 1 and the remaining substituents are as
def$ned initially for the compounds of Formula I.
,,f
- 1 3300')0
5010P/1286A - 17 - 17358IA
Table 1
Novel TetrahYdrocarbazole
Alkanoi~ Esters
Rl 5 4 R7
8 /
R8-CH R9--C-CO2R1
RS~R6
1 330090
5010P/1286A - 18 - 17358IA
Compound Rl R2 R R6 R9 R9' R7 R8 R10
1 (Ex.l) 6-F H 4'-Cl H H, H H H Et
2 (Ex. 4) 6~Me H 4'~1 H H, H H H Et
3 (Ex. 18) 6-F H 4'-Cl H H, H H H Me
(-) i somer
10 4 (Ex. 19) 6-F H 4'-Cl H H, H H H Me
(~) i somer
5 (Ex. 5) 6-F H H H H, H H H Et
6 (Ex. 6) 6-F H 4'~0Mo H H, H H H Et
7 (Ex. 7) 6-F H 3'-C1 4'-Cl H, H H H Et
20 8 (Ex. 8) 6-F H H H H, H H Mo Et
_ _
9 (Ex 9) H H 4'-Cl H H, H H H Et
10 (Ex. 10) 6-Cl H 4'-Cl H H, H H H Et
11 (Ex. 11) 8-Me H 4'-Cl H H, H H H Et
12 (Ex. 12) 6-3r H 4'-Cl H H, H H H Et
30 13 (Ex. 13) 6-Me H 4'-Cl H H, H H H Et
.,
~ 3300qO
5010P/1286A - 19 - 17358IA
Co~pound R R2 R5 R6 R , RR R R
14 (Ex. 15) 8-F H 4'-Cl H H, H H H Et
6-F H 4'-Cl H H, H3-t-5u H CH2C6H5
16 5-F H 4'-Cl H H, H H H CH2OAc
CH2 CH3
17 7-F H 4'-Cl H H, H H H ~
0~0
o
18 (Ex. 16) 5-C1 7-C1 4'-Cl H H, H H H Et
19 (Ex. 17) 6-C1 8-C1 4'-Cl H H, H H H Et
o
~ N-Me
6-F H 4'-Cl H Me, H H H CH2-N
o
21 6-F H 4'-Cl, H Me, Me H H
0
~L,
22 6-F H 4'-Cl H H, H l-Me H CH2-N ~
_ _ _
1 3300~3
5010P/1286A - 20 - 17358IA
23 (ex 20) 6-CH~Me)2 H 4'-Cl H H,H H H Et
24 (ex 21) 6-C(Me)3 H 4'-Cl H H,H H H Et
25 (ex 22) 6-CF3 H 4'-Cl H H,H H H Et
26 (ex 23) 6-SMe H 4'-C1 H H,H H H Et
27 (ex 24) 6-SOMe H 4'-Cl H H,H H H Et
28 (ex 25) 6-SO2Me H 4'-Cl H H,H H H Et
29 (ex 26~ 8-CH(Me)2 H 4'-Cl H H,H H H Et
30 (ex 27) 8-SMe H 4'-Cl H H,H H H Me
10 31 (ex 28) 8-SOMe H 4'-Cl H H,H H H Me
32 (ex 29) 6-F H 4'-Cl H H,H 3-Me H Et
33 (ex 30) 6-F 8-F 4'-Cl H H,H H H Me
34 (ex 31) 6-Me 8-M~ 4'-C1 H H,H H H Me
35 ( ex 32) 6-OMe 8-Me 4 ' -Cl H H, H H H Me
15 36 (ex 33) 6-F(-)Isomer 8-F 4'~1 H H,H H H Me
37 (ex 34) 6-F(~)Isom~r 8-F 4~-Cl H H,H H H Me
38 (ex 35) 8-Me(-)Isomer H 4'-Cl H H,H H H Me
39 tex 36) 8-MQ(~)Isomer H 4'-Cl H H,H H H Me
40 (ex 37) 8-F(-)Isomer H 4'-Cl H H,H H H Me
20 41 (ex 38) 8-F(~)Isomer H 4'-Cl H H,H H H Me
42 6-F 8-F 3'-C1 4'-Cl H,H H H Me
43 6-F 8-F 2'-C1 4'-Cl H,H H H Me
44 6-F 8-F 4'-OMe H H,H H H Me
6-F 8-F 4'-OH H H,H H H Me
25 46 6-F . 8-F 4 ' -SMe H H, H H H Me
47 6-F H 4'-S(O)Me H H,H H H Me
48 6-F 8-F 4 ' -NHCOMe H H, H H H Me
49 6-F H 4'-S(O)2Me H H,H H H Me
6-F H 4 ' -F H H, H H H Me
1 330090
5010P/1286A - 21 - 17358IA
51 6-F H 4'-Br H H,H H H MQ
52 6-F 8-Me 4'-Cl H H,H H H Me
53 6-F H 4'-C02H H H,H H H Me
54 6-F H 4'~02Me H H,H H H Me
6-F 8-F 4'-n-C3H7 H H,H H H Me
56 6-F 8-F 3'-I 4'4H H,H H H Me
S7 6-F 8-F 4'-I H H,H H H Me
58 6-N3 H 4'-Cl H H,H H H Me
59 6-F H 4'-N3 H H,H H H Me
-` ~ 330~9
5010P/1286A - 22 - 17358IA
The following reaction schemes illustrate
the preparation of the compounds of the present
invention:
5 Scheme I PreParation of Formula I Compounds
Rl
R2 ~ R7 lower
R ~ ~ alkanol
R N - NH2 ~ ~ ~ >
¦ . HCl R7 ~ R7 reflux
~--\ R8 \(C) r-C02R14
R5 R6 III
II
RZ~/~;
I 7
~1~ \(C) r-Co2R14
R5 ~ 0 ~ R6 R8
~/
2S ,,Ia
The reaction can be conveniently carried out
in an alcohol solvent such as t-butanol, i-butanol, i-propanol .
and the like.
The following ketones (1,2,4) of structure
III are known in the art, and ketone 3 is readily
prepared by procedures analogous to those for the
known ketones.
1 3300~0
5010P/1286A - 2~ - 17358IA
TABLE 2
Ketones of Formula III
No Structure Reference
Ethyl 2-cyclohexanone
1. ~ acetate; commercially
~ ~ available (Aldrich)
O~y
C2Et
Methyl 2-cyclohexanone
2. ~ propionate; J.A.C.S. 85
~ ) 207 (1963)
0~' y G. Stork, A. Brizzolara,
H. Landesman,
J. Scmuszkovicz and
CO2Me R. Terrell
3. Methyl 4-t-butyl-2-
~ cyclohexanone acetate
O
C2Me
4. Methyl 2-(2-cyclohexanone)
~ propionate
~ ) J.A.C.S. 85 207 ~1963)
O y G. Stork, A. Brizzolara,
H. Landesman,
/ CO2ME J. Scmuszkovicz and
R. Terrell
1 330~ql~
5010P/1286A - 24 - 17358IA
TABLE 2 (continued)
KETONES OF FORMULA III
5 No. Structure Reference
5. Ethyl 4-methyl-2-cyclo-
~ / hexanone acetate
C2ET
The sequence described above is an
application of the Fischer Indole Synthesis. Numerous
indole syntheses are described in reviews, such as,
for example "Heterocyclic Compounds~ Volume 25, Parts
I, II, III, W. J. Houlihan (Ed.), Interscience,
J. Wiley & Sons, N. ~., 1979. Appropriate
manipulations of functional groups using sequences
described in such reviews will lead to the compounds
of the present invention.
.... . .
. .
~ - .
. . .
.' "
1 330090
5010P/1286A - 25 - 17358IA
Scheme IIPreparation of Hydrazine Derivatives (II)
Rl CHR8
R2 ¦` 1
~ \ 5 ~ toluene >
R4 N-NH2 ~ R R6 reflux
H .HCl
(IV) (v)
Rl
R2 ~ Z is a leaving group such as
15 R3 ~ ~ Cl, ~r, I, mesylate or tosylate.
O I
R4~ ~ N-NH2
1 .HCl
~ R8
20 R5 ~ R6
II
With regard to Scheme II, the preparation of
hydrazine starting materials is illustrated by the
preparation of 1-(4-chlorobenzyl)-1-(4-methoxyphenyl)-
hydrazine. A mixture of 10 g of p-methoxyphenyl-
hydrazine hydrochloride, 75 ml of toluene and 11.5 ml
of triethylamine was heated at reflux ~or 60 minutes.
Then, 7.1 g of p-chlorobenzyl chloride was added.
After stirring 16 hours at reflus, triethylamine
hydrochloride was filtered off and washed with ethyl
1 33009~
5010P/1286A - 26 - 17358IA
ether. The filtrate and washing were concentrated in
vacuo and chromatographed on a silica gel column
(hesane-ethyl acetate, 9:1) to give 6.64 9 of 1-(4-
chlorobenzyl)-1-(4-methosyphenyl)hydrazine. Other
hydrazines, similarly prepared, are also shown in
Table 3, below.
1 33~0qO
5010P/1286A - 27 - 17358IA
TABLE 3
H~draz ines
4 ~
X~
N - NH2 HCl
4 ~ R8
Compound
No. X Y R Compound Name
1. 4-F 4-C1 H 1-(4-chlorobensyl)-1-(4-
20 ~luorophenyl) hytrazlne hydro-
chlorldo
2. 3,5-C12 4-Cl H 1-(4-chlorobenzyl)-1-~3,5-
dichlorophenyl)hydrazine
hydrochlorlde
3. 4-OMe 4-Cl H 1-(4-chlorobenzyl)-1-(4-
metho~yphenyl) hydrazine
- hydrochlorido
4~ 2-Mo 4-Cl H 1-(4-chlorobenzyl)-1-(2-
methylphenyl) hydrazine
hydrochlorido
.. ,, . ., : ,
~` 1 330~0
5010P/1286A - 28 - 17358IA
TABLB 3 (Cont'dl
5. 4-Me 4-Cl H 1-~4-chlorobenzyl)-1-(4-
methylphenyl) hydrazine
hydrochloride
6. 4-C1 4-Cl H 1-(4-chlorobenzyl)-1-(4-
chloropher,yl) hydrazine
hydrochloride
7. H 4-Cl H 1-(4-chlorobenzyl)-1-(phenyl)
hydrazine hydrochloride
8. 4-Br 4-Cl H 1-(4-chlorobenzyl)-1-(4-
bromophenyl) hydrazine
hydrochloride
9. 3-F 4-Cl H 1-~4-chlorobenzyl)-1-(3-
fluorophenyl) hydrazine
hydrochlorlde
10. 2,4-C12 4-Cl H 1-(4-chlorobenzyl)-1-~2,4-
dichlorophenyl)hydrazine
hydrochloride
11. 4-F H H l-(benzyl)-1-~4-fluorophenyl)
hydrazine hydrochloride
12. 4-F 4-OMe H 1-~4-metho~ybenzyl)-1-~4-
fluorophenyl) hydrazine
hydrochlorlde
,.. ,.. ~,. . . .
. ~ .
.: ~
-
1 330090
5010P/1286A - 29 - 17358IA
TABLE 3 ~Cont'd~
13. 4-F 3,4-C12 H 1-~3,4-dichlorobenzyl)-1-
(4-fluoro-phenyl) hydrazine
hydrochloride.
14. 4-F H CR3 1-tl-(phenyl)ethyl]-1-(4-
fluorophenyl) hydrazine
hydrochlorlde
15. 2-F 4-Cl H 1-(4-chlorobenzyl)-1-(2-
fluorophenyl) hydrazine
hydrochloride.
15 16. 4-CH(Me)2 4-Cl H 1-(4-chlorobenzyl)-1-(4-lso-
propylphenyl)hydrazine hydro-
chloride
17. 4-C(Me)3 4-Cl H 1-(4-chlorobenzyl)-1-(4-tert-
butylphanyl)hydrazine)hydro-
chloride
18. 4-CF3 4-Cl H 1-(4-chlorobenzyl)-1-(4-tri-
fluoromethylphenyl)hydrazine
, hydrochloride
19. 4-SMe 4-Cl H 1-(4-chlorobenzyl)-1-(4-methyl-
thiophenyl)hydrazine hydro-
chloride
20. 2-CH(Me)2 4-Cl H 1-(4-chlorobenzyl)-1-(2-i~o-
propylphenyl)hydrazine hydro-
chloride
~` 1 3300qn
5010P~12~6A - 30 - 17358IA
To prepare certain esters representative of the
Formula I compounds, it may be advantageous to first
prepare the lower alkyl esters as illustrated in
Scheme I. Hydrolysis of these lower alkyl esters by
conventional means, such as by using NaOH or XOH in
agueous ethanol, followed by acidification, then
produces the corresponding carbo~ylic acids VI, as
illustrated in Scheme III. The carbo~ylic acid is
then reacted with an alkylating agent, R10-Z, in
the presence of a suitable base and solvent
combination, such as for e~ample, Na2CO3 in
acetone or triethylamine in dimethylformamide, to
produce the esters Ib. Compounds 15, 16, 17, 20, 21
and 22 of Table I are conveniently prepared by this
method.
Another method of preparing the esters of
Formula I from the acid, VI consists of treating the
latter with a diazoalkane (such as diazomethane) in a
suitable non-reactive solvent such as ether or
methanol to obtain an ester Ic. Other methods are
shown in Ogliaruso and Wolfe in Patai, The Chemistry
of Acid Derivatives, Supplement ~, Wiley, New York,
1979, pp. 411-436).
5010P~1286A 3~ - 17358IA 1 3 3 0 0 9 O
SChem~ III PneD~rat;On ~f FOnnU1a I COmDOUndS
R7 ~14 ~17
LR (C)n~O2 J R (C)n-CO2H
R5~6 R9 R ~R
Ia VI
2 2 / ¦ Rlû z
solvent / basQ
~ sol~ont
R2~R7 R~)~R7
~N~ R ~N~)R
,~ (C),-C02M~ ~ (C~r-COzM10
IC . / Ib
In tho~e instances whon asymmetric centers
are pre~ent, mor- than on~ ~tereoisomer i~ pos~ible,
and all po~siblo isomeric forms are deemea to be
included within the planar structural representations
shown. Optically active (R) and (S) isomer~ may bo
resolqed using conqentional technigues known to the
skilled artisan.
~,
.
1 3300~0
5010P~1286A - 32 - 17358IA
Among the resolved isomers in Table 1, the
esters of the minus ~-) acids, compounds 3,36,38 and
40 are preferred.
Schem- IV Alt~rnat~vo PreDarat1on of Fonnula I CornDound~
low-r ~lk~nol R R
~ IV r-flw~ ~R~ 14
H ~C)r-C02Q
Est~r 1ntermediatQ
1Hydrolys1 s
~7
~as per Scheme III R
lb, lc H (C)r~C2H
Acld 1 nt~ d1at~
Scheme IV illustrate~ an alternative synthesis
of the compound~ of Formula I. In this Scheme a
Fischer indole synthesis i~ carried out using a
phenylhydrazine IV and the ketone IIr, followed by
hydrolysi~. The acid intermediata is then N-benzylated
with ths reagent V, preferably using a strong basa such
as potas~ium t-buto~ide to effect the reaction.
~.,
.. ... .
..
`~ `, .i, .:
1 3300qO
5010P/1286A - 33 - 17358IA
Acidification of the reac~ion misture then yield~ the
acid VI which can be converted to compounds of Formula
I as indicated in Schem~ III.
S ~chem~ V PreDarat~on of Sulfox1des and Sulfones of Fonnul~ I comDourlds
7 ~ TC b-low.
R2'~\ /~ R is replacod by SR 1, S(O)Rll or
R3~ l 11 J S~)2Rll
~\N/~ R2-R6 ;~5Rll 0~ S(o)Rll
~L R ~C) ~--
R5--~_R6
~SRl 1
TC-C02R
Id
1.5 ~9. o~
` ,~P~ .
- S(O)R 2 o9. mCPB~ ~ ~ )2
TC-C02R , TC-C02R
In Scheme V ig illustrated a method of preparing
derivative~ of Formula I in which one of the substi-
tuents among Rl-R4 is a sulfo~idc or a sulfone.
It will be obviou~ to one s~illed in the art that a
... .,, .~ .
1 3300qO
5010P~1286A - 34 17358IA
sulfoside or sulfone deri~ative of R5 or R6 could be
prepared in the ~ame way.
E~ter I~ ~a representative of I) is prepared
according to Scheme I or Scheme III. Treatment of I~
wit~ a limited amount of an osidizing agent such as
m-chloro-perbenzoic acid yields the sulfoside ester
Ie. Further treatment of Ie with the osidizing agent,
or treatment of Id with an esress (>2 eg.) of the
osidizing agent yields the sulfone ester lf.
Sch~ VI , Prec~r~t~on of Hvtra21n~ D~n~t~v~s IV
~ 1 ) N~N2 ~ lz-lCl
NH2^HCl ) 2 2 4
SV
With regard to Scheme VI, th0 preparation of
hydrazine ~tartlng mater~al~ is ~llustrate~ by prepara-
tion of 4-methylthiophenyl hy~razine hydrochloride.
4-Methylthio~niline ~13.9 9) wa~ added dropwise to cold
HCl (6N) (50 mL) and st~rred for 5 min in an ice ~ath.
A solution of NaN02 in water ~7.25 g, 15 mL) was then
ad~ed dropwise and stirred for 15 min. The cold
diazonium salt wa~ then cannulatea into a stirred cold
solution of Na2S204 in water (50 9, 250 mL).
After 20 min, ether (200 mL) wa~ added an~ the reaction
misture basified with NaOH(lON). The ether layer was
decanted, washed with brine, dried over Na2S04 and
HCl gas wa~ passed through the ether solution to form
.. ,, , , ~ ~ , , _
5010P~1286A 1 3 3 0 0 9 ~ l7358IA
the hydrochloride salt which precipitated out. After
filtration, there was obtained 7.0 9 of pure final
product. Other hydrazines, similarly prepared, are
also shown in ~able 4, below.
s
TABLE 4
HYDRAZ INES
R2~\
R ~
R ~ ~NHNH2-HCl
IV
R~ ~ Compound Name
1 4-SMe H 4-methylthiophenyl
hydrazine
hydrochloride
2 2-CH(Me)2 H 2-isopropylphenyl
hydrazine
hydrochloride
3 2-SMe H 2-methylthiophenyl
hydrazine
hydrochloride
4 2-Me 4-Me 2,4-dimethylphenyl
hydrazine
hydrochloride
2-Me 4-OMe 4-metho~y-2-methyl-
phenyl hydrazine
hydrochloride
R3~R4~
1 33009~
5010P/1286A - 36 - 17358IA
The prostaglandin antagonist properties of
the compositions and compounds of the present
invention can be demonstrated by the biological assay
described below.
s
Agonist-induced Bronchonconstriction
in Anesthetized Guinea Pias (Konzett-Rossler~
Male Hartley strain guinea-pigs (350-500 g)
are anesthetized wtih urethane (1.5 g/kg i.p.), given
succinylcholine chloride (5 mg/kg s.c.) to suppress
spontaneous respiration and artificially ventilated
at 60 breaths/minute. Changes in insufflation
pressure (resistance to inflation) are measured using
a Statham PM-5E differential pressure transducer and
recorded on a Beckman Type R-Dynograph. Increases in
insufflation pressure are induced at 20 minute
intervals with bolus intravenous injections of
arachidonic acid (0.5 mg/kg), or U-44069 (15(S)-
hydrosy-9, lla-(eposymethano)-prosta-5-cis,
13-trans-dienoic acid) (a stable prostaglandin
endoperoside analogue) (2 ~g/kg). After obtaining
at least two reproducible control responses to the
agonist under study, the antagonist activity of the
test compound is determined. In order to assess
antagonist activity, the test compound (in solution
or suspension) or drug vehicle (1 ml/kg) is
administered intravenously in cumulative doses 5
minutes prior to each subsequent agonist challenge.
Reductions of the agonist response are calculated as
a percent of the control response which immediately
preceded the initial antagonist or vehicle dose.
~. .
1 330090
5010P/1286A - 37 - 17358IA
ED50 values (dose requir~d to inhibit the
insufflation pressure increase by 50 percent) are
calculated by regression analysis.
In a variation of the assay, the test
compound is administered as a single dose intraduo-
denally (injected into the duodenum which has been
previously esposed through a ventral mid-line
incision) 10 minutes before challenge with arachidonic
acid ~0.5 mg/kg) or U-44069 ~2 ~g/kg). The agonist
challenge is continued 10 minutes after adminis-
tration of the antagonist and every 20 minutes
thereafter for a period of up to 2 hours. Reduction
of the post drug response is calculated as a percent
of the pre-drug control response. This variation of
the assay also permits the evaluation of antagonists
with a delayed onset of action.
Compounds of Formula I can be tested using
the following assay to determine their mammalian
leukotriene biosynthesis inhibiting activity.
Rat Peritoneal Polymorphonuclear (PMN)
LeukocYte Assay
Rats under ether anesthesia are injected
(i.p.) with 8 ml o a suspension of sodium caseinate
(6 grams in ~. 50 ml water). After 15-24 hr. the
rats are sacrificed (CO2) and ~he cells from the
peritoneal cavity are recovered by lavage with 20 ml
of buffer (Eagles MEM containing 30 m~ HEPES adjusted
to pH 7.4 with NaOH). The cells are pelleted (350 x
g, 5 min.), resuspended in buffer with vigorous
shaking, filtered, through lens paper, recentrifuged
and finally suspended in buffer at a concentration of
, ... ., . " , ~ ... .. ..
- `
1 3300~0
5010P/1286A - 38 - 17358IA
10 cells/ml. A 500 ~1 aliquot of PMN suspension
and test compound are preincubated for 2 minutes at
37C, followed by the addition of lO ~M A-23187.
The suspension is stirred for an additional 4 minutes
then bioassayed for LTB4 content by adding an
aliquot to a second 500 ~1 portion of the PMN at
37C . The LTB4 produced in the first incubation
causes aggregation of the second PMN, which is
measured as a change in light transmission. The size
of the assay aliquot is chosen to give a subma~imal
transmission change (usually -70%) for the untreated
control. The percentage inhibition of LTB4
formation is calculated from the ratio of transmission
change in the sample to the transmission change in
the compound-free control.
The cytoprotective activity of a compound
may be observed in both animals and man by noting the
increased resistance of the gastrointestinal mucosa
to the nosious effects of strong irritants, for
e~ample, the ulcerogenic effects of aspirin or
indomethacin. In addition to lessening the effect of
non-steroidal anti-inflammatory drugs on the gastro-
intestinal tract, animal studies show that cyto-
protective compounds will prevent gastric lesions
induced by oral administration of strong acids,
strong bases, ethanol, hypertonic saline solutions
and the like.
Two assays can be used to measure cyto-
protective ability. These assays are; (A) an ethanol-
induced lesion assay and ~B) an indomethacin-induced
ulcer assay and are described in EP 140,684.
,
~;' . .
1 3300q
5010P/1286A - 39 - 17358IA
The magnitude of a prophylactic or thera-
peutic dose of a compound of Formula I will, o~
course, vary with the nature or the severity of the
condition to be treated and with the particular
compound of Formula I and its route of administration.
In general, the daily dose range for anti-asthmatic,
anti-allergic, or anti-thrombotic use lies within the
range of from about 0.01 mg to about 100 mg per kg
body weight of a mammal.
The esact amount of a compound of Formula I
to be used as a cytoprotective agent will depend on,
inter ~li~, whether it is being administered to heal
damaged cells or to avoid future damage, on the nature
of the damaged cells (e.g., gastro-intestinal
lS ulcerations vs. nephrotic necrosis), and on the nature
of the causative agent. An e~ample of use of a
compound of Formula I to avoid future damage is co--
administration with a non-steroidal anti-inflammatory
drug (for esample, indomethacin).
The effective daily dosage level for
compound8 of Formula I inducing cytoprotection in
mammals, especially humans, will generally range from
about 0.002 mg/kg to about 100 mg/kg, preferably from
about 0.02 mg/kg to about 30 mg/kg. The dosage may
be administered in single or divided individual doses.
Any suitable rou~e of administration may be
employed for providing a mammal, especially a human
with an effective dosage of a compound of Formula I.
For e~ample, oral, rectal, topical, parenteral,
ocular, nasal, buccal, intravenous and the like may
be employed. Dosage forms include tablets, troches,
~ 330090
5010P/1286A - 4g - 17358IA
dispersions, suspensions, solutions, capsules, creams,
ointments, aerosols and the like.
The pharmaceutical compositions of the
present invention comprise a compound of Formula I as
an active ingredient or a pharmaceutically acceptable
salt thereof, and may also contain a pharmaceutically
acceptable carrier and optionally other therapeutic
ingredients. The term ~pharmaceutically acceptable
salts~ refers to salts prepared from pharmaceutically
acceptable non-to~ic bases including inorganic bases
and organic bases. The compositions include
compositions suitable for oral, rectal, ophthalmic,
pulmonary, nasal, dermal, topical or parenteral
(including subcutaneous, intramuscular and
intravenous) administration, although the most
suitable route in any given case will depend on the
nature and severity of the conditions being treated
and on the nature of the active ingredient. They may
be conveniently presented in unit dosage form and
prepared by any of the methods well-known in the art
of pharmac~.
For use where a composition for intravenous
administration is employed, a suitable dosage range
for anti-asthmatic, or anti-allergic use is from
about 0.01 mg to about 20 mg (preferably from about
0.1 mg to about 10 mg) of a compound of Formula I per
kg of body weight per day and for cytoprotective use
from about 0.002 mg to about 100 mg (preferably from
about 0.02 mg to about 30 mg and more preferably from
about 0.1 mg to about 10 mg) of a compound of Formula
I per kg of body weight per day. In the case where
an oral composition is employed, a suitable dosage
1 330090
SOlOP/1286A - 41 - 17358IA
range for anti-asthmatic, or anti-allergic use is,
e.g. from about 1 to about 100 mg of a compound of
Formula I per kg of body weight per day, preferably
from about 5 mg to about 40 mg per kg and for cyto-
protective use from about 0.01 mg to about 100 mg
(preferably from about 0.1 mg to about 30mg and were
preferably from about 0.1 mg to about 10 mg) of a
compound of Formula I per kg of body weight per day.
For administration by inhalation, the
compounds of the present invention are conveniently
delivered in the form of an aerosol spray presenta-
tion from pressurized packs or a nebuliser, or a
powder which may be formulated as a cartridge from
which the powder composition may be inhaled with the
aid of a suitable device. The preferred delivery
system for inhalation in a metered dose inhalation
(MDI) aerosol, which may be formulated as a
suspension or solution in fluorocarbon propellants.
Suitable topical formulations of compound I
include transdermal devices, aerosols, creams,
ointments, lotions, dusting powder, and the like.
In practical use, a compound of Formula I
can be combined as the active ingredient in intimate
admisture with a pharmaceutical carrier according to
conventional pharmaceutical compounding techniques.
The carrier may take a wi~de variety of forms depending
on the form of preparation desired for administration,
e.g., oral or parenteral (including intravenous). In
preparing the compositions for oral dosage form, any
of the usual pharmaceutical media may be employed,
such as, for esample, water glycols, oils, alcohols,
flavoring agents, preservatives, coloring agents and
. ", , :
1 3300q0
5010P~128~A - 42 - 17358IA
the like in the case of oral liquid preparation~,
such aæ, for e~ample, suspensions, eli~irs and
solutions; or carriers such as ætarches, sugars,
microcrystalline cellulose, diluents, granulating
agents, lubricants, binder~, disintegrating agents
and the like in the case of oral solid preparations
such aæ, for e~ample, powder~, capsules and tablets.
Because of their ease of administration, tablets and
capsules represent the most advantageous oral dosage
unit form, in which case solid pharmaceutical carriers
are obviously employed. If desired, tablets may be
sugar coated or enteric coated by standard techniques.
In addition to the common dosage forms set
out above, the compounds of Formula I may also be
administered by controlled release means and/or
delivery devices such as those described in U.S.
Patent Nos. 3,845,770; 3,916,899; 3,536,809;
3,598,123; 3,630,200 and 4,008,719.
Pharmaceutical composltions of the present
invention suitable for oral administration may be
presented as discrete units such as capsules, cachets
or tablets each containing a predetermined amount of
the active ingredient, as a powder or granules or as
a solution or 3 suspension in an aqueous liquid, a
non-aqueous liquid, an oii-in-water emulsion or a
water-in-oil liquid emulsion. Such compositions may
be prepared by any of the methods of pharmacy but all
methods include the step of bringing into association
the acti~e ingredient with the carrier which consti-
tutes one or more necessary ingredients. In general,
the compoæitions are prepared by uniformly and
~,~
-`` 1 330090
SOlOP~1286A - 43 - 17358IA
intimately admi~ing the active ~n~redient with liquid
carriers or finely d~vided solid carrier~ or both,
and then, if necessary, shaping the product into the
desired presentat~on. For e~ample, a tablet may be
S prepared by compression or molding, optionally with
one or more accessory ingredients. Compressed tablets
may be prepared by compressing in a suitable machine,
the active ingredient in a free-flowing form such as
powder or granules, optionally mi~ed with a binder,
lubricant, inert diluent, surface active or dispersing
agent. Molded tablets may be made by molding in a
suitable machine, a mi~ture of the powdered compound
moistened with an inert liquid diluent. Desirably,
each tablet contains from about 2.5 mg to about 500
lS mg of the active ingredient and each cachet or capsule
contains from about 2.5 to about S00 mg of the active
ingredient.
The following are e~amples of representative
pharmaceutical dosage forms for the compounds of
Formula I:
In;ectable Suspension (I.M.) mg/ml
Compound of Formula I 2.0
Methylcellulose S.0
25 Tween-80 O.S
8enzyl alcohol - 9.0
~enzalkonium chloride 1.0
Water for injection to a total volume of 1 ml
1 330090
5010P/1286A - 44 - 17358IA
Tablet ma/t2blet
Compound of Formula I 25.0
Microcrystalline Cellulose 415.0
Providone 14.0
5 Pregelatinized Starch 43.5
Magnesium Stearate 2.5
500
Capsule ma/capsule
10 Compound of Formula I 25.0
Lactose Powder 573.5
Magnesium Stearate 1.5
600
In addition to the compounds of Formula I,
the pharmaceutical compositions of the present
invention can also contain other active ingredients,
such as non-steroidal anti-inflammatory drugs
(NSAIDs), peripheral analgesic agents such as
zomepirac, diflunisal and the like, cycloosygenase
inhibitors, leukotriene antagonists, leukotriene
biosynthesis inhibitors, H2-receptor antagonists,
antihistaminic agentæ, prostaglandin antagonists, ACE
inhibitors, and thrombo~ane synthetase inhibitors.
The weight ratio of the compound of the Formula I to
the second active ingredient may be varied and will
depend upon the effective dose of each ingredient.
Generally, an effective dose of each will be used.
Thus, for example, when a compound of the Formula I
is combined with a second active ingredient the
weigh~ ratio of the compound of the Formula I to the
second ingredient will generally range from about
- 1 3300qO
- 45 - 17358IA
looo:l to about l:looo, preferably from 200:1 to
1:200. Combinations of a compound of the Formula I
and other active ingredients will generally be within
the aforementioned range, but in each case, an effec-
tive dose of each active ingredient should be used.
NSAIDs can be characterized into five groups:
(l) the propionic acid derivatives;
(2) the acetic acid derivatives;
(3) the fenamic acid derivatives;
(4) the biphenylcarboxylic acid derivatives;
and
(5) the oxicams
or a pharmaceutically acceptable salt thereof.
NSAIDS which are within the scope of this invention
are those disclosed in EP 140,684.
Pharmaceutical compositions comprising the
Formula I compounds may also contain other inhibitors
of the biosynthesis of the leukotrienes such as are
disclosed in EP 138,481 (April 24, 1985), EP 115,394
(August 8, 1984), EP 136,893 (April 10, 1985), and EP
140,709 (May 8, lg85).
The compounds of the Formula I may also be
used in combination with leukotriene antagonists such
as those disclosed in EP 106,565 (April 25, 1984) and
EP 104,885 (April 4, 1984), and others known in the
art such as those disclosed in European Patent Appli-
cation Nos. 56,172 and 61,800; and in U.K. Patent
Specification No. 2,058,785.
.
' ':- .
1 330093
5010P/1~86A - 46 - 17358IA
Pharmaceutical compositions comprising the
Formula I compounds may also contain as the second
active ingredient, other prostaglandin antagonists
such as those disclosed in Eusopean Patent Application
11,067 (May 28, 1980) or other thrombosane antagonists
such as those disclosed in U.S. 4,237,160. They may
also contain histidine decarbosyase inhibitors such
as a-fluoromethyl-histidine, described in U.S.
4,325,961. The compounds of the Formula I may also
be advantageously combined with an Hl or
H2-receptor antagonist, such as for instance
benadryl, dramamine, histadyl, phenergan, terfenadine,
acetamazole, cimetidine, ranitidine, famotidine,
aminothiadiazoles disclosed in EP 40,696 ~December 2,
1981) and like compounds, such as those disclosed in
U.S. Patent Nos. 4,283,408; 4,362,736; ~,39~,S08.
The pharmaceutical compositions
may also contain a ~+~H+ ATPase inhibitor such as
omeprazole, disclosed in U.S. Pat. 4,255,431, and the
like. Compounds of I may also be usefully combined
with most cell stabilizing agents, such as 1,3-bis(2-
carbo~ychromon-5-ylosy)-2-hydrosypropane and related
compounds described in ~ritish Patent Specifications
1,144,905 and 1,144,906. Another useful pharma-
ceutical composition comprises the Formula I
compounds in combination with serotonin antagonists
such as methysergide, the serotonin antagonists
described in Nature, Vol. 316, pages 126-131, 1985,
and the like.
1 33009~
5010P/1286A - 47 - 17358IA
When the second active ingredient in
compositions of this invention is a thromboxane
synthetase inhibitor, such inhibitor can be as
described in UK 2,038,821 (e.g., UK 37248 and
dazosiben hydrochloride), U.S.P. 4,217,357 (e.g., UK
34787), U.S.P. 4,444,775 (e.g., CGS 13080), U.S.P.
4,226,878 ~e.g., ONO 046), U.S.P. 4,495,357 te.g.,
U63557A) U.S.P. 4,273,782 (e.g., UK-38485), or EP
98,690 (e.g., CV-4151).
An embodiment of the invention is a cardio-
vascular composition useful for treating arterial
thrombosis which comprises an antithrombotic compound
of the Formula I.
A further embodiment of the invention is a
cardiovascular composition useful for treating
arterial thrombosis which comprises: (1) the
antithrombotic Formula I compound defined above; and,
(ii) an angiotensin converting enzyme ~ACE) inhibitor
compound which is a member of the group: carboxyalkyl
dipeptide derivatives; captopril 11-(3-mercapto-2-
methyl-l-osopropyl)-L-proline~; 2-tN-(S)-l-ethosy-
carbonyl-3-phenylpropyl)-S-alanyl~ ,endo-2-azabi-
cyclol3,3,0]octane-3(S)-carbosylic acid; N-((S)-l-
ethosycarbonyl-3-phenylpropyl)-L-alanyl-N~(2-indanyl)-
glycine; l-(N-t(S)-l-ethosy-carbonyl-3-phenylpropyl~-
L-alanyl)-cis,~yn-octahyd~o-(H-indole-2-S)-carboxylic
acid; 2-(N-t~S)-l-ethosy-carbonyl-3-phenylpropyl]-L-
alanyl)-1,2,3,4-tetrahydro-iso-isoguinoline-3(S)-
carbosylic acid; and, l-carbosy-methyl-3(S)-(l(S)-
ethosycarbonyl-3-phenylpropylamino)-2,3,4,5-tetrahydro-
lH~l]-benzazepine-2-one.
1 330090
5010P~1286A - 4~ - 17358I~
In particular the clas~ o~ ACE inhibitors
which have been found to have a potentiating effect
when used in combination with the Formula I compounds
are those disclosed in U.S. Patent 4,374,829, which
also discloses methods for their preparation.
Of
the carboxyalkyl dipeptides disclosed in U.S. Patent
4,374,829, those of particular interest in this
invention are N-[l~S)-ethosycarbonyl-3-phenyl-
propyl]-L-alanyl-L-proline, also ~nown and referred
to herein as enalapril; N-tl(S)-carbo~y-3-phenyl-
propyl]-L-alanyl-L-proline, also know and referred to
herein as enalapril diacid; and, N-tl(S)-carbo~y-
3-phenylpropyl]-L-lysyl-L-proline, also known and
referred to herein as lisinapril.
The combination composition of the invention
can contain varying amounts of (i) the Formula I
antithrombotic compound and (ii) ACE inhibitor
antihypertensive compounds. The weight ratio of
(i):(ii) can range from about 25 to 1; preferably
from about 10 to 1. In addition to the active
ingredients of (i) alone or of (i) and (ii) in
combination, the compositions of the invention can
also contain other conventional pharmaceutically
acceptable compounding ingredients, as necessary or
desire~. Such ingredient~ are generally referred to
as carriers or diluents. Conventional procedures for
preparing such compositions in appropriate dosage
forms can be utilized. Whatever the dosage form, it
will contain a pharmaceutically effective amount of
the present composition.
r~4i ~
1 330090
5010P/1286A - 49 - 17358IA
The combination compositions can be
administered orally or other than orally; e.g.,
parenterally, by insufflation, topically, rectally,
etc.; using appropriate dosage forms; e.g., tablets,
capsules, suspensions, solutions, and the like, for
oral administration; suspension emulsions, and the
like, for parenteral administration; solutions for
intravenous administration; and ointments, transdermal
patches, and the like, for topical administration.
These compositions are formulated similarly to the
compositions discussed above.
Treatment dosage for human beings for
cardiovascular use can be varied as necessary.
Generally, daily dosages of the composition of the
invention can range from about 6000 to about 10 mg;
preferably, from about 3000 to about 20 mg.
The amount of active ingredient that may be
combined with the carrier materials to produce a
single dosage form for cardiovascular use will vary
depending upon the host treated and the particular
mode of administration. For example, a formulation
intended for oral administration may contain from 5
mg to 5 gm of active agents compounded with an
appropriate and convenient amount of carrier material
which may vary from about 5 to about 95 percent of
the total composition. josage unit forms will
generally contain between from about 20 mg to about
500 mg of active ingredients.
It will be understood, however, that the
specific dose level for any particular patient will
depend upon a variety of factors including the
activity of the specific compound employed, the age,
.. . . .
1 330090
5010P/1286A - 50 - 17358IA
body waight, general health, se~, diet, time of
administration, route of administration, rate of
e~cretion, drug combination and tho severity of the
particular disease undergoing therapy.
S The composition of this invention inhibits
platelet accumulation at the damaged endothelial
surface via the Formula I compound. This inhib~tory
effect i~ potentiated ~y the presence of th~
antihypertensive compound.
Thus, the compositions of the invention are
useful in treating thrombosis and are also of value
in the management of acute and chronic congestive
heart failure, and limitation of myocardial infarct
damage.
In ViVQ testing of the composition of this
invention in test animals (rabbits) can be used to
demonstrate that this composition is pharmaceutically
effective in decreasing platelet-related arterial
thrombic formation.
To demonstrate the potentiation of the
antihypertensive compound on the anti-thrombotic
Formula I compound comprising the combination
composition of the invention, the effect of these
compounds on test animals ~rabbits) can be determined
separately and then in combination. The effect of a
different class of antihypertensive agents singly and
in combination with the Formula I compound of the
invention can also be determined for comparative
purposes. The methods employed are described in U.S.
Patent 4,558,037.
fA~
5010P~1286A - 51 - 1 3 3 0 9 l7358IA
The following esamples illustrate th~
preparation of the compounds of the present invention
without, however, limiting the æame thereto.
All temperature are in degrees Celsius.
E~ample 1
Ethyl 9-p-chlorobenzyl-6-fluoro-1,2,3,4-tetrahydro-
s ~ ~azo~ ~ acetate _ _
To 3.50 9 of 1-(4-chlorobenzyl)-1-(4-
fluorophenyl)hydrazine hy~rochloride in 70 cc of
-propan~ ~was added 2.23 g of ethyl-2-cyclo-
he~anone acetate. The reaction was reflused under
nitrogen for 16 hours. The resulting reaction
misture was then evaporated to dryness and the
residue 6uspended in ether. The solid material was
then filtered. The ether filtrate was washed with
water, dried and evaporated. The resulting syrup was
chromatographed on silica gel to give 2.8 g (42%) of
the title compound.
H NMR S :1.25 (t, 3H, C02CH2~
1.80-2.00 ~m, 4H); 2.35-2.85 (m, 4H); 3.38 (m, lH);
4.10 (q, 2H, C02~CH3); 5.28 (2d, 2H, Ar
~2); 6.80-7.30 (m, 7H, Ar).
EsamDle 2
Methyl 3-(9-p-chlorobenzyl-6-fluoro-1,2,3,4-tetra-
hYdrocarbazol-l-rl~-proDanoate
Following the procedure o Esample 1, but
u8ing 1-(4-chlorobenzyl)-1-(4-fluorophenyl)hydrazine
hyarochlo~ide and methyl 2-cyclohesanone propionate
as starting material~, the title compound is prepared.
~ .,
: .
~ .. I
.
1 330090
5010P/1286A - 52 - 17358IA
ExamPle 3
Methyl 3-(9-p-chlorobenzyl-6-methoxy-1,2,3,4-tetra-
hydrocarbazol-l-yl)-propanoate
Following the procedure of Example 1, but
using 1-(4-chlorobenzyl)-1-(4-methoxyphenyl)hydrazine
hydrochloride and methyl 2-cyclohexanone propionate
as startinq materials, the title compound is prepared.
Example 4
Ethyl 9-p-chlorobenzyl-6-methosy-1,2,3,4-tetrahydro-
carbazol-l-yl-acetate
Following the procedure of E~ample 1, but
using 1-(4-chlorobenzyl)-1-(4-metho~yphenyl)hydrazine
hydrochloride and ethyl 2-cyclohe~anone acetate as
starting materials, the title compound is prepared.
E~am~le 5
Ethyl 9-benzyl-6-fluoro-1,2,3,4-tetrahydrocarbazol-
l-rl-acetate
Following the procedure of E~ample 1, but
using l-(benzyl)-1-(4-fluorophenyl)hydrazine
hydrochloride and ethyl 2-cyclohe~anone acetate as
starting materials, the title compound was prepared.
HNMR~ : 1.15 (t, 3H, CO2CH2~3);
1.80-2.00 (m, 4H); 2.38-2.85 (m, 4H); 3.45 (m, lH);
4-10 (q, 2H, CO2~CH3); 5.30 (2d, 2H, Ar
Ç~2); 6.78-7.25 (m, 8H, Ar).
_.,-,. . .
.
1 330090
5010P/1286A - 53 - 17358IA
Example 6
Ethyl 9-p-methosybenzyl-6-fluoro-1,2,3,4-tetrahydro-
carbazol-l-yl-acetate
Following the procedure of Example 1, but
using 1-(4-methosybenzyl)-1-(4-fluorophenyl)hydrazine
hydrochloride and ethyl 2-cyclohe~anone acetate as
star~ing materials, the title compound was prepared.
H NMRS: 1.15 (t, 3H, CO2,CH2CH3);
1.80-1.95 (m, 4H); 2.40-2.85 (m, 4H); 3.63 (m, lH);
3.75 ~s, 3H, OCH3); 4.13 (q, 2H, CO2CH2CH3);
5.25 (2d, 2H, Ar Ç~2); 6.75-7.25 (m, 7H, Ar).
ExamDle 7
Ethyl 9-(3,4-dichloro)benzyl-6-fluoro-1,2,3,4-tetra-
hYdrocarbazol-l-yl-acetate
Following the procedure of Example 1, but
using 1-(3,4-dichlorobenzyl)-1-(4-fluorophenyl)
hydrazine hydrochloride and ethyl 2-cyclohexanone
acetate as starting materials, the title compound was
prepared.
H NMR~: 1.15 (t, 3H, CO2CH2~3~;
1.80-2.00 (m, 4H); 2.30-2.85 (m, 4H); 3.35 (m, lH):
4.1S (q, 2H, CO2CH2CH3); 5.25 ~2d, 2H, Ar
CH2); 6.70-7.45 (m, 6H, Ar).
Esa~Dle 8
Ethyl 9-[1-(1-phenyl)ethyl]-6-fluoro-1,2,3,4-tetra-
hydrocar~azol-l-yl-acetate _
Following the procedure of Esample 1, but
using 1-tl-(1-phenyl)ethyl~-1-(4-fluorophenyl)-
hydrazine hydrochloride and ethyl 2-cyclohesanone
acetate as starting materials, the title compound was
prepared.
.,
1 3300~0
5010P~1286A - 54 - 17358IA
H NMR S : 1.15 (t, 3H, CO2CH2CH3);
1.80-2.00 (m, 4H); 2.05 (d, 3H, Ar - CH - Me); 2.50-
2.85 (m, 4H); 4.55 (m, lH); 4.20 (Q, 2H,
CO2CH~CH3); 5.65 (q, lH, Ar - CH-CH3); 6.60-
7.40 (m, 8H, Ar).
Example 9
Ethyl 9-p-chlorobenzyl-1,2,3,4-tetra~ydrocar~azol-
l-yl-acetate
Following the procedure of Example 1, but
using 1-(4-chlorobenzyl)-1-(phenyl)hydrazine
hydrochloride and ethyl 2-cyclohesanone acetate as
starting materials, the title compound was prepared.
H NMR ~ : 1.25 (t, 3H, CO2CH2CH~);
1.80-2.00 (m, 4H); 2.38-2.88 (m, 4H); 3,40 (m, lH);
4.15 (q, 2H, CO2C_~ H3); 4.30 (2d, 2H, Ar
CH2); 6.85-7.55 (m, 8H, Ar).
E~am~le lQ
Ethyl 9-p-chlorobenzyl-6-chloro-1,2,3,4-tetrahydro-
carbazol-l-yl-acetate
Following the procedure of Example 1, but
using 1-(4-chlorobenzyl)-1-(4-chlorophenyl)hydrazine
hydrochloride and ethyl 2-cyclohexanone acetate as
stating materials, the title compound was prepared.
H NMR ~ : 1.25 ~t, 3H, CO2CH2Ç~
1.80-2.00 (m, 4H); 2.35-2.85 (m, 4H); 3.40 (m, lH);
4.15 (q, 2H, Ar CH2); 6.82-7.50 (m, 7H, Ar).
1 3300qO
5010P/1286A - 55 - 17358IA
E~amPle 11
Ethyl 9-p-chlorobenzyl-8-methyl-1,2,3,4-tetrahydro-
carbazol-l-yl-acetate
Following the procedure of E~ample 1, but
S using 1-(4-chlorobenzyl)-1-t2-methylphenyl)hydrazine
hydrochloride and ethyl 2-cyclohe~anone acetate as
starting materials, the title compound was prepared.
lH NMR ~: }.30 (t, 3H, CO2CH2CH3);
2.00-2.25 (m, 4H); 2.60-3.15 (m, 4H); 3.58 (m, lH);
4.40 (q, 2H, CO2~2CH3); 5.78 (2d, 2H, Ar
~2); 6.95-7.65 (m, 7H, Ar).
E~ample 12
Ethyl 6-bromo-9-p-chlorobenzyl-1,2,3,4-tetrahydro-
carbazol-l-Yl-acetate
Following the procedure of E~ample 1, but
using 1-(4-chlorobenzyl)-1-(4-bromophenyl)hydrazine
hydrochloride and ethyl 2-cyclohesanone acetate as
starting materials, the title compound is prepared.
E~am~le 13
Ethyl 9-p-chlorobenzyl-6-methyl-1,2,3,4-tetrahydro-
carbazol-l-yl-acetate
Following the procedure of E~ample 1, but
using 1-(4-chlorobenzyl)-1-(4-methylphenyl)hydrazine
hydrochloride and ethyl 2 cyclohexanone acetate as
starting materials, the title compound is prepared.
1 3300qO
5010P/1286A - 56 - 17358IA
E~ample 14
Methyl 2-(9-p-chlorobenzyl-6-fluoro-1,2,3,4-tetra-
hydrocarbazol-l-yl~-Dropanoate
Followinq the procedure of Esample 1, but
using 1-(4-chlorobenzyl)-1-(4-fluorophenyl)hydrazine
hydrochloride and methyl 2-(2-cyclohexanone)
propionate as starting materials, the title compound
was prepared.
lH NMR S 0.98 ~d, 3H, CH3-CH-CO2 Me);
1.72-2.00 (m, 4H); 2.60-2.85 ~m, 3H); 3.35 ~m, lH~;
3.64 (s, 3H, CO2~); 5.27 (2d, 2H, Ar-~2);
6.75-7.25 ~m, 7H, Ar).
ExamDle 15
Ethyl 9-p-chlorobenzyl-8-fluoro-1,2,3,4-tetrahydro-
carbazol-l-Yl-acetate
Following the procedure of Example 1, but
using 1-~4-chlorobenzyl)-1-~2-fluorophenyl)hydrazine
hydrochloride and ethyl 2-cyclohexanone acetate as
starting materials, the title compound was prepared.
lH NMR~ : 1.25 ~t, 3H, CO2 CH2 Ç~
1.50-2.00 (m, 4H); 2.38-2.95 ~m, 4H); 3.38 ~m, lH);
4.15 ~q, 2H, CO2~2CH3); 5.45 (2d~ 2H~ Ar
CH2); 6.78-7.40 ~m, 7H, Ar).
ExamDle 16
Ethyl 9-p-chlorobenzyl-5,7-dichloro-1,2,3,4-tetra-
hydrocarbazol-l-yl-acetate
Following the procedure of Example 1, but
using 1-(4-chlorobenzyl)-1-(3,5-dichlorophenyl)-
hydrazine hydrochloride and ethyl 2-cyclohexanone
acetate as starting materials, the title compound is
prepared.
~ . ,
1 3300qo
5010P/1286A - 57 - 17358IA
Example 17
Ethyl 9-p-chlorobenzyl-6,8-dichloro-1,2,3,4-tetra-
hydrocarbazol-l-yl-acetate
Following the procedure of Example 1, but
using 1-(4-chlorobenzyl)-1-(2,4-dichlorophenyl)-
hydrazine hydrochloride and ethyl 2-cyclohexanone
acetate as starting materials, the title compound is
prepared.
Esample 18
(-)9-p-chlorobenzyl-6-fluoro-1,2,3,4-tetrahydro-
carbazol-l-Y1-acetic acid. methyl ester
Step I
To 1.59 g of ethyl ester from Esample 1, in
10 cc of methanol was added 10 cc of water and 420 mg
of potassium hydro~ide. The resulting solution was
refluxed for 4 hours. Upon cooling the reaction
misture was then acidified with HCl (lN). The
resulting precipitate was filtered and washed with
water. Analytically pure material was prepared by
triturating the solid with a misture of hesane/ethyl
acetate (9:1) followed ~y filtration and drying on a
high vacuum pump to give 1.24 g of the racemic acid.
(89%).
Analysis calculated for C21HlgNClFO2
C H N Cl F
Calculated 67.83 5.15 3.77 9.53 5.11
Found 67.88 5.47 3.63 9.52 5.12
: .
~ - "
1 3300'10
5010P/1286A -- 58 -- 17358IA
Step II
10.0 g of 9-p-chlorobenzyl-6-fluoro-1,2,3,4-
tetrahydro~arbazol-l-yl-acetic acid from Step I was
dissolved in a mixture of hot (reflusing) acetonitrile
(150 cc~ and ethanol (25cc), and 4.4 g of d(+)
ephedrine was added. The reflux was continued for 15
minutes and the hot solution was filtered and allowed
to cool to room temperature. Crystals separated from
the solution and were separated by filtration. After
three recrystallizations form acetonitrile 3.9 g of
the pure salt was obtained.
Step III
3.9 g of pure salt from Step II was
dissolved in 200 cc of methanol and acidified using
lN hydrochloric acid. Water was added and the
crystals were separated by filtration and dryed under
vaccum. Upon trituration with he~ane-ethyl acetate
~9:1) the resolved acid was prepared.
~D = -42.5 (methanol) m.p. 151 - 151.5C.
Step IV
To a solution of 1.0 g of the acid from Step
III in 50 ml of ether is added a solution of
diazomethane in ether until a slight escess of
diazomethane is present. The escess diazomethane is
destroyed by addition of a few drops of acetic acid.
The reaction mixture is washed with 50 ml of 5
Na2C03 solution, water, and dried over MgS04.
Filtration and evaporation of the ,solvent leaves the
title compound.
,.
.. .~., . , ;
1 330~qO
5010P~1286A - 59 - 17358IA
E~am~le 19
(+) 9-p-chlorobenzyl-6-fluoro~1,2,3,4-tetrahydro-
carbazol-l-yl-acetic acid. methyl ester
Following the method of Example 18, but
using 1~-) ephedrine in Step II, there is obtained
the title compound.
EsamPle 20
Ethyl 9-p-chlorobenzyl-6-isopropyl-1-2,3,4-tetra-
hydrocarbazol-l-yl-acetate
Following the procedure of Example 1, but
using l-~4-chlorobenzyl)-1-(4-isopropylphenyl)
hydrazine hydrochloride and ethyl 2-cyclohexanone
acetate as starting materials, the title compound was
prepared as a misture of ethyl and isopropyl esters.
Example 21
Ethyl 9-p-chlorobenzyl-6-tert-butyl-1-2,3,4-tetra-
hydrocarbazol-l-yl-acetate
Following the procedure of Esample 1, but
using 1-(4-chlorobenzyl)-1-(4-tert-butylphenyl)
hydrazine hydrochloride and ethyl 2-cyclohesanone
acetate as starting materials, the title compound was
prepared as a misture of ethyl and isopropyl esters.
Esa~ple 22
Ethyl 9-p-chlorobenzyl-6-trifluoromethyl-1,2,3,4-
tetrahydrocarbazol-l-vl-acetate
Following the procedure of Esample 1, but
using 1-~4-chlorobenzyl)-1-~4-trifluoromethylphenyl)
hydrazine hydrochloride and ethyl 2-cyclohesanone
acetate as starting materials, the title compound was
prepared as a mixture of ethyl and isopropyl esters.
...
1 330090
5010P/1286A - 60 - 17358IA
Example 23
Ethyl 9-p-chlorobenzyl-6-methylthio-1,2,3,4~tetra-
hYdrocarbazol-l-y~-acetate
Following the procedure of Example 1, but
using 1-(4-chlorobenzyl)-1-(4-methylthiophenyl)
hydrazine hydrochloride and ethyl 2-cyclohexanone
acetate as starting materials, the title compound was
prepared as a mi~ture of ethyl and isopropyl esters.
The pure title compound was obtained by purification
on a flash chromatogram.
E~ample 24
Ethyl 9-p-chlorobenzyl-6-methylsulfinyl-1,2,3,4-tetra-
hydrocarbazol-l-yl-acetate
To 498 mg of ethyl 9-p-chlorobenzyl-6-
methylthio-1,2,3,4-tetrahydrocarbazol-1-yl-acetate
from Esample 23 in 10 cc of methylene chloride was
added 300 mg of m-chloro perbenzoic acid. The
resulting mixture was stirred for 1.5 hours at room
temperature. The reaction mixture was diluted with
ether and consecutively washed with a solution of
sodium bicarbonate, water and brine. The crude
product obtained after evaporation of the organic
layer was purified on silica gel by flash
chromatography eluting with 20% hesane/ethyl acetate
and yielded 420 mg (82%) df the pure title compound.
Esample 25
Ethyl 9-p-chlorobenzyl-6-methylsulfonyl-1,2,3,4-tetra-
hYdroearbazol-l-yl-acetata
~ o 439 mg of ethyl 9-p-chlorobenzyl-6-methyl-
sulfinyl-1,2,3,4-tetrahydrocarbazol-1-yl-acetate from
Esample 24, in 10 cc of methylene chloride was added
1 330090
5010P/1286A - 61 - 17358IA
353 mg of m chloro perbenzoic acid. The resulting
mixture was stirred for 18 hours at room temper-
ature. The reaction misture was diluted with ether
and washed consecutively with a solution of sodium
bicarbonate, water and brine. The crude product
obtained after evaporation of the organic layer was
purified on silica gel by flash chromatography
eluting with 30% hexane/ethyl acetate and yielded 200
mg (42~) of ths pure title compound.
Example 26
Ethyl 9-p-chlorobenzyl-8-isopropyl-1,2,3,4-tetra-
hYdrocarbazol-l-yl-acetate
Following the procedure of Esample 1, but
using 1-(4-chlorobenzyl)-1-~2-isopropylphenyl)
hydrazine hydrochloride and ethyl 2-cyclohexanone
acetàte as starting materials, the title compound was
prepared as a misture of ethyl and isopropyl esters.
EsamPle 27
9-p-Chlorobenzyl-8-methylthio-1,2,3,4-tetrahydro-
carbaæol-l-yl-acetic acid, methyl ester
Following the procedure of Esample 30, but
using 1-(2-methylthiophenyl) hydrazine hydrochloride
and ethyl 2-cyclohexanone acetate as starting
materials, the ti*le compound was prepared.
E~am~le 28
9-p-Chlorobenzyl-8-methylsulfinyl-1,2,3,4-tetrahydro-
carba~ol-l-Yl-acetic acid, methYl_e~ter
Following the procedure of Example 24, but
using methyl ester from E~ample 27, there is obtained
the title compound.
- ~ 3300q
5010P/1286A - 62 - 17358IA
Example 29
Ethyl 9-p-chlorobenzyl-6-fluoro-3-methyl-1,2,3,4-
tetrah~droca~bazol-l-yl-acetate
Following the procedure of Example 1, but
using 1-(4-chlorobenzyl)-1-(4-fluorophenyl) hydrazine
hydrochloride and ethyl 4-methyl-2-cyclohexanone
acetate as starting materials, the title compound was
prepared as a misture of ethyl and isopropyl esters.
Esample 30
9-p-Chlorobenzyl-6,8-difluoro-1,2,3,4-tetrahydro-
carbazol-l-yl-acetic acid. methyl ester
SteD I
To 114 g of 1-(2,4-difluorophenyl) hydrazine
hydrochloride in 350 cc of 2-propanol containing 40
cc of acetyl chloride was added 138 g of ethyl
2-cyclohesanone acetate. The reaction was refluxed
under nitrogen for 2 days. After cooling, 200 cc of
ether was added and the precipitate filtered. The
filtrate was evaporated to dryness. The resulting
residue was dissolved in a (1:1) mixture of
ether~ethyl acetate and consecutively ashed with
water, sodium bicarbonate solutin and brine. The
organic iayer was dried over sodium sulfate and
evaporated to dryness. The crude product was passed
through a silica gel bed eluting with 5% ethyl
acetate/hesane to yield 84 g of a 1:2 mi~ture of
ethyl and isopropyl esters.
.
:
.. ,~ .
1 330090
5010P/1286A - 63 - 17358IA
Step II
84 9 of esters from Step I was dissolved in
250 cc of methanol and 400 cc of sodium hydroside
(lN) was added and reflused 4 hours. After cooling,
the reaction misture was washed with a (1:1) misture
of
ether~hesane and the aqueous layer was acidified with
HCl ~lN). The resulting precipate was filtered,
washed with water and air dried to afford 50 9 of
6,8-difluoro-1,2,3,4-tetrahydrocarbazol-1-yl-acetic
acid.
Ste~ III
A solution of 11.1 9 of acid from Step II in
100 cc of THF was added portionwise 10.3 9 of
potasslum tert-butoside. The resulting misture was
stlrred for 45 min at room temperature and 10.3 9
p-chlorobenzyl bromide was added portionwise. The
reaction misture wa~ stirred 18 hour~ at room
.20 temperature. The resultlng mlsture was diluted with
; 100 cc of water and washea with hesane. The agueous
layer wa~ acidified with HC1 (lN) and the resulting
precipitate filteredi, washed with water and air-dried
to afford 9.4 g of 9-p-chlorobenzyl-6,8-difluoro-
1,2,3,4-tetrahydrocarbazol-1-yl-acetic acid.
SteD IV
Following the method of Esample 18, Step IV
but using the acid of Step III, there was obtained
the title compound.
~.i.. ,
J ~ ~
. ~ .
.:. ~ :: :
. . `
: -
.,~` `` , ,
~: . `
; - ' "`:` " :
1 330090
5010P/1286A - fi4 - 17358IA
lH NMR (CDC13) ~ 1.75-2.00 (m, 4H, (CH2)2)
2.38-2.78 (m, 4H, CH2CO2Me, CH2-C=C), 3.35-3.45
(m, lH, CH-C=C), 3.70 (s, 3H, CO2Me), 5.40 (dd, lH,
CH2-Ar), 6.60 (ddd, lH, H7), 6.92 (dd, lH, H5),
6.82 and 7.22 (2d, 4H, Ar).
Esample 31
9-p-Chlorobenzyl-6,8-dimethyl-1,2,3,4-tetrahydro-
carbazol-l-Yl-acetic acid. methvl ester
Following the procedure of Example 30, but
using 1-(2,4-dimethylphenyl) hydrazine hydrochloride
and ethyl 2-cyclohexanone acetate in Step I, as
starting materisl, the title compound is prepared.
EsamDle 32
9-p-Chlorobenzyl-6-methosy-8-methyl-1,2,3,4-tetrahydro-
carbazol-l-yl-acetic acid. methYl ester
Following the procedure of Esample 30, but
using 1-(4-methosy-2-methylphenyl) hydrazine
hydrochloride and ethyl 2-cyclohesanone acetate in
Step I, as starting material, the title compound is
prepared.
Esample 33
~-) 9-p-Chlorobenzyl-6,8-difluoro-1,2,3,4-tetrahydro-
- carbazol-l-yl-acetic acid. methyl ester
Following the method of Example 18, Step II
to Step IV, but using 9-p-chlorobenzyl-6,8-difluoro-
1,2,3,4-tetrahydrocarbazol-1-yl-acetic acid from
Esample 30, Step III and using d(~)ephedrine in Step
III, there was obtained the title compound.
~ 330090
5010P/1286A - 65 - 17358IA
H NMR (CDC13) ~ 1.75-2.00 (m, 4H, (CH2)2)
2.38-2.78 (m, 4H, C~2C02Me, CH2-C=C), 3.35-3.45
(m, lH, CH-C=C), 3.70 (s, 3H, C02Me), 5.40 (dd, lH,
Ç~2Ar), 6.60 (ddd, lH, 47), 6.92 (dd, lH, H5),
6.82 and 7.22 (2d, 4H, ~r).
~sample 34
(~) 9-p-Chlorobenzyl-6,8-difluoro-1,2,3,4-tetrahydro-
carbazol-l-yl-acetic a~id, methyl ester
Following the method of Example 18, Step II
to Step IV, but using 9-p-chlorobenzyl-6,8-difluoro-
1,2,3,4-tetrahydrocarbazol-1-yl-acetic acid from
Example 30, Step III and using l(-~ephedrine in Step
III, there is obtained the title compound.
Example 35
(-) 9-p-Chlorobenzyl-8-methyl-1,2,3,4-tetrahydro-
carbazol-l-Yl-acetic acid methyl ester
Following the method of Esample 18, but
using the ethyl ester from esample 11 in Step I and
using d(~)ephedrine in Step I~, there i8 obtained the
title compound.
Esample 36
~) 9-p-Chlorobenzyl-8-methyl-1,2,3,4-tetrahydro-
carbazol-l-Yl-acetic acidJ methyl ester
Following the method of Example 18, but
using ethyl ester from Esample 11 in Step I and using
l(-)ephedrine in Step II, there is obtained the title
compound.
1 3300qo
5010P/1286A - 66 - 17358IA
E~ample 37
(-) 9-p-Chlorobenzyl-8-fluoro-1,2,3,4-tetrahydro-
carbazol-l-yl-acetic acid. methyl ester
Following the method of Esample 18, but
using the ethyl ester from Esample 15 in Step I and
using d(+)ephedrine in Step II, there is obtained the
title compound.
E~ample 38
(~) 9-p-Chlorobenzyl-8-fluoro-1,2,3,4-tetrahydro-
carbazol-l-yl-acetic acid. methyl ester
Following the method of Esample 18, but
using the ethyl ester from Example 15 in Step I and
using l(-)ephedrine in Step II, there is obtained the
title compound.