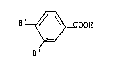Note: Descriptions are shown in the official language in which they were submitted.
1 3300q4
INTERMEDIATES F~R PR~PARATION OF OE PHALOSPORIN
DERIVATIV~S WITH IMPROVED PHAKMACOKINETICS
The present invention-relates to novel
intermediates used in the preparation of cephalosporin
derivatives with improved pharmacokinetics.
This application is a division of copending
Canadian patent application Serial No. 579,575 filed
October 7, 1988. The parent application desGribes
certain novel cephalosporin derivatives with improved
pharmacokinetics, procedures for the preparation of the
same and pharmaceutical compositions containing the
same.
In French patent application no. 84 14 878
published on 28/03/86 under the number 2 570 702, the
Applicant Company described a family of cephalosporin
derivatives possessing a broad activity against both
Gram-negative germs and Gram-positive germs.
These derivatives include, in particular, compounds
substituted in the 3-position by a group:
-CH20CO~
NH-CO -Allc-NH2
in which Alk represents an optionally substituted lower
alkylene group and in which the substituent NH-CO-Alk-
NH2 is located in the 3-position or 4-position.
It has been found, surprisingly, that by slightly
modifying the nature of the substituent in the 3-
position, compounds are obtained which preserve thegood activity of the compounds described in the prior
art but additionally have greatly improved
pharmacokinetic properties and, in particular, very high
plasma concentrations which persist over a very long
period.
Modifying the pharmacokinetic parameters in this
way is important inasmuch as it opens up the possibility
1 3300q~
of reducing the dosage and the num~er of doses of the
product required for the same therapeutic effect.
The cephalosporin derivatives according to the
invention claimed in the parent application have the
general formula
H2N S~
N 1~ 0 S
C-R; ~ C~20CO ~ A
R3 COOH
in which:
- Rl, R2 and R3 each denote a hydrogen atom, or Rl and
R2 each denote a hydrogen atom or a methyl group and R3
denotes a carboxyl group, or alternatively R1 and R2,
taken together with the carbon atom to which they are
bonded, form a cyclobutyl ring and R3 denotes a
carboxyl group, and
- the two groups A are different, one representing a
group OH and the other denoting a group -NHS02-Alk-NH2,
in which Al~ denotes a C2-C4 lower alkylene group. Also
included are the pharmaceutically-acceptable salts,
including inner salts, and pharmaceutically-acceptable
esters of such compounds.
As a consequence of the presence of an oxime group
in their formula, the compounds (I) can exist in 2
isomeric forms: syn and anti. The syn isomers, which
have the greater therapeutic activity, are the
preferred compounds.
It i5 understood that the compounds (I) indicated
below can exist
- either in the form indicated in formula (I)
- or in the tautomeric form (I'):
1 330094
~Z C; NH ~ C~2oCo ~ A (I ~
R3 COOH
in which R1, R2, R3 and A are as defined above.
Salts of the compounds of formula I (or I') can be
either salts with pharmaceutically acceptable acids
which can be formed with the amino groups of the
molecule, or alkali metal salts, alkaline earth metal
salts or salts of amino acids or amines, such as
triethylamine or ethanolamines, which can be formed wi~h
the carboxyl group in the 4-position of the compound
(I) or, i~ it exists, with the carboxyl group present in
the substituent of the oxime, or with both these
carboxyl groups.
They can also be inner salts which can be formed
between the carboxyl group (or groups) carried by the
molecule and the primary amine groups present in the
molecule on the substituent A, on the one hand, and the
thiazole ring, on the other.
The readily hydrolyzable or metabolically labile
esters derived from one or other or both of the carboxyl
groups which may be present in the molecule, include, in
particular, the phthalidyl esters:
~0
1 33ooq4
CH3
the l-acetoxyethyl esters: -CH-O-COCH3 ,
CH3
the l-ethoxycarbonyloxyethyl esters: -CH-O-CO-OC2H5
and
the (4-methyl-2-oxodioxol-4-en-5-yl)methyl esters:
-CH2-C = C-CH3
O O
C
o
The invention claimed in the parent application
further relates to a process for the preparation of the
compounds of formula I (or I') represented by the
following reaction scheme:
N\ R~
R2 OOtBu
~'
io
COOH
________
Tr-l~S\
o 8-R ~ o \~ ~
~2 OCtBu
H~ 3
_________> ~T'~
1 330094
In these formulae, Tr represents a protecting group
for the amine group, preferably the trityl group, tBu
represents the tert-butyl group and R'3 denotes hydrogen
Gr a readily labile ester group, preferably a group
COOtBu.
The 2 groups A' are different, one of them
representing a group OH and the other representing a
group derived from the group -NHS02AlkNH2 by blocking
its amino group with a labile group.
Finally, Rl, R2 and Alk have the meanings defined
above.
The iodine compound 1 is reacted with the acid 2
(or one of its active derivatives) in which the amine
group has been protected beforehand, according to a
known method, by a group such as tert-butoxycarbonyl or
trichloroethoxycarbonyl.
In general, the reaction takes place in solution in
a suitable solvent (aprotic, polar), preferably
dimethylformamide, in the presence of potassium
bicarbonate or a tertiary amine of low nucleophilicity,
such as diisopropylethylamine.
The reaction is carried out at low temperature: 0
to 20C.
The resulting protected compound 3 is used to
prepare the compounds (I) by elimination of the protec-
ting groups carried by the amines and the ester or
esters, according to a known process, in particular by
hydrolysis in an acid medium using, for example, tri-
fluoroacetic acid or a mixture of formic acid with a
strong acid such as hydrochloric acid or methanesulfonic
acid.
Under these conditions, the compound (I) is
isolated directly in the form of a salt of the amino
group with the strong acid used for deprotection, i.e.
in the form of the trifluoroacetate, hydrochloride,
methanesulfonate etc.
1 33009~
If desired, these salts can be converted into
other salts of strong acids by passing a solution of
this salt over a basic ion exchange resin in the form of
the salt of a weak acid (for example the formate or
acetate).
The strong acid whose salt it is desired to o~tain
is added to the resulting solution and the salt obtained
is isolated, for example by lyophilization.
The inner salts are prepared by desalification of
the salts of strong acids obtained on deprotection,
either by reaction with a base in an anhydrous medium
or by passage through a column of ion exchange resin.
The iodine derivatives 1 used as starting materials
are known or can be prepared by a known process, in
particular as indicated in German patent application no.
3 311 300.
The protected amino acids 2 are prepared from the
corresponding hydroxyamino acids according to the
equation:
~OOH ~ R4NH-Alk-S02Cl ~ A'_~OOH
4 5 2
in which the groups B are different, one representing OH
and the other representing NH2, and the groups A' are
different, one representing OH and the other
representing a group R4NH-Alk-SO2NH-, in which R4
represents a protecting group for the amine group and in
which Alk has the meaning indicated above.
The reaction is carried out in solution, for
example in methylene chloride, in the presence of an
acid acceptor and a reagent which activates the amine
1 3300q4
group and at the same time protects the carboxylic acid
group. This is preferably done using trimethylchloro-
silane.
The compounds 2 in which R4 represents a given
protecting group can be ~onverted by acid hydrolysis
into the product 2 in which R4 is hydrogen, and then a
compound 2 in which the amine group is protected by a
- different protecting group from the initial protecting
group can be obtained by a known process.
In accordance with the present invention, there is
provided a novel compound of the formula:
B'
B ~OOH
in which the two groups B' are different, one of them
representing a group OH and the other representing a
group X-NH-Alk-SO2NH-, in which X represents hydrogen
or a protecting group for the amine group and in which
Alk represents a C2 to C4 lower alkylene group.
The carboxylic acid esters and salts of the
compounds (I) of the invention are obtained from the
compounds (I) by reactions known per se.
Thus the inorganic salts are obtained by reacting
the compounds (I) with an inorganic base, such as sodium
hydroxide, potassium hydroxide or sodium bicarbonate, in
equimolecular amounts; the salification reaction is
carried out in a solvent such as water or ethanol, and
the salt obtained is isolated by evaporation of the
solution.
The salts of organic bases are obtained by reacting
a solution of the acid I with an equimolecular amount
of the organic base in a solvent or a mixture of
suitable solvents. The salt is isolated by
precipitation with ether. The esters are obtained by
13300q4
the known esterification processes; for example, the
reaction of halogen derivative with a salt of the acid,
such as the sodium salt, will advantageously be used;
the reaction will preferably be carried out in a solvent
s which is capable of dissolving the starting acid
derivative, for example in dimethylformamide.
The syn and anti isomeric forms are obtained
through an appropriate choice of reagents.
The following Examples will provide a clearer
understanding of the scope of the invention without
however implying a limitation.
As is usual in this family of compounds, the
products do not have sharp melting points, but only
decomposition points by which they cannot be
characterized.
The products will therefore be characterized by
their nuclear magnetic resonance spectra. Unless
indicated otherwise, the spectra are run at 250 MHz
with hexamethyldisiloxane as the internal standard.
The spectra are run in deuterated dimethyl
sulfoxide: 10 mg in 0.5 ml.
The chemical shifts are measured to + 0.01 ppm and
the coupling constants to + 0.5 Hz.
The following abbreviations will be used:
- S : singlet
- D : doublet
- D of D : doublet of doublets
- S.b. : broadened singlet
- M : multiplet
- Q : quadruplet
- T : triplet
- AB : AB system
- J : coupling constant
Elemental microanalyses were also carried out in
each case and are in agreement with the formulae
indicated.
1 33009~
EXAMPLE 1
7-[2-(2-Aminothiazol-4-yl)-2-methoxyiminoacet-
amido]-3-[[4-(2-aminoethylsulfonamido)-3-hydroxy-
benzoyl]-oxymethyl]-3-cephem-4-carboxylic acid
bis-trifluoroacetate, syn isomer (SR 44337)
(I) Rl = R2 = R3 = H; A = -OH (3);
A = -NHSO2CH2CH2NH2 (4)
A) 3-Hydroxy-4-[(2-tert-~utoxycarbonylaminoethyl)sul-
fon-amido]benzoic acid
1 - 3-Hydroxy-4-[(2-benzyloxycarbonylaminoethyl)-
sulfonamido]benzoic acid
84 g of 4-amino-3-hydroxybenzoic acid are sus-
pended in 1.8 liters of methylene chloride, 228.5 ml of
triethylamine are then added under a nitrogen atmosphere
and the mixture is cooled to 10~C.
228.5 ml of trimethylchlorosilane are added in 1
hour, with stirring.
The temperature is allowed to return to 20C and
the mixture is stirred for 2 hours 30 minutes at this
temperature.
168 g of 2-benzyloxycarbonylaminoethane sulfo-
chloride are then added with protection from the light
and the reaction mixture is stirred at room temperature
for 6 days. It is poured into 2 liters of sulfate
buffer of pH 2 and 2 liters of ethyl acetate. The
organic phase is separated off, washed with water and
dried over magnesium sulfate. The solvent is evaporated
off to dryness and the residue is taken up in 500 ml of
ether. The solid is filtered off, rinsed with isopropyl
ether and then dried. Weight: 64.5 g.
A second crop of the same product (54 g) is
isolated by concentration of the mother liquor to
dryness and treatment of the residue with isopropyl
ether to which a small amount of ether has been added.
The 2 crops are combined and dissolved in 470 ml of
ethanol. 1.5 liters of water are added slowly and the
lo 1 3300':)4
solution is stirred at room temperature for 30 minutes
and then cooled in ice.
The product is filterëd off, rinsed with water and
dried under vacuum over phosphorus pentoxide.
Weight: 102.2 g. Melting point: 194C.
2 - 3-Hydroxy-4-(2-aminoethylsulfonamido)benzoic
acid trifluoromethylsulfonate
102 g of the product obtained in A) are added, with
stirring, to a mixture of 1 liter of trifluoroacetic
acid, 110 ml of trifluoromethanesulfonic acid and 150
ml of thioanisole, cooled to 10C.
The temperature rises to 20C and the mixture is
stirred for l hour at this temperature.
The product crystallizes and the mixture has to be
diluted with 4 liters of methylene chloride. Stirring
is continued for 15 minutes, after which the solîd is
filtered off, rinsed with methylene chloride and then
dried under vacuum. Weight: 103.8 g. Melting point:
240C (decomposition).
3 - 3-Hydroxy-4-~(2-tert-butoxycarbonylamino-
ethyl)sulfonamido~benzoic acid
lO0 g of the trifluoromethanesulfonate obtained
above are dissolved in 1 liter of water, the pH is
adjusted to 7.5 by the addition of a concentrated
solution of sodium hydroxide, and 500 ml of dioxane are
then added.
A solution of 65 g of ditert-butyl dicarbonate in
300 ml of dioxane is added in 15 minutes, the pH being
~ept at 7.5 by the addition of sodium hydroxide.
After 30 minutes, the pH remains stable. The
reaction mixture is washed with 2.5 liters of ether and
the a~ueous phase is then poured into 2.5 liters of
sulfate buffer of pH 2.
The pH is ad~usted to 2 by the addition of a
solution of potassium bisulfate and extraction is then
carried out with 2 liters of ethyl acetate. A second
1 3300q4
11
extraction is carried out with 1 liter of ethyl acetate
and the organic extracts are combined. The combined
extracts are washed with 1 liter of a saturated
solution of sodium chloride and the organic solution is
dried over magnesium sulfate. It is evaporated to
dryness and the solid residue is taken up in 1 liter of
acetonitrile. The solution is stirred for 15 hours at
room temperature and then cooled in ice and the solid
is filtered off, washed with isopropyl ether and dried.
10Weight: 78 g. Melting point: 228-230C (decomposition).
B) Tert-butyl 7-[2-(2-tritylaminothiazol-4-yl)-2-
methoxyiminoacetamido]-3-[[4-(2-tert-butoxycar-
bonyl-aminoethylsulfonamido)-3-hydroxybenzoyl]-
oxymethyl]-3-cephemcarboxylate syn isomer
1537.5 g of the protected acid prepared in A) and 15
ml of diisopropylethylamine are dissolved in 200 ml of
dimethylformamide. The mixture is cooled to a
temperature of between 0 and +5C and 60 g of tertbutyl
7-[2-(2-tritylaminothiazol-4-yl)-2-methoxyimino-
acetamido]-3-iodomethyl-3-cephem-4-carboxylate, syn
isomer, are added.
The reaction mixture is stirred at 0-5C for 5
hours and then poured into 2 liters of iced water. The
precipitate is filtered off and rinsed with water. The
solid is dissolved in methylene chloride, the solution
i8 dried over magnesium sulfate and the solvent is
evaporated off to dryness. The residue is chromato-
graphed on a column of silica H (1 kg). Impurities are
removed by elution with a 99.1/0.9 vol/vol mixture of
methylene chloride and methanol and the expected product
is then obtained with a 98/2 vol/vol mixture of the same
solvents.
30.7 g are obtained after evaporation of the
solvents and washing with isopropyl ether.
1 3300q4
12
C) SR 44337
35 g of the protected product prepared above in B)
are added, with stirring, to a mixture of 330 ml of
trifluoroacetic acid and 33 ml of anisole, cooled in an
ice bath.
The temperature is allowed to rise to 20C and the
mixture is stirred for 1 hour at this temperature.
It is poured into 1.5 liters of isopropyl ether
cooled to ooc.
The precipitate is filtered off, washed with ether
and then dried under vacuum over phosphorus pentoxide.
26.8 g of the expected product are obtained.
This is dissolved in 100 ml of methanol and the
solution is poured into 1.25 l of ether. The
precipitate is filtered off, rinsed with ether and dried
under vacuum and 22 g of the title product are finally
obtained.
NMR SPECTRUM
1 H at 10.7 ppm (S.b., NH-SO2) 1 H at 9.6 ppm (D, J
= 8 Hz, -N_CO-) - 1 H at 9.5 ppm (S.b., -OH) - 9 H
between 7 and 8.2 ppm (M, 3 H aromatic and 2N~I3) - 1 H
at 6.7 ppm (S, H thiazole) - 1 H at 5.75 ppm (D of D, J1
8 Hz~ J2 = 4 Hz, H7) - 1 H at 5.15 ppm (D, J = 4Hz,
H6) - 2 H at 4.9 and 5.2 ppm (AB, JAB = 14 Hz, CH20CO) -
3 H at 3.8 ppm (S, NOCH3) - 6 H between 3.1 and 3.75 ppm
(M, CH2NH2, CH2SO2, CH2S).
EXAMPLE 2
7-[2-(2-Aminothiazol-4-yl)-2-methoxyimino-
acetamido]-3-[[4-(2-aminoethylsulfonamido)-3-hydroxy-
benzoyl]oxymethyl]-3-cephem-4-carboxylic acid bis-
hydrochloride, Ryn isomer
10.53 g of the protected product obtained in
Example 1 B are dissolved in 60 ml of 98~ formic acid
and the solution is stirred for 2 hours at 20C. 50
ml of a 10 N aqueous solution of hydrochloric acid are
1 33009~
13
added and the mixture is stirred for a further 2 hours
at 20C.
The reaction mixture is poured into 500 ml of ethyl
ether. The bis-hydrochloride is filtered off, washed
with ether and dried under vacuum.
7 g of the expected product are obtained.
Melting point: 145C (decomposition).
NMR SPECTRUM
1 H at 9.8 ppm (D, J = 8 Hz, NHCO) - 6 H at 8.2 ppm
(M, 2NH3+) - 3 H between 7.2 and 7.6 ppm (M, H aromatic)
- 1 H at 6.9 ppm (S, H thiazole) - 1 H at 5.8 ppm (D of
D, Jl = 8 Hz, J2 = 4 Hz, H7) - 1 H at 5.2 ppm (D, J = 4
Hz, H6) - 2 H between 4.9 and 5.2 ppm (A8, JAB = 14 Hz,
CH2OCO) - 3 H at 3.9 ppm (S, -N-OCH3) - 2 H at 3.7 ppm
(M, C_2-S) - 4 H between 3.2 and 3.5 ppm (M,
-CH2CH2NH2 ) -
EXANPLE 3
Inner salt of 7-[2-(2-aminothiazol-4-yl)-2-
methoxyiminoacetamido]-3-t[4-(2-aminoethylsulfonamido)
3-hydroxybenzoyl]oxymethyl]-3-cephem-4-carboxylic acid,
syn isomer
1 g of the bis-trifluoroacetate obtained in Example
l C is dissolved in 10 ml of anhydrous dimethylforma-
mide, the solution is cooled to 5 D C and a solution of
0.24 9 of diethanolamine in 2 ml of methanol is then
added. The mixture is stirred for 15 minutes at 5C and
then poured into 100 ml of ether cooled to 5~C. The
solid is filtered off and dried under vacuum in the
presence of phosphorus pentoxide.
0.800 g of inner salt is obtained.
NMR SPECTRUM
1 H at 9.5 ppm (D, J = 8 Hz, NHCO) - 1 H between
7.5 and 11 ppm (S.b., OH) - 3 H between 7 and 7.5 ppm
(M, H aromatic) - 1 H at 6.7 ppm (S, H thiazole) - 1 H
at 5.6 ppm (D of D, J1 = 8 Hz, J2 = 4 Hz, H7) - 1 H at
5.05 ppm (D, J = 4 Hz, H6) - 2 H at 4.85 and 5.15 ppm
1 3300q4
14
(AB, JAB = 14 Hz, CH2OCO) - 3 H at 3.80 ppm (S, NOCH3) -
2 H at 3.6 ppm (S, C_2S) - 2 H at 3.12 ppm (M,
CH2SO2NH) - 2 H at 2.95 ppm (M, C_2NH2).
EXAMPLE 4
7-[2-(2-Aminothiazol-4-yl)-2-(2-carboxyprop-2-
yloxyimino)acetamido]-3-[[4-(2-aminoethylsulfonamido)-3-
hydroxybenzoyl]oxymethyl]-3-cephem-4-carboxylic acid
bis-trifluoroacetate, syn isomer (SR 44338)
(I) Rl = R2 = CH3; R3 = COOH; A = OH (3);
A = -NHSO2CH2CH2NH2 (4)
This product is prepared in the same way as the
product of Example 1, the tert-butyl 7-[2-(2-trityl-
aminothiazol-4-yl)-2-methoxyiminoacetamido]-3-iodo-
methyl-3-cephem-4-carboxylate, syn isomer, being
replaced in step B with an equivalent amount of
tert-butyl 7-[2-(2-tritylaminothiazol-4-yl)-2-(2-tert-
butoxycarbonyl-prop-2-yloxyimino)acetamido]-3-
iodomethyl-3-cephem-4-carboxylate, syn isomer.
The product SR 44338 is obtained after deprotection
as indicated in Example 1 C.
NM~ SPECTRUM
1 H at 9.4 ppm (D, J = 8 Hz, CON~) - 11 H between 7
and 11 ppm (M, 3 H aromatic, N~SO2, O~, 2NH3) - 1 H at
6~7 ppm (S, _ thiazole) - 1 H at 5.8 ppm (D of D, Jl = 8
25 Hz, J2 = 4 Hz, H7) - 1 H at 5.2 ppm (D, J = 4 Hz, H6) -
2 H at 4.9 and 5.25 ppm (AB, JAB = 14 Hz, C_2OCO) - 6 H
between 3.1 and 3.75 ppm (M, C_2NH2, C_2SO2), C_2S) -
~ C_3
6 H at 1.4 and 1.42 ppm (2S, O-C- ).
C~3
The products were studied for their pharmacological
properties.
The bacteriostatic action was deter~ined in vitro
by the dilution method. The study was carried out on
both Gram-positive strains and Gram-negative strains.
~ 33009~
The results are expressed as minimum inhibitory
concentrations (MIC - ~g/ml).
By way of example, the results obtained with SR
44337 (Example 1) are collated in Table 1.
TABLE 1
STRAIN SR 44337
Staph. aureus Smith 0.25
Escherichia coli 0.06
Cl/Col
El :: Tn3
Escherichia 0.5
coli SOL RL so
Klebsiella 4
pneumoniae R30
Proteus vulgaris
GN 76/C-l
Providencia
155
Pseudomonas 2
aeruginosa
NCTC 8203
These results show that the products according to
the invention have a broad spectrum of activity and
possess a very good intrinsic activity.
Furthermore, the pharmacokinetic behavior of the
products according to the invention was studied on
baboons after admini~tration at a dose of 20 mg/kg by
intravenous injection.
The plasma concentration of the product studied
is determined by microbiological assay using blood
samples taken at various times after administration.
This makes it possible to draw the curve showing
the plasma concentration as a function of time and to
determine different pharmacokinetic parameters of the
compound studied:
1 3300q~
16
- the elimination half-life (t~ B) is calculated by
the formula ln 2 , in which B represents the
elimination slope;
- the area under the curve (AUC) is determined by the
trapezium method.
Furthermore, the protein binding is obtained by
comparing two standard series, one being carried out in
baboon plasma and the other in phosphate buffer (pH 7,
0.03 M).
The plasma concentrations obtained with the product
of Example 1 are collated in Table 2. They are
expressed in ~g/ml.
By way of comparison, this Table includes the
results obtained with a similar product of the formula
H2N~ S
CI - CO ~
OCH3 ~ cH2oco ~ -NHso2cH2cH2NH2
COO~
.2CF3COOH
Compound A
1 330094
17
TABLE 2
TIME ~g/ml
(hours~
SR 44337 Compound
(Example 1) A
0.08 265.5 232.8
0.16 250.9 212.6
0.25 237.5 189.7
0.33 206.1 170.4
0.5 194.3 124.2
0.75 190.0 120.7
1 182.5 119.8
1.5 163.8 112.4
2 152.7 98.4
3 151.7 72.9
4 146.7 68.7
141.1 62.1
6 120.1 54.4
24 39.0 7.7
48 14.4 0.6
Table 3 below collates, for the product of Example
25 1 and the comparison product, the pharmacokinetic
parameters determined according to the same experiments.
1 330094
18
TABLE 3
.
Parameter Product
SR 44337 Compound
(Example 1) A
.
Maximum plasma concentration 265.5 232.8
(~g/ml)
Plasma concentration (~g/ml)
at 24 h 39.0 7.7
at 48 h 14.4 0.6
t~ ~ (h) 13.9 6.3
AUC 0~ g/ml x h) 3316 1210
Excretion in urine (% dose) at
6 h 14 18
The results given in Tables 2 and 3 show that the
plasma concentrations of the product of the invention
are extremely high and long-lasting.
When compared with reference product A, the plasma
concentrations at 24 and 48 hours obtained for the
compound of Example 1 are respectively 5 and 24 times
higher. Likewise, as regards the area under the curve,
the increase is 2.7-fold relative to the reference
product.
The compounds therefore have very valuable
pharmacokinetic parameters which can make it possible
~: substantially to reduce the amount of active principle
1 330094
19
used and the number of daily administrations required
for a given therapeutic effect.
Finally, the toxicity of the products is
sufficiently low to enable them to be used in therapy.
The products of the parent application can
therefore be employed as antibiotics in human or
veterinary medicine. They have a broad spectrum and can
be used for all bacterial infections caused by
sensitive germs.
The products can be administered by a general
route (parenteral, oral, rectal) or topically.
The pharmaceutical compositions are prepared from
the compounds (I) in a soluble form obtained by
salification of at least one of their acid groups or
amine groups.
The pharmaceutical compositions which contain the
antibiotic claimed in the parent application, as the
active ingredient, in combination with a
pharmaceutically acceptable vehicle can be solid or
liquid and can take the form of, for example, injectable
preparations, tablets, gelatin capsules, granules,
ointments, creams, gels or suppositories. The dosage
can vary within wide limits, in particular according to
the type and severity of the infection to be treated
and according to the mode of administration.
The adult dosage is most frequently between 0.250 g
and 4 g per day, administered by injection.
As an example of a pharmaceutical preparation,
it is possible to prepare an injectable solution
30 containing the following in each ampoule:
SR 44337 1 g
Water for injectable preparations 5 ml
Sodium carbonate q.s. for pH - 6.3