Note: Descriptions are shown in the official language in which they were submitted.
1 334781
METHOD OF ~IlNG A NO~w~v~N WEB FROM A
SURFACE-SEGREGATABLE THERMOPLASTIC COMPOSITION
Back4Loulld of the Invention
The present invention relates to a method of forming
a nonwoven web from a surface-segregatable, melt-extrudable
thermoplastic composition. More particularly, the present
invention relates to a method of forming a nonwoven web
from a thermoplastic composition which surface segregates
in a controllable manner upon melt extrusion to form
fibers having modified surface characteristics.
Polymers are used widely throughout the world to make
a variety of products which include blown and cast films,
extruded sheets, injection molded articles, foams, blow
molded articles, extruded pipe, monofilaments, and nonwoven
webs. Some of such polymers, such as polyolefins, are
naturally hydrophobic, and for many uses this property is
either a positive attribute or at least not a disadvantage.
20There~are a number of uses for polyolefins, however,
where their hydrophobic nature either limits their useful-
ness or requires some effort to modify the surface charac-
teristics of the shaped articles made therefrom. By way of
example, polyolefins are used to manufacture nonwoven webs
which are employed in the construction of such disposable
absorbent articles as diapers, fe~in;ne care products,
incontinence products, and the like. Frequently, such
nonwoven webs need to be wettable. Wettability can be
obtained by spraying or coating the web with a surfactant
solution during or after its formation. The web then must
be dried, and the surfactant which remains on the web is
removed upon exposure of the web to aqueous media.
Alternatively, a surfactant can be included in the
polymer which is to be melt-processed, as disclosed in
U.S. Patent Nos. 3,973,068 and 4,070,218. In that case,
however, the surfactant must be forced to the surface of
-1- ~
1 334781
the fibers from which the web is formed. This typically
is done by heating the web on a series of steam-heated
rolls or "hot cans". This process, called "blooming", is
expensive and still has the disadvantage of ready removal
of the surfactant by aqueous media. Moreover, the surfac-
tant has a tendency to migrate back into the fiber which
adversely affects shelf life, particularly at high storage
temperatures. In addition, it is not possible to incor-
porate in the polymer levels of surfactant much above 1
percent by weight; surfactant levels at the surface appear
to be limited to a maximum of about 0.33 percent by weight.
Most importantly, the blooming process results in web
shrinkage in the cross-machine direction and a significant
loss in web tensile strength.
Two common methods of preparing nonwoven webs are
meltblowing and spunbonding. When using a surface-segre-
gatable, melt-extrudable thermoplastic composition to
prepare a nonwoven web or fabric by either method as
disclosed herein, it was found that certain problems could
be encountered. Such problems typically were dependent
upon the level of additive present in the composition
prior to melt extrusion. For example, at additive levels
less than about 1 percent by weight, there often was
insufficient additive present at the surfaces of the
fibers comprising the web to impart to the surfaces a
characteristic of the additive. In addition, at levels of
~additive of from about 1 to about 2 percent by weight, the
amount of additive at the fiber surfaces often was not as
high as desired. The present invention addresses both
problems.
Moreover, it was found in spunbonding processes that
at additive levels equal to or greater than about 1 percent
by weight, the formed web lacked coherency. That is, upon
attempting to remove the web from the foraminous collecting
surface, the web simply fell apart. The present invention
addresses this problem and permits the use of spunbonding
1 334781
processes for the formation of nonwoven webs which have
the neceææAry coherency for further processing and/or
incorporation into products when the additive levels
present in such thermoplastic composition are equal to or
5 greater than about 1 percent by weight.
As is well known in the art, nonwoven webs may be
formed by meltblowing in accordance with U.S. Patent Nos.
3,016,599, 3,704,198, 3,755,527, and 3,849,241; or by
spunbonding in accordance with U.S. Patent Nos. 3,341,394,
10 3,655,862, 3,692,618, 3,705,068, 3,802,817, 3,853,651,
4,064,605, 4,340,563, and 4,434,204; or by coforming in
accordance with U.S. Patent Nos. 4,100,324 and 4,118,531
to E. R. Hauser. See also U.S. Patent No. 4,663,220.
In addition to those already described, other methods
15 of imparting wettability to, or otherwise affecting the
surface characteristics of, fibers or other shaped articles
made from polyolefins and other hydrophobic polymers are
known. Representative examples of a number of such methods
are described in the paragraphs which follow.
U.S. Patent No. 4,578,414 describes wettable olefin
polymer fibers. The fibers are formed from a composition
comprising a polyolefin resin and one or more defined
surface-active agents. Such agents may be present in an
amount of from about 0.01 to about 5 percent by weight.
25 The surface-active agents can be (1) an alkoxylated alkyl
phenol in combination with a mixed mono-, di-, and/or
-triglyceride; (2) or a polyoxyalkylene fatty acid ester;
or (3) a combination of (2) with any part of (1). The
preferred polyolefin is polyethylene, and all of the
30 examples employed an ethylene/l-octene copolymer, the
latter apparently being a minor component. The surface-
active agents are stated to bloom to the fabricated fiber
surfaces where at least one of the surface-active agents
remains partially embedded in the polymer matrix. The
35 patent further states that the permanence of wettability
1 33478 1
can be controlled through the composition and concentration
of the additive package.
Polysiloxane/polyoxazoline block copolymers are
discIosed in U.S. Patent No. 4,659,777. The copolymers
are stated to be useful as surface-modifying additives for
base polymers. Such use apparently has primary reference
to personal care products where the surface properties to
be imparted include glossiness, smoothness, and lubricity.
However, incorporation of the copolymers into fibers is
stated to impart surface stain resistance, antistatic
properties, flame retardancy, and wettability by both
polar and nonpolar solvents. Such incorporation preferably
is in the range of from about 1 to 5 parts by weight.
Suitable base polymers include some vinyl polymers, acrylate
15 polymers, polyurethanes, cellulose derivatives, and poly-
ethylene, polypropylene, ethylene-propylene copolymers,
and copolymers of ethylene with, for example, vinyl acetate.
However, the single example illustrating incorporation of
the disclosed copolymers into a base polymer employed as
20 the base polymer poly(vinyl chloride), and the resulting
mixture was used to cast films from solution.
U.S. Patent No. 4,672,005 describes a process for
improving the hygroscopic, soil release, and other surface
properties of a polymer substrate. The process involves
25 contacting the substrate with an aqueous mixture containing
a water-soluble vinyl monomer and a hydrophobic vinyl
-monomer. Polymerization of the water-soluble vinyl monomer
then is initiated by a polymerization initiator, thereby
forming a vinyl polymer on the surface of the polymer
substrate.
U.S. Patent No. 4,698,388 describes a method for
modifying the surface of a polymer material by means of a
block copolymer. The block copolymer consists of a hydro-
philic polymer portion formed from a vinyl monomer and a
- 35 polymer portion which is compatible with the polymer
material, also formed from a vinyl monomer. The block
--4--
1 33~
copolymer is added to the polymer material by, for example,
coating the material with a solution or suspension of the
block copolymer, mixing the block copolymer with the
polymer material during formation of the article, forming
5 a film from the block copolymer which then is melt-pressed
or adhered to the surface of the polymer material, and
coating the surface of the polymer material with powdered
block copolymer.
Polymer compositions having a low coefficient of
10 friction are described by U.S. Patent No. Re. 32,514.
The compositions comprise a blend of at least 80 percent
by weight of a polymer and at least 0.35 percent by weight
of a crosslinked silicone polycarbinol. The polymer
preferably is a blend of cellulose nitrate and a hydrophobic
15 acrylate polymer. The silicone polycarbinol in general is
a hydroxy-terminated polysiloxane or hydroxy-substituted
polysiloxane. The compositions typically are prepared by
dissolving the polymer or polymer blend, silicone polycar-
binol, and crosslinking agent in a suitable solvent and
20 casting a film from which the solvent is allowed to evapor-
ate.
Canadian Patent No. 1,049,682 describes the inclusion
in a thermoplastic polymer of from 0.1 to 10 percent by
weight of a carboxy-functional polysiloxane. Suitable
25 thermoplastic polymers include polyolefins. Such inclusion
is stated to enhance the properties or characteristics of
-the thermoplastic polymer in one or more ways. By way of
illustration, products or articles made from the polymer
mixture were stated to have self-lubricating properties and
increased resistance to wear. For molded articles, less
friction during transfer, injection or extrusion molding
was observed, and better release of parts from the molds
was obtained. See, also, German Published Patent Applica-
tion (Offenlegungschrift) No. 2,506,667 tChem. Abstr.,
35 84:91066z (1976)].
1 334781
other, similar references which may be of interest
include R. H. Somani and M. T. Shaw, Macromolecules, 14,
886 (1981), which describes the miæcibility of polydimethyl-
siloxane in polystyrene; and S. N. Pandit et al., PolYm.
5 Compos., 2, 68 (1981), which reports the use of a vinyltri-
ethoxysilane polymer as a coupling agent in glass fiber-
reinforced polypropylene.
Also for the sake of completeness, it may be noted
that polysiloxanes have been utilized in the production of
10 nonwoven webs or fabrics, or products made therefrom, as
illustrated by the references which follow.
U.S. Patent No. 3,360,421 describes a bonded nonwoven
backing material having perforate selvage which is used in
the manufacture of carpet. In the production of the
15 nonwoven backing material, a nonwoven web is produced from
a polyolefin such as polyethylene or polypropylene. The
resulting web then is subjected to bonding conditions,
followed by applying to the web a lubricant which can be,
among other things, methyl hydrogen polysiloxane and
20 dimethyl polysiloxane.
A finish composition for application to a continuous
filament polypropylene sheet is disclosed in U.S. Patent
No. 3,766,115. The composition comprises a mixture of two
polysiloxane components, the first of which is a dyeable
25 component comprising a primary or secondary aminoalkyl- or
aminoalkoxyalkylpolysiloxane fluid having an amine function-
ality in the range of 4-7 percent and being substantially
free of other reactive groups. The second component is a
lubricant component comprising a polydialkyl/arylsiloxane
fluid having hydroxy end groups and being substantially
free of other reactive groups. The polypropylene sheet
typically is a spunbonded sheet made from isotactic poly-
propylene.
U.S. Patent No. 3,867,188 relates to a spunbonded
35 nonwoven fabric which is especially useful as a carpet
1 334781
backing. The fabric has on it a silicone-glycol copolymer
having the general formula:
(CH3)3sio{(CH3)2sio}x{(CH3)GSio}ysi(cH3)3
in which G is a radical of the structure -R(C3H6)zOH, R is
an alkylene radical containing from 1 to 18 carbon atoms,
x has an average value of from 40-90, y has an average value
of from 1-10, and z has an average value of from 1-10.
U.S. Patent No. 3,929,509 describes a hydrophilic
microporous film which is useful as a battery separator.
The film comprises a hydrophobic microporous film coated
with a silicone glycol copolymer surfactant, preferably at
a level of from 2 to 20 percent by weight, based on the
uncoated film. In preferred embodiments, the surfactant
coating comprises a mixture of a silicone glycol copolymer
surfactant and a second surfactant which preferably is an
imidazoline tertiary amine. The silicone glycol copolymer
surfactant preferably is a polyoxyethylene polymethyl-
siloxane.
A yarn finish formulation is disclosed in U.S. PatentNo. 4,105,569. In preferred embodiments, the formulation
contains a hydrocarbon-soluble, long molecular chain
polymeric viscosity improver, such as polyisobutylene, and
a polysiloxane. Preferably, the polysiloxane is an alkoxy-
lated polysiloxane, such as a dimethylpolysiloxane with
-substituted polyethylene glycol or polypropylene glycol
side chains or mixed polyethylene/polypropylene glycol
side chains.
U.S. Patent No. 4,563,190 describes a siloxane/oxy-
alkylene copolymer as an optional component of a dyeing
assistant for dyeing or printing polyamide fiber material
with anionic dyes. See also U.S. Patent Nos. 4,444,563 and
4,426,203.
U.S. Patent No. 4,645,691 describes a method for
treating materials with organopolysiloxane compounds. The
1 33478 1
method involves applying to the material a composition
containing a silicone compound which has one or more
alkoxysilylalkyl groups and one or more polyoxyalkylene
groups. The materials to be treated preferably are fibers
and fiber-containing materials.
For a limited review of similar applications of sili-
cones, see A. J. Sabia and R. B. Metzler, Nonwovens Ind.,
14, 16 (1983). Also note British Patent No. 1,273,445
[Chem. Abstr., 76: 89559z (1972)], which describes the use
of a block polysiloxane, among other materials, in the
preparation of a leather substitute.
It may be noted that the above review briefly discusses
polysiloxanes which have been modified by inclusion of a
poly(oxyalkylene) moiety; such modified polysiloxanes can
15 be employed in the composition of the present invention as
an additive.
A modified polysiloxane in which the poly(oxyalkylene)
moiety is a poly(oxypropylene) is described in U.S. Patent
No. 3,867,188. The modified polysiloxane apparently is
20 employed as a lubricant which coats a spunbonded nonwoven
fabric. The fabric, in turn, is employed as a carpet
backing. The addition of the modified polysiloxane to the
backing is stated to reduce damage to the backing which
results from the tufting process used to manufacture the
25 carpet.
Additionally, polysiloxanes have been used in the
- manufacture of films. For example, U.S. Patent No.
4,652,489 describes a sealable, opaque polyolefinic multi-
layer film. The film is composed of a polypropylene base
layer, a nonsealable surface layer, and a sealable surface
layer. The nonsealable layer is a combination of a propyl-
ene homopolymer and a slip agent which preferably is a
polydiorganosiloxane. The polydiorganosiloxane is used in
an amount of from about 0.3 to about 2.5 percent by weight
and preferably comprises a polymethylphenylsiloxane or a
polydimethylsiloxane.
- 1 334781
Finally, several references are known which are or
may be of interest in relation to the additive when it
contains a disubstituted siloxane. Such references are
described below.
Siloxane-oxyalkylene block copolymers are disclosed
in U.S. Patent No. 3,629,308. The copolymers are stated
to be particularly useful as a foam stabilizer in the
production of polyurethane resin foams. The copolymers
are represented by the formula:
R0 R
R3SiO(RSiO)r[R~(ocmH2m)noR li 0 ]p 3
15 in which R is a monovalent hydrocarbon group, R0 is hydrogen
or a monovalent hydrocarbon group, R' is hydrogen or a
monovalent hydrocarbon group, R" is a divalent hydrocarbon
group, r nas a value of at least 0, m is an integer that
has a value of at least 2, n is a number that has a value
20 of at least 1 (preferably at least 4), p is a number that
has a value of at least 1, there are not more than three
hydrogen atoms represented by R0 in the copolymer (prefer-
ably less than one or none), and at least 25 weight-percent
of the groups represented by (OCmH2m) are oxyethylene
25 groups.
U.S. Patent No. 4,150,013 describes melt-processible
-tetrafluoroethylene copolymers containing organopolysilox-
anes which are useful as wire insulation coatings. The
organopolysiloxane is present in an amount of between
30 about 0.2 and 5 percent by weight, based on the weight of
the resulting copolymer composition. Representative
organopolysiloxanes include polyphenylmethylsiloxane,
polydimethylsiloxane, polymethylsiloxane, a copolymer of
phenylmethylsiloxane and dimethylsiloxane, and the like.
- 35 A high viscosity silicone blending process is disclosed
in U.S. Patent No. 4,446,090. The blends produced by the
~ 3~17~11
process are stated to have engineering properties and
flame retardance superior to known blends. The process
involves (a) melting a solid thermoplastic composition
comprising one or more thermoplastic polymers within an
extruder, (b) injecting a high viscosity silicone fluid
into the molten thermoplastic composition within the
extruder, and (c) blending said molten thermoplastic
composition with said high viscosity silicone fluid within
the extruder. The thermoplastic compositions include
polyethylene and polypropylene. The silicone fluid typi-
cally is a polydimethylsiloxane. The blend can contain
such additives as reinforcing fillers, antioxidants,
lubricants, flame retardants, and the like. The additives
can be introduced by means of the thermoplastic polymers,
the silicone fluid, or both. Typical flame retardants
include magnesium stearate, calcium stearate, barium
stearate, antimony oxide, and decabromodiphenyloxide.
Siloxane-containing polymers are described in U. S.
Patent Nos. 4,480,009 and 4,499,149. The properties of
polymeric compositions are stated to be improved by the
presence of a polysiloxane unit having a defined formula.
The listing of polymers, however, does not include polyole-
fins. The disclosed compositions apparently are useful as
protective coatings and as molding, extruding, laminating,
and calendaring compositions. Solutions of the composi-
tions can be used to prepare films and fibers.
- U.S. Patent No. 4,500,659 relates to extrudable,
curable polyorganosiloxane compositions. The compositions
are similar to those of U.S. Patent No. 4,585,830, described
below. In the present case, the compositions comprise (A)
a liquid triorganosiloxy end-blocked polydimethylsiloxane
wherein the triorganosiloxy units are dimethylvinylsiloxy
or methylphenylvinylsiloxy; (B) a reinforcing silica
filler which has been reacted with a liquid or solubilized
treating agent, at least one component of which is a
liquid hydroxy end-blocked polyorganosiloxane wherein at
--10--
-- 1 334781
least S0 percent of the silicon atoms are bonded to a
fluorine-substituted hydrocarbon radical; (C) a liquid
methylhydrogensiloxane having an average of at least three
silicon-bonded hydrogen atoms per molecule; and (D) a
platinum-containing catalyst. The bonded treating agent
for the silica filler would be incompatible, i.e., in-
soluble, with the polydimethylsiloxane component if it
were not bonded to the silica.
Olefin polymer compositions containing silicone
additives are described in U.S. Patent No. 4,535,113.
The compositions apparently can be extruded through rela-
tively narrow die gaps at commercial extrusion rates to
provide films having improved optical and mechanical
properties. The silicone additives have the formula,
(R)(R)(R)Si-o-[si(R)(R)-o]x-[si(R)(Rl)-o]y-si(R)(R)(R)~
in which each R, which can be the same or different, is an
alkyl radical preferably having from one to six carbon
atoms, Rl is a monovalent organic radical containing at
least one ethyleneoxide group, vicinal epoxy group, or
amino group, and x and y, which can be the same or differ-
ent, each have a value of at least 1 and generally have a
value of from about 4 to about 5,000. The silicone addi-
tives typically are present in the compositions in anamount of from about 0.01 to about 5 percent by weight.
-- U.S. Patent No. 4,585,830 describes polyorganosiloxane
compositions useful for preparing unsupported extruded
profiles. Such compositions are stated to include a
triorganosiloxy end-blocked polydiorganosiloxane containing
at least two vinyl radicals per molecule, in which at
least 50 percent of the silicon-bonded organic radicals
are methyl; and an organohydrogensiloxane containing at
least two silicon-bonded hydrogen atoms per molecule, in
35 which said hydrogen atoms are bonded to different silicon
atoms. Examples of such two types of compounds are di-
--11--
1 33478 1
methylvinylsiloxy end-blocked polydimethylsiloxanes and
trimethylsiloxy end-blocked dimethylsiloxane/methylhydro-
gensiloxane copolymers, respectively.
From the foregoing, it is evident that surfactants
5 have been added to polymers to impart a hydrophilic charac-
ter to the surface of the shaped article made from the
polymer. These efforts appear to fall into either of two
categories. In the first category, the surfactant is
compatible with the polymer at melt-extrusion temperatures,
10 in which the shaped article must be bloomed or heated after
formation thereof to bring the surfactant to the surface.
However, the surfactant is incompatible at melt-extrusion
temperatures. In the second, the surfactant moves spon-
taneously to the surface of the shaped article because it
15 is incompatible with the polymer at any temperature. Such
incompatibility at melt-extrusion temperatures prevents
the use of such surfactants in the formation of melt-
extruded fibers because the surfactant prevents the con-
tinuous formation of fibers.
Although surface-segregatable, melt-extrudable thermo-
plastic compositions are a significant advance in the art
of modifying the surface characteristics of fibers prepared
from a thermoplastic polymer, there is a need to overcome
the aforementioned problems associated with the use of
25 such compositions in the formation of nonwoven webs by
such processes as meltblowing, spunbonding, and coforming.
Summary of the Invention
Accordingly, the present invention provides a method
of forming a nonwoven web from a surface-segregatable,
melt-extrudable thermoplastic composition which comprises
at least one thermoplastic polymer and at least one silox-
ane-containing additive having at least two moieties, A
35 and B, which method comprises the steps of:
-12-
1334781
(A) forming fibers by extruding a molten thermoplastic
- composition through a die;
(B) drawing said fibers;
(C) collecting said fibers on a moving foraminous
surface as a web of entangled fibers, which
fibers have less than about 0.35 percent by
weight, based on the weight of said fibers, of
solvent-extractable additive at their interfacial
surfaces and have surface properties charac-
teristic of said at least one thermoplastic
polymer; and
(D) heating said web at a temperature of from about
27 to about 95 C for a period of time sufficient
to provide at least about 0.35 percent by weight,
based on the weight of said fibers, of solvent-
extractable additive at the interfacial surfaces
of the fibers, which fibers- have a surface
property characteristic of said at least one
additive as a consequence of said heating;
in which:
(1) said additive is compatible with said polymer at
melt extrusion temperatures but is incompatible at tempera-
tures below melt extrusion temperatures, but each of said
moiety A and moiety B, if present as separate compounds,
25 would be incompatible with said polymer at melt extrusion
temperatures and at temperatures below melt extrusion
--temperatures;
(2) moiety B has at least one functional group which
imparts to said additive said at least one characteristic;
30(3) the molecular weight of said additive is in the
range of from about 400 to about 10,000; and
(4) said additive is present in said thermoplastic
composition at a level of from about 0.5 to about 2 percent
by weight, based on the weight of said polymer.
35The present invention further provides a method of
forming a nonwoven web from a surface-segregatable, melt-
1 33478 1
extrudable thermoplastic composition which comprises at
least one thermoplastic polymer and at least one siloxane-
containing additive having at least two moieties, A and B,
which method comprises the steps of:
S (A) forming fibers by extruding a molten thermoplastic
composition through a die;
( B) drawing said fibers;
(C) collecting said fibers on a moving foraminous
surface as a web of entangled fibers, which
fibers have at least about 0.35 percent by
weight, based on the weight of said fibers, of
solvent-extractable additive at their interfacial
surfaces and have a surface property charac-
teristic of said at least one additive; and
(D) heating said web at a temperature of from about
27 to about 95~ C for a period of time sufficient
to increase the amount of solvent-extractable
additive at the interfacial surfaces of the
fiber to at least about 0.75 percent by weight,
based on the weight of said fibers;
in which:
(1) said additive is compatible with said polymer at
melt extrusion temperatures but is incompatible at tempera-
tures below melt extrusion temperatures, but each of said
25 moiety A and moiety B, if present as separate compounds,
would be incompatible with said polymer at melt extrusion
temperatures and at temperatures below melt extrusion
temperatures;
(2) moiety B has at least one functional group which
30 imparts to said additive said at least one characteristic;
(3) the molecular weight of said additive is in the
range of from about 400 to about 10,000; and
(4) said additive is present in said thermoplastic
composition at a level of from about 0.5 to about 2 percent
- 35 by weight, based on the weight of said polymer.
1 33478 1
The present invention also provides a method of
forming a nonwoven web comprising the steps of:
(A) forming continuous filaments by extruding a
molten thermoplastic composition through a die;
(B) quenching said continuous filaments to a solid
state;
(C) drawing said filaments;
(D) collecting said continuous filaments on a moving
foraminous surface as a web of entangled fila-
ments; and
(E) passing said web between a pair of compacting
rolls, at least one of which is heated, before
removing said web from said moving foraminous
surface, said compacting rolls applying heat and
pressure to said web sufficient to impart coher-
ency thereto;
wherein said thermoplastic composition comprises a surface-
segregatable, melt-extrudable thermoplastic composition
which comprises at least one thermoplastic polymer and at
least one additive having at least two moieties, A and B,
in which:
(1) said additive is compatible with said polymer at
melt extrusion temperatures but is incompatible at temper-
atures below melt extrusion temperatures, but each of
25 moiety A and moiety B, if present as separate compounds,
would be incompatible with said polymer at melt extrusion
~-temperatures and at temperatures below melt extrusion
temperatures;
(2) moiety B has at least one functional group which
imparts to said additive at least one desired character-
istic;
(3) said additive is a siloxane-containing compound;
(4) the molecular weight of said additive is in the
range of from about 400 to about 10,000; and
(5) the weight ratio of said polymer to said additive
is in the range of from about lO to about lOO.
- ~ 33478 1
In preferred embodiments, moiety A comprises at
least one tetrasubstituted disiloxanylene group, optionally
associated with one or more groups selected from the group
consisting of trisubstituted silyl and trisubstituted
siloxy groups, the substituents of all such groups being
independently selected from the group consisting of mono-
valent alkyl, cycloalkyl, aryl, and heterocyclic groups,
each of which is substituted or unsubstituted, and moiety
B.
In still other preferred embodiments, the additive
contains a plurality of groups selected from the group
consisting of the following general formulae:
(1) Bl-,
(2) B2-O-,
(3) R1-,
(4) R2-Si-,
(5) (R3)(R4)(R5)Si-,
(6) (R6)(R7)(R8)si--~
(7) t-Si(Rg)(R1o)-O-]a~ and
(8) [-Si(Rl1)(B3)--]b;
in which each of R1 and R2 independently is a monovalent
group selected from the group consisting of hydrogen,
alkyl, cycloalkyl, aryl, and heterocyclic groups, each of
which, except for hydrogen, is substituted or unsubsti-
tuted; each of R3-R5, inclusive, independently is a mono-
valent group selected from the group consisting of alkyl,
~-cycloalkyl, aryl, and heterocyclic groups, each of which
is substituted or unsubstituted, and B4; each of R6-Rl1,
inclusive, independently is a monovalent group selected
from the group consisting of alkyl, cycloalkyl, aryl, and
heterocyclic groups, each of which is substituted or
unsubstituted; each of a and b independently represents an
integer from 0 to about 70 which indicates only the quantity
of the respective group present in the additive without
indicating or requiring, in instances when an integer is
greater than 1, that such plurality of the respective
1 ~3~ 7~ 1
group are connected to one another to form an oligomer or
polymer or that all of such groups have identical sub-
stituents; and each of B1-B4, inclusive, independently is
a moiety which imparts to the additive at least one desired
5 characteristic; with the proviso that such plurality of
groups results in at least one tetrasubstituted disilox-
anylene group.
In still other preferred embodiments, the additive is
a compound having the general formula,
lRl2
B5-0-(-Si-O-)C-B6
R13
in which each of R12 and R13 independently is a monovalent
group selected from the group consisting of hydrogen,
alkyl, cycloalkyl, aryl, and heterocyclic groups, each of
which, except for hydrogen, is substituted or unsubsti-
20 tuted; each of B5 and B6 independently is a monovalentgroup having a desired characteristic; and c represents an
integer from 2 to about 70.
In yet other preferred embodiments, the additive is a
compound having the general formula,
IR15 IR17 IRlg lR2 o
R14-Si--(-Si--)d-(-Si-O-)e~li R
Rl6 Rl8 B7 R22
in which each of R14-R22, inclusive, independently is a
monovalent group selected from the group consisting of
hydrogen, alkyl, cycloalkyl, aryl, and heterocyclic groups,
each of which, except for hydrogen, is substituted or
unsubstituted; B7 is a monovalent group having a desired
35 characteristic; d represents an integer from O to about
70; and e represents an integer from 1 to about 70.
-17-
-- 1 33478 1
In yet other preferred embodiments, the additive is a
compound having the general formula,
R24
R23-Sl[(-0-lsl-)f-B8]3
R25
in which each of R23-R25, inclusive, independently is a
10 monovalent group selected from the group consisting of
hydrogen, alkyl, cycloalkyl, aryl, and heterocyclic groups,
each of which, except for hydrogen, is substituted or
unsubstituted; B8 is a monovalent group having a desired
characteristic; and f represents an integer from l to
15 about 70.
The process of the present invention is particularly
useful for the preparation of nonwoven webs, the fibers
of which have at least one surface characteristic which is
different from the surface characteristics of the polymer
20 component of the thermoplastic composition. Such webs, in
turn, are useful in the construction of such disposable
absorbent products as diapers, feminine care products,
incontinence products, and the like.
Brief Description of the Drawings
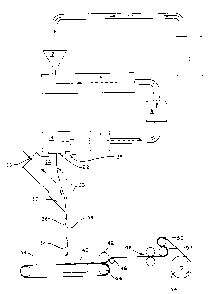
Figure lA is a generalized flow diagram illustrating
one process of the present invention.
Figure lB is a generalized flow diagram illustrating
30 another process of the present invention.
Figure 2 consists of two hand-drawn representations of
photomicrographs of a composition of the present invention,
i.e., the fibers of Example 325, taken through a hot-stage
microscope at two different temperatures and a magnification
of 350X.
Figure 3 consists of two hand-drawn representations of
photomicLoyraphs of the polymer component only of the fibers
-18-
1 33478 1
of Example 325, taken through a hot-stage microscope at two
different temperatures and a magnification of 350X.
Figure 4 consists of two hand-drawn representations of
photomic~Gy~aphs of the composition of Example 38 consisting
of the polymer component of the fibers of Example 325 and
an incompatible silicon-containing compound, taken through
a hot-stage microscope at two different temperatures and a
magnification of 350X.
Figure 5 consists of two hand-drawn representations
of photomicrographs of the composition of Example 43,
taken through a hot-stage microscope at two different
temperatures and a magnification of 350X.
Figure 6 is a diagrammatic representation of a section
of melt-pressed film prepared from a composition of the
present invention, as described in Examples 129-173,
inclusive.
Figure 7 is a diagrammatic representation of a scanning
electron mic~Gy~aph~ using a silicon x-ray probe, of a
sample of the film of Example 169, superimposed on the
diagrammatic representation of Figure 6, which film was
prepared from a composition of the present invention in
which the additive was a silicon-containing compound.
Figure 8 is a plot of silicon concentration in atom
percent versus depth in A below the interfacial surface
for a sample of the film of Example 169, the data for the
plot having been obtained by Rutherford back scattering
-spectrometry.
Figure 9 is a diagrammatic representation of a scanning
electron micrograph, using a silicon x-ray probe, of a
section of the spunbonded nonwoven web of Example 361
prepared from a composition of the present invention, in
which the additive was a silicon-containing compound.
Figures 10 and 11 are plots of silicon concentrations
in atom percent versus depth in A below the interfacial
- 35 surface for the fibers of two spunbonded nonwoven webs
made in accordance with the present invention, in which
--19--
1 33478 1
the additive was a silicon-containing compound, the data
for the plots having been obtained by Rutherford back
scattering spectrometry.
Figure 12 consists of two hand-drawn representations
of photomic~G~aphs of a composition consisting of the
polymer component of the fibers of Example 325 and a
surfactant commonly used in a blooming process to render
polypropylene fibers wettable, taken through a hot-stage
microscope at two different temperatures and a magnification
of 350X.
Detailed Description of the Invention
One method of the present invention applies to any
nonwoven process, e.g., to meltblowing, spunbonding, and
coforming processes. The other method of the present
invention applies only to a spunbonding process. For con-
venience, the two methods will be discussed separately as
the general method and the spunbonding method, respectively.
The nature of the thermoplastic composition employed in
both methods then will be described.
The General Method
In the first step of the general method of the present
invention, fibers are formed by extruding a molten thermo-
plastic composition, hereinafter defined, through a die.Although the nature of the die is not known to be critical,
-it most often will have a plurality of orifices arranged
in one or more rows extending the full machine width.
Such orifices may be circular or noncircular in cross-
section. The fibers extruded may be either continuous ordiscontinuous.
The fibers then are drawn, typically by entraining
them in a fluid stream having a sufficiently high velocity.
When continuous fibers are produced, the fibers first are
cooled in a quenching fluid which usually is low pressure
air. The fluid stream which draws the fibers, usually air,
-20-
1 334781
can be a stream of high velocity air separate from the
quenching fluid, or it can be a portion of the quenching
fluid which is accelerated by passage into a narrow nozzle.
In the production of discontinuous fibers, on the other
s hand, the fluid stream usually is a heated, high velocity
stream of air which draws the fibers while they are in an
at least partially molten or softened state.
The drawn fibers then are collected on a moving
foraminous surface as a web of entangled fibers. The
foraminous surface can be, by way of example only, a
revolving drum or a continuous belt or wire screen; the
latter is most commonly used on commercial-scale equipment.
In some cases, the collected fibers will have at
their interfacial surfaces less than about 0.35 percent by
weight, based on the weight of the fibers, of solvent-
extractable additive. Such fibers typically will have
surface properties characteristic of the at least one
thermoplastic polymer component of the thermoplastic
composition from which the fibers were prepared. When the
collected fibers have at their interfacial surfaces at least
about 0.35 percent by weight, based on the weight of the
fibers, of solvent-extractable additive, the fibers typical-
ly will have surface properties characteristic of the
additive.
As used herein, the term "solvent-extractable additive"
refers to additive which is on or sufficiently close to
the interfacial surfaces of the fibers to be removed by a
mild extraction procedure that does not result in fiber
swelling. An example of such a procedure is soaking or
agitating the fibers in isopropanol for 5-15 minutes. The
amount of additive present in the extract then is readily
determined by known means, such as by either gravimetric
or chromatographic analysis.
Finally, the web of entangled fibers is heated at a
temperature of from about 27 to about 95 C for a period
of time sufficient to cause additional additive to move to
1 334781
the surfaces of the fibers. As a general rule, heating
times of from about 1 to about 30 seconds will accomplish
the desired movement of additive to the surfaces of the
fibers. However, longer or shorter times can be used,
5 depending upon the level of additive in the thermoplastic
composition, the average molecular weight and molecular
weight range (polydispersity) of the additive, and the
desired additive level at the fiber surfaces. Preferably,
heating times of from about 1 to about 5 seconds at a
10 temperature of from about 65 to about 85 C will be employ-
ed.
When the web before the heating step has surface
properties characteristic of the at least one thermoplastic
polymer component of the composition from which the fibers
15 were made, the fiber surfaces typically have at their
interfacial surfaces an amount of solvent-extractable
additive which is less than about 0.35 percent by weight,
based on the weight of fibers. In this context, the
heating step will cause the amount of solvent-extractable
additive at such surfaces to increase to at least about
0.35 percent by weight, which in turn results in the
fibers having a surface property characteristic of the at
least one additive present in the melt-extruded composition.
Preferably, the heating step will increase the amount of
surface-extractable additive at the fiber surfaces to at
least about 0.75 percent by weight, and most preferably to
at least about 1 percent by weight, based on the weight of
the fibers.
On the other hand, when the fibers of the web have,
before the heating step, a surface property characteristic
of the at least one additive present in the composition,
the amount of solvent-extractable additive at the surfaces
of the fibers usually is greater than about 0.35 percent
by weight, based on the weight of fibers. However, such
amount usually is less than about 0.75 percent by weight.
Consequently, the heating step is intended to increase the
-22-
1 334781
amount of solvent-extractable additive at the fiber surfaces
to at least about 0.75 percent by weight and preferably to
at least about 1 percent by weight, based on the weight of
the fibers.
The heating step can be accomplished by any known
means. For example, the web can be irradiated with infrared
or microwave radiation, passed through an oven, or passed
over one or more heated rolls. If heated rolls are used,
such rolls in turn may be heated by any convenient means.
Thus, such rolls can be heated with steam or by a circulat-
ing heated oil or other heat-exchange medium. Alternative-
ly, the surfaces of the rolls can be irradiated with, e.g.,
infrared radiation. In general, heated rolls are preferred
for continuous processes. However, it is not necessary
that the heating step immediately follow the formation of
the web. That is, the web may be formed as described,
then wound up as a roll of fabric and stored or set aside
temporarily. The stored roll of fabric then can be unwound
and subjected to the heating step.
Some aspects of the method of the present invention
are described in more detail in earlier-referenced U.S.
Patent Nos. 3,016,599, 3,704,198, 3,755,527, 3,849,241,
3,341,394, 3,655,862, 3,692,618, 3,705,068, 3,802,817,
3,853,651, 4,064,605, 4,340,563, 4,434,204, 4,100,324,
4,118,531, and 4,663,220.
The general method is further described by reference
to Figure lA which is a generalized flow diagram illustrat-
ing a preferred embodiment. Although Figure lA illustrates
a typical spunbonding process, it should be understood by
those having ordinary skill in the art that meltblowing or
other methods may be used.
In discussing Figure lA, the term "filaments" is used
to emphasize the continuous nature of the fibers produced
3s by the spunbonding process. For the purposes of the present
invention, however, the terms "filaments" and "fibers" are
-23-
,~ .
1 33478 1
,
used synonymously. Thus, the use of either term should
not be construed as in any way limiting the scope of the
present invention.
Turning now to Figure lA, the thermoplastic composi-
tion is fed from supply 10 to hopper 12, then throughextruder 14, filter 16, and metering pump 17 to die head
18 having die face 22 with a plurality of orifices arranged
in one or more rows generally in the cross-machine direc-
tion. As the continuous filaments emerge from die face
22, they form a curtain of filaments 20 directed into
quench chamber 24. In the quench chamber 24, filaments 20
are contacted with air or other cooling fluid through
inlet 26. The quenching fluid is maintained at a tempera-
ture which is lower than the temperature of the filaments
20, typically at ambient temperature, e.g., in the range of
from about 4 to about 55 C. The quenching fluid is
supplied under low pressure, i.e., less than about 12 psi,
and preferably less than about 2 psi, and a portion prefer-
ably is directed through the curtain of filaments 20 and
removed as exhaust through port 28. The proportion of
quenching fluid supplied that is discharged as exhaust
will depend upon the composition being used and the rapidity
of quenching needed to give the desired filament charac-
teristics, such as denier, tenacity, and the like. In
general, the greater the amount of fluid exhausted, the
larger the resulting filament denier and, conversely, the
lower the exhaust fluid ratio, the lower the filament
denier.
As quenching is completed, the curtain of filaments
20 is directed through a smoothly narrowing lower end 30
of the quenching chamber into nozzle 32 where the quenching
fluid attains a velocity of from about 45 to about 245
meters per second. Nozzle 32 extends the full width of
the machine, equivalent to the width of die 22. Nozzle 32
preferably is formed by a stationary wall 34 and a movable
wall 36, both of which also span the width of the machine.
-24-
1 334781
The function of movable wall 36 is described in said U.S.Patent No. 4,340,563.
After exiting nozzle 32, filaments 20 are collected
on a moving foraminous surface such as an endless screen
or belt 38 to form a nonwoven web 40. Before being removed
from belt or screen 38, web 40 is passed under compaction
roll 42, optionally in conjunction with guide roll 46.
Compaction roll 42 conveniently is opposed by the forward
drive and/or support roll 44 for the continuous foraminous
belt or wire screen 38. Compaction roll 42 typically is
not heated, although it could be, if desired. Upon exiting
compaction roll 42, the web is bonded at roll nip 48. The
web then is passed over two steam-heated rolls 51 and 52
having a surface temperature of about 85 C, after which
the web is wound on take-up roll 54. Combined or total
residence times of the web on rolls 51 and 52 typically is
in the range of from about 1 to about 5 seconds, although
longer or shorter times can be used, depending upon the
nature of the additive, the extent to which additive
already is located at the surfaces of the fibers, and the
desired final amount of additive at the fiber surfaces.
Rolls 51 and 52, as already noted, may be heated by any
convenient means (not shown). For example, a heated fluid
may be circulated through them as described in the Examples.
Alternatively, the surface of rolls may be irradiated by
infrared heaters or lamps with appropriate surface temper-
~ature monitors in order to control the surface temperatures
of the rolls.
The Spunbonding Method
In the first step of the spunbonding method of the
present invention, continuous filaments are formed by
extruding a molten thermoplastic composition, hereinafter
defined, through a die. Although the nature of the die is
not known to be critical, it most often will have a plural-
ity of orifices arranged in one or more rows extending the
-25-
1 33478 1
full machine width. Such orifices may be circular or
noncircular in cross-section.
The continuous filaments thus formed then are quenched
by means of a quenching fluid, usually air, which is at a
5 temperature lower than that of the filaments as they
emerge from the die. The purpose of the quenching fluid
is to cool the filaments to a solidified state. Most
often, low pressure air is used.
The solidified filaments are drawn or attenuated,
10 typically by entraining the filaments in a fluid stream
having a sufficiently high velocity. The fluid stream,
which usually also is air, can be a stream of high velocity
air separate from the quenching fluid or a portion of the
quenching fluid which is accelerated by passage into a
15 narrow nozzle.
The drawn continuous filaments then are collected on
a moving foraminous surface as a web of entangled filaments.
The foraminous surface can be, by way of example only, a
revolving drum or a continuous belt or wire screen; the
20 latter is most commonly used on commercial-scale equipment.
Finally, the web of entangled fibers is passed between
a pair of compacting rolls, at least one of which is
heated, before removing the web from the foraminous surface,
said compacting rolls applying heat and pressure to the
25 web sufficient to impart coherency thereto. If desired,
one of the compacting rolls can be the forming drum or the
forward drive and/or support roll for the continuous
foraminous belt or wire screen, as appropriate.
The amount of pressure which is required generally is
30 small, typically in the range of from about S to about 20
psi. Preferably, the pressure applied by the compacting
rolls will be in the range of from about 7 to about 13
psi. These pressure ranges, however, are given by way of
suggestion only because the pressure employed is in part
35 dependent upon the temperature of the at least~one heated
-26-
1 334781
compaction roll. The use of higher temperatures usually
permits lower pressures and vice versa.
The temperature of the at least one heated compaction
roll typically will be in the range of from about 27 to
about 150 C. The preferred temperature range is from
about 27 to about 70 C.
The at least one compaction roll can be heated by any
means known to those having ordinary skill in the art.
For example, a heated fluid may be circulated through the
at least one compaction roll. Alternatively, the surface
of the at least one compaction roll may be irradiated by
infrared heaters or lamps with appropriate surface temper-
ature monitors in order to control the temperature of the
roll.
Some aspects of the method of the present invention
are described in more detail in earlier-referenced U.S.
Patent Nos. 3,692,618 and 4,340,563.
The present invention is further described by reference
to Figure lB which is a generalized flow diagram illustrat-
ing a preferred embodiment of the spunbonding process of
the present invention.
Turning now to Figure lB, the thermoplastic composi-
tion is fed from supply 10 to hopper 12, then through
extruder 14, filter 16, and metering pump 17 to die head
18 having die face 22 with a plurality of orifices arranged
in-one or more rows generally in the cross-machine direc-
tion. As the continuous filaments emerge from die face
22, they form a curtain of filaments 20 directed into
30 quench chamber 24. In the quench chamber 24, filaments 20
are contacted with air or other cooling fluid through
inlet 26. The quenching fluid is maintained at a tempera-
ture which is lower than the temperature of the filaments
20, typically at ambient temperature, e.g., in the range of
35 from about 4 to about 55~ C. The quenching fluid is
supplied under low pressure, i.e., less than about 12 psi,
-27-
~",,~
- 1 33478 1
and preferably less than about 2 psi, and a portion prefer-
ably is directed through the curtain of filaments 20 and
removed as exhaust through port 28. The proportion of
quenching fluid supplied that is discharged as exhaust
5 will depend upon the composition being used and the rapidity
of quenching needed to give the desired filament charac-
teristics, such as denier, tenacity, and the like. In
general, the greater the amount of fluid exhausted, the
larger the resulting filament denier and, conversely, the
10 lower the exhaust fluid ratio, the lower the filament
denier.
As quenching is completed, the curtain of filaments
20 is directed through a smoothly narrowing lower end 30
of the quenching chamber into nozzle 32 where the quenching
15 fluid attains a velocity of from about 45 to about 245
meters per second. Nozzle 32 extends the full width of
the machine, equivalent to the width of die 22. Nozzle 32
preferably is formed by a stationary wall 34 and a movable
wall 36, both of which also span the width of the machine.
20 The function of movable wall 36 is described in said U.S.
Patent No. 4,340,563.
After exiting nozzle 32, filaments 20 are collected on
a moving foraminous surface such as an endless screen or
belt 38 to form a nonwoven web 40. Before being removed
25 from belt or screen 38, web 40 is passed between compaction
rolls 42 and 44, optionally in conjunction with guide roll
46. Compaction roll 44 conveniently is the forward drive
and/or support roll for the continuous foraminous belt or
wire screen 38. Heat and pressure are applied to web 40
30 by means of compaction roll 42. Upon exiting compaction
rolls 42 and 44, web 40 now has sufficient coherency to
permit further processing such as bonding at roll nip 48
and winding at 50.
Compaction roll 42, as already noted, may be heated by
35 any convenient means (not shown). For example, a heated
fluid may be circulated through the at least one compaction
-28-
- 1 33478 1
roll as described in the Examples. Alternatively, the
surface of the at least one compaction roll may be ir-
radiated by infrared heaters or lamps with appropriate
surface temperature monitors in order to control the
5 temperature of the roll.
The Thermoplastic ComDosition
Fibers formed from a thermoplastic composition describ-
ed herein have a differential, increasing concentration
of the additive from the center to the surface thereof,
10 such that the concentration of additive in at least one of
the interfacial surface, effective surface, and subsurface
of the fiber is greater than the average concentration of
additive in the core of the fiber, thereby imparting to
the surface of the fiber at least one desired characteristic
15 which otherwise would not be present.
As used herein, the term "surface" consists of the
interfacial surface and effective surface. The interfacial
surface in essence is the monomolecular layer of the fiber
which is at the air/polymer (or nonfiber/fiber) interface.
20 The effective surface begins at the interfacial surface
and extends into the fiber a distance of about 15 A. The
subsurface lies below the effective surface and extends
into the fiber to a depth of about 1,000 A; thus, the
subsurface has a thickness of about 985 A.
The term "core" has reference to the remainder of the
fiber which is not included in the surface and subsur-
-face, i.e., that portion of the fiber which is below the
subsurface. The term "bulk" refers to all of the fiber,
i.e., the surface, subsurface, and core. The latter term
30 typically is used in reference to elemental analyses of
the fiber.
The surface-segregatable, melt-extrudable thermoplas-
tic composition employed in the present invention compris-
es at least one thermoplastic polymer and at least one
35 additivè.
-29-
1 33478 1
The term "melt-extrudable" is equivalent to "melt-
procecs~hle" and is not intended to be limited in any way.
That is, the term is intended to encompass the use of the
composition in any melt-extrusion process which is or may
5 be employed to prepare fibers, provided the process meets
the limitations imposed by the claims. Thus, the term
includes the use of the composition in melt-spinning of
continuous filaments; meltblowing, spunbonding, and coform-
ing of nonwoven webs; and the like.
In general, the term "thermoplastic polymer" is used
herein to mean any thermoplastic polymer which can be used
for the preparation of filaments (fibers) by melt extrusion.
Examples of thermoplastic polymers include, by way of
illustration only, end-capped polyacetals, such as poly(oxy-
15 methylene)orpolyformaldehyde,poly(trichloroacetaldehyde),
poly(n-valeraldehyde),poly(acetaldehyde),poly(propionalde-
hyde), and the like; acrylic polymers, such as polyacryl-
amide, poly(acrylic acid), poly(methacrylic acid), poly-
( ethyl acrylate), poly(methyl methacrylate), and the like;
20 fluorocarbon polymers, such as poly(tetrafluoroethylene),
perfluorinated ethylene-propylene copolymers, ethylene-
tetrafluoroethylene copolymers, poly(chlorotrifluoroethyl-
ene), ethylene-chlorotrifluoroethylene copolymers, poly-
(vinylidene fluoride), poly(vinyl fluoride), and the like;
25 polyamides, such as poly(6-aminocaproic acid) or poly(~-
caprolactam), poly(hexamethylene adipamide), poly(hexa-
-methylene sebacamide), poly(11-aminoundecanoic acid), and
the like; polyaramides, such as poly(imino-1,3-phenylene-
iminoisophthaloyl) or poly(_-phenylene isophthalamide),
30 and the like; parylenes, such as poly-~-xylylene, poly-
(chloro-~-xylylene), and the like; polyaryl ethers, such
as poly(oxy-2,6-dimethyl-1,4-phenylene) or poly(~-phenylene
oxide), and the like; polyaryl sulfones, such as poly(oxy-
1,4-phenylenesulfonyl-1,4-phenyleneoxy-1,4-phenylene-
isopropylidene-1,4-phenylene),poly(sulfonyl-1,4-phenylene-
oxy-1,4-phenylenesulfonyl-4,4'-biphenylene), and the like;
-30-
1 334781
-
polycarbonates, such as poly(bisphenol A) or poly(carbonyl-
dioxy-1,4-phenyleneisopropylidene-1,4-phenylene), and the
like; polyesters, such as poly(ethylene terephthalate),
poly(tetramethyleneterephthalate),poly(cyclohexylene-1,4-
5 dimethylene terephthalate) or poly(oxymethylene-1,4-cyclo-
hexylenemethyleneoxyterephthaloyl), and the like; polyaryl
sulfides, such as poly(~-phenylene sulfide) or poly(thio-
1,4-phenylene), and the like; polyimides, such as poly-
(pyromellitimido-1,4-phenylene), and the like; polyolefins,
10 such as polyethylene, polypropylene, poly(l-butene),
poly(2-butene), poly(l-pentene), poly(2-pentene), poly(3-
methyl-l-pentene), poly(4-methyl-1-pentene), 1,2-poly-1,3-
butadiene,l,4-poly-1,3-butadiene,polyisoprene,polychloro-
prene, polyacrylonitrile, poly(vinyl acetate), poly(vinyl-
15 idene chloride), polystyrene, and the like; copolymers ofthe foregoing, such as acrylonitrile-butadiene-styrene
(ABS) copolymers, and the like; and the like.
The preferred polymers are polyolefins and polyesters,
with polyolefins being more preferred. Even more preferred
20 are those polyolefins which contain only hydrogen and
carbon atoms and which are prepared by the addition polymer-
ization of one or more unsaturated monomers. Examples of
such polyolefins include, among others, polyethylene,
polypropylene, poly(l-butene), poly(2-butene), poly(1-
25 pentene), poly(2-pentene), poly(3-methyl-1-pentene),
poly(4-methyl-1-pentene), 1,2-poly-1,3-butadiene, 1,4-
pol-y-1,3-butadiene, polyisoprene, polystyrene, and the
like. In addition, such term is meant to include blends
of two or more polyolefins and random and block copolymers
30 prepared from two or more different unsaturated monomers.
Because of their commercial importance, the most preferred
polyolefins are polyethylene and polypropylene.
Broadly stated, the additive must have at least two
moieties, A and B, in which:
(A) said additive is compatible with said polymer at
melt extrusion temperatures but is incompatible at temper-
-31-
1 334781
atures below melt extrusion temperatures, but each of
moiety A and moiety B, if present as separate compounds,
would be incompatible with said polymer at melt extrusion
temperatures and at temperatures below melt extrusion
5 temperatures; and
(B) moiety B has at least one functional group which
imparts to said polymeric material at least one desired
characteristic.
Because the additive is compatible with the polymer
10 at melt extrusion temperatures, the additive is miscible
with the polymer and the polymer and the additive form a
metastable solution. The solution formed by the additive
and the polymer at temperatures above melt extrusion
temperatures is referred to herein as a metastable solution
15 since the solution is not stable at temperatures below
melt extrusion temperatures. As the temperature of the
newly formed fiber drops below melt extrusion temperatures,
the polymer begins to solidify which contributes to additive
separating from the polymer phase. At the same time, the
20 additive becomes less compatible with the polymer. Both
factors contribute to the rapid migration or segregation
of additive toward the surface of the newly formed fiber
which occurs in a controllable manner.
This preferential migration or segregation is control-
25 lable because the extent or degree of migration is, atleast in part, a function of the molecular weight of the
-additive, the shear rate, and the throughput. While the
mechanism of additive migration or segregation is not
fully understood, it appears that the rate of migration or
30 segregation is:
(1) indirectly proportional to the additive molecular
weight - the higher the additive molecular weight, the
slower the rate of segregation;
(2) directly proportional to the shear rate - the
35 higher the shear rate, the faster the rate of segregation;
and
-32-
1 334 78 ~
(3) indirectly proportional to throughput - the
higher the throughput, the slower the rate of segregation.
There are at least three very surprising and unexpected
aspects to the se~re~ation phenomenon. The first is that
5 the additive as defined herein is compatible with the
polymer at melt extrusion temperatures, given the fact
that moieties A and B, if present as separate compounds,
would be incompatible with the polymer at any temperature.
The second is that lower molecular weight additives perform
10 better than higher molecular weight additives; this is
contrary to the conventional wisdom of polymer additives
which favors higher molecular weights. The third and
perhaps most startling aspect is the rapidity with which
such segregation takes place.
As just noted, the effect of additive molecular
weight on the rate of segregation was surprising, especially
in view of past experiences with polydimethylsiloxane.
Upon reflection, it now appears that the movement of lower
molecular weight additives through the gradually solidifying
20 polymer is roughly analogous to the movement of small
particles through a viscous fluid - the larger the parti-
cles, the greater the resistance to movement through the
fluid. This analogy seem appropriate since it has been
demonstrated that the additive exists as small globules in
25 the polymer, which globules become smaller as the tempera-
ture of the molten composition increases. By imposing shear
forces on the molten composition, the globules are broken
down into smaller globules far more quickly than would
have occurred in the absence of shear. Thus, shear is a
30 contributing factor which enhances the segregation of the
additive to the surface of the newly formed filament.
In general, the shear rate will be in the range of from
about 50 to about 30,000 sec~1. Preferably, the shear
rate will be in the range of from about 150 to about 5,000
35 sec~1, and most preferably from about 300 to about 2,000
sec~l .
1 33478 1
It perhaps should be mentioned at this point that the
compatibility requirement is critical. That is, if the
additive is not compatible with the polymer at melt-extru-
sion temperatures, the composition cannot be melt processed
5 to give satisfactory filaments.
By way of clarification, it already has been noted that
com~oullds such as polydimethylsiloxane have been incorpor-
ated in polymers which were extruded, but not melt process-
ed to give fibers. Such compounds migrated to the surface
10 of the extruded article to provide a lubricated surface to
aid further processing or removal from a mold. Because
extrusion times were very slow compared to the melt process-
ing times typically experienced in fiber formation, migra-
tion or segregation rates were not an issue. However, the
incompatibility of the added compounds prevents acceptable
melt-processing because of discontinuities in fiber forma-
tion. In addition, such compounds often reduce friction
within the extruder to the point that the molten mixture
rotates essentially as a plug with no downstream movement
20 taking place.
Finally, throughput is of importance because it
affects the time the newly formed filament is in a suffi-
ciently molten or fluid state to allow migration or segre-
gation of the additive to the newly formed surfaces, even
25 though throughput also affects the shear rate. Stated
differently, it is possible to control the rate of migration
or segregation by controlling the rate of cooling of the
newly formed filament. Thus, for any given molecular weight
additive, the extent of migration can be reduced by rapidly
30 cooling the filament. Alternatively, migration can be
enhanced by reducing the rate of cooling.
Throughput typically will be in the range of from
about 0.01 to about 5.4 kg/cm/hour. Preferably, throughput
will be in the range from about 0.1 to about 4.0 kg/cm.hour.
- 35 The throughput most preferably will be in th~ range of
from about 0.5 to about 2.5 kg/cm/hour.
-34-
1 33478 1
As used herein, the phrase "molten state" does not
nececc~rily mean "flowable". Rather, the term is used to
denote a condition of the thermoplastic composition in
which the additive molecules still are capable of migrating
5 or segregating to the surface of the newly formed filament.
Thus, the term is somewhat imprecise and not readily
subject to accurate measurement. Consequently, this
composition fluidity factor preferentially is described or
accounted for by the term "throughput".
The controlled migration or segregation of additive
toward the surface of the filament results in a controllable
differential concentration of additive in the filament. If
measurable migration is allowed to occur, the concentration
of the additive in the filament will increase with increas-
ing distance from the center thereof. By the proper
selection of additive, additive molecular weight, shear
rate, and throughput (or rate of cooling), a substantial
amount, or perhaps even all, of the additive can be found
in the surface. Because the concentration of additive in
20 the core of the filament typically will vary nonlinearly
from the concentration of the additive in the surface,
this concentration difference is referred to herein as a
differential concentration.
While the additive can be either a liquid or a solid,
25 a liquid is preferred. It also is preferred that a liquid
additive have a surface tension which is less than that of
~virgin polymer; the lower surface tension assures that the
additive will be more likely to completely "wet" or cover
the surface of the filament as the segregation process
30 proceeds to completion, especially under conditions favoring
a large concentration differential.
As already noted, additive surface segregation is
influenced by the molecular weight of the additive. More
specifically, the lower the molecular weight of the addi-
35 tive, the more rapid is the rate of segregation of theadditive to the surface of the filament at any given
- 1 334781
temperature at which the filament still is in a sufficiently
molten state.
It should be apparent that the additive can be monomer-
ic, oligomeric, or polymeric. Indeed, polymeric additives
5 are required in order to achieve the higher additive
molecular weights permitted by the present invention.
Because lower additive molecular weights are preferred,
the preferred additives perhaps are properly referred to
as oligomers. However, such nomenclature can be misleading
10 and reliance instead should be placed on the molecular
weight of the additive and the other parameters already
described. It is for this reason that the additive is not
referred to as a polymeric additive, even though in many
instances the additive will be oligomeric or polymeric in
15 nature.
As already stated, the additive molecular weight will
be in the range of from about 400 to about 10,000. This
range encompasses suitable additive molecular weights,
regardless of whether the additive is to be used by itself
20 or in a mixture of additives; the additive molecular
weight range depends in part on whether or not an additive
will be used by itself.
Accordingly, the molecular weight range for additives
which are to be used individually in compositions for
25 filament formation and not as part of a mixture of additives
typically is from about 400 to about 3,000. Preferably,
-this range is from about 500 to about 2,000, and more
preferably from about 500 to about 1,500. The most prefer-
red range is from about 500 to about 1,000.
When additives are intended to be used in a mixture,
however, higher molecular weights can be employed. Although
the reasons for this are not clearly understood, mixtures
of additives frequently are more compatible with the
polymer at melt-extrusion temperatures than are the in-
35 dividual additives. Although the selection of additive
mixtures is somewhat empirical, in general such mixtures
-36-
1 33478 1
can utilize additives having molecular weights in the
range of from about 400 to about 10,000 and preferably
from about 400 to about 8,000.
In this regard, some clarification of the term "used
sllcceRRfully" is necessary. The successful use of an
additive or a mixture of additives has reference to two
factors. First, the additive or additive mixture must
segregate to the target zone in order to achieve the
intended properties. For example, if water-wettable
filaments are desired, the additive or additive mixture must
segregate to either or both of the interfacial surface and
the effective surface of the filaments. Second, the
composition containing the additive or additive mixture must
process well enough in commercial-scale spunbonding equip-
15 ment to give a web or fabric having the required aestheticand physical properties.
It should be noted that the foregoing molecular weight
ranges are based on the assumption that oligomeric or
polymeric additives will have relatively broad polydisper-
20 sities, e.g., of the order of about 1.2. While narrowpolydispersities certainly are achievable, usually at a
higher cost, they are not necessary, even if relatively
low molecular weight additives are to be employed. As a
guideline, it may be noted that for a given additive, the
25 average molecular weight of an additive having a narrower
polydispersity usually should be slightly lower than the
~-average molecular weight of an additive having a broad
polydispersity. While this guideline is not precise and
is somewhat empirical in nature, one skilled in the art
30 will be able to properly select an additive of any polydis-
persity without undue experimentation.
The term "additive" is used broadly herein to encompass
the use of two or more additives in a given composition.
Such two or more additives may have the same or similar
- 35 moieties B, or different moieties B having the same charac-
teristic, e.g., water wettability. On the other hand, two
-- 1 334781
or more additives may be used which have different charac-
teristics, which characteristics may be related or unre-
lated. Such two or more additives may be present in
similar or significantly different amounts. Moreover, the
5 additives may have the same or similar molecular weights
in order to s6~L6~ate in the filament to approximately the
same region. Alternatively, different molecular weight
additives may be employed in order to effectively layer
the additives in the surface.
The use of different molecular weight additives is
especially attractive for some characteristics which
reinforce each other, an example of which is the use of a
first additive having a moiety B which is an absorber of
ultraviolet radiation and a second additive having a light
15 stabilizing or degradation inhibiting moiety B which
functions by deactivating excited oxygen molecules or
terminating free radicals. The first additive normally
will have a lower molecular weight than the second. While
both additives segregate to the surface, the first additive
20 migrates primarily to the effective surface, while the
second additive migrates primarily to the subsurface.
Thus, actinic radiation which is not absorbed by the first
additive is effectively nullified by the second additive.
The result is a complimentary or even synergistic effect
25 which is greater than that which would be achieved if the
two additives were commingled in the same region.
- The additive is a material which will be referred to
herein loosely as a siloxane. Hence, moiety A will comprise
at least one tetrasubstituted disiloxanylene group, option-
30 ally associated with one or more groups selected from thegroup consisting of trisubstituted silyl and trisubsti-
tuted siloxy groups, the substituents of all such groups
being independently selected from the group consisting of
monovalent alkyl, cycloalkyl, aryl, and heterocyclic
35 groups, each of which is substituted or unsubstituted.
As a practical matter, moiety A often will consist of all
-38-
1 334781
three groups. Moreover, more than one tetrasubstituted
disiloxanylene group often will be present, particularly
when the additive has an appreciable molecular weight.
As used herein, the term "tetrasubstituted disiloxanyl-
5 ene group" means a group having the following general
formula:
R33 IR35
--S i--O--S i--
l l
R34 R36
in which each of R33-R36, inclusive, is a monovalent group
independently selected from the group consisting of alkyl,
cycloalkyl, aryl, and heterocyclic groups.
As noted, the substituents of the groups comprising
moiety A can be alkyl, cycloalkyl, aryl, or heterocyclic
~ou~s which are the same or different and which in turn
are substituted or unsubstituted. Other than the obvious
requirement that such substituents not adversely affect
additive stability or other properties, there are no known
limitations to such substituents. However, for reasons
relating primarily to commercial availability and ease of
synthesis, such substituents preferably are alkyl groups
and more preferably are unsubstituted alkyl groups having
from 1 to 3 carbon atoms. Most preferably, such sub-
stituents are methyl groups.
More specifically, the additive preferably contains a
plurality of groups selected from the group consisting of
the following general formulae, it being understood that
not all groups need to be present and that the presence of
some groups precludes the presence of others:
(1) Bs~~
(2) B6-O-,
- 35 (3) R13-,
(4) R14-Si-
(5) (Rls)(R16)(R17)Si ,-39-
1 33478 1
(6) (R18)(Rl9)(R20)Si--~
(7) ~-si(R2l)(R22)-o-]a~ and
(8) ~-si(R23)(B7)-o-]b;
in which each of R13 and R14 independently is a monovalent
5 group selected from the group consisting of hydrogen,
alkyl, cycloalkyl, aryl, and heterocyclic groups, each of
which, except for hydrogen, is substituted or unsubsti-
tuted; each of R15-R17, inclusive, independently is a mono-
valent group selected from the group consisting of alkyl,
10 cycloalkyl, aryl, and heterocyclic groups, each of which
is substituted or unsubstituted, and B8; each of R18-
R23, inclusive, independently is a monovalent group selected
from the group consisting of alkyl, cycloalkyl, aryl, and
heterocyclic groups, each of which is substituted or
15 unsubstituted; each of a and b independently represents an
integer from 0 to about 70 which indicates only the quantity
of the respective group present in the additive without
indicating or requiring, in instances when an integer is
greater than 1, that such plurality of the respective
20 group are connected to one another to form an oligomer or
polymer or that all of such groups have identical sub-
stituents; and each of B5-B8, inclusive, independently is
a moiety which imparts to the additive at least one desired
characteristic; with the proviso that such plurality of
groups results in at least one tetrasubstituted disiloxanyl-
ene group.
- Molecular weight limitations, if desired, are readily
achieved by limiting the sum of a and b to the extent
required to achieve the desired molecular weight.
In general, the preparation of the siloxane moiety is
well known to those having ordinary skill in the art.
Siloxanes that have reactive groups, such as H-Sia, R0-
sia, and Cl-sia~ are used as starting products. Such
materials are prepared either by hydrolysis of, e.g.,
methylchlorosilanes or by copolymerization of cyclic or
linear polymethylsiloxanes with functional siloxanes.
-40-
1 334 78 ~
See, for example, W. Noll, "Chemistry and Technology of
Silicones," Academic Press, New York, 1968; and R. Meals,
"Encyclopedia of Chemical Technology," Vol. 18, 2nd Edition,
1969, p.221.
Turning now to moiety B, it is this moiety which must
have at least one functional group which imparts to the
additive at least one desired characteristic. Because the
additive rapidly migrates or segregates toward the surface
of the filament upon its formation, it is the presence of
10 moiety B in the surface of the filament which results in
such surface acquiring the at least one characteristic of
moiety B. Such at least one characteristic clearly would
not be found in the surface of the filament in the absence
of the additive. Examples of such characteristics incIude,
15 by way of illustration only and without limitation, wett-
ability by water or other polar solvents, preferential
wettability by alcohols, enhanced hydrophobicity which
contributes to a nonst~ining surface, and stability to
actinic radiation, especially ultraviolet radiation.
It perhaps should be noted at this point that the term
"functional group" refers to that portion of moiety B
which imparts the desired at least one characteristic; the
term is not to be equated to "reactive", although a group
which also is reactive is not precluded by the term "func-
25 tional group".
Moiety B need not be limited to a single desired
~-characteristic. Alternatively, the additive can contain
two or more moieties B which have different characteristics.
For example, a moiety B may have a wettable group and a
30 group which is stable to actinic radiation or a group
which absorbs ultraviolet radiation and a group which
- inhibits actinic radiation-induced degradation, or one
moiety B may have a wettable group while a second moiety B
is stable to actinic radiation.
The point of attachment of moiety B to ~oiety A is
not known to be critical. For example, when moiety A is a
1 33478 1
.
siloxane, moiety B can be a substituent of any one or more
of the tetrasubstituted disiloxanylene, trisubstituted
silyl, and trisubstituted siloxy groups which may be
present.
Those having ordinary skill in the art, upon determin-
ing the characteristic or characteristics desired for any
given additive, will know what functional group or groups
may be required for moiety B. In other words, the selection
of functional groups is well within the abilities and
understanding of one having ordinary skill in the art in
view of the teaching herein. In order to illustrate the
principle involved, though, a preferred embodiment for
moiety B when the desired characteristic is water wett-
ability will be described in detail.
To obtain a filament having a surface which is water
wettable, moiety B preferably is a poly(oxyalkylene)
moiety. More preferably, the alkylene- portion of such
moiety will contain from 2 to about 6 carbon atoms. Most
preferably, moiety B is a poly(oxyalkylene) moiety in
which the oxyalkylene repeating units are oxyethylene or
oxypropylene or a mixture thereof.
References which disclose polysiloxanes containing
one or more poly(oxyalkylene) moieties suitable for use as
the additive include, among others, U.S. Patent Nos.
2,836,748, 2,917,480, 2,991,300, 2,991,301, 3,168,543,
3,172,899, 3,236,252, 3,278,485, 3,280,160, 3,299,113,
3,356,758, 3,402,192, 3,480,583, 3,505,377, 3,509,192,
3,530,159, 3,600,418, and Re. 27,541; Belgian Patent No.
627,281; British Patent Nos. 892,819, 954,041, 963,437,
981,811, 981,812, 1,073,368, and 1,098,646; French Patent
Nos. 1,259,241, 1,356,962, 1,411,757, 1,413,125, 1,482,133,
1,511,661, 1,520,444, and 1,179,743; German Published
Specification (Offenlegungschrift) Nos. 1,495,927,
1,570,656, 1,595,730, 2,045,360, and 2,555,053; German
Patent Nos.1,235,594, 1,257,433, 1,301,576, 1,570,647, and
1,195,953
-42-
1 33478 1
By way of illustration only, three types of additives
for imparting water wettability to the surfaces of fila-
ments, referred to hereinafter as types A, B, and C,
respectively, are described below with reference to the
plurality of preferred groups described earlier. In each
case, moiety B is an oxyalkylene-containing moiety which
is represented by the following general formula:
- (CH2 ) x-- (C2H40) y (C3H60) z~R
in which R26 is a monovalent group selected from the group
consisting of hydrogen and lower alkyl; x represents an
integer from 0 to about 3; and each of y and z independently
represents an integer from o to about 70 which indicates
only the quantity of the respective group present in the
additive without indicating or requiring, in instances
when an integer is greater than 1, that such plurality of
the respective group are connected to one another to form
an oligomer or polymer.
Type A Additives
The first type, which is most preferred, consists of
groups of formulae 1, 2, and 7, in which each of Rg and
R1o independently is an alkyl group containing from 1 to 3
carbon atoms; R26 is an alkyl group containing from 1 to 4
carbon atoms; a is in the range of from 3 to about 60; x
is 0; y is in the range of from about 5 to about 25; and z
.is in the range of from about 0 to about 25.
Specific examples of type A additives, by way of
illustration only, include materials having the following
general formula:
H3
Bg~O~(~Si~o~)g~B9
CH3
-43-
~ 334 78 1
in which Bg is -(C3H6O)htC2H40)i-R27 and R27 is hydrogen or
a lower alkyl group.
Commercially available additives of this type include
*TEGOPREN BC-1781, in which g has an average value of 5.5,
R27 is _-butyl, and the ethylene oxide/propylene oxide
weight percent ratio in Bg is 40/60; TEGOPREN D-985, in
which g has an average value of 4.3, R27 is methyl, and the
ethylene oxide/propylene oxide weight percent ratio in Bg
is 70/30; and TEGOPREN V-337, in which g has an average
10 value of 4, R27 is methyl, and the ethylene oxide/propylene
oxide weight percent ratio in Bg is 100/0.
Type A additives in general are prepared by heating
silicon with, e.g., chloromethane in the presence of a
copper catalyst at about 300~ C to give dichlorodimethyl
silane (see, e.g., U.S. Patent No. 2,380,995 to E. G.
Rochow) which, when reacted with water, gives a polymer
having the following general formula:
CH3 CH3 CH3
Cl-Si-O-(Si-o-)j-Si-Cl,
CH3 CH3 CH3
where j is an integer representing the number of repeating
units in the molecule. See, for example, B. B. Hardman
and A. Torkelson, "Encyclopedia of Chemical Technology,"
3rd Edition, John Wiley & Sons, Inc., New York, 1982, pp.
922-962. The polymer then is reacted in the presence of
trifluoroacetic acid with an oxyalkylene-containing compound
having the general formula,
HO-(c3H6o)y(c2H4o)z-R26
in which R26, y, and z are as already defined, to give the
additive. See U.S. Patent No. 2,836,748. See also U.S.
Patent No. 2,917,480, U.S. Patent No. 3,505,377, and
German Patent No. 1,259,241.
-44-
* - Trade-mark
,~,.
- 1 33478 1
Type B Additives
The second type of additives consists of groups of
formulae 5-8, inclusive, in which each of R3-R11, in-
clusive, independently is an alkyl group containing from 1
to 3 carbon atoms; R26 is an alkyl group containing from 1
to 4 carbon atoms; a is in the range of from about 3 to
about 30; b is in the range of from about 1 to about 10; x
is 3; y is in the range of from about 5 to about 25; and z
is in the range of from about 0 to about 25.
Specific examples of type B additives, also by way of
illustration only, include materials having the following
general formula:
ICH3 ICH3 ICH3 ICH3
H3C-S i-o- ( -s i-o- ) k-(-S i-o- ) l-s i-CH3
CH3 CH3 B1o CH3
in which Blo is ~(cH3)3-o-(c2H4o)m(c3H6o)nR28 and R28 is
hydrogen or a lower alkyl group.
Commercially available examples of this type include
*SILWET L-77, SILWET L-7500, and SILWET L-7602 (Union Car-
bide Corporation, Danbury, Connecticut). Other commer-
cially available examples include TEGOPREN 5843, in which
the k/l value is 13/5, R28 is hydrogen, and the ethylene
oxide/propylene oxide weight percent ratio in Blo is 100/0;
TEGOPRENr 5847, in which the k/l value is 0/1, R28 is
hydrogen, and the ethylene oxide/propylene oxide weight
percent ratio in B1o is 80/20; TEGOPREN 5852, in which
the k/l value is 20/5, R28 is hydrogen, and the ethylene
oxide/propylene oxide weight percent ratio in B1o is 20/80;
TEGOPREN 5863, in which R28 is hydrogen and the ethylene
oxide/propylene oxide weight percent ratio in B1o is 40/60;
TEGOPREN 5873, in which the k/l value is 20/5, R28 is
35 hydrogen, and the ethylene oxide/propylene oxide weight
percent ratio in Blo is 35/65; and TEGOPREN 5878, in which
R28 is hydrogen and the ethylene oxide/propylene oxide
-45-
A * Trade-mark
1 334~8 1
,
weight percent ratio in Blo is 100/0 (Th. Goldschmidt AG,
Essen, Federal Republic of Germany).
The synthesis of the type B additives begins with a
reactive silicon fluid, prepared by known methods, such as
5 that represented by the following formula:
fH3 fH3 fH3 fH3
H3C-Si-o-(-Si-o-) k-(-Si--)l-Si-CH3
CH3 CH3 H CH3
in which k and 1 are as already defined. The fluid is
reacted with a compound having the general formula,
CH2=cHcH2-o-(c2H4o)m(c3H6o)nR28
in which R28, m and n are as already defined, to give the
additive. The reaction is carried out in the presence of
a platinum/r-aluminum oxide catalyst at a temperature of
20 the order of 150 C. See, e.g., U.S. Patent No. 3,280,160,
U.S. Patent No. 3,172,899, and U.S. Patent No.3,505,377.
The compound which is reacted with the silicone fluid is
obtained by the condensation of ethylene oxide and propylene
oxide with allyl alcohol in the presence of a catalytic
25 amount of potassium hydroxide, a well-known reaction.
Type C Additives
The third, and last, type of additives consists of
groups of formulae 2, 4, and 7, in which each of R2, Rg,
and R1o independently is an alkyl group containing from 1
30 to 3 carbon atoms; R26 is an alkyl group containing from 1
to 4 carbon atoms; a is in the range of from 0 to about
50; x is 0; y is in the range of from about 5 to about 25;
and z is in the range of from about 0 to about 25.
Specific examples of type C additives, again by way of
illustration only, include materials having the following
general formula:
-46-
1 33478 1
.
ICH3
R 9-si~(-o-si-)g-(oc2H4)p(oc3H6)q-oR3o]3
CH3
in which R29 and R30 are lower alkyl groups, g is as
already defined, and each of p and q represents an integer
from 0 to about 70.
A specific commercially availabIe example is SILWET
L-720 (Union Carbide Corporation, Danbury, Connecticut).
When the desired characteristic of the additive is
ultraviolet light absorption, moiety B is a chromophore,
especially a chromophore having a sufficiently high
efficiency for the absorption of ultraviolet radiation.
Preferably, moiety B is a benzotriazolyl group, most
preferably a 2-(substituted-phenyl)benzotriazolyl group.
Moiety B is a degradation inhibitor when the desired
characteristic of the additive is light stabilization.
Preferably, such inhibitor contains a piperidyl group. Most
preferably, such inhibitor contains a polyalkyl-substituted
piperidyl group.
When a nonstaining or low surface energy filament is
desired, i.e., a filament having a hydrophobicity which is
higher than that of the virgin polymer component of the
composition, moiety B conveniently can be a perfluoro-
hydrocarbon group, any number of which are known to those
having ordinary skill in the art. Also known to those
having ordinary skill in the art are groups which can be
use as moiety B in order to impart a buffering capacity to
the filament, such as a buffering capacity against hydrogen
ions. In view of the teachings herein, other possible
characteristics of moiety B will be readily apparent.
In general, the weight ratio of polymer to additive
can vary from about 10 to about 100. That-is, the amount
of additive in the surface-segregatable, melt-extrudable
thermoplastic composition of the present invention can
1 334 78 1
range from about 10 percent by weight to about 1 percent
by weight.
The thermoplastic composition can be prepared by any
number of methods known to those having ordinary skill in
the art. For example, the polymer in chip or pellet form
and the additive can be mixed mechanically to coat the
polymer particles with additive. If desired, the additive
can be dissolved in a suitable solvent to aid the coating
process, although the use of a solvent is not preferred.
The coated polymer then can be added to the feed hopper of
the extruder from which the filaments will emerge. Alterna-
tively, the coated polymer can be charged to a heated
compounder, such as a heated twin-screw compounder, in
order to disperse the additive throughout the bulk of the
polymer. The resulting thermoplastic composition typically
is extruded as rods which are fed to a chipper. The
resulting chips then serve as the feed stock for a melt-
processing extruder. In another method, the additive can
be metered into the throat of the hopper which contains
the polymer in particulate form and which feeds the ex-
truder. In yet another method, the additive can be metered
directly into the barrel of the extruder where it is
blended with the molten polymer as the resulting mixture
moves toward the die.
The present invention is further described by the
examples which follow. Such examples, however, are not to
-be construed as limiting in any way either the spirit or
scope of the present invention, especially since the
experimental work concentrated on (but is not limited to)
imparting wettability to polyolefin filaments. In the ex-
amples, all temperatures are in degrees Celsius and all
parts are by weight unless stated otherwise.
.
-48-
1 33478 1
,
Examples
For convenience, the examples are divided into six
sections describing (1) the additives and polymers employed;
(2) the preparation ofsurface-segregatable, melt-extrudable
thermoplastic compositions; (3) the preparation of melt-
pressed films from the thermoplastic compositions; (4) the
preparation of fibers from the thermoplastic compositions;
(5) evaluation of a known material as an additive by way
of comparison; and (6) a hot-stage microscope study of a
composition described in U.S. Patent No. 4,070,218.
I. Descriptions of Additives and Polymers
A. Additives
Each of the additives employed in the examples was a
type A, B, or C additive. The structures imparting water
wettability are identified in Tables 1, 3! and 5 ("MW"
represents molecular weight); if an additive were commer-
cially available, the material designation or catalog
number is given in the column labeled "I.D." and a manufac-
turer code is given in the column labeled "Source". Theproperties of the additives identified in Tables 1, 3,
and 5 are summarized in Tables 2, 4, and 6, respectively.
The structures of additives imparting characteristics other
than water wettability are given in Table 7 and their
properties are summarized in Table 8.
Table 1
Type A Additives Imparting Water Wettability
CH3
30R27-o-(c2H4o)i(c3H6o)h-(-si-o-)g-(c3H6o)h(c2H4o)i R27
CH3
Additive
R27_ q h i MW I.D. Source
A01 CH3 3 0 3 516 V-363 Ga
A02 CH3 3 o 3 516 V-360 G
-49-
1 33478 1
A03 CH3 4 0 3 590 V-361 G
A04 CH3 3 0 4 604. V-336 G
A05 CH3 4 0 4 678 KC-V2b G
A06 CH3 4 0 4 678 V-337 G
A07 CH3 3 1.5 3 690 V-362 G
A08 CH3 4 1 3 706 V-3003 G
A09 CH3 3 1.5 4 778 V-338 G
A10 CH3 4 1 4 794 KC-V3b G
All CH3 4 1.5 4 852 T-3004 G
A12 CH3 4 1.5 4 852 V-339 G
A13 CH3 4 1.5 4 852 V-335 G
A14 CH3 4 0 6 854 KC-V4 G
A15 CH3 4.31.5 5 1023 D-985 G
A16 CH3 5.71.5 5 1127 D-984 G
A17 CH3 4.31.5 7.5 1130 D-979 G
A18 NAc NA 0 NA 1200 PS-071 ucd
Al9 CH3 5.51.5 7.5 1200 D-978 G
A20 B-C4Hg 5.5NA NA 1450 BC-1781 G
A21 NA NA NA NA 2400 PS-555 UC
A22 CH3 6 NA NA NA . V-284 G
A23 NA6 NA NA NA V-290 G
A24 H60 17 16 7922 T-5830 G
aTh. Goldschmidt AG, Essen, Federal Republic of Germany.
bSynthesis utilized a purer polyether.
CNot available.
dUnion Carbide Corporation, Danbury, Connecticut.
Table 2
Properties of the Type A Additives of Table 1
Cloud Surface
Code Viscositya Pointb TensionC
A01 7 NAd 24.9
A02 10 1 24.4
A03 11 1 22.5
A04 16 7 24.2
-50-
1 33478 1
A05 13 <0 23.5
A06 15 2 23.4
A07 18 7 26.0
A08 15 <0 NA
A09 17 4 25.2
A10 24 <0 24.3
All 23 <3 25.2
A12 16 2 22.8
A13 18 2 24.3
A14 22 15 23.9
A15 26 22 NA
A16 31 21 NA
A17 58 45 25.8
A18 20 20 NA
Al9 59 40 24.0
A20 40 0 24.9
A21 320 NA NA
A22 38 4 22.8
A23 44 4 24.3
A24 2400 Te 21.0
aIn centistokes at 25 C.
bIn degrees C, of a 1 percent by weight aqueous
solution.
CIn dynes/cm, + 1.5, of a 1 percent by weight
aqueous solution.
dNot available.
eTurbid
-51-
1 33478 1
Table 3
Type B Additives Imparting Water-Wettability
ICH3 ICH3 ICH3 ICH3
H3c-7i-o-(-7i-o-)k-(-si-o-)l-7i-cH3
CH3 CH3 CH3
(CH3)3-o-(c2H4o)m(c3H6o)nR28
Additive
Code R28- k 1 _m n MW I.D. Source
B01 CH3 NAaNA NA NA 600 L-77 UCb
B02 H 0 1 10 2 836 T-5847Gc
B03 CH3 0 2 10 2 850 T-5878 G
B04 CH3 NA NA NA NA 3000 L-7602UC
B05n-C4Hg NA NA NA NA 3000 L-7500UC
B06 H 18 5 12 0 4724 T-5842 G
B07 H 20 5 3 10 5792 T-5852 G
B08 H 20 5 13 3 5962 T-5851 G
B09 H 18 5 16 2 6184 T-5857 G
B10 H 20 5 8 12 7472 T-5873 G
Bll H 43 5 22 23 15,444 T-5863 G
aNot available.
bUnion Carbide Corporation, Danbury, Connecticut.
CTh. Goldschmidt AG, Essen, Federal Republic of Germany.
Table 4
- Pro~erties of the Ty~e B Additives of Table 3
CloudRefractive Surface
30 Code Viscositya Pointb IndexC Tension
B01 20 10 NAd 2le
B02 100 45 NA 23f
B03 25 Tg 1.446 20f
B04 100 0 NA 22e
B05 175 Ih NA NA
B06 560 80 1.450 30f
-52-
1 334781
B07 290 10 1.444 NA
B08 430 65 1.450 30f
B09 580 84 1.449 28f
B10 440 30 1.449 28f
B11 2700 42 1.450 30f
aIn centistokes at 25 C.
bIn degrees C, of a 1 percent by weight aqueous solution.
cAt 20 C, i 0.005-
dNot available.
eIn dynes/cm, i 1.5, of a 0.1 percent by weight aqueous
solution.
fIn dynes/cm, i 1.5, of a 1 percent by weight aqueous
solution.
gTurbid .
hInsoluble.
Table 5
TY~e C Additive Imparting Water WettabilitY
CH3
R29~sit(~~Si~)g~(OC2H4)p(OC3H6)q~OR30]3
CH3
Add.
25 CodeR29- R30_ q p g MW I.D. Source
C01n-C4Hg NAa NA NA NA 8000 L-720 ucb
aNot available.
bUnion Carbide Corporation, Danbury, Connecticut.
Table 6
Properties of the Type C Additive of Table 3
Cloud Refractive Surface
Code ViscositYa Pointb IndexC Tensiond
C01 1100 42 NAe 29
aIn centistokes at 25 C.
bIn degrees C, of a 1 percent by weight aqueous solution.
-53-
1 33478 1
cAt 20 C, + 0-005-
dIn dynes/cm, + 1.5, of a 0.1 percent by weight aqueous
solution.
eNot available.
Table 7
Additives Imparting Characteristics
Other Than Water Wettability
10 Additive
Code Structure Source
CH3 CH3
W 1a,b (CH3)3Si-(o-si-)4-o-si-o-si(cH3)3 Ex. 1
CH3 (1CH2)3
CHOH
CH2R3 1
CH3 CH3
Do2c,d (CH3)3Si-(o-si-)4-o-si-o-si(cH3)3 Ex. 2
CH3 (1CH2)3
CHOH
CH2R32
CH3
D03e (CH3)3Si-o-(si~-)4-si(cH3)3 Gf
(CH2)3
O
H2
CH-CH2-N[CH(cH3)2]2
OH
1 334781
fH3
D04g (CH3)3Si-o-(si-o-)32-si(cH3)3 ph
ICH2
CH2
I
CF3
CH3
Do5i (CH3)3si-o-(si-o-)l22-si(cH3)3 Pi
CH3
aImparts ultraviolet radiation absorption.
bR31 is 2-(2-hydroxy-3-t-butyl-5-methylphenyl)-2H-benzo-
triazol-5-yl, lithium salt.
CImparts light stabilization by deactivating excited
oxygen molecules or terminating free radicals.
dR32 is poly(N-~-hydroxyethyl-2,2,6,6-tetramethyl-4-
hydroxypiperidyl succinate) covalently coupled through
an ether linkage via the 4-hydroxy group of the terminal
piperidyl moiety.
eImparts buffering capacity against hydrogen ions.
fD-1059, Th. Goldschmidt AG, Essen, Federal Republic of
Germany.
gImparts a low surface energy.
hPS-182, Petrarch Systems, Bristol, Pennsylvania.
- iA control additive which lacks a moiety B.
iPS-042, Petrarch Systems, Bristol, Pennsylvania.
Table 8
Properties of the Additives of Table 7
Refractive Surface
Code Viscositya Indexb TensionC
35 D01 NAd NA NA
D02 NA NA NA
-55-
~ 1 334781
D03 NA NA NA
D04 1,000 1.382 NA
D05 500 1.403 21.1
aIn centistokes at 25 C.
bAt 20 C, + 0.005.
cIn dynes/cm~ + 1-5-
dNot available.
Example 1
Preparation of Additive DO1
A 100-ml, three-necked, round-bottomed flask was
fitted with a pressure-equalized side arm addition funnel,
condenser, and rubber septum. The addition funnel and
15 condenser also were fitted with rubber septa. The flask
was purged continuously with dry nitrogen (Matheson extra
dry grade) which was introduced via a syringe needle
inserted through the rubber septum fitted on one of the
three necks of the flask; the nitrogen exited via another
20 syringe needle inserted through the condenser-mounted
rubber septum. Using a syringe, the flask was charged
with 0.5 g (1.56 mmole) of 2-(2-hydroxy-3-t-butyl-5-methyl-
phenyl)-5-chlorobenzotriazole (*TINUVIN 326, Ciba-Geigy
Corporation, Hawthorne, New York) dissolved in 30 ml of
25 dry tetrahydrofuran (THF)(*Gold Label, 99.9 percent, Aldrich
Chemical Company, Inc., Milwaukee, Wisconsin). The result-
ing solution was cooled in a dry ice/acetone bath to a
temperature of about -78 while being stirred with a
magnetic stirrer. To the cold solution was slowly added
30 dropwise 0.48 g of lithium diisopropylamine (Aldrich
Chemical Company, Inc.) in approximately 5 ml of THF which
had been added via a syringe to the addition funnel. The
resulting mixture was stirred for one hour, after which
time 0.91 g (1.56 mmole) of a compound having the following
formula (TEGOPREN 3010, Th. Goldschmidt AG, Essen, Federal
Republic of Germany), dissolved in about 5 ml of THF, was
-56-
* - Trade-marks
1 33478 1
added dropwise by means of the addition funnel (charged by
syringe injection), over a 20-minute period:
CH3 ICH3
(CH3)3Si- (o-si-) 4-o-si-o-si (CH3)3
CH3 (1CH2)3
HC~
H2 1
The resulting mixture was allowed to warm to ambient
temperature, with stirring. The mixture was allowed to
stir for four hours, after which time the solvent was
removed under reduced pressure by means of a rotating
15 evaporator (*Buchi RotovaprModel RE 120). The residue was
a pale yellow wax. Infrared analysis of the material
showed absorption maxima at 3600 and 3100 cm~1.
Example 2
Preparation of Additive D02
The procedure of Example 1 was repeated, except that
the 2-(2-hydroxy-3-t-butyl-5-methylphenyl)-5-chlorobenzo-
triazole was replaced with 10 g (4 mmole) of poly(N-~-
25 hydroxyethyl-2,2,6,6-tetramethyl-4-hydroxypiperidylsuccin-
ate) having a molecular weight of approximately 2300
(TINW IN 622 LD, Ciba-Geigy Corporation, Ardsley, New
York), the lithium diisopropylamine was replaced with 0.26
g (4 mmole) of butyl lithium (Aldrich Chemical Cbmpany,
Inc.), and the amount of TEGOPREN 3010 was increased to
2. 4 g (4 mmole). The yield of additive was 9.6 g (77
percent).
B. Polymers
The polymers employed are summarized in Table 9 which
is based on data supplied by the manufacturers. In the
table, the melt flow rate is given in the column labeled
-57-
* - Trade-mark
- 1 33478 1
"MFR" and was determined in accordance with ASTM Test
Method D1238-82, "Standard Test Method for Flow Rates of
Thermoplastics by Extrusion Plastometer." The polydisper-
sity, PD, is the ratio of the weight-average molecular
5 weight, Mw, to the number-average molecular weight, Mn.
Table 9
Summary of Polymers Employed
10 Polymer Temp.
Code MFR PD Mn- M~ Ranqea
ppAb 35 2.7 52,000 140,000 293-316
ppBc 400 4.0 17,000 68,000 254-304
ppcd 400 4.0 17,000 68,000 254-304
PPDe 60 4.0 30,000 NAf NA
PPEg NA NA NA NA 204-260
ppFh NA NA NA NA NA
PEAi NA NA NA NA NA
PEBj NA NA NA NA NA
20 psAk NA NA NA NA 245
aDegrees C.
bType PC-973 polypropylene, Himont Incorporated, Wilm-
ington, Delaware.
CType PF-441 polypropylene, Himont Incorporated.
dType PF-015 polypropylene, Himont Incorporated; the
polymer is type PF-441 to which has been added 500 ppm
of Lubrizol 101 (Lubrizol, Inc., Wickliffe, Ohio).
eType PF-444 polypropylene, Himont Incorporated.
fNot available.
gType 5A08 polypropylene, Shell Chemical Co., Houston,
Texas; melt index, 3.0 g/10 min.; and specific gravity,
0.903.
hType WRS-5-144 polypropylene, Shell Chemical Co.,
Houston, Texas.
iType 61800.06 low density polyethylene, Dow Chemical
Co., Midland, Michigan.
-58-
1 33478 1
Type 3404 low density polyethylene, Norchem, Inc.,
Rolling Meadows, Illinois; melt index, 1.8 g/10 min.;
and density, 0.922 g/cm3.
kType PET 7352 poly(ethylene terephthalate), Eastman
Chemical Products, Inc., Kingsport, Tennessee; melt
index, 1.2 g/10 min.; and specific gravity, 1.4.
lRecommended melt processing temperature.
II. Preparation of Compositions
Surface-segregatable thermoplastic, melt-extrudable
compositions as provided by the present invention were
prepared by several methods. However, only those methods
are described below which permitted isolation of the
composition prior to a melt-processing step; i.e., a
bench-scale method and a pilot-scale method. The prepara-
tions of compositions simultaneously with melt-processing
are described in conjunction with such melt-processing.
Examples 3-49
A. Bench-Scale Method
Approximately 10 g of a polymer in pellet form was
mixed in a beaker with the desired amount of additive.
The resulting mixture was poured into the hopper of a
small compounding unit (Max Mixing Extruder, No. CS-194-
FA-093, Custom Scientific Instruments, Inc., New York, New
York). The mixture was heated in the extruder of the
compounder to a temperature of 180 and extruded through a
die having a single, approximately 4-mm diameter, orifice.
The extruded composition was collected either on aluminum
foil or in a glass evaporating dish. The cooled material
was cut manually into approximately 6-mm long pieces.
The compositions prepared are summarized in Table 10.
-59-
1 334781
Table 10
Summary of Bench-Scale Prearations of Comositions
CompositionPolymer Additive(s)
5 Example Code Code Code(s) Wt. Percent
3 PP01-1 PPA A13 2
4 PP02-1 PPA A18
PP03-1 PPA A18 3
6 PP04-1 PPA A20
7 PP05-1 PPA A20 3
8 PS01-1 PSA A20 2
9 PS02-1 PSA A20 5
PP06-1 PPA A21
11 PP07-1 PPA A21 3
12 PE01-1 PEA A21
13 PE02-1 PEA A21 3
14 PS03-1 PSA A23 2
PP08-1 PPA B01
16 PP09-1 PPA B01 2
17 PP10-1 PPA B01 3
18 PE03-1 PEA B01
19 PE04-1 PEA B01 3
PP11-1 PPA B04
21 PP12-1 PPA B04 3
22 PE05-1 PEA B04
23 PE06-1 PEA B04 3
-- 24 PP13-1 PPA B05
PP14-1 PPA B05 3
26 PE07-1 PEA B05
27 PE08-1. PEA B05 3
28 PP15-1 PPA B06 3
29 PP16-1 PPA B09 3
PP17-1 PPA B10 3
31 PP18-1 PPA C01
32 PP19-1 PPA C01 3
33 PE09-1 PEA C01
-60-
1 33478 1
34 PE10-l PEA C01 3
PEll-l PEA D01
36 PE12-1 PEA D01 3
37 PE13-1 PEA D02 3
38 PEl4-1 PEA D03 3
39 PP20-1 PPA D04 3
PP21-1 PPA D05 3
41 PP22-2 PPA B02 1.5
Bll 1.5
42 PP23-2 PPA B06 1.5
B10 1.5
43 PP24-2 PPA B10 1.5
Bll 1.5
44 PP25-3 PPA B04 0.33
B05 0.33
C01 0.33
PP26-3 PPA B04
B05
C01
46 PP27-3 PPA B04 1.67
B05 1.67
C01 1.67
47 PE15-3 PEA B04 0.33
B05 0.33
C01 0.33
48 PE16-3 PEA B04
B05
C01
49 PE17-3 PEA B04 1.67
B05 1.67
C01 1.67
-61-
1 334781
Examples 50-130
B. Pilot-Scale Method
To a weighed amount of polymer, typically from about
5 13 to about 45 kg, in a plastic-lined fiber drum was added
the desired amount of additive. The components then were
mixed mechanically in a paddle mixer (Banbury, Ann Arbor,
Michigan). The hopper of a twin-screw compounding unit
(Egan Machinery Company, Sommerville, New Jersey) was
10 charged with the resulting mixture. The mixture was
gravity-fed to the compounding screws. Compounding was
accomplished at a temperature of from about 180 to about
250, depending on the polymer employed. The resulting
composition was extruded though a die having six orifices
15 with diameters of about 3 mm. The extruded filaments
were passed through a ten-foot water bath and then a
forced-air blower. The dried filaments were pelletized in
a rotary pelletizer (Cumberland Company, New York, New
York) and stored in 23-kg lots in plastic-lined boxes.
20 The resulting compositions are summarized in Table 11. In
some cases, an elemental analysis was carried out on the
composition by Galbraith Laboratories, Inc., Knoxville,
Tennessee. The results of the elemental analyses are
summarized in Table 12.
Table 11
- Summary of Pilot-Scale Pre~arations of Com~ositions
Composition Polymer Additive(s)
30 Example Code Code Code(s) Wt. Percent
PP28-1 PPA A21
51 PP29-1 PPA A21 3
52 PP30-1 PPA A21 5
53 PP31-1 PPA A21 12
54 PE18-1 PEA A21
PEl9-1 PEA A21 3
-62-
- - I 334781
56 PE20-1 PEA A21 5
57 PP32-1 PPA B01 3
58 PP33-1 PPA B01 5
59 PP34-1 PPB B01 3
PP35-1 PPB B01 5
61 PP36-1 PPC B01 3
62 PP37-1 PPC B01 5
63 PE21-1 PEA B01 3
64 PE22-1 PEA B01 5
PP38-1 PPA B02 3
66 PP39-1 PPA B02 5
67 PP40-1 PPC B02 3
68 PP41-1 PPC B02 5
69 PP42-1 PPA B03 3
PP43-1 PPA B03 5
71 PP44-1 PPC B03 3
72 PP45-1 PPC B03 5
73 PP46-1 PPA B04 3
74 PP47-1 PPA B04 5
PE23-1 PEA B04 3
76 PE24-1 PEA B04 5
77 PP48-1 PPA B05 3
78 PP49-1 PPA B05 5
79 PE25-1 PEA B05 3
PE26-1 PEA B05 5
81 PP50-1 PPA B06 3
- 82 PP51-1 PPA B06 5
83 PP52-1 PPC B06 3
84 PP53-1 PPC B06 5
PP54-1 PPA B07 . 3
86 PP55-1 PPA B07 5
87 PP56-1 PPC B07 3
88 PP57-1 PPC B07 5
89 PP58-1 PPA B08 3
PP59-1 PPA B08 - 5
91 PP60-1 PPC B08 3
-63-
1 334781
- 92 PP61-1 PPC B08 5
93 PP62-1 PPA B09 2
94 PP63-1 PPA B09 3
PP64-1 PPA B09 - 5
96 PP65-1 PPC B09 3
97 PP66-1 PPC B09 S
98 PP67-1 PPA B10 3
99 PP68-1 PPA B10 5
100 PP69-1 PPC B10 3
101 PP70-1 PPC B10 5
102 PP71-1 PPA Bll 3
103 PP72-1 PPA Bll 5
104 PP73-1 PPC Bll 3
105 PP74-1 PPC Bll 5
106 PP75-1 PPA C01
107 PP76-1 PPA C01 3
108 PP77-1 PPA C01 5
109 PE27-1 PEA C01
110 PE28-1 PEA C01 3
111 PE29-1 PEA C01 5
112 PP78-1 PPA D03 3
113 PP79-1 PPA D04 3
114 PP80-1 PPA D05 3
115 PP81-2 PPA B02
Bll
116 PP82-2 PPA B02 1.5
Bll 1.5
1-17 PP83-2 PPA B06
B10
118 PP84-2 PPA B06 1.5
B10 1.5
119 PP85-2 PPA B10
Bll
120 PP86-2 PPA B10 . 1.5
` Bll 1.5
-64-
1 334781
121 PP87-3 PPA B06
B09
B10
122 PP88-3 PPA B06
B09
Bll
123 PP89-3 PPA B09 0.67
B10 0.67
Bll 0.67
124 PP90-3 PPA B04 0.33
B05 0.33
C01 0.33
125 PP91-3 PPA B04 0.67
B05 0.67
C01 0.67
126 PP92-3 PPA B04
B05
C01
127 PP93-3 PPA B04 1.67
B05 1.67
C01 1.67
128 PE30-3 PEA B04 0.33
B05 0.33
C01 0.33
129 PE31-3 PEA B04
B05
C01
130 PE32-3 PEA B04 1.67
B05 1.67
C01 1.67
-65-
1 33478 1
Table 12
Elemental Analyses of Selected Compositions
Composition Elemental Analysis
5 Example Code % C % H % Si % F
PP28-1 85.60 13.96 0.23
52 PP30-1 84.28 13.54 0.77
PP38-1 84.36 13.83 0.50
74 PP47-1 84.44 13.50 0.47
78 PP49-1 84.51 13.47 0.36
81 PP50-1 84.90 13.79 0.77
93 PP62-1 83.56 13.39 0.42
98 PP67-1 84.49 13.65 0.47
102 PP71-1 83.86 13.55 0.42
108 PP77-1 84.05 13.58 0.38
112 PP78-1 83.83 13.49 1.06 0.93
121 PP87-3 84.30 13.70 0.45
122 PP88-3 82.70 13.50 0.64
123 PP89-3 84.36 13.74 0.33
124 PP91-3 85.04 13.58 0.27
126 PP92-3 85.11 13.59 0.52
It is evident from the data in Table 12 that each
composition analyzed contained additive. However, the
effectiveness of the additive remained to be demonstrated.
C. Hot-Stage Microscope StudY
-- A hot-stage microscope study was conducted on several
polymer-additive combinations in an effort to gain an
insight into the compatibility aspect of the additive with
the polymer. Although the study actually was done later
in the program, it is reported here for convenience,
except for one part which will be described in Section VI.
Briefly, polymer, either in the form of small granules
or fibers, both with and without additives, was observed
- 35 under a hot-stage microscope at two temperatures-, 160 and
220, at a magnification of 350x. The equipment consisted
-66-
1 33478 1
of a Mettler hot-stage and a Zeiss Universal optical
microscope equipped with transmitted light optics. The
presence of additive globules at either temperature was an
indication of the incompatibility of the additive with the
5 polymer at the temperature of observation. The study was
conducted by Ricerca, Inc., Painesville, Ohio.
The first material studied was the web of Example 327
which was prepared from a composition of the present
invention consisting of polymer PPA and 3 percent by
10 weight of additive All. Figure 2A is a representation of
the photomicrograph at 160 and Figure 2B is a representa-
tion of the photomicrograph at 220. In Figure 2A, additive
globules 21 clearly are present. Also present are what
appear to be a few particles 22 of debris or foreign
15 matter. At 220, as seen in Figure 2B, a few additive
globules 21 seem to be present, but they appear to be
slightly smaller in size. Again, some debris particles 22
are present.
The existence of a large number of additive globules
20 at 160 demonstrates that the additive is incompatible
with the polymer at that temperature. Moreover, the fact
that the number of globules decreases significantly at
220 indicates that additive compatibility with the polymer
has increased substantially. Since melt-extrusion tempera-
25 tures for polymer PPA typically are in the range of fromabout 250 to about 300, the additive clearly will be
compatible with the polymer at melt extrusion temperatures.
The second material consisted of polymer PPA alone as
a negative control. Figures 3A and 3B are representations
of the hot-stage photomic~G~Laphs at 160 and 220, respect-
ively. In Figure 3A, crystallites 31 are seen. While not
apparent from the Figures, such crystallites 31 differ in
appearance and are distinguishable from additive globules,
such as additive globules 21 in Figure 2A. Upon heating
to 220, as shown by Figure 3B, most of the crystallites
31 have disappeared; some debris 32 is present.
-67-
1 334781
As a positive control, composition PP21-1 from Example
40 was studied under the same conditions. Representations
of the photomi~LG~Laphs are shown as Figures 4A and 4B.
In both figures, numerous globules 41 of additive D05 are
5 apparent. Some of such globules apparently have coalesced
at the higher temperature to form droplets 43 (Figure 4B).
At least one debris particle 42 is seen in Figure 4A.
The incompatibility of additive D05 in polymer PPA at
both 160 and 220 is striking, especially when Figure 4B
10 is compared with Figure 2B. Moreover, it is clear that
the additive becomes less compatible with the polymer as
the temperature of the polymer increases.
This discussion of the hot-stage microscope study
concludes with the results obtained with composition PP26-
15 3 from Example 45. That composition, it will be recalled,consists of polymer PPA and a mixture of additives having
molecular weights of 3,000, 3,000, and 8,000, respectively.
The presence of additive globules 51 is seen in Figure 5A
which represents the hot-stage photomicrograph at 160.
20 Such globules appear to be nearly gone at 220 (Figure 5B).
Thus, Figures 5A and 5B are similar to Figures 2A and
2B, respectively, and demonstrate that the additive mixture
changes from incompatible to compatible as the temperature
of the polymer is raised from 160 to 220.
Several other compositions of the present invention
were included in the hot-stage microscope study with
results similar to those shown in Figures 2A, 2B, 5A, and
5B.
From the foregoing, it is apparent that the use of the
30 hot-stage microscope as just described can be used as a
simple method for determining whether or not any given
additive or additive mixture is likely to segregate in a
controlled manner to the surface of a fiber or film as
described herein. If the additive or additive mixture
forms globules which remain at both 160 and 220, the
probability is that such additive or additive mixture
-68-
1 33478 1
will not segregate to one or more of the interfacial
surface, effective surface, and subsurface. In addition,
the melt-processing of a composition incorporating therein
such additive or additive mixture probably will not be
5 successful. On the other hand, if the additive or additive
mixture does not form globules at 160, the additive or
additive mixture is compatible with the polymer at tempera-
tures below melt-extrusion temperatures and probably will
remain distributed throughout the bulk of the resulting
10 fiber or film without any controlled segregation toward the
surface.
III. Pre~aration of Melt-Pressed Films
Examples 131-176
As an initial screening method, films were pressed from
various of the compositions prepared and described in
Section II, above. The apparatus employed was a Carver
Laboratory Press, Model 2518 (Fred S. Carver, Inc., Meno-
20 monee Falls, Wisconsin) having heated plates. From about1 to about 10 g of a composition was placed between two
sheets of aluminum foil and the resulting assembly was
placed on the bottom plate of the press, the plates having
been preheated to about 180. Pressure up to about 10,000
25 psig was applied and maintained for no more than about 5
seconds. The pressure was released and the foil sandwich
was removed from the press. The foil was removed and the
film thus obtained was stored in a plastic bag. Film
thicknesses of from about 1 to about 5 microns typically
30 were obtained. The wettability of each film made with a
type A, B, or C additive was qualitatively estimated by
simply placing a drop of water on the surface and observing
whether or not the drop wet the surface of the film. The
films obtained and the results of the wettability screen
are summarized in Table 13.
-69-
- 1 334781
Table 13
Summary of Melt-Pressed Films Pre~ared
from Compositions Prepared in Section II
Composition
Example ExamDle CodeWettabilit
131 3 PP01-1Positive
132 4 PP02-1Positive
133 5 PP03-1Positive
134 6 PP04-1Positive
135 7 PP05-1Positive
136 8 PS01-1Positive
137 9 PS02-1Positive
138 10 PP06-1Positive
139 11 PP07-1Positive
140 12 PE01-1Positive
141 13 PE02-1Positive
142 14 PS03-1Positive
143 15 PP08-1Positive
144 16 PP09-1Positive
145 17 PP10-1Positive
146 18 PE03-1Positive
147 19 PE04-1Positive
148 20 PPll-lPositive
149 21 PP12-1Positive
150 22 PE05-1Positive
- 151 23 PE06-1Positive
152 24 PP13-1Positive
153 25 PP14-1Positive
154 26 PE07-1Positive
155 27 PE08-1Positive
156 28 PP15-1Positive
157 29 PP16-1Positive
158 30 PP17-1Positive
159 31 PP18-1Positive
160 32 PP19-1Positive
-70-
1 334781
161 33 PE09-1 Positive
162 34 PE10-1 Positive
163 35 PEll-l N/Aa
164 36 PE12-1 N/A
165 37 PE13-1 N/A
166 38 PE14-1 N/A
167 39 PP20-1 N/A
168 40 PP21-1 N/A
169 41 PP22-2 Positive
170 42 PP23-2 Positive
171 43 PP24-2 Positive
172 44 PP25-3 Positive
173 45 PP26-3 Positive
174 46 PP27-3 Positive
175 47 PE15-3 Positive
176 48 PE16-3 Positive
177 49 PE17-3 Positive
aNot applicable, since the additive was not designed
to impart water wettability.
In an effort to obtain some indication of the prefer-
ential segregation of additive(s) to the surface of the
melt-pressed films, a sample of the film of Example 173
was subjected to scanning electron microscopy in conjunction
with a silicon x-ray probe (Si-SEM) in accordance with
standard procedures. The scanning electron microscope was
-manufactured by Cambridge Instruments, Cambridge, England,
and the x-ray probe was manufactured by Princeton Gamma
Tech, Princeton, California.
30The sample of the film of Example 173 is represented
diagrammatically by Figure 6, in which film sample 60 has
top surface 61 and front end surface 62. Figure 7 is the
diagrammatic representation of Figure 6 on which has been
superimposed the results of the Si-SEM. In Figure 7, film
35sample 70 has top surface 71 and front end surface 72.
Each of dots 73 represents the presence of silicon atoms.
-71-
1 334781
.
It is clear that the additives included in the compo-
sition from which the film of Example 1?3 was prepared have
segregated preferentially to the surface region of the
film. The absence of silicon in the core region of the
5 film is striking. The irregular distribution of silicon
along top surface 71 (Figure 7) is believed to have resulted
from the irregularities present in the surf~ce of the top
plate of the press. Such irregularities include the
generally streaked orientation of silicon atoms along
10 surface 71.
Water contact angles were measured for several of the
melt-pressed films. The apparatus employed was an NRL
Goniometer, Model No. 100-00-115 (Ramé-Hart, Inc., Mountain
Lakes, New Jersey. The water used was HPLC Grade water
(Fisher Scientific, Pittsburgh, Pennsylvania). The results
of the measurements are summarized in Table 14.
Table 14
Water Contact Angles for Selected
20Melt-Pressed Films
Film Exam~le Contact Angle.
131 <2
144 <2
25 156 10
157 12
158 10
171 7
Controla 98
30 167 105
168b 115
aFilm pressed from virgin polymer (PPA)
without any additive.
bFilm pressed from the composition consisting
35of polymer PPA and additive D05 as a positive
control.
-72-
1 334~8 1
The presence of either an additive intended to impart
water wettability or an additive intended to increase the
surface energy of the film clearly changed the contact
angle measurement of the film relative to the control film
5 which did not contain additive. Additives of the former
type decreased the contact angle, as expected, and the
additive of the latter type increased the contact angle,
also as expected.
With respect to the two films which contained an
10 additive which absorbed ultraviolet radiation, i.e., the
films of Examples 163 and 164, they showed a broad, strong
absorption band from 220 to 360 nm when analyzed on a
ultraviolet spectrophotometer.
Samples of both films were subjected to electron
spectroscopy for chemical analysis (ESCA). The ESCA data
were collected by Surface Science Laboratories, Inc.,
Mountain View, California, using a Hewlett-Packard 5950 B
spectrometer with a monochromatic aluminum K-alpha x-ray
source. The scans were done with the open aperture setting
for high sensitivity (low resolution). The x-ray power
setting was 600-800 watts and charge neutralization was
accomplished with a flood gun setting of 13 electron
volts. The vacuum utilized was lo~8 Torr. The area
analyzed was about 1 x 4 mm and the sampling depth was
about 100 A.
In addition, each film was subjected to bulk elemental
analysis. The ESCA data and the results of the elemental
analyses are summarized in Table 15.
1 33478 1
.
Table 15
SummarY of ESCA Data and Elemental AnalYses
on Melt-Pressed Films Containinq a W Absorber
ESCA Data Bulk Elemental Analyses
Example % C % O ~ N % Si % C % H % N % Si
163 64 12 12 6 85.30 14.10 0.13 0.26
164 61 11 14 7 85.10 14.37 0.10 0.33
Because ESCA analyses are limited to a depth of about
loo A, two film samples were submitted for analysis by
Rutherford back scattering (RBS) spectrometry. The analyses
were carried out by Charles Evans & Associates, Redwood
City, California. The apparatus employed was a General
Ionics Model 4110 Tandem Accelerator (General Ionics
Corporation, Newburyport, Massachusetts) using an Evans
End Station (Charles Evans & Associates). A 2.275 MeV
He++ ion probe was used, with a detection angle of 160
degrees. Typical beam currents were 1-20 nanoamps. Ions
were detected by surface barrier detectors. Data analysis
involved the TOS source code written by Charles Evans &
Associates and owned by General Ionics Corporation. The
energy losses of the scattered helium nuclei give informa-
tion on the nature and depth of the target atoms in the
polymer matrix. The results are summarized in Table 16.
- Table 16
SummarY of RBS AnalYses on
Melt-Pressed Films
Atomic Concentration Atom %
Example De~th, A C o si Ti
144 0-500 30 0.3 o.og <0.01a
>500 30 0.1 0.03 <0.01a
1 334781
173 0-500 30 1.0 0.56 <O.Ola
- 500-1000 30 0.6 0.15 <O.O1a
>1000 30 0.1 0.04 <O.01a
aThis concentration was at or near the detection
limit; the actual concentration may be considerably
lower.
The RBS data from Table 16 for the film of Example
173 were plotted as the atomic concentration of silicon
10 in atom percent (y-axis) versus depth in A (x-axis); the
plot is shown as Figure 8. In this and all subsequent
plots of RBS data, the silicon concentrations were drawn
parallel to the x-axis as lines which correspond to the
depth field and the midpoints of such lines then were
15 connected to obtain the curve shown in the plot. It is
evident from Figure 8 that most of the additives have
segregated to the interfacial surface, effective surface,
and subsurface of the film. Below a depth of around 1000-
1250 A, the concentration of silicon is very low, i.e.,
20 no more than about 0.04 atom percent.
The films from Examples 144 and 173 also were submitted
for ESCA and bulk elemental analyses. The results of these
analyses are shown in Table 17.
Table 17
Summarv of ESCA Data and Elemental Analyses
-- for the Films of Examples 144 and 172
ESCA Data Bulk Elemental Anal.
Example% C % 0 % Si % C % H % Si
144 94 4.4 1.3 84.21 13.32 0.24
173 62 25 12 85.11 13.59 0.52
.
It is apparent that the ESCA data and the RBS data
cannot be correlated, partly because of the differences
in the depths of measurements and partly because of the
1 3347&1
nonlinear concentration gradient which exists from the
interfacial surface to the core of the film. Taken togeth-
er, however, the data clearly establish the controlled
segregation of additive toward the surface of the film.
The evaluation of the film from Example 165 which
contained additive D02 consisted of an accelerated ultravio-
let radiation exposure trial. A sample of film measuring
3.8 x 10 cm, along with a control film pressed from virgin
polymer, was suspended 0.91 m in front of a 400-watt
mercury arc lamp (Hanovia 674A10). Both films were exposed
continuously for 12 hours. The films then were moved to a
distance of 0.30 m from the lamps and exposed continuously
for an additional 8 hours. Upon examining both films, it
was found that the film of Example 165 appeared to be un-
changed, whereas the control film was brittle and could
not be bent without breaking.
Before evaluating the film of Example 166 which
contained buffering additive D03, the additive itself was
examined for its buffering capabilities. This was done by
charging a 50-ml beaker with 15 ml of deionized water and
a small magnetic stirring bar. The beaker was placed on
top of a magnetic stirrer and fitted with a calibrated pH
electrode. The beaker then was charged with 0.032 g (1
drop) of *TRITON X-102 (Rohm and Haas Co., Philadelphia,
Pennsylvania) and the pH of the resulting solution measured.
To the solution in the beaker then was added 0.032 g (1
-drop) of additive D03, followed by the measurement of the
solution pH. Three additional, equal amounts of additive
D03 were added sequentially, with the solution pH- being
measured after each addition. The results are presented
in Table 18.
-76-
* - Trade-mark
~., "
1 334781
Table 18
SummarY of pH Measurements of
Aqueous Additive D03 Solutions
Solution Composition Solution ~H
Water and 1 drop TRITON 5.50
Water, 1 drop TRITON, 1 drop D036.25
Water, 1 drop TRITON, 2 drops D038.30
Water, 1 drop TRITON, 3 drops D038.72
The solution containing 1 drop of TRITON X-102 and 3
drops of additive D03 (0.096 g) then was titrated with
0.01 N hydrochloric acid. That is, incremental volumes of
hydrochloric acid were added, with the pH of the solution
15 being measured after each addition. The results are
summarized in Table lg, which shows the cumulative volume
of acid added.
Table 19
Titration of Additive D03 Solution
Volume (ml) HCl Added Solution pH
- 8.72
0.2 6.55
0.5 6.91
1.0 6.73
-- 2.0 6.74
3.0 6.70
4.0 6.62
It is clear that additive D03 is capable of acting as
a buffer. The sharp drop in pH with the first addition of
acid was expected, since a buffer system consists of a
weak acid or base and its salt; consequently, buffering
35 behavior could not be seen until acid had been added to
form the salt of additive D03.
1 33478 1
.
Having verified the buffering capability of additive
D03,- the procedure which provided the data for Table 19
was repeated, except that the three aliquots of additive
D03 were replaced with a sample of the film of Example
5 166 weighing 0.211 g and only three 0.5-ml additions of
hydrochloric acid were done. The results are summarized
in Table 20; again, the cumulative volume of acid is shown.
Table 20
Titration of 0.211-g Sample of Film 166
Volume (ml) HCl AddedSolution pH
None (sample absent) 5.71
None (sample present)5.91
0.5 5.90
1.0 5.90
1.5 5.75
The titration of a sample of the film of Example 166
20 was repeated, except that the film sample weighed 0.474 g.
The results are shown in Table 21 which shows the cumulative
volume of acid added.
Table 21
Titration of 0.474-g Sample of Film 166
Volume (ml) HCl AddedSolution PH
None (sample absent) 5.60
None (sample present)6.70
0,5 6.69
1.0 6.69
1.5 6.69
2.0 6.60
2.5 6.40
3.0 4.60
-78-
1 33478`1
Additive D03 not only retains its buffering capability
when incorporated into a composition from which a film is
formed, but also clearly is on the interfacial surface;
otherwise, the additive could not buffer the solution in
5 which the film was placed since the solution could not
swell the film under the conditions of the test.
While the additives clearly segregated to the surfaces
of the melt-pressed films and in general were effective in
imparting to the film surfaces the desired characteristics,
10 the critical test remained to be conducted; namely, the
preparation of melt-processed fibers or films to determine
whether or not additive segregation will occur under the
conditions encountered during fiber and continuous film
formation. Thus, the preparation of fibers is the subject
15 of the next section.
IV. Preparation of Fibers
ExamDles 178-239
A. Meltblown Fibers from Bench-Scale Apparatus
As a simple screening method, fibers were formed by
means of a bench-scale apparatus having a single orifice
in the die tip. The apparatus consisted of a cylindrical
steel reservoir having a capacity of about 15 g. The
25 reservoir was enclosed by an electrically heated steel
jacket. The temperature of the reservoir was thermo-
statically controlled by means of a feedback thermocouple
mounted in the body of the reservoir. The extrusion
orifice had a diameter of 0.016 inch (0.41 mm) and a length
30 of 0.060 inch (1.5 mm). A second thermocouple was mounted
near the die tip. The exterior surface of the die tip was
flush with the reservoir body. Composition extrusion was
accomplished by means of a compressed air piston in the
reservoir. The extruded filament was surrounded and
35 attenuated by a cylindrical air stream exiting a circular
0.075-inch (l.9-mm) gap. Attenuating air pressures typical-
-79-
1 33478 1
ly were of the order of 5-90 psig. The forming distance
was approximately 10 inches (25 cm). The attenuated
extruded filament was collected on the clear plastic film
of an 8.5 x 11 inch (21.6 x 27.9 cm) loose leaf protector
5 having a black paper insert.
In each case, the material extruded consisted of a
simple mixture of a polymer and the desired additive(s) in
the desired amount(s). The mixtures extruded (meltblown)
are summarized in Table 22.
Table 22
Summary of Compositions Meltblown
on Bench-Scale A~aratus
Polymer Additive
Example CodeCode Wt. Percent
178 PPA A01 3
179 PPC A01 3
180 PPA A02 3
181 PPC A02 3
182 PPA A03 3
183 PPC A03 3
184 PPA A04 3
185 PPC A04 3
186 PPA A05 3
187 PPC A05 3
~ 188 PPA A06 3
189 PPC A06 3
190 PPA A07 3
191 PPC A07 3
192 PPA A08 3
193 PPC A08 3
194 PPA A09 3
195 PPC A09 3
196 PPA A10 2
197 PPA A10 3
-80-
,1334781
198 PPC A10 3
199 PPA All 3
200 PPA All 5
201 PPB All 3
202 PPB All 5
203 PPA A12 3
204 PPC A12 3
205 PPA A13 2
206 PPA A13 3
207 PPC A13 3
208 PPA A14 3
209 PPC A14 3
210 PPA A15 2
211 PPA A15 3
212 PPC A15 3
213 PPA A16 3
214 PPC A16 3
215 PPA A17 2
216 PPC A17 3
217 PPA A18 2
218 PPA A18 3
219 PPC A18 3
220 PPA Al9 3
221 PPC Al9 3
222 PPA A20 2
223 PPA A20 3
-. 224 PPB A20 3
225 PPC A20 3
226 PPA A22 3
227 PPC A22 3
228 PPA A24 2
229 PPA . A24 3
230 PP8 A24 3
231 PPC A24 3
- 35 232 PPA B01 2
233 PPA B02 2
-81-
1 33478 1
234 PPA B03 2
235 PPA B04 2
236 PPA B11 2
237 PPA B04 0.33
B05 0.33
C01 0.33
238 PPA B04 0.67
B05 0.67
C01 0.67
- 10 239 PPA B04
B05
C01
Meltblowing conditions for any given composition
depended primarily on the polymer component. Consequently,
standardized conditions were utilized for each of the
three polymers as summarized in Table 23.
Table 23
Summary of Meltblowinq Conditions
Using the Bench-Scale ApParatusa
Polymer Die Air
Code Temp... Temp.,
PPA 260 228
PPB 249 249
- PPC 240 230
aThe conditions given are approximate
only and typically may vary by as
much as + 30.
The wettability of each web was estimated by placing
a drop of water on a sample of the nonwoven material and
measuring the time required for complete penetration of
the water drop into the fabric (referred to hereinafter as
"wetting time"). Each sample was tested with a minimum of
-82-
1 334781
five drops of water placed in five different locations.
If all of the drops wet the web within three seconds, the
web was considered to be immediately wettable (i.e.,
wettable). If the wetting times of the drops were greater
5 than three seconds and equal to or less than 30 seconds,
the web was considered to be slowly wettable. If wetting
times were greater than 30 seconds, the web was considered
to be nonwettable.
Of the webs obtained in Examples 178-239, inclusive,
10 those from Examples 178-227, 232-234, and 237-239, in-
clusive, were immediately wettable, although in some cases
wettability was dependent upon fiber diameter. Those from
Examples 228-231, inclusive, 235, and 236 were nonwettable.
It is seen from Table 16 that Examples 228-231 employed
15 additive A24, Example 235 employed additive B04, and
Example 236 employed additive Bll. According to Table 1,
additive A24 has a molecular weight of about 7,900. From
Table 3, it is seen that additive B04 has a molecular
weight of about 3,000 and additive Bll has a molecular
20 weight of about 15,000. All three molecular weights are
high enough to prevent the rapid segregation of the additive
to the effective and/or interfacial surface region of the
fibers. Consequently, the fibers were not wettable.
It should be noted, however, that webs made from a
25 composition containing a mixture of additives having
molecular weights equal to or greater than about 3,000,
-i.e., the webs of Examples 237-239, inclusive, were wett-
able, while webs made from a composition containing any
one of the additives used in the mixture were not wettable
(i.e., the web of Example 235). This illustrates the
apparent synergistic effect which can result from combining
additives, even though such additives individually do not
segregate under similar melt-processing conditions above the
subsurface of the fibers or films.
-83-
1 334781
Some qualitative observations on web quality and
wettability as a function of fiber diameter are appropriate
at this point, at least for webs made with polymer PPA.
Web quality was based on visual inspection or inspec-
tion under a low-power optical microscope and was rated on
a scale of from 1 to 4 as follows:
4 - fibers having uniform diameters with no shot
present;
3 - fibers having a small amount of fiber diameter
nonuniformity, with small amounts of shot present (fiber
diameter nonuniformity refers to variations of fiber
diameter, i.e., the presence of varying large and small
fiber diameters);
2 - moderate fiber nonuniformity and a moderate
amount of shot present; and
1 - substantial fiber nonuniformity and a large
amount of shot present.
Fiber diameters also were estimated visually or under
the microscope and were simply classed as small, medium,
or large. As will be described in greater detail later,
fiber diameter is a function of attenuating air pressure-
the higher the pressure, the smaller the fiber diameters.
A number of the webs obtained in Examples 178-239,
inclusive, were evaluated for web quality and fiber diame-
ter. The results of this evaluation and the wettabilities
of the webs evaluated are summarized in Table 24.
Table 24
Summary of Evaluations of
30Web Oualitv and Fiber Diameters
Additive Cloud Primary Web
Code MW Pointa AirbRating WettabilityC
A06 678 2 25-90 4 WS, WM, WL
35 All 852 3 25-90 4 WS, WM, WL
A13 852 2 25-90 4 WS, WM, WL
-84-
- 1 334781
A17 1130 45 27 1 WL
A19 1200 40 30 1 WL
A20 1450 0 26-90 4 WS, WM, WL
A22 NAd 4 25-85 4 WS, WM, WL
A23 NA 4 25-90 4 WS, WM, WL
B01 600 10 30-90 4 WS, WM, WL
B04 3000 0 30-80 4 WL
B05 3000 Ie 25 1 Nonwettablef
B07 5792 10 25-45 3 WL
B08 5962 65 25 1 SlowlyWett.f
B11 15,444 42 25 1 Nonwettablef
C01 8000 42 25 2 Nonwettablef
aIn degrees C.
bIn psig.
CCode: WS = small diameter fibers wettable;
WM = medium diameter fibers wettable: and
WL = large diameter fibers wettable.
dNot available.
eInsoluble.
fOnly large fibers were produced.
The data in Table 24 substantiate the already-observed
decrease in wettability associated with increasing additive
molecular weight. In addition, however, the data suggest
that there is a correlation between web quality and additive
cloud point. That is, when the cloud point of the additive
- is above about 20O C, web quality declines significantly.
Thus, the cloud point of additives employed to impart
water wettability to the surface of fibers or films prefer-
ably will be no more than about 20 C and most preferablyno more than about 10 C.
Examples 240-261
In order to more fully understand the segregation
phenomenon, three series of the bench-scale meltblowing
-85-
1 334781
experiments were repeated under somewhat more carefully
controlled conditions. The first series employed either
polymer PPA or PPB and additive levels of two percent by
weight; the process and product details are summarized in
Table 25. Fiber diameters were estimated from scanning
electron photomicrographs taken by Surface Science Labora-
tories, Inc., Mountain View, California. The instrument
employed was a Camscan Series 4 Scanning Electron Micro-
scope. The accelerating voltage was 24 keV, the working
distance was 20 mm, and the spot size was 5. The instrument
was calibrated with 0.76-micron diameter National Bureau
of Standards latex spheres. Each sample was gold coated
(loo-A thickness) to increase conductivity under the
electron beam.
Table 25
Summary of First Series of Additional
Bench-Scale Meltblowing Experiments
Additive Air Fiber
Examplea Code MW Press.b Dia.C
240 B01 600 40 15
241 B01 600 80 3
242 B02 836 20 12
243 B02 836 80 3
244 B03 850 40 12
- 245 B03 850 80 4
246 A13 852 35 12
247 Al3 852 80 4
248 B04 3000 25 12
249 B04 3000 40 5
250d B04 3000 12 20
B05 3000
C01 8000
-86-
1 33478~
251d B04 3000 20 6
B05 3000
C01 - 8000
252d B04 3000 25 5
B05 3000
C01 8000
253d B04 3000 40 2-3
B05 3000
C01 8000
aPolymer PPA was employed in every case, except for
Examples 250-253, inclusive, which utilized polymer
PPB.
bIn psig.
CIn micrometers.
dThe polymer contained a mixture of all three addi-
tives in equal concentrations; the total of all
three additives still was two percent by weight.
In each case, a coherent web was obtained. Each web
was subjected to ESCA analysis. Additionally, each web
was subjected to bulk elemental analysis and the water
drop test. The ESCA data and the results of the elemental
analyses and water drop tests are summarized in Table 26.
Table 26
Summary of Analytical Data And Water DroP Test
-- for Webs from Experiments 240-253. Inclusive
Additive Fiber ESCA Bulk
30 Exam~le MW Dia a sib SicWettabilitY
240 600 15 1.80.006Wettable
241 600 3 2.00.007Wettable
242 836 12 1.90.017Wettable
243 836 3 1.50.018Wettable
244 850 12 2.60.008Wettable
245 850 4 1.70.009Wettable
-87-
1 334781
246 852 12 4.3 0.011Wettable
247 852 4 4.5 0.011Wettable
248 3000 12 13.0 0.017Nonwettable
249 3000 5 6.3 0.016Nonwettable
2503-8 x 103d 20 8.5 0.010Wettable
2513-8 x 103d 6 5.8 0.010Slowly Wett.
2523-8 x 103d 5 5.9 0.010Slowly Wett.
2533-8 x 103d 2-3 4.8 0.010Slowly Wett.
aIn micrometers.
bAverage concentration in atom-percent to a depth of
approximately 100 A.
CAverage concentration in atom-percent throughout the
bulk of the fibers.
dThe polymer contained three additives having molecular
weights of 3,000, 3,000, and 8,000, respectively.
From Table 26, it is seen that only two webs were not
wettable; both webs were made with additive B04 which has
a molecular weight of about 3,000. Interestingly, the
20 fibers of both webs had higher bulk silicon concentrations
and higher surface silicon concentrations than any of the
webs which were wettable. Indeed, the fibers of the web
from Example 248 had from three to nine times as much
silicon in the top loo-A layer of the surface as the
fibers of webs which were wettable. Notwithstanding such
high concentrations, it is evident that there was insuffi-
--cient additive in the effective surface to render the webs
wettable. Thus, while the higher molecular weight additives
will segregate to some extent, additive molecular weights
30 of less than about 3,000 are required in order for additive
to migrate to the interfacial surface or effective surface
in concentrations sufficient ~o impart wettability to the
fibers, at least for fibers having diameters in the 3-15
micrometer range.
- 35 In order to demonstrate the effect of fiber diameter
on surface silicon concentration, a second series of
-88-
~ 3 ~ 3 ~
bench-scale meltblowing experiments was carried out. In
this series, the polymer was PPB and the additive was A10
at a level of two percent by weight (the additive molecular
weight is 794 - see Table 1). ESCA analyses were carried
5 out on the webs, all of which were wettable. The results
are summarized in Table 27.
Table 27
Summary of Second Series of Additional
Bench-Scale Meltblowing Ex~eriments
Air Fiber ESCA DataC
Example Press.a Dia.b % C ~ Si
254 40 6 84 4.7
255 50 4 87 4.1
256 60 2 88 3.9
aIn psig.
bIn micrometers, estimated from scanning electron
photomi~LG~Laphs as already described.
CAverage concentration in atom-percent to a depth
of approximately 100 A; the bulk silicon concen-
tration as determined by elemental analysis was
0.01 atom-percent.
From the discussion earlier regarding the factors
influencing the segregation of the additive, it is apparent
-that there are two competing factors in the segregation of
additive during fiber formation. First, as the diameter
of the fiber is diminished, the distance to the surface
30 also is diminished, thereby contributing to higher additive
concentrations in the surface region. Second, as the
diameter of the fiber is diminished, the time the fiber
remains in a molten state also is diminished, thereby
shortening the time during which the additive can migrate
35 toward the surface. From the data in Table-27, it is
evident that the second factor was controlling since
-89-
1 334781
the additive concentration was reduced as the fiber diameter
decreased.
As already pointed out, the higher molecular weight
additives segregate toward the surface of the fiber or
5 film, but typically do not reach either the interfacial
surface or the effective surface. In cases where the
additive has segregated to the subsurface and is sufficient-
ly close to the effective surface, the additive can be
"coaxed" to the effective surface by the application of
10 relatively mild heating conditions. This phenomenon is
illustrated by a third series of bench-scale meltblowing
experiments.
The third series of experiments involved the incorpor-
ation of two weight percent of an additive in PPA polymer
15 essentially as described in Examples 178-239, inclusive.
An ESCA and elemental analysis was obtained for each web.
The wettability of each web also was estimated by the
water drop test. A sample of each web then was heated in
an oven at 120 degrees for 20 seconds. An ESCA analysis
20 was obtained on the heated web and its wettability estimated
as before. The results are summarized in Tables 28 and 29.
Table 28
SummarY of Third Series of Additional
25Bench-Scale Meltblowing Experiments
- Additive Bulk
Example Code MW % Sia
257 A15 1023 0.005
258 A18 1200 0.014
259 A20 1450 0.014
260 A23 NAb 0.008
261 Bll 15,444 0.006
aAverage concentration in atom-percent throughout
the bulk of the fibers.
bNot available.
--90--
1 33~781
Table 29
- SummarY of ESCA Data and Wettability Testing
for Third Series of Bench-Scale Meltblowing
ExPeriments Before and After Heatinq the Webs
s
Before Heating After Heating
Example % Sia Wettability % Sia Wettability
257 3.2Nonwettable 5.8Slowly Wett.
258 1.9Nonwettable 2.7Wettable
259 6.9Wettable 7.4Wettable
260 4.3Nonwettable 3.3Nonwettable
261 4.7Nonwettable 5.3Nonwettable
aAverage concentration in atom-percent to a depth of
approximately 100 A.
While the heat treatment did not convert every nonwett-
able web into a wettable one, the procedure was successful
for the two lowest molecular weight additives. Whether or
not such treatment can be used depends, at least in part,
on whether or not the additive has segregated to the
subsurface sufficiently close to the effective surface to
permit a gentle heat treatment to move the material into
the effective surface region. Such segregation in turn is
in part dependent upon the diameter of the fibers, i.e.,
25 the time the fibers remain in a molten state. Thus, the
choice of additive and heat treatment conditions is, of
-necessity, somewhat empirical.
The ability of additive to be moved from the subsurface
to either the effective surface or the interfacial surface,
or both, expands the types of products based on nonwoven
webs prepared in accordance with the present invention. A
few examples in the area of household and industrial wipes
will serve by way of illustration:
(1) a wipe consisting of a single polyolefin nonwoven
web prepared in accordance with the present invention, in
which additive is present in either or both of the effective
--91--
1 334781
surfaces and the interfacial surfaces of the fibers - the
wipe is hydrophilic or water wettable and is suited for
washing or cleaning tasks using aqueous cleaning solutions;
(2) a wipe consisting of a single polyolefin nonwoven
5 web prepared in accordance with the present invention, in
which additive is present in the subsurface of the fibers-
the web is hydrophobic or oleophilic and is suited for
cleaning oily surfaces, but on washing the wipe is converted
to a hydrophilic wipe because the heat of the washing or
10 drying environment causes additive to migrate from the
fiber subsurface to either or both of the fiber effective
surface and interfacial surface, which conversion aids in
the removal of oily residues from the wipe; and
(3) a wipe consisting of two polyolefin nonwoven
15 layers, one prepared from virgin polymer and the other
consisting of a web as described in either (1) or (2)
above - in the first instance, the wipe will be effective
for both water-soluble or water dispersible substances and
oily substances, depending on which layer is used as the
20 wiping layer, and in the second instance, the wipe can be
converted to a wipe of the first instance by laundering.
B. Meltblown Fibers from Pilot-Scale ApParatus
ExamPles 262-297
Since the above bench-scale meltblowing experiments in
general were successful, meltblowing trials were conducted
on a pilot-scale meltblowing apparatus essentially as
described in U.S. Patent No. 4,663,220. Briefly, such
30 meltblowing was accomplished by extruding a composition
(or a simple mixture) through a 0.75-inch (19mm) diameter
- Brabender extruder and then through a meltblowing die
having nine extrusion capillaries per linear inch
(approximately 3.5 capillaries per linear cm) of die tip.
Each capillary had a diameter of about 0.0145 inch (about
0.37 mm) and a
-92-
- 1 334781
length of about 0.113 inch (about 2.9 mm). The process
variables in general were as follows:
polymer extrusion rate, 2.5-3.5 g per capillary
per minute;
polymerextrusiontemperature,250-300, depending
upon the polymer employed;
extrusion pressure, 490-510 psig;
die tip temperature, 270-275;
attenuating air temperature, 304-310;
attenuating air pressure, 8-11 psig; and
forming distance, 20-40 cm.
The collecting arrangement consisted of a rotating
15.2-cm wide drum having a diameter of 76.2 cm. The
surface of the drum was a screen.
The polymer and additive typically were mixed by one
of several methods before introducing the mixture to the
feed hopper of the extruder. In the first (method A), a
standard portable cement mixer was charged with 50 pounds
of the polymer in pellet form. The mixer then was started
and charged with the desired amount of additive. Mixing
was allowed to continue for 20 minutes, after which time
the mixture was removed from the mixer and stored in
plastic-lined boxes. In a variation of that method, the
additive was used in an amount higher than that intended
for melt-processing to give a stock mixture. The stock
mixture then was mixed in a similar fashion with additional
~polymer in a ratio calculated to give the desired final
additive concentration (method B). In the third (method
C), a metered stream of additive was pumped into the feed
hopper about 15 cm above the feed screws as polymer pellets
flowed downward by gravity into the screws. All three
methods worked equally well, although method C was used
with only one additive.
In each case, a coherent web was obtained which had a
- 35 basis weight in the range of from about 20 to about 50
g/m2. Wettability was estimated by means of the water
-93-
1 334781
drop test. The trials are summarized in Table 30, along
with the results of the water drop test.
Table 30
SummarY of Pilot-Scale Meltblowing Trials
PolYmer Additive
Exam~le Code Code Wt. % Wettability
262 PPA All 2 Wettable
263 PPA All 3 Wettable
264 PPA All S Wettable
265 PPB All 2 Wettable
266 PPB All 3 Wettable
267 PPB All 5 Wettable
268 PPA A18 1 Wettable
269 PPA A18 3 Wettable
270 PPA A18 5 Wettable
271 PPB A18 1 Wettable
272 PPB A18 3 Wettable
273 PPB A18 5 .Wettable
274 PPA A21 1 Wettable
275 PPA A21 3 Wettable
276 PPA A21 5 Wettable
277 PPC A21 1 Wettable
278 PPC A21 3 Wettable
279 PPC A21 5 Wettable
- 280 PPA B01 1 Wettable
281 PPA B01 3 Wettable
282 PPA B01 5 Wettable
283 PPB B01 1 Wettable
284 PPB B01 3 Wettable
285 PPB B01 5 Wettable
286 PPC B01 1 Wettable
287 PPC B01 3 Wettable
288 PPC B01 5 Wettable
289 PPA B04 1 Nonwettable
-94-
1 3347~i
290 PPA B04 3 Nonwettable
291 PPA B04 5 Nonwettable
292 PPA B05 1 Nonwettable
293 PPA B05 3 Nonwettable
294 PPA B05 5 Nonwettable
295 PPA C01 1 Nonwettable
296 PPA C01 3 Nonwettable
297 PPA C01 5 Nonwettable
The results obtained are consistent with the bench-
scale meltblowing experiments. Single additives having
molecular weights of the order of 3,000 or higher do not
segregate to the interfacial surface or effective surface
when fiber diameters are relatively small, as they are in
typical meltblowing processes.
C. Spunbonded Fibers from Pilot-Scale APparatus
Examples 298-365
Spunbonded trials were conducted on a pilot-scale
apparatus essentially as described in U.S. Patent No.
4,360,563.
The polymer and additive typically were mixed by one
of the methods described above with respect to Examples
262-297, inclusive.
In each case, a web was obtained which had a basis
- weight in the range of from about 14 to about 60 g/m2. In
some cases, webs of different basis weights were made
during a trial by changing the velocity of the forming
wire. Typical basis weights thus prepared were 14, 19,
36, 47, and 59 g/m2. Wettability was estimated by means
of the water drop test.
Unlike the meltblown trials, however, it was dis-
covered that when the additive level was greater than 1
percent by weight, there was no web integrity;- that is,
the web simply fell apart upon attempting to remove it
-95-
1 33478 1
.
from the forming wire, even when excellent fiber formation
was obtained. The problem was overcome by running the web
under a heated compaction roll before removing it from the
forming wire. Thus, all of the spunbonded examples in
which additive levels were greater than 1 percent by
weight utilized a heated compaction roll. While a compac-
tion roll temperature of about 66 was employed, lower or
higher temperatures can be used.
The trials are summarized in Table 31, along with the
results of the water drop test; because wettability was
independent of web basis weight, the latter is not included
in the table.
Table 31
Summary of Pilot-Scale S~unbonding Trials
Polymer Additive
Example Code Code Wt. % Wettability
298 PPA A05 1 Wettable
299 PPA A05 3 Wettable
300 PPC A05 1 Wettable
301 PPC A05 3 Wettable
302 PPD A05 1 Wettable
303 PPD A05 3 Wettable
304 PPA A080.75 Wettable
305 PPA A08 1 Wettable
~306 PPA A08 3 Wettable
307 PPD A080.75 Wettable
308 PPD A08 1 Wettable
309 PPD A08 3 Wettable
310 PPE A08 1 Wettable
311 PPE A08 - 3 Wettable
312 PPA A10 0.5Slowly Wett.
313 PPA A100.75 Wettable
314 PPA A10 1 Wettable
315 PPA A10 1.5 Wettable
-96-
1 33478 1
316 PPA A10 2 Wettable
317 PPA A10 3 Wettable
318 PPE A10 0.5Slowly Wett.
319 PPE A100.75Wettable
320 PPE A10 1 Wettable
321 PPE A10 1.5Wettable
322 PPE A10 2 Wettable
323 PPE A10 3 Wettable
324 PPE All 0.5Slowly Wett.
325 PPE All0.75Wettable
326 PPE All 1 Wettable
327 PPE All 1.5Wettable
328 PPA All 2 Wettable
329 PPA A11 3 Wettable
330 PPD All 0.5Slowly Wett.
331 PPD All0.75Wettable
332 PPD A11 1 Wettable
333 PPD A11 1.5Wettable
334 PPD All 2 Wettable
335 PPD All 3 Wettable
336 PPE All 0.5Slowly Wett.
337 PPE All0.75Wettable
338 PPE A11 1 Wettable
339 PPE A11 1.5Wettable
340 PPE A11 2 Wettable
341 PPE A11 3 Wettable
-- 342 PPA A14 1 Wettable
343 PPA A14 3 Wettable
344 PPD A14 1 Wettable
345 PPD A14 3 Wettable
346 PPA B01 1 Wettable
347 PPA B01 3 Wettable
348 PPA B01 5 Wettable
349 PPD B01 0.5Wettable
- 35 350 PPD B01 1 Wettable
351 PPD B01 2 Wettable
-97-
1 334781
352 PPD B01 3 Wettable
353 PPD B01 5 -Wettable
354 PPA B04 1 Wettable
355 PPA B04 3 Wettable
356 PPA B04 5 Wettable
357 PPA B05 1 Wettable
358 PPA B05 3 Wettable
359 PPA B05 5 Wettable
360 PPA C01 1Nonwettable
361 PPA C01 3Nonwettable
362 PPA C01 5Nonwettable
363a PPA B04 0.33Wettable
B05 0.33
C01 0.33
364a PPA B04 0.67Wettable
B05 0.67
C01 0.67
365b PPA B04 1 Wettable
B05
C01
aThe composition also contained 2.5 percent by
weight titanium dioxide.
bThe composition also contained 2 percent by weight
titanium dioxide.
Because spunbonded fibers typically have larger
diameters on the average than meltblown fibers, the spun-
bonded webs were wettable or slowly wettable with additives
having molecular weights up to about 3,000. However, the
30 use of an additive having a molecular weight of about
8,000 did not produce a wettable web.
In order to further investigate the ability of a
gentle post-formation heat treatment to bring additive to
the effective surface and/or interfacial surface, ESCA
35 analyses were carried out on three of the spunbonded webs.
The webs then were heated at 110 for 1 minute in a labora-
-98-
1 33478 1
tory oven and the heated webs were subjected to ESCA
analyses. The results of the ESCA analyses before and
after heating are summarized in Table 32.
Table 32
Summary of ESCA Analyses
Before and After Heatinq
ESCA AnalYses Before and After Heatinga
Before Heating After Heatinq %
Example % C % O ~ Si % C % O % Si Inc.b
325 95 3.2 1.6 91 6.6 2.8 75
326 95 3.9 1.6 79 15 6.5 306
327 ~34 11 5.0 76 17 7.4 48
aIn atom percent.
bPercent silicon increase in first 100 A of surface.
The data in Table 32 clearly show the remarkable
increase in silicon concentration within the first 100 A
of the surface upon exposing a web to a mild heat treat-
ment, especially at an additive level of 1 percent by
weight.
Because spunbonded webs commonly are employed as
liners in disposable diapers, the mild heat treatment
phenomenon was investigated by two different methods in
conjunction with a simple diaper run-off test. The diaper
run-off test involved removing the liner from a standard
*KIMBEE diaper. The linerless diaper was mounted on a
plate which was inclined at a 45 angle, the back edge of
the diaper being at the top of the plate. The test fabric
was laid over the diaper. A reservoir containing 100 ml
of 0.85 percent (weight per volume) saline (cat. no. SS-442-
lo, Fisher Scientific, Pittsburgh, Pennsylvania) at 37O was
located at the top of the plane 2 inches (5.1 cm) above
the uppermost edge of the diaper's absorbent~pad. The
saline then was allowed to run out of the reservoir in a
_99 _
~ ~ * - Trade-mark
1 33478 1
~, .
steady stream. Fluid which was not retained by the diaper
was collected and measured, the volume of which was the run-
off value.
In the first method, samples of a spunbonded nonwoven
S web made from a composition of the present invention and
having a basis weight of 27 g/m2 were heated in an oven at
two different temperatures. Run-off measurements were
made on samples which had not been heat treated and those
which had. In every case, the additive was All and the
10 polymer was PPE. The results are summarized in Table 33.
Table 33
SummarY of Results of Run-Off Test
After First Heat Treatment Method
Web Add. Oven Heating Run-Off
Exam~le Levela Temp... Time Test ml
324 0.5 - - loob
0.5 80 3 min. 20-30
0.5 110 30 sec. 30-40
325 0.75 - - 70-80b
0.75 80 3 min. 0-1
0.75 110 30 sec. 40-50
326 1 - - 20-30b
1 80 3 min. 0
1 110 30 sec. 0
aIn weight percent.
bControl .
The efficacy of the heat treatment in each case is
readily apparent. It appears that 80 for 3 minutes is
more effective than 110 for 30 seconds, at least for the
webs having the two lowest concentrations of additive.
Either temperature treatment, however, converts the web
containing 1 percent by weight of additive into a highly
wettable, highly efficient transfer layer.
--100--
1 334781
In the second method, samples in continuous roll form
of the same webs used in the first method were passed over
two steam cans in series which were heated by steam at a
pressure of 5 psig. The surfaces of the cans were at
5 about 85. Each sample was passed over the cans at two
different line speeds, after which the run-off test was
performed. The results are summarized in Table 34.
Table 34
Summary of Results of Run-Off Test
After Second Heat Treatment Method
Web Add. Line Run-Off
Example Levela Speed. m/minTest. ml
324 0.5 - loob
0.5 9 80-90
0.5 4.5 80-90
325 0.75 - 70-80b
0.75 9 50
0 75 4-5 50
326 1 - 20-30b
1 9 5-10
1 4.5 0-5
aIn weight percent.
bControl.
- The results from the second method were similar to
those of the first method in that the concentration of
additive leading to the most efficient transfer layer was
1 percent by weight; the slower line speed gave slightly
better results at that concentration.
Because of the success with the Si-SEM procedure with
a melt-pressed film, a similar effort was carried out with
spunbonded fibers prepared from a composition containing a
35 mixture of additives in polymer PPA, i.e., Example 365.
In this case, a bundle of fibers was collected before they
--1 0 1--
1 33478 1
reached the forming wire. The bundle was cut and inserted
into a small plastic tube about 19 mm long and having an
inside diameter of about 3 mm, thereby packing the tube
with fibers. The packed tubing was placed in liquid
5 nitrogen, removed, and cut with a razor blade. The sample
was placed on the SEM mount and sputtered with carbon
before carrying out the analysis. A diagrammatic represen-
tation of the results of the analysis is shown by Figure
9. In Figure 9, the fibers 50 are bilobal in cross-section.
10 As with the film analysis, each of dots 51 represents the
presence of silicon atoms.
It is clear that the additives included in the compo-
sition from which the fibers of Example 365 were prepared
have segregated preferentially to the surface region of the
15 film. While the core region is not as devoid of silicon
as was the core region of the film, there clearly is a
lower concentration of the additives in the core region
than in the area at or near the surfaces of the fibers.
This result was expected, however, because of the relatively
20 rapid formation of the fibers as compared to the film
formation time. That is, the fibers remained in a molten
state for a time which was much shorter than the time the
film remained in a molten state. The fact that the addi-
tives segregated to the surfaces of the fibers in such a
25 short time is, as already pointed out, a result of the
influence of shear during the extrusion process.
- Two samples of fibers from the spunbonded trials
were submitted for analysis by RBS. The results are
summarized in Table 35.
-102-
1 334781
. ,
Table 35
SummarY of RBS Analyses on
Spunbonded Fibers
Atomic Concentration. Atom %
ExamPleDePth, A C O Si Ti
329 0-1000 30 0.7 0.28 O.01a
1000-3000 30 0.2 0.06 0.02
>3000 30 0.2 0.03 0.03
329b 0-1000 29 0.3 0.13 O.01a
1000-2000 29 0.1 0.02 0.02
>2000 30 0.1 0.02 0.02
364 0-250 28 3.6 1.94 0.02
250-900 28 2.2 0.90 0.02
900-1600 29 1.5 0.45 0.05
1600-2900 29 1.0 0.37 0.05
2900-4900 29 0.8 0.26 0.05
>4900 29 0.8 0.12 0.05
aThis concentration was at or near the detection
limit; the actual concentration may be considerably
lower.
bA second analysis was carried out on the same sample.
From the data for the two analyses on the same sample,
25 it appears that the RBS procedure causes some loss of
additives as evidenced by the decreased silicon concentra-
tion values. Thus, it is probable that the concentration
values are lower than the actual concentrations. Neverthe-
less, the procedure is helpful because it gives at least a
30 qualitative view of the segregation of the additives in
the surface region and the core region adjacent thereto.
The RBS data from Table 35 for the webs of Examples
329 and 364 were plotted as already described. The plots
for the two analyses of the web of Example 329 are shown
35 as Figures lOA and lOB. The plot for the analysis of the
web of Example 364 is shown as Figure 11.
-103-
1 334781
The plots are similar to that for the RBS analysis of
the film of Example 173. Figures 8 and lOA are especially
similar, although in the latter the concentration of
silicon diminishes to the minimum concentration at around
2,000 A, rather than at around 1,000 A. In Figure 11, it
is seen that the silicon concentration diminishes more
slowly with depth, although all of the plots resulted in
curves having similar shapes.
The webs from Examples 329 and 364 also were submitted
for ESCA and bulk elemental analyses. The results of
these analyses are shown in Table 36.
Table 36
Summary of ESCA Data and Elemental AnalYses
for the Webs of Exam~les 329 and 364
ESCA Data Bulk Elemental Anal.
Example % C % 0 % Si % C % H % Si
329 77 17 6.6 83.84 13.23 0.35
364 62 27 11 82.23 13.40 0.89
D. Meltblown Fibers from Pilot-Scale Coformin~
Apparatus
Examples 366-439
- A number of larger-scale meltblowing runs were carried
out on a coforming apparatus of the type described in U.S.
Patent Nos. 4,100,432 and 4,663,220, in regard to Examples
262-297, inclusive.
Meltblowing was accomplished by extruding the composition
from a 1.5-inch (3.75-cm) Johnson extruder and through a
meltblowing die having 15 extrusion capillaries per linear
inch (about 5.9 extrusion capillaries per linear
-104-
1 334781
cm) of die tip. Each capillary had a diameter of about
0.018 inch (about 0.46 cm) and a length of about 0.14 inch
(about 3.6 mm). The composition was extruded through the
capillaries at a rate of about 0.5 g per capillary per
minute at a temperature of about 184. The extrusion
pressure exerted on the composition in the die tip was in
the range of from about 180 to about 200 psig. The composi-
tion viscosity in the die tip under these conditions was
about 500 poise. The die tip configuration was adjusted
to have a positive perpendicular die tip distance of about
0.01 inch (about 0.25 mm). The air gaps of the two attenu-
ating air passageways were adjusted to be about 0.067 inch
(about 1.7 mm). Forming air for meltblowing the composi-
tion was supplied to the air passageways at a temperature
of about 209 and a pressure of about 2 psig. The fibers
thus formed were deposited on a forming screen drum which
was approximately 18 inches (46 cm) below and 20 inches
(51 cm) back from the die tip.
The more significant process variables generally were
as follows:
barrel temperature, 280-300;
die temperature, 285-316;
melt temperature in die, 275-316;
barrel pressure, 220-570 psig;
die pressure, 55-130 psig;
primary air temperature, 235-349;
primary air pressure, 3-4.5 psig;
throughput, 7-360 g per cm of die width per hour;
forming distance, 36 cm; and
basis weight, 27-85 g/m2, with the more typical
basis weights being 27, 51, and/or 85 g/m2.
The compositions which were meltblown were prepared
by melt-blending polymer and additive(s) as described in
Examples 50-130, inclusive. Coherent webs were formed in
each case. As with previous trials, wettability of the
formed webs was estimated by the water drop test as appro-
-105-
-- 1 334781
priate. The compositions meltblown and the results of the
water drop test are summarized in Table 37.
Table 37
Summary of Meltblowinq Trials on
Pilot-Scale Coforming Apparatus
Comp. Polymer Additive(s)
Exam~le Code Code Codefs) Wt. % Wettability
366 PP28-1 PPA A21 1 Wettable
367 PP29-1 PPA A21 3 Wettable
368 PP30-1 PPA A21 5 Wettable
369 PP31-1 PPA A21 12 Wettable
370 PE18-1 PEA A21 1 Wettable
371 PE19-1 PEA A21 3 Wettable
372 PE20-1 PEA A21 5 Wettable
373 PP32-1 PPA B01 3 Wettable
374 PP33-1 PPA B01 5 Wettable
375 PP34-1 PPB B01 3 Wettable
376 PP35-1 PPB B01 5 Wettable
377 PP36-1 PPC B01 3 Wettable
378 PP37-1 PPC B01 5 Wettable
379 PE21-1 PEA B01 3 Wettable
380 PE22-1 PEA B01 5 Wettable
381 PP38-1 PPA B02 3 Wettable
382 PP39-1 PPA B02 5 Wettable
383 PP40-1 PPC B02 3 Wettable
384 PP41-1 PPC B02 5 Wettable
385 PP42-1 PPA B03 3 Wettable
386 PP43-1 PPA B03 5 Wettable
387 PP44-1 PPC B03 3 Wettable
388 PP45-1 PPC B03 5 Wettable
389 PP46-1 PPA B04 3Nonwettable
390 PP47-1 PPA B04 5Nonwettable
391 PE23-1 PEA B04 3Nonwettable
392 PE24-1 PEA B04 5Nonwettable
-106-
1 334781
393 PP48-1 PPA B05 3Nonwettable
394 PP49-1 PPA B05 5Nonwettable
395 PE25-1 PEA B05 3Nonwettable
396 PE26-1 PEA B05 5Nonwettable
397 PP50-1 PPA B06 3Nonwettable
398 PP51-1 PPA B06 5Nonwettable
399 PP52-1 PPC B06 3Nonwettable
400 PP53-1 PPC B06 5Nonwettable
401 PP54-1 PPA B07 3Nonwettable
402 PP55-1 PPA B07 5Nonwettable
403 PP56-1 PPC B07 3Nonwettable
404 PP57-1 PPC B07 5Nonwettable
40S PP58-1 PPA B08 3Nonwettable
406 PP59-1 PPA B08 5Nonwettable
407 PP60-1 PPC B08 3Nonwettable
408 PP61-1 PPC B08 5Nonwettable
409 PP62-1 PPA B09 2Nonwettable
410 PP63-1 PPA B09 3Nonwettable
411 PP64-1 PPA B09 5Nonwettable
412 PP65-1 PPC B09 3Nonwettable
413 PP66-1 PPC B09 5Nonwettable
414 PP67-1 PPA B10 3Nonwettable
415 PP68-1 PPA B10 5Nonwettable
416 PP69-1 PPC B10 3Nonwettable
417 PP70-1 PPC B10 5Nonwettable
418 PP71-1 PPA Bll 3Nonwettable
~. 419 PP72-1 PPA B11 5Nonwettable
420 PP73-1 PPC Bll 3Nonwettable
421 PP74-1 PPC Bll 5Nonwettable
422 PP75-1 PPA C01 1Nonwettable
423 PP76-1 PPA C01 3Nonwettable
424 PP77-1 PPA C01 5Nonwettable
425 PE27-1 PEA C01 1Nonwettable
426 PE28-1 PEA C01 3Nonwettable
427 PE29-1 PEA C01 5Nonwettable
428 PP78-1 PPA D03 3Wettable
-107-
- 1 3:~4781
429 PP79-1 PPA D04 3 N/Aa
430 PP80-1 PPA D05 3 N/A
431 PP82-2 PPA B02 1.5 Wettable
Bll 1.5
432 PP84-2 PPA B06 1.5 Wettable
B10 1.5
433 PP86-2 PPA B10 1.5 Wettable
Bll 1.5
434 PP90-3 PPA B04 0.33 Wettable
B05 0.33
C01 0.33
435 PP92-3 PPA B04 1 Wettable
B05
C01
436 PP93-3 PPA B04 1.67 Wettable
B05 1.67
C01 1.67
437 PE30-3 PEA B04 0.33 Wettable
B05 0.33
C01 0.33
438 PE31-3 PEA B04 1 Wettable
B05
C01
439 PE32-3 PEA B04 1.67 Wettable
B05 1.67
C01 1.67
aNot applicable.
The results of the meltblowing trials on the coforming
30 apparatus with additives which impart water wettability to
the surfaces of the fibers were consistent with those of
the previous meltblowing trials.
In order to verify the presence of additive D04 on
the surfaces of the fibers, ESCA and bulk elemental analyses
-35 were run on the web from Example 429. Similar analyses
-108-
- 1 334781
were carried out with the web from Example 430 as a control.
The results of these analyses are summarized in Table 38.
Table 38
sSummary of ESCA and Bulk Analyses
on the Webs from Exam~les 429 and 430
ESCA Data Bulk Elemental AnalYses
Example % C % F % Si % C % F % Si
429 73 11 6.9 83.66 0.99 0.50
430 69 - 16 84.72 - 1.06
Controla 100 - - 98
aPolymer PPA which did not contain any additive.
According to the analytical data for the web from
Example 429, it is evident that additive D04 has segregated
to the surface region; i.e, the first 100 A of the surface
as measured from the interfacial surface. The web from
Example 430 also contained a substantial amount of additive,
in this case D05, in the same surface region.
As already pointed out, however, additive D05 moved
to the surface of the fibers because it is incompatible
with the polymer. Such incompatibility resulted in poor
web formation; that is, the web was characterized by
nonuniform fiber diameters, an unusually high proportion
of discontinuous fibers, and a substantial amount of shot.
The process was characterized by a frequent, almost ex-
plosive, expulsion of polymer from the die orifices which
is potentially hazardous to the operators.
E. Coformed Webs from Pilot-Scale Coforming Apparatus
Exam~les 440 and 441
Two fibrous coformed nonwoven webs were formed by
35 meltblowing a composition of the present invention and
incorporating polyester staple fibers therein.
--109--
1 334 78 1
Meltblowing was accomplished as described for Examples
366-439, inclusive. In each case, the polymer was PPA and
the additive was B01 at a level of 3 percent by weight.
The more significant meltblowing process conditions
were approximately as follows:
die tip temperature, 296O;
primary air temperature, 2840;
primary air pressure, 3.5 psig;
throughput, 179 g per cm of die width per hour;
horizontal forming distance, 51 cm; and
vertical forming distance, 43 cm.
Following the procedure illustrated by Figure S of
said U.S. Patent No. 4,663,220 and described therein, 3-
inch (7.6-cm) long, 40 denier per filament polyester staple
(type 125, E. I. Du Pont de Nemours & Co., Inc., Wilmington,
Delaware) was incorporated into the stream of meltblown
fibers prior to deposition upon the forming drum. The
polyester fibers were first formed by a Rando Webber mat-
forming apparatus into a mat having a basis weight of
about 100 g/m2. The mat was fed to the picker roll by a
feed roll which was positioned about 0.13 mm from the
picker roll surface. The picker roll was rotating at a
rate of about 3,000 revolutions per minute and fiber
transporting air was supplied to the picker roll at a
pressure of about 2.5 psig. While actual measurement of
the position of the nozzle of the coform apparatus with
respect to the stream of meltblown fiber was not made, it
was estimated to be about 5.1 cm below and about 5.1 cm
away from the die tip of the meltblowing die.
Two coformed webs were prepared, both of which had a
width (cross-machine direction) of about 51 cm. The first
web was composed of about 70 percent by weight of the
polyester staple fibers and about 30 percent by weight of
the meltblown fibers and the second web was composed of
3s-about 50 percent by weight of each of the two types-of
--110--
1 33478 1
fibers. Each web had a basis weight of about 100 g/m2 and
wet immediately when subjected to the water drop test.
Although not described in detail here, other coformed
webs were similarly prepared with staple fiber:meltblown
fiber ratios of 85:15, 75:25, 65:35, and 15:85. In addi-
tion, webs utilizing other sources of polyester staple
fibers were prepared at each of the foregoing ratios.
Such other polyester staple fibers were as follows:
3.25-inch (8.3-cm) x 25 denier (Eastman Chemical
Products, Inc., Kingsport, Tennessee);
type ES 1.5-inch (3.8-cm) x 1.5 denier (Chisso
Corporation, Tokyo, Japan); and
type 41-D 1.5-inch (3.8-cm) x 1.5 denier (Eastman
Chemical Products, Inc.).
Example 441
The procedure of Examples 440 and 441 was repeated,
except that the composition was 3 percent by weight of
additive B01 in polymer PEA, the secondary fibers were
wood pulp fibers, and a dual meltblowing die/center secon-
dary fiber duct arrangement was employed. Thecomposition
was meltblown through one die at a throughout of either
179 or 894 g per cm per hour. In either case, the melt
25 temperature was about 288. The die tip pressure was
either 90 or 220 psig, depending upon the throughput.
Polymer PPC was meltblown through the other die at a
throughput of from about 179 to about 716 g per cm per
hour. The melt temperature was in the range of from about
246 to about 274 and the primary air temperature was in
the range of from about 280 to about 302. The primary air
pressure was in the 2-5 psig range.
Coformed webs containing pulp:polymer ratios of 70:30
and 90:10 were prepared. The webs wet immediately and the
35 composition did not impede the absorbency of the web.
--111--
I 334781
V. Evaluation of Known Material
In conclusion, an additive of the type described in
U.S. Patent No. 4,659,777 was evaluated both in melt-
pressed films and fibers from the bench-scale meltblowing
5 apparatus. The additive was a poly(2-ethyloxazoline)-
polydimethylsiloxane-poly(2-ethyloxazoline)blockcopolymer,
each of the blocks having a molecular weight of about 3,000.
Example 446
A melt-pressed film was prepared successfully as
described for Examples 131-176, inclusive. The material
contained 10 percent by weight of the additive in polymer
PPA.
The surface energy of the film was estimated by means
of Pillar wetting agents (Pillar Corporation, West Allis,
Wisconsin) to be 34-35 dynes per cm. The value for virgin
polymer is about 30. The film then was subjected to ESCA
analysis. None of the additive was found to be in the
20 first 100 A below the interfacial surface.
Example 447
Meltblown fibers were prepared with a bench-scale
25 apparatus as described for Examples 178-239, inclusive.
The composition consisted of 3 percent by weight of the
-additive in polymer PPA. Meltblowing was conducted at an
air pressure of 35 psig and melt temperatures of 264, 285,
and 308. Although webs were obtained in each case, web
30 quality was poor and decomposition of the additive occurred
at each melt temperature. Decomposition was especially
severe at the highest temperature. No analyses of the
webs were attempted since the additive obviously is unsuited
for melt-processing procedures and does not segregate to
- 35 the surface.
-112-
1 33478 1
VI. Hot-Stage Microscopy StudY of a Composition Described
in U.S. Patent No. 4 070.218
One last hot-stage microscope analysis needs to be
described. The composition consisted of polymer PPA with
5 3 percent by weight of Triton X-102 (Rohm and Haas Co.,
Philadelphia, Pennsylvania), a surfactant which is commonly
used to make polypropylene wettable by means of the blooming
technique already described. The representations of the
photomicLGyLaphs are shown in Figures 12A and 12B. Globules
121 of the surfactant are seen in both Figures; some
debris 122 in Figure 12A also is apparent. The most
noteworthy fact about the two Figures is that the surfactant
not only is incompatible with the polymer at 160, but is
even less compatible at about 220. In view of Figures
15 12A and 12B, it is easy to understand why a blooming
process is required to bring the surfactant to the surface
of the fiber or film and why the material migrates back
into the polymer.
It now should be evident that the additives described
20 herein and the compositions of the present invention
function in a manner which is different from the materials
previously added to thermoplastic polymers to alter the
surface characteristics of shaped articles, such as fibers
and films, made therefrom. Moreover, the compositions of
25 the present invention permit the control of the segregation
phenomenon, which control was not possible with prior art
procedures. Thus, the method of the present invention is,
in reality, very different from that of said U.S. Patent
No. 4,070,218. Moreover, HLB terminology is not applicable
30 to the additives employed in the present invention.
Having thus described the invention, numerous changes
and modifications thereof will be readily apparent to
those having ordinary skill in the art without departing
from the spirit or scope of the invention. For example,
35 the compositions useful in the present invention also can
contain fillers, colorizers, stabilizers, and the like.
-113-