Note: Descriptions are shown in the official language in which they were submitted.
- 1 - 1 33 5 6 ~ O
This invention relates to a process for producing
ammonia (NH3) and sulfur dioxide (SO2) from ammonium
sulfate. More specifically, it relates to a process for
producing NH3 and SO2 which comprises decomposing
ammonium sulfate at a temperature of not more than 200
C to NH3 and a metal sulfate and decomposing the
metal sulfate in the presence of a reducing agent to form
a metal oxide and SO2.
A great quantity of ammonium sulfate is formed
in various industries, for example the chemical industry
involving treatment of an ammoxidation product of organic
matter with sulfuric acid (such as the production of
epsilon-caprolactam, methyl methacrylate by the acetone
cyanohydrin method, and acrylonitrile by the ethylene
cyanohydrin method~, an industry in which a liquor result-
ing from ammonia neutralization of a desulfurization
waste liquor formed by treatment of crude oil with sulfuric
acid is discharged, and an industry in which desulfuriza-
tion of exhaust gases is carried out by using aqueous
ammonia. This ammonium sulfate is used mainly as a
fertilizer. Since, however, the supply of ammonium
sulfate is excessive for its demand, its price decreases
to cause a marked economic disadvantage. Furthermore, a
very great amount of ammonia is required for neutralizing
sulfuric acid remaining in the waste liquor, and this
becomes a great burden. Furthermore, since ammonium
sulfate contains impurities, much work is needed for
purifying it to a form suitable for use. Thus, in the
industries where ammonium sulfate occurs, it is an import-
ant problem to recover it as an economically feasibleform. If, therefore, ammonium sulfate can be recovered
efficiently as NH3 and sulfuric acid by decomposition,
it would become a promising solution to the above problem.
Many methods have been known for decomposing
- 2 _ 1 33 5 69 0
ammonium sulfate and recovering it as NH3 and SO2 (or
sulfuric acid). For example, "Bessatsu Kagaku Kogyo"
(Chemical Industry, Separate Volume), vol. 15, No. 2
(1971), pages 119 to 128 discloses a method which compris-
5 es causing magnesium oxide ~MgO) to absorb SO2 contain-
ed in a waste gas thereby to form magnesium sulfite,
converting magnesium sulfite into magnesium sulfate using
manganese oxide, etc. as a catalyst, decomposing it into
MgO and SO2 at more than 750 C using carbon, and
10 recovering SO2.
Japanese Patent Publication No. 8251/1962
describes a process for producing NH3 and SO2 which
comprises reacting ammonium sulfate with zinc oxide at a
temperature of about 300 to 500 C to generate NH3
15 and form zinc sulfate, and then reacting the residue from
this first reaction with a coke at a temperature of at
least 600 C to form SO2.
Japanese Laid-Open Patent Publication No.
30599/1972 describes a process for obtaining SO2 by
20 thermally decomposing ammonium sulfate at 300 to 500 C
and then burning and decomposing the decomposition product
at 850 to 1250 C.
Japanese Laid-Open Patent Publication No.
73619/1981 discloses a process for producing an aqueous
25 solution of highly pure ammonium sulfite and acid ammo-
nium sulfite which comprises thermally decomposing ammo-
nium sulfate at a temperature of at least 250 C to
form NH3 and a composition containing acid ammonium
sulfite, thermally decomposing the composition at a
30 temperature of 250 to 440 C to form a decomposition
gas composed of SO2 and NH3, purifying the decomposi-
tion gas, causing the purified decomposition gas to be
absorbed by water or an aqueous ammonia solution to form
an aqueous solution of ammonium 3U~I,C and acid ammonium
= 35 sulfite.
Japanese Laid-Open Patent Publication No.
- 3 - t 3~ 6~ 0
101294~1975 discloses a process for obtaining a gaseous
mixture of NH3 and SO2 which comprises reacting
ammonium sulfate with carbon at 480 to 630 C.
Japanese Laid-Open Patent Publication No.
5 125300/1978 discloses a process for treating ammonium
sulfate, which comprises adding a solid form or slurry of
magnesium oxide or magnesium hydroxide to an aqueous
solution of ammonium sulfate, almost coanpleting the
reaction of the mixture by elevating the reaction
10 temperature to form ammonia and magnesium sulfate, then
concentrating the aqueous magnesium sulfate solution to
separate and recover MgSO4.nH2O (n=l, 2, 3, 4, 6, or 7),
and eeding MgS04.nE~20 into a heat decomposition furnace
to form sulfur dioxide.
In the conventional ammonium sul~ate decompos-
ing techniques described above, ammonium sulfate is
decomposed at high temperatures, and the reaction is
carried out in a high ammonium sulfate conversion.
Accordingly, the recovered NH3 or SO2 inevitably
20 contain impurities formed at the time of decomposition.
This tendency is particularly pronounced when the start-
ing ammonium sulfate contains organic materials such as
diacetonesulfonic acid or its polymer. For use in appli-
cations requiring high purity, the recovered NH3 or
25 SO2 must be purified by a complex purifying step. The
conventional techniques, therefore, are not economically
advantageous.
On the other hand, it i8 known in the field of
desulfurization of exhaust gases to decompose ammonium
30 sulfate at low temperatures. This technique involves
reacting ammonium sulfate obtained by exhaust gas de-
sulfurization with calcium hydroxide and recovering
gypsum. NH3 is obtained as ammonium hydroxide. The
ammonium hydroxide is recycled to the absorbing step for
35 reuse, and does not require so high a purity. This
technique does not involve an idea of recovering it as
1 3356~
NH3 gas. It may be possible to add slaked lime or
quick lime to ammonium sulfate and heat the mixture to
volatilize NH3. This method, however, would usually be
feasible in pollution control. In the field of pollution
control, for example desulfurization of exhaust gases
which almost disregards economy, the purpose is to remove
SO2. It has no idea of efficiently recovering NH3
and SO2 and re-using them, much less its commercializa-
tion. Recovery of NH3 from a liquid containing SO2
absorbed therein might be conceivable from the techniques
used in the field of pollution control in which the
significance of recovering NH3 from ammonium sulfate
quite differs from that in the industry which forms
ammonium sulfate. However, an idea of using, in combina-
`,f;~ 15 tion,~ a step of decomposing a metal sulfate and efficient-
ly producing NH3 and SO2 of high purity cannot be
easily derived from the techniques used in the field of
pollution control.
It is an object of this invention therefore to
provide a process for efficiently producing NH3 and
S2 of good purity by decomposing ammonium sulfate
under relatively mild operating conditions.
The present inventors made extensive investiga-
tions in order to achieve the above object, and have
found that NH3 and SO2 of good purity can be efficient-
ly produced by a relatively simple and convenient process
which comprises reacting ammonium sulfate occurring in
various chemical industries with a metal oxide or hydro-
xide at a temperature of not more than 200 C to form
NH3 and a metal sulfate and decomposing the metal
sulfate in the presence of a reducing agent to form a
metal oxide and SO2.
According to this invention, there is provided
a process for producing NH3 and SO2 from ammonium
sulfate, which comprises (i) reacting ammonium sulfate
with a metal oxide or hydroxide at a temperature of not
-- 5 --
more than 200 C to form NH3, water and a metal
sulfate and recovering NH3, (ii) decomposing the metal
sulfate in the presence of a reducing agent to form a
metal oxide and SO2 and recovering S02, and (iii)
recycling the metal oxide to step (i) as such or after it
is converted to a metal hydroxide.
The term ~ammonium sulfate" as a starting
material used in this invention dnotes not only ammonium
~ t
sulfate ~ also acid ammonium sulfate, acid ammonium
~ 10 sulfate containing sulfuric acid, and mixtures of these.
These starting materials may include impurities in a
concentration of up to 10 % by weight. The impurities
include, for example, non-combustible carbon, ash, epsilon-
caprolactam, acetonedisulfonic acid, p-toluenesulfonic
acid, polymers of these and sulfur which are contained in
an epsilon-caprolactam waste liquor, a methyl meth-
acrylate waste liquor from the acetone-cyanohydrin
method, an acrylonitrile waste liquor from the ethylene-
cyanohydrin method and in a crude oil refining waste
liquor
In step (i) of the method of this invention,
the ammonium sulfate is reacted, usually in a reaction
medium such as water, with the metal oxide or hydroxide
at a temperature of not more than 200 C, preferably in
the range of 60 to 120 C.
Since in the process of this invention, the
metal oxide or hydroxide is re-used, it may be an oxide
or hydroxide of a relatively expensive metal. Generally,
however, the use of an oxide or hydroxide of a metal
selected from Mg~ Ca and Ba is preferred because it
produces a good effect and is practicable. The use of
magnesium oxide or hydroxide is especially preferred
because the reduction temperature used in the decomposi-
tion of the resulting metal sulfate in the presence of a
reducing agent can be lowered. These metal oxides or
hydroxides may be used singly or as a mixture. The metal
1 335690
oxides are preferably used in this invention.
The oxide or hydroxide (A) of a metal selected
from Mg, Ca and Ba may be used in combination with at
least one oxide or hydroxide (B) of a metal selected from
Mn, Cu, Ni and Zn. In this case, the oxide or hydroxide
(B) is used preferably in an amount of generally not more
than 50 mole % based on the metal oxide or hydroxide tA).
A combination of these metal oxides or hydroxides in
which the mole ratio of the metal oxide or hydroxide (A)
to the metal oxide or hydroxide (B) is generally from
1:0.01 to 1:0.5, particularly from 1:0.1 to 1:0.3, is
preferred because it aids in the decomposition of organic
compounds contained as impurities and the metal sulfate
and makes it possible to lower the decomposition tempera-
ture.
This reaction can usually be carried out inaqueous solution, and the pH of the aqueous solution is
desirably maintained at about 8 to 13, preferably at 9 to
10, in order to inhibit corrosion of the reactor and
increase the ratio of recovery of ammonia. Accordingly,
it is generally preferred to adjust the mole ratio of the
metal oxide or hydroxide to the sulfate ion to 1 - 2,
preferably 1.03 - 1.4.
There is no particular restriction on the
reactor used in the reaction, and it may be, for example,
a stirred tank or a stirred tank equipped with a distil-
lation column. The material of which the reactor is
made is not particularly limited, but stainless steel is
preferred in view of corrosion resistance.
The ammonium sulfates reacted with the metal
oxide or hydroxide to form NH3, water and the metal
sulfate. NH3 is recovered by known means such as
distillation.
Preferably, from the standpoint of the increase
of the ratio of NH3 recovery or energy consumption, the
reaction between ammonium sulfate and the metal oxide or
_ 7 _ ~33569~
hydroxide is carried out in two or more successively
aligned reactors, and the reaction in the first-stage
reactor is carried out at a reaction temperature of 60 to
100 C, preferably 70 to 90 C. It is more preferred
to carry out the reaction of ammonium sulfate with the
metal oxide or hydroxide in at least two successively
aligned reactors in accordance with a so-called ~multiple-
effect evaporation method" by which heat is given to a
reactor desired to be set at the highest reaction tempera-
ture and the vapor generated from it is supplied as aheat source to a reactor to be set at the next highest
reaction temperature, and the vapor generated in this
reactor is then supplied as a heat source to a reactor to
be set at the third highest reaction temperature, and the
vapor is successively supplied from one reactor to another
in this manner. The first-stage reactor, as referred to
herein, denotes a reactor in which the starting ammoniu~
sulfate, a reaction medium such as water and the metal
oxide or hydroxide are first put. Heat is given to the
reactor to be desired to be set at the highest reaction
temperature, by introducing a heat medium such as steam
into an inside or an outside tube or a jacket in the
reactor.
In carrying out the process of this invention
2S in two or more successively aligned reactors, it is
important that the reaction temperature in the first-
stage reactor be 60 to 100 C. The reaction tempera-
tures in the second-stage and subsequent reactors may be
not more than 200 C.
If, in the process of this invention, the
ammonium sulfate is reacted with the metal oxide or
hydroxide while maintaining the conversion of ammonium
sulfate at 60 to 85 mole %, preferably 65 to 80 mole %,
ammonia can be obtained in a high purity. This pro-
cedure, therefore, is a preferred embodiment.
When the metal sulfate obtained in the above
1 335690
reaction is a water soluble compound such as MgSO4,
impurities insoluble in the reaction medium, such as
carbon and sulfur compounds can be removed. The carbon
can be re-used as a fuel or a reducing agent, and the
sulfur compounds can be recovered as SO2~ If the metal
sulfate is a water-insoluble compound such as CaSO4, a
water-soluble impurity such as acetonedisulfonic acid can
be removed. The impurity can be concentrated and recover-
ed for use as a reducing agent. In either case, the
impurities can be separated, recovered and re-used, and
the industrial wastes can be drastically decreased.
The resulting metal sulfate is concentrated or
crystallized by a difference in solubility, and separated
by a solid-liquid separator if it is water soluble. If
it is insoluble in water, it can be separated by a solid-
liquid separator and recovered as a solid metal sulfate.
If the mother liquor resulting from separating
and recovering of the metal sulfate and the unreacted
metal oxide or hydroxide from the reaction product ob-
tained by the reaction of ammonia sulfate with the metaloxide or hydroxide is recycled to step (i) in the proces~
of this invention, the ratio of ammonia recovery can be
increased. Hence, this is a preferred embodiment.
The reaction product obtained by the reaction
f ammonium sulfate with the metal oxide or hydroxide in
which the metal is magnesium is usually an aqueous solution
or slurry of magnesium sulfate in a concentration of at
least 15 % by weight. If this aqueous solution or slurry
i8 fed to the next step as such, the energy for dehydra-
tion is consumed greatly. This is not industriallydesirable. Usually, anhydrous magnesium sulfate and
magnesium sulfate monohydrate are obtained by dehydrating
magnesium heptahydrate by heating it in an open system,
hot air drying, etc. It iæ well known that the energy
consumed for evaporating and removing the water of crystal-
lization is very great.
1 335690
The present inventors noted that if water can
be removed in the form of a liquid, the energy required
to obtain 1 kg of magnesium sulfate monohydrate can be
decreased by 422.5 kcal/mole in accordance with the
following formulae.
MgS04 7H20 --~ MgS04 H20 ~ 6H20(Q) - 17.1 kcal/mol
MgS04 7H20 -~ MgS04 H20 + 6H20(g) - 75.4 kcal/mol
Further investigations on this basis led to the
discovery that if the above aqueous solùtion or slurry of
magnesium sulfate is heated in a closed system to 100 to
200 C, preferably 120 to 170 C to separate and
recover-a solid product, ~ater can be removed in the form
of a liquid, and a solid product having a water content
of 17 to 30 ~ by weight and composed mainly of magnesium
sulfate monohydrate can be obtained with a low energy
consumption, and that if this solid product is supplied
to the next step, the thermal load decreases.
If the above aqueous solution or slurry is
heated to a temperature of more than 200 c, the energy
for temperature elevation becomes very great, and the
effect of the invention undesirably tends to be reduced.
A solid product having a water content of 17 to
30 % by weight can be obtained with better efficiency by
heating magnesium sulfate heptahydrate or an aqueous
solution or slurry of magnesium sulfate in a concentra-
tion of at least 15 % by weight in a closed system to 100
to 200 C, preferably 120 to 170 C and maintaining
it at 100 to 200 C, preferably 120 to 170 C, for at
least 60 minutes to separate and recover the solid product;
or by first heating the aqueous solution or slurry to 120
to 170 C and then quickly lowering the pressure to
atmospheric pressure to separate and recover the solid
~ ~ product; orAYheating the aqueous solution or slurry to 120
^~ to 170 C, maintainng it at this temperature for at
1 335690
-- 10 --
least 60 minutes, and then quickly lowering the pressure
to atmospheric pressure to separate and recover the solid
product. Separation and recovery of the solid after
heating may be effected under pressure, but it is practi-
s cable from the viewpoint of operability to separate and
recover the solid product after the pressure is returned
to atmospheric pressure.
The term "quickly" as used above refers to the
time within which magnesium sulfate monohydrate is not
converted back to magnesium sulfate heptahydrate. Usual-
ly, it means 5 minutes or less. It is pre~erred to add
an excess of magnesium sulfate monohydrate or magnesium
sulfate anhydride salt at a time when the aqueous solu-
tion or slurry containing magnesium sulfate in a concen-
tration of at least 15 % by weight is heated to 100 to200 C, preferably 120 to 170 C, or when it is
maintained at this temperature for at least 60 minutes,
and when thel~ressure is lowered to atmospheric pressure,
~i ~ because a ~ product having a water content of 17 to 30
~ by weight can be obtained with better efficiency. The
time during which the above solution or slurry is main-
tained at 100 to 200 C, preferably 120 to 170 C,
may be at least 60 minutes. Usually, it is practical to
perform this operation for 1 to 3 hours. When a solid of
magnesium sulfate heptahydrate is to be heated, the above
method can be applied because if it is heated to 100 to
200 C, liquid water exists in it.
When water is not used in the previous step,
the resulting metal sulfate is directly fed to the next
step. This metal sulfate may permissibly contain organic
matter.
The metal sulfate is then decomposed in the
presence of a reducing agent to form a metal oxide and
SO2. Examples of the reducing agent include carbon
such as coal, activated carbon, coke, petroleum coke,
graphite and carbon black, carbon monoxide, hydrogen,
1 335690
fuel decomposition gases, and an oxo gas obtained by
decomposition of, for example, methanol.
In the present invention, the metal sulfate is
decomposed in the presence of a reducing agent in step
(ii) to form a metal oxide and sulfur dioxide. The
specification o~ the above-cited Japanese Laid-Open
Patent Publication No. 125300/1978 describes a process in
which MgSO4.nH2O is fed into a heat decomposition
furnace. Investigations of the present inventors have
shown that even at the same reaction temperature, the
decomposition reaction of MgSO4.nH2O in the presence of
a reducing agent can give a higher decomposition rate.
For example, when the thermal decomposition was performed
by the method disclosed in Japanese Laid-Open Patent
Publication No. 125300/1978 at a reaction temperature of
800, 900 and 1000 C, the decomposition rate of MgSO4
anhydrous salt was less than about 2 mole %, about 3
mole % and about 6 mole % respectively according to the
reaction temperature. In contrast, when the MgSO4
anhydrous salt was decomposed in the presence of carbon
as the reducing agent, the decomposition rate was 30
mole %, 88 mole % and about 100 mole %, respectively.
Thus, in the presence of the reducing agent, the decom-
position reaction of the metal sulfate can be efficiently
carried out.
The reactions involved in the reduction of the
metal sulfate can be shown by the following three reac-
tion equations in which M represents a metal.
SO4 + H2 --~ MO + S02 + H2O (1)
MSO4 + CO > MO ~ SO2 + CO2 (2)
MSO4 + ~C ~ MO + SO2 + ~ 2 (3)
Those skilled in the art would easily think that
to decrease the amount of gases formed by the reaction,
it is advantageous to carry out the decomposition of the
1 33~69~
- 12 -
metal sulfate in accordance with the reaction equation
(3). However, the reaction in accordance with equation
(3) is a solid-solid reaction, and investigations of the
present inventors have shown that its reducing efficiency
is not entirely satisfactory.
In order to achieve the above object of this
invention, the present inventors paid their attention to
equation (3), and extensively worked for a method of
decomposing the metal sulfate by a solid-solild reaction
with a high reducing efficiency. As a result, they have
found that when the metal sulfate is submitted to decompo-
sition reaction as a mixture with carbon, the decomposi-
tion reaction can be performed with a high reducing
efficiency.
Examples of the carbon to be mixed with the
metal sulfate are coal, petroleum coke, activated carbon
and carbon black. Preferably, carbon with a low content
of hydrogen is used. From the standpoint of its price,
coal or petroleum coke is preferred. A mixture of carbons
may also be used equally.
In the present invention, the metal sulfate is
subjected to decomposition reaction as a mixture with
carbon. The mixture may be obtained by simply mixing the
metal sulfate and carbon having much the same particle
diameter in a kneader or the like, or by mixing particu-
late carbon with the metal sulfate. When the starting
ammonium sulfate contains the aforesaid organic im-
purities, these impurities also act as a reducing agent.
Preferably, the reduction temperature is low,
and the decomposition reaction of the ammonium sulfate is
fast. When the reduction temperature is more than about
2,000 C, the selection of a material of which a reduc-
tion furnace is made is a problem, and will cause economic
disadvantage. If it is too low, the decomposition of
organic compounds as impurities is suppressed. It is
generally desirable to carry out the reduction at a
1 335690
- 13 -
temperature of 650 to 1,800 C, preferably 750 to 1,600
C, more preferably 800 to 1000 C.
The reduction furnace may be, for example, an
ordinary furnace of the fluidized bed, fixed bed or
moving bed type (such as a rotary kiln). The fluidized
bed-type furnace is advantageous in operating efficiency
and cost in dealing with a large-scale process.
In carrying out the decomposition reaction of
the metal sulfate in a fluidized bed, the mixture of the
metal sulfate and carbon may be used in the form of a
slurry or granules. For practical purposes, the granules
preferably have a particle diameter of about 0.1 to 50
mm. The granules may be in the form of pellets.
In this step, water ~including water of crystal-
lization) remaining in the treated product from theprevious step is gasified, and the metal sulfate is
decomposed into a metal oxide and SO2. From an indust-
rial viewpoint, it i8 preferred to recover the metal
oxide entrained in the discharge gas by using a cyclone,
cottrel, etc. in a customary manner, cooling it together
with the metal oxide recovered from the bottom of the
reduction furnace, mixing the metal oxide with water, and
to use the mixture again in the step of decomposing
ammonium sulfate.
In the present invention, it is more efficient
and preferable to carry out the decomposition step by a
method comprising decomposing the metal sulfate while a
mixture of it with carbon is being fed into fluidized
solid particles, and carrying the resulting metal oxide
on a gas current away from the system.
The solid particles used in this method may be
any solid particles which have heat resistance and do not
scatter easily. Generally, these solid particles prefer-
ably have a knoop hardness of at least 600 kg.f/mm2.
In particular, solid particles of silica, alumina or a
mixture of both are preferably used because they produce
1 335690
- 14 -
particularly good results. Such silica and alumina
particles may contain other particles which do not impede
the effects of the present invention. For good abrasion
resistance, spherical solid particles are preferred.
Usually, these solid particles have an average particle
diameter of preferably 0.1 to 5 mm, more preferably 0.5
to 1.5 mm, because too large particles are difficult of
flowing and too small particles tend to scatter.
Since the decomposition reaction of the metal
sulfate in the present invention is an endothermic reac-
tion, it is necessary to heat the reaction system by, for
example, burning the carbon in order to maintain a suitable
reaction temperature. To maintain a suitable reaction
temperature and a suitable reducing atmosphere, it is
preferred to supply air so that the mole ratio of oxygen
to the reducing agent is generally from 0~6 to 1.05,
preferably from 0.7 to 0.9.
Air is blown into the S02-containing gas
recovered to burn the reducing gas and increase the yield
Of S02. It may then be used as a material for various
reactions. Alternatively, it may be recovered as sulfuric
acid by a known technique and sold on the market or again
used in the process of producing epsilon-caprolactam or
in the process of producing methyl methaxrylate by the
acetone cyanohydrin method. Thus, sulfuric acid can be
utilized effectively. Particularly, if the process of
this invention is carried out in the production of methyl
methacrylate by the acetone cyanohydrin method using
ammonia and sulfuric acid as materials, the entire process
can be carried out completely in a closed system, and the
effects of the invention are great.
The process of this invention can be most
efficiently performed when the metal sulfate is magnesium
sulfate.
The process of this invention will now be
described more specifically by referring to Figure 1
1 33~690
accompanying this application which is a flow sheet
showing one embodiment of the process of this invention.
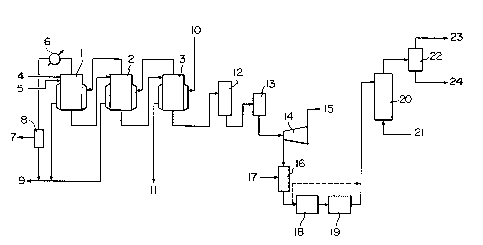
The reference numerals have the following means.
1: first-stage reactor
2: second-stage reactor
3: third-stage reactor
4: line for feeding the metal oxide or
hydroxide
5: line for feeding an aqueous solution or
slurry of ammonium sulfate
6: cooler
7: pressure reduction system
8: gas-liquid separator
9: aqueous ammonia
10: incoming heat medium
11: outgoing heat medium
12: temperature and pressure elevator
13: temperature and pressure lowering device
14: centrifugal liquid-solid separator
15: mother liquor
16: carbon mixer
17: carbon
18: solidifying device
19: pulverizing and classifying device
20: fluidized reduction furnace
21: air
22: cyclone
23: sulfur dioxide
24: metal oxide
The reactors 1, 2 and 3 are for carrying out
the reaction of ammonium sulfate with the metal oxide or
hydroxide. The metal oxide or hydroxide is fed from the
line 4, and an aqueous solution or slurry of ammonium
sulfate is fed from the line 5. In the embodiment shown
in Figure 1, three reactors are used, and the third-stage
reactor 3 is heated with a heat medium 10 such as steam.
1 335690
- 16 -
As can be clearly seen from Examples to be given herein-
below, various modes of the reaction can be taken, for
example, in regard to the sequence of supplying a heat
source. In Figure 1, the vapor generated from the third-
stage reactor 3 is supplied to the second-stage reacto~ 2
as a heat source, and the vapor generated from the second-
stage reactor 2 is supplied to the first-stage reactor 1
as a heat source. When the heat medium used as a heat
source in the third-stage reactor is steam, it is dis-
charged as waste water 11, or as required, recovered.When it is an oil, it is re-heated and used as the heat
source. The vapor generated from the third-stage reactor,
after it is used as the heat source in the second-stage
reactor 2, is condensed as aqueous ammonia which is mixed
with aqueous ammonia resulting from co~densation of the
vapor used as the heat source in the flrst ~s~t~ reactor
1. The mixture is then recovered as ammonia from 9 in a
customary manner. The vapor generated from the first-
stage reactor 1 is condensed under reduced pressure via
the cooler 6, and mixed with the aforesaid aqueous am-
monia. 8 is a gas-liquid separator. The reactors used
may usually be reactors equipped with a jacket and a
stirrer. The reaction mixture obtained from the third-
stage reactor 3 consists mainly of magnesium sulfate and
water when magnesium oxide or magnesium hydroxide is used
as the metal oxide or hydroxide. It is converted mainly
into magnesium sulfate monohydrate in the temperature and
pressure elevator 12, the temperature and pressure lower-
ing device 13 and the centrifugal liquid-solid separator
14. The mother liquor lS obtained from the centrifugal
liquid-solid separator 14 is recycled to the first-stage
reactor 1. If desired, the mother liquor may be further
treated and discarded. However, to increase the recovery
ratio of ammonia, the mother liquor is desirably recycled
to step (i). Thereafter, the metal sulfate is mixed with
carbon 17 by the carbon mixer 16, and introduced into the
1 335690
- 17 -
reduction furnace 20 having a fluidized bed, etc. via the
solidifying device 18 effecting solidification by cooling
or drying and the pulverizing and classifying device 19.
The metal sulfate mixed with carbon in the carbon mixer
16 may be directly introduced into the reduction furnace
20 without going through the solidifying device 18 and
the pulverizing and classifying device 19. Air 21 is fed
from the lower part of the reduction furnace, and the
metal sulfate is reduced. By the cyclone 22, the metal
oxide (such as MgO) is collected from the gas current
from the reduction furnace. The metal oxide (such as MgO)
collected mainly from 24 and SO2 is recovered from
~p ~ As required, the metal oxide, etc. may be withdrawn
from the bottom of the reduction furnace.
The following examples illustrate the present
invention more specifically. It should be understood
however that the scope of the invention is not limited by
these examples.
EXAMPLE 1
A l-liter four-necked flask equipped with a
stirrer and a reflux condenser was charged with 40 g of
MgO and 250 g of water, and the mixture was stirred at 80
C for 60 minutes. Thereafter, 110 g of ammonium
sulfate powder ~extra pure reagent made by Wako Pure
Chemicals Industries, Ltd.) was added from the top of the
flask, and reacted at 80 C for 60 minutes. After the
reaction, the reaction mixture was cooled, and filtered
under pressure to separate the unreacted MgO and Mg(OH)2.
The mother liquor was distilled to give NH3 in a yield
f 97 %.
The MgSO4 solution after removal of NH3 was
concentrated, cooled and subjected to solid-liquid separa-
tion to recover 200 g of MgSO4.7H2O. It was dried by
a hot air dryer at 100 C to give 115 g of a powder.
The powder was charged into a quartz column having an
inside diameter of 40 mm. Hydrogen was passed through it
1 335690
~ 7566-1115
- 18 -
at a rate of lS0 N ml/sec , and reduction was performed at
800 C for 30 minutes. The resulting powder (33 g)
was analyzed and was found to contain 98% MgO. This MgO
could be re-used in the step of reacting ammonium sulfate
with the metal oxide or hydroxide.
NH3 recovered was reacted with methane to
form hydrogen cyanide, and further reacted with acetone
to form acetone cyanohydrin. SO2 was recovered as
sulfuric acid. These compounds could be fully used in
the production of methyl methacrylate by the acetone
cyanohydrin method.
EXAMPLE 2
Example l was repeated except that the reaction
of the MgO slurry with ammonium sulfate was carried out
lS at 102 C using a reactor having a distillation column
at its upper part. As aqueous ammonia, 97 ~ of ammonia
could be recovered from the top of the distillation
column. The recovered MgO could be re-used. As in
Example l, NH3 and SO2 could be fully re-used.
EXAMPLE 3
Example l was repeated except that CO was used
as the reducing agent. Almost the same results as in
Example l were obtained.
EXAMPLES 4-8
Example l was repeated except that a mixture of
60 g of ammonium sulfate and 37 g of sulfuric acid
~Example 4), a mixture of 60 g of ammonium sulfate, 37 g
of sulfuric acid and 50 g of water (Example 5), a mixture
of 60 g of ammonium sulfate, 37 g of sulfuric acid, 50 g
of water and 5 g of p-toluenesulfonic anhydride (extra
pure reagent, Wako Pure Chemicals Industries, Ltd.)
(Example 6), a mixture of 60 g of ammonium sulfate, 37 g
of sulfuric acid, 50 g of water and 6.33 g of acetone-
disulfonic acid (extra pure reagent, Wako Pure Chemicals
Industries, Ltd.) (Example 7) and a mixture of 60 g of
ammonium sulfate, 37 g of sulfuric acid, 50 g of water
~. .
- 19 _ 1 335690 67566-1115
and 3.29 g of epsilon-caprolactam (extra pure reagent,
Wako Pure Chemicals Industries, Ltd.) (Example 8) were
each used instead of the ammonium sulfate in Example l.
The ratio of NH3 recovery was 96 %, 98 ~, 96 %, 96 %
and 96 %, respectively, and the ratio of SO2 recovery
was 98 %, 98 %, 102 %, 102 %, and 102 %, respectively.
Other results were almost the same as in Example l.
EXAMPLE 9
Six grams of a commercial carbon (activated
carbon, coke, petrocoke, graphite or carbon black) was
used as the reducing agent in Example l, and mixed with
115 g of the dry powder obtained in the first step. The
mixture was treated at 800 C for 30 minutes in a
stream of nitrogen at a flow rate of 150 Nml/min. The
resulting powder (33.7 g) was analyzed, and the formation
of 96 % MgO could be determined. The MgO with residual
carbon was again used. The steps could be performed
without any trouble. The residual carbon could be sepa-
rated together wit~ MgO and Mg~OH)2 at the time of
filtration under pressure. Twenty grams of sulfuric acid
was added to the separated solid, and the mixture was
stirred for 20 minutes and subjected to solid-liquid
separation. The liquid was recycled to the first step.
The solid could be re-used as the reducing agent after
drying
EXAMPLE 10
A mixture of 30 g of MgO and 17.7 g of MnO2
was used instead of 40 g of MgO in Example l. Otherwise,
the same treatment as in Example l was carried out, and
the reduction was carried out at 750 C for 30 minutes
in a hydrogen stream at a flow rate of 150 Nml/sec
There was recovered 46.3 g of a mixture of MgO and MnO2.
Other results were the same as in Example l.
EXAMPLE 11
A l-liter four-necked flask equipped with a
stirrer and a reflux condenser was charged with 56 g of
1 335690
.
- 20 - ~7566-1115
CaO and 275 g of water, and the mixture was stirred at
80 C for 60 minutes. Then, 110 g of ammonium sulfate
powder was added from the upper part of the flask, and
reacted at 80 C for 60 minutes. After the reaction,
the reaction mixture was cooled, and filtered under
pressure to separate it into a mixture of CaO, Ca(OH)2
and CaSO4 and the mother liquor. The mother liquor was
distilled to drive off NH3 and 80 % of NH3 was reused.
The remainder (20 %) was evaporated to dryness and used
as a reducing agent. The ratio of NH3 recovery was 98 %.
The separated solid product was dried by a hot air dryer
at 100 C to give 155 g of a powder. The powder was
mixed well with 6 g of carbon, charged into an amumina
tube having inside diameter of 40 mm, and reduced at
1100 C for 30 minutes in a stream of nitrogen at a flow
rate of 150 Nml/min. The yield of the reduction product
as a sulfur compound was 95 %. When CaO obtained by
reduction was recycled to the process, all the steps
could ~e carried out without a trouble.
EXAMPLE 12
In a l-liter powder mixer, 40 g of MgO, 110 g
of ammonium sulfate powder and 60 g of water were mixed
at 100 C for 90 minutes. Nitrogen was passed from the
lower part of the powder mixer at a rate of 50 Nml/minute.
Ammonia recovered from the upper part of the powder
mixer was recovered by a cooler. The ratio of recovery
of ammonia at this time was 90 %. The powder was dried
at 150 C for 30 minutes in the powder mixer, then
introduced into a quartz tube having an inside diameter
Of 40 mm, and reduced at 800C for 30 minutes in a
stream of hydrogen at a flow rate of 150 Nml/sec^ 98 %
of the resulting powder l33 g) was MgO. The off-gas
contained 8 % of NH3 gas.
EXAMPLE 13
A l-liter glass four-necked flask equipped with
a stirrer and a reflux condenser was charged with 154 g
1 3 3 5 6 9 0 ~7566-1115-
- 21 -
of BaO (cxtra pure)reagent, Wako Pure Chemicals Industries,
Ltd.) and 250 g of water, and the mixture was stirred at
80 C for 60 minutes. Thereafter, 110 g of ammonium
sulfate powder was added from the upper part of the
flask, and reacted at 80 C for 60 minutes.
After the reaction, the reaction mixture was
cooled and filtered under pressure to separate it into a
mixture of BaO, Ba(OH)2, and BaSO4, and the mother
liquor. The mother liquor was distilled to drive off
NH3- The ratio of recovery of NH3 was 97 %. The
separated solid was dried in a hot air dryer at 100 C
to give 220 g of a powder. The powder was well mixed
with 6 g of carbon, introduced into an alumina tube
having an inside diameter of 50 mm, and reduced at 1,600
C for 30 minutes in a nitrogen stream at a flow rate
of 150 Nml/min. The yield of the product as a sulfur
compound was 80 %. When BaO obtained by the reduction
was recycled to the process and again used, the indivi-
dual steps could be performed without a trouble. The
recovered NH3 did not poison an ammoxidation catalyst
for methane and cyclohexane.
EXAMPLE 14
Example 10 was repeated except that 26 g of CaO
was used instead of 17.7 g of MnO2. When the reduction
was carried out at 820 C for 30 minutes in a stream of
hydrogen at a flow rate of 150 Nml/sec , 55.4 g of a
mixture of MgO and CuO could be recovered. Other results
were the same as in Example 10.
EXAMPLE lS
Example 10 was repeated except that 22 g of NiO
was used instead of 17.7 g of MnO2. When the reduction
was carried out at 800 C for 30 minutes in a stream of
hydrogen at a flow rate of 100 Nml~ec., 51.4 g of a
mixture of MgO and NiO could be recovered. The results
were the same as in Example 10.
~,. =
, .. ~ .
~ 1 335690
- 22 - 67566-1115
EXAMPLE l~
Example 10 was repeated except that 12.2 g of
ZnO was used instead of 17~7 g of MnO2. When the
reduction was carried out at 800 C for 30 minutes in a
stream of a mixture of CO ~50 Nml/sec:) and nitrogen (100
Nml/min.), 41 g of a mixture of MgO and ZnO could be
recovered. Other results were the same as in Example 10.
COMPARATIVE EXAMPLE 1
ZnO ~40.68 g), 55 g of ammonium sulfate powder
and 6 g of activated powder were mixed, introduced into a
quartz tube having an inside diameter o 40 mm, and
heated to 410 C. During the reaction, nitrogen was
passed at a flow rate of 150 Nml/min. for 20 minutes.
The discharged gas was cooled and condensed and caused to
be absorbed by water. The NH3 recovery ratio from the
- recovered water was 9S %. When the recovered ammonia was
again used in the process, the catalyst for ammoxidation
of methane and cyclohexane was poisoned.
COMPARATIVE EXAMPLE 2
Comparative Example l was repeated except that
3 g of acetonedisulfonic acid was further added as a raw
material. The recovered aqueous ammonia contained acetone
and SO2. When the recovered aqueous ammonia was again
used, the activity of the ammoxidation reaction catalyst
was reduced.
EXAMPLE 17
Three 10-liter stainless steel reactors equip-
ped with a stirrer and a jacket were connected to each
other in series. MgO preheated to 80 C and a 35.3 %
by weight aqueous solution of ammonium sulfate were fed
by a screw feeder and a supply pump to a first-stage
reactor at a flow rate of l.S kg/hr and 13.3 kg/hr,
respectively. Steam under 4 kg/cm2-G was fed to the
jacket of a second-stage reactor at a rate of 2.1 kg/hr.
The vapor generaed from the second-stage reactor wa~ fed
to the jacket of the first-stage reactor, and the vapor
1 335690
- 23 -
generated from the first-stage reactor was supplied to
the jacket of a third-stage reactor. The temperatures in
the reactors we~e set at 80, 100 and 60 C, and the
residence time in each reactor was maintained at 20
minutes. The third-stage reactor and its jacket were
maintained under reduced pressure, and the vapor was
cooled and condensed and mixed with aqueous ammonia
condensed through the jacket of the first-stage reactor
to recover aqueous ammonia. The concentration of ammonia
was 17.4 % by weight, and the aqueous ammonia was further
purified by a customary method. From the third-stage
reactor, MgS04, H20, ammonium sulfate and MgO were
obtained at a rate o~ 4.3 kg/hr, 3.34 kg/hr, O.OS kg/hr
and 0.09 kg/hr, respectively. When the mother liquor
lS obtained by solid-liquid separation was recycled to the
first-stage reactor snd the reaction was carried out, the
ratio of ammonia recovery was more than 99 mole ~, and
the amount of steam consumed was 0.525 kg/kg-MgS04.
When the recovered ammonia was used in the ammoxidation
reaction o~ metane, no problem such as catalyst poisoning
arose. When the reaction was carried out without recycl-
ing the mother liquor, the ratio of ammonia recovery was
94 mole %. When the recovered ammonia was used in the
ammoxidation of methane, no problem such as catalyst
poisoning arose. From the third-stage reactor,
MgS04, H20, ammonium sulfate and MgO were obtained at
a rate of 4.0 kg/hour, 3.9 kg/hour, 0.28 kg/hour and 0.16
kg/hour, respectively. The amount of steam consumed was
0.525 kg/kg-MgS04.
EXAMPLE 18
One reactor of the type used in Example 17 was
used, and MgO pre-heated to 80 C and a 35.3 % by
weight aqueous s,olution of ammonium sulfate were fed at a
rate of 0.75 ~ .r- and 6.65 kg/hour. Steam under 4
kg/cm2-G was supplied to the jacket of the reactor at a
rate of 3.15 kg/hour. The reactor was maintained under
- 24 - I 3 3 5 6 9 0
500 torr, and the vapor was cooled and condensed. The
average residence time in the reactor was set at 60
minutes. The reaction mixture obtained by the reaction
was subjected to solid-liquid separation. When the
mother liquor was recycled to the reactor and the re-
action was carried out, the ratio of ammonia recovery was
97 mole %, and the concentration of ammonia was 16.2 % by
weight. The amount of steam consumed was 1.575 kg/kg-
MgSO4. When the reaction was carried out without
recycling the mother liquor, the ratio of ammonia recovery
was 85 mole %.
EXAMPLE 19
In Example 17, steam was supplied to the jacket
of the second-stage reactor and maintained at 100 C.
The vapor generated from the second-stage reactor was
used as a heat source for the third-stage reactor. The
vapor generated from the third-stage reactor set at a
reaction temperature of 60 C was used as a heat source
for the first-stage reactor. The first-stage reactor was
operated at 40 C. The ratio of ammonia recovery was
98 mol %, and the concentration of ammonia was 16.6 % by
weight. When the reaction was carried out without recycl-
ing the mother liquor, the ratio of ammonia recovery was
87 mole %.
EXAMPLE 20
In Example 17, steam was fed to the jacket of
the third-stage reactor. The vapor generated from the
third-stage reactor was fed to the jacket of the second-
stage reactor. The vapor generated from the second-stage
reactor was used as a heat source for the first-stage
reactor. The process was operated while maintaining the
first-stage, second-stage and third-stage reactors at
110, 130 and 150 C. The ratio of ammonia recovery was
90 mole % and the concentration of ammonia was 15 ~ by
weight. When the reaction was carried out without re-
cycling the mother liquor, the ratio of ammonia recovery
was 78 mole %.
- 25 _ 1 3 3 5 6 9 0
EXAMPLE 21
A l-liter glass reactor equipped with a stir-
rer, a thermometer and a heater and having a port for
liquid withdrawal and a port for gas discharge was
charged with 500 g of magnesium sulfate heptahydrate and
20 g of water. With stirring, the temperature was raised
to 140 C. After temperature elevation, the tempera-
ture was maintained for 60 minutes. The mixture was
filtered through a pressure-resistant filter under an
inside pressure of 2 kg/cm2-G and a differential pres-
sure of 1 kg/cm2. It was further filtered under a
nitrogen pressure (differential pressure 1 kg/cm2), and
the pressure was lowered to atmospheric pressure. There
was obtained 195 g of a solid product. The solid product
contained 18 % by weight of water, and 65.5 % of magne-
sium sulfate could be recovered.
EXAMPLE 22
As in Example 21, the temperature was raised to
140 C, and this temperature was maintained for 60
minutes. Then, the pressure was lowered to atmospheric
pressure over ~he course of 5 minutes, and immediately
then, the mixture was filtered through a filter under
reduced pressure. There was obtained 210 g of a solid
product containing 27 % by weight of water. Furthermore,
63 % of magnesium sulfate could be recovered.
EXAMPLE 23
Example 21 was repeated except that the tempera-
ture maintained was changed to 170 C. There was
obtained 250 g o~ a solid product containing 18 % by
weight of water. Furthermore, 84 % of magnesium sulfate
could be recovered.
EXAMPLE 24
Example 21 was repeated except that the tempera-
ture maintained was changed to 120 C. There was
obtained 165 g of a solid product containing 22 % by
weight of water. Furthermore, 53 ~ of magnesium sulfate
could be recovered.
1 335690
- 26 -
EXAMPLE 25
Example 21 was repeated except that the tem-
perature maintained was changed to 170 C, and after
temperature elevation, the pressure was immediately
lowered. There was obtained 154 g of a solid product
containing 21 % by weight of water. Furthermore, 50 % of
magnesium sulfate could be recovered.
EXAMPLE 26
Example 21 was repeated except that the tem-
perature maintained was changed to 90 C, and solid-
liquid separation was carried out at 85 C. There was
obtained 61 g of a solid product containing 42 % by
weight of water. Furthermore, 20 % of magnesium sulfate
could be recovered.
EXAMPLE 27
Example 21 was repeated except that the tem-
perature maintained after temperature elevation was
changed to 220 QC. There was obtained 183 g of a solid
product containing 20 % by weight of water. Some magne-
sium sulfate tended to adhere to the heated surface.
EXAMPLE 28
Example 17 was repeated except that the tem-
peratures in the first-stage, second-stage and third-
stage reactors were set at 80, 100 and 120 Cs steam
under 4 kg/cm2-G was supplied to the jacket of the
third-stage reactor at a rate of 2.1 kg/hour; the vapor
generated from the third-stage reactor was supplied to
the jacket of the second-stage reactor; and the vapor
generated from the second-stage reactor was supplied to
the jacket of the first-stage reactor.
From the third-stage reactor, MgSO4, H2O,
ammonium sulfate and MgO were obtained at a rate of 4.3
kg/hr, 3.34 kg/hr, 0.05 kg/hr and 0.09 kg/hr, respec-
tively.
Then, 140 kg of the liquid of the above compo-
sition obtained ~rom the third-stage reactor was charged
1 335690
- 27 -
into a temperature and pressure elevating machine con-
sisting of a 200 liter stainless steel tank equipped with
a stirrer and a heater, heated to 140 C and maintained
at this temperature for 60 minutes. Then, quickly, the
liquid was lowered in pressure in a pressure lowering
machine composed of another tank. It was subjected to a
centrifugal liquid-solid separator ~Model HS-204 LS, made
by Ishikawajima Harima Heavy Industries Co., Ltd.) at a
temperature of 105 C, an acceleration speed of 3,000 G
and a feed rate of 300 liters/hour to separate magnesium
sulfate monohydrate. The recovered magnesium sulfate
contained 20 % by weight of water, and 70 % of magnesium
sulfate could be recovered.
The ratio of ammonia recovery was 99 mole %,
and the concentration of ammonia was 17.4 ~ by weight.
When the recovered ammonia was used in the ammoxidation
reaction of methane, no particular problem arose.
When the reaction was carried out without
recycling the mother liquor, the ratio of ammonia re-
covery was 92 mole %.
The slurry of magnesium sulfate monohydrate wasmixed with petrocoke in a mole ratio to magneisum sulfate
monohydrate of 1.4 by using a carbon mixer (a double arm
kneader S5-3 type made by Moriyama Mg. Works, Ltd.).
The mixture was fed to a stainless steel solidifying
machine (of the type in which cooling water was passed
through a rotating drum having a diameter o 500 mm and
an inner capacity of 500 liters and the slurry of magne-
sium sulfate added dropwise on the surface of the drum is
scraped by a scraper~ by means of a screw feeder equipped
with a hopper, and cooled and solidified. The solidified
product was pulverized by a hammer mill (Hammer mill H-12
type made by Hosokawa Micron Corporation) to obtain black
magnesium sulfate having an average particle diameter of
0.75 mm
- 28 _ 1 33~690
Alumina particles having an average particle
diameter of 0.7 mm (0.8 liter) was introduced into a
stainless steel fluidized bed fully kept at an elevated
temperature and ahving an inside diameter of 100 mm, and
fully heated with a combustion gas of LPG. When the
inside temperature reached 950 C, the supply of LPG
was stopped. The above black magnesium sulfate was fed
to the fluidized bed at a rate of 1.57 kg/hour. At the
same time, air was supplied from the lower part of the
fluidized bed at 1160 Nl/hour (the ratee 0.83 times the
amount required for burning carbon). About 3 hours later
when the reaction was stabilized, a cyclone was set up at
the exit of the fluidized bed and scattering MgO was
collected. The magnesium collected was 99 ~ by weight of
the magnesium fed. The collected MgO had a purity of 95
mole ~.
The ratio of SO2 recovery from the gas current
flowing from the cyclone was 94 %. This SO2 was recover-
ed as H2SO4 in a customary manner. The recovered
2~ ammonia and sulfuric acid could be fully used as in
Example 1. MgO could be fully re-used in the reaction of
ammonium sulfate in accordance with this invention.
According to this invention, NH3 and SO2 of
good purity can be efficiently produced from ammonium
sulfate. Since they can be re-used as starting materials,
the process of this invention is useful not only as a
measure of pollution control, but also in industries
yielding large quantities of ammonium sulfate, for example
in the production of methyl methacrylate by the acetone
cyanohydrin method or by the ethylene cyanohydrin method,
and in the production of epsilon-caprolactam.