Note: Descriptions are shown in the official language in which they were submitted.
- GC155f
-1- 1 3 3 8 6 7 0
B-LAcTAM ANTIBIOTICS
This invention is directed to a novel
family of B-lactam antibiotics, and to the use
of such compounds as antibacterial agents.
It has been discovered that the B-lactam
nucleus can be biologically activated by a
sulfonic acid salt substituent attached to the
nitrogen atom in the nucleus.
B-Lactams having a sulfonic acid, or a salt
chereof, substituent in the l-position and an
acylamino substituent in tne 3-position exhibit
activity against a range of gram-negative and
gram-positive bacteria.
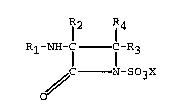
15The preferred members of the novel family
of B-lactam antibiotics of this invention are
those encompassed by the formula
I _2 -4
20Rl-NH-C- -C-R3
// N-SO3M
o
In addition to the above described B-lactams
having a sulfonic acid, ~r a salt thereof, substi-
tuent in the l-position and an acylamino substituent
in the 3-position, this invention also encompasses
~-lactams naving a sulfonic acid, or a salt thereof,
substituent in the l-position and an amino substi-
tuent in the 3-position.
,,, ~
G(~-I55f
_ 2 1 338670
The preferred compounds of this type have the
formula
Ia R2 _4
NH -C C-R3
0// N-S03M
These compounds are intermediates useful for
the preparation of corresponding 3- acylamino
compounds.
As used in formulas I and Ia, and through-
-out the specification, the symbols are as defined
below:
Rl is acyl;
R2 is hydrogen or alkoxy of 1 to 4 carbons;
R3 and R4 are the same or different and each
is hydrogen, `alkyl, cycloalkyl, phenyl or sub-
stituted phenyl, or one of R3 and R4 is hydrogen
and the other is alkoxycarbonyl, alken-l-yl,
alkyn-l-yl, 2-phenylethenyl or 2-phenylethynyl;
~4 is hydrogen or a cation.
1 338670
The terms "alkyl" and "alkoxy" refer to both straight and
branched chain groups. Those groups having 1 to 10 carbon atoms
are preferred.
The terms "cyclalkyl" and "cycloalkenyl" refer to cyclo-
alkyl and cycloalkenyl groups having 3, 4, 5, 6, or 7 carbonatoms.
The term "alkenyl" refers to both straight and branched
chain groups. Those groups having 2 to 10 carbon atoms are
preferred.
The term "halogen" refers to fluorine, chlorine, bromine
and iodine.
The term "substituted phenyl" refers to a phenyl group
substituted with one, two or three amino, halogen, hydroxyl,
trifluoromethyl and lower alkyl or alkoxy groups.
The term "protected carboxyl" refers to a carboxyl group
which has been esterified with a conventional ester protecting
group. These groups are well known in the art; see, for ex-
ample, United States patent 4,144,333, issued ~larch 13, 1979.
The preferred protected carboxyl groups are benzyl, benzhydryl
and t-butyl esters.
The term "acyl" includes all organic radicals derived
from an organic acid (i.e., a carboxylic acid) by removal of
the hydroxyl group. Certain acyl groups are, of course, pre-
ferred, but this preference should not be viewed as a limita-
tion of the scope of this invention. Exemplary acyl groupsare those acyl groups which have been used in the past to
acylate ~-lactam antibiotics including 6-aminopenicillanic
acid and derivatives and 7-aminocephalosporanic acid and der-
ivatives; see, for example, Cephalosporins and Penicillins,
edited by Flynn, Academic Press (1972), German Offenlegungs-
schrift 2,716,677 published October 10, 1978, Belgian patent
867,994, published December 11, 1978, United States patent
4,152,432, issued
--4--
1 338670
-
May 1, 1979, United States patent 3,971,778,
issued July 27, 1976, United States patent
4,172,199, issued October 23, 1979, and British
patent 1,348,894, published March 27, 1974.
The following list of acyl groups is presented
to further exemplify the term "acyl"; it should
not be regarded as limiting that term. Exemplary
acyl groups are:
(a) Aliphatic groups having the formula
R -C-
wherein R5 is alkyl; cycloalkyl; alkoxy; alkenyl;
cycloalkenyl; cyclohexadienyl; or alkyl or alkenyl
substituted with one or more halogen, cyano, nitro,
amino, mercapto, alkylthio, or cyanomethylthio groups.
(b) Carbocyclic aromatic groups having the
formula
R7
~\~(CH2 ) n~C~ '
R7
6\ ~ f H-CI -
Rg
R7
6~CH2_o_C_
GCi55f
_5_ 1 338670
6 ~ O-CH2-C-
R
~6 ~ S-CH2-C- or
R
8
~ CH -S-C-
wherein n is 0, 1, 2 or 3; R6, R7, and R8 each
is independently hydrogen, halogen, hydroxyl,
nitro, amino, cyano, trifluoromethyl, alkyl
of 1 to 4 carbon atoms, alkoxy of 1 to 4 carbon
atoms or aminomethyl; and Rg is amino, hydroxyl, a
carboxyl salt, protected carboxyl, formyloxy,
a sulfo salt, a sulfoamino salt, azido, halogen,
hydrazino, alkylhydrazino, phenylhydrazino, or
[(alkylthio)thioxomethyl]thio.
Preferred carbocyclic aromatic acyl groups
include those having the formula
H ~
~ C 2
H2NH2
1 33867~ GC155f
HO ~ CH-C- (Rg is preferably
a carboxyl salt or sulfo salt) and
. ~ CH-C- (Rg lS preferably
a carboxyl salt or sulfo salt).
(c) lleteroaromatic groups having the
formula O
Rlo (CH2)n
O
~oCH~C~
Rg O
Rlo-o-cH2 -C-
Rlo-S-CH2-C- , or
It 11
Rlo--C--C-
wherein n is 0, 1, 2 or 3; Rg is as defined
above; and Rlois a substituted or unsubstituted
5-,6-or ~membered heterocyclic ring containing
1, 2, 3 or 4 (preferably 1 or 2) nitrogen,
oxygen and sulfur atoms. Exemplary heterocyclic
GC155f
--7
1 338670
rings are thienyl, furyl, pyrrolyl, pyridinyl,
pyrazinyl, thiazolyl, morpholinyl, pyrimidinyl
and tetrazolyl. Exemplary substituents are
halogen, hydroxyl, nitro, amino, cyano, trifluoro-
methyl, alkyl of 1 to 4 carbon atoms, or alkoxyof 1 to 4 carbon atoms.
Preferred heteroaromatic acyl groups
include those groups of the above formulas
wherein Rlois 2-amino-4-thiazolyl, 2-amino-5-halo-
4-thiazolyl, 4-aminopyrimidin-2-yl, 5-amino-1,2,4-
thiadiazol-5-yl, 2-thienyl or 2-furanyl.
(d) 1[(4-Substituted-2,3-dioxo-1-piperazinyl)-
carbonyl~amino]arylacetyl groups having the formula
l l ~
-C-CH-NH-C-N N-R12
11 ~
O O
wherein R~ is an aromatic group (including
carbocyclic aromatics such as those of the formula
and heteroaromatics as included within the definition
of Rld;and R12 is alkyl, substituted alkyl
(wherein the alkyl group is substituted with one
or more halogen, cyano, nitro, amino or mercapto
gro-ups), arylmethyleneamino (i.e., -N=CH-Rll
wherein Rll is as defined above), arylcarbonylamino
O
(l.e., -NH-C-Rll wherein Rllis as defined above)
or alkylcarbonylamino.
GC155f
_ -8- 1 3 3 8 6 7 0
Preferred [[(4-substituted-2,3-dioxo-1-
piperazinyl)carbonyl]amino~arylacetyl groups
include those wherein R12 is ethyl, phenylmethylene-
amino or 2-furylmethyleneamino.
(e) (Substituted oxyimino)arylacetyl groups
having the formula
o
-C-C=N-O-R13
Rll
wherein Rllis as defined above and R13 is hydrogen,
alkyl, cycloalkyl, alkylaminocarbonyl, arylamino-
o
carbonyl (i.e., -C-NH-Rll wherein Rllis as defined
above) or substituted alkyl (wherein the alkyl
group is substituted with 1 or more halogen,
cyano, nitro, amino, mercapto, alkylthio,
aromatic group (as defined by Rl~, carboxyl
(including salts thereof), amido, alkoxycarbonyl,
phenylmethoxycarbonyl, diphenylmethoxycarbonyl,
hydr~oxyalkoxyphosphinyl, dihydroxyphosphinyl,
hydroxy (phenylmethoxy)phosphinyl, or dialkoxy-
phosphinyl substituents).
Preferred (substituted oxyimino)aryl-
acetyl groups include those wherein Rllis
2-amino-4-thiazolyl. Also preferred are those
groups wherein R13 is methyl, ethyl, carboxy-
methyl, or 2-carboxyisopropyl.
(f) (Acylamino)arylacetyl groups having
the formula
O O
Il ll
-C-CH-NH-C-R14
Rll
wherein Rllis as defined above and R14 is
- GC155f
_g_
1 338670
R7
R ~ (CH2)n~~, amino, alkylamino,
(cyanoalkyl)amino, amido, alkylamido, (cyanoalkyl)-
NH NH2 O
amido, -CH2-NH-C ~ , -CEI-CH2-C-NH-CH3 ,
~ so2-N(C~2 CH2 OH)2 ~ ~ CH3
OH
OH OH
~ ~ N~ ~ N N -Cll
Preferred (acylamino)arylacetyl groups
of the above formula include those groups
wherein R14 is amino, or amido. Also preferred
are those groups wherein Rllis phenyl or 2-thienyl.
(g) [[[3-Substituted-2-oxo-1-imidazoli-
dinyl]carbonyl]amino]arylacetyl groups having the
formula o
o O C
Il 11 / ~
-C-CH-NH-C-N N-R15
Rll CH CH
GC155f
-lo- 1 3 3 8 6 7 0
wherein Rllis as defined above and R15 is hydrogen,
alkylsulfonyl, arylmethyleneamino (i.e.,
-N=CH-~lwherein Rllis as defined above),
o
-C-R16 (wherein R16 is hydrogen, alkyl or
halogen substituted alkyl), aromatic group (as
defined by Rllabove), alkyl or substituted alkyl
(wherein the alkyl group is substituted with one
or more halogen,cyano, nitro, amino or mercapto
groups).
Preferred [[3-substituted-2-oxo-1-imidazoli-
dinyl]carbonyl]amino]arylacetyl groups of the
above formula include those wherein Rllis phenyl
or 2-thienyl. Also preferred are those groups
wherein Rls is hydrogen, methylsulfonyl, phenyl-
methyleneamino or 2-furylmethyleneamino.
The term "cation", as used throughout the
specification, refers to any positively charged
atom or group of atoms. The "-SO3M~" substituent
on the nitrogen atom of the ~-lactams of this
invention encompasses all sulfonic acid salts.
Pharmaceutically acceptable salts are, of
course, preferred, although other salts are also
useful in purifying the products of this invention
or as intermediates for the preparation of
pharmaceutically acceptable salts. The cationic
portion of the sulfonic acid salts of this
invention can be obtained from either organic
or inorganic bases. Such cationic portion
includes, but is not limited to, the following
ions: ammonium; substituted ammonium, such as
alkylammonium (e.g., tetra-n-butylammonium,
referred to hereinafter as tetrabutylammonium);
alkali metal, such as lithium, sodium and potassium;
alkaline earth metal, such as calcium and magnesium;
- 11 - 1 338~70
pyridinium; dicyclohexylammonium; hydrabaminium; benzathinium;
N-methyl-D-glucaminium.
As set forth in formula I, and in the definitions
following formula I, M~ can be hydrogen.
As is described hereinafter, the ~-lactams of this
invention can be prepared by synthetic means. The non-alkoxy-
lated 4-unsubstituted ~-lactams of formula I, l.e., those
compounds of formula I wherein R2, R3 and R4 are hydrogen can
be prepared using 6-aminopenicillanic acid or a 6-acylamino-
penicillanic acid as a starting material. The ~-lactams of
formula I wherein R2 is alkoxy can be prepared from the corres-
ponding non-alkoxylated ~-lactam. Some of the compounds of
this invention may be crystallized or recrystallized from sol-
vents containing water. In these cases water of hydration
may be formed. This invention contemplates stoichiometric
hydrates as well as compounds containing variable amounts of
water that may be produced by processes such as lyophiliza-
tion.
Some of the ~-lactams of formula I have also been
prepared by biological means. Cultivation of a strain of
the microorganism Chromobacterium violaceum SC 11,378, yields
a salt of (R)-3-(acetylamino)-3-methoxy-2-oxo-1-azetidine-
sulfonic acid. Cultivation of various acetic acid bacteria
e.g., Gluconobacter species SC 11,435, yields a salt of (R)-
3-[[N-(D-~-glutamyl)-D-alanyl]amino]-3-methoxy-2-oxo-1-aze-
tidinesulfonic acid.
GC155f
-12- 1 3 3 8 6 7 0
B-Lactams having a sulfonic acid salt
substituent in the l-position and an amino or
acylamino substituent in the 3-position contain
at least one chiral center the carbon atom
(in the 3-position of the B-lactam nucleus) to
which the amino or acylaminosubstituent is
attached. This invention is directed to those
B-lactams which have been described above, wherein
the stereochemistry at the chiral center in
the 3-position of the B-lactam nucleus is the
same as the configuration at the carbon atom
in the 6-position of naturally occurring penicillins
(e q., penicillin G) and as the configuration at
the carbon atom in the 7-position of naturally
occurring cephamycins (e.g., cephamycin C).
With respect to the preferred B-lactams
of formulas I and Ia, the structural formulas
have been drawn to show the stereochemistry
at the chiral center in the 3-position. Because
of the nomenclature convention, those compounds
of formulas I and Ia wherein R2 is hydrogen
have the S configuration and those compounds
of formulas I and Ia wherein R2 is alkoxy have
the R configuration.
Also included within the scope of this
invention are racemic mixtures which contain
the above-described B-lactams.
- ~ 338670
-12a-
The invention as claimed herein is a process
for preparing a ~-lactam having a sulfonic acid sub-
stituent -SO3M wherein M is hydrogen or a cation in
the l-position and an amino substituent -NH2 which
may optionally be acylated or protected with a read-
ily removable protecting group in the 3-position with
the proviso that when the ~-lactam ring is unsubsti-
tuted in the 4-position, the second valency at the
3-position is not hydrogen or methoxy, said process
being characterized by sulfonating a corresponding
~-lactam which is unsubstituted in the l-position or
acylating a corresponding ~-lactam which has the amino
group in the 3-position.
A preferred substituent in the 4-position in
the above process is an alkyl radical, particularly
a methyl radical, and the process is especially val-
uable when used to prepare a ~-lactam of the for-
mula
R2 R4
R ~ - R3
~ N-SO3H
or a pharmaceutically acceptable salt thereof wherein:
R is an amino group which may optionally be
acylated or protected with a readily remov-
able amino protecting group;
R2 is hydrogen or alkoxy of 1 to 4 carbon atoms;
R3 and R4 are the same or different and each is
Cl-C10 alkyl, C3-C7 cycloalkyl, phenyl or
phenyl substituted with 1 to 3 amino, halo-
gen, trifluoromethyl or Cl-C4 alkyl or al-
koxy groups or one of R3 and R4 is hydrogen
and the other is Cl-C10 alkyl, C3-C7 cyclo-
,
-12b- 1 3 3 ~
alkyl, phenyl, optionally bearing substi-
tuents as hereinbefore defined, Cl-C10 al-
koxycarbonyl, C2-C10 alken-l-yl, C2-C10
alkyn-l-yl, 2-phenylethenyl or 2-phenyl-
ethynyl.
The invention as claimed herein is further de-
fined as a process for preparing a compound of the
formula
R2 R4
R - ~ R3
~ N-S03H
o
or a pharmaceutically acceptable salt thereof wherein:
R is an amino group which may optionally be
acylated or protected with a readily remov-
able amino protecting group;
R2 is hydrogen or alkoxy of 1 to 4 carbon atoms;
one of R3 and R4 is hydrogen and the other is
g , Cl C10 alkyl, C3-C7 cycloalkyl
phenyl optionally substituted with 1 to 3
amino, halogen~ trifluoromethyl or Cl-C4
alkyl or alkoxy groups, Cl-C10 alkoxycarbonyl,
C2-C10 alken-l-yl, C2-C10 alkyn-l-yl, 2-
phenylethenyl or 2-phenylethynyl;
characterized by cyclizing a compound of the formula
R .~2 ~ R4
R3
oD NH-SO3H
~ -12c- 1 3 3 8 6 7 0
or a salt thereof wherein:
V is a methanesulfonyl, benzenesulfonyl,
toluenesulfonyl, chloro, bromo or iodo
type leaving group; and
R is an amino group which is acylated or
protected and removing said protecting
group to form a compound wherein R is
amino and acylating said compound to form
a product wherein R is acylated amino.
Preferred substituents in the process immed-
iately above are those wherein R2 is hydrogen and
one of R3 and R4 is hydrogen and the other is an
alkyl radical, particularly a methyl radical.
GC155f
_ -13-
1 338670
~ -Lactams having a sulfonic acid salt
substituent in the l-position of the ~-lactam
nucleus and an amino or acylamino substituent in
the 3-position of the ~-lactam nucleus have activity
against a range of gram-negative and gram-positive
organisms. The sulfonic acid salt substituent is
essential to the activity of the compounds of this
invention. The compounds wherein R3 and/or R4 are
- hydrogen or alkyl, especially methyl, exhibit
especially useful activity.
The compounds of this invention can be used as
agents to combat bacterial infections (including
urinary tract infections and respiratory infections~
in m~mm~ 1 ian species, such as domesticated animals
(e.g., dogs, cats, cows, horses, and the like) and
humans.
For combating bacterial infections in mammals
a compound of this invention can be administered to
a mammal in need thereof in an amount of about
1.4 mg/kg/day to about 350 mg/kg/day, perferably
about 14 mg/kg/day to about 100 mg/kg/day. All
modes of administration which have been used in the
past to deliver penicillins and cephalosporins to
the site of the infection are also contemplated for
use with the novel family of ~-lactams of this
invention. Such methods of administration include
oral, intravenous, intramuscular and as a suppository.
GC155f
,
-14- 1 3 3 8 6 7 0
The ~-lactam products of this invention are
generally prepared by the introduction
of a sulfonic acid substituent (a sulfo group -SO3-)
onto the nitrogen atom in the l-position of the
~-lactam nucleus. This sulfonation reaction is readily
effected by treating the ~-lactam with a sulfur trioxide
complex or with an e~uivalent sulfonating reagent such
as a chlorosulfonate.
The sulfur trioxide complexes most commonly used
are pyridine-sulfur trioxide; lutidine-sulfur trioxide;
dimethylformamide-sulfur trioxide; and picoline-sulfur
trioxide. Instead of using a pre-formed complex, the
complex can be formed in situ, e.g., using chloro-
sulfonyl-trimethylsilyl ester and pyridine as reagents.
Alternatively, sulfonation can be effected by way of an
intermediate compound as for example first silylating
the nitrogen atom of the ~-lactam nucleus and then
subjecting the silated compound to a silyl interchange
reaction with trimethylsilylchlorosulfonate or a similar
reagent. Exemplary silylating agents are monosilyltri-
fluoroacetamide, trimethylsilylchloride/triethylamine
and bis-trimethylsilyl trifluoroacetamide.
Generally, the sulfonation reaction is carried out
in the presence of an organic solvent such as pyridine or
a mixture of organic solvents, preferably a mixture of a
polar solvent such as dimethylformamide and a halogenated
hydrocarbon such as dichloromethane.
The product initially formed in the sulfonation
reaction is a salt of the sulfonated ~-lactam.
When pyridine-sulfur trioxide is the sulfonating complex
the product initially formed is the ~-lactam-sulfonated
pyridinium salt of the sulfonated ~-lactam wherein M+ in
the formula below is the pyridinium ion:
/ ~-SO3 M
_ GC155f
-15- 1 3 3 8 6 7 ~
These complexes can be converted to other sulfonic
acid salts using conventional techniques (e.g., ion-
exchange resins, crystallization or ion-pair extraction.
These conversion techniques are also useful in purifying
the products. Conversion of the pyridine salt to the
potassium salt using potassium phosphate or potassium
ethyl hexanoate; to the tetrabutylammonium salt using
tetrabutyl ammonium hydrogen sulfate; or to a zwitterian
(M+ = hydrogen) using formic acid; are particularly
useful.
It should be appreciated that the sulfonation
reaction which introduces the sulfo group onto the
nitrogen atom of the ~-lactam nucleus can be effected
at various stages of the synthesis, including inser-
tion prior to the formation of a ~-lactam nucleus,
where such procedure is followed as outlined below.
The sulfonation reaction is effected in the presence
of solvents previously described and usually at room
temperature. Where the amino function is present it is
preferably conducted with the amino function protected.
Using a benzyloxycarbonyl protecting group as an
example, the sulfonation reaction can be visualized as
follows:
'~2 ~4 ~2 R~
C6H5CH2OCO R3 ~H2N R3
0~ ~ 2)d~L~ ~on ~ N-SO3 M
II III
Other protecting groups can be used to protect the
amine function, for example, a t-butyloxycarbonyl group,
a simple acyl grou~ such as acetyl or benzoyl of phenyl-
acetyl, a triphenylmethyl group, or having the amino
function in the form of an azide group. The desired acyl
group (Rl) can then be affixed by a conventional acylation
xeaction.
- GC155f
-16-
1 338670
Exemplary acylation techniques for converting a
compound of formula IIIto a product of formula I
include reaction with a carboxylic acid ~Rl-OH), or
corresponding carboxylic acid halide or carboxylic
acid anhydride. When R2 is alkoxy, acylcation is
best effected by use of an acid chloride or acid
bromide. The reaction with a carboxylic acid proceeds
most readily in the presence of a carbodiimide such as
dicyclohexylcarbodiimide and a substance capable of
forming an active ester in situ such as N-hydroxybenzo-
triazole. In those instances when the acyl group (Rl)
contains reactive functionality (such as amino or
carboxyl groups) it may be necessary to first protect
those functional groups,then carry out the acylation
reaction, and finally deprotect the resulting product.
Alternatively, the sulfonation reaction can be effected
with the acyl group already in place, i.e.,
~2 R4 ~2 R4
Rl-NH - R3 ti ~ R -NH -- R3
sulfona ~ -S03M
O O
IV V
In the case where R2 is lower alkoxy, there can
be an additional variation in that the R2-lower alkoxy
group can be inserted after the sulfonation as well as
before the sulfonation using the conventional procedure
of chlorinating the acylated nitrogen atom in the 3-
position followed by reaction with a lower alkoxide
1 3 3 8 6 7 0 (;C155f
--.17--
Cl H R=4 (R2)alkoxide R2 R4
acyl-N l R3 ~ acyl-NH l R3
~-S03 M s5: S03
VI . . V
The acyl groups in the above reactionalsoin~ easily
removable groups which function as a protecting group
and which can be removed after the reaction to afford
the "deacylated" product (-NH2).
In addition, the ~-lactam ring can be formed via
a cyclization reaction and the sulfonation reaction
can be éffected prior to cyclization as well as
subsequent thereto, i.e.,
2 0 R4 > '~
acyl-NH ~R cyclization acyl-NH ~3
S03 M C 3
VII
wherein V is a leaving group such as methanesulfonyl,
benzenesulfonyl, toluenesulfonyl, chloro, bromo or
iodo. The acyl group in this reaction can also be an
easily removable group which functions as a protect-
ing group and which on removal affords the -NH2 pro-
duct.
GC155f
-18- 1 338670
The azetidinone starting materials wherein R2
is hydrogen and at least one of R3 and R4 is hydrogen
can also be prepared from amino acids having the formula
~H
NH2 ¦ / 4
XII H C
R3
~C OH
o
(at least one of R3 and R4 is hydrogen). The amino
group is first protected with a classical protecting
group, e.g., t-butoxycarbonyl (referred to hereinafter
as "Boc"). The carboxyl group of the protected amino
acid is then reacted with an amine salt having the
formula
XIII Y-O-NH3Cl
wherein Y is alkyl or benzyl, in the presence of a
carbodiimide to yield a compound having the formula
OIH~ R4
XIV BOC-NH-CH C
¦ 3
O"C NH-O-Y
GC155f
-19- 1 3 3 8 6 7 o
(at least one of R3 and R4 is hydrogen). The hydroxyl
group of a compound of formula XIV is converted to a
leaving group (V) with a classical reagent, e.g.,
methanesulfonyl chloride (methanesulfonyl is referred
to hereinafter as "Ms"). Other leaving groups (V)
which can be utilized are benzenesulfonyl, toluene-
sulfonyl, chloro, bromo and iodo.
The fulIy protected compound having the formula
BOC-NH- H
XV ~ R3
O~C NH-O-Y
(at least one of R3 and R4 is hydrogen) is cyclized
by treatment with base, e.g., potassium carbonate.
The reaction is preferably carried out in an organic
solvent such as acetone, under reflux conditions,
and yields a compound having the formula
R4
BOC-NH I = R3
XVI
~ N-O-Y
(at least one of R3 and R4 is hydrogen).
Alternatively, cyclization of a compound of
formula XIV can be accomplished without first
converting the hydroxyl group to a leaving group.
Treatment of a compound of formula XIV with triphenyl-
phosphine and diethylazodicarboxylate, yields a
compound of formula XVI wherein at least one of R3
and R4 is hydrogen.
GC155f
-20- 1 3 3 8 5 7 0
Removal of the protecting group from the 1-
position of an azetidinone of formula XVI can be
accomplished via sodium reduction when Y is alkyl,
and yields an intermediate having the formula
R4
BOC-NH - R3
XVII
~ - NH
(at least one of R3 and R4 is hydrogen). If Y is
benzyl, catalytic (e.g., palladium on charcoal)
hydrogenation will initially yield the corresponding
N-hydroxy compound, which upon treatment with titanium
trichloride yields an intermediate of formula XVII
wherein at least one of R3 and R4 is hydrogen.
Synthesis involving ring closure of the type
described above results in inversion of the stereo-
chemical configuration of the R3 and R4 substituents.
As previously mentioned, the above azetidinone can
be sulfonated to form a com?ound having the formula
-4
XVIII BOC-NHI 3
o~ - N-S03 M
(at least one of R3 and R4 is hydrogen).
An alternative procedure for preparing a compound
of formula I wherein R2 is hydrogen and at least one
of R3 and R4 is hydrogen, utilizes as a starting
material an amino acid amide having the formula
OH
1 / R4
NH2-fH C R
XIX 3
o~,C NH2
(at least one of R3 and R4 is hydrogen).
GC155f
21
- - 1 3 3 8 6 7 0
Protection of the amino group with a classical
protecting group, e.g., benzyloxycarbonyl (reffered
to hereinafter as Z) or Boc, and conversion of the
hydroxyl group to a leaving group (V) such as Ms
yields a compound having the formula
OMs
XX ANH ICH C ~ R4
o~,C NH2 3
(at least one or R3 and R4 is hydrogen), wherein A is a
protecting group.
Sulfonation of a compound of formula XX yields a
compound having the formula
QMs
ANH CH C ",R4
XXI ¦ 3
~C NHSO3 M
(at least one of R3 and R4 is hydrogen).
Cyclization of a compound of formula XXI is
accomplished with base, e.g., potassium carbonate. The
reaction is preferably carried out in a mixture of water
and an organic solvent (e.g., a halogenated hydrocarbon
such as 1,2-dichloroethane) under reflux conditions and
yields a compound having the formula
_4
ANH
XXII
C~ N-S03 M+
(at least one of R3 and R4 is hydrogen).
Deprotection of a sulfonated azetidinone of formula
XXII , wherein A is a protecting group, as well as the
counterpart compounds previously described having an R2-
alkoxygroup by catalytic hydrogenation yields a compoundhaving the formula
- GC155f
~ -22- 1 3 3 8 6 7 0
2 _4
H2N I R3
XXIII N-SO3 M
(at least one of R3 and R4 is hydrogen), wherein R2
is hydrogen or alkoxy which can be converted to the
corresponding zwitterion having the formula
R2 _4
XXIV NH3 = 3
O~ ~-SO3
(at least one of R3 and R4 is hydrogen) by treatment
with an acid such as formic acid.
Deprotection of a sulfonated azetidinone of
formula XXII wherein A is a Boc protecting group
using acidic conditions (e.g., using formic acid)
yields the corresponding zwitterion of formula XXIV.
An excellent source for the ~lactam starting
materials are the 6-aminopenicillanic acids and the
7-aminocephalosporanic acids which can bear an optional
6 alkoxy or 7-alkoxy substituent respectively. These
compounds are of the formulae
~2~ CH3
Rl-NH ~ ~ -CH3
XXVa
O" C-COOH
~ and
R3
XXVb R ~ S ~
N ~ CH3
COOH
_ ~C155f
-23~ 1 3 3 8 6 7 0
respectively wherein R2 is hydrogen or alkoxy and Rl
is hydrogen or acyl. By adapting procedures described
in the literature, 3-amino-2-azetidinones can be
prepared; see for example, Chem. Soc. Special
Publication No. 28, pg. 288(1977); The Chemistry of
Penicillins, Princeton Univ. Press, pg. 257, and
Synthesis, 494(1977).
The 6-amino-penicillanic acid or 7-amino-
cephalosporanic acid is first desulfurized by reduction
using Raney nickel. The reaction can be run in water
under reflux conditions; the resulting compound has
the structural formula
~2
Rl-NH
XXVI
-fH-CH(CH3)2
COOH
Replacement of the carboxyl group of the compound
of formula XXVI with an acetate group followed by
hydrolysis yields a 3-amino-3-alkoxy-2-azetidinone of
formula IV wherein Rl is hydrogen or acyl, R2
is hydrogen or lower alkoxy and R3 and R4 are hydrogen.
Treatment of a compound of formula XXVI with cupric
acetate and lead tetraacetate in an organic solvent
(e.g., acetonitrile) replaces the carboxyl group with
an acetate group. Hydrolysis of the resul-ting compound
can be accomplished using potassium carbonate in the
presence of sodium borohydride.
Introduction of a sulfo group in the l-position
of the above 3-amino-3-alkoxv-2-azetidinone compound
can be accomplished by reacting the intermediate with
a complex of dimethylformamide and sulfur trioxide.
GC155f
-
-24-
1 338670
A 3-azido-2-azetidinone starting material can
be prepared by first reacting an olefin having the
formula
R14
XXVII CH C - R
with a halosulfonylisocyanate (preferably chloro-
sulfonylisocyanate) having the formula
XXVIII O = C = N - S02-halogen
to yield an azetidinone having the formula
R4
R3
XxIx
~ N-S02-halogen
Reductive hydrolysis of an azetidinone of formula
XXIX yields an N-unsubstituted ~-lactam having the
formula R4
R
XXX
O" NH
For a more detailed description of the above described
reaction sequence reference can be made to the
literature; see, for example, Chem. Soc. Rev., 5, 181
(1976) and J. Org. Chem., 35, 2043 (1970).
An azido group can be introduced in the 3-
position of an azetidinone of formula XXX (or the
sulfonated counterpart) by reaction of the compound
with an arylsulfonyl azide (such as toluenesulfonyl
azide) to obtain a starting azetidinone having the
formula
- GC155,f,
-25- 1 3 3 8 6 70
N3 = R3
XXXI
o~ NH
The reaction proceeds best by first protecting the
azeti'dinone nitrogen with a silyl residue (e.g.,
t-butyldimethylsilyl, or t-butyldiphenylsilyl), then
generating the anion at the 3-position of the nucleus
with a strong organic base (e.g., lithium diisopropyl-
amine) at a low temperature, and then treating the
anion with toluenesulfonyl azide. The resulting
intermediate is quenched with trimethylsilyl chloride,
and subsequent acid hydrolysis or fluoride solvolysis
of the N-protecting group yields the compound of
formula XXXI.
Alternatively, the compound of Formula XXXI can
be obtained by first reacting a primary amine having
the formula
H2N-CH2 ~ O-alkyl or 2N ~ O-alkyl
O-alkyl
with an aldehyde of the formula R3CH=O to yield the
corresponding Schiff base. A[2+2]cycloaddition with
an activated form of -azido acetic acid yields a 3-5 azido-2-azetidinone having the formula
R4
N ~ R3
N - Q
GC155f
-26- 1 33867
wherein Q is
-CH2 ~ O-alkyl ~ O-alkyl
or
O-alkyl
Oxidative removal of the Q-substituent yields the
compound of formula XXXI.
The 3-acylamino-2-azetidinones can be obtained
by first reducing a 3-azido-2-azetidinone of formula
XXXI to obtain the corresponding 3-amino-2-azetidinone
and then acylating the 3-amino-2-azetidinone.
As previously mentioned in the case where R2 is
lower alkoxy, the product can be prepared from the
counterpart product wherein R2 is hydrogen.
Clorination of the amide nitrogen of a nonalkoxylated
compound yields an intermediate having the formula
Cll H R4
R -N R
XXXII 1 3
~ N-SO3 M
Reagents and procedures for N-chlorinating amides are
known in the art. Exemplary reagents are tert.-butyl
hypochlorite, sodium hypochlorite, and chlorine. The
reaction can be run in an organic solvent (e.g., a
lower alkanol such as methanol) or in a two phase
solvent system (e.g., water/methylene chloride) in the
presence of a base such as sodium borate decahydrate.
The reaction is preferably run at a reduced temperature.
GC155f
-27- 1 33867~
Reaction of an intermediate of formula XXXI with
an alkoxylating agent, e.g., an alkali metal alkoxide
yields a product of formula I wherein R2 is alkoxy, in
combination with its enantiomer. The reaction can be
run in an organic solvent, e.g., a polar organic solvent
such as dimethylformamide, at a reduced temperature.
An alternative synthesis for preparing the
compounds of formula I wherein R2 is alkoxy
comprises first alkoxylating an intermediate of
formula VI wherein RlNH is a carbamate (e.g., Rl is
benzyloxycarbonyl) and R2 is hydrogen and then
introducing a sulfo group in the l-position of the
resulting compound. Chlorination of a compound of
formula VI using the procedure described above (for
chlorination of a non-alkoxylated compound of formula
I to yield a compound of formula XXXII yields an
intermediate having the formula
Cl R4
6 5 2 CO N
XXXIII 3
~ N-Cl
Using the alkoxylation procedure described above
(for converting a compound of formula XXXII to a
product of formula I), and subsequently adding a
reducing agent such as trimethylphosphite, the com-
pound of formula XXXIII can be converted to an
intermediate having the formula
O-alkyl R4
C H O-C0-NH = R
XXXIV 6 5 3
~ NH
in a combination with its enantiomer.
GC155f
28 1 338670
The above procedures yield those products of
formula I wherein R2 is alkoxy as a racemic mixture.
If desired the enantiomer having the R configuration
can be isolated from the racemic mixture using
conventional techniques such as fractional
crystallization of a suitable salt with an optically
active organic amine or by ion-paired chromatography
utilizing an optically active cation.
~C155f
_ -29- 1 338670
Biological Production of the Antibiotic ~M5117
Salts of (R)-3-(acetylamino)-3-methoxy-2-oxo.
l-azetidinesulfonic acid (formula I, Rl is acetyl
and R2 is methoxy; referred to hereinafter as
~5117) can also be prepared by cultivation of
a strain of the microorganism Chro~obacterium
violaceum SC 11,378, which has been deposited
in the American Type Culture Collèction as
A.T.C.C. No. 31532.
The Microorganism
The microorganism used for the production
of EM5117 is a strain of Chromobacterium violaceum
SC 11,378. A subculture of the microorganism
can be obtained from the permanent collection of
the American Type Culture Collection, Rockville,
~aryland. Its accession number in the repository
is A.T.C.C. No. 31532. In addition to the
specific microorganism described and characterized
herein, it should be understood that mutants of
the microorganism (e.g., mutants produced through
the use of x-rays, ultraviolet radiation or
nitrogen mustards) can also be cultured to
produce E~15117.
Chromobacterium violaceum SC 11,378, A.T.C.C.
~o. 31532 can be isolated from a moist soil sample
containing the microorganism by first stamping
- 1 338670
GC155f
-30-
the soil sample on a medium containing:
Soil Extract - 400 ml
Distilled Water- 600 ml
Yeast Extract - 5.0 g
Glucose - 10.0 g
Agar - - 17.5 g
The medium is adjusted to p~l 6.0 and sterilized
in an autoclave at 121 C for 20 minutes. After
24 to 72 hours incubation at 25C colonies of the
Chromobacterium violaceum SC 11,378 are isolated
from the plated soil. These isolated colonies
are then grown on a medium containing:
Yeast Extract - 1 g
Beef Extract - 1 g
NZ amine A - 2 g
Glucose - 10 g
Agar - 15 g
Distilled water to 1 liter
The medium is adjusted to pH 7.3 and autoclaved
at 121C for 30 minutes.
Chromobacterium violaceum SC 11,378 is a
gram negative rod often showing barring, bipolar
staining and lipid inclusions. It is motile
by a single polar flagellum with occassional
1 338670
--GC155f
--31--
lateral flaqella which are smaller in size.
On nutrient agar Chromobacterium violaceum
SC 11,378 produces violet colonies. The pigment
is enhanced on media rich in tryptophane and
S yeast extract. In nutrient broth it produces a
violet ring on the wall of the tube but no
confluent pellicle. The violet pigment is
soluble in ethanol but insoluble in water and
chloroform.
Chromobacterium violaceum SC 11,378 is
mesophilic, growing over a range of 15-37C;
no growth occurs at 4C or above 37C. Casein
is hydrolyzed strongly by the microorganism,
which is oxidase positive. In the presence of
Chromobacterium violaceun SC 11,378 glucose,
fructose and trehalose are fermented (method
of Hugh ~ Leifson, 1953). L-Arabinose is not
- utilized by the microorganism either fermentatively
or oxidatively. Hydrogen cyanide is produced by
20 the microoryanism and aesculin hydrolysis is
negative.
The above described key characteristics
provide the basis for the identification of the
microorganism as Chromobac terium violaceum as
25 distinguished from Chromobacterium lividum,
the only other species of the yenus Chromobacterium
recognized in the 8th edition of Bergey's Manual
of Determinative Bacteriology.
Additional strains of Chromobacterium
30 . violaceum can also be cultured to produce EM5117.
-
---ÇC155f
--32--
The Antibiotic
To obtain the antibiotic EMS117, Chromobacterium
violaceum SC 11,378 A.T.C.C. No. 31532 is grown
at, or about, 25C under submerged aerobic conditions
- 5 on an aqueous nutrient medium containing an
assimilable carbon and nitrogen source.
The fermentation is carried out until substantial
antibiotic activity is imparted to the medium,
usually about 18 to 24 hours, preferably about
10 20 hours.
Using the following procedure, EM5117 can
be separated from the fermentation medium and
purified. After the fermentation-has been completed
the broth can be centrifuged to remove the mycelium
lS or, alternatively, filtration can be used to remove
the mycelium from the broth. After removing the
mycelium from the broth E~.5117 can be extracted
f-rom the broth. Preferably the broth is extracted
at pH 5 by ion-pair extraction with cetyldimethyl-
20 - benzylammonium chloride in methylene chloride and
back-extracted into aqueous sodium iodide (adjusted
to pH 5 with acetic acid). After concentration of
the sodium iodide extract in~-vacuo, an
aqueous solution of the residue can be washed with
25 butanol. Concentration of the resulting aqueous
solution to dryness yields a residue whi~h is
dissolved, to the extent possible, in methanol.
Centrifugation can be used to separate insolubles
which are washed with methanol and then discarded.
.
1 338670 GC155f
-33-
Purification of the antibiotic can be
accomplished by dissolving the methanol concentrate
in methanol/water ~1:1) and applying it to a
chromatography column, e.q., one comprising
Sephadex G-10 in the same solvent mixture.
After elution with the same mixture active
fractions are combined and concentrated to
dryness. The residue is mixed with methanol
and insoluble material is filtered out and
discarded.
Further purification is achieved by applying
the methanol soluble material to a column of
DEAE cellulose. The column can be eluted
with a linear gradient.prepared from pH 5
sodium 0Ø M phosphate buffer and pH 5 sodium
0.1 M phosphate buffer. Active fractions are
combined, concentrated and methanol insoluble
materials are removed and discarded.
Still further purification of EM5117 is
accomplished by dissolving this material in
water and placing the solution on a column of
*
Sephadex LH-20, a~d eluting the column with water.
Active fractions are combined and concentrated.
Further purification of EM5117 is then acco;nplished
by dissolving the material in water, placing the
solution on a column of Diaion HP-20AG and
eluting with water. Active fractions are combined
and concentrated. The concentrate is dissolved
in water and passed through a column of ~owex 50W-X2,
potassiurn form, washing with water. The effluent
can be concentrated to yield a crystalline material
*Trade Mark
GC155f
~34~ 1 3 3 8 6 7 0
which is relatively pure potassium salt of EM5117.
The above described isolation and purification
process yields the potassium salt of EM5117. Other
salts can be prepared corresponding to the form
of the ion exchange resin utilized in the final
purification step.
Biological Pr~oduction of the Antibiotic EM5210
Salts of (R)-3-[[_-(D-y-glutamyl)-D-alanyl]-
amino]-3-methoxy-2-oxo-1-azetidinesulfonic acid
(formula I, Rl is D-~-glutamyl-D-alanyl and R2
is methoxy; referred to hereinafter as EM5210)
can also be prepared by cultivation of various
acetic acid bacteria.
The Microorganism
The microorganism Gluconobacter species
- SC11,435 can be used for the production of
EM5210. A subculture of the microorganism can
be obtained from the permanent collection of the
American Type Culture Collection, Rockville,
Maryland. Its accession number in the repository
is A.T.C.C. No. 31581. In addition to the specific
microorganism described and characterized herein,
it should be understood that mutants of the
microorganism (e.g., mutants produced through
the use of x-rays, ultraviolet radiation or
nitrogen mustards) can also be cultured to
produce EM5210.
GC155f
-35- 1 33 8 6 70
Gluconobacter species SC11,435, A.T.C.C.
No.31581 can be isolated from ground moss
containing the microor~anism by first incubating
the ground moss in a 10% aqueous pectin solution
(pH 2.5~ for 7 days at 25 C. The organism can
- then be isolated ~y plating on a medium containing:
*Extract of Spartina patens Grass 400 ml
Distilled Water 600 ml
Yeast Extract 5 g
Glucose 10 g
Crude Flake Agar 17.5 g
*Extract of Spartina patens grass is prepared
by adding 500 g of chopped, dried grass to 3 liters
of tap water, brought to a boil and simmered for
30 minutes.
The medium is adjusted to p~ 6.0 and sterilized
in an autoclave at 121C for 20 minutes. After
- about 18ho~rs incubation at 25 C, colonies of the
Gluconobacter species SC11,435 are isolated from the
plated pectin enrichment solution. These isolated
colonies are then grown on a medium containing:
Yeast Extract 1 g
Beef Extract l-g
NZ amine A 2 g
Glucose 10 g
~gar 15 g
Distilled Water to 1 liter
Tne medium is adjusted to pl~ 7.3 and autoclaved
at 121C for 30 minutes.
Gluconobacter species SC11,435 is a
pleiomorphic gram negative rod motile by means
GC155f
-36-
1 338670
of one to three polar Flagella. It is obligately
aerobic, catalase positive and oxidative. It is
differentiated from Pseudomonas in that it is
cytochrome oxidase negative and tolerant of
extremely acid conditions. These characteristics
indicate that the microorganism is more closely
related to the acetic acid bacteria than to true
Pseudomonads.
On BBL Trypticase soy agar Gluconobacter
species SC11,435 growfi a~ a mixture of rough
and smooth colony types. The rough type is
associated with a faint yellow soluble pigment
whereas the smooth type is mucoid and pigmentless.
Dissociation between the two types is influenced
by media, temperature and storage conditions.
EM5210 activity tends to decrease in cultures
- where the rough component is dominant.
On BBL Wart agar (pH 4.8) Gluconobacter
species SC11,435 grows luxuriantly as heaped up
mucoid slimy colonies. Similar growth is obtained
on ~alt-yeast extract agar (1% each) adjusted to
pH 4.5.
On medium containing 1% yeast extract, 10%
glucose, 3~ calcium carbonate and 2.5~ agar,
sufficient acid is produced from the glucose to
produce a zone of clearing of the calcium carbonate
around the growth. This is a characteristic
feature of Acetobacter and Gluconobacter.
In the presence of Gluconobacter species
SC11,435:(i) a brown soluble pigment is produced
on yeast extract-glycerol and yeast extract-calcium
GC155f
-37-
1 338670
lactate agar plates, (ii) acid is pro~ucea on
glucose, fructose, galactose, mannose, xylose,
mannitol and arabinose, but no growth or acid
production occurs on rhamnose, lactose, sucrose
or maltose, when these sugars are incorporated
into ~ugh and Leifson's medium.
The biological production of E~15210 is not
- limited to Gluconobacter species SC11,435, but
is broadly distributed throughout the acetic
bacteria. The following cultures can each be
used for the production of E~15210:
Acetobacter pasteurianus subsp. pasteurianum
A.T.C.C. 6033
Acetobacter aceti subsp. aceti A.T.C.C. 15973
Gluconobacter oxydans subsp. oxydans
A.T.C.C. 19357
Gluconobacter oxydans subsp. suboxydans
A.T.C.C. 23773
Gluconobacter oxydans subsp. oxydans
A.T.C.C. 15178
Acetobacter aceti suvsp. liquefaciens
A.T.C.C. 23751
Acetobacter peroxydans A.T.C.C. 12874
Gluconobacter oxydans subsp. suboxydans
A.T.C.C. 19441
Acetobacter sp. A.T.C.C. 21780
Gluconobacter oxydans subsp. industralis
A.T.C.C. 11894
GC155f
-
-38-
1 338670
The ~ntibiotic
To obtain the antibiotic EM5210, Gluconobacter
species S~11,435 A.T.C.C. No. 31581 or one of the
microorganisms listed above, can be grown at, or
about, 25C under submerged aerobic conditions on
an aquèous nutrient medium containing an assimilable
carbon and nitrogen source. The fermentation is
carried out until substantial antibiotic activity
is imparted to the medium, usually ab~ut 16 to 24
hours, preferably about 20 hours.
Using the following procedure EM5210 can
be separated from the fermentation medium and
purified. After the fermentation has been completed
the broth is centrifuged to remove the bacteria
and the antibiotic is removed from the broth
supernateat pH 3.7 by absorption onto an anion
exchange resin e , Dowex l-X2. Antibiotic is
eluted from the resin with sodium chloride at about
pH 4, and the eluate is concentrated and passed
through a charcoal column. EM5210 is then eluted
from the charcoal with methanol:water,l:l.
The active fractions are collected and dried,
and then placed on an anion exchange column, e g.,
*
Dowex l-X2, and eluted with a gradient of sodium
chloride buffered at pH 4. Active fractions are
concentrated and desalted on a macroreticular
styrene-divinylbenzene copolymer resin (Dianion
HP20AG) column. Active fractions eluted with
water are concentrated and freeze dried and this
material represents the sodium salt of EM5210.
* Trade Mark
GC155f
~39~ 1 3 3 8 6 7 0
The above described purification procedure
will yield the sodium salt of EM5210. Other salts
can be obtained by changing the salt used to elute
EM5210 in the ion-exchange chromatography described
above, e.g., using potassium chloride to give
the potassium salt.
The following examples are specific embodiments
of this invention.
G~155f
_ -40-
1 338~70
Example 1
(S)-N-(2-Oxo-l-sulfo-3-azetidinyl)-2-phenylacetamide,
ootassium 5 alt
.lethod I:
A) l-[(lR)-carboxy-2-methyl(propyl)]-2-oxo-(3S)-
~phenyl[acet-yl(amino)~azetidine
Raney nickel is washed with water by
decantation for several hours until the pH of the
water (5-6 times volume of that of the Raney
nickel)is 7.6.
To a solution of 9.0 g of penicillin G
(Na ) in 500 ml of water is added 54 g (90 ml)
of Raney nickel. The flask, fitted with a
reflux condenser, is immersed in a bath at
155C. ~7hen refluxing begins, it is conti~e~
for 15 minutes. The flas~ is immediately cooled
in an ice-water bath, and the Raney nickel is
removed by filtration through Celite. The pH is
adjusted to 3 using dilute HCl, and the aqueous
solution is concentrated to ca. 150 ml and cooled.
The oily layer crystallizes upon scratching.
After washing with water and drying in vacuo
for 3 hours at 50 C there is 3.83 g of the title
compound.
B) l-[Acetyloxy-2-methyl(propyl)]-2-oxo-(3S){phenyl-
[acetyl(amino)]]azetidine
Nitrogen is bubbled for 15 minutes through
a stirred suspension of 608 m~ (2 mmol) of the
above compound in 20 ml of dry acetonitrile.
A water bath at 40-45C is used for several
minutes to dissolve all of the acid. The water
~ GC155f
_ ~ -41- 1 338670
bath is removed, and powdered cupric acetate
monohydrate (182 mg, 1 mmol~ is added, followed
after 1 minute of stirring, by 886 mg (2 mmol)
of lead tetracetate. The mixture is stirred
at room temperature for 20 minutes. The acetonitrile
solution is decanted from the precipitate, and
the solids are washed with ethyl acetate. The
combined acetonitrile-ethyl acetate solution
is evaporated to a residue, which is taken up
in ethyl acetate-water. The ethyl acetate layer
is washed sequentially with water (3 times),
aqueous sodium bicarbonate (pH 7), and water.
The ethyl acetate layer is dried over sodium
sulfate and evaporated to a residue (515 mg),
lS which is used without further purification in
the next reaction.
C) 2-Oxo-(3S)-[phenyl[acetyl(amino)]]azetidine
To a solution of 911 mg (2.86 mmol) of
the above compound in 21 ml of methanol is
added 3.5 ml of water followed by 383 mg
- (2.86 mmol) of potassium carbonate. The mixture
is stirred under nitrogen for 1 minute, and then
1~0 mg (4.30 mmol) of sodium borohydride is
added. The reaction is stirred at room temp-
erature for 20 minutes. The methanol is removedin vacuo, and the residue is taken up in ethyl
acetate and a small amount of water. This is
adjusted to pH 2.5. The ethyl acetate layer
is washed at pH 7.0 with aqueous sodium bicar-
bonate and then a small volume of water and
~CI-~5~
-
-42-
~ 338670
finally dried over sodium sulfate and evaporated
to give the crude product (493 mg). Addition of
a small amount of ethyl acetate gives 250 mg
(43% yield) of the desired crystalline product.
Further quantities of product can be obtained
by crystallization or chromatography.
D) (S)-N-(2-Oxo-l-sulfo-3-azetidinyl)-2-phenyl-
acetamide, potassium salt
Pyridine SO3 complex (215 mg, 1.35 mmol) is
added to a stirred solution of 251 mg (1.23 mmol)
of the above product in 2 ml of dry dimethyl-
formamide and 2 ml of dry methylene chloride
under nitrogen at room temperature. The mixture
is stirred for 3 hours. The solvents are
removed _ vacuo, and the residue is taken up
in methylene chloride-water. The pH is adjusted
to 6.5 using 2N potassium hydroxide. The aqueous
layer is washed with methylene chloride (3 times)
filtered and evaporated to a residue. The
residue is stirred with 20 ml of methanol, and
the potassium sulfate is removed by filtration.
The filtrate is evaporated to a residue, which
is stirred with 10-15 ml of methanol. The
solids are collected to give 49 mg of the title
compound, melting point 189C, dec.
Anal. Calc'd for CllHllN2O5SK: C, 40.99; H, 3.44;
N, 8.69; S, 9.93
Found: C, 45.96; H, 3.83;
N, 9.86; S, 8.99
The compound has identical spectral characteristics
to the product obtained in Method II.
GC155f
1 338670
Method II:
A solution of 660 mg of (S)-2-oxo-3- tI (phenyl-
methoxy)carbonyl]amino]-l-azetidinesulfonic acid,
potassium salt (see Example 3) in 13 ml of water
is stirred in an atmosphere of hydrogen for 2
hours with 200 mg of 10% palladium on charcoal.
The catalyst is filtered and the filtrate diluted
with an equal volume of acetone and cooled in an
ice bath. Over 30 minutes, phenylacetyl chloride
(eight 40 ~1 portions) and 10% potassium bicarbonate
solution are added tpH kept between 5.2-5.8). After
40 minutes, the solution is concentrated in vacuo
to remove acetone and applied to a 200 ml HP-20
column. Elution with water and then water-acetone
(9:1) gives 160 mg of crude product after T~C
eY~m;n~tion of Rydon positive fractions, followed
by pooling and evaporation. Crystallization
from methanol-ether gives 101 mg of the title
compound, melting point 210 C, dec.
A-nal. Calc'd for CllHllN2O5SK /2 H2O: C, 39.86;
H, 3.65; N, 8.45; S, 9.68; K, 11.80
Found: C, 40.01; H, 3.37; N, 8.59; S, 9.59;
K, 11.98
NMR(D20) 3.66 (s,3), 3.67(d of ~, )=6.4), 3.90
(t,)=)), 4.90 (d of d, )=6.4), 7.36 ppm (m.5).
- - GC155f
-44-
Method III: 1 338670
To a solution of (S)-3-amino-2-oxo-1-
azetidinesulfonic acid, tetrabutylammonium salt
(121 mg; see Example 6A) in dry methylene chloride
S (3 ml) is added 40 mg of phenylacetic acid and
61 mg of dicyclohexylcarbodiimide. The mixture
is stirred for 48 hours at room temperature, and
filtered to remove dicyclohexylurea. The solvent
is removed in vacuo and the compound residue is
taken up in acetone, filtered and the title
compound precipitated by the addition of 5 ml
of acetone saturated with potassium iodide.
The supernatant is decanted, the residue washed
with acetone (3 times) and, after drying, 48 mg
of product is obtained,having spectral characteristics
in agreement with the products of Methods I and II.
Method IV:
A solution of 2.83 g of (S)-2-oxo-3[~(phenyl-
me~hoxy)carbonyl~]amino]-l-azetidinesulfonic acid,
pyridine salt (see Example 2) in 36 ml of water
is stirred in an atmos~here of hydrogen with
707.5 mq of 10% palladium on charcoal (175 ml
of hydroqen taken llp). After 2 hours the slurry
is filtered, and the filtrate cooled to 0C
and diluted with 46 ml of acetone (initial pH=
4.25 adjusted to pH 6.7 with cold 10% potassium
bicarbonate solution). A solution of 2.4 ml
of phenylacetyl chloride in 10 ml of acetone is
added dropwise over 15 minutes. The pH is
maintained between 5.2 to 5.8 by the simultaneous
addition of cold 10% potassium bicarbonate
solution. After 45 minutes the slurry is
GC155f
_45- 1 3 3 8 6 7 0
diluted with 93 ml of O.SM potassium phosphate
(pH=4.2) and concentrated to remove acetone.
The slurry is filtered and washed with water.
The filtrate and washings are combined and applied
S to a 450 ml HP-20 column. Elution with 1 liter
of 0.5M potassium phosphate (pH=4.2), 1 liter
of water, and then 2.5 liters of 9:1 water-acetone
gives 1.285 g of the title compound in fractions
14-19 (fraction 1-15 were 200 ml, fractions
16-21 were 100 ml). Spectral data is in agreement
with that obtained in the foregoing methods.
~C155f
-46- 1 3 3 8 6 7 0
Example 2
(S)-2-Oxo-3-~[(phenylmethoxy)carbonyl]amino]-
l-azetidinesulfonic acid, pyridine (1:1) salt
S Method I:
A) l-~(lR)-Car~oxy-2-methyl(propyl)]-2-oxo-(3SL-
~(phenylmethoxy)carbonyl]amino]azetidine
A slurry of 6-aminopenicillanic acid
(12.98 g, 0.06 mole) in 140 ml of water containing
5.18 g of sodium bicarbonate (stirred for ca.
10 minutes without camplete solution) is added
in one portion to a well-stirred (mechanical
stirrer) suspension of Raney nickel (washed
with water to pH 8.0, 260 ml of slurry=130 g)
in a 70 C oil bath. After 15 minutes the slurry
is cooled, filtered, and the filtrate treated
with 5.18 g of sodium bicarbonate and a solution
of 11.94 g (0.07 mole) of benzyl chloroformate
in 12 ml of acetone. After 30 minutes, the
solution is acidified to pH 2.5 and extracted
with methylene chloride. The organic layer is
dried, evaporated, and triturated with ether-
hexane to give a total of 6.83 g of the title
compound.
B) l-~(Acetyloxy)-2-methyl(propyl)]-2-oxo-(3S)-
[[(~henylmethoxy)carbonyl]amino~azetidine
A solution of 6.83 g (0.0213 mole) of the
above acid in 213 ml of acetonitrile is treated
-30 with 1.95 g (0.Q107 mole) of cupric acetate
monohydrate and 9.5 g (0.0213 mole) of lead
GC155f
-47~ 1 3 3 8 6 7 0
tetraacetate. The slurry is immersed in a 65C
oil bath and stirred with a stream of nitrogen
bubbling through the slurry until the starting
material is consumed. The slurry is filtered
and the solids washed with ethyl acetate. The
combined filtrate and washings are evaporated
_ vacuo and the residue taken up in 100 ml each
- of ethyl acetate and water and adjusted to pH 7.
The ethyl acetate layer is separated, dried, and
evaporated to give 6.235 g of the title compound.
C) (S)-(2-Oxo-3-azetidinyl)carbamic acid,
phenylmethyl ester
A solution of 3.12 g (0.0093 mole) of the
above acetate in 70 ml of methanol and 7 ml of
water is cooled to -15C and 1.33 g of potassium
carbonate and 349 mg of sodium borohydride are
added. The reaction mixture is stirred at
-15C - 0C. After the reaction is complete
(ca. 2 hours), the mixture is neutralized to
pH 7 with 2N HCl and concentrated in vacuo.
The concentrate is adjusted to pH 5.8, saturated
with salt and extracted with ethyl acetate (3
times). The organic layer is dried and evaporated
in vacuo. The residue is combined wit~ material
from a similar experiment and triturated with
ether to give 3.30 g of the title compound.
GC155f
_ -48-
1 338670
D) (S)-2-Oxo-3-~(phenyLmethoxy)carbonyl]amino]-
l-azetidinesulfonic acid, pyridine salt
Method I: -
A solution of 440 mg (0.002 mole) of the
above azetidinone in 2 ml each of dry methylene
chloride and dry dimethylform~ e is stirred
for 2 hours under nitrogen with 35Q mg (0.0022 mole)
of pyridine-sulfur trioxide complex. The bulk
of the solvent is then removed in vacuo and
the residue triturated with ethyl acetate to
give 758 mg of solid, which is primarily the
title compound.
NMR(D20-CD30D) 3.63(1H, d of d~ )~6-4)~ 3-90 (lH~ t~
)=6), 4.85 (lH, d of d, )=6.4), 5.10(2~,S) 7.27 (5H,S)
8-0-9.oppm(m's 5H).
Method II:
Chlorosulfonyltrimethylsilyl ester (18.87 g)
is added dropwise at -20C to 7.9 g of anhydrous
pyridine with stirring under a nitrogen atmosphere.
When the addition is complete, stirring is
continued for 30 minutes at room temperature,
and trimethylchlorosil-ane is then removed in
vacuo. A solution of 20 g of the above azetidinone
(Method I, part C) in 120 ml of dimethylform~m;~e
and 120 ml of methylene chloride is added and
stirring at ambient temperature is continued for
3.5 hours. The solvent is distilled off in vacuo
and the oily residue crystallized by addition
of ethyl acetate, yielding 31 g of the title
compound. NMR data is identical to that of the
Method I product.
GC155f
_ _49_
1 338670
Example 3
. ( S ) -2-Oxo- 3-~l(phenylmethoxy)carbonyl]amino~-
l-azetidinesulfonic acid, potassium salt
Method I:
(S) -2-Oxo-3- ~ I (phenylmethoxy~carbonyl~amino]-
l-azetidinesulfonic acid, pyridine salt (13S mg;
see Example 2 ) is dissolved in 2 ml of 0 . 5 M
monobasic potassium phosphate (adjusted to pH S . 5 .
with 2N potassium hydroxide) and applied to a
25 ml HP-20AG column. The column is eluted
with 100 ml of buffer, 200 ml of water and 100 ml
of 1:1 acetone-water. Fractions (25 ml) 14-15
are highly Rydon positive. Evaporation yields
lS 80 mg of material which is primarily the title
compound(spectral data identical to that prepared below).
Method II:
(S ) -2-Oxo-3- [ [ (phenylmethoxy)carbonyl~amino]-
20 - l-azetidinesulfonic acid, pyridine salt (600 mg;
see Example 2 ) is dissolved in 2 ml of water and
mixed with 15 ml of pH 5 . S monobasic potassium
phosphate buffer. A solid forms and the slurry
is cooled to 0C, filtered, washed with cold
buffer, cold 50% ethanol, ethanol and ether to
give 370 mg of the title compound (containing
excess potassium ion by analysis). A solution
of 280 mg of the salt in 10 ml of water is applied
to a 100 ml HP-20 column. The column is eluted
with 200 ml of water and then water-acetone (9:1).
GC155f
~5~-
1 338670
Fractions (5Q ml) are collected; evaporation of
fraction 7 gives a solid. Trituration with acetone,
filtration, and drying in vacuo gives 164 mg of
the title compound, melting point 193-196 C.
Anal. Calc'd for Cl~HllN2O6SK 1/2H2O: C, 38.02;
H, 3.48; N, 8.06; S, 9.23; K, 11.25
Found: C, 38.19; H, 3.24; N, 8.15; S, 2.12;
K, 11.53
NMR(D20) 3.69(1H, d, of d, )=6.4), 3.91(1~, t, J=6)~
4.76(lH, m), 5.16(2H,S), 7.43ppm (5H,S)
Method III:
(S)-(2-Oxo-3-azetidinyl)carbamic acid,
phenylmethyl ester (20.0 g, see Example 2C) is
suspended in 200 ml of acetonitrile, 21.6 ml
of monotrimethylsilyltrifluoroacetamide (25.3 g)
is added and the mixture is heated to 50C with
stirring for 1 hour. After cooling in an ice
bath to 0 C, 17.2 g of trimethylsilyl chlorosul-
fonate is dropped in and the solution is stirred
at ambient temperature for 6 hours. To the
solution is added 24.2 g of potassium ethyl
hexanoate in 100 ml of butanol and stirring is
continued for an additional 1 hour. The slurry
is poured into 1 liter of dry diethyl ether and
the precipitate is filtered off and dried in
vacuo. The compound is dissolved in 500 ml of
water, the pH is adjusted to 5.0 with potassium
carbonate, insoluble material is filtered off
and the mother liquor is freeze dried. The yield
of crude compound is 19.4 g. The compound contains
small amounts of potassium chloride which is
removed by chromatography,spectral data ide~tical
to that of Method II.
~ GC155f
-51- 1 3 3 8 6 7 0
Example 4
(S)-2-Oxo-3-~[(phenylmethoxy)carbonyl]amino~-
l-azetidinesulfonic acid, tetrabutylammonnium
salt (1:1)
S ..
Method I:
(S)-2-Oxo-3-t I (phenylmethoxy)carbonyl~amino]-
l-azetidinesulfonic acid, pyridine salt (1:1~
(34.3 g; see Example 2) is dissolved in 800 ml of
water. The solution is cleared with charcoal,
30.7 g of tetrabutylammonium hydrogen sulfate
in 80 ml water is added and the pH is adjusted to
5.5 with lN-potassium hydroxide. The solvent
is removed in vacuo until a volume of about 200 ml
lS is reached. The precipitated tetrabutylammonium
salt is filtered off and dried in vacuo. The
compound can be recrystallized from water or
dissolved in methylene chloride, filtered and
precipitated by addition of ether. Yield 34.3 g
melting point 108-110C.
Method II:
(S)-2-Oxo-3-l~(phenylmethoxy)carbonyl]amino~-
l-azetidinesulfonic acid, potassium salt (1:1)
(20.2 g; see Example 3) is dissolved in 500 ml
of water, filtered and 20.3 g of tetrabutyl-
ammonium hydrogen sulfate in 100 ml of water is
added. The pH is brought to 5.5 with lN potassium
hydroxide.~ The volume is reduced in vacuo to
about 100 ml and the precipitated tetrabutyl-
ammonium salt is filtered off. The compound is
GC155f
-52- 1 3 3 8 6 7 0
dissolved in 30 ml of methylene chloride, filtered
and precipitated by addition of ether, yielding
21 g of the title compound, melting point 109-111C.
- Example 5
(3S~ t(2-Oxo-l-sulfo-3-azetidinyl)amino~-
carbonyl]benzeneacetic acid, phenylmethyl ester,
- potassium salt (1:1)
A) (S)-3-~mino-2-Azetidinone
(S)-(2-Oxo-3-azetidinyl)carbamic acid,
phenylmethyl ester (3 g; see Example 2C) is
hydrogentated in 100 ml of methanol in the
presence of 1 g of palladium on charcoal catalyst.
When the theoretical amount of hydrogen is
absorbed, the catalyst is filtered off and the
filtrate evaporated to dryness. On st~n~ing,
1.1 g of the title compound crystallizes.
B) (3S)--[~(2-Oxo-3-azetidinyl)amino~carbonyl]-
~20 benzeneacetic acid, phenylmethyl ester
The above azetidinone (3.0 g) is dissolved
in 100 ml of dimethylformamide. The solution is
cooled to 0C and 4.5 g of N-methylmorpholine is
added followed (dropwise) by 10.8 g of
a-(chlorocarbonyl)benzeneacetic acid, phenyl-
methyl ester in 50 ml of acetonitrile withstirring. The mixture is stirred for about
16 hours at 5C. The solvent is distilled off
in vacuo and 100 ml of water is added to the
residue. The aqueous suspension is extracted
twice with 100 ml portions of methylene chloride.
GC155~
~53~ 1 3 3 8 6 7 0
The organic layers are combined, washed with
sodium bicarbonate, 2N phosphoric acid and water,
dried with sodium sulfate, filtered and evaporated
to dryness. The residue is crystallized with
ethyl acetate and petroleum ether, yielding 8.7 g,
melting point 164-166C.
C) (3S)-a-11(2-Oxo-l-sulfo-3-azetidinyl)amino~-
carbonyl]benzeneacetic acid, phenylmethyl ester,
potassium salt (1:1~
The aboYe compound (6.9 g3 is suspended in
150 ml of acetonitrile. Monotrimethylsilyl-
trifluoroacetamide (5.7 g)is added and the
solution is heated for 30 minutes at 50C with
stirring. The solution is cooled to 0C and
3.9 g ~rimethylsilyl chlorosulfonate is
added dropwise. When the addition is complete
the mixture is heated to 50C for 5 hours.
After cooling to 20 C, 7.6 g of potassium ethyl
hexanoate in 10 ml of butanol is added and
stirring is continued for 30 minutes. On
addition of 300 ml of ether the ti~tle compound
precipitates and is filtered off. The crude
product is stirred with 100 ml of dry acetonitrile
for 30 minutes and filtered off, yielding 4.5 g
of the title compound, melting point 118-120C.
Further purification of the crude product by HP20
chromatography-followed by freeze-drying yields pure
material havinq a meltina point of 188-190C.
- GC155f
-54-
1 338670
Example 6
(S)-3-~[(2-Amino-4-thiazolyl)acetyl]amino~-2-
oxo-l-azeti~;nesulfonic acid,- potassium salt
A~ (S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt
(S)-2-Oxo-3-[~(phenylmethoxy)carbonyl~amino~-
l-azetidinesulfonic acid,tetrabutylammonium salt
(2 g; see Example 4) is dissolved in 100 ml o~
di~ethylforr~ e and hydrogenated for about 30
minutes with 1 g of palladium on charcoal (10%)
as catalyst. The catalyst is filtered off and
the dimethylformamide is removéd leaving the
title compound as an oil. NMR (CDC13) 3.82 (lH, t,)=
5.5),4.05(d. lH, d of d, J= 5.5, 2.5 cps).
B) (S)-3-[[(2-Amino-4-thiazolyl)acetyl]amino]-
2-oxo-1-azetidinesulfonic acid, potassium salt
The above compound (2 g), 0.5 g of amino-
thiazole acetic acid and 0.4 g of hydroxybenzo-
triazole are stirred at 0 C in 100 ml of dry
dimethylformamide, while a solution of 0.7 g
of dicyclohexylcarbodiimide in 10 ml of dimethyl-
forr-mi~e is added dropwise. After the addition
is complete, stirring is continued for 12 hours
at 20C. Insoluble urea is- filtered off and
the solvent is evaporated in vacuo. The oily
residue is treated with a solution of potassium
perfluorobutane sulfonate in 20 ml of acetone at
room temperature for 15 minutes. After the
addition of 200 ml of dimethyl ether the title
compound precipitates, and is filtered off,
dried and purified via a 300 ml HP-20 chroma-
tography column using water as eluent. The yield
~C155f
-55-
1 338670
is 8 sa mg of the title compound, melting point
>300C.
Example 7
[3S(~)]-3-Lt(Formyloxy)phenylacetyl]amino]-2-
oxo-l-azetidinesulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (1.5 g; see
Example 6A) in 100 ml of dimethylformamide
and 2 ml Qf propylene oxide are cooled to 0C.
A solution of O-formylmandelic acid chloride in
10 ml of acetonitrile is added dropwise with
stirring. The temperature is maintained for
1 ho-lr and the solvent is then distilled off
in vacuo. The oily residue is treated with a
solution of 2 g of potassium perfluorobutane
sulfonate in 15 ml of acetone. After adding 200 ml
of ether, the title compound crystallizes and
is filtered off yielding 1.5 g of product. The
product is purified by HP-20 chromatography, m.p.
180-185C. with decomposition.
Example 8
[3S(+)]-3-tt(Formyloxy)phenylacetyl]amino]-
2-oxo-1-azetidinesulfonic acid, potassium salt
Following the procedure of F.~mpl e 7, but
substituting D-O-formylmandelic acid chloride
for O-formylmandelic acid chloride, yields the
title compound, melting point 120-125C, after
freeze drying.
GC155f
-56- ~ 3 3 8 6 7 0
Example 9
[3S(-)~-3-[~(Formyloxy)phenylacetyl]amino-2-
oxo-l-azetidinesulfonic acid, potassium salt
~ollowing the procedure of Example 7,
but substituting L-O-formylmandelic acid chloride
for O-formylmandelic acid chloride, yields the
title compound, containing 1 mole of water,
melting point, 203-205C. After careful drying
the product melts at 228-230C.
Example 10
(S)-2-Oxo-3- I (l-oxooctyl)amino]-l-azetidine-
sulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (1.5 g; see Example 6A)
in 100 ml of dimethylformamide and 2 ml of
propylene oxide are cooled to 0C. At this
temperature a solution of 0.8 g of caprylic acid
chloride in 20 ml of dry acetone is added dropwise
and stirring is continued for 30 minutes. The
solvent is evaporated in vacuo, and the oily
residue is treated with 2 g of potassium perfluoro-
butane sulfonate in 15 ml of acetone. The
acetone is distilled off in vacuo. The residue
is dissolved in S ml of water and chromatographed
using 300 ml of HP-20 resin and water/acetone
(9:1) as eluent, yielding 0.9 g of the title
compound, melting point 173-180C, after freeze-
drying.
GC155f
~57~ 1 3386 70
Example 11
13S(Z)]-3-[[~2-Amino-4-thiazolyl)[[[hydroxy(phenyl-
methoxy)phosphinyl]methoxy]imino]acetyl]amino]-
2-oxo-1-azetidinesulfonic acid, potassium salt
S (S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (0.8 g; see Example 6A)
in 30 ml of dimethylfo~r~ e~ 0.9 g of (Z)-2-
amino-a-[[[hydroxy(phenylmethoxy)phosphinyl]-
methoxy]imino]-4-thiazoleacetic acid, 0.3 g of
hydroxybenzotriazole and 0.7 g of dicyclohexyl-
carbodiimide are stirred for 24 hours at room
temperature. The precipitated urea is filtered
off and the solvent is removed in vacuo. The
reamining oil is treated with an equivalent amount
of potassium perfluorobutane sulfonate in 10 ml
of acetone. The title compound is filtered off
and purified using HP-20 resin and water as
eluent, yielding 500 mg, melting point 210-215C,dec.
Example 12
[3S(Z)]-3-[~(2-Amino-4-thiazolyl)(ethoxyimino)-
acetyl]amino]-2-oxo-1-azetidinesulfonic acid,
potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic acid
tetrabutylammonium salt (1.5 g; see Example 6A)
in 100 ml of dimethylfor~ e, 0.6 g of hydroxy-
benzotriazole, 1 g of dicyclohexylcarbodiimide
and 0.8 g of (Z)-2-amino-~-(ethoxyimino)-4-
thiazoleacetic acid are stirred at room temperature
for 24 hours. The solvent is distilled off and
the residue is dissolved in 30 ml o~ acetone.
- 58 33 8 6 70 GC155f
Urea is filtered off and the mother liquor is
treated with a solution of 2 g of potassium
perfluorobutane sulfonate in 20 ml of acetone.
After the addition of 200 ml of ether the title
S compound precipitates, is filtered off and
dried. Purification is accomplished by chroma-
tography using anHP-20 column and water as
eluent, yielding 1.1 g of the title compound,
melting point 180-185C, dec.
Example 13
[3S(E)]-3-[l(2-Amino-4-thiazolyl)(ethoxyimino)-
acetyl]amino]-2-oxo-1-azetidinesulfonic acid,
potassium salt
Following the procedure of Example 12,
but substituting (E)-2-amino--~ethoxyimino)-
4-thiazoleacetic acid for (Z)-2-amino-a-(ethoxy-
imino)-4-thiazoleacetic acid, yields the title
compound, melting point 160-170C after freeze
drying.
Example 14
[3S(Z)-3-[[(2-Amino-4-thiazolyl)](2,2,2-trifluoro-
ethoxy)imino~acetyl]amino]-2-oxo-1-azetidinesulfonic
acid, potassium salt
Following the procedure of Example 12, but
substituting (Z)-2-amino-a-[(2,2,2-trifluoro-
ethoxy)imino]-4-thiazoleacetic acid for (z)-2-amino-
-(ethoxyimino)-4-thiazoleacetic acid, yields the
title compound melting point 160-170C after
freeze-drying.
....
- . 1 3 3 8 6 7 ~
-59-
Example 15
(S)-2-Oxo-3-l(l-oxopropyl)amino]-1-azetidine-
sulfonic acid, potassium salt
Method I:
(S)-3-Amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (1.5 g; see Example
6A) in 100 ml of dry dimethylformAm;~e and 4 ml
of propylene oxide are cooled to 0C with stirring.
At this temperature 0.5 g of propionic acid
chloride in 10 ml of acetonitrile is added
dropwise and the mixture is stirred for 2 hours.
The solvent is distilled off in ~acuo and the
oily residue is treated with an equivalent
amount of potassium perfluorobutane sulfonate in
5 ml of acetone. On addition of ether the title
compound crystallizes and is filtered off, yielding
0.8 g of product, melting point 135-140 C after
freeze-drying.
Method II:
(S)-2-Oxo-3-tl(phenylmethoxy)carbonyl~amino~-
l-azetidinesulfonic acid, tetrabutylammonium salt
(4 g; see Example 4) is hydrogenated in 100 ml
of diglyme with 1 g of palladium on charcoal.
The hydrogenation is complete after 2.5 hours.
The catalyst is filtered off and 2 ml of propylene
oxide is added. After cooling to 0 C,0.5 g
propionic acid chloride in 10 ml dry diglyme is
added with stirring. After 30 minutes the solvent
is removed in vacuo and the oily residue is treated
GC155f
1 338670
-60-
with an equivalent amount of potassium perfluoro-
butane-sulfonate in 20 ml of acetone. After
the addition of ether, the title compound
crystallizes, is filtered off and recrystallized
from water/acetone, yielding 0.9 g of product,
melting point 156-160C (dec.).
Example 16
[3S(+)~-3-[(Hydroxyphenylacetyl)amino]-2-oxo-
l-azetidinesulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (1.5 g; see
Example 6A) in 100 ml of dry dimethylformamide
is stirred for about 16 hours with 1.5 g of
15- dicyclohexylcarbodiimide, 0.5 g of hydroxy-
benzotriazole and 0.6 g of mandelic acid. The
solvent is removed in vacuo and the residue is
dissolved i~ 20 ml of acetone. The precipitated
urea is filtered off and the mother liquor is
treated with an equivalent amount of potassium
perfluorobutane sulfonate. After the addition
of ether the title compound precipitates and is
filtered off, yielding 1.4 g of crude product.
After recrystallization from water, the product
has a melting point of 138-140C.
Example 17
(S)-3-[[[(Cyanomethyl)thio]acetyl]amino]-2-oxo-
l-azetidinesulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (1.5 g; see
Example 6A) and 0.72 g of [(cyanomethyl)thio]acetic
acid are dissolved in 70 ml of acetonitrile and
a solution of 1.04 grams of dicyclohexylcarbodiimide
GC155f
-
-61-
1 338670
in acetonitrile is added dropwise. The mixture
is stirred for about 16 hours at 0C and the
precipitated dicyclohexylurea is filtered off,
the filtrate evaporated and the oily residue
dissolved in acetone. On the addition of a
saturated solution of potassium-iodide in acetone,
the title compound precipitates, yielding 1.1 g of
product, melting point 150-155C.
Example 18
(S)-2-Oxo-3-[(lH-tetrazol-l-ylacetyl)amino]-l-
azetidinesulfonic acid, potassium salt
To a solution of 0.005 mole of (S)-3-amino-
2-oxo-1-azetidinesulfonic acid, tetrabutylammonium
salt (see Example 6A) in 70 ml of dimethylforr~m;~e
is added 0.77 g of lH-tetrazole-l-acetic acid and
a solution of 1.13 g of dicyclohexylcarbodiimide
in 5 ml of dimethylformamide. The mixture is
stirred for about 16 hours at room temperature
and the solvent is removed in vacuo. The
remaining oil is dissolved in 20 ml of acetone
and treated with 0.006 mole of a solution of
potassium perfluorobutane sulfonate in acetone,
yielding 1.5 g of the title compound, melting
point 170-175C, dec.
Example 19
(S)-2-Oxo-3-[(2H-tetrazol-2-ylacetyl)amino]-1-
azetidinesulfonic acid, potassium salt
Following the procedure of Example 18,
but substituting 2H-tetrazole-2-acetic acid for
lH-tetrazole-l-acetic acid, yields the title
compound, melting point 175-177 C, dec.
GC155f
._
-62- 1 3 3 8 6 7 0
Example 20
(S)-2-Oxo-3-~(2-thienylacetyl)amino]-1-azetidine-
sulfonic acid, potassium salt
Following the procedure of Example 18,
but substituting 2-thiopheneacetic acid for
l~-tetrazole-l-acetic acid, yields the title
compound, melting point 180-190C, dec.
Exa~ple 21
~3S(Z)]-3-f[(2-Amino-4-thiazolyl)(methoxyimino)-
acetyl~amino]-2-oxo-1-azetidinesulfonic acid,
potassium salt
(S)-3-Amino-2-oxo-1-azeti~;nesulfonic
acid, tetrabutylammonium salt (prepared as
described in Example 6A from 7.9 g of (S)-2-oxo-
3-~(phenylmethoxy)carbonyl]amino]-1-azetidine-
sulfonic acid, tetrabutylammonium salt) is cooled
to 0 C and 3.53 g of (Z)-2-amino-a-(methoxyimino)-
4-thiazoleacetic acid is added, followed by a
- 20 solution of 3.27 g of dicyclohexylcarbodiimide
in 10 ml of dimethylformamide. The mixture is
stirred for 16 hours at 5C, filtered and the
solvent removed in vacuo. The residue is
dissolved in acetone and filtered. On addition
of 60 ml of a 10% solution of potassium perfluoro-
butane sulfonate in acetone, 4.7 g of crude
product crystallizes. The crude product is
purified by chromatography on HP-20, 100-200 mesh
to yield 3.0 g of the title compound, melting
point 235C.
GC155f
-63- 1 3 3 8 6 7 0
Éxample 22
(3S~-a~ 2-Oxo-1-sulfo-3-azetidinyl)amino~-
carbonyl~benzeneacetic acid, dipotassium salt
(3S)~ (2-Oxo-l-sulfo-3-azetidinyl)amino~-
carbonyl]benzeneacetic acid, phenylmethyl ester,
potassium salt (100 mg; see Example 5) is dissolved
in 20 ml of anhydrous methanol. Palladium on
charcoal (10 mg, 10%) is added and the mixture
is treated with hydrogen for 15 minutes. The
catalyst is filtered off, and the methanol evap-
orated in vacuo. me residue is dissolved in5 ml of water and the pH adjusted to 6 with
lN potassium hydroxide. After freeze drying
crude product is obtained. The crude material
is chromatographed over HP 20 resin (water as
eluent), yielding 60 mg of the title compound
melting point 80-85C.
Example 23
(S)-3-(Acetylamino)-2-oxo-1-azetidinesulfonic
acid, potassium salt
Method I:
A) (S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
potassium salt
(S)-3-(Benzyloxycarbonylamino)-2-oxo-1-
azetidinesulfonic ~acid, potassium salt (169 mg;
see Example 3) is dissolved in 4.0 ml water
and hydrogenated over 37 mg of 10% palladium
on charcoal for 1 hour 40 minutes. The catalyst
is removed by filtration and washed with 1 ml of
50% aqueous acetone.
_ ~C155~
-64~ 1 3 3 8 6 7 0
B) (S)-3-(Acetylamino)-2-oxo-1-azetidinesulfonic
acid, potassium salt
The above solution of the free amine is
diluted with 3.5 ml of acetone and stirred in
an ice bath. Acetyl chloride (320 ~1) is added
over ca. 15 minutes in small portions and alternated
with solid potassium bicarbonate to maintain pH
6.5-7.2. After 30 minutes a silica gel TLC in
acetone:acetic acid (19:1) and the ~ydon test in~
the reaction to be essentially finished. Six ml
of 0.5M pH 5.5 monobasic potassium phosphate
buffer are added and the solution is acidified
to pH 4.8 with 2N hydrochloric acid. The acetone
is removed in vacuo and the aqueous solution
thus obtained is passed through a 50 ml HP-20 AG
column to give 2.197 g of solid. This is digested
with methanol to yield 282 mg of extractable
material which still contains some salt. The
product is further purified by passage through
an IRC-S0 column, followed by acidification to
pH 3.8 with subsequent stripping to dryness
in vacuo. Trituration with acetone gives 64 mg
of desired product containing ca. 0.5 equivalent
of inorganic potassium salts. Final passage
through a 200 ml HP-20 AG column, followed by
lyophilization from .5 ml of water gives 22 mg
of product as amorphous powder which is dried
ln vacuo for 2 hours at 50C, melting point
170-180C after softening at 100C.
Anal. for C5H7O5N2SK, Calc'd: C, 24.38; H, 2.87;
N, 11.37; K, 15.9
Found: C, 26.06; H, 3.14;
N, 9.96; K, 18.04
GC155f
_ , .
-65-
1 338670
~ethod II:
A solution of 2.0 g of (S)-2-oxo-3-I[(phenyl-
methoxy)carbonyl]amino]-l-azetidineuslfonic acid,
pyridine salt (see Example 2) in 25 ml of water
is hydrogenated over 500 mg of 10% palladium on
charcoal. After 2 hours the solution is filtered,
cooled to 0C and 40 ml of acetone is added.
The pH of the solution is kept between 5.2-5.8 by
simultaneous addition of acetyl chloride and cold
10% potassium bicarbonate solution. The pH of
the solution is adjusted to 4.2 with acetyl
chloride and the solution is concentrated on
the rotary evaporater to remove acetone.
Chromatography on a 300 ml HP-20 AG column
lS (water eluant -25 ml fractions) gives 900 mg
of the title compound in fractions 13 and 14
contaminated with some potassium acetate.
Rechromatography on HP-20 AG gives an analytical
sample, melting point 205-210C.
Anal.calc'd for C5H7N2O5SK: C,24.38; H,2.86;
N,11.38; S,13.02; K,15.88
Found: C,24.23; H,2.81; N,11.25; S,12.86;
K,15.74
- GC155f
-66- 1 338670
~xA~ple 24
(S~-2-Oxo-3-l(phenoxyacetyl)amino]-1-azetidine-
sulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (1.5 g; see
Example 6A) in 100 ml of dry dimethylformamide
is cooled to 0C. Propylene oxide (2 ml) is
added and 1 g of phenoxyacetyl chloride
is added dropwise with stirring. The reaction is
completed within 1 hour. The solvent is removed
in vacuo and the residue is treated with an
equivalent amount of potassium perfluorobutane
sulfonate in 20 ml of acetone. On addition of
ether the title compound (1 g) crystallizes
and is filtered off and dried. After recrystal-
lization from boiling water the title compound
has a melting point of 176-180C.
Example 25
[3S(R*)~-3-[[[[[3-~(2-Furanylmethylene)amino]-2-
oxo-l-imidazolidinyl]carbonyl]amino]phenylacetyl]-
amino]-2-oxo-1-azetidinesulfonic acid, potassium
- salt
(S)-2-Oxo-3-l[(phenylmethoxy)carbonyl]amino]-
l-azetidinesulfonic acid, tetrabutylammonium
salt (3 g; see Example 4) is hydrogenated in
100 ml of dimethylformamide with 1.5 g of
palladium on charcoal. After 0.5 hours, the
catalyst is filtered off and 1.8 g of dicyclo-
hexylcarbodiimide, 2 g of (R)-~-lL[3-~(2-furanyl-
methylene)amino]-2-oxo-1-imidazolidinyl~carbonyl~-
GC155f
-67-
1 338670
amino~benzeneacetic acid and 0.9 g of hydroxy-
benzotriazole are added. After 3 hours the
reaction mixture is evaporated to dryness,
dissolved in 50 ml of dry acetone and the
precipitated urea remo~ed by filtration~ An
equivalent amount of potassium perfluorobutane
sulfonate in 20 ml of acetone;is added and the
title compound precipitates. Crystallization
is completed by addition of 200 ml ether.
After filtration the title compound is recrystal-
lized from water, yielding 2 g, melting point -
220-225C, dec.
Example 26
[3S(R*)]-2-Oxo-3-[[[~(2-oxo-1-imidazolidinyl)-
carbonyl]amino]phenylacetyl]amino]-l-azetidine-
sulfonic acid, potassium salt
(S)-2-Oxo-3-l[(phenylmethoxy)carbonyl]amino]-
l-azetidinesulfonic acid, tetrabutylammonium
salt (3 g; see Example 4) is hydrogenated in
100 ml of dry dimethylformamide with 1.5 g of
palladium on charcoal; the catalyst is filtered
off after 30 minutes. (R)-~-[[(2-Oxo-l-imidazoli-
dinyl)carbonyl~amino]benzeneacetic acid (1.8 g),
1.3 g of dicyclohexylcarbodiimide and 0.9 g
hydroxybenzotriazole are added and the solution
is stirred for 2.5 hours. The solvent is removed
in vacuo and theresidue is dissolved in 50 ml of
acetone. The precipitated urea is filtered off
and the mother liquor is treated with an equivalent
GC155~
_
1 338670
amount of potassium perfluorobutane sulfonate
in 20 ml of acetone. The title compound crystal-
lizes and is filtered off after the addition
of 200 ml of ether, yielding 1.8 g of product,
melting point 210-215C after recrystallization
from water/acetone.
Example 27
[3S(Z)]-3-~[(Methoxyimino)phenylacetyl]amino~-2-
oxo-l-azetidinesulfonic acid, potassium salt
A) [3S(Z)~-3-[l(Methoxyimino)phenylacetyl~amino~-
2-azetidinone
(Z)-a-(Methoxyimino)benzeneacetic acid
(3.58 g) is dissolved in methylene chloride,
cooled to 5C, and treated with a solution of
4.53 g of dicyclohexylcarbodiimide in 50 ml
of methylene chloride. The mixture is stirred
for 30 minutes at 5C and a solution of 1.72 g
of 3-amino-2-azetidinone (see Example 5A) in
100 ml of methylene chloride is added. The
reaction mixture is kept at 5 C for 1 hour and
for 2 hours at room temperature. After removal
of the dicyclohexylurea by filtration, the filtrate
is evaporated, yielding 6.6 grams of crude product.
This material is purified by chromatography on
750 grams of silica gel, using a mixture of
methylene chloride/ethyl acetate (7:3) as eluent,
yielding 2.9 g of product.
GC155f
-
-69- t 3 3 8 6 7 0
B) [3S(Z))]-3-¦ r (Methoxyimino)phenylacetyl~-
amino~-2-oxo-1-azetidinesulfonic acid, potassium
salt
Pyridine (0.5 ml) is dissolved in 5 ml of
absolute methylene chloride, cooled to -3QC
and a solution of 0.93 ml of trimethylsilyl
chlorosulfonate in 5 ml of methylene chloride
is added. The mixture is stirred at room
temperature for 30 minutes and evaporated in vacuo
to dryness. The residue is dissolved in 10 ml
of dimethylformamide and treated at room temper-
ature with a solu~inn of l.23 ~ of the above azetidinone
in 10 ml of dimethylformamide. After stirring for two
hours at room t~m~rature, the solution is
evaporated to dryness to yield 2.1 g of crude
[3S(Z)]-3-[[(methoxyimino)phenylacetyl]amino]-
2-oxo-1-azetidinesulfonic acid, pyridine salt.
Treatment of the pyridine salt with
tetrabutylammonium hydrogen sulfate yields
the corresponding tetrabutylammonium salt
which is extracted with methylene chloride
and remains as an oil after evaporation.
Treatment of the tetrabutylammonium
salt with an equimolar amount of potassium
perfluorobutane sulfonate in acetone,evaporation,
and treatment of the residue with ether, yields
1.6 grams of the title potassium salt which is
purified by chromatography on HP-20. Elution
is carried out with water/acetone 90:10 and
yields a product having a melting point of 220C,dec.
~ GC155f
-70-
1 338670
Example 28
[3S(Z)~-3-~[(2-Amino-4-thiazolyl)~2-(diphenyl- -
methoxy)-l,l-dimethyl-2-oxoethoxy]imino]acetyl]-
amino~2-oxo-1-azetidinesulfonic acid, potassium
salt (1:1)
A solution of 0.005 mole of (S)-3-amino-
2-oxo-1-azetidinesulfonic acid, tetrabutyl-
ammonium salt (see Example 6A) and 0.006 mole of
(Z)-2-amino-a- I 12-(diphenylmethoxy)-1,1-dimethyl-
2-oxoethoxy]imino]-4-thiazoleacetic acid in 60 ml
of dimethylformamide is treated with 0.7-g of
hydroxybenzotriazole and 1.13 g of dicyclohexyl-
carbodiimide. The mixture is stirred for about
16 hours at room temperature, filtered and the
filtrate evaporated. The residue is dissolved
in 30 ml of acetone, filtered and treated with
20 ml of a solution of 10~ potassium perfluoro-
butane sulfonate in acetone. After the addition
of petroleum ether the title compound precipitates
- 20 and is treated with ether and filtered to yield
3.8 g of product,melting point 190C, dec.
Example 29
[3S(Z)]-3-[[(2-Amino-4-thiazolyl)[(l-carboxy-1-
methylethoxy)imino]acetyl]amino]-2-oxo-1-azetidine-
sulfonic acid, dipotassium salt
(3S(Z)]-3-~(2-Amino-4-thiazolyl)[ r 2-
(diphenylmethoxy)-l,l-dimethyl-2-oxoethoxy~imino]-
acetyl]amino~-2-oxo-1-azetidinesulfonic acid,
potassium salt (2 g; see Example 28) is suspended
in 5 ml of anisole and 25 ml of trifluoroacetic
GC15~f
-71- 1 338670
acid is added at -10C. The mixture is stirred
for 10 minutes at -10C. Ether (100 ml) is added
slowly at -10C and subsequently ~0 ml of petrol-
eum ether is added. Ihe preci?itate is filtered to yield
1.6 g of the trifluoroacetic acid salt. This is
suspended in 20 ml of water at 0C, adjusted to
pH 5.5 with diluted potassium hydroxide and
purified on an HP-20 column. The title compound
is eluted with water and has a melting point of
225C,dec.
Example 30
~3S(Z)~-3-~[(2-Amino-4-thiazolyl)~2-(diphenyl-
methoxy)-2-oxoethQxy]imino]acetyl]amino~-2-oxo-
l-azetidinesulfonic acid, potassium salt
Following the procedure described in
Example 28, but substituting (z)-2-amino-~-[[2-
(diphenylmethoxy)-2-oxoethoxy]imino]-4-thiazole-
acetic acid for (Z)-2-amino-~-[[2-(diphenylmethoxy~-
1,1-dimethyl-2-oxoethoxy]imino]-4-thiazoleacetic
- acid, yields the title compound, melting point
180 C, dec.
GC155f
-72~
1 338670
Example 31
[3S(+)~-3-~(Azidophenylacetyl)amino]-2-oxo-1-
azetidinesulfonic acid, potassium salt
Method I:
A) (+,S)-a-Azido-N-(2-oxo-3-azetidinyl)benzene-
acetamide
(S)-3-Amino-2-azetidinone (2.15 g; see
Example 5A) and 2.1 g of sodium bicarbonate
are dissolved in 50 ml of acetone/water (2:1).
(+)-~-Azidobenzeneacetyl chloride (5 g) dissolved
in 10 ml of acetone is added dropwise whi~e
maintaining the temperature at 0-5C and the pH
at 6.8 with sodium bicarbonate. After stirring
for 1 hour, the acetone is distilled off and
the r~m~i n; ng aqueous solution is adjusted to
pH 8 with sodium carbonate and extracted with
three 50 ml portions of methylene chloride.
Evaporation of the sodium sulfate dried organic
layers yields 3.6 g of the title compound as
an oil which crystallizes after triturating
with ether. Recrystallization from methylene
chloride/ether, yields the title compound,
melting point 97-100C.
~;C155f
-73- 1 33867~
B) ~3S (+)]-3- [(Azidiophenylacetyl)amino~-2-oxo-
l-azetidinesul fonic acid-, potassium salt
A solution of 2.45 g of the above azetidinone
and 3 g of monosilyltrifluoroacetamide in 20 ml
5 of acetonitrile are kept for 1 hour at 4ac.
After cooling to 0C the solution is treated
with 1.88 g of trimet}~ylsilyl c~lorosulfonate
and stirred for 5 hours under argon.
Finally 6.12 ml of a 2N solution of potassium-2--
10 ethyl hexanoate in n-butanol are added and
stirring is continued for 45 minutes. The
- solution is pol~red into 300 ml of ether and the
precipitate is filtered off. A filtered solution
of 1.2 g of precipitàte in phosphate buffer
(pH 5.5) is chromatographed on 100 ml of HP-20.
Elution is performed with: 1) 20 ml buffer;
2) 200 ml H2O; 3) 200 ml water:acetone (9:1);
4) 200 ml water:acetone (3:1). The elution
is monitored with TLC (~ydon test on SiO2). 25 ml
20 fractions are taken and from fractions 15 and 16
280 mg of the title compound are obtained. A
second column chromatography of this material
yields 120 mg of the title compound, melting
point 148C, dec.
Method II:
(S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (2.03 g; see Example 6A)
and 0.9 g of (+)-~-azido-benzeneacetic acid are
30 dissol~ed in 30 ml of acetonitrile and a solution
of 1.03 g of dicyclohexylcarbodiimide in 10 ml
GC155f
- ~74 1 33867~
of acetonitrile is added. The temperature is
maintained for one hour at 0C and for lO`hours
at room temperature. After filtering off the
resultant precipitate, the solvent is distilled
off in vacuo and the oily residue is dissolved
in 20 ml of acetone and treated with 1.70 g
potassium perfluorobutane sulfonate in acetone.
The addition of 10 ml of ether crystallizes the
title compound, melting point 149C, dec.
Method III:
~-Azidiophenylacetyl chloride (2.5 g) is
added to a solution of 4.06 g of 3-amino-2-oxo-
l-azetidinesulfonic acid, tetrabutylammonium
salt (see Example 6A) and 5 g of propylene oxide
in 30 ml of acetonitrile. After stirring for
two hours the solvent is distilled off in vacuo
and the oily residue is treated with one equivalent
amount of potassium perfluorobutane sulfonate
in acetone. After the addition o ether the title
compound crystallizes and is filtered off, melting
point 148-149C dec.
Example 32
[3S(D)] -3-[[~[[(4-Methoxyphenyl)methoxy]carbonyl]-
amino]phenylacetyl~amino]-2-oxo-1-azetidinesulfonic
acid, potassium salt
To a solution of 2.03 g of 3-amino-2-oxo-
l-azetidinesulfonic acid, tetrabutylammonium salt
(2.03 g; see Example 6A) and 1.58 g of D-~- r L [ (4-
methoxyphenyl)methoxy~carbonyl]amino~benzeneacetic
GC155f
~75~ 1 33 8 6 70
acid in 50 ml of acetonitrile, a solution of
1.03 g of dicyclohexylcarbodiimide in 10 ml of
acetonitrile is added dropwise. The temperature
is maintained at 5C for one hour and for 6
S hours at room temperature. Separation of the
formed dicyclohexyl urea and distilling off the
solvent yields the title compound as an oily
residue. Treating this oil in acetone with
potassium perfiuorobutane sulfonate and ether
yields 2.4 g of product, melting point 108-111C
dec.
Examples 33-37
Following the procedure of Example 32,
lS but substituting the compound listed in column I
for (D)-~-~[~(4-methoxyphenyl)methoxy]carbonyl~-
amino]benzeneacetic acid, yields the compound
listed in column II.
GC155f
- -76- 1 3 3 8 6 7 0
U
_ _ 1
_, , , " . .
~ ~ ~ ~ , O ~ ~ ~ ~ ~ u U a~
s ~ ~ ~ U t~ ~ s ~ _, U s ~ ~ ,
~S ~ d a) ,~ ~ o ,r, ~ I c I ~ ~ ~ ~ ~
~ ~ ~ 0 ~ , ~c ~ u o ~ ~--I ~ ~ a) ~ 0 ~ ~ I ~ 0
'C I ~ - S 0 ~ ~ 1,~ x ~ ~`
s e ~ ~ N e ~ o ' e
J 1~ U ~ ~ ~J C ~ r ~ c,) r~ _~ r ~ C~
O ~ 1 ~~ ~ O ~ ~ -- O I ~ ~ O ~
a~ c U ~D I e ~ J ~ _1 U u~ U C~
e-~ U~ O^ ~ OJJ ~X~ U E~.-l I U l~ e-,l I u ~r
I E~ I r _l C ~ C 0 1 ~0 ~1 e o ~ _, I e o ~ -
~r ~ O ~ S O ~ ~ ~ X ~ I~r a x I I
--_x o~ e ~ ~ e ~ --_o 0~ --_0 O~D
~ _I O ~ 1 0 ~ o ~ _I I P.l` --_~ I ~
~ I s~ ~ e c ~
~ c~ ~~ ~o ~ ~ o ~ c I ' --c I
--c ~ ~ ~ o--~ ~ ~ ~
~ --X~ ~ o~ C--_ O
I ~ ~ u-,, --o e ~ 0 ~ u ,~ ~ ~ c u ,l
0 I 5 n~ ~ I Cl-- 0 ~ ~ ~ O ~ r ~ a O
I 'J e ~
I a~ _I N ~0 1 ~ V ~ U --~ a U
~ X_I C C ~ I C 1 0~ _ X_~ ~ X_I ~ C ~ X~
-- O ~1 0 "I +~ e~ C _1 0 N + I O O ~ ~ -- O ~1 0 ~ O ~1 0
tn s ~ I Id I ~ ~ C I ~ S ~ ~~ S ~ ~1
U ~ X o ~ ~ a~ ~ r
--~ ~ u~ ~ --~, O ~ 0 --e,
l l l l l
U _I _ >1 ~1 ~ ~ t,
~ '1 ~ o ~ U X ~l X ~
o~ ~ ~ o ~ o u o--
a ~
~ ~ o ~ ~ u
e ~
_1 4. o ~r ~I cJ ~ I J
~-- s 1 >., r
t _ ~ ~ t t .. t
O
t _ ~ s ~ rl
lC I ~ ~ lC I ~C t ~C I
o ~ ~--. o ~ o a~ o
S I ~_I s I s ~ ~ I
a) o I c ~ o ~ o ~ o
e ~c ~ o e ~ e c e t
o~ ~ ~ e
~ ~0 t r ~
I I x ~ I t
O ~I o ,l 2~ o
1: ~ I S ~) I R 1: 1:
-- - ~ -- ~ a) _ s, _ _ , ,1
~ +1 0 C) +I r~
~ --e ~
a
_I
k ~ ~r ~ ~ I~
GC15~f
_77- 1 3 3 8 6 7 3
Example 38
(3S)-3- L [ 11 (Methylthio)thioxomethyl]thio]~henyl-
acetyl]amino]-2-oxo-1-azetidinesulfonic acid.,
tetrabutylammonium salt
A solution of 1.03 g of (+)-~ (methylthio)-.
thioxomethyl~thio]benzeneacetic acid and 1.63 g of
3-amino-2-oxo-1-azetidinesulfonic acid, tetrabutyl-
ammonium salt (see ~xample 6A) in 30 ml of acetoni-
trile is treated with 0.8 g of dicyclohexyl-
}0 carbodiimide dissolved in 10 ml of acetonitrile
at -5C. After stirring for about 16 hours the
formed dicyclohexyl urea is filtered off and the
mother liquor is evaporated. The remaining oily
residue is chromatographed on a column of 400 g
of silica (ethyl/acetate/methanol/water (8.5:1:0.5)
is the eluent), yielding 1.3 g of the title compound.
Example 39
3(S)~3-~[(Methylthio)thioxomethyl~thio]phenyl-
acetyl]amino]-2-oxo-1-azetidinesulfonic acid,
potassium salt
(3s)-3-t~[[(Methylthio)thioxomethyl]thio]-
phenylacetyl]amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (1.3 g; see Example 38)
is dissolved in acetone and treated with an
equivalent amount of potassium perfluorobutane
sulfonate. The addition of ether c~ystallizes
the title compound, which is filtered off, yield
0.18g of product, melting point 157C, dec.
GC155f
-78- 1 338670
Example 40
[3S(D)]-3-[[~1~4-Ethyl-2,3-dioxo-1-piperazinyl)-
carbonyl]amino]-2-thienylacetyl]amino]-2-oxo-1-
azetidinesulfonic acid, potassium salt
S (D)--[1(4-Ethyl-2,3-dioxo-1-piperazinyl)-
carbonyl]amino}2-thiopheneacetic acid (3.25 g),
4.20 g of 3-amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (see Example 6A~ and 1.3 g
of N-hydroxybenzotriazole are dissolved in 25 ml
of acetonitrile. Dropwise addition of 2.06 g
of dicyclohexylcarbodiimide dissolved in 10 ml
of acetonitrile at -10C takes 20 minutes and
stirring is continued for about 16 hours. Formed
urea is filtered off, and the solvent is evaporated
in vacuo. The remaining oil is dissolved in
50 ml of acetone and after treatment with an
equivalent amount of potassium perfluorobutane
sulfonate, the title compound crystallizes,
melting point 185-187C, dec.
Example 41
[3S(+)]-3-[(Bromophenylacetyl)amino]-2-oxo-1-
azet;~;~esulfonic acid, potassium salt
~-Bromophenylacetyl chloride (1.4 g) in
10 ml of acetonitrile is added dropwise to a
solution of 5mM 3-amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt, and 3 g of propylene
oxide in acetonitrile at 0C. After three~hours
stirring, the solvent is distilled off and the
remaining oily residue is dissolved in 30 ml of
acetone. The equivalent amount of potassium
perfluorobutane sulfonate in acetone is added.
The addition of ether crystallizes the title
1 338670 GC155f
-79-
compound, melting point 135-137C dec.
- Example 42
[3S(+)]-3-~[~(Aminocarbonyl)amino~-2-thienylacetyl]-
amino]-2-oxo-1-azetidinesulfonic acid, potassium salt
Nethod I:
To a solution of 2 g of .(S)-3-amino-2-oxo-1-
azetidinesulfonic acid, tetrabutylammonium salt
and 1.5 g of propylene oxide in acetonitrile is
added 1.1 g of (+)-2-amino-4-(2-thienyl)-5(4H)-
oxazolone, hydrochloride. After three hours
stirring at 0 C and one hour at room temperature,
the solvent is distilled off and the oily residue
is dissolved in 30 ml of acetone. By adding the
equivalent amount of potassium perfluorobutane
sulfonate in acetone, the title compound crystallizes
out. Purification by column chromatography on
HP-20 using water as eluent yields the title
compound, melting point 218-222 C, dec.
GC155f
-80- 1 3 3 8 6 7 0
Method II:
To a suspension of 2 g of (+)--~(amino-
carbonyl)amino]-2-thiopheneacetic acid, lOmM of (S)-
3-amino-2-oxo-1-azetidinesulfonic acid, tetrabutyl-
ammonium salt and lOmM of N-hydroxybenzotriazole
in 50 ml of acetonitrile at 0C is added (with
stirring) lOmM of dicyclohexylcarbodiimide
dissolved in 15 ml of acetonitrile. Stirring
is continued for one hour at -15C and for about
16 hours at room t~mr~rature. After filtering
off the dicyclohexyl urea, the solvent is
evaporated and the oily residue is dissolved
in 50 ml of acetone. Adding the equi~alent amount
of potassium perfluorobutane sulfonate in
acetone yields crystalline product. Purification
by column chromatography on HP-20, using water
as an eluent, yields the title compound, melting
point 220-223C.
Example 43
[3S(+)]-3-[[[~(Methylamino)carbonyl]amino]-2-
thienylacetyl]amino]-2-oxo-1-azetidinesulfonic
acid, potassium salt
(+)--[[(Methylamino)carbonyl]amino~-2-
thiopheneacetic acid (0.54 g) and 1.00 g of
(S)-3-amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (see Example 6A) are
dissolved in 20 ml of acetonitrile and 0.5 g of
dicyclohexylcarbodiimide is added at a temperature
of O C. After stirring for 8 hours the dicyclo-
hexylurea is filtered off and after distilling off
GC155f
-81- 1 338670
the solvent the oily residue is dissolved in
50 ml acetone and the equivalent amount of
potassium per~luorobutane sulfonate is added.
Crystalline product is filtered off and purification
is performed on HP-20 using water as an eluent,
melting point 205C, dec.
Example 44
13S (+~ ] -3-1~ mi nooxoacetyl)amino]-2-thien
acetyl]amino]-2-oxo-1-azetidinesulfonic acid,
potassium salt
Following the procedure of Example 42,
method II, but substituting (+)-~-[(aminooxoacetyl)-
amino]-2-thiopheneacetic acid for (+)-a-[(amino-
carbonyl)amino]-2-thiopheneacetic acid, yields
the title compound, melting point 218-222 C.
Example 45
[3s(R*)]-3-[[~[(4-Ethyl-2~3-dioxo-l-piperazinyl)-
carbonyl]amino]phenylacetyl]amino]-2-oxo-1-azetidine-
sulfonic acid, potassium salt
Following the procedure of Example 40, but
substituting (R)-a-1~(4-ethyl-2,3-dioxo-1-pipera-
zinyl)carbonyl]amino]benzeneacetic acid for
(D)-~-[1(4-ethyl-2,3-dioxo-1-piperazinyl)carbonyl]-
amino]-2-thiopheneacetic acid, yields the title
compound, melting point 155-157C, dec.
GC155f
. , .
-82- 1 3386~
Example 46
3-(Acetylamino)-3-methoxy-2-oxo-1-azetidinesulfonic
acid, potassium salt
Method I:
A) 3-[~N-Acetyl-N-chloro)amino]-2-oxo-1-azetidine-
sulfonic acid, mixed sodium and potassi~m salt
To a solution of 3-(acetylamino)-2-oxo-1-
azetidinesulfonic acid,potassium salt (172 mg,
see Example 23) in methanol (17 ml) containing
4% sodium borate decahydrate cooled to -15 to
-10C is added tert-butyl hypochlorite (110 ~1).
The mixture is stirred in the cold for 1 hour,
45 minutes, poured into 0.5 M monobasic potassium
phosphate solution (50 ml), and the pH is lowered
to 5.5. Solvent is removed in vacuo, and the
residue is dissolved in a mi n;mum amount of water
and chromatographed on HP-20AG, 100-200 mesh (140 ml).
Elution with water yields an oil (63 mg) which
crystallizes on standing. Trituration with
methanol/ether and then ether gives a solid
(53 mg) melting point 124C,slow dec.
GC155f
-83-
1 338670
B) 3-(Acetylamino)-3-methoxy-2-oxo-1-azetidine-
sulfonic acid, potassium salt
3-[(N-Acetyl-N-chloro~amino]-2-oxo-1-
azetidinesulfonic acid mixed salt (37 mg) is
dissolved in l.S ml of dry dimethylformamide
and added to a solution of lithium methoxide (50 mg)
inl.~L~l (1 ml) at -78&. After stirring for 15 minutes
at -78C, a 0.5 M solution of monobasic potassium
phosphate (10 ml) is added, and the solution is
then acidified to pH 4 with lN hydrochloric acid.
To the solution is added tetrabutylA~mQn;um hydrogen
- sulfate (70 mg) and the solution is extracted
four times with methylene chloride. The combined
extracts are dried ~sodium sulfate) and solvent
is removed in vacuo to give 55 mg of an oil.
Chromatography on 5.5 g of silica gel yields the
oily product (41 mg) as the tetrabutylammonium
salt (eluted with 8% methanol:92% methylene
chloride). The oil (31 mg) is dissolved in water
and passed through an ion-exchange column (5 ml
of AG 50W-X2,K form, 200-400 mesh, 0.6 meq/ml).
Removal of water in vacuo gives an oil which
crystallizes from methanol-ether. Trituration
twice with ether gives the product as a colorless
powder (11 mg); melting point 182-183C, dec.
- GC155f
-84- i 338670
Method II:
A) 3-Amino-3-methoxy-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt
A 4% sodium borate decahydrate in methanol
solution (100 yl) is added to a suspension of
10% palladium on charcoal (30 mg) in methanol
(2 ml), and the mixture is stirred under an
atmosphere of hydrogen for 15 minutes. 3-Methoxy-
2-oxo-3-.[[(phenylmethoxy)carbonyl]amino~-1-
azetidinesulfonic acid, tetrabutylammonium salt(60mg;see Example 49) in methanol (2 ml) is added and th.e
mixture is vigorously stirred for 15 minutes
under a hydrogen atmosphere. Catalyst is removed
by filtration through Celite on a millipore
filter (0.5 m~), and solvent is removed from
the filtrate in vacuo, and the residue is extracted
with methylene chloride. Removal of solvent
under reduced pressure yields 35 mg of the
title compound, as an oil.
B) 3-(Acetylamino)-3-methoxy-2-oxo-1-azetidine-
sulfonic acid, potassium salt
To a solution of 3-amino-3-methoxy-2-oxo-
l-azetidinesulfonic acid, tetrabutylammonium
salt (35 mg) in methylene chloride (10 ml) at
0C is added propylene o~ide (2 ml) and acetyl
chloride (74 ~1). After 2 hours the solvent
is removed under reduced pressure and the residual
oil is chromatographed on silica gel (4 g).
Elution with 6-8% methanol in methylene chloride
gives an oil (18 mg) which is dissolved in water
and passed through an ion-exchange resin (3 ml
GC155f
_.
-85- 1 3 3 8 6 7 0
of AG 50W-X2, K form, O.6 meq/ml). RemoYal of
water from the eluate in vacuo yields the desired
product (10 mg).
Example 47
N-.(3-Methoxy-2-oxo-1-sulfo-3-azetidinyl)-2-phenyl-
acetamide, tetrabutylammonium salt
A) N-Chloro-N-(2-oxo-1-sulfo-3-azetidinyl)-2-
phenylacetamide, potassium salt
To a solution of (S)-N-(2-oxo-1-sulfo-3-
azetidinyl)-2-phenylacetamide, potassium salt
(50 mg; see Example 1) in methanol containing
4% sodium borate decahydrate (5 ml) cooled in
a -5C bath is added tert-butyl hypochlorite
(20 ~1). After stirring for 32 minutes, the
mixture is poured into 0.5 M pH 5.5 potassium
phosphate buffer at 0C. The resultant solution
(pH 5.9) is adjusted to pH 4.5, concentrated
_ vacuo to remove methanol, and chromatographed
on HP-20AG, 100-200 mesh (100 ml). After washing
the column Wit}l 0.5 M pH 5.5 buffer (100 ml)
and water the desired product is eluted with 9:1
water:acetone. Concentration in vacuo gi~es
50 mg of the title compound.
B) N-(3-Methoxy-2-oxo-1-sulfo-3-azetidinyl)-2-
phenylacetamide, tetrabutylammonium salt
To a stirring solution of lithium methoxide
(160 mg) in 5 ml of methanol cooled to -78C is
added a solution of N-chloro-N-(2-oxo-1-sulfo-
3-azetidinyl)-2-phenylacetamide, potassium salt
GC155f
-86- 1 338670
(149 mg) in 10 ml of dry dimethylformamide. After
addition is complete, the mixture is stirred
for 15 minutes at -78 C, poured into 0.5 N
monobasic potassium phosphate solution (100 ml),
and washed three times with methylene chloride.
Tetrabutylammonium bisulfate (213 mg) is added
to the aqueous layer, which is then extracted
three times with methylene chloride. The
combined extracts are dried (sodium sulfate),
and solvent is removed in vacuo giving an oil
(271 mg). Chromatography of the oil on silica
gel (25 g) and elution with 4% methanol:96%
methylene chloride yields 149 mg of the product
as an oil.
Example 48
N-(3-Methoxy-2-oxo-1-sulfo-3-azetidinyl)-2-
phenylacetamide, potassium salt
N-(3-Methoxy-2-oxo-1-sulfo-3-azetidinyl)-
2-phenylacetamide, tetrabutylammonium salt
(91 mg; see Example 47) is dissolved in water
and passed through an ion-exchange column
(10 ml of AG 50W-X2, K~ form). The eluate is
concentrated in vacuo and the ~sidual oil solid-
ifies on scratching with a methanol-acetone-ether
mixture. After triturating twice with ether,
the product is obtained as a solid (53 mg):
umax1762, 1665 cm ; NMR (CD30D) ~ 3.41 (S, 3H, OCH3),
3.59 (S, 2H, CH2), 3.84 (ABq, J=6.3 Hz, 2H, H4),
7.30 (m, 5H, aromatic).
Analysis: Cal- for C12Hl3~06~l/2H2o
Calc.: C,39.88; H,3.62; N,7.75
Found : C, 39.62; H,3.65; N,7.60
GC155f
-87- 1 338670
Example 49
3-Methoxy-2-oxo-3- I [ (phenylmethoxy)carbonyl]amino]-
l-azetidinesulfonic acid, tetrabutylammonium salt
Method I:
A) 2-Oxo-3-~N-chloro-N-~(phenylmethoxy)carbonyl]-
amino]-l-azetidinesulfonic acid, tetrabutylammonium
salt
(S)-2-Oxo-3-[[(phenylmethoxy)carbonyl~- -
amino]-l-azetidinesulfonic acid, tetrabutylammonium
salt (0.9g; see Example 4) dissolved in 8Q ml of
methylene chloride is added to a mixture (cooled
to 0-5C) of 3.17 g of sodium borate decahydrate
and 11.8 ml of a 5.25% sodium hypochlorite
solution in 70 ml of water. The reaction mixture
is vigorously stirred for 55 minutes while
cooling in an ice bath. After diluting the
mixture with 0.5 M monobasic potassium phosphate
solution, the product is extracted with methylene
chloride (three 150 ml portions). Combination
of the extracts, drying (sodium sulfate), and
removal of solvent in vacuo yields the title
compound as an oil (0.94 g).
B) 3-Methoxy-2-oxo-3-[[(phenylmethoxy)carbonyl]-
amino]-l-azetidinesulfonic acid, tetrabutyl-
ammonium salt
To a stirring solution of lithium methoxide
(667 mg) in anhydrous methanol (10 ml) at -78C
is added a solution of 2-oxo-3-[N-chloro-N-[(phenyl-
methoxy)carbonyl~amino]-l-azetidinesulfonic acid,
tetrabutylammonium salt (0.94 g) in dry dimethyl-
GC155f
-88- 1 3 3 8 6 7 0
formamide (10 ml). After stirring the mixture
at -78C for 1 hour, it is poured into 0.5 M
monobasic potassium phosphate solution and
extracted with methylene chloride (three 150 ml
portions). The combined extracts are dried
(sodium sulfate) and solvent is removed in vacuo
to give an oil (0.83 g). The desired product
is obtained by chromatographing the oil on silica
gel (100 g) and eluting with 4-5~ methanol in
methylene chloride to give an oil (513 mg):
(neat) 1767, 1720 cm 1; NMR (CDC13) ~ 3.40
(S,OCH3), 3.03 (ABq, J=6.5 Hz, H4), 5.08 (S,CH2),
6.00 (S,NH), 7.27 (S, aromatic).
Method II:
To a solution of 3-benzyloxycarbonylamino-
3-methoxy-2-oxo-1-azetidinesulfonic acid, potassium
salt (400 mg) in water is added a 0.1 M tetra-
butylammonium bisulfate solution (10.9 ml, adjusted
to pH 4.3 with potassium hydroxide). The mixture
is extracted three times with methylene chloride,
the extracts are combined, dried (Na2S04) and
solvent is removed in vacuo to give a foam (625 mg)~
having spectral characteristics approximating those
of the product of Method I.
- - GC155f
-89-
1 338670
Example 50
3-Methoxy-2-oxo-3-~[(phenylmethoxy)carbonyl]amino]-
l-azetidinesulfonic acid, potassium salt
Method I:
A)- 2-Oxo-3-[N-chloro-N-[(phenylmethoxy)carbonyl]-
amino]-l-azetidinesulfonic acid, potassium salt
To a solution of (S)-2-oxo-3-l~(phenyl-
- methoxy)carbonyl]amino]-l-azetidinesulfonic acid,
potassium salt (1.00 g; see ~YAmple 3) in methanol
(90 ml) containing 4% sodium borate decahydrate
at -10C is added tert-butyl hypochlorite (420 ~1).
After stirring for 2 hours at -10 C, 0.5 M mono-
basic potassium phosphate solution (100 ml) is
added and the pH is adjusted to 6 with 1 N hydro-
chloric acid. After concentration of the solvent
in vacuo to 30 ml, the aqueous solution is
chromatographed on HP-20AG, 100-200 mesh (200 ml).
After passage of a solution of monobasic potassium
phosphate (50 g) in water (1000 ml), followed
by water (2000 ml), the product is eluted with
10% acetone-90~ water. Solvent is removed
in vacuo and the title compound is crystallized
from water to give a solid (530 mg), melting
point 173-175C.
GC155f
-go- 1 338 6 70
B) 3-Methoxy-2-oxo-3-[l(phenylmethoxy)carbonyl]-
amino]-l-azetidinesulfonic acid, potassium salt
To a solution of lithium methoxide (874 mg)
in dry methanol (10 ml) at -78 C is added 2-oxo-
3-[N-chloro-N-[(phenylmethoxy)carbonyl]amino]-
l-azetidinesulfonic acid, potassium salt (857 mg)
in dry dimethylformamide (13 ml). After 15
minutes at -78C the mixture is poured into
0.5 M monobasic potassium phosphate solution
(200 ml) and the pH is adjusted to 5.5 with lN
hydrochloric acid. The aqueous mixture is washed
with methylene chloride (three 100 ml portions),
and tetrabutylammonium bisulfate (1.169 g) is added.
The product is extracted with methylene chloride
(three 200 ml portions), dried (sodium sulfate),
filtered, and concentrated in vacuo. The
residual oil is chromatographed on silica gel
(150 g) and the product is eluted with 2-4~
methanol in methylene chloride yielding the
- 20 tetrabutylammonium salt of the product as an
oil (701 mg). A portion of the oil (51 mg)is
dissolved in water and passed through an ion-
exchange column (3 ml of AG 50W-X2, 200-400 mesh,
K~ form, 0.6 meq/ml). Concentration of the
eluate in vacuo yields an oil (30 mg), which is
crystallized by scratching with acetone:
vmax(KBr) 1760, 1725 cm ; NMR (D20) ~ 3.48
(S, 3H, OCH3), 3.92 (S, 2H, H4); 5.20 (S, 2H, CH2),
7.42 (S, 5H, aromatic), m.p. 196-198.
GC155f
1 338670
Method II: `
A) l-Chloro-3-1N-chloro-N-l(phenylmethoxy)carbonyl]-
2-azetidinone
A solution of 3-~[(phenylmethoxy)carbonyl]-
amino]-2-azetidinone (440 mg.; see Example 2C)
in 40 ml of 4% methanolic borax is cooled to
0C and 0.5 ml of t-butyl hypochlorite is added.
After 30 minutes at 0C, the solution is poured
into 200 ml of cold water and extracted with two
100 ml portions of ethyl acetate. The combined
ethyl acetate layer is washed with water, dried,
and evaporated in vacuo to give 546 mg of the
title compound as an oil.
B) 3-Methoxy-3-[[(phenylmethoxy)carbonyl~amino~-
2-azetidinone
A solution of 730 mg (0.0025 mole) of l-chloro-
3-[N-chloro-N-[(phenylmethoxy)carbonyl~amino]-2-
azetidinone in 5 ml of tetrahydrofuran is cooled
to -78C and 4 ml of methanol containing 285 mg
of lithium methoxide is added. After 20 minutes at
-78 C, 0.6 ml of acetic acid and 0.6 ml of
trimethylphosphite are added. The solution is
stirred for 5 minutes at -78C, allowed to warm
to ambient temperature and stirred for 30 minutes.
The resulting solution is diluted with ethyl
acetate, washed with 5% sodium bicarbonate,
water, 5% potassium bisulfate, water, saturated
salt solution, and dried. Solvent removal gives
an oil that is applied to four 20 x 20 cm xl mm
Çcl55f
-92 1 3 3 8 6 7 0
silica gel plates. Development with benzene-
ethyl acetate (1:1) and isolation of the major
W-active band of Rf=0.25 gives 91 mg of oil
that crystallizes from ether to give a solid.
Recrystallization from ether gives the title
compound, melting point 112-114C.
C) 3-Methoxy-2-oxo-3-[~(phenylmethoxy)carbonyl~-
amino~ azetidinesulfonic acid, potassium salt
~ solution of 25 mg of 3-methoxy-3-[[(phenyl-
methoxy)carbonyl]amino]-2-azetidinone in 0.175 ml
each of dichloromethane and dimethylformamide
is stirred for 24 hours with 55.4 mg of a complex
of pyridine-sulfur trioxide. The resulting
slurry is diluted with 5 ml of cold 0.5 M monobasic
potassium phosphate (adjusted to pH 4.5) and
extracted with ethyl acetate. The aqueous layer
is applied to a 40 ml HP-20 AG column. Elution
with additional buffer, water, and water-acetone
(9:1~ gives 32 mg of the title compound as an
oil that slowly solidifes. Crystallization from
acetone gives the title compound, melting point
196-198C, dec.
- G~155f
-93-
1 338670
Example 51
3-lll,3-Dioxo-2-phenyl-3-(phenylmethoxy)propyl]amino]-
3-methoxy-2-oxo-l-azetidinesulfonic acid, potassium
salt
S A) 3-[l1,3-Dioxo-2-phenyl-3-(phenylmethoxy)propyl~-
amino]-3-methoxy-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt
Crude 3-amino-3-methoxy-2-oxo-1-azetidine-
sulfonic acid, tetrabutylammonium salt (431 mg,
see Example 74), containing borax, is dissolved
in 30 ml of dry acetonitrile. Dry pyridine
( 317 I~l) is added and the solution stirred well
at -lO C under dry nitrogen. ~-(Benzyloxycarbonyl)-
phenylacetyl chloride (568 mg) in 3 ml of dry
acetonitrile is added dropwise. Thin layer
chromatography indicates the reaction to be
complete after 15 minutes. Potassium phosphate
buffer (0.5 M, pH 5.5, 17 ml) is added and most
of the acetonitrile is removed in vacuo. The
2~0 residue is diluted with water and extracted
three times with equal volumes of methylene
chloride. The extract is dried over anhydrous
sodium sulfate and evaporated in vacuo to give
1.032 g of crude product, which is dissolved in
3 ml of methylene chloride and chromatographed
on a silica column using methylene chloride-
methanol to give 470 mg of the title compound.
- 1 3 3 8 6 70 GC155f
-94-
B) 3-ltl~3-Dioxo-2-phenyl-3-(phenylmethoxy)-
propyl~amino]-3-methoxy-2-oxo-1-azetidinsulfonic
acid, potassium salt
3-[[1,3-Dioxo-2-phenyl-3-(phenylmethoxy)-
propyl]amino]-3-methoxy-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (470 mg) is dissolved
in 15 ml of 30% acetone: water and put through a
Dowex 50~X2(K form) column using the same solvent
as eluent. The total eluate is evaporated in
vacuo to yield 345 mg of an amorphous solid
which is lyophilized to an amorphous powder,
melting point 100-120C.
Anal. for C20H19O8N2SK Calc'd: C,49.37; H,3.94
N,5.76; S,6.59
lS Found: C,49.08; H,4.00; N,5.58; S,6.29
Example 52
(+)-3-tL~(Cyanomethyl)thio]acetyl]amino~-3-methoxy-
2-oxo-1-azetidinesulfonic acid, potassium salt.3
To a solution of 3-amino-3-methoxy-2-oxo-
l-azetidinesulfonic acid, tetrabutylammonium
salt (414 mg, see Example 74) in dry acetonitrile
(50 ml) at -20C is added diethylaniline (210 yl)
and cyanomethylthioacetyl chloride (169 mg).
After 10 minutes the solvent is removed under
reduced pressure and a 0.LM solution of tetrabutyl-
ammonium sulfate (22.8 ml) adjusted to pH 4.3
with potassium hydroxide is added and the product
extracted with three 50 ml portions of methylene
chloride, dried, filtered and concentrated under
reduced pressure. The oily residue is purified
on silica gel (60 g) and the product (294 mg)
is eluted with 4% methanol-methylene chloride.
The purified product is passed through an ion
1 338670 GC155f
--95--
exchange resin (16 ml AG 50W-X2(K form), 0.6
mequiv./ml 200-400 mesh). Removal of water
gives 164 mg of partially purified product.
The product is further purified on Diaion AG HP 20
(100 ml) using water as eluent. The solvent is
removed under reduced pressure to give 126 mg of
product which is triturated with ether to
give 76 mg of the title compound, melting point
110-125C.
Anal. Calc'd for C8HloN3S2O6K: C,27.66; H,2.88;
N,12.10; S,18.44
Found: C,27.25; H,3.00; N,10.84; S,17.53
Examples 53-56
Following the procedure of Example 42, Method
II, but substituting the compound listed in
Column I for (+)-a-~ (aminocarbonyl)amino]-2-
thiopheneacetic acid, yields the compound listed
in Column II.
1 338670
GC155f
-
--96--
I ~ I
0 ~ ~ ~
O ~ _I
O ~ ,~ ,1 ~ ~ ~ .,1 ~
c _I U O E ~ E~ e
,, ~ e ~
. E ~ ~ N ' I~ N '
~ e u ~ O ~
,1 ~ c a) ~ ~ c E c E
c e o o ~ ~ O
_, o ~ e ,~ ,~ x , - ,
1 0 S ~ ~ O N U' '.~ N Ul
1 ~ O I U O I O~
H G
o ,., ~n ~U ~ N a) I~ ''I O ~lf ''I ~
, e ~" N U~ e ~ o ~E C: o
e ~,~ ~ ~ o I e --8 C.) --e
~c ~ 1~ ~ ~--I ~-
0 --~ ~ C l I~ N
I r e ~ ~ ~
~ N ~0 1 ~N OQ _ ~ O O ~ U N
+1 0 0 U ~ +1 '~ O ~ O ~ ~ ~1 0 JJ
~ e I o -l ~ X ~c ~ O ~ ~ O
c`~ l u c
o-~ I a
x J
O o ~ o
c - _ c) c o
o ~ ~1 c
.,, ~ nJ e
e ~
--I ~ s _~ _
~ _ ~ >1 _I
H J _I O J ~,,
~11 C
C ' S ~- C ~ o
u
Y ~ e~ ~ ~
O~ U O I
C ~ : ~ O U C
~ O C.~l C
~ U ~ C,-,, ~
F ~ e ~ ~ ~
~ --e ~u ~u
I _ I U
+ l ~ + l U ~ U
_,, _ " -- -- ~a
,
~ ~ In ~D
Ul u~ Ln U~
1 3386~0 GC155f
-97-
Example 57
[3S(+)]-3-[[2-~ethylthio)-1-oxopropyl~amino~-2-
oxo-l-azetidinesulfonic acid, potassium salt
Following the procedure of Example 31,
S Method II, but substituting (+)-2-(methylthio)-
propanoic acid for (+)--azido-benzeneacetic acid,
yields the title compound, melting point 173C,dec.
Example 58
[3S(~)~]-3-t~[[~2,3-Dioxo-4-I(phenylmethylene)amino]-
l-piperazinyl]carbonyl]amino]phenylacetyl]amino]-2-
oxo-l-azetidinesulfonic acid, potassium salt
(R)--~[~2,3-Dioxo-4-l(phenylmethylene)amino~-
l-piperazinyl]carbonyl]amino]benzeneacetic acid
(0.7 g) and (S)-3-amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (0.8 g; see Example
6A) are dissolved in 20 ml of acetonitrile.
While stirring, 0.4 g of dicyclohexylcarbodiimide
is added dropwise at 0C. Stirring is continued
for 18 hours and, after filtration, the solvent
is distilled off leaving an oily residue. The
residue is dissolved in acetone and treated with
potassium perfluorobutane sulfonate to precipitate the
title compound. Purification by column
chromatography on HP-20 using water as eluent
yields the title compound, melting point 193-194 C.
GC155f
- 1338670
-98-
Example 59
~3S(Z)]-3-[[~2-Amino-4-thiazoly~[(2-methoxy-2-
oxoethoxy)imino]acetyl]amino]-2-oxo-1-azetidine-
- sulfonic acid, potassium salt
(z)-2-Amino--t(2-methoxy-2-oxoethoxy)imino]-
4-thiazoleacetic acid (1.3 g) and (S)-3-amino-2-oxo-
l-azetidinesulfonic acid, tetrabutylammonium salt
(2.03 g; see Example 6A) is dissolved in 50 ml
.of acetonitrile and 1.03 g of dicyclohexyl-
carbodiimide dissolved in 5 ml of acetonitrile
is added dropwise at 0C. A~ter stirring for
15 hours and filtering off dicyclohexyl urea the
solvent is distilled off. The remaining oily
residue is dissolved in acetone and treated with
the equivalent amount of potassium perfluoro-
butane sulfonate. The title compound is isolated
and purified by column chromatography on HP-20
using water as eluent, yielding the title compound
melting point 195-198C.
Example 60
[3S~R*)~-3-1[1[[3-~(4-Chlorophenyl)methylene~-
amino]-2-oxo-1-imidazolidinyl]carbonyl]amino]-
phenylacetyl]amino]-l-azetidinesulfonic acid,
potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (1.5 g; see Example
6A) in 100 ml of absolute diglyme (diethylene-
glycoldimethyl ether), 1.5 g of (R) -a- [ r [3-~(4-
chlorophenyl)methylene]amino~-2 oxo-1-imidazolidinyl]-
carbonyl]aminolbenzeneacetic acid, an equivalent
amount of dicyclohexylcarbodiimide, and Q~5 g of
1 338670 GC155f
_99_
hydroxybenzotriazole are stirred together for
12 hours. The solvent is removed in vacuo
and the residue is dissolved in 50 ml of acetone
and filtered. Acetone is distilled off and
the residue is dissolved in 200 ml of methylene
chloride. The solution is washed with aqueous
sodium bicarbonate and then with aqueous sodium
chloride solution. The methylene chloride
layer is dried over sodium sulfate, evaporated
to dryness and crystallized by the addition of
ether. The compound is recrystallized from
acetone-ether. The resulting white crystalline
powder is dissolved in acetone and treated with
an equivalent amount of potassium perfluoro-
butane sulfonate in acetone. The title compoundprecipitates and is filtered off, yielding
1.4 g of product melting point 217-222C.
Example 61
[3S(R*)]-3-[[~[12-Oxo-3-[(phenylmethylene)amino]-
l-imidazolidinyl]carbonyl~amino]phenylacetyl]-
amino]-l-azetidinesulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylAmmon;um salt (2.25 g; see Example
6A) in 100 ml of dry dimethylformamide (DMF),an
equivalent of dicyclohexylcarbodiimide, 2.5 ~ of
(R) -a- [ [ [2-oxo-3-[(phenylmethylene)amino~-1-
imidazolidinyl]carbonyl]amino]benzeneacetic acid
and 0.85 g of hydroxybenzotriazole are stirred
together at ambi~nt temperature for 12 hours.
After this time, DMF is removed in vacuo and
the residue is dissolved in 50 ml of acetone.
GC155f
._
lOO 1 338670
The precipitated urea is filtered off and the
mother liquor is treated with an equivalent
amount of potassium perfluorobutane sulfonate
in 20 ml of acetone. After addition of 100 ml
of ether, the title compound precipitates and
is filtered off. Purification is achieved by
dissolving the compound in DMF/acetone and
precipitating with water, yielding 1.5 g of
product, melting point 224-226C, dec.
Example 62
~3S(R*)]-3-t~[~13-(Methylsulfonyl)-2-oxo-1-
imidazolidinyl]carbonyl~amino]phenylacetyl]-
amino}2-oxo-1-azetidinesulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (2.25 g; see Example 6A)
in 60 ml of dimethylformamide is stirred for 12
hours with 1.9 g of (R)-~-[~[3-(methylsulfonyl)-
2-oxo-1-imidazolidinyl]carbonyl]amino]benzeneacetic
acid, 0.75 g of hydroxybenzotriazole and 2 3 g
of dicyclohexylcarbodiimide. The solvent is
removed in vacuo and the residue is dissolved
in 20 ml of acetone and filtered. The mother
liquor is treated with 1.87 g of potassium
perfluorobutane sulfonate in 20 ml of acetone.
After the addition of ether, the title compound
precipitates, yielding 2.0 g of material. The
compound is purified by recrystallization from
water and has a melting point of 240-245C, dec.
~ GC155f
-lol- I 3 3 8 6 7 0
Example 63
[3S(R*)]-3-[(Hydroxyphenylacetyl)a~inoJ
azetidinesulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (1.5 g; see Example 6A)
in 100 ml of dry dimethylformamide is stirred
for about 16 hours with 1.5 g of dicyclohexyl-
carbodiimide, 0.5 g of hydroxybenzotriazole
and 0.6 g-of R-~-hydrox~benzeneacetic acid.
The solvent is removed in vacuo and the residue
is dissolved in 20 ml of acetone. The precipitated
urea is filtered off and the mother liquor is
treated with an equivalent amount of potassium
perfluorobutane sulfonate. After addition of
ether, the title compound precipitates and is
filtered off, yielding 1.3 g of crude product.
The product is purified by chromatography
using HP-20 and water/acetone (9:1) as eluent,
and has a melting point of 145-150C, dec.
Example 64
[3S(S*)]-3-~(Hydroxyphenylacetyl)amino]-2-oxo-
l-azetidinesulfonic acid, potassium salt
Following the procedure of Example 63,
but substituting (S)-~-hydroxybenzeneacetic acid
for (R)--hydroxybenzeneacetic acid, yields
the title compound, melting point 195-197 C.
~C15s~
.
-1~2~ 1 33 8 6 70
Example 65
[3S(+)]-2-Oxo-3-[(phenylsulfoacetyl)amino]-1-
azetidinesulfonic acid, potassium salt (1:2)
(S)-3-Amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (2.25 g; see
~Y~mrle 6A) in 100 ml of dry diglyme, 2.4 g
of triethylamine and 0.3 g of dimethylaminopyridine
are cooled to 0C. (R)-~-(Chlorocarbonyl)-
benzenemethanesulfonic acid (1.8 g) in 20 ml
of diglyme is added dropwise. The temperature
is maintained for 2 hours, the solvent is
distilled off in vacuo and the residue is
dissolved in acetone. The insoluble precipitate
is filtered off and the mother liquor is treated
with an equivalent amount of potassium perfluoro-
butane sulfonate. After the addition of ether,
the title compound precipitates and is filtered
off, yielding 1.4 g of crude product; melting point
240-245C dec., after recrystallization from
water/acetone-
Example 66[3S(Z)-3-tt(2-Amino-4-thiazolyl)lt(diethoxy-
phosphinyl)methoxy]imino]acetyl]amino]-2-oxo-
l-azetidinesulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt (2.25 gJ see
Example 6A) in 100 ml of dry dimethylformamide
is treated with 1.87 g of (Z)-2-amino-~-[[(diethoxy-
phosphinyl)methoxy]imino]-4-thiazoleacetic acid,
0.75 g of hydroxybenzotriazole and 2.29 g of
dicyclohexylcarbodiimide for 12 hours with stirring.
The precipitated urea is filtered off and the solvent
- GC155f
-103- 1 33 8 6 70
removed in vacuo. The remaining oil is treated
with an equivalent amount of potassium perfluoro-
butane sulfonate in 20 ml of acetone. After the
addition of ether, the title compound precipitates
and is filtered off, yielding 2.77 g of crude
product. Purification of this crude product
by column chromatography using HP-20 and water/
acetone (9:1) as eluent yields the title compound,
melting point 155-160C, dec.
Example 67
[3S(Z)]-3-~(2-Amino-4-thiazolyl)[12-(1,1-dimethyl-
ethoxy)-2-oxo-1-phenylethoxy]imino~acetyl~amino]-2-
oxo-l-azetidinesulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (2.25 g; see Example 6A)
in 60 ml of dimethylformamide is stirred at room
temperature with 2.4 g of (Z)-2-amino--~[2-
(l,l-dimethylethoxy)-2-oxo-1-phenylethoxy]imino~-
4-thiazoleacetic acid, 1 g of hydroxybenzotriazole
and 1.5 g of dicyclohexylcarbodiimide for 12 hours.
The solvent is removed in vacuo and the residue
is dissolved in 50 ml of acetone. The precipitated
urea is filtered off and the mother liquor is
treated with an equivalent amount of potassium
perfluorobutane sulfonate. After the addition
of ether, the title compound crystallizes and
is filtered off. Purification of the compound
is achieved by HP-20 column chromatography using
water/acètone (7:3) as eluent, yielding 1 g of
product, melting point >250 C,dec.
GC155f
-104- 1 3 3 8 6 7 0
Example 68
[3S(Z)]-3-11(2-Amino-4-thiazolyl)[(lH-tetrazol-
5-ylmethoxy)imino]acetyl~amino]-2-oxo-1-azetidine-
sulfonic acid,potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (1.9 g; see Example 6A)
in 60 ml of dimethylformamide is treated with 1.4 g
of (Z)-2-amino-~-[(lH-tetrazol-5-ylmethoxy)imino]-
4-thiazoleacetic acid, 0.7 g of hydroxybenzotriazole
and 1.4 g of dicyclohexylcarbodiimide with stirring
for 24 hours. Afte~ the solvent is removed in vacuo,
the residue is dissolved in acetone and the pre-
cipitated urea filtered off. The mother liquor is
treated with an equivalent amount of potassium
perfluorobutane sulfonate in 10 ml of acetone.
The title compound is precipitated by the addition
of 200 ml of ether. Purification is achieved by
HP-20 column chromatography using HP-20 resin and
water as eluent and yields 1.05 g of product,
melting point 250C, dec.
Example 69
[3S(Z)]-3-[{(2-Amino-4-thiazolyl)~(phenylmethoxy)-
imino]acetyl]amino]-2-oxo-1-azetidinesulfonic acid,
potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (1.5 g; see Example 6A),
1.23 g of (Z)-2-amino-~-[(phenylmethoxy)imino]-4-
thiazoleacetic acid, 0.57 g of hydroxybenzo-
triazole and 1.14 g of dicyclohexylcarbodiimide
are stirred in 60 ml of dimethylformamide at
room temperature for 24 hours. The precipitated
urea is filtered off, the solvent removed and
GC155f
-105-
1 338670
the residue treated with an equivalent amount of
potassium perfluorobutane sulfonate in 10 ml of
acetone. After the addition of 200 ml of ether,
the title compound precipitates, is filtered
off and is purified by HP-20 column chroma-
tography using water/acetone (9:1) as eluent,
yielding 1 g of material, melting point 200 C, dec.
- Example 70
[3S(Z)]-3-[~(2-Amino-4-thiazolyl)[(carboxymethoxy)-
imino]acetyl]amino~-2-oxo-1-azetidinesulfonic acid,
potassium salt (1:1)
[3S(Z3]-3-lt(2-Amino-4-thiazolyl)t~2-(diphenyl-
methoxy)-2-oxoethDxy]imino]acetyl]amino]-2-oxo-1-
lS azetidinesulfonic acid, potassium salt (1:1) (1.3 g;see Example 30) is mixed with 5 ml of anisole.
At -15C 25 ml of trifluoroacetic acid is added
and the mixture is stirred for 10 minutes. Ether
(100 ml) is added slowly at -10C and subsequen~ly
50 ml of petroleum ether. The precipitate is
suspended with cooling in 20 ml of water and
adjusted to pH 5.0 with diluted potassium
hydroxide. The produc~ is purified by chroma-
tography on an HP-20 column, yielding 3.0 g of
the title compound, melting point 230-235C, dec.
- GC155f
-106- 1 3 3 8 6 7 0
Example 71
[3S(Z)]-3-~[(2-Amino-4-thiazolyl)1[2-oxo-2-(phenyl-
methoxy)ethoxy]imino]acetyl]amino]-2-oxo-1-azetidine-
sulfonic acid, potassium salt
Following the procedure of Example 28,
but substituting- (Z)-2-amino-a-[[2-oxo-2-(phenyl-
methoxy)ethoxy]imino]-4-thiazoleacetic acid for
(Z)-2-amino-~-1[2-(diphenylmethoxy)-1,1-dimethyl-
2-oxoethoxy]Lmino]-4-thiazoleacetic acid, yields
the title compound, melting point ca. 170C, dec.
Example 72
[3S(Z)]-3-[1~(2-Amino-2-oxoethoxy)imino](2-amino-
4-thiazolyl)acetyl]amino}2-oxo-1-azetidinesulfonic
acid, potassium salt
Following the procedure described in Example
28, but substituting (Z)-2-amino-~-[(2-amino-
2-oxoethoxy)imino]-4-thiazoleacetic acid for
(Z)-2-amino-~-[12-(diphenylmethoxy)-1,1-dimethyl-
2-oxoethoxy]imino~-4-thiazoleacetic acid, yields
the title compound melting point 205-210C, dec.
Example 73
[3S(Z)]-3-[1(2-Amino-4-thiazolyl)(hydroxyimino)-
acetyl]amino]-2-oxo-1-azetidinesulfonic acid,
potassium salt
A solution of 0.6 grams of 90% hydroxy-
benzotriazole in 100 ml of dimethylformamide
is stirred for one hour with 10 grams of 4A
molecular sieves, filtered,and the filtrate
added to a solution of 0.004 mole of (S)-3-amino-
2-oxo-1-azetidinesulfonic acid, tetrabutylammonium
GC155f
-107- 1 338670
salt (see Example 6A) in dimethylformAm;~e.
(Z)-2-Amino-a-(hydroxyimino)-4-thiazoleacetic
acid (0.89 g) is added, followed by the addition
of 0.91 g of dicyclohexylcarbo~ e. The
mixture is stirred for about 16 hours, evaporated
in vacuo and the residue dissolved in 20 ml of
acetone and filtered. The addition of a solution
of potassium perfluorobutane sulfonate causes
the title compound to Precipitate. Chromatography
on HP-20 resin yields 0.44 g of p~oduct, melting
point >240C. - ~
Example 74
lS 3-Methoxy-2-oxo-3-[(2-thienylacetyl)amino]-1-
azetidinesulfonic acid, potassium salt
A) 3-Amino-3-methoxy-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt
(+)-3-Methoxy-3-[[(phenylmethoxy)carbonyl]-
aminoJ-2-oxo-1-azetidinesulfonic acid, tetra-
butylammonium salt (143 mg, see Example 49) is
- dissolved in 15 ml of dry methanol. Na2B4O7 10 H2O
(12 mg, 0.1 equiv.) is added, followed by 10%
palladium on carbon (72 mg). The mixture is
hydrogenated at one atmosphere pressure for 15
minutes. The catalyst is removed by filtration
and the filtrate evaporated in vacuo, yielding
114 mg of the title compound.
GC155f
-
-108-
- 1 338670
B) 3-Methoxy-2-oxo-3-[(2-thienylacetyl)amino~-1-
azetidinesulfonic acid, tetrabutylammonium salt
3-Methoxy-3-amino-2-oxo-1-azetidinesulfonic
acid, tetra~utylammonium salt (102 mg) is dissolved
S in 10 ml of dry acetonitrile. Dry pyridine
(56.~ ~1) is added and the solution is stirred
well at -10 C under dry nitrogen. Thienylacetyl
chloride (44 ~1) in 1 ml of dry acetonitrile
is added dropwise. ~n 15 minutes the reaction
is complete as shown by thin layer chromatography.
Potassium phosphate buffer (0.5 M, pH 5.5, 4.2 ml)
and tetrabutylammonium sulfa*e (8.5 mg, 0.1 equiv.)
are added and most of the acetonitrile is removed
_ ~acuo. The residue is diluted with water and
extracted with three 20 ml portions of methylene
chloride. The extract is dried over anhydrous
sodium sulfate and evaporated in vacuo to yield
107 mg of a gum. The crude product is purified
by chromatography on silica gel using methylene
chloride-methanol, yielding 66 mg of the title
compound.
C) 3-Methoxy-2-oxo-3-1(2-thienylacetyl)amino]-
l-azetidinesulfonic acid, potassium salt
3-Methoxy-2-oxo-3-[(2-thienylacetyl)amino-
l-azetidinesulfonic acid tetrabutylammonium
salt (154 mg) is dissolved in 3 ml of 30%
acetone-water, passed through a column of
Dowex 50W-X2 (K+ form) and eluted with the
30 same solvent. The total eluate is evaporated
in vacuo to yield 95 mg of product which is
GC155f
-109- 1338670
lyophilized to gi~e an amorphous powder, melting
point 120-135C.
Anal. Calc'd for CloHllN2O6S2K: C,33.51; H,3.09;
N,7.82; S,17.89
Found: C,33.46; H,3.08; N,7.92; S,17.64
Example 75
[3S(Z)]-3-~(2-Amino-4-thiazolyl)[(carboxymethoxy)-
imino]acetyl~amino]-2-oxo-1-azetidinesulfonic acid,
potassium salt
[3S(Z)]-3-[ I (2-Amino-4-thiazolyl)[[2-oxo-2-
(phenylmethoxy)ethoxy]imino]acetyl]amino]-2-oxo-
l-azetidinesulfonic acid, potassium salt (0.1 g;
see Example 71) is dissolved in a mixture of
5 ml of ethanol and 5 ml of water and hydrogenated
at room temperature in the presence of 0.2 g
of 10% palladium on charcoal. After 2 hours
the catalyst is filtered off and the remaining
solution is freeze-dried yielding the title compound.
m.p. 235(dec.).
Example 76
3-[~(S)-[(Aminocarbonyl)amino~-2-thienylacetyl~-
amino]-3-methoxy-2-oxo-1-azetidinesulfonic acid,
potassium salt, isomer A
To a solution of 3-amino-3-methoxy-2-oxo-1-
azetidinesulfonic acid, tetrabutylammonium salt
(277 mg; see Example 46, method II, part A) in
15 ml of dry acetonitrile at -20C is added
pyridine (71 ~1) 0.888 mmole) and (D)-2-amino-4-
(2-thienyl)-5-(4H)-oxazoline hydrochloride (166 mg).
The mixture is stirred for 10 minutes and a second
GC155f
-llo- 1 3 3 8 6 7 0
aliquot of pyridine ~71 ~1) and oxazoline (166 mg)
is added. After another 10 minutes the solvent is
removed under reduced perssure and the residue is
dissolved in a water-acetone mixture and passed
through an ion-exchange resin (20 ml AG 50W-X2,K+
form, 200-400 mesh). Removal of the water from fractions
2-3 gives a mixture of diastereomers (248 mg).
The product is purified and the diastereomers
separated on Diaion AG HP 20 (130 ml) and eluted with
water. Isomer A is eluted in fractions 23-28 (30 mg),
and isomer B in fractions 35-45 (30 mg) (8 ml fractions).
The middle fractions (10 mg) are combined with fractions
from other runs and a total of 35 mg of isomer A and
59 mg of isomer B is isolated.
Anal. for isomer A, m.p. 158-165C.
C CllH13 4 7 2 K / H2O
C, 31.05; H, 3.29; N, 13.18
Found:C, 30.95; H, 2.97; N, 12.98
Anal. for isomer B, m.p. 160-170C(dec).
Calc'd for CllH13N4O7S2 / 2
C, 31.05; H, 3.29; N, 13.18; S, 15.05
Found:C, 31.17; H, 3.09; N, 13.13; S, 15.09
Example 77
3-Methoxy-2-oxo-3-[(phenylsulfoacetyl)amino]-1-
azetidinesulfonic acid, dipotassium salt
To a stirred solution of crude 3-amino-3-
methoxy-2-oxo-1-azetidinesulfonic acid, tetrabutyl-
ammonium salt (366 mg; see Example 74A) and 38 mg
of borax in 35 ml of dry acetonitrile at -10C
under nitrogen is added 0.53 ml of dry pyridine
followed by a two minute addition of a solution
of ~-sulfophenylacetyl chloride monoetherate
GC155f
-111- 1 3 3 8 6 7 0
(348 mgt in 8 ml of acetonitrile. After 20 minutes
the solvent is remo~red in vacuo and the residue is
treated with 35 ml of 0.5 ~1 pH 5.5 potassium
phosphate buffer. Tetrabutylammonium hydrogen
sulfate (383 mg) is added and the mixture is
extracted three times with methylene chloride.
The methylene chloride is dried (sodium sulfate)
and evaporated to give 685 mg of crude product.
The crude product is combined with 115 mg
io of crude from a second run and the combined material
is purified on a col-umn of SiliCAR CC-4 using
methylene chloride and then 2,4,6,8 and 10% methanol
in methylene chloride as eluent. The product, a
mixture (about 6:1) of racemic diastereomers in
the tetrabutylalr~nonium salt form, is converted to
the potassium salt by passage through Dowex 50W-X2
(K+ form) resin using 20~ acetone in water as
solvent. The product is lyophilized yielding
171 mg of the title compound, melting point
205-210C, dec.
Anal. Calc'd for C12H12N2OgS2K2 2
N,5.74; S,13.10
Found: C,29.45; H,2.74; N,5.51; S,12.82
Example 78
3-[(Carboxyphenylacetyl)amino]-3-methoxy-2-oxo-
1-azetidinesulfonic acid, dipotassium salt
3- [[1,3-Dioxo-2-phenyl-3-tphenylmethoxy)-
propyl]amino]-3-methoxy-2-oxo-1-azetidinesulfonic
acid, potassium salt (39 mg; see Example 51) is
dissolved in methanol (5 ml). Anhydrous potassium
GC155f
-112-
1 338670
carbonate (3.9 mg) and 10% palladium on carbon
(19 mg) are added and the mixture is hydrogenated
at atmospheric pressure for 20 minutes. The catalyst
is removed by filtration and the filtrate is
S evaporated in vacuo to yield a glassy residue
(34 mg) which is lyophilized to an amorphous
powder, melting point 178-190C,dec.
C13H12O8N2S K2- 0 5 H2O: C,35.20; H 3 18;
N,6.32; S,7.23
Found: C,35.51; H,2.96; N,6.29; S,6.92
Example 79
[3S(Z)]-3-[[(2-Amino-4-thiazolyl)[[2-(1,1-dimethyl-
ethoxy)-l-(methylthio)-2-oxoethoxy]imino]acetyl]-
amino]-2-oxo-1-azet;~;nesulfonic acid, potassium salt
Following the procedure of Example 73,
but substituting (Z)-2-amino-~-[[2-(1,1-dimethyl-
ethoxy)-l-(methylthio)-2-oxoethoxy]imino]-4-
thiazoleacetic acid for (Z)-2-amino-a-(hydroxy-
imino)-4-thiazoleacetic acid, yields the title
compound, melting point 130 C, dec.
GC155f
_
-113-
1 338670
Example 80
(+)-3-Butoxy-3-l~(phenylmethoxy)carbonyl]amino]-2-
oxo-l-azetidinesulfonic acid, potassium salt
A solution of 185 mg of 2-oxo-3-[N-chloro-N-
- 5 [(phenylmethoxy)carbonyl~amino]-l-azetidinesulfonic
acid, tetra~utylammonium salt (see Example 49A) in
1 ml of dimethylfor~ e is cooled to -78C and
0.73 N lithium n-butoxide (3.8 ml) in n-butanol
is added at -78C. After 15 minutes, 0.5 M monobasic
potassium phosphate buffer is ~ and the-product
extracted into dichloromethane (three 40 ml
portions), dried over sodium sulfate, filtered
and concentrated in vacuo to give 179 mg of
the corresponding tetrabutylammonium salt of
the title compound.
To a solution of 109 mg of the tetrabutyl-
ammonium salt in acetone is added perfluoro-
butylsulfonic acid, potassium salt (60 mg)
in acetone. The solvent is removed in vacuo
and ethyl acetate is added. The product
. crystallizes and is collected and dried-to
yield 66 mg of the title compound, melting
point 186.5-187.5C, dec.
Example 81
[3+(E)]-3-~ethoxy-3-[[(methoxyimino)[2-[[(phenyl-
methoxy)carbonyl]amino]-4-thiazolyl]acetyl]-
amino-2-oxo-1-azetidinesulfonic acid, potassium salt
A suspension of 3-amino-3-methoxy-2-oxo-
l-azetidinesulfonic acid, tetrabutylammonium salt
(Example 46, Method II, part A) (0.175 mmole)
and sodium borate (0.0175 mmole) in 2 ml of
GC155f
~114~ 1 3 3 8 ~ 70
dichloromethane at 0C is treated with 28 ~1
of pyridine and 0.175 mmole of (E)-~-(methoxy-
imino)-2-[l(phenylmethoxy)carbonyl]amino]-4-
thiazolylacetyl chloride. After 1 hour, the
mixture is diluted with dichloromethane and
quenched with water. The organic layer is washed
with water, saturated salt, dried, and evaporated
in vacuo. The residue is purified on Mallinckrodt
SilicAR CC-4 silica gel (20 g) to give 43 mg
of the corresro~ing tetrabutylammonium salt of
the title compound.
The tetrabutylammonium salt (43 mg) is
dissolved in 0.5 ml of acetone and 20 mg of
potassium perfluorobutane sulfonate in 0.5 ml
of acetone is added. After addition of 3 ml of
ether the solid is collected and dried in vacuo
to give 28 mg of the title compound melting
point 144-146C, dec.
Example 82
I3+(Z)]-3-Methoxy-3-~(methoxyimino)~2-~(phenyl-
methoxy)carbonyl~amino~-4-thiazolyl]acetyl]amino~-
2-oxo-1-azetidinesulfonic acid, potassium salt
Following the procedure of Example 81, but
substituting (Z)-~-(methoxyimino)-2-[I(phenyl-
methoxy)carbonyl]amino]-4-thiazolylacetyl chloride
for (E)-~-(methoxyimino)-2-[~(phenylmethoxy)-
carbonyl]amino]-4-thiazolylacetyl chloride,
yields the title compound, melting point 168-172C,
dec.
GC155f
-
-115- 1 3 3 8 6 7 o
Example 83
3-[l(R)--[[(4-Ethyl-2,3-dioxo-1-piperazinyl)-
carbonyl]amino]phenylacetyl~amino~-3-methoxy-2-
oxo-l-azetidinesulfonic acid, potassium salt
To a stirred solution of 0.69 mmole of
3-amino-3-methoxy-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (Example 46, ~ethod II,
part A) in 30 ml of dry acetonitrile at -20 C
under nitrogen is added 242 ~1 of dry pyridine
followed by a solution of 352 mg of (R)-~-[1(4-
ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino]-
phenylacetyl chloride in 4 ml of acetonitrile.
After 1 hour 84 yl of pyridine is added followed
by an additional 117 mg of the above named acid
chloride in 1 ml of acetonitrile. The reaction
is stirred for 20 minutes, diluted with 24 ml
of 0.5 M pH 5.5 monobasic potassium phosphate
buffer and concentrated in vacuo to remove
acetonitrile. The aqueous remainder is extracted
three times with methylene chloride and the combined
extracts are dried (Na2SO4) and evaporated
leaving 546 mg of residue. Passage of the residue
through a silica gel column using methylene
chloride and then2%, 4% and 6% methanol in
methylene chloride, provides two fractions
(285 mg and 173 mg) of the corresponding
tetrabutylammonium salt of the title compound.
Passage of the 173 mg fraction through
4.5 g of Dowex 50-X2(K+) resin using acetone-
water as eluent yields 119 mg of the title
compound. A 104 mg portion of this material is
GC155f
1 338670
applied to a column of HP 20-AG re-sin in water.
Sequential elution with water,5% acetone in
water and 10% acetone in water yields 60 mg
of product as a mixture (ca. 1:1) of diastereomers.
Lyophilization of the 60 mg fraction yields a
solid, metling point 171-172C, dec.
Example 84
N-(3-Butoxy-2-oxo-1-sulfo-3-azetidinyl)-2-phenyl-
acetamide, tetrabutylammonium salt
Following the procedure of Example 80, but
substituting N-chloro-N-(2-oxo-1-sulfo-3-azetidinyl)-
2-phenylacetamidej tetrabutylammonium salt (see
Example 47A for preparation of the corresponding
potassium salt) for 2-oxo-3-lN-chloro-N-~(phenyl-
methoxy)carbonyl]amino]-l-azetidinesulfonic
acid, tetrabutylammonium salt, yields the title
compound as an oil: nmr (CDC13) 3.62 (s,2H,C6H5CH2),
4.03 (ABq,2H,v=7cps, C-4 CH2), 6.98 (s,lH,NH)
and 7.30 ppm (S,SH,C6H5).
Example 85
~R)-3-Methoxy-2-oxo-3-[(phenylacetyl)amino]-1-
azetidinesulfonic acid, potassium salt
A lM solution of dimethylformamide-sulfur
trioxide complex is prepared by the slow addition
of trimethylsilyl chlorosulfonate to dimethyl-
formamide at 0C followed by evacuation at 0.1 mm
for 30 minutes at 0-25C. Under an argon atmosphere,
50 mg of (R)-N-(3-methoxy-2-oxo-1-azetidinyl)-
phenylacetamide is dissolved in 0.2 ml of anhydrous
dimethylformamide and cooled to 0C. Cold lM
_ GC155f
-117-
1 338670
dimethylformamide-sulfur trioxide solution
(0.428 ml) is added, the mixture is stirred
for 2 hours and poured into 15 ml of 0.5 N
monobasic potassium phosphate. The solution
is extracted twice with dichloromethane (discard)
and 73 mg of tetrabutylammonium bisulfate is
added. Extraction with dichloromethane (three
10 ml portions) gives a viscous oil after drying
and evaporation in vacuo. Chromatography on
Mallincrodt CC-4 silica gel (50:1i~usingi2~ me~hanol
in dichloromethane as eluant gives 34 mg of
(R)-3-methoxy-2-oxo-3-~(phenylacetyl)amino~-1-
azetidinesulfonic acid, tetrabutylammonium
salt. Ion exchange on Dowex 50W-X2 (K+, 10
equivalents) give the title potassium salt after
lyophilization of the aqueous eluate: melting
point 130 C, dec., [~D=+52 (C=0.5, water).
Example 86
(S)-3-[[[[(1-Ethyl-4-hydroxy-3-methyl-lH-pyrazolo-
[3,4-b]pyridin-5-yl)-carbonyl]amino]phenylacetyl]-
amino]-2-oxo-1-azetidinesulfonic acid, potassium
salt
Following the procedure of Example 73, but
substituting ~-[[(1-ethyl-4-hydroxy-3-methyl-lH-
pyrazolo[3,4-b]pyridin-5-yl)carbonyl]amino]benzene-
acetic acid for (Z)-2-amino-~-(hydroxyimino)-4-thia-
zoleacetic acid, yields the title compound, melting
point 233-236C, dec.
_ GC155f
-118- 1 338670
Example 87
(R)-3-Acetylamino-3-methoxy-2-oxo-1-azetidine-
sulfonic acid, potassium salt
~) 3-Acetylamino-l-~l-carboxy-2-methyl(propyl)]-
(3R)-3-methoxy-2-oxoazetidine
To a solution of (6R-cis)-7-acetylamino-7-
methoxy-3-methyl-8-oxo-5-thia-l-azabicyclo[4.2.o]oct
2-ene-2-carboxylic acid (650 mg) and sodium
bicarbonate (191 mg) in water is added a slurry
(11 ml) of commercial grade Raney nickel (0.6 g/ml),
which has been washed to neutrality with water.
The mixture is lowered into an oil bath preheated
to 170 C and proceeds to reflux in 2-3 minutes,
while maintaining the bath t~mperature at
150-170 C. After refluxing for 15 minutes,
the reaction is quenched by cooling in an ice
bath. Catalyst is removed by filtration through
Celite and the filtrate (pH 11) is adjusted to
pH 2 with 1 N hydrochloric acid. After extracting
the aqueous solution five times with ethyi acetate,
the combined extracts are dried (Na2SO4) and
solvent is removed in vacuo to give an oil
(487 mg). Chromatography on silica gel yields
the product (eluted in chloroform) as an oil
(381 mg).
B) 3-Acetylamino-l-[-l-(acetyloxy)-2-methyl(propyl)]-
(3R)-3-methoxy-2-oxoazetidine
The above azetidinone (464 mg) is dissolved
in dry acetonitrile (15 ml) and the solution is
purged with argon for 15 minutes. Copper acetate
(359 mg) is added, stirred for one minute to
dissolve the salt, and the lead tetraacetate
GC155f
-- --119-- .
1 338670
(797 mg) is added. ~hile argon continues to
bubble through the mixture, the temperature
of the reaction is raised by lowering the flask
into an oil bath, preheated and maintained at
55-65C for 15 minutes. The mixture is allowed
to cool to room temperature, filtered through
Celite and the filter pad is washed well with
acetonitrile. Solvent is removed in vacuo
from the combined filtrate and w~shings. The
residue is taken up in water, and extracted
four times with ethyl acetate. The combined
extracts are dried (Na2SO4) and solvent is
removed in vacuo to yield the desired product
as an oil (382 mg).
C) (R)-N-(3-Methoxy-2-oxo-1-azetidinyl)acetamIde
The above oil is dissolved in a methanol
(10 ml):water (1 ml) mixture, cooled in an
ice-methanol bath at -10 to -15C and potassium
carbonate (194 mg) is added followed by sodium
borohydride (53 mg). After stirring at -15 to
-8C for 110 minutes, solvent is removed in vacuo,
the residue is taken up in water and the solution
is adjusted to pH 6 with 1 N hydrochloric acid.
Exhaustive extraction with ethyl acetate, drying
(Na2SO4), and removal of solvent in vacuo yields
an oil (224 mg). Chromatography of the oil on
silica gel, eluting with 5% methanol:95% methylene
chloride giving an oil (169 mg). The title
compound crystallizes from ether-pentane to give
131 mg of material, melting point 106-112C
(sintering 103.5C).
GC155f
-120- 1 338670
D) (R)-3-Acetylamino-3-methoxy-2-oxo-1-azetidine-
sulfonic acid, potassium salt
Under an argon atmosphere, S0 mg of
(R)-3-acetylamino-3-methoxy-2-oxo-1-azetidine
is placed in a flask and cooled to 0C. A 1 M
solution of dimethylf orm~m; ~-sul fur trioxide
complex in dimethylformamide (.95 ml) is then
added and the solution is stirred for 15 minutes.
The contents of the flask are then poured into
40 ml of 0.5 N K2HP04 soultion and extracted
- twice with 10 ml of methylene chloride. Tetrabutyl-
ammonium sulfate (1.2 equivalents) is added to
the aqueous solution and the resulting mixture
is extracted with four 10 ml portions of methylene
chloride. The extracts are then dried over
Na2SO4 and concentrated to afford 39 mg of
product. The tetrabutylammonium salt is
converted to the title potassium salt by passing
it through a column of Dowex 50-X2 (K+).
Concentration of the aqueous fraction gives
19 mg of the potassium salt, with identical NMR
spectra to that prepared in EX46Ib and by isolation
from natural sources (Ex. 165).
GC155f
-121-
1 338670
Example 88
(+) -3- ~ (Azidophenylacetyl~ amino-] -3-methoxy-2-o~-1-
azetidinesulfonic acid, potassium salt
A) (+)-3-~(Azidophenylacetyl~amino]-3-methoxy-2-
oxo-l-azet~ esulfohic acid, tetrabutylammonium s-alt
3-Amino-3-methoxy-2-oxo-1-azetidinesulfonic
acid tetrabutylammonium salt (.2Q2 mgs; see Example
74A2 is dissolved in 20 ml of dry acetonitrile.
To the well stirred solution at -20C under dry
nitrogen are added dry pyridine (167 pl) and
a-azidophenylacetyl chloride (96 yl). After 20
minutes, 0.5M pH 5.5 monobasic potassium phDsphate
buffer (12 ml) is added and the acetonitrile
removed in vacuo. The aqueous residue is
extracted three times with methylene chloride.
The extract is dried over anhydrous sodium
sulfate and evaporated in vacuo to give 281 mg
of crude product as a gum. This is purified
by chromatography through a column of silica
gel (30 g) using methylene chloride and mixtures
of methylene chloride-methanol up to 6% methanol,
and yielding 231 mg o the title compound.
B) (+)-3-[(Azidophenylacetyl~amino]-3-methoxy-2-
oxo-l-azetidinesulfonic acid, potassium salt
(+)-3-~(Azidophenylacetyl~amino~-3-methoxy-
2-oxo-1-azetidinesulfonic acid, tetrabutylammonium
salt (231 mg) in 30~ acetone: water (15 ml) is
passed through a column of Dowex 50w-X2 (K form; 3 ml)
and eluted with water. The total eluate is
evaporated in vacuo to give a colorless glass
GC155f
_ ,
-122- 1 3 3 8 6 7 0
(168 mg~ as a 1:1 mixture of diastereomers.
Passage of this through a 6Q ml column of
~P2Q-AG using water and water:10% acetone gives
71 mg of a-l:l mixture of racemic diaste eu crs
and 70 mg of a 1:3 mixture of racemic diastereQmers.
The 1:1 mixture is lyophilized and dried in vacuo
at 4QC to gi~e t~e dried product as a hemihydrate,
melting point dec. 130C.
Analysis for C12H12N56-K 5 H2O Calc~d C~ 35-~;
H, 3.26; N, 17.45; S, 7.98
Found: C, 35.~4; H, 3.Q7; N, 17.24; S, 8.Q2
EXample 89
3-~(Azidophenylacetyl~amino]-3-methoxy-2-oxo-1-
azetidinesulfonic, potassi-um salt, isomer A
The 1:3 mixture of racemic diastereomers
obt~;ne~ in Example 88B is allowed to stand in
deuterated water at room temperature, and isomer A
crystallizes. After refrigeration, the mother
liquor is removed, and the crystals (28 mg) are
dried in vacuo at 40C, melting point 130C, dec
as a mono-deuterate.
Example 90
[3+(R*)]-3-[~1 I (4-Ethyl-2,3-dioxo-1-piperazinyl~-
carbonyl]amino]phenylacetyl]amino]-3-methoxy-2-
oxo-l-azetidinesulfonic acid, potassium salt
To a stirred solution of 3-amino-3-methoxy-
2-oxo-1-azetidinesulfonic acid, tetrabutylammonium
salt (0.69 mmol; see Example 74A) in 30 ml of dry
acetonitrile at -20C under nitrogen is added
242 ~1 of dry pyridine followed by a solution of
GC155f
-123- 1338670
352 mg of (R)~ (4-ethyl-2,3-dioxo-1-piperazinyl)-
carhonyl~amino~phenylacetyl chloride in 4 ml of
acetonitrile. After 1 hour, 84 ~1 of pyridine
is added followed by 117 mg more of the acid
chloride in 1 ml of acetonitrile. The reaction
is stirred for 20 minutes, diluted with 24 ml
of 0.5 M pH 5.5 mono~asic potassium phosphate
buffer, and concentrated in vacuo to remove
acetonitrile. The aqueous remainder is extracted
three times with methylene chloride and t~e
combined methylene chloride extract is dried
(Na2SO4) and evaporated to a residue (546 m~.
Passage of this material through a column of
~ilicAR CC-4, using methylene chloride and then
2%, 4%, and finally 6% methanol in methylene
chloride provides two fractions (285 mg and
173 mg) of purified product in the tetrabutyl-
ammonium salt form.
Passage of the 173 mg portion through 4.5 g
of Dowex 50-X2 (K ) resin using acetone-water
yields 119 mg of potassium salt.~ A 104 mg portion
of this material is applied to a column of HP20-AG
resin in water. Sequential elution with water,
5% acetone in water, and finally 10% acetone
in water provides 60 mg of product as a mixture
(ca. 1:1) of diastereomers and 21 mg of product as
a mixture (ca. 9:1) of diastereomers. Lyophili-
zation of the 60 mg fraction yields the title
compound, melting point 171-172C, dec.
Analysis for Cl9H22NsgSK H2O: Calc~d C, 41.23;
H, 4.37; N, 12.65; S, 5.78
Found: C, 41.39; H, 4.12; N, 12.58; S, 5.63
GC155f
--124- -1 338670
Lyophilization of the 21 mg fraction yields the
title compound, melting point 171-172 C, dec.
-Analysis for ClgH22NsOgSK-H2O: Calc~d C, 41.23
H, 4.37; N, 12.65
Found: C, 41.43; H, 4.11; N, 12.28
Treatment of the 285 mg fraction of tetra-
butylammonium salt with Dowex 50-X2(R ) provides
145 mg of potassium salt, which is combined with
the remaining 15 mg of the aforementioned 119 mg
portion of Dowex resin derived potassium salt.
Passage of this material through HP2Q-AG, as
already described, provides an additional 31 mg
of product as a mixture ~ca. 1:11 of d;astereomers
and an additional 42 mg of product as a mixture
(ca. 9:1) of diastereomers. The total amount
of (1:1) mixture of diastereomers is 91 mg, and
the total amount of (9:1) mixture of diastereomers
is 63 mg.
Example 91
~[3S(Z)]-3-l~(Methoxyimino)~2-~l(phenylmethoxy)-
carbonyl]amino]-4-thiazolyl]acetyl]amino~-2-oxo-
l-azet;~;~esulfonic acid, potassium salt
To a solution of (S)-3-amino-2-oxo-1-
azetidinesulfonic acid, tetrabutylammonium
salt (0.170 mmol; see Example 6A) and sodium
borate (0.170 mmol) in 2 ml of methylene chloride
at 0C is added pyridine (62 ~1) and (Z)-~-(methoxy-
imino)-2-[~(phenylmethoxy)carbonyl~amino]-4-
thiazoleacetyl chloride (0.51 mmol). The reactionmixture, after 40 minutes, is diluted with
methylene chloride and water, followed by 0.1 M
GC155f
-
-125- 1338670
tetrabutylammonium suifate buffered to pH 4
~5.1 ml). The organic layer is separated and
washed with water, adjusted to pH 2, water
adjusted to pH 7, water saturated with sodium
chloride, dried over sodium sulfate, filtered,
and concentrated in Ya-cuo. The residue is
purified on SilicAR CC-4 silica gel ~10 g) and
the product eluted with 10% methanol/methylene
chloride.
The tetrabutylammonium salt, after dissolution
in acetone/water, is passed through an ion-~YrhAn~e
resin (8 ml, AG 50W-X2, R form lQ0-20Q mesh~.
Removal of ~h~ water in v-acuo from fractions
1-2 give 40 mg of the title c~mpound,
melting point 172-174C, dec.
Analysis for C17H16N58S2K H2
H, 3.33; N, 12.99; S, 11.87
Found: C, 37.95; H, 3.30; N, 12.73; S, 11.53
Example 92
(+)-3-Butoxy-2-oxo-3-[(phenylacetyl)amino~-1-
azeti~;~esulfonic acïd, potassium salt
A) 3-tChloro(phenylacetyl)amino]-2-oxo-1-azetidine-
sulfonic acid tetrabutylammonium salt
A solution of (S)-2-oxo-3-[t(pheny~methoxy)-
carbonyl]amino]-l-azetidinesulfonic acid, tetra-
butylammonium salt (350 mg; see Example 4) in
methylene chloride (3 ml) is added to a suspension
of sodium borate (1.27 g) in a 5.25% solution of
sodium hypochlorite (4.72 ml) and water (20 ml~
GC155f
-126-
1 338670
at 0 C. After 1 hour 0.5 M monobasic potassium
phosphate (25 ml2 is added and the mixture is
extracted three times with methylene chloride
~50 ml portions). The organic extracts are dried
over sodium sulfate, filtered and concentrated
in vacuo to give 344 mg of the title compound.
B) (+)-3-Butoxy-2-oxo-3~ henylacetyl).-amino~
azetidinesulfonic aci.d', tetrabu'tylamm~h'ium salt
A solution of 3-~chloro(~henylmethoxy2~
carbonyl~amino]-l-azetidinesulfonic acid,
tetrabutylammonium salt (344 mg~ in dimethyl-
formamide (5 ml2 îs added to 0.73 N n-lithium
butoxide in n-butanol ~6 mll in dimethylfonmamide
(1 ml) at -78C under an inert atmosphere. After
10 minutes the mixture is diluted with 0.5 M
monobasic potassium phosphate solution (175 ml).
After extracting three times with methylene
chloride, the organic extract is dried over sodium
sulfate, filtered, and the solvent removed in vacuo.
The residue is purified on SilicAR CC-4 silica
gel (80 g) and the title compound (130 mg) is
eluted with 4-8% methanol in methylene chloride.
C) (+)-3-Butoxy-2-oxo-3-1(phenylacetyl)amino]-
l-azetidinesulfonic acid, potassium salt
(+)-3-Butoxy-2-oxo-3-[(phenylacetyl~amino]-
l-azetidinesulfonic acid, tetrabutylammonium salt
(43 mg) is dissolved in a water-acetone mixture
~9:1) and placed on a cation exchange column
(Dowex AGMP 50w-X2, 100-200 mesh, 5 g, K form).
GC155f
-127- 1 338 6 ~o
The product is eluted with water and the eulate
concentrated 'in vacuo to give 2Q mg of the title
compound, melting point 122-125C.
- Analysis calc'd for C15H12N2O6SK 1/2 H2O: C, 44.66;
H, 4.96; N, 6.q5; S, 7.94
Found: C, 44.77; H, 4.76; N, 6.76î S, 7.75
EXample g3
(~)-3-Ethoxy-2-oxo-3-~(phen~lacetyllamino3-1-
aze-~ n~sulfonic''acid, pot'assium salt
3-IChloro~phenylacet~l)amino~-2-oxo-l-
azet;~in~culfonic acid, tetrabutylammonium salt (200 mg;
see~example 92A~ in dimeth~lformamide ~4 ml) is added
to Q.5 N lithium ethoxide in ethanol (12.20 ml)
at -78C under an inert atmosphere. After 10
minutes the mixture is diluted with 0.5 M
monobasic potassium phosphate solution (15 ml).
After extracting three times with methylene
chloride, the organic layer is dried over sodium
sulfate, filtered and the solvent removed
in vacuo. The residue is purified on SilicAR
CC-4 silica gel (20 g) and the tetrabutylammonium
salt of the product (40 mg~ is eluted with 2%
methanol in methylene chloride.
The tetrabutylammonium salt is dissolved
in a water-acetone mixture (9:1) and placed
on a cation exchange column (Dowex AGMP 50W-X2,
100-200 mesh (5 g, K form)). The product is
eluted with water and the eluate concentrated
in vacuo to give 25 mg of the title compound,
melting point 94-96C.
Analysis calc'd for C13H15N2O6
H, 4.10; N, 7.65; S, 8.74
Found: C, 40.36; H, 3.66; N, 6.77; S, 8.44
GC155f
-128- 1 338670
Exa~ple 94
I 3+(Z)]-3-~C2-Amino-4-thiazol~(methoxyimino~_
acetyl~amino~-3-methoxy-2-oxo-1-azetidines~lfonic
acid, potassium salt
3-Amino-3-methoxy-2-oxo-1-azetidinesulfonic
acid, tetrabutylammonium salt ~see Example 46,
method II, part Al is dissolved in acetonitrile
(20 ml~ and pyridine a ml~ and adde~ to a
vigorously stirring suspension of (Zl-a-Cmeth
imino~-2-amino-4-t~iazolPacetyl chloride in
acetonitrile (2a ml~ cooled to Q-5C. After
stirring cold for 1 hour the mixture is diluted
with Q.5 M mono~asic potassium p~osphate solution
(100 ml~ CpH of the mixture is 4.8) and solvent
is removed in vacuo. The residue is taken up
in a min;mAl amount of water containing a small
amount of acetone. Chromatography on ion exchAnge
resin (AG 50W-X2, 100-200 mesh, K form 200 ml)
gives the crude product as the potassium salt upon
elution with water. Further purification on
HP-20 resin (200 ml) using water as the eluant
gives S9 mg of the product as a powder after
trituration with acetonitrile-ether and then
twice with ether. The product is an amorphous
powder that melts slowly and decomposes above
150C.
Analysis calc'd for CloH12N5O7SK: C, 28.77;
H, 2.90; N, 16.78; S, 15.36; K, 9.37
Found: C, 27.77; H, 2.82; N, 15.87; S, 13.63;
K, 10.11
GC155f
.
-129- 1 3 3 ~ 6 7 0
Exa~nple 9S
[3R(R*) and 3S CS*~-3~ '(p~inocArbonyllam-ino~-
phenylacetyl] amino~'-3-methoxy-2-oxo-1-azeti'dine-
sulfohic acid, potassi~n salt
A) (+~-3-~ (Aminopheh~lacety~l~amino~ -3-me-thQX5r-2-
oxo-1-azetidinesulf'onic acid, inne~' salt
(~)-3-1(AzidopElenylacetyl)amino]-3-methoxy-
2-oxo-1-aze~i~inesulfonic acid, potassium salt
(209 mg; see Example 88)~is dissolved in 6a ml
of dry me~hAnol. Anhydrous trifluoroacetic acid
(0.6 ml) and ln% palladilml on carbon ClQ5 mg~
are added and the mixture is hydrogenated for
hour. The catalyst is removed by fîltration
15 and the filtrate is evaporated ln vacuo to
yield 271 mg of crude product.
B) ~3R(R*) and 3S(S*)~ -3-I[I(Aminocarbonyl)amino]-
phenylacetyl]amino~ -3-methoxy-2-oxo-1-azetidine-
20 sulfonic acid, potassium salt
(+)-3-l(Aminophenylacetyl)amino]-3-methoxy-
2-oxo-1-azetidinesulfonic acid, potassium salt
(271 mg) is dissolved in 6.5 ml of water.
Potassium cyanate (87 mg) is added and the
25 mixture is stirred at room temperature for 3 hours.
The solution is concentrated in vacuo to approxi-
mately 2 ml and chromatographed on a 100 ml
column of HP20-AG using water as eluant. Isomer A
(29 mg) is isolated after lyophilization, melting
30 point 160C, dec.
Analysis for C13H15N4O7SK monohydrate: Calc'd: C, 36.44;
H, 3.99; N, 13.07; S, 7.48
Found: C, 36.35; H, 3.79; N, 12.81; -S, 7.32
GC155f
-130- 1 338670
Exa~ple ~6
~3R(S*) and 3s(R*)l-3~ cAminocarbonyl~amih
- phenylacetyl]amiho]-3-methoxy-2-oxo-l-azetidine
sulfonic acid, po*assium salt
Along with l3R(R*) and 3S(S*)~-3-tl~amino-
carbonyl~amino]phenylacetyl]amino-3-methoxy-2-
oxo-l-azetidinesulfonic acid, potassium salt
produced in Example ~5, there are produced
21 mg of l3R(S*1 and 3SCR*II-3~ (aminocarbonyl~-
amino~phenylacetyl]amino]-3-methoxy-2-oxo-1-
azeti~inesulfonic acid, potassium salt as a
lyophilate, melting point 16QC dec.
Analysis for C13H15N4O7SK sesquihydrate:
Calc'd: C, 35.6q; H, 4.14; N, 12.81; S, 7.33
Found: C, 35.98; H, 3.87; N, 12.5Q; S, 7.32
Example 97
13-(s*)]-3-Methoxy-3-~ 2-oxo-3-I(phenylmethylene)
amino~-l-imidazolidinyl]carbonyl]amino]-2-thienyl-
acetyl]amino~-2-oxo-1-azetidinesulfonic acid,
potassium salt
To a stirred solution of 3-amino-3-methoxy-
2-oxo-azetidinesulfonic acid, tetrabutylammonium
salt (306 mg of crude material assumed to contain
274 mg of organic material, prepared as described
in Example 74) in 20 ml of dry acetonitrile at
-20C under nitrogen is added 0.30 ml of dry
- pyridine (3.72 mmol) followed by 484 mg of
(S)-~[[2-oxo-3-phenylmethylene)amino]-1-imidazolidinyl]-
carbonyl]amino]-2-thienylacetyl chloride partially
dissolved and suspended in 10 ml of dry acetonitrile.
GC155f
-131- 1 3 3 8 6 7 o
The reaction is stirred and allowed to rise to
QC over the course of 1 hour.
The reaction was diluted with a large volume
of methylene chloride and then treated with
22 ml of 0.5 M pH 5.5 monobasic potassium phosphate
buffer. The aqueous layer is washed with methylene
chloride and the combined meth,ylene chloride
extract is washed ~ith water, dried Q~a2SO41,
and evaporated to a residue (5Q8 mg2. This residue
is chromatographed on 5Q g of SilicAR CC-4, using
methylene chloride and then 2% and 4% methanol
in methylene chloride to give 251 mg of the
tetrabutylammonium salt of the title ccYmpound.
To this salt (251 mg) in acetone is added
a solution of 107 mg of perfluorobutanesulfonic
acid potassium salt in several milliliters of
acetone. Ethyl acetate is added, and the precipitate
is washed three times with ethyl acetate by
centrifugation, and dried in vacuo at 40 C/lmm
for 2 hours to give 95 mg of desired potassium
salt as a mixture (ca. 1:2) of diastereomers --
having a melting point 20ac, dec.
Calc, d for -C21H21N6OgS2K: C, 42.85; H, 3.60;
N, 14.28; S, lQ.87
Found: C, 43.02; H, 3.~4; N, 13.94; S, 10.71
GC155f
-132- 1 338670
Ex'ample 98
(+--~L~)-4-Methyl-'2-oxo-3-I'['(phehylmethoxy]carbonyl~-
amino~-l-azeti~in~sulfonic'acid'r potassium 'salt
A~ N-Benzyloxy-t-~oc*-~llothreonine amide
A solution of 6.9 g of d,l-t-boc-allothreonine
and the free amine from 5.3 g of o-benzylhydroxyl-
amine HCl (~0.033 mole, et~yl acetate-sodium
bicarbonate liberation~ in 80 ml of tetrahydro-
furan is treated with 4.82 g of N-hydroxy-
benzotriazole and 6.5 g of dicyclohexylcar~odiimide
in 20 ml of tetra~ydrofuran. After stirring for
about 16 hours at room temperature the slurry is
fîltered, concentrated in vacuo and chromatographed
lS on a 400 ml of silica gel column. Elution with
5-10% ethyl acetate in chloroform gives 6.8 g
of the title compound in fractions (200 ml
each) 7-22.
B) (+-c ~N-Benzyloxy-3-t-butoxycarbonylamino-4-
methylazetidinone
A solution of 6.8 g of N-benzyloxy-t-
boc-allothreo~ine amide in 200 ml of tetrahydrofuran
is stirred for about 16 hours with 5.24 g of
triphenylphosphine and 3.2 ml of diethylazodicarboxylate.
The solvents are evaporated in vacuo and the
residue is chromatographed on a 500 ml silica gel
column. Elution with methylene chloride followed
by crystallization from ether gives a total of
2.65 g of azetidinone. Rechromatography of
the mother liquors and mixed fractions give an
* "boc" is used to describe butoxycarbonyl
GC155f
-133- 1 3 3 8 6 7 0
additional 0.6 g. Crystallization of a portion
twice from ether (-2Q C~ gives the analytical
sample of the title compound melting point 140-142C.
Cl (+-cis~3-t-~uLo~ycarbonylamino-l-hydroxy-4-
methylazeti~ino~e
A solution of 3.2 g of cis-N-benzyloxy
3-t-butoxycar~onylamino-4-methylazeti~;no~e in
200 ml of 95% ethanol is stirred in an atmosphere
of hydrogen wit~ 0.7 g of 10% palladium on
charcoal. After 4Q minutes the slurry is
filtered (uptake 249 ml) and th~ filtrate is
evaporated and triturated with ether to give, in
two crops, 2.05 g of solid, melting point 134-136C.
D) (+-cis-3-t-~utoxycarbonylamino-4-methylazetidinone
A solution of 2.05 g of -c -3-t-butyloxy-
carbonylamino-l-hydroxy-4-methylazetidinone in
60 ml of methanol is treated with a total of
90 ml of 4.5 M ammonium acetate (40, 20 and 30 ml
portions) and 45 ml of 1.5 M titanium trichloride
(20, 10 and 15 ml portions2 the second and third
additions are made after 15 and 120 minutes,
respectively. After 135 minutes the solution is
diluted with an equal volume of 8% sodium chloride
and extracted with three 300 ml portions of ethyl
acetate. The combined or~anic layer is washed
with a mixture of 100 ml each of 5% sodium
bicarbonate and saturated salt, dried, and
evaporated. Trituration with ether ~ives, in
two crops, 1.65 g of solid. A portion of the
first crop is recrystallized from ether to give
the analytical sample, melting point 176-178.5C.
GC155f
_
-134- 1 338670
E) (+ -cis~3-Benz~ carLo~ m;no-4~met~ylazetidinone
A solution of 1.55 g of- cis-3-t-butoxy-
carbonylamino-4-methylazeti~ino~e in 4 ml each of
methylene chloride and anisole is cooled to 0C
and 5Q ml of cold trifluoroacetic acid is added.
After 9Q minutes the solvents are e~aporated
in vacuo (~en7ene added and evaporated three timesl.
The residue is dissol~ed in 25 ml of acetone,
the initial pH (2.51 i8 raised to 7 wi.th 5%
sodium bicarbonate, and 2 ml of benzylchloroformate
is added. The solution is kept at QC and pH 7
for 4 hours and the acetone is r~o~ed in vacuo
to give a slurry that is filtered. The filtrate
is saturated with salt and extracted ~ith methylene
chloride. The solid is dissolved in methylene
chloride and dried. The organic layers are
combined, concentrated, and the residue chromato-
graphed on a 200 ml silica gel column. Elution
with 3:1 chloroform, ethyl acetate gives 850 mg
of the title compound in fractions (100 ml each)
4-11. Crystallization of a small sample from
ether gives the analytical sample, melting point
165-166C.
F) (~-cis)-4 ~e~h~1-2-oxo-3-[~(.phenylmethoxy)-
carbonyl]amino]-l-azetidinesulfonic acid, potassium salt
To a suspension of -cis-3-benzyloxycarbonyl-
amino-4-methylazetidinone (0.75 g) in 7 ml each of
dimethylformamide (dried with 4A sieves activated
at 320C for 15 hours under argon flow) and
methylene chloride (dried through basic A12O3)
GC155
-135- 1 3 3 8 6 7 0
is added 1.66 g of pyridine-sulfur trioxide complex.
After 3- hours stirring at room temperature under
nitrogen, an additional amount of pyridine-sulfur
trioxide complex (1.66 g) is added. The reaction
mixture is then stirred at room temperature under
nitrogen for about 16 hours. The dimethyl-
fonmamide is removed in vacuo to give 4.6 g of
residue which is ~isFolved in 300 ml of Q.5 ~
monobasic potassium phosphate solution C4QC for
10-15 minutes~. T~e solution is cooled, passed
through a column of HP-20 resin (3 cm x 60 cm)
with 400 ml of 0.5 M ~n9h~sic potassium phosphate 1 L of
distilled water and C14:1l water:acetone to give
280 mg of ~loduct in fractions 13 to 26 ~l~Q ml
eachl. Crystallization from MeOH: petroleum ether
gives 757.5 mg of an analytical sample, melting
point 214-215.5C, dec.
Analysis calc'd for C12H13N2SO6K: C, 40.90; H, 3.72
N, 7.95; S, 9.10; K, 11.10
Found: C, 40.43; H, 3.60; N, 7.89; S, 8.69;
K, 10.82
Example 99
(3 S-trans~-4-Methyl-2-oxo-3-~ I ~pheny~methoxy)car~onyl~-
amino]-l-azetidinesulfonic acid, potassium salt
Following the procedure of Example 98,
but substituting l-t-boc-threonine for d,l-t-boc-
allothreonine, yields the title compound, melting
point 133-135C.
Analysis calc'd for C12H13N2O6SK: C, 40.90; H, 3.72;
N, 7.95; S, 9.10; K, 11.10
Found: C, 40.72; H, 3.60; N, 7.99; S, 8.80;
K, 10.82
GC155f
-136- 1 3 3 8 6 7 0
Example laO
~S-trans~-4-Methyl-2-oxo-3- I (phenylacetyl2amino~-
l-azetidinesulfonic acid-, potassium salt
A2 (3S~-3-Amino-4-methyl-2-oxo-1-azetidinesulfonic
acid, tetrabutyl`ammoni~m salt
(4S-*rans~-4-Methyl-2-oxo-3-[ I (phPnylmethoxy2 -
carbonyl]amino]-l-azeti~ineculfonic acid, potassium
salt (352.4 mg; see Example 92~ is dissolved in
20 ml of distilled water and treated with 373.5 mg
(1 mmole) of tetra~ut~l ammonium hydrogen sulfate.
After lQ minutes s~tirring at room temperature,
the solution is extracted three times with lQ ml
portions of methylene chloride after saturation
with sodium chloride. The methylene chloride is
dried over sodium sulfate and evaporated in vacuo
to give 536 mg of the tetrabutylammonium salt
which is hydrogenated with 270 mg of 10% palladium
on charcoal in 25 ml of dimethylformamide. The
mixture is filtered through Celite and washed
twice with 2.5 ml portions of dimethylformamide
to yield the title compound in solution.
B) (Bs-tran~4-Met~yl-2-oxo-3-~(phenylacetyl2-
amino]-l-azetidinesulfonic acid, potassium salt
The crude (3S)-3-amino-4-methyl-2-oxo-1-
azetidinesulfonic acid, tetrabutylammonium salt
from part A (the combined filtrate and washings)
are treated at 0C with 206 mg of dicyclohexyl- -
carbodiimide, 153 mg of N-hydroxybenzotriazole,
and 138 mg of phenylacetic acid. The reaction
GC155f
-137- 1 3 3 8 6 7 0
mixture i5 stirred at QC for 1 hour and then
at room temperature for 2 hours. The resulting
precipitate is ~iltered, the filtrate is evaporated
in vacuo, the residue is dissolved in 10 ml of
acetone and filtered. The filtrate is treated
with 25 ml of acetone saturated with potassium
iodide and then 2Q0 ml of ether. The resulting
solid (752.7 mg~ i5 a mixture of the potassium
and tetra~utylammonium salts of the title compound.
The solid is dissol~ed in 5Q ml of Q.5 mono~asic
potassium phosphate and applied to an HP-2Q column.
Elution with water and t~en water-acetone gi~es
several fractions that are combined and evaporated
to give the purified tetrabutylammonium salt. An
aqueous solution of this material is passed
through Dowex 50W-X2 (R form) to give the title
potassium salt (121.4 mg). Trituration with
acetone-hexane yields lQ4.6 mg of the title
compound, melting point 211-213C.
Analysis calc'd from C12H13N2O5SK 1/2 H2O: C, 41.72;
-~ H, 4.09; N, 8.11; S, 9.28; K, 11.32
Found: C, 41.70; H, 4.01; N, 8.Q7; S, 9.01;
K, 11.02
Exa~ple lQl
(cis)-4-Methyl-2 o~o 3-~(phenylacetyl)amino~
azetidinesulfonic acid, potassium salt
A solution of 320 mg of (+ -cis)-4-methyl-
2-oxo-3-~(phenylmethoxy)carbonyl]amino]-l- azetidine-
sulfonic acid, potassium salt (see example 98) isErepared
in 20 ml of water containing 483 mg of tetrabutyl-
ammonium hydrogen sulfate and adjusted to pH 5.5.
GC155f
-138- - 1 3 ~ 8 6 7 0
E~ctraction with 8iX 25 ml portions of methylene
chloride giv~;517.3 mg of oil. A solution of
this material in 15 ml of dimethylfor~ e is
stirred with 4QQ mg of 10% palladium on charcoal
5 in an atmosphere of hydrogen for 90 minutes.
The catalyst is filtered and the filtrate stirred
with 150 mg of phenylacetic acid, 16~ mg of
N-hydroxybenzotriazole and 247 mg of dicyclo}~xyl-
carbodiimide for 7.5 hours. The solvent is removed
10 in vacuo and the residue is dissol~ed in 2Q ml
of acetone and filtered. The filtrate is treated
with 25 ml o~ Q.Q44 M potassium iodide in acetone.
Dilution with an equal volume of ether gives a
solid (330 mg~ which is applied to a 5Q ml HP-2Q
15 column in 20 ml of O.Q5 M monobasic potassium
phosphate. Elution with 2QQ ml of water followed
by 1:9 acetone-water giYes Rydon positive material
in fractions (~50 ml) 6-10. Evaporation of fractions
7-9 gives 81 mg of solid. Recrystallization from
20 acetonitrile-water gives 46 mg of the title compound,
which decomposes at >205C. A second crop (6 mg)
is obtained from the filtrate. A further 5 mg
is obtA; n~ from fractions 6 and 10 by e~raporation
and recrystallization.
Analysis calc'd for C12H13N2O5SK: C, 42.84; H, 3.89
N, 8.33; S, 9.53; K, 11.62
Found: C, 42.75; H, 3.82; N, 8.32; S, 9.26
K, 11.63
GC155f
-139-
1 338670
EXa~ple lQ2
~3S-~3~ (z),4~]~-3~ 2-Amino-4-thiazolyl) (methoxyimino]-
acetyl] amino~-4-met~y'1-2'-oxo-1-azetidine'sulfohic a-cid,
potassium salt
s
A~ (~S-trans~-4-Meth~1--2-oxo--3-~l(pheny-Lmeth~xyl-
carbonyl]amino~ azet'id'inesul'fonic ac'id','-'tetra-
butylammonium salt
(3S-trans~-4-Methyl-2-oxo-3-~1Cphenylmethox~)-
10 carbonyl]amino]-l-aze~ neculfonic acid, potassium
salt (352.4 mg; see Example 99~ is dissolYed in
20 ml of water and tetral~utylammonium hy~oyen
sulfate (373.5 mgl is added. The aqueous solution
is extracted three times with methylene c~loride
15 and the combined extracts are dried over sodium
sulfate. After removal of the solvent, 534.6 mg
of the title compound is obtained.
B) [3S-[3a(Z),4~]]-3-~(2-Amino-4-thiazolyl) (methoxy-
20 imino)acetyl]amino3-4-methyl-2-oxo-1-azetidinesulfonic
acid, potassium salt
A solution of 534.6 mg of ~3'S-trans)-4-methyl-
2-ox,o-3-~1(phenylmethoxy)carE~onyl]amino]-l-azeti~in~-
sulfonic acid, tetrabutylamn~onium salt in 20 ml of
25 dimethylformamide is hydrogenated with 220 mg of
10% palladium on charcoal at atmospheric pressure
for 2.75 hours; the uptake of hydrogen is 26.3 ml.
The mixture is filtered and washed twice with 2.5 ml
of dimethylformamide. The filtrate and washings
30 (.total ca. 25 ml) are stirred under nitrogen with
161 mg of (Z)-~-(methoxyimino)-2-amino-4-thiazole-
acetic acid, 136 mg of N-hydroxybenzotriazole
GC155f
~~ -140- 1 3 3 8 6 7 0
and 164.8 mg of dicyclohexylcarbodïimide. The
mixture is stirred under nitrogen for about
16 hours. The dimethylformamide is removed
in vacuo and the gummy residue is dissolved
in acetone and filtered to ~ove urea. To
the filtrate is added a solution contA; ni ng
272 mg (0.8 mmole) of perfluorobutanesulfonic
acid, potassium salt in a .8 ml of acetone. The
- slurry is diluted with an equal Yolume of et~er
and filtered to give 325.5 mg of crude product
which is purified through c~romatography on
75 ml of EP-20AG. Elution with 400 ml of water
and 4ao ml of (9:11 water: acetone mixture (5Q ml
fractions) gives 33S mg in fractions 3 to 10.
After trituration with acetone-hexane, 97.3 mg
of an analytical sample is obtA;ne~ from fractions
3-5. S;m; 1 ~r trituration of fractions 6-lQ gives
an additional 90.4 mg of product as a solid.
Analysis calc'd for CloH12N5O6S2K: C, 29.92; H, 3.01;
N, 17.45; S, 15.97; K, 9.74
Found: C, 30.32; H, 3.49; N, 15.82; S, 13.95;
R, 10.45
NMR(D20) 1.57 (3H, d,)=7), 3.97 (3H,S), 4.30(1H, d of q,
)=7.3), 4.70(1H, d. )=7), 6.95ppm(1H,S).
~Amp1e lQ3
I~S-~3a (Z), 4~]-3-l[~2-Amino-4-thiazolyl)~ carboxy-
l-methylethoxy)imino~acetyl~amino]-4-methyl-2-oxo-
l-azetidinesulfonic acid, dipotassium salt
A) N-Benzyloxy-t-boc-threonine amide
A solution of 8.76 g of t-boc-threonine and
the free amine from 6.4 g of D-benzylhydroxylamine
HCl (ethyl acetate-sodium bicarbonate liberation)
in 100 ml of tetrahydrofuran is treated with 6.12 g
GC155f
-141- 1 3 3 8 6 7 0
of N-hydroxybenzotriazole and 8.24 g of dicyclo-
hexylcarbodiimide in 2Q ml of tetrahydrofuran.
The mixture is stirred under nitrogen for
26 hours, filtered, and evaporated in vacuo.
The residue is chromatographed on a 300 g silica
gel column ~elution with chloroform and chloroform-
ethyl acetate (3:1~1 yielding 7.2 g of com~ound,
Crystallization from ether-h~Y~n~ gives 4.18 g of
the title compound.
B) (3S-trans) N ~ loxy-3 L ~tox~carbonylamino-
4-methylazetidinone
A solution of 12.67 g of N-benzyloxy-t-boc-
threo~;ne amide, 11.5 g of triphenylphosphine,
and 6.23 ml ofd~thylazodicarboxylate in 380 ml
of tetrahydrofuran is stirred under nitrogen
for about 16 hours. The solution is evaporated
- and chromatographed on a 900 gram silica gel
column. Elution with chloroform-ethyl acetate
(3:1) gives 13.69 g of compound that crystallizes
from ether-hexane to yield 9.18 g of the title
compound.
C) (3S-trans~)-3-t-B~toxycarbonylamino~ ydroxy-
4-methylazetidinone
A solution of 9.18 g of (3S-trans)-N-benzyloxy-
3-t-butoxycarbonylamino-4-methylazetidinone in
300 ml of 95% ethanol is stirred in an atmosphere
of hydrogen with 1.85 g of 10% palladium on charcoal.
After 141 minutes the slurry is filtered and
evaporated in vacuo. The residue is recrystallized
from ether-hexane to yield 5.12 g of the title compound.
GC155f
-142- 1 338670
D) (3S-trans~-3-t-ButoxycarbonylAmino-4-~ethyl-
azetidinone
A solution of 4.98 of (3S-trans~-3-t-butoxy-
carbonylamino-l-hydroxy-4-methylazetidinone in 2QQ ml
of methanol is treated with 132 ml of 4.5 M ammonium
acetate and then 66 ml of l.S M titanium trichIoride
and stirred for 4.5 hours. m e aqueous solution
is diluted with an equal volume of 8% sodium
chloride and extracted with ethyl acetate to give
3.48 g of crude product. Recrystallization from
ether-hexane yields 3.3 g of the title compound.
E~ (3s-trans)-3-Benzyloxycarbohylamino-4-meth
azet;~ino~e
A solution of 3.3 g of ~3S-trans)-3-t-butoxy-
carbonylAmino-4-methylazetidinone in 10 ml each
of dichloromethane and anisole is cooled to 0C
and 112 ml of trifluoroacetic acid is added. The
solution is stirred for 90 minutes and evaporated
in vacuo (benzene added and evaporated three times~.
The residue is dissolved in 70 ml of acetone and
the solution is adjusted to pH 7 with 5~ sodium
bicarbonate solution. A total of 5.33 g of benzyl
chloroformate is added over 1 hour at pH 6.5-7.5.
The mixture is stirred for 30 minutes at pH 7,
diluted with lOO ml of saturated salt, and
extracted with ethyl acetate (three 400 ml portions).
The residue obtained by evaporation is chromatographed
on a 1 liter silica gel column. Elution with
chloroform-ethyl acetate (4:1) gives 2.19 g of
compound. Crystallization from ether-hexane yields
1.125 g of the title compound.
~143~ 6 G&l55f
F) (3S-trans)-4 ;~eL~y~1-2-oxo-3-~l(p~en~methoxy~-
carbohyl]amino]-l-azetidinesulfonic acid', tetra-
butylammonium salt
A solution of 600 mg of ~3S-trans~-3-
benzyloxycar~onylamino-4-methylazetidinone in
2 ml of dimethylformamide is cooled to ~C and
4 ml of 0.8 M sulfur trioxide in dimethylformamide
is added. The solution is stirred at room temperature
under nitrogen for 1 hour and poured.into 8Q ml
of cold 0.5 N monobasic potassium phosphate
(adjusted to pH 5.5~. The solution is extracted
with three 50 ml portions of methylene chloride
(discarded) and 868 mg of tetrabutylammonium
bisulfate is added. The resulting solution is
extracted with four 75 ml portions of methylene
chloride. The combined organic layer is washed
with 8% aqueous sodium chloride, dried, and
evaporated in vacuo yielding 1.54 g of the title
compound.
G~ ~3S-~3a(Z),4~]}3-lI(2-Amino-4-thiazolyl)-
[(l-diphenylmethoxycarbonyl-l-methylethoxylimino~-
acety~-amino~-4-methyl-2-oxo-1-azetidinesulfonic
acid, potassium salt
A solution of 1.54 g of (3S-trans)-4-methyl-
2-oxo-3-~I(phenylmethox,y)carbonyl]amino~-l-azetidine-
sulfonic acid, tetrabutylammonium salt in 45 ml
of dimethylformamide is stirred in an atmosphere
of hydrogen with 800 mg of 10% palladium on
charcoal for 2 hours. The catalyst is filtered
and the filtrate stirred for about 16 hours with
GC155f
~144- 1 3 3 8 6 7 0
1.24 g of (Z)-2-amino-~-~(1-diphenylmethoxycarbonyl-1-
methylethoxy)i~ino]-4-thiazoleacetic acid, 0.4 g
of N-hydroxybenzotriazole, and 580 mg of dicyclo-
hexylcarbodiimide. The slurry i8 evaporated
in vacuo and the residue is triturated with
20 ml of acetone and filtered. The filtrate
~plus 2 ml of w~shings~ is treated with 868 mg of
potassium perfluoro~utanesul~onate in 3 ml of
acetone. Dilution wi~h 75 ml of ether gives a
solid thatis isolated ~y decantation of the mother
liquor, trituration with ether, and filtration
to give 0.91 g of the title oompound. The mother
liquor is diluted with a further 100 ml of ether
to give a second crop, Q.45 g, of the title compound.
H~ [3S-[31(z),4~]-3-I~(2-Amino-4-thiazolyl)~(1-
carboxy-l-methylethoxy)imino]-acetyl]amino~-4-
methyl-2-oxo-1-azetidinesulfonic acid, dipotassium
salt
A slurry of 140 mg of ~3S-r3~(z),4~]-3-
~1(2-amino-4-thiazolyl)[(l-diphenylmethoxycarbonyl-
l-methylethoxy)imino]acetyl]amino]-4-methyl-2-
oxo-l-aze~i~in~sulfonic acid, potassium salt (first
crop) in 0.5 ml of anisole is stirred at -12C
under nitrogen and 2.5 ml of cold (-10C) trifluoro-
acetic acid is added. After 10 minutes, 10 ml of
ether and 5 ml of hexane are added and the resulting
slurry is stirred for 5 minutes at -12C, and allowed
to warm to room temperature. The solid is isolated
by centrifugation and washed twice with ether. A
solution of this solid in 5 ml of cold water is
GC155f
-145- 1 3 3 8 6 70
immediately adjusted to pH S.5 with 0.4 N potassium
hydroxide and then applied to an 80 ml HP-20AG
column. Elution with water gives 72 mg of the
title compound in fractions (10 ml~ 7-11 after
evaporation Cacetonitrile added and evaporated
three times~ and trituration with ether,m.p.,-250(dec.).
Analysis calc'd for cl3~lsNso8s2K2 C~ 30-51; H~ 2-gS;
N, 13.6~; S, 12.53; R, 15.28
Found: C, 29.63; H, 3.2Q; N, 12.96; S, 11.~4;
X, 12.78
NMR(D20) 1.46(S, 6H), 1.58(1H, d, J=7) 4.28(1H, d of q,
J=7~ 2.5), 4.67 (lH, d, J=2), 6.95ppm(S, lH).
The remaining 1.22 g of I3S-I3a(Z~,4~ 3-
~l(2-amino-4 -~h i ~ zolyl 2 I Cl-diphenylmethoxycarbonyl-
l-methylethoxy~imino]acetyl]amino-4-methyl-2-
oxo-l-azeti~inesulfonic acid, potassium salt ~crops
1 and 2) are treated as above (4.2 ml anisole,
16 ml of trifluoroacetic acid, 13 minutes at
23 -15 C). Chromatography on a 300 ml HP-20AG
column gives 694 mg of the title compound in
fractions (60 ml) 6-9 after treatment as above.
Examples 104-133
Following the procedure of Example 11, but
substituting the acid listed in column I for
(Z)-2-amino--[ I lhydroxy(phenylmethoxy)phosphinyl~-
methoxy]imino]-4-thiazoleacetic acid, yields the
compound listed in column II.
GC155f
-- -146- '1 338670,
-- C
~; U , - ~ o
8 ~
.~ ~ , C ~ C . `, ~
~ S ~ ~ q-l 1~ ~ - rA ~ rn
~ ~ ~ S ~ -I ~
~ ~ C I ~ . C --
5 ~ c
N ~ _ ~ C & ~r O ~ t U¦ ~ ~ t
O~ O ~ ~ --I ~ .
. ~ C~ I . C ~ ,~ _ I ~ ' U C~ I C U
I . ., o rr) ~
I _ 0 _ U a~ I U ~ ~ u 0 ~ ~ o
-i U O ~ N U G) ~ N ~ ~'7 ~ ' O ~ ~1 0
-t~a ~ a ~ ~ ~ s I ~
.~ ~a
~ I -.~ I I .
_ U _ _
'C5 , t r~ ,-1 ,-1 r
u ~ c
~a .-- .-- . . , ~ ~
r-l . ; r~ O ~t
I ~ t ,t nl
_ .-- ~ - ~ 'l
Al ~ t ~ .,. ._ ~r
`' t ~ ~ ~ r
r- U I ~ r_
I ~ X
rO _ ~ r ~ ~ r
O ~r ' S
C~ r-l _ N I N N ~ ~
o , o o ~ ._
~, E~ ~r ~ ~ r~ N ~-
r t ~
. ~:
I . ~5 1 ._ I
t -- ;,~ ~
r~ _ , r~
.
O O O O O
- GC155f
-147- 1 3 3 8 6 7 0
- ~ Iu l I
D ~ .1 ~aC ~_ U ~ ~
_1 ~ e _1 ,I c ~ 0
U ~ O
O ~ u U c
' " 4 ~ 50~D ~ e
0 ~ ~ ~ o .
~ ~ 0 ~ I ~ ~ 04 ,.
-- C4 1-- U~ O rl ~ 4 U
o.q ~a c _
--I ~ U ~J C4 -- C
,~ ,C, ~ I C I ~ _I ~ U U
Q ' -- ~ O--~ -- X 'a
H ' :--~ L4 ~ ' ~ --I N O _I , O
H -- ~ C ~ . S --
~ U
C ~ ~ r ~ o
O ~ ~ m ~
U ~ 0 ~ 8, O
I N ~~ I E El I I~ ~
N ~, ~ N ~ ~ ~ O C ~ N '3
U ~ 0 1 ~ O 8 ~ - C
~ u I ~ a u I ~ o
I q ._ N ._ C 4 U '~ ~r~ I C4
N X U N ~ P O ~ ~. O U U N N ` -
--O ~ O --~1--e~a--S C ~0 ~ I
cn s _ Ul ~ I C o u~ o
~ ~ ~ a ~ ~ 8 U~ ~ 8 0 ~ ~ *
'4 n~ ~ 8 ~ ~ ~ O N
_~
~1
_ U
.~ r n~
-- ~ ~
_ I _ _I n~
-- C _IC
c a~
O
C n
--
~ O
--~ >1 C C
r C --I ~
r~ ~,8d 1 ~
'~ ~-- ~U
.~ ~ ni . ,_
U 8 ~ ' " ~ ~ . C
_l u ~ I _l e
-- n~
~ --I --I ~.
8 -
r~ ~ ~'I ., r`
C I ~_
~ O ~
I n I ~ I C I ~ I
N .~ N U ~ 6 ~ N
-- I -- nl -- ni ~-
.
cr~ o_I
O _l_l _l _~
GC155f
-148- 1 3 3 8 6 7 0
,~ ~
."
" U ~ . .,
, ~ ~o o,~ ~ ~o
JW I _ U 'O W '~
~ C OOu,0 _ rl ~. W I ~,, ~
~1 N ~ . W C ~
-- -- r l ~ ~ O A ~ ~ ~ O ~
~ -- ~ r~ CC: ~_ _ C WO
-- Q q Q ,_ ~ o
_ l,~ 4 _ o l
~ n ,~ ~ 1 ~ ~ C u~
~ ~ 'O 'O C O r~ I ~ 'O 'O ~ ~ ~ o ~ ~n
O ~ C ~ ~ o ~
,~ 0 v ~ q ~ _w o ~ . ,-- w m J~
. Q ~ O ~ w N IL I O ~ ^~ ~ O 0 C
U ~ O O ~ ~ O ~ I ~_ C ~
w ,_ ~ O I ~ l o
N 8 c JJ w -- ~ ~, c
-- ,~ c .4 ~ ~ ~ _ c~ c t~ w ~o
o ~ e ' 0~
N ~ ' - ~; 2 i W ~ , "~, Cl: ' '--
_, .. .,~ I I ._ . _ _ ~
n ~ ~_ c _ u~ cn - ~1
' ~ ~ Ou~ ~ C ~ , ~ ~ I n5
0 --r~ m
C
,~
~0 r~5 .,
~rl ~
~ --a ,~, I
,, .. ~ o
_, , ,' ~s ~
F ~, C , '~ q E;
r-l ~ 0 N
O S ~
~
~ nl I ~ , rr~
n5 ,0 ,_
8 '~ X ~ U C r
J O r l
n~ ~ n ~ _
N ~ 8 0 8
n I ._
~ I ~ n5 ~ -- _-
.
~ ~ 0
Column I Column II
119. a-1[[3-[(2-furanylmethylene)aminol-2-oxo-1- (3s)-3-tl[[t3-1(2-furanylmethylene)amino]-2-
imidazolidinyl]carbonyl]amino]-4-hydLo~y- oxo-l-imidazolldinyl]carbonyl]amino](4-
benzeneacetic acid l,~d.vA~hel.yl)acetyl]amino]-2-oxo-l-
azet~Ain~ulfonicOacid, potassium saltt
meltlng polnt 244 C, dec.
120. (R)-a-[[[3-[[3-(2-furanyl)-2-propenylidene]- ¦3S(R*)]-3-[[[[t3-[[3-(2-furanyl)-2-
aminol-2-oxo-1-i ;AA7olidinyl]carbonyl]- ~ ope,.ylidene]amino]-2-oxo-1-i ~AA7olidinyl]-
amino]benzeneacetic acid carbonyl]amino]phenylacetyl]amino]-2-oxo-
l-azetiA;no~ulfonoc acid, potassium 8altt
melting point 195 C, dec.
121. (Z)-2-amino-a-[[[(ethylamino)carbonyl]oxy]- [3S(Z)]-3-[[(2-amino-4-thiazolyl)[[[(ethyl-
imino]-4-thiazoleacetic acid amino)carbonyl]oxy]imino]acetyl]amino]-2-
oxo-l-azet~di n~ l fonic acid, potassium
saltt melting point 230 C, dec.
122. (R)-a-[[[2,3-dloxo-4-(phenylmethyl)-1- 13S(R*)]_3{[1[ t 2~3-dioxo-4-(phenylmethyl)-
piperazinyl]carbonyl]amino]benzeneacetic acid l-piperazinyl]carbonyl]amino]phenylacetyl]-
amino]-2-oxo-1-azetiAin~sulfonic acid
potassium saltS melting point 158-159C,
dec.
123. (R)-a-[[[4-(1-methylethyl)-2,3-dioxo-1- t3S(R*)]-3-[[[[[4-(1-methylethyl)-2,3-
piperazinyl]carbonyl]amino]benzeneacetic acid dioxo-l-piperazinyl]carbonYl]aminO]phenyl- CO ~
acetyl]amino]-2-oxo-1-azetidinesulfonic C~ ~n
acid, pOtassium salt~ melting point ~~
185-187 C
GC155f
, 1 338670
- U
I ~ N l C
~ C O ~r C C r~
_~ O--I ; r~
H ~ AUJ V N _ ~ 0
H ~ ~ r_A ~A
~ U U~
C O ~ ~O r-~ I ~ N m
0 . ~ c ~ I ~ ~ 8 0
8 . ¦ 1~ , u
A , e ~ .~ ,AJ _ ~AJO
A r-- r'~ ~ r-- O r_ r~
C\ U~ S ~ o ~ U~ ~ a I u~ C C
~) U ~ X~AJ _ E u ~ ~ S J ~
~J r_l ~ O ~ ,AJ ~
r_ r_~ r
~1) `1 ~-
aJ ~ ~ O
a
~ .r~
r_ ,AJ
r-i l- ,~ ~_1 _
~ ~ ~a ,~ ` c
u
e;) r_
r~
.. ._
_ _I s o
c ~--
_ _~
~ N ~ ~
82~ 0 0 25 ~ O
I C C I . I C
6 6 î~ 6
~ ui ~D t--
Column I Column II
128. (R)-a-1[[2-oxo-3-[[(phenylmethoxy)carbonyll- [3S(R*)]-2-oxo-3-t[[[[2-oxo-3-[[(phenyl-
aminol-l-imidazolidinyl]carbonyllaminol- methoxy)carbonyllamino]-l-~ l~A ~olidinyl]-
benzeneacetic acid carbonyl]amino]phenylacetyl]amino]-l-azet~ne~ulfonic acid, potassium salt;
meltlng polnt 175 & , dec.
129. (Z)-2-amino-a-[(2-amino-1,1-dimethyl-2- [3S(Z)]-3-[[[(2-amino-1,1-dimethyl-2-oxo-
oxoethoxy)iminol-4-thiazoleacetic acid ethoxy)-imino]~2-amino-4-thiazolyl)acetyl]-
amino]-2-oxo-1-azet1d~n~sulfonic ocid,
potassium salt~ melting point 210 C, dec.
130. (R)-a-[[[4-(1-methylethyl)-2,3-dioxo-1- [3S~R*)]-3-[1[[[4-(1-methylethyl)-2,3-
piperazinyllcarbonyllamino]-4-hydroxybenzene- dioxo-1-piperazinyl]carbonyl]amino](4-
acetic acid l,~dLoAy~henyl)acetyl]amino]-2-oxo-1- I Co
azet;~ne~ulfonic aciO, potassium salt; C~
melting point 201-203 C, dec. -J
o
131. (R)-a-[[[3-(1-methylethyl)-2-oxo-1- [3S(R*)]-3-[[[[[3-(1-methylethyl)-2-oxo-1-
imidazolidinyl]carbonyllamino]benzeneacetic acid t ~A 7Olidinyl]C~L~ 11amino]phenyl-
acetyl]amino]-2-oxo-1-azet~nesulfonic
acid, poOtassium salt; melting point
195-200 C, dec.
132. (Z)-2-amino-~-[[2-(diphenylmethoxy)-1-methyl- [3S(Z)]-3-[[(2-amino-4-thiazolyl)[[2- g
- 2-oxoethoxy]imino]-4-thiazoleacetic acid (diphenylmethoxy)-1-methyl-2-oxoethoxy]-
imino]acetyl]amino]-2-oxo-1-azetidine-
sulfonic acidO potassium salt; melting
point 145-150 C
1 338670
$C155f
-
-`152-
I CJ
_ _
,~ .
, - ~
,
H ~1 '
~D ~1 6
_I ~ ~1
O I ~11
~'0 ~ Ul .
6 C'
1 6
O
'
I . ~ I
O
~ .
r~
r
O
~) I
r~
rl
Ul
_1
1 338670 GC155f
~153~
Example 134-135
Following the procedure of Example 70, but
substituting the compound listed in column I for
[3S(Z)]-3-[[(2-amino-4-thiazolyl)[[2-(diphenyl-
methoxy)-2-oxoethoxy]imino]acetyl]amino]-2-oxo-
l-azetidinesulfonic acid, potassium salt, yields
the compound listed in column II.
1 3 3 8 6 7 0 ~C155f
~154~
~, .
` .
. ~ _I
- m
-- N
_ ~ . O ~
~ C
_ .~ ~
~ ~D I _ I
H n. --I ~ --I --I
H- _ U I '`
- 3 t ~ o ~ ~1
~ ~ t, ~
o. ~ , ,.
o C C~ V
,S4~oO
~ 8- e ' c ~ ~
I ~ c
~ r-~
1 C O
N ~ ~1 1 0
Ul ~ O ~1 U~
~ E X ~ -- nl
_ _ o m ~ C) -~
I ~ a ' _
_I , _
~ I ~
~ . , .~ _I
- ~ ' a) ~ _ -' a
t~
C r ~ ~_
H
n . .-1 n5 O
r .r . I ~n x _ I
-- '' X q --
o ~ w C
c qr 1- ~.a_l--
q
E w
_ I .~ w
~ ', ' - o
_ .~
. ~ U
~a ~ w r
~ a) I _ a) -
I _ C) ~.: _ .~ s ~
_ '. w -- _ ~. Q~ I
._ ~ _
N ~~ à
~- E ,~
. .,.~ ~,, ~ W
n
r~
.~ .
GC155f
-155- 1 3 3 8 6 7 0
Example 136
[3a(Z),4a]-3-[t(~-~mino-4-thiazolyl)(methoxy-
imino-)acetyl]amino]-4--methyl--2-oxo-1-azetidine-
sulfonic acid, potassium salt
A solution of 51.8 mg of (cis)-4-methyl-
2-oxo-3-[[(phenylmethoxy)carbonyl]aminol-l-
azetidinesulfonic acid, potassium salt and 51 mg
of tetra-n-butyl-ammonium bisulfate in 5 ml
of water is extracted with methylene chloride
(four 10 ml portions) to give 81 mg of oil.
This is stirred in an atmosphere of hyd~o~en
for 2 hours with 40 mg of 10% palladium on
charcoal in 4 ml of dimethylformamide. The
catalyst is filtered and washed with 1 ml of
dimethylformamide. The filtrate and washings
are combined and stirred for about 16 hours
with 31 mg of (Z)-2-amino-~-(methoxyimino)-4-
thiazoleacetic acid, 27 mg of N-hydroxybenzo-
triazole, and 31.5 mg of dicyclohexyl-
carbodiimide. The solution is evaporated in
vacuo and the residue triturated with 3 ml of-
acetone. The resulting slurry is centrifuged
and the liquid treated with 51 mg of potassium
perfluorobutanesulfonate. Dilution with 5 ml
of ether and filtration gives a solid. Chroma-
tography on HP-20 AG (40 ml) gives Rydon
positive material in fractions (20 ml) 3-5
(elution with water). Evaporation and ether
trituration gives 23 mg of product, as a hygroscopic solid.
Analysis calc'd for CloHl2N5o6s2K
C,29.91; H,3.01; N,17.44
Found: C,29.30; H,3.31; H,16.66
NMR(D20) 1.40(3H, d, )=7), 3.97 (3H,S), 4.46 (lH,
apparent pentet, )=7), 5.37(1H, d, )=7), 6.97 ppm(lH,S).
GC155f
-156- 1 3 3 8 6 7 0
Exampl-e 137
(3S-cis)-3-Amino-4-methyl-2-oxo-1-azetidinesulfonic
acid
A) t-Boc-1-allothreonine
A suspension of 6.72 g of l-allothr~o~
in 70 ml of 50% aqueous ~ioY~e is treated with
9.45 ml of triet~ylamine and 18.1 g of t-~utyl-
pyrocarbonate. The resul~ing mixture isstirred at room temperature for 4 hours, and
then diluted with 70 ml of water and 14Q ml
of ethyl acetate. After thoroug~ ~h~ ki n~,
the layers are separated and the organic layer
is washed with 30 ml of 2:1 water:brine. Com~ined
aqueous layers are then back-extracted with
70 ml of ethyl acetate. The aqueous layer is
cooled in an ice bath and 10% potassium bisulfi~e
solution is added to pH 2 3. The acidified
2~ solution is èxtracted with ethyl acetate
~four 1~0 ml portions). Combined organic layers
are dried over anhydrous sodium sulfate and
stripped of solvent to give 9.13 g of the title
compound.
B) N-Methoxy-t-boc-l-allothreonine amide
t-Boc-l-allothreonine (9.13 g) is dissolved
in 85 ml of water and 41 ml of lN potassium
hydroxide solution. Methoxyamine hydrochloride
(5.22 g) and 8.67 g of 1-ethyl-3,3-(dimethyl-
aminopropyl)carbodiimide HCl are added. The
GC155f
-157- 1 3 3 8 6 7 0
mixture i5 stirred at room temperature for 4 hours
and then saturated with sodium potassium tartarate.
The resulting mixture is extracted with ethyl
acetate (four 150 ml portions) and the organic
layer is dried over anhydrous sodium sulfate and
stripped of solvent to give 7.38 g of the title
compound as a solid.
C) -C-Methanesulfonyl-N-methoxy-t-boc-l-allothreonine
amide
N-Methoxy-t-boc-l-allothreonine amide ~7.32 g~
is dissolved in 4Q ml of pyridine and cooled to
-20 C under nitrogen. MethAneculfonyl chloride
(3 ml~ is added dropwise by syringe over a
5-minute period. The resulting mixture is slowly
warmed to 0C and stirred at that temperature
for 3 hours. Ethyl acetate (500 ml) is added
and the solution washed with 250 ml of ice-cold
3N HCl solution, then 100 ml of 5% NaHCO3 solution.
The ethyl acetate layer was dried over anhydrous
sodium sulfate and stripped of solvent to give -
8.64 g of the title compound as a white solld.
D) (3S-cis)-3-t-R~ltoYycarbony1~;no-1-meth~xy-
4-methylazetidinone
O-Methanesulfonyl-N-methoxy-t-boc-l-allo-
threonine amide (8.64 g) is dissolved in 530 ml
of acetone and 11 g of solid potassium carbonate
is added. The mixture is slowly heated to 65C
under nitrogen and stirred at this temperature
for one hour. The reaction mixture is then
GCl55f
-158- l 3 3 8 6 7 0
filtered through Celite and the filter cake is
washed with ethyl acetate. The filtrate i5
concentrated and the residue is taken up in
25Q ml of ethyl acetate. The ethyl acetate
solution is washed with lO0 ml of lN hydrochloric
acid solution and lO0 ml of 5% sodium
bicarbonate solution. The ethyl acetate layer
is dried over anhydrous sodium sulfate and
stripped of solvent to give 6.63 g of crude
product.
E~ (3S-cis)-3-t-Butox~carbonylamino-4~m~thyl-
azetidi none
Sodium (1.35 g2 is dissolved in about 3QQ ml
of liquid ~luuonia at -50C and 5.87 g of (~S-cis)-
3-t-Butoxycarbo~ylamino-l-metihoxy-4-methylazetidinone
in 35 ml tetrahydrofuran is added dropwise via
syringe. An additional 10 ml of tetrahydrofuran
is used for rinsing. Near the end of the addition
about 100 mg of additional sodium is added. The
mixture is stirred for five more minutes, then
qllenchP~ by adding 3.35 g of solid ammonium
chloride in one portion. Ammonia is ~lown off
with a nitrogen stream and 250 ml of ethyl acetate
is added to the residue. After filtration and
washing the solid with ethyl acetate, the combined
filtrate is stripped of solvent to give 4.82 g
of the title compound.
- -
GC155f
-
-159- 1 3 3 8 6 7 0
F~ (3~-cis~-3-t-Butoxycarbonylamino-4-methyl-
2-oxo-1-azetidinesulfonic acid,- tetrabutyl
ammonium salt
~3S,4R]-3-t-8utoxycarbonylamino-4-methyl-
azetidinone (4.98 g~ is dissolved in 30 ml of
dimethylformamide. Pyridine-sulfur trioxide
complex 11.9 g is added and the mixture is
stirred at room temperature under nitrogen.
After 14 hours ~tirring, an additional 1.8 g
of pyridine-sulfur trioxide complex is added
and stirring is continued for 8Q hours. The
reaction mixture is poured into 70Q ml of Q.5 M
monobasic potassium phosphate solution and
washed with methylene chloride (three 30Q ml
portions). tetra-n-Butylammonium bisulfate
(8.45 g) is added to the aqueous solution and
the mixture is extracted with methylene chloride
(four 300 ml portions). The combined methylene
chloride layers are dried over anhydrous sodium
sulfate and stripped of solvent to give 10.76 g
of the title compound as a gum.
G) (3S-cis)-3-Amino-4-methyl-2-oxo-1-azetidine-
sulfonic acid
(3~-cis)-3-t-ButoxycarbonylAmino-4-methyl-
2-oxo-1-azetidinesulfonic acid, tetrabutyl
ammonium salt (10.76 g) is dissolved in 50 ml
of 95-97% formic acid and stirred for 4 hours
under nitrogen. A small amount of product from
a previous reaction is added as seed and the
mixture is stirred for one more hour. The
338670
GC155f
-l6a-
mixture is stored in the freezer for about 16 hDursand the frozen mixture i8 warmed to room temperature
and stirred for an additional hour. The solid
formed is filtered and washed with methylene
chloride to yield ~82 mg of the title compound.
The filtrate is diluted with 1 liter of methylene
chloride and kept at -20C for 4 hours. The
precipitate that forms is recrystallized from
water-methanol-acetone to give an additional
167 mg of title compound.
NMR(D20) 1.63 (3H, d, J=6.5 cps), lR(nujol)1775 cm
Example 138
I3s- I~.(Z) ,4~J]-3-Ir2-Amiho-4-thiazolYl)-
methoxyimino~acetyl]-amino-4-methyl-2-oxo-1-
azetidinesulfonic acid, potassium salt
A solution of 201 mg of (Z)-2-amino-a-
~methoxyimino)-4-thiazoleacetic acid and 153 mg
of N-hydroxy-benzotriazole monohydrate in 3 ml
of dimethylformamide is treated with 206 mg
of dicyclohexylcarbodiimide. The mixture is
stirred at room temperature for 20 minutes under
nitrogen and a solution of 180 mg of (3S-cis)-
3-amino-4-methyl-2-oxo-1-azetidinesulfonic acid
and 0.14 ml of triethylamine in 2 ml of dimethyl-
formamide is added (.an additional 1 ml of
dimethylformamide is used for rinsing) and the
mixture is stirred for about 16 hours. The
slurry is evaporated in vacuo, triturated with
12 ml of acetone, centrifuged and the liquid
treated with 338 mg of potassium perfluorobutane-
sulfonate. Dilution with 10 ml of ether andfiltration gives a solid product which is chroma-
tographed on 200 ml of HP-20 resin eluting with
1 338670 GC155f
-161-
water. Fractions (20 ml each) 18-30 are combined and
lyophilized to give 274 mg of the title compound as a
hygroscopic solid.
Anal. calc'd. for CloH12N5O6S2
N,17.44
Found: C,30.03; H,3.21; N,17.06
NMR(D20) 1.40 (3H, d, J=6.5)~ 3.98(3H,S), 4.48 lH, d of t,
)=6.4~ 5.5), 5.36(1H, d, J=5.5) 6.97 (lH,S).
Example 139
(3S-trans)-3-Amino-4-methyl-2-oxo-1-azetidine-sulfonic
a _
A) Threonine, methyl ester, hydrochloride
Under an atmosphere of nitrogen, a flask containing
500 ml of methanol is cooled to -5C (ice/brine) and
130 ml (excess) of thionyI chloride is added at such a
rate as to maintain the reaction temperature between
0 and 10C. After recooling to -5C, 59.5 g of 1-
threonine is added and the mixture is allowed to reach
room temperature and stirred for 16 hours. The mixture
is concentrated and evacuated at 10 1 torr for 2 hours
to yield a vis-cous oil. This material is used directly
in the following step.
~ GC~55f
~ -162- 1 338670
B) Threonine amide
The crude product from part A is dissolved
- in 2.5 1 of methanol and cooled to -5C Cice/brine).
The solution is saturated with ammonia gas, the
cooling bath is removed and the sealed vessel is
allowed to stand for 3 days. After removing
the bulk of the unreacted ammonia via aspirator,
100 g of sodium ~icar~onate and 50 ml of water
is added and t~e mixture is stripped to a viscous
oil.
C2 Benzyloxycar~onylthréonine amide
The crude product from part B (already
cont~i n i ng the requisite amount of sodium
bicarbonate is diluted to a volume of 1 liter
with water. To this rapidly stirring solution,
94 g (88 ml of 90% pure material) of benzyloxy-
carbonyl chloride is added as a solution in 80 ml
of tetrahydrofuran over a 1 hour period. The
reaction mixture is then stirred for an additional
16 hours and extracted with ethyl acetate (one
500 ml portion, two 250 ml portions). The
combined extracts are dried over magnesium
sulfate and concentrated. The crystalline
residue is then dissolved in 250 ml of hot
ethyl acetate and 300 ml of hexane is added
followed by boiling until a clear solution is
reached. Cooling and filtration of the crystalline
mass give, after drying, 104 g of the title
compound.
GC155f
~ -163- 1 338670
Dl Benzyloxycarbon~lthreonine amide~ mesylate
Under an atmosphere of argon, 100 g of
benzyloxycarbonylthreonine amide is dissolved
in 400 ml of anhydrous pyridine and cooled in
an ice/salt bath. To this stirring solution,
36.8 ml (54.5 gl of meth~n~sulfonyl chloride
is added over a 15 minute period. After 2 hours
of stirring an additional 0.3 equivalents of
me~h~nesulfon~l c~Ioride is added. The reaction
is then stirred for 1 ~our and poured into a
mixture of 1.5 1 of ice and 2 1 of water. The
resulting slurry is stirred for about 3Q minutes
and filtered. Drying of the crude product at
60C for about 16 hours in a vacuum oven gives
109 g of the title compound.
E) N-Sulfonyl benzyloxycarbonylthreonine amide,
o-mesylate, tetrabutylammonium salt
A solution of 2-picoline (17.8 ml) in
90 ml of methylene chloride is cooled to -5C
(ice-brine) and chlorosulfonic acid 5.97 ml
is added at such a rate as to maintain the
internal reaction temperature below 5C. The
resulting solution is added via canula, to a
suspension of 7.56 g of benzyloxycarbonylthreonine
amide, ~-mesylate in 120 ml of methylene chloride. The
resulting heterogeneous mixture is refluxed
for about 16 hours yielding a clear solution.
The solution is poured into 500 ml of pH 4.5
phosphate buffer (0.5 M) and further diluted
with 120 ml of methylene chloride. The separated
GC155f
1 338670
~164~
organic layer is then washed once with lOQ ml
of buffer solution and the combined aqueous
phases are treated with 10.2 g of tetra-n-butyl-
ammonium hydrogensulfate and extracted with
methylene chloride ~one 3QQ ml portion and
two 150 ml portionsl. After drying the combined
organic extracts over sodium sulfate, the solution
is concentrated to yield 12.7 g of a foam.
F~ (3S-trans-~-3-Ami;no 4~meth~1-2-oxo--1-azeti~;~e-
sulfonic acid
A mixture consisting of 5.52 g of potassium
carbonate in 20 ml of water and 160 ml of 1,2-
dichloroethane is brought to reflux and 15.5 mmole
of N-sulfonyl benzyloxycarbonylthreonine amide,
O-mesylate, tetrabutylammonium salt is added in
20 ml of 1,2-dichloroethane (20 ml used as a
rinse). After refluxing for 30 minutes, the
mixture is poured into a separatory funnel,
diluted with 50 ml of water and 100 ml of
methylene chloride and the phases split. The
resulting organic ph.ase is dried over sodium
su-lfate and co~c~ntrated to yield crude ~3S-trans)-
3-benzyloxycarbonylamino-4-methyl-2-oxo-1-azetidine-
sulfonic acid, tetrabutylammonium salt. The
crude azetidinone is treated in 250 ml of ethanol
with 0.8 g of 5~ palladium on charcoal catalyst
and hydrogen is bubbled through the solution.
After 90 minutes the mixture is filtered through
Celite with 50 ml of ethanol used as a rinse.
GC155f
-165- 1 3 3 8 6 7 o
The addition of 1.2 ml of formic acid to this
solution causes an immediate precipitation of
the title zwitterion which is filtered after
stirring for 1 hour to yield, after drying at
10 1 torr for 1 hour, 1.1 g of product. A
second crop of product is obtained upon
concentration of the filtrate and addition of more
formic acid to give 1.3 g of the title zwitterion.
m.p. ~218dec., [a]D = -41.1(C=l, H2O).
NMR(D20) 1.58(3H, d )=7), 4.80(2H,M).
Examples 140-143
Following the procedure of Example 138, but
substituting (3S-trans)-3-amino-4-methyl-2-oxo-
l-azetidinesulfonic acid for (3S-cis)-3-amino-4-
methyl-2-oxo-1-azetidinesulfonic acid and the acid
listed in column I for (Z)-2-amino-~-(methoxyimino)-
4-thiazoleacetic acid, yields the compound listed in
column II.
140. (R)-a-[[[3-[(2-furanylmethylenelamino]- [3S-t3d(R*),4~]]-3-[[t[t3-[(2-furanyl-
2-oxo-1-imidazolidinyl]-carbonyl~amino]- ~ methylene)amino~-2-oYo-l-~ ;dA7olidinylJ-
benzeneacetic acid carbonyl]amin~phenylacetyl~amino~-4-methyl-
2-oxo-1-azeti~lnPsul~onOc acid, potassium
salt; melting point 213 C, dec.
141. (R)--[[(4-ethyl-2,3-dioxo-1-piperazinyl)- [3S-[3a(R*),4~]~-3-[[[[(4-ethyl-2,3-dioxo-
carbonyl]amino]benzeneacetic acid l-piperazinyl)carbonylJaminoJphenylacetyl]-
amino]-4-methyl-2-oxo-1-aze~i~ine~ulfonic
acid, potassium salt; melting point 177 C,dec.
142. (Z)-2-amino--(hydroxyimino)-4-thiazole- [3S~[3a(Z),4~]-3-[[(2-amino-4-thiazolyl)-
acetic acid (hydroxyimino)acetyl]amino]-4-methyl-2-oxo- 1 ~
l-azetidinesulfonic acid, potassium salt; W
melting point 230C. Co
143. (+)-~-[(amïnooxoacetyl)amino]-2-thio- 13S-[3at+)~4~]-3-ltt~aminQnyo7-eLyllamino]-2- ~J
pheneacetic acid thienylacetyl~emino]-4-methyl-2-oxo-1-azetidine-
sulfgniC acid, pota~sium salt; melting paint
135 C, dec.
1 33s6~o
GC155f
-167-
Example 144
[3S-[3~(Z),4~]]-3-[[(2-Amino-4-thiazolyl)[tl,l-
dimethyl-2-[(4-nitrophenyl)methoxy]-2-oxoethoxy]-
imino]acetyl]amino]-4-methyl-2-oxo-1-azetidine-
sulfonic acid, potassium salt
To a slurry of (3S-trans)-3-amino-4-methyl-
2-oxo-1-azeti~;ne-sulfonic acid
(0.36 g; see example 139) in dry dimethylformamide
(30 ml) under nitrogen at 26C is added triethyl-
amine (309 ~1). After about 5 minutes a clear
solution is obt~ine~ and (~)-2-amino-a--[~
~ethyl-2-I(4-nitrophenyl)methoxy~-2-oxoethQxy~-
imino]-4-thiazoleacetic-acid (Q.816 g~ is added
followed by N-hydroxybenzotriazole (0.334 g)
and dicyclohexylcarbodiimide (0.453 g). The
mixture is stirred for twelve hours at 26C,
whereupon the solvent is removed in vacuo, and
the residue is triturated with acetone (30 ml).
After stirring five minutes, the solids are
removed and the filtrate is treated with potassium
perfluorobutane sulfonate (3.680 gj in acetone,
(5 ml). Addition of ether (approximately 40 ml)
affords a precipitate which is collected and
dried in vacuo (1.073 g; second crop 0.066 g;
total of 1.14 g).
Anal. calc'd. for C20H21N6Olo 2 2
H, 3.70; N, 13.41; S, 10.23; K, 6.24
Found: C, 38.30; H, 3.63; N, 13.41; S, 9.88;
K, 5.98
1 3 3 8 6 7 0 GC155f
.
-168-
Example 145
[ 3a ( Z ), 4~]- 3- [1(2-Amino-4-thiazolyl)[(l-carboxy-
l-methylethoxy)iminolacetyl]amino~-4-methyl-2-
oxo-l-azetidinesulfonic acid, potassium salt (1:2)
s
A) [3a (Z) ,4a]-3-[~2-Amino-4-thiazolyl)[(l-
~ nylmethoxycarbonyl-l-methylethoxy)imino]-
acetyl~amino]-4-methyl-2-oxo-1-azetidinesulfonic
acid, potassium salt
A solu~ion of (cis)-4-methyl-2-oxo-3-[l(phenyl-
methoxy)carbonyl~amino]-l-azetidinesulfonic acid,
tetrabutylammonium salt (201 mg.; prepared from
the correspo~ potassium salt of example 98
as described $n example 136) in 5 ml of dimethyl-
form~m;~e is stirred with 90 mg of 10% palladium
on calcium carbonate in an atmosphere of hydrogen
for 2 hours. The slurry is filtered and the
filtrate is stirred for about 16 hours with 146 mg of
~Z)-2-ami~-[1-dipheny~-~PUluxy~L~lJyl~ ~UIyletho~y3imino~-
4-thiazoleacetic acid, 73 mg of dicyclohexy~ rl~l;;m;~P
and 51 mg of N-hydroxybenzotriazole under nitrogen.
The slurry is evaporated in vacuo and triturated
with 4 ml of acetone. The slurry is filtered
and the solid washed twice with 2 ml portions
acetone. The filtrate and washings are combined
and treated with 113 mg of potassium perfluoro-
butanesulfonate. Dilution with 24 ml of ether
gives a solid that is isolated by centrifugation
and washed three times with ether yielding 186 mg
of the title compound.
1 338670 GC155f
-
-169-
B) 13~(Z),4~]-3-[~(2-Amino-4-thiazolyl)~l-carboxy -
l-methylethoxy]imino]acetyl]amino]-2-methyl-4-oxo-
l-azet;~;nesulfonic acid, potassium salt (1:2)
A slurry of 186 mg of [3(Z),4a]-3-1[2-amino-
4-thiazolyl)[(l-diphenylmethoxycarbonyl-1-
methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-
l-azetidinesulfonic acid, potassium salt in
O.6 ml of distilled anisole is cooled to -12C
and 3.0 ml of distilled trifluoroacetic acid
io (at -10C) is added. The solution is stirred
for 10 minutes and 12 ml of ether followed by
6 ml of hexane are added. After 5 minutes at
-10C and stirring for 15 minutes at ambient
temperature, the solid is isolated by centrifugation
and washed four times with ether to give 141 mg
of material. This is dried in vacuo, powdered,
dissolved in 5 ml of cold water and immediately
adjusted to pH 5.6 with 0.4 N potassium hydroxide.
The solution is applied to a 100 ml HP-20AG column
and eluted with water. Fractions (10 ml) 8-12
are combined and evaporated in vacuo (acetonitrile
is added three times and evaporated. The residue
is triturated with ether to give 101.7 mg of
~ G~u~t~ as a hygroscopic solid.
Anal. Calc'd for C13N15N5O8S2
N, 13.69; S, 12.53; K, 15.28
Found: C, 30.11; H, 3.26; N, 13.35; S, 12.12;
K, 15.02
1 338670
GC155f
--170--
Example 146
[3S-[3a(Z),43]]-3- [[(2-Amino-4-thiazolyl)-~(1-
carboxy-l--methylethoxy)imino]acetyl]amino]-4-
methyl-2-oxo-1-azetidinesulfonic acid
[3S-[3~(Z),4B]]-3-[[(2-Amino-4-thiazolyl)-
[(l-carboxy-l-methylethoxy) imino]acetyl]amino]-
4-methyl-2-oxo-1-azetidinesulfonic acid,
dipotassium salt (87.3 mg; see example 103)
is dissolved in 1.38 ml of water, cooled to 0C,
treated with 0.34 ml of lN hydrochloric acid and
the resulting crystals separated by centrifugation.
The wet solid is dissolved in methanol, filtered,
concentrated to about 0.5 ml and mi ~re~ with 1 ml
of water, giving 55.9 mg of the title compound.
Example 147
~3S-[3(Z),4~]]-3-[~(2-Amino-4-thiazolyl) ~(1-
carboxy-l-methylethoxy)imino]-4-methyl-2-oxo-1-
azetidinesulfonic acid, sodium salt
A 99.7 mg sample of ~3S-[3~(Z),4~-3-
[[(2-amino-4-thiazolyl) [(l-carboxy-l-methyl-
ethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-
azetidinesulfonic acid is mixed with
0.207 ml of lN sodium hydroxide and
the resulting mixture is gently wanned to dissolve
the r~mA;ning solid. Water is removed
azeotropically with acetonitrile and the residue
is crystallized from a mixture of 0.5 ml of
methanol (to dissolve the residue) and 1 ml of
acetonitrile, giving 81.8 mg of solid. A second
recrystallization from 0.8 ml of methanol gives
47.9 mg, a third from 0.24 ml of methanol and
0.24 ml of absolute ethanol gives 44.8 mg, and
- 1 3 3 8 6 7~,~
GC155f
-171-
a fourth from 0.225 ml of methanol and 0.225 ml
of absolute ethanol gives 38.8 mg. The solid
is dried at 20C and 0.01 mm of Hg for 18 hours
and then equilibrated with atmospheric moisture
for 24 hours, giving 40.9 mg of the title compound.
Example 148
[3S-[3a(Z),4B]]-3-[[(2-Amino-4-thiazolyl)~(1-
carboxy-l-methylethoxy)imino]-4-methyl-2-oxo-1-
azetidinesulfonic acid, disodium salt
A 3.00 g sample of ~3S-[3(Z),4~]]-3-
[L(2-Amino-4-th;~zolyl)-[(l-carboxy-l-meth
ethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-
azetidinesulfonic acid, (see exam~le 146),
is suspended in 30 ml of water and titrated with
lN sodium hydroxide requiring 12.0 ml to give
the title disodium salt. The pH is reduced to
6.5 by the addition of a little Dowex 50W-X2 (H+).
The mixture is filtered and the filtrate diluted
to 66.3 g with water. A 6.63-g portion is
removed for other purposes. The remaining filtrate
is lyophilized, giving 2.38 g of solid. Partial
equilibration (24 hours) with atmosphereic
moisture gives 2.54 g of the title cG.~ul-d.
~Y~rles 149-151
Following the procedure of Example 138,
but substituting the compound listed in column I
for (Z)-2-amino--(methoxyimino)-4-thiazoleacetic
acid, yields the compound listed in column II.
Column I Cblumn II
149. (R)-a-[[[2,3-dioxo-4-1[(phenylmethoxy)- ~3S-~3a(R*),4a]~4-[1l2-l(4-methyl-2-oxo-1-
carbonyl]amino]-l-piperazinyl]carbonyl]- sulfo-3-azetidinyl)amino]-2-oxo-1-amino]benzeneacetic acid phenylethyl]amino]carbonyll-2,3-dioxo-
l-piperazinyl]carbamic acid, phenylmethyO
` ester, potassium 6alt, melting point 191 C,dec.
150. (R)--111[(2-furanylmethylene)amino]-2-oxo- [3S-¦3a(R*),4]~-3-[1[¦[3-¦(2-furanylmethylene)-
imidazolidinyl]carbonyl]amino]benzeneacetic amino]-2-oxo-1-~ olidinyl]carbonyl]-
acid amino]phenylacetyl]amino]-4-methyl-2-oxo-
l-azetidinesulfonic acid, potassium salt
151. (R)-a-[[(4-ethyl-2,3-aioxo-1-piperazinyl)- [3S-[3a(R*),4a~]-3-~[~[l4-ethyl-2,3-dioxo-
carbonyl]amino]benzeneacetic acid l-piperazinyl)carbonyl~amino]phenylacetyl~- ~
amino]-4-methyl-2-oxo-1-azeti~ine~ulfonic 1 _,
acid, potassium salt
'
- - 1 338670 GC155f
-173-
Example 152
~3S-13~(Z),4]]-3-I~(2-Amino-4-thiazolyl)~
carboxy-l-methylethoxy)imino]acetyl]amino~-4-methyl-
2-oxo-1-azetidinesulfonic acid, potassium salt (1:2)
s
A) [3S-13a(Z),4~]-3-~1(2-Amino-4-thiazolyl)-
I(l-diphenylmethoxycarbonyl-l-methylethoxy)imino]-
acetyl]amino]-4-methyl-2-oxo-1-azet;~;nesulfonic
acid, potassium salt
A solution of 440 mg of (Z)-2-amino--
[(l-c~rhoYy-l-methylethoxy)imino]-4-thiazoleacetic
acid and 153 mg of N-hydroxybenzotriazole mono-
hydrate in 3 ml of dimethylform~m;~e is treated
with 206 mg of dicyclohexylcarbodiimide. The
mixture is stirred at room temperature for 30
minutes under nitrogen and a solution of 180 mg
of (3S-c )-3-amino-4-methyl-2-oxo-1-azetidine-
sulfonic acid, (see example 137) and
0.14 ml of triethylamine in 2 ml of dimethylformamide
is added (an additional 1 ml of dimethylfor~-m;~e
is used for rinsing) and the mixture is stirred
for about 16 hours. The slurry is evaporated
in vacuo and triturated with 12 ml of acetone.
The slurry is filtered and the solid washed
with acetone (two 3 ml portions). The combined
filtrate and washings are treated with 338 mg of
potassium perfluorobutanesulfonate. Dilution
with 30 ml of ether gives a gummy solid, which
slowly solidifies. The solid is filtered and
washed with ether to give 656 mg of the title
compound.
1 338670 GC155f
--17 4--
B) [3S- [3~ (Z), 4a] ] -3~ t~(2-Amino-4-thiazolyl)-
[(l-carboxy-l-methylethoxy) imino]acetyl]amino]-4-
methyl-2-oxo-1-azetidinesulfonic acid, potassium
salt (1:2)
A slurry of 656 mg of [3S-[3~(Z),4]]-3-
[[(2-Amino-4-thiazolyl) ~(l-diphenylmethoxycarbonyl-
l-methylethoxy) imino] acetyl]aminol-4-methyl-2-
oxo-l-azet;~linesulfonic acid, potassium salt in
2.3 ml of distilled anisole is cooled to -12C
and 11.5 ml of trifluoroacetic acid (precooled
to -10C) is added. The solution is stirred for
15 minutes and 46 ml of ether followed by 23 ml
of hPYAn~ are added. After 5 minutes at -10 C
and stirring for 15 minutes at room temperature,
the solid is filtered and washed with ether to
give 457 mg of a very hydroscopic gum. This is
dissolved in 6 ml of cold water and i~nediately
adjusted to pH 5.6 with 0.4N potassium hydroxide
solution. The solution is applied to a 200 ml
HP-20 resin and eluted with water. Fractions
(50 ml each) 7-11 are co~ined and lyophilized
to give 239 mg of t~e title compound as a solid.
Anal- Calc'd. for C13H15O8N5S2R2-1/2 H2O C~ 29-99;
H, 3.10; N, 13.45; S, 12.32
Found: C, 29.94; H, 3.30; N, 13.30; S, 11.93
NMR(D20) 1.44 (3H d, )=75), 1.46(6H,S), 4.48(1H,d of t
)=75, 5.5), 5.34(1H, d, )=5.5) 6.96ppm(1H,S).
1 33 8 6 70 GC155f
-175-
Example 153
(i)-3-Amino-4,4-dimethyl-2-oxo-1-azetidinesulfonic
acid
A) (+)-4,4-Dimethyl-2-oxo-1-azetidine-tertbutyl-
diphenylsilane
A solution of 40.5 ml of t-butylchlorodiphenyl-
silane in 112 ml of dimethylformamide is cooled
to 0C. To this is added 22 ml of triethylamine.
A solution of 12.87 g of 4,4-dimethyl-2-azeti~ino~e
in 25 ml of dimethylformamide is added dropwise
over 10 minutes to the cooled triethylamine solution.
The resulting cloudy solution is stirred for 18
hours at 5C under argcn. This mixture is poured
into 400 ml of ice water and extracted with three
150 ml portions of 2:1 ether:ethyl acetate. The
combined extracts are washed with four 100 ml
portions of 0.5M monobasic potassium phosphate
buffer, one 150 ml portion of sodium bicarbonate
solution, two 150 ml portions of water and one
150 ml portion of saturated sodium chl~ride
solution. The solution is dried over sodium
sulfate and concentrated in vacuo to yield 33.03 g
of the title compound as a solid.
B) (i)-3-azido-4~4-dimethyl-2-oxo-l-azetidine
tertbutyldiphenylsilane
A solution of 4.25 ml of 1.6M (in hexane)
n-butyllithium and llml of dry tetrahydrofuran
is prepared at -50C under argon in a 100 ml
three-necked flask. A solution of 0.083 g of
triphenylmethane in 1 ml of tetrahydrofuran is
1 33~670
_ ~C155
~176~
added. The resulting solution is cooled to -60 & ,
and 1.0 ml of diisopropylamine is added dropwise ~
by syringe. This is stirred for 15 minutes and
then cooled to -78C. A solution of 2.3 g of
(I)-4,4-dimethyl-2-oxo-1-azetidine-tertbutyl-
diphenylsilane in 8 ml of tetrahydrofuran is added
slowly by syringe. The resulting solution is
stirred for 20 minutes at -78C, during which time
heavy precipitation occurs and uniform stirring
becomes difficult. A solution of 1.33 g of
p-toluenesulfonyl azide in 5 ml of tetrahydrofuran
is added dropwise. The resulting mixture is
allowed to stir at -78C for 20 minutes, and 2 ml
of trimethylsilyl chloride is added dropwise.
The reaction mixture is warmed to ambient temperature
and stirred for 1 hour. Then the mixture is cooled
to 0C and poured into 150 ml of 0C ethyl acetate.
Enough 0.5 M monobasic potassium phosphate buffer
is added to make both the aqueous and organic
layers clear. The two layers are separated and
the organic layer is washed with three 150 ml
portions of 0.5 M monobasic potassium phosphate
solution, one 150 ml portion of sodium chloride
solution, one 150 ml portion of saturated sodium
chloride solution and dried over sodium sulfate.
The solution is concentrated in vacuo to 2.83 g
of oil, which upon trituration with hexane yields
1.67 g of the title compound as a solid.
1 3 3 8 6 7 0 GCli5f
-177-
C) (+)-3-Azido-4,4-dimethyl-2-oxo-1-azetidine
In a 50 ml three-necked flask, 1.52 g of
(+)-3-azido-4,4-dimethyl-2-oxo-1-azetidine-
tertbutyldiphenylsilane is dissolved in 25 ml
of acetonitrile. To this stirred solution is
added 0.25 ml of 48% hydrofluoric acid. This
is stirred at ambient temperature, and 0.5 ml
portions of 48% hydrofluoric acid are added every
60 minutec until, after 6.5 hours, a total of
3.25 ml of 48% hydrofluoric acid has been added.
The reaction mixture is then cooled to 0C,
neutralized with saturated sodium bicarbonate,
and extracted with 120 ml of ethyl acetate. The
organic layer is then washed with 100 ml of
water, 100 ml of saturated sodium chloride solution,
and dried over sodium sulfate. The dry solution
is concentrated in vacuo to yield 1.34 g of oil.
This impure oil is chromatographed on 27 g of
silica gel with hexane, followed by 33% ethyl acetate
in hexane, to yield 0.358 g of the title compound
as a solid.
D) (+)-3-Azido-4,4-dimethyl-2-oxo-1-azetidine-
sulfonic acid, tetrabutylammonium salt
To 0.100 g of (+)-3-azido-4,4-dimethyl-2-
oxo-l-azetidine at 0C is added under argon
2.8 ml of 0.5M dimethylformamide-sulfur trioxide
complex. This mixture is allowed to warm to
ambient temperature and stir for 45 minutes.
The solution is then poured into 20 ml of 0.5 M
pH 5.5 monobasic potassium phosphate buffer.
1 338670
GC155f
-178-
This is washed with three 20 ml portions of
methylene chloride (~discarded~ and Q.237 g of
tetrabutylammonium hydrogen sulfate is added to
the aqueous solution. This was extracted with
four 20 ml portions of methylene chloride and
the combined organic extracts are washed with
20 ml of 8% sodium chloride solution. The
methylene chloride solution is dried Csodium
sulfate) and co~centrated in vacuo to yield 0.31 g
of oil, which appeared by nmr to be 50~ dimethyl-
formamide and 50% of the title compound.
E2 (+)-3-Amino-4,4-dimethyl-2-oxo-1-azetidi-ne-
sulfonic acid
A solution of 0.155 g of (+~-3-azido-4,4-
dimethyl-2-oxo-1-azetidinesulfonic acid, tetra-
butylammonium in 0.6 m 1 of methanol is hydrogenated
over 10% palladium on charcoal for 20 minutes at
1 atmosphere. The catalyst is filtered off and
rinsed with methylene chloride which is combined
with the methanol solution. This clear solution
is treated with 0.123 ml 97% formic acid. Upon
addition of the acid the solution immediately
becomes cloudy. After s~An~i~g for 1 hour at 5C,
the solid is filtered off to yield 0.0664 g of
the title compound, m.p. 200-202C(dec.).
NMR(D20) 1.64(3H,S), 1.68(3H,S), 4.42(1H,S),
IR(KBr) 1765cm
1 338670 GC155f
-179-
Example 154
[3+(Z)3-3-1[(2-Amino-4-thiazolyl)(methoxyimino)-
acetyl]aminol-4,4-dimethyl-2-oxo-1-azeti~;neculfonic
acid, potassium salt
A solution of N-hydroxybenzotriazole hydrate
(~0 mg) and (Z)-2-amino-~-(methoxyimino)-4-
thiazoleacetic acid (0.323 mmole) in 0.5 ml of
dimethylformamide is treated with 67 mg of
dicyclohexylcarbodiimide under argon at ambient
temperature. The resulting mixture is stirred
for 1 hour at which time (i)-3-amino-4,4-dLmethyl-
2-oxo-1-azetidinesulfonic acid
(57 mg; see exam~le 153) is added as a solid
followed by triethy~ e dropwise (0.05 ml).
The reaction is stirred at ambient t~mperature
for 16 hours. The dimethylformamide is removed
under high vacuum at 30C, and the residue is
slurried in 4 ml of acetone and filtered. The
filter cake is washed with an additional 4 ml of
acetone, and potassium perfluorobutanesulfonate
(85 mg) is added to the filtrate, followed by
ether. Trituration of the resulting gum with
ether gives 40 mg of a tan solid which is chromato-
graphed on a 70 ml HP-2QAG column. Elution with
2S water gives 20 mg of the title compound in
fractions (5 ml) 16-40 after evaporation,
trituration with 1:1 acetone-hexane and drying.
m.p. 225(dec).
Anal. Calc'd. for CllH14N5O6S2 K
N, 16.86; S, 15.43
Found: C, 29.47; H, 3.48: N 14.98
- 1 338670 GC155f
-180-
Example 155
(+)-4,4-Dimethyl-2-oxo-3-[(phenylacetyl)amino]-
l-azetidinesulfonic acid, potassium salt
A solution of N-hydroxybenzotriazole hydrate
(45 mg) and phenylacetic acid (40 mg) in 0.5 ml
of dimethylforr-m;~ is treated with dicyclohexyl-
carbodiimide (61 mg) under argon at ambient
temperature. The resulting mixture is stirred
for 1 hour and (+)-3-amino-4,4-dimethyl-2-oxo-
l-aze~i~ineculfonic acid, (52 mg; see
example 153) is added as a solid, followed by
triethylA~ine dropwise (0.04 ml). The reaction
is stirred at ambient temperature for 24 hours.
The dimethylformamide is removed under high
vacuum at 30C and the residue is slurried in
acetone and filtered. Potassium perfluorobutane-
sulfonate is added to the filtrate, ether~ is
added and the mixture is cooled. The resulting
solid is washed with acetone, hexane, and dried
yielding the title compound as a powder.
Anal. calc d. for C13H15N205SK: C, 44.55; H, 4.32;
N, 8.00; S, 9.15; K, 11.16
Found: C, 43.83; H, 4.16; N, 7.96; S, 8.76;
K, 11.43
NMR(D20) 1.33(S,3H), 1.58(S,3H), 3.68(S,3H), 4.70
(S,lH), 7.56ppm (broad S, 5H).
EXample 156
(3S-trans)-3-~[(2-Amino-4-thiazolyl)oxoacetyl]-
amino~-4-methyl-2-oxo-1-azetidinesulfonic acid,
potassium salt
To a solution of diphenylphosphinyl chloride
(1.85 g) in dry dimethylformamide (15 ml) cooled
in an ice-methanol bath (-15 ~o -20C) is added
(2-amino-4-thiazolyl)glyoxylic acid, triethylamine
1 338670
GC155f
-181-
salt (2.14 g). After stirring for 0.5 hour a
solution of (3S-trans)-3-amino-4-methyl-2-oxo-
l-azetidinesulfonic acid (1.08 g;
see example 139) and triethylamine (1.92 ml)
in dry dimethylformamide (5 ml) is added to the
cold mixed anhydride solution and the reaction
mixture is stirred at 5C for 24 hours. Solvent
is removed in vacuo, the residual dark oil is
dissolved in water, and chromatographed on
Dowex 50 X 2-400 resin (~ form, 200 ml). Upon
elution with water ~15 ml fractions~ the crude
product is collected in fractions 13-27 (3.37 g).
Chromatography on EP-20 resin (200 ml~, eluting
with water (15 ml fractions), gives the desired
product in fractions 18-26. Removal of water
in vacuo gi~es the title compound as an amorphous
powder.
Anal. Calc'd for CgHgN4O6S2K (372.42): C, 29.02;
H, 2.44; N, 15.04; S, 17.22; K, 10.50
Found: C, 28.87; H, 2.62, N, 14.85; S, 15.09;
K, 10.81
Example 157
[3S(R*)]-3-[1[(Aminoacetyl)amino]phenylacetyl]amino]-
2-oxo-1-azetidinesulfonic acid, potassium salt,
trifluoroacetate (1:1) salt
The deprotection of r3s(R*)~-3-I~ L(4-
methoxyphenyl)methoxy]carbonyl]amino]acetyl]amino]-
phenylacetyl]amino]-2-oxo-1-azetidinesulfonic
acid potassium salt (see example 127) using
trifluoroacetic acid and anisole yields the title
compound, melting point 165C, dec.
1 338670 GC155f
- -182-
Example lg8
(3S-tFans)-3-Methoxy-4-methyl-2-oxo-3-'[1(phenyl-
methoxy)carbonyl~amino-] l-azetidinesulfonic acid,
potassium salt
A) (3~-trans)-4-Methyl-3-methoxy-2-oxo-4-
[[(phenylmethoxy)carbonyl-~amino~aze~i~ine
A solution of 2.5 g (0.0106 mole) of
(3R-trans)-4-methyl-2-oxo-3[~(phenylmethoxy)-
carbonyl~amino~azeti~ine (prepared from d-
threo~in~ in 12.6% yield essentially asdescribed for the racemic cis isomer in
Example 98C) in 112 ml of 4% borax in methanol
is cooled to 0C and 3.5 ml of t-butyl hypo-
chlorite is added. After 20 minutes the
solution is poured into 1 liter of cold water
and extracted with two 750 ml portions of
cold ethyl acetate. The organic layer is
washed with cold water (two 750 ml portions),
saturated salt, dried and evaporated to give
3.05 g of crude N-,N'-dichloroamide.
A solution of 426 mg of lithium methoxide
in 20 ml of dry methanol is cooled to -78C
and diluted with 40 ml of dry tetrahydrofuran.
Over 30 seconds a solution of the above chloro-
amide in 20 ml of tetrahydrofuran (-78C) are
added via syringe. After 20 minutes at -78C,
2 ml each of acetic acid and trimethyl phosphite
are added. After 40 minutes at room temperature
the solution is poured into 500 ml of water
and extracted with ethyl acetate (two 300 ml
portions). The organic layer is washed with
338670
GC155f
-183-
water, dried, and evaporated to give an oil.
Chromatography on a 200 ml silica gel column
eluting with 3:1 chloroform-ethyl acetate
gives a total of 1.25 g of the title compound.
B) (3S-trans)-3-Methoxy-4-methyl-2-oxo-3-
~l(phenylmethoxy)carbonyl]amino-1-azetidinesulfonic
acid, potassium salt
A solution of sao mg (0.00303 mole) of
(3S-trans)-4-methyl-3-methoxy-2-oxo-4- I [ (phenyl-
methoxy)carbonyl]amino]azetidine in 2 ml of dimethyl-
formamide is cooled to 0C and 4 ml of dimethyl-
formamide-sulfur trioxide complex is added. After
1 hour at 0C and 4 hours at room temperature the
solution is poured into 80 ml of 0.5 M monobasic
potassium phosphate (adjusted to pH 5.5) and
extracted with methylene chloride (two 50 ml
portions, discard). The aqueous layer is treated
with 1.04 g of tetrabutyl ammonium sulfate and
extracted with dichloromethane to give 1.42 g of
oil. This is dissolved in acetone and treated
with 1.04 g of potassium perfluorobutanesulfonate
in 10 ml of acetone. Dilution with 250 ml of ether
and extensive trituration of the oily solid gives
584 mg of crude product. Chromatography on HP-20 AG
(200 ml) gives 418 mg of purified product in fractions
(100 ml) 13-16 (elution with 1 liter of water and then
9:1 water-acetone). Trituration of 114 mg of this
material with ether gives 104 mg of an analytical
sample.
Analysis calc'd for C13H14N2O7SK H2O: C, 39.06;
H, 4.04; N, 7.01; S, 8.03; K, 9.78
Found: C, 38.91; H, 3.62; N, 6.91; S, 8.06;
K, 9.51
NMR(D20) 1.33(3H, d, )=7), 3.46(3H,S), 4.22(2H, d of d,
) =6), 5.18 (2H,S), 7.43ppm (5H,S) .
GC155f
--184--
1 338670
Exa~ple l59
(3S-trans)-3 ~clhoxy-4-methyl-2-oxo-3-~(phenylacetyl)-
amino]-l-azetidines~lfonic acid, potassium sa-lt
(3S-trans)-3-am~no-3 ~U~y 4 1~Ll~yl-2-X~l~
5 azetidinesulfonic acid, tetrabutylammonium salt
is prepared by the catalytic hydrogenation of
(3S-trans)-3-methoxy-4-methyl-2-oxo-3-I[(phenyl-
methoxy)carbonyU amino~-l-azeti~ineæulfonic acid,
potassium salt (see example 158) after conYersion -to
lO the tetrabutyla~onium salt. Following the procedure
of example 88,hlt u~ ;n~ (3s-trans)-3-arrllno-3
4-methyl-2-oxo-l-azetidinesulfonic acid, tetra-
butyla~onium salt and phenylacetyl chloride yields
the title co...yound.
Anal. calc'd for Cl3Hl5N2O6SX: C,42.61; H,4.31; N,7.65
Found: C,39.67; H,4.09; N,7.30
r~R (D20) 1.29(BH, d, )=7) 3.45(3H,S), 3.73(2H,S),
4.36 (2H, d of d, )=6), 7.38ppm (5H,S).
Example 160
[3S-13a(Z),4~]]-3- [I(2-Amino-4-thiazolyl) 1[2-
(diphenylmethoxy)-2-oxoethoxy]imino]acetyl]-
amino]-2-methyl-4-oxo-l-azetidinesulfonic acid,
potassium salt
Following the procedure of example 138,
but substituting (Z)-2-amino-a- [12-(diphenylmethoxy)-
2-oxoethoxy]imino]-4-thiazoleacetic acid for
(Z)-2-amino-a-(methoxyimino)-4-thiazoleacetic
acid and first treating the (3S-cis)-3-amino-
4-methyl-2-oxo-l-azetidinesulfonic acid,
with triethylamine, yields the title compound,
melting point 155-160C, dec.
GC15-5f
--185--
1 338670
Example 161
13S_~3~(Z),4~]] -3-I[~(Carboxymethoxy) imino] (2-amino-
4-"hiazolyl)acetyl-]amino]-4-methyl-2-oxo-1-
azetidinesulfonic acid, dipotassium salt
The deprotection of l3S-~3c~(Z1~4 ~ ~ -3-Il(2-
amino-4-thiazolyl)ll2-(diphenylmethoxy)-2-oxoethoxy]-
imino]acetyl]amino]-2-methyl-4-oxo-1-azetidine-
sulfonic acid, potassium salt (see example 160)
using trifluoroacetic acid and anisole yields
the title compound, which decomposes a.~.>250C.
Example 162
(S)-3-1[~(2,6-Dichloro-4-pyridinyl) thio~ acetyl]-
amino]-2-oxo-1-azetidinesulfonic acid, potassium salt
Acylation of (S)-3-amino-2-oxo-1-azetidine-
sulfonic acid, tetrabutylammonium salt (see example
6A) with ~(2,6-dichloro-4-pyridinyl)thio]acetic
acid, 4-nitrophenyl ester, followed by treatment
with potassium perfluorobutane sulfonate, yields
the title compound, melting point 212-214C.
Example 163
I3S (R*)] -3-I I ~ I ( 4-~mino-2,3-dioxo-1-piperazinyl~-
carbonyl] ami-no]phenylacetyl~amino]-4-methyl-2-
25 oxo-l-azetidinesulfonic acid, potassium salt
[3S(R*)]-I4-~I[2-~(4-methyl-2-oxo-1-sulfo-
3-azetidinyl)amino]-2-oxo-1-phenylethyl]amino] -
carbonyl]-2,3-dioxo-1-piperazinyl]carbamic acid,
phenylmethyl ester, potassium salt (see example 149)
30 is hydrogenated using gaseous hydrogen and 10%
palladium on charcoal as catalyst, yielding the
title compound, melting point 165C, dec.
GC155f
_ -186~
- 1 338670
Example 164
[3S(Z)]-3-[f(2-Amino-4-thiazolyl)~[(l-carboxy-1-
methylethoxy)imino]acetyl]amino]-2-oxo-1-azetidine-
sulfonic acid, sodium salt ~1:2)
[3S(Z)]-3-~1(2-Amino-4-thiazolyl~[~2-(diphenyl-
methoxy)-l,l-dimethyl-2-oxoethoxy]imino~acetyl]amino]-
2-oxo-1-azeti~ sulfonic acid, tetrabutylammonium salt
(see example 28) is deprotected using trifluoroacetic
acid and anisole to yield the title compound,
melting point 185C, dec~ after conversion to the
disodium salt with aqueous sodium hydroxide and
purification on HP-20.
Examples 165-168
Following the procedure of Example 11, but
substituting the acid listed in column I for
~Z~-2-amino--~lhydroxy(phenylmethoxy)phosphinyl]-
methoxy]imino~-4-thiazoleacetic acid, yields
th compound li~ted in column II.
Column I Column II
165. 2,6-dimethoxybenzoic acid (S)-3-[(2,6-dimethoxybenzoyl)amino]-2-oxo-l-azetidinesulfon~c acid, potassium salt;
melting point 180 C, dec.
166. (Z)-2-amino-a-[~4-(diphenyl- [3S(Z)1-2-[1(2-amino-4-thiazolyl)~[4-methoxy-)-4-oxobutoxy]imino]-4- (diphenylmethoxy)-4-oxobutoxy]imino]-
thiazoleacetic acid acetyllamino]-2-oxo-1-azetidinesulfonic
aciOd, potassium salt; melting point 125-
130 C, dec.
167. 2-t~lphenylmethoxy)carbonyl]- (S)-2-oxo-3-~[2-t[~(phenylmethoxy)carbonyl]-
amino]methyl]benzoic acid amino]methyl]benzoyl~amino~-l-azetidine-
sulfonic acOd, potassium salt; melting
point 234.5 C, dec.
168. ~-~[l5-hYdroxy-2-(4-formyl-1- (3S)-3-[~[~5-hydroxy-2-(4-formyl-1-
piperazinyl)pyrido[2,3-d]- piperazinyl~pyrido~2,3-d~pyrimidin- 6-
pyrimidin-6-yl]carbonyl~amino~- yl]carbonyl]amino]phenylacetyl]amino]-2-
benzeneacetic acid oxo-l-azetidinesulfonic aci~, potassium
salt; melting point 265-270 C, dec.
oo ~
`J ~n
GC155f
-188-.
1 338670
Example 169
(trans~-3-A~ino-4-eth~l-2:-oxo-1-azat;~in~sulfonic acid
A) t-Boc-N-methoxy-B-threoethylserinamide
threo-D,L-~-Ethylserine (1.33 g) is dissolved
in 10 ml of 2N potassium hydroxide and 5 ml of
t-butanol. After t~e addition of 2.46 g of
di-t-butylpyrocar~onate the Lw3 ~hase mixture
is stirred for 4 hours at am~ient temperature.
O-Methylhydroxylammonium chloride (1.25 g) is
added and the pH is adjusted to 4 with lN
hydrochloric acid. l-Ethyl-3-(3-dimet~ylamino-
propyl)carbodiimide, hydrochloride (1.92 g) is
added and the pH is again adjusted to 4. After
stirring for 1 hour the reaction mixture is
saturated with sodium chloride and extracted
with four 50 ml portions of ethyl acetate.
The ethyl acetate extracts are combined and
dried over MgSO4. Removal of the solvent
in vacuo yields 1 g of the title compound.
B) t-Boc-O-me~Anesulfonyl-N-methoxy-B-threo-
propionamide
t-Boc-N-methoxy-~-threoethylserinamide (10.5 g)
is dissolved in 65 ml of pyridine. Methanesulfonyl
chloride (4.65 ml) is added dropwise at 0C.
After stirring for 3 h.ours at ambient temperature,
the reaction mixture is poured into 200 g of
~ ice and 300 ml of lN h.ydrochloric acid. The pH
is adjusted to 4 with concentrated hydrochloric
acid. After extraction with three 85 ml portions
GC155f
-189-
1 338670
of ethyl acetate the comh;ned extracts are dried
over MgSO4 and concentrated in vacuo. The residue
is treated with carbon tetrachloride and
concentrated again. Stirring with ether followed
by filtration yields 6.9 g of the title compound.
C) (trans~-3-t-Butox-ycar~onylamino-4-eth-yl-1-
methoxy-2-azeti-~ino~
Anhydrous potascium car~onate ~4.15 g)
and 125 ml of dry acetone are brought to reflux
and 3.4 g of t-Boc-Oqmethanesulfonyl-N-methoxy-
~-threo-propionamide in 25 ml of acetone is added.
After 1 hour the reaction mixture is cooled
and filtered, and t~e filtrate is concentrated
in vacuo. The oily residue is stirred with
hexane to yield 2.2 g of the title compound.
D) (trans)-3-t= Butoxycarbonylamino-4-ethyl-2-
azetidinone
(transl-3-t-Butoxycarbonylamino-4-ethyl-1-
methoxy-2-azetidinone (3 gl is added to 170 ml
of liquid ammonia at -78C under nitrogen and
1.68 g of sodium is added in 5 portions with
stirring over a S minute period. Stirring is
continued for 30 minutes. Ammonium chloride is
then added slowly until the blue color of the
reaction mixture disappears. After removal of
the ammonia under nitrogen the solid is extracted
with two 100 ml portions of ethyl acetate.
Removal of the solvent followed by drying in vacuo
yields 2.7 g of the title compound.
GC155f
-19Q~
1 33867a
E) (trans)-3-t-~uLGx~ca ~.onylam-ino-4 et~ 2-
-
oxo-l-azetidinesulfoni-c acid, tetr-abutylammonium
salt
To a 2 ml of ab.solute pyridine in 20 ml
of dry dichlor~methane is added trimethylsilyl-
sulfonyl chloride (~3.7 ml~ in 5 ml of dry
dichloromethane. The addition is acoomplished
at -30C, under nitrogen, over a 10 minute
period. After stirring at ambient temperature
for 30 minutes, the flask is evacuated to yield
a pyridine-sulfur trioxide complex. (:trans~-3-
' t-Butox~rcarbonylamino-4-ethyl-2-azeti~ino~e
(2.67 g) and 20 ml of dry pyridine are added
to the flask which. is then placed in an oil
bath preheated to 90C. After 15 minutes a
clear solution is obtained and poured into 200 ml
of a lM solution of dibasic potassium phosphate.
After the addition of 27 g of dibasic .potassium
phosphate and lQ0 ml of water a clear solution
is obtained. The solution is extracted with
two 60 ml portions of ethyl acetate. Tetrabutyl-
ammonium hydrogensulfate is added to the aqueous
layer, and the aqueous solution is extracted
with three 100 ml portions of dichloromethane
and the combined organic layers are dried over
MgSO4. Concentration in vacuo yields 6.9 g
of the title compound.
GC155f
-191- 133~
F2 (trans)-3-Amino-4-ethyl-2-oxo-l-azetidinesulfonic
acid
(trans)-3-t-Butoxycarbonylamino-4-ethyl-2-
oxo-l-azetidinesulfonic acid, tetrabutylammonium
salt (6.75 g2 in 4Q ml of 98% formic acid is
stirred for 3 hours at am~ient temperature.
Dichloromethane C60 ml) is added and the mixture
is refrigerated for about 16 hours. The resulting
precipitate is separated ~y filtration and then
dried in vacuo to yield 0..85 g of the title compound,
melting point 185C, dec.
Example 170
(.trans,Z)-3-rf(2-Amino-4-thiazolyl)r(l-car~oxy-
1-methylethoxy)imino]acetyl]amino]-4-ethyl-2-
oxo-l-azetidinesulfonic acid, dipotassium salt
A) (trans,Z)-3-I~(2-Amino-4-thiazolyl)[(l-
diphenylmethoxycarbonyl-l-methylethoxy)imino]-
acetyl]amino]-4-ethyl--2-oxo-1-azetidinesulfonic
acid, dipotassium salt
(trahs1-3-Amino-4-ethyl-2-oxo-l-azet;~;ne-
sulfonic acid (Ø55 g2 and 335 mg of triethylamine
are dissolved in 50 ml of dry dimethylformamide.
(Z)-2-Amino-~-r(l-diphenylmethoxycarbonyl-l-
methylethoxy)imino]-4-thiazoleacetic acid (1.14 g)
is added with stirring at 0C followed by 450 mg
of hydroxybenzotriazole and then 0.69 g of
dicyclohexylcarbodiimide. After stirring for
about 16 hours at 0C the flask is evacuated.
Dry acetone (25 ml) is added to the s~olid with
_ GC155f
-192- 1 3 3 8 6 7 0
stirring. The mi~ture is filtered and Q.94 g
of potassium perfluoro~utanesulfonate is added
to the filtrate followed ~y 100 ml of ether.
After standing for 1 hour at 0C the solid is
filtered, washed with ether and dried -in vacuo
yielding 1.58 g of the title c~mpound.
B) (trans,Zl-3-I-~(2-Amino-4-thiazol~ C1-carboxy-
l-methyle-thoxy)imino]acetyl~amino`]-4-ethyl-2-
oxo-l-azetidinesulfonic acid,-dipotàssium salt
To a suspension of 1.31 g of (transl,Z)-3-
[~2-amino-4-thiazolyl~](l-diphenylmethoxycarbonyl-
l-methylethox~imino~acetyl~amino]-4-ethyl-2-
oxo-l-azetidinesulfonic acid, dipotassium salt
in 10 ml of anisole is added 5 ml of trifluoro-
acetic acid over a 10 minute period at -15C.
After stirring for 2 hDurs at -10 C a clear
solution is obtained. At -30C 80 ml of dry
ether is added and the resulting precipitate
is filtered and then treated with 5 ml of water.
The pH is adjusted to 5.5 with lN potassium
hydroxide at 0C, and the mixture is filtered
to remove unconverted starting material. The
filtrate is chromatographed on HP-20 with water
as eluent. Lyophilization yields 185 mg of
the title compound, melting point 160C, dec.
GC155f
-193-
1 338670
Exa~ple''171
~3S(.Z)]-3-~['('2-Amino-4-thiazolyl')[t4-hydroxy-4-
oxobutoxy)imino]ace-tyl]amino]-2-oxo-1-azetidine-
sulfonic acid, potassium' salt
The deprotection of I 3S(Z)]-3-~[(2-amino-4-
thiazolyl)[~4-(diphenylmethoxy)-4-oxobutox~]imino]-
acetyl]amino]-2-oxo-1-azetidinesulfonic acid,
potassium salt Csee exæmple 1661 using trifluoro-
acetic acid and anisole yields t~e title compound,
melting point ~2Q0C.
EXam'ple 172
~S2-3-I [2-(Amihomethyl~benzoyl]amino]-2
azetidinesulfohic ac'id','inher salt
The deprotection of (S)-2-oxo-3-[[2-~l[(phenyl-
methoxy)carbonyl]amino]methyl]benzoyl]amino]-l-
azetidinesulfonic acid, potassium salt using
hydrogen gas, palladium on charcoal, and hydrochloric
acid, yields the title compound, melting point
162-165C.
Example 173
(S)-3-[~2-(4-Formyl-l-piperazinyl)-5-hydroxy-
pyrido[2,3-d~pyrimidin-6-yl]carbonyl]amino]-2-
oxo-l-azetidinesulfonic acid, potassium salt
(S)-3-Amino-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (see example 6A) is
coupled with 2-(4-formyl-1-piperazinyl)-5-hydroxy-
6-L(4-nitrophenoxy)carbonyl]pyrido[2,3-d]pyrimidine
and treated with potassium perfluorobutanesulfonate
in acetone to yield the title compound, melting
point 290 C, dec.
GC155f
-194-
1 338670
F~ le 174
(3S-trans~ l I (4-Methyl-2-oxo-1-~ul~o-3-azetidiny-1)-
amino]carbonyl]benzeneacetic acid', d'ipo'tassium salt
(3S-trans)-3-Amino-4-methyl-2-oxo-1-azetidine-
sulfonic acid (see example 139) is coupled with
~-(carboxyllbenz~ne~cPtyl chloride and treated
with triethyl~mine and potassium perfl~or~utane-
sulfonate to yield the title compound, melting point
147 C, dec.
Ex'ampl'e''1'75
~3S-trans~-3-hmin~-4-~y~lohexyl-2-oxo-1-azetidine-
sulfonic acid
A) -(t-Butoxycar~onylamino)-~-cyclohexyl--~-
hydroxy-threo-propionic acid
B-Cyclohexyl-~-amino-B-hydroxy-threo-propionic
acid (15 g) is suspended in 150 ml of acetonitrile
and 70 ml of water. Triethylamine (17.8 g) is
added and the mixture is heated with stirring
to 60C. At this temperature a clear solution
is ob~ P~ and 21.0 g of di-t-butylpyrocarbonate
is added and stirring at 60C is continued for
1.5 hours. The solvent is ~ oved in vacuo
and 50 ml of water is added. The aqueous layer
is extracted with ethyl acetate at a pH of 2,
which is adjusted by addition of 3N HCl. The
organic layer is separated, dried over Na2SO4
and evaporated to dryness. The remaining
crystalline material is filtered with petrol
ether, yielding 20.4 g of the title compound,
melting point 113-115C.
_ GC155f
-195-
1 338670
B~ -(t-Butoxycarhony-lamino)-B-cyclohexyl-~-
hydroxy-N-methoxy--threo-propionamide
~ -(t-Butoxycarbon~l A~; no) -B-CyClohexyl-B-
hydroxy-threo-propionic acid (20.2 g) and 7.6 g
of O-methylhydroxylamine hydroch.loride are
suspended in 350 ml of water and 175 ml of
t-butanol. Th~ pH of the mixture is adjusted
with potassium carbonate to 4.1-Ethyl-3-~.3-dimethyl-
aminoprop~l)carbodiimide C16 .4 gl is added and
the pH is maintained at 4 with stirring for 1.5
hours. t-Butanol is removed in vacuo and the
remaining aqueous solution is saturated with
sodium chloride and extracted twice with. 100 ml
portions of ethyl acetate. The organic layers are
combined, dried with Na2SO4 and evaporated to
dryness. The remaining crystals are filtered
off with petrol ether yielding 18.6 g of the
title compound, melting point 125-127C.
C) a-(t-Butoxycarbonylamino)-~-cyclohexyl-~-
(methanesulfonyloxy)-N-methoxy-threo-propionamide
a- (t-Butoxycarbonyl ~m; no) -B-cyclohexyl-B
hydroxy-N-methoxy-threo-propionamide (18.3 g)
is dissolved with stirring in 100 ml of dry
pyridine. The solution is cooled with stirring
to 0 C and 9.3 g of methanesulfonyl chloride
is dropped in. After one hour at 0C an additional
3.3 g of methanesulfonyl chloride is added and
stirring is continued for one more hour. The
solution is poured into 300 ml of ice water,
1 33867~
-
GC155f
-196- --
2Q0 ml of ethyl acetate is added and the pH is
adjusted to 3 with dilute sulfuric acid. The
organic layer is separated, dried with Na2SO4
and the solvent is removed ln vacuo. The
- 5 remaining solid is collected with petrol ether
yielding 1~.0 g of the title compound, melting
point 150-152C.
D) [3S-trans]-3~ Bu-toxycarbonyl~minol-4-
cyclohexyl~ methoxy-'2-azetid'inone
~-(t-Butoxycarbonylamino)-~-cyclohexyl-B-
(methanesulfonyloxy)-N-methoxy-threo-propionamide
(18.7 g) is dissolved in 50Q ml of dry acetone.
Potassium carbonate (9.8 g) is added and the
suspension is heated to reflux temperature with
stirring for 5 hours. The insoluble inorganic
material is filtered off and the solvent removed
_ vacuo and the remaining oil is dissolved in
30 ml of ethyl acetate. Upon the addition of
petrol ether, the title compound precipitates
and is filtered off (12.9 g), melting point
110-112C.
E) ~3S-trans]-3-('t-Butoxycarbonylamino)-4-
cyclohexyl-2-azetidinone
~3S-trans]-3-(t-Butoxycarbonylamino)-4-
cyclohexyl-l-methoxy-2-azetidinone (1 g) is
added to 50 ml of liquid ammonia with stirring.
Sodium (0.154 g) is added in 5 to 6 portions
within 5 minutes. After this time an additional
_ GC155f
-197- 1 338670
amount of 0.025 g sodium is added and stirring
- is continued for 5 minutes. Ammonium chloride
(Q.89 ~ is added and t~ ammonia i5 removed.
The residue is extracted with warm ethyl acetate.
The organic extract is evaporated to dr~ness
and the remaining crystals of the title compound
are filtered with petrol et~er, yielding Q.5 g,
melting Point 13Q-132C.
F) ~3S-transl-3-(t-Bu'toXyc'ar~oh'yl'aminO'1-4-
cyclohexyl-2-oxo-1-azetidinesulfohic ac'id,
pyridine salt
~ 3S-trans]-3-(_-Butoxycarbonyl ~mi no~ -4-
cyclohexyl-2-azetidinone (5.3) is dissolved in
20 ml of methylene chloride and 80 ml of
dimethylformamide. After the addition of
60 mmole of pyridine-sulfur trioxide complex
the solution is stirred for ~ hours at room
temperature. Removal of the solvent in vacuo
yield 11.3 g of the title compound as an oil.
G) [3S-trans]-3-(t-Butoxycarbonyl-amino~-4-
cyclohexyl--2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt
L3s-trans]-3-(t-Butoxycarbonylamino)-4
cyclohexyl-2-oxo-1-azetidinesulfonic acid,
pyridine salt (11.3 g) is dissolved in 250 ml
of water. Tetrabutylammonium hydrogensulfate
(9.0 g) is added with stirring and the pH is
adjusted to 6.5 with lN potassium hydroxide.
The aqueous solution is extracted twice with
GC155f
-198- 1 338670
2Q0 ml portions of methylene c~loride. The
organic portions are dried with Na2SO4, filtered
and the solvent is distilled off, yielding 8 g
of the title c~.~o~l.d, melting point 135-138C.
H) ~3S-trans~-3-Amino-4-cyclohexyl-2-oxo~1-
azetidinesulfonic acid
13S-trans]-3-(t-Butoxycarbonylaminol-4-
cyclohexyl-2-oxo-1-azetidinesulfonic acid,
tetrabutylammonium salt (3.8 g) is stirred in
20 ml of formic acid for 3 hours, followed ~y
the addition of 2~ ml of methylene chloride.
The precipitated title compound (1.0 g) is
filtered off, melting point 217-219C.
Exam~le 176
[3S-~3(Z),4B]]-3-[[(2-Amino-4-thiazolyl)-
(methoxyimino)acetyl]amino]-4-cyclohexyl-2-
oxo-l-azetidinesulfonic acid, potassium salt
~3S-transJ-3-Amino-4-cyclohexyl-2-oxo-
l-azetidinesulfonic acid (0.25 g) is dissolved
in 30 ml of dry dimethylformamide and 0.12 g
of triethylamine with stirring. When a clear
solution is obtained, (Z)-2-amino-~-(methoxy-
imino)-4-thiazoleacetic acid (0.2 g), 0.16 g
of hydroxybenzotriazole and 0.42 g of dicyclo-
hexylcarbodiimide are added. Stirring is
continued for 48 hours at ambient temperature.
The precipitated urea is filtered off and the
solvent is removed in vacuo. The residue is
_ GC155f
--1 9 9--
1 338670
dissolved in 10 ml of acetone and Q.41 g of
potassium perfluoro~utanesulfonate is added.
After the addition of 50 ml of ether, the
title compound precipitates and is filtered.
Column chromatograph~ using HP-20 and water~
acetone (9:11 as eluent, yields ~.36 g of product,
melting point 200-2Q5C Cafter lyophilization~.
EXample' 177
[3S-[3~(Z)~,4B~-3-[I~2-Amino-4-thiazol~l~ I (l-
carboxy-l-methylethoxyJimino]acetyl~amino~-4-
cyclohexyl-2-oxo-1-a'zetidinesulfonic'acid,
dipotassium salt
A) [3S-~3~(Z),4~]]-3-~[(2-Amino-4-thiazolyl)[(l-
diphenylmethoxycarbonyl-l-methylethoxy)imino]-
acetyl]amino]-4-cyclohexyl-2-oxo-1-azetidinesulfonic
acid, potassium salt
[3S-trans]-3-Amino-4-cyclohexyl-2-oxo-1-
azetidinesulfonic acid (0.2 g; see example 175)
is dissolved in 30 ml of dimethylformamide and
0.09 g of triethylamine with stirring. Hydroxy-
benzotriazole (0.12 g), 0.30 g of (Z)-2-amino-~-
l(l-diphenylmethoxycarbonyl-l-methylethoxy)imino]-
4-thiazoleacetic acid and 0.33 g of dicyclohexyl-
carbodiimide are added and stirring at ambient
temperature is continued for 12 hours. The
precipitated urea is filtered off and the mother
liquor is evaporated to dryness. The remaining
oil is dissolved in 5 ml acetone, treated with
0.3 g of potassium perfluorobutanesulfonate and
- ' GC155f
-200-
1 338670
poured into lQ0 ml of ether wit~ stirring.
~3S-~3CZ~,4~]]-3-~(2-Amino-4-thiazolyl~-
~(l-diphenylmethoxycar~onyl-l-methylethoxy2-
imino]acetyl]amino]-4-cyclohexyl-2-oxo-1-azetidine-
sulfonic acid, potassium salt ~0.61 g~ precipitatesand is filtered off.
B) l3S-[3~(.Z~,4~-3-~C2-Amino-4-thiaZolyl)t(l-
carboxy-l-meth.yleth~xy~i~ino~acetylJamino~-4-
cyclohexyl-2-oxo-1-azetidine'sul'fo'nic''acid,
dipotassium salt
[3S-13a~Z),4B]]-3-[ I (2-Amino-4-thiazolyl)-
[(l-diphenylmethoxycar~onyl-l-methylethoxy~imino]-
acetyl]amino]-4-cyclohexyl-2-oxo-1-azetidinesulfonic
acid, potassium salt (0.61 g) is suspended in
6.ml of anisole, cooled to -15C and 5 ml of
trifluoroacetic acid is dropped in with stirring.
The temperature is maintained for one hour and
then lowered' to -30C. About 100 ml of dry
ether is added at such a rate, that the temperature
does not exceed -10C. The precipitated compound
is filtered off and chromatographed using HP-20
resin and water~acetone (9:lJ as eluent, yielding
0.3 g of the title compound, melting point
11-12QC, dec. (after lyophilization).
-- GCl55f
.. -201-
1 338670
Example 178
13S-~3~,4~]-4-Cyclohexyl-3-I[I I 13- ~ C'2-furanyl-
methylene~amino]-2-oxo-l-imidazolidinyl'~car~onyl~-
amino]phenylacetyl]amino]-2-oxo-l-azetidinesulfohic
acid, potassium salt
3S-trans~-3-Amino-4-cyclohexyl-2-o
azetidinesulfonic acid (O.l g; see example 175
is dissolved in a mîxture of 3~ ml of dry
dimethylformamide and 0.~5 g of triethylamine
with stirring. I 1 I ~C2-Furanylmethylene)amino]-
2-oxo-l-imidazolidinyl]carbonyl~amino~phenylacetic
acid (.Q.14 g), 0.06 g hydroxybenzotriazole
and 0.17 g of dicyclohexylcarbodiimide are added
and the solution is stirred for 5 days at room
temperature. The solvent is removed in vacuo,
the residue is dissolved in lO ml of acetone
and the precipitated urea is filtered off.
The mother liquor is agitated with 0.15 g of
potassium perfluorobutanesulfonate and diluted
with 50 ml of ether. The precipitate is
filtered, and chromatographed with HP-20 resin
using water/acetone (9:l) as eluent yielding
0.14 g of product, melting point 195-200C, dec.
(after lyophiliaztion).
GC155f
-202
1 338670
EXample 17~
. ~ 3S - 13 a. (R* 2 34.~ ] ] -4 ~ Q~ P~yl -3~ 3 -. ~4.-.e.thyl.-2., .3 -
dioxo-l-piperaz`inyl'~-1.'3'-dioxo-2-phenylpropyl]-
amino]--2-oxo--1-azet;dinesulfonic 'ac'id','potassium salt
~3S-trans]-3-Amino-4-cyclohexyl-2-oxo-1-
- azeti~inesulfonic acid (0.1 g; see example 175)
is dissol~ed in 30 ml of dimethylformamide and
O.5 g of triethylamLne wit~ stirring. (R~--
[t(4-ethyl-2~3-dioxo-l-piperazinyl~car~onyl]
amino]~enzeneacetic acid, Q.06 g of hydroxy-
benzotriazole and n. 17 g of dicy~lohexylcar~odiimide
are added, and the mixture is stirred for a~out
16 hours at room temperature. The solvent is
distilled off in vacuo and the remaining oil
is dissolved in 10 ml of acetone. The precipitated
urea is filtered off and the mother liquor is
agitated with 0.15 g of potassium perfluoro-
butanesulfonate and diluted with 50 ml of ether.
.The precipitate is filtered off and chromatographed
with HP-20 resin, using water/acetone (9:1) as
- eluent, yielding 0.15 g of the title compound,
melting point 17~180C (after lyophilization~.
EXample 180
(.trans,Z)-3-~(2-Amino-4-thiazolyl)(me-thoxyimino)-
acetyl]amino2-4-ethy1-2-oxo-1-azetidinesulf~nic
acid, potassium salt
Following the procedure of example 170,
part A, but substituting (Z)-2-amino-~-(methoxy-
imino)-4-thiazoleacetic acid for (z)-2-amino-~-
[(l-diphenylmethoxycarbonyl-l-methylethoxy)imino~-
4-thiazoleacetic acid, yields the title compound,
melting point 190C, dec.
-
- GC155f
-203-
1 338670
Example 181
(i)-(trans2-3-Amino-2-oxo-4-phenyl-l-azeti~;~e
sulfonic acid
S A~ (~)-(trans)-2-oxo-4-p~enyl-1-azet;~i~e LerL-
- butyldiphenylsilane
A solution of tert-~utylchlorodip~enylsilane
(20.56 gl in dimethylformamide C45 ml~ is cooled
to 0C under argon and treated with triethylamine
(10.4 ml2 and then (~-2-oxo-4-phenyl-l-azet;~ine.
After several h~urs at 0C, the resulting mixture
is treated with additional trieth~lamine (1 mll
and tert-butylchlorodiphenylsilane (2.11 g),
and allowed to stir for 65 hours at 5C. The
reaction mixture is poured into ice water (300 ml~
and extracted with 3:1 ether-ethyl acetate
(three 125 ml portions). The organic extracts
are washed with pH 4.5 phosphate buffer (three
50 ml portions), saturated sodium bicar~onate
solution (50 ml), water (two 50 ml portions)
saturated sodium chloride solution and dried
(Na2SO4). Filtration, and concentration
in vacuo yields a~solid which is washed with
hexane to give after drying (high vacuum)
15 g of the title compound as a solid.
~~ GC155f
-204- 1 3 3 8 6 7 0
B) (~ (transl-3-azido-2-oxo-4-phsnyl-1-azetidine-
tert-~u*yldiphenylsilane
A 50 ml flask equipped with stirring bar,
gas inlet, and septum is flame dried under argon
and charged with n-butyl lithium ~0.65 ml of a
1.6 N solution in heYAn~) whic~ is cooled to
-4QC and dissolved in tetrahydrofuran ~2 ml).
Diisopropylamine ~0.16 mll is added dropwise,
the resulting mixture is stirred for 30 minutes
and cooled to -78C. A solution of C~-(trans)-
2-oxo-4-phenyl-1-azetidine-tert-~utyldiphenylsilane
(40Q mg1 in tetrahydrofuran n.5 ml) is added
dropwise over a~out 5 minutes. After stirring
an additional 20 minutes the solution is treated
with p-toluenesulfonyl azide (204 mg) in
tetrahydrofuran (0.5 ml). The resulting mixture
is stirred 10 minutes at -78C and treated dropwise
with chlorotrimethylsilane (0.4 ml). After an
additional 10 minutes of stirring, the cooling
bath is removed and the reaction mixture is
stirred at ambient temperature for 2.5 hours.
Then, while cooling at 0C, ethyl acetate (20 ml)
is added followed by pH 4.5 phosphate buffer
(8 ml). The organic layer is washed with additional
buffer (two 8 ml portions), 5% sodium bicarbonate
solution (three 10 ml portions), 50% sodium
chloride solution (10 ml)l saturated sodium
chloride solution (10 ml) and dried (~a2SO4).
Filtration and concentration ln vacuo yields
500 mg of oil which is flash chromatographed
with 5% ethyl acetate-hexane, yielding the
title compound (253 m~).
GC155f
-205- ~ 338670
Cl L+~-(trans)~3-azi.do-2-oxo-4-phen~l-1-aze*idine
A solution of 17 g of crude C+~-(trans)-3-
- azido-2-oxo-4-ph.enyl-1-azetidine-tert butyl-
diphenylsilane is dissolved in methanol (240 ml)
and treated dropwise at ~C with concentrated
- HCl ~35 ml~. The cooling bath is removed, the
reaction stirred at ~m~ient temperature for
1 hour, and recooled to OC whereupon saturated
- sodium bicar~onate solution is added to neutrality.
The resulting mixture i5 extracted Wit~ et~l
acetate (one 300 ml portion and four lOQ ml
portions) and the organic extracts are washed
with 1:1 5% sodium ~icar~onate 50% sodium
chloride solution, saturated sodium chloride
lS solution, and dried (Na2SO4). Filtration and
concentration in vacuo yields;15 g of a heavy
oil which is chromatographed on 100 g of silica
gel with 20~ ethyl acetate-hexane, yielding
460 mg of the title compound.
D) (.+)-(trans)-3-azido-4-phenyl~ azetidine-
sulfonic acid, tetrabutylammonium salt
A solution of (+)-(transl-3-azido-2-oxo-
4-phenyl-1-azetidine (300 mg) in dimethyl-
formamide (3 ml) is cooled to 0C under argon
and treated dropwise with a complex of dimethyl-
formamide and sulfur trioxide (4.78 ml of a
0.5 M solution in dimethylformamide). The
cooling bath is removed, the reaction mixture
is stirred at ambient temperature for 2 hours
GC155f
-206- 1 3 3 8 6 7 0
and poured'into 80 ml of a.5 M monobasic potassium
phosphate (pH 5.5~. The 'solution is extracted
' with dichloromethane '(:discarded2 and 541 mg
of tetrabutylAm~non;um bisulfate is added. The
5 resulting mixture is extracted with dicElloromethane
and the organic extracts are washed with 10% sodium
chloride solution and dried CNa2SO41. Filtration
and concentration in vacuo affords 8Q0 mg of
oil; approximately 4Q% the desired product,
10 the remainder dimethylformamide. This mixture
is used without purification in the next step.
E) (~)-(trans)-3--amino-4-phenyl-1-azetidine-
sulfonic acid
A solution of (+)-(trahs)-3-amino-4-phenyl-
l-azetidinesulfonic acid, tetrabutylammonium salt
in 4 ml of methanol is hydrogenated over 30 mg
of platinum oxide at 1 atmosphere and room
temperature. After 15 minutes, the system is
20 evacuated and fresh hydrogen is introduced.
After an additional 45 minutes the reaction
is complete, and the system is flashed with
nitrogen. After several days at room temperature
in dichloromethane-methanol (4:1, 200 ml)
25 catalyst aggregation is complete and filtration
is accomplished. The filtrate is concentrated
_ vacuo to 18 ml and 0.2 ml of 97~ formic
acid is added. /After cooling at 5C for several
hours the resulting solid is filtered and
30 washed with dichloromethane to afford, after
drying, 150 mg of the title compound as a solid.
Analysis calctd for CgHlgN2O4S: C, 44.62; H, 4.17;
N, 11.57; S, 13.23
Found: C, 43.36; H, 4.31; N, 11.09; S, 13.02
_ ' GC155f
-207-
1 338670
Example 182
~ (trans)-2-~xo-4-phenyl-3-E(:phenylacetyl)amino~-
l-azetidihesulfonic' acid, potassium salt
Al (+)-trans-2-ox~-4-p~.enyl-3-I(phenylacetyl~amino]-
~aze~i~inesulfonic acid tetra~ut~lammonium salt
A solution of N-hydroxy~enzotriazole mono-
hydrate (52 mg) and phenylacetic acid C46 mg)
in dimethylformamide (0.3 ml) is treated with
solid dicyclohexylcar~odiimide (~7Q mg~ under
argon at 0C. T~e cooling bath. is removed and
the resulting mixture stirred at ambient temperature
for 1 hour. After dilution~with additional dimethyl-
formamide (0.3 ml), (+)-(trans)-3-amino-2-oxo-4-
phenyl-l-azetidinesulfonic acid is added as a
solid (75 mg, see example 181) followed by
triethylamine (0.05 ml) dropwise. The reaction
is stirred at ambient temperature for 23 hours,
filtered and the filter cake washed with dimethyl-
formamide. The filtrate was added to 20 ml0.5 monobasic potassium phosphate (pH 4.5),
the mixture washed with three 8 ml portions of
ethyl acetate (discard), and 105 mg of tetrabutyl-
ammonium bisulfate (0.31 mmole) is added. The
resulting mixture is extracted with dichloromethane
(three 15 ml portions). The extracts are washed
with 10% sodium chloride solution (two 15 ml portions),
saturated sodium chloride solution (10 ml) and
dried (Na2SO4). Filtration and concentration
in vacuo yields (after heating at 32C under high
vacuum) 165 mg of oil; approximately 40~ is the
desired compound, and 60% dimethylformamide.
GC155f
-208- 1 3 3 8 6 7 0
B2 (+)- -trans-2-~xo-4-p~enyl-3-I (~11~ nylacetyl~
amino~-l-azetidinesulfohic acid', p'otassium'salt
A solution of ~)-*rans-2-oxo-4-phenyl-3-
~(phenylacetyl~amino]-l-azetidinesulfonic acid
tetrabutylammonium salt in 1.5 ml acetone is
treated with 41 mg (a.121 mmole~ of potassium
perfluorobutanesulfonate.~ Dilution with 12 ml
of ether affords a glass, whic~ upon trituration
with ether affords 43 mg of solid which contains
about 20% of an Lmpurity contAining a tetrabutyl-
ammonium moiety. The solid is dissol~ed in
5Q% aqueous acetone and passed through a
Dowex 50W-X2 ~ ion-~h~nge resin (1 ml).
Removal of solvent yields a solid whïch is
washed with acetone, hexane and dried (60C,
high vacuum). The yield of the title compound
is ]5 mg.
Analysis calc'd for C17H15N2O5S K: C, 51.23; H, 3.80;
N, 7.03; S, 8.05; K, 9.81
Found: C, 50.44; H, 4.20; N, 7.01; S, 7.59;
K, 9.40
Example 183
(i)-(trans,Z)-3-~12-Amino-4-thiazolyl)(methoxyimino)-
acetyl]amino]-2-oxo-4-phenyl-1-azetidinesulfonic
acid, potassium salt
A solution of N-hydroxybenzotriazole hydrate
(52 mg) and (Z)-2-amino-~-(methoxyimino)-4-
thiazoleacetic acid (69 mg) in dimethylformamide
(Q.3 ml) is treated with solid dicyclohexyl-
carbodiimide (7Q mg) under argon, at ambient
GC155f
-209- 1 3 3 8 6 7 0
temperature. The resulting mixture is stirred
for 1 hour, ~ (trans)-3-amino-2-oxo-4-phenyl-
l-azetidinesulfonic acid (75 mg; see example 1812
is added as a solid, followed by triethyl~mi ne
dropwise (0.05 ml~. The reaction is stirred at
ambient temperature for 23 hour5 . The dimethyl-
formamide is removed under high vacuum at 30C,
and the residue triturated wlt~ 2 ml of acetone
and filtered. The filter cake is was~ed with
additional acetone (two 3 ml portions~ and potassium
perfluorobutanesulfonate (86 mg) is added to the
filtrate. Dilution with 10 ml of ether produces
a gummy solid which is triturated, washed with
acetone and hexane to yield, after drying, 82 mg
of the title compound as a solid.
Analysis for C15H14N5O6S2
Calc'd: C, 40.26; H, 3.16; N, 15.65; S, 14.33;
K, 8.74
Found: C, 38.60; H, 3.19; N, 15.07; S, 13.87;
K, 7.5
Example 184
(cis)-2-Oxo-4-phenyl-3-l(phenylacetyl)amino]-1-
azetidinesulfonic acid, potassium salt
A) N-Benzylidene-2,4-dimethoxybenzylamine
12.0 g of 2,4-dimethoxybenzylamine hydrochloride
is added to 100 ml of lN sodium hydroxide solution
and the mixture is extracted with 125 ml of ethyl
acetate. The organic layer is dried over anhydrous
sodium sulfate and stripped of solvent to give
GCl55f
-
-210- 1 3 3 8 6 7 0
10.2 g of 2~4-dimethDxybenzy~ `n~ as an oil.
~his amine is dissolved in 150 ml of ~enzene;
6.47 g of benzaldehyde and 0.6 g of ~-toluene-
sulfonic acid monohydrate are added. The mixture
is heated under reflux removing water wit~ a
Dean-Stark 5eparator and in two hours the
calculated amount of water Cl .1 ml~ separates
out. The mixture is cooled to ro~m temperature.
Upon further cooling thB ~enzene solutîon
deposits some precipitate. Benzene is lemoved
under reduced pressure and 60 ml of petroleum
ether is added to the residue. An oily- layer
separates out with more precipitate. Benzene
(lO ml) is added to make the layers homogeneous
and the remaining precipitate is filtered. The
filtrate is stripped of solvent to give 14.2 g
of the title compound as an oil.
B) (+)-(cis)-4-Phenyl-1-(2,4-dimethoxybenzyl)-
2-oxo-3-azidoazetidine
~ -Azidoacetic acid (1.62 g) is dissolved
in 25 ml of methylene chloride under nitrogen.
To this solution are added 3.24 g of triethyl-
amine and 1.02 g (4.0 mmole) of the imine
N-benzylidene-2,4-dimethoxybenzylamine dissolved
in 10 ml of methylene chloride. The mixture
is cooled in an ice-bath and 3.36 g o~ trifluoro-
acetic anhydride is added slowly; the solution
becomes dark colored. After stirring for l hour
- 30 in an ice-bath, the mixture is warmed to room
temperature and stirred an additional 15 minutes.
- GC155f
-211-
1 338670
The solution is then was~ed with ~ater ~60 ml),
5% NaHCO3 solution Ctwo 50 ml portionsl, and lN
HCl solution ~60 ml~. The organic layer is dried
over anhydrous sodium ~ulfate and stripped of
solvent to give 1.72 g of crude product as dark
gum. The gum i5 treated with charcoal several
times and the resulting ~rown mixture is chromato-
graphed on 40 g silica gel eluting with 1:1
petroleum ether:ethyl acetate. T~e com6ined
fractions yield crystal upon quick-freezing
in a dry ice-acetone ~ath. Using this as a
seed the product i5 recrystallized'from petroleum
ether-ethyl acetate to give 817 mg of the title
compound as needles which melt upon warming to
room temperature.
C) (+7-(cis)-4-Phenyl-2-oxo-3-azidoazetidine
(+)-(cls)-4-Phenyl-1-(2,4-dimethoxybenzyl)-
2-oxo-3-azidoazetidine (737 mg) is dissolved in
25 ml of acetonitrile and heated to 80 -83C
under nitrogen. To the resulting solution are
added over a 1 hour period 943 mg of potassium
persulfate,and 570 mg of potassium monohydrogen
phosphate, both dissolved in 25 ml of water.
After the addition, the mixture is further heated
at 80-83C for 7 hours. The mixutre is cooled
and the pH is adjusted to 6-7 by adding solid
potassium monohydrogen phosphate. Most of the
acetonitrile is removed under reduced pressure
and the resulting mixture is extracted with
GC155f
1 338670
~212~
60 ml of chloroform. The chlorofor.m layer is
washed with water (60 mll, dried over anhydrous
sodium sulfate and stripped of solvent to gi~e a
crude product as an oil. Th.e crude product is
chromatogrpahed on 40 g of silica gel eluting
with 1:1 petroleum ether-ethyl acetate. The
combined fractions y-ield crystals and the
product is recrystallized from petroleum ether-
ethyl acetate to give 267 mg of t~e title compound.
D) (+)-(cis~-4-Pheny1--2-ox~-3-azido l-azetid-ine-
sulfonic acid, tetra~utyl ~ onium salt
(+)-(cis)-4-Phenyl-2-oxo-3-azidoazetidine
(162 mg) is cooled to 0C under nitrogen and
3.5 ml of caD . SM dimethylformamide-sulfur trioxide
complex solution in dimethylformamide is added
dropwise via syringe. The resulting clear
solution is stirred at 0C for 15 minutes. The
mixture is then poured into 50 ml of 0.5M
monobasic potassium phosphate solution and
washed with methylene chloride (three 50 ml)portions).
tetra-n-Butylammonium bisulfate (292 mg) is
added to the aqueous solution and the mixture is
extracted with methylene chloride (six 5Q ml
portions). The combined methylene chloride
layers are dried over anhydrous sodium sulfate
and stripped of solvent to give 272 mg of the
title compound as a gum.
_ GC155f
-213-
1 338670
E) (~-(cis)-2-oxo-4-phenyl-3- I Cphenylacetyl)ami-no~-
l-azetidinesulfonic acid, potassium ~alt
C~ - (Ci5) -4-Phenyl-2-oxo-3-azido-1-azetidine-
sulfonic acid, tetra~utyl ammonium salt ~2~3 mg)
is disaolved in 4 ml of ethanol and hydrogenated
with 8Q mg of platinum oxide catalyst at one
abmosphere. After 1 ~our stirring t~P catalyst
iS filtered through a Millipore filter with
Celite; some catalyst particles pass throug~ the
1 filter to give a ~lack filtrate. Et~anol is
removed under reduced pressure and the residue
is dissolved in 4 ml of dLmethylformamide.
N-Hydroxybenzotriazole -m~nohydrate (81 mg~,
78 mg of phenylacetic acid, and 117 mg of
dicyclohexylcarbodiimide are added and the
mixture is stirred for about 16 hours under
nitrogen. The slurry is evaporated in vacuo
and triturated with 10 ml of acetone. The
resulting slurry is filtered through a Millipore
filter with a Celite top and the brown filtrate
is treated with 193 mg of potassium perfluoro-
butane sulfonate. Upon adding 20 ml of ether
a gum separates out. Liquid is removed and the
gum is washed with ether. The gum is dissolved
in 10 ml of methanol. Upon adding ether, a
small amount of precipitate is formed. The
mixture is filtered and the colored filtrate
is treated with more ether. The precipitate
formed is filtered and recrystallized twice
from ether-methanol to give 26 mg of the title
compound.
Analysis calc'd for C17H1505N2 2
C, 46.99; H, 4.41; N, 6.45
Found: C, 47.24; H, 4.19; N, 6.34
GC155f
-214- 1 3 3 8 6 7 0
Example 185
(cis,ZI-3-~I-(-2-Amino-4-t~iazolyl) ~methoxyimino)-
acetyl] amino] -2-oxo-4-phenyl-1-a'zetidinesul'fohic
acid, potassium salt
(cis~-2-Oxo-4-phenyl-3-I(phenylacetyllamino]-
l-azetidinesulfonic acid, potassium salt ~560 mg;
see example 184, part DI is d;ssol~ted in 5 ml of
etlianol and hydrogenated wi~h llQ mg of platinum
oxide catalyst a~ one atmosphere. After one
hour stirring the catalyst is filtered through a
Millipore filter with Celite; catal~st particles
pass through the filter to give a black filtrate.
Ethanol is removed under reduced pressure and
the residue is dissolved in 4 ml of dimethyl-
formamide. N-Hydroxybenzotriazole monohydrate
(168 mg), 221 mg of the (Z)-2-amino-a-(methoxyimino)-
4-thiazoleacetic acid and 227 mg of dicyclohexyl-
carbodiimide are added and the mixture is stirred
for about 16 hours under nitrogen. The slurry
is evaporated in vacuo and triturated with 15 ml
of acetone. The resulting slurry is filtered
through a Millipore with Celite and the filtrate
is treated with 372 mg of potassium perfluoro-
butane sulfonate. Upon adding 15 ml of ether a
gum separates out. Liquid is removed and the
gum is washed with ether. The gum is dissolved
in 5 ml of water and applied on 150 ml of HP-20
resin eluting with water. Fractions (30 ml each)
16-34 are combined and lyophilized to give 201 mg
30 of the title compound as a solid.
Analysis calc'd for C15H14O6N5S2 / 2
C, 36.73; H, 3.49; N, 14.28; S, 13.07;
K, 7.97
Found: C, 36.65; H, 3.00; N, 13.~9; S, 13.48;
K, 8.30
, GC155f
-215-
1 338570
Example '186
(cis)-3-Amino-2--oxo-4-(2-phen~letfienyll-1-
azetidinesulfonic acid
A~ N-(,3-phenyl-2-prope-nylidene~'-4-me'thoxyaniline
~ -Anisidine a2.32 g) is dissolved in 160 ml
of methylene c~loride and 2Q g of anhydrous
magnesium sulfate is- added. The mixture is
cooled in an ice bat~ and 13.22 g of' trans-
cinnamaldehyde is added. The mixture is stirredunder nitrogen for 2 hours and then filtered.
The filtrate is e~aporated to give a solid.
- The crude product is recrystallized from met~ylene
chloride-petroleum ether to give 20.96 y of the
title compound as a solid.
B) (+)-~cls)-3-Azido-1-(4-Methoxyphenyl)-2-oxo-
4-(2-phenylethenyl)azetidine
2-Azi~oacetic acid (24.26 g) is dissolved
20 ih 100 ml of methylene chloride and cooled in an
ice bath. To this solution is added 48.57 g
of triethylamine and 14.24 g of N-(3-phenyl-2-
propenylidene)-4-methoxyaniline dissolved in
250 ml of methylene chloride. To the resulting
solution is added 50.41 g of trifluoroacetic
anhydride dropwise over a one hour period. After
stirring for one hour in an ice bath, tne mixture
is warmed to room temperature and stirred for
about 16 hours. The mixture is then diluted
with 250 ml of methylene chloride and washed
_ GC155f
-216- 1 3 3 8 6 7 0
with water (750 mll, 5% sodium bicar~onate
solution (two 75Q ml portions), and lN HCl
solution (75Q ml~. The organic layer is dried
over anhydrous sodium sulfate and stripped of
solvent to give a solid. The crude product is
recrystallized from ethyl acetate to give 11.39 g
of the title compound a5 a solia.
C~ (+)--(C1S~-3 - AZidO-2 OAO ~-~2-p~enylet~en~l)
azetidine
To a solution of 10.22 g of ceric ammonium
nitrate in 13 ml of water at 0C is added 1.99 g
of (+)-(cis~-3-azido-1-(4-methoxyphenyl)-2-oxo-
4-(2-phenylethenyl)azetidine is dissolved in
65 ml of acetonitrile during a 15 minute period
(additional 10 ml of acetonitrile is used for
rinse). The mixture is stirred for an additional
15 minutes at 0 C, diluted with 750 ml of ethyl
acetate, washed with water (six 600 ml portions),
dried over anhydrous sodium sulfate, and stripped
of solvent to give an oil. The crude product
is chromatographed on 90 g of silica gel, eluting
first with 250 ml of 30% ethyl acetate/petroleum
ether, then 50% ethyl acetate/petroleum ether.
Fractions (50 ml each) 11-16 are combined and
evaporated to give 802 mg of the title compound
as an oil.
GCl55f
-217- ~ 338670
D) (+)-(cis)-3-Azido-2 o~o q-~2-phenylet~enyl)-l-
azetidinesulfonic ac:id, tetra-n ~.uLylammonium salt
(~)-(cis~-3-Azido-2-oxo-4-(2-phenylethenyl)-
azetidine (334 mgl is dissolved in 3 ml of
dimethylformamide and 868 mg of pyridine-sulfur
trioxide is added. The mixture is stirred at
room temperature for 40 hours under nitrogen and
then poured into 20~ ml of n.5 M monobasic potassium
phosphate solution and was~ed wit~ 30 ml of
methylene chloride. tetra-n-Butyl ammonium
bisulfate (530 mg~ is added to the aqueous
solution and the mixture is extracted with
methylene chloride (four 50 ml portions). The
combined organic layers are back-washed with water
(two lO0 ml portions), dried over anhydrous
magnesium sulfate and stripped of solvent to
give 824 ma of the title c~mpound as a gum.
E) (+)-(cis)-3-Azido-2-oxo-4-(2-phenylethenyl)-1-
azetidinesulfonic acid
(+)-(cis)-3-Azido-2-oxo-4-(2-phenylethenyl)-
l-azetidinesulfonic acid, tetra-n-butylammonium salt
(300 mg) is dissolved in 4 ml of tetrahydrofuran
and stirred rapidly. To the mixture is added
600 mg of zinc dust followed by 0.8 ml of lN
monobasic potassium phosphate solution. The
mixture is heated to 45C and stirred at this
temperature for 3 hours. The mixture is then
~ filtered and the filtrate is taken in 4n ml of
methylene chloride and 10 ml of water. The
_ GC155f
1 33867~
-218- -
aqueous layer is further extracted with methylene
chloride (three 40 ml portions2 and t~e com~ined
methylene chloride layers are stripped of solvent
to give 256 mg of a foam. This crude product
is dissolved in a small amount of ca. 3~%
acetone~water and applied on 7.5 ml of Dowex CR 2
resin (0.7 meq./vnl~ eluting wlt~ 4a ml of
water. The eluent is evaporated to give 151 mg
of foam, which is dissolved in 2 ml of water
and acîdified to p~ 2 with lN HCl solution.
A small amount of acetonitrile is added to
dissolve the precïpitate and the resulting solution
is applied on 15 ml of HP-20 resin, eluting with
150 ml of water, then 10% acetone/water. Fractions
(15 ml each) 2-13 are combined and evaporated
to give 101 mg of the title compound as a foam.
Example 187
(+)-tcis,Z)-3-[[(2-Amino-4-thiazolyl)(methoxyimino)-
acetyl]amino]-2-oxo-4-(2-phenylethenyl)-1-azetidine-
sulfonic acid, potassium salt
A solution of 68 mg of (Z)-2-amino-~-
(methoxyimino)-4-thiazoleacetic acid and 51 mg
of N-hydroxybenzotriazole monohydrate in 2 ml
of dimethylformamide is treated with 69 mg of
dicyclohexylcarbodiimide. The mixture is stirred
at room temperature for 30 minutes under nitrogen.
(cls)-3-Amino-2-oxo-4-(2-phenylethenyl)-1-
azetidinesulfonic acid (90 mg; see example 186)
and 34 mg of triethylamine are added and the
mixture is stirred for 20 hours under nitrogen.
GC155f
-219- 1 3 3 8 6 7 0
The slurry is evaporated in' vac~o and triturated
with lQ ml of acetone. TEle slurry îs filtered
and the filtrate is; treated w~th 113 mg of
potassium perfluorobutanesulfonate. Dilution with
5 30 ml of ether and filtration gives 169 mg of
a solid product, which is dissolved in a small
amount of ca. 109~ acetonitrile/water and applied
on 34 ml of ~P-20 resin, eluting with 15Q ml
of water, thsn lQ~ acetone~water. Fractions
(15 ml each) 16-1~ are cam~iined and stripped of
solvent to gi~re llQ mg of the title cG...~ound
as a solid.
Analysis calc'd for C17H16O6N5S2K H2O: C, 40.23;
H, 3.57; N, 13.80; S, 12.63; K, 7.70
Found: C, 40.03; H, 3.05; N, 13.61;
S, 12.31; K, 7.56
Example 188
(cis)-3-Amino-4- (methoxycarbonyl)-2-oxo-1-
azetidinesulfonic acid
A) ~(4-Methoxyphenyl)imino]acetic acid, me'hyl ester
A dry 3-necked, 1 liter flask equipped with a
nitrogen inlet and stirring bar is charged with
56.88 g of MgSO4 followed by a solution of
recrystallized anisidine ('19.43 g) in dichloromethane
(250 ml). After cooling to 0 C a solution of
methyl glyoxylate hemiacetal (19.92 g) in
dichloromethane (250 ml) is added over 1.5 hours.
30 After stirring an additional 20 minutes at 0 C,
GC155f
,
-220
- 1 338670
the reaction mixture is suction filtered, dried
over sodium sulfate, filtered and concentrated
in vacuo to one-quarter volume. ~Y~ne C30~ ml)
is added, and t~e solution is concentrated to an
oil which semi-solidifies on stAn~ing under high
vacuu~ at 5C.
B) (cisl-3-(-1,3-~ihydro-1,3-dioxo-2H-isoind~l-
2-yl)-4-methoxycar~onyl-2-oxo-1-t:4-methoxyph.enyl)
azetidine
A dry 3-necked 500 ml flask equipped with
stirring bar, addition funnel, septum and nitrogen
inlet is charged with a solution of [(4-Metho~y-
phenyl)imino]acetic acid, methyl ester (21.09 g)
in dichloromethane (150 ml) and cooled to 0 C.
Triethylamine (19.2 ml) 0.14 mole is added
dropwise followed by a solution of (N-phthalimido)-
acetyl acid chloride (28.4 g) in dichloromethane
(150 ml) over 1 hour. The resulting mixture is
stirred for 1.5 hours at 0C, and diluted with
2.5 1 of dichloromethane. The organic solution
is washed with pH 4.5 monobasic potassium phosphate
(two 500 ml portions), 5~ sodium bicarbonate
(two 500 ml portions), saturated sodium chloride
solution (500 ml), and dried over sodium sulfate.
Filtration and concentration in vacuo yields
a solid which is washed with ethyl acetate,
cold acetone and hexane to yield 18.65 g of
product.
-. GC155f
-221- 1 3 3 8 6 7 0
C) (cisl-4-~Methoxycarbon~1)~ 4-methoxyphenyl)-
2-oxo-3-~l(pheny-lmethoxylcarbonyl~amino~azetidine
A dry 50Q ml flask equipped with. nitrogen
inlet, stirring bar, and septum was charged with
18.65 g of (.cis1-3-(1,3-dioxo-2H-isoindol-2-yl)-
4-methoxycar~onyl-2-oxo-1-~4-methoxyphenyl)azet;~;ne
and 325 ml of dichloromethane. Th.e res~lting
sl-cpension i8 cooled to -3aC, and metAyl
hydrazine C3.52 mlI is added dropwise. The
reaction is warmed to ac and stirred for l hour.
An additional Q.4 ml of methy-l h~ydrazine is added
and the mixture is stirred for 10. minutes. Th.is
sequence is repeated with a total of 2.9 equivalents
of methyl hydrazine (7.7 ml) has been added.
The solvent was removed in vacuo; 200 ml of
fresh dichloromethane is added, and the mixture
is again concentrated. This sequence was
repeated t~o additional times, the resulting foam
. was dried under high volume for 20 minutes,
redissolved in 225 ml dichloromethane, and allowed
to stand at ambient temperature for about 16 hours
during which time a considerable amount of solid
precipitates. The mixture is filtered under
nitrogen, the filtrate cooled to 0C (nitrogen
atmosphere) and treated with diisopropylethyl
amine (17 ml) followed by benzyl chloroformate
- (7 ml) dropwise. The reaction is stirred at
0C for 30 minutes, then at ambient temperature
for 1.5 hours. The mixture is washed with two
300 ml portions of pH 4.5 monobasic potassium
phosphate buffer, 5% sodium bicarbonate (two 300 ml
portions), saturated sodium chloride (300 ml),
~ GC155f
-222- 1 3 3 8 6 7 0
dried (sodium sulfatel, and filtered. Concentration
in ~acuo, yields a foam which on tr;turation with
ether yields 2.~ g of the title compound as a solid.
D) (cis)-4-(IIe~lG~carbonyl)-2-oxo-3~ henyl-
methoxy)carb~nyl]amino~ azetidine
A solution of ceric ~wnium nitrate
(8.59 g) in 6Q ml of 1:1 acetonitrile-water
is treated with a slurry of 2 g (cls~-4-
(methoxycarbonyll-1-C4-methoxyphenyll-2-oxo-3-
I[(Phenylmethoxy2car~onyl~amino]azetidine in
50 ml acetonitrile over 10 minutes. The reaction
mixture is stirred an additional 10 minutes
at ambient temperature and diluted with ethyl
acetate (100 ml). The separated aqueous layer
is washed with ethyl acetate (three 40 ml portions)
and the combined organic extracts are washed with
50% sodium bicarbonate (three 70 ml portions).
The basic washings are back-washed with ethyl
acetate (50 ml) and the combined organic extracts
are washed with aqueous sodium sulfite, 5%
aqueous sodium car~onate (100 ml), 5% sodium
chloride solution (two 100 ml portions), saturated
sodium chloride (two 50 ml portions), and stirred
over Darco G-60 charcoal for 30 minutes. Sodium
sulfate is added, and the mixture is filtered
and concentrated ln vacuo to yield an oil which
on trituration with ether yields 685 mg of the
title compound as a solid.
GC155f
---223-
1 338670
E) (cis)-4-(Met-hoxyca~ yl~ -2-oXD-3-~ (phenyl-
methoxy)car~onyl]amino~ azetidinesulfonic acid,
tetrabutylammonium salt
A mixture of (cis)-4-(methoxycarbonyl)-2-
oxo-3-l~(phenylmethoxy~carbonyl]amino]-1-azetidine
(100 mg) and 172 mg of a pyridine-sulfur trioxide
complex in 1 ml of pyridine is stirred under
argon for 3 hours at 80C. The reaction mixture
is poured into 7~ ml of Q.5 M monobasic potassium
phosphate (pH 5.51 and extracted wit~ four 30 ml
portions of dichloromethane (discard~. Tetrabutyl-
ammonium hydrogen sulfate ~122 mg) is added to
the aqueous layer which is then extracted with
dichloromethane (four 30 ml portions). The organic
lS extracts are washed with 8% sodium chloride solution,
dried over sodium sulfate and filtered. Concentration
in vacuo yields 186 mg of the title compound as
a viscous oil.
F) (cis)-3-Amino-4-~methoxycarbonyl)-2-oxo-1-
azetidinesulfonic acid
A solution of 186 mg of (cis)-4-(methoxy-
carbonyl)-2-oxo-3- I ~ (:phenylmethoxy~carbonyl]amino]-
l-azetidinesulfonic acid, tetrabutylammonium salt
in 2 ml of methanol is hydrogenated over 10%
palladium on charcoal (95 mg) for 1.5 hours at
1 atmosphere. The catalyst is filtered off and
rinsed with dichloromethane-and the filtrate is
treated with 97% formic acid and cooled to
-50 C (the presence of a seed crystal at this
staqe is necessary to induce crystallization).
After crystallization commences, the mixture is
allowed to stand for about 16 hours at 10C.
- GC155f
-224- 1 3 3 ~ 6 7 0
The resulting solid is ~ashed with dichloromethane,
hexane, and dried in wacuo, ~ielding 50 mg of
the title c~ d.
Example -182
(cis)-3-~2-Amino-4-thiazol~ (diphenyl-
methoxycarbo-nyl)-l-methylethoxyJimino]acetyl]-
amino]-4-Cmethoxyca-r~onyl)-2-oxo-1-azetidinesulfonic
acid, potassium salt
A solution of N-hyd~o~y~enzotriazole hydrate
(34 mg) and lQl mg of 2-amino-a-IIl-(dip~enyl-
methoxycar~onyll-l-methylethoxy~imino~-4-
thiazoleacetic acid in 0.5 ml of dimethylform~m;~e
is treated with solid dicyclohe~ylcarbodiimide
(45 mg) and the mixture is stirred under argon
for 45 minutes (ambient temperature). (cls)-3-
Amino-4-(methoxycarbonyl)-2-oxo-1-azetidinesulfonic
acid (45 mg; see example 188) is then added as
a solid followed by triethylamine (0.03 ml)
dropwise. The reaction is stirred at ambient
temperature for about 16 hours. The dimethyl-
formamide is removed under high vacuum at 30C
and the residue is triturated with acetone.
The supernatant is treated with potassium
perfluorobutanesulfonate (67 mg). Dilution
with ether produces a solid which is washed
with ether and dried in vacuo to yield 93 mg
of the title compound.
GC155f
-225- 13386 a
Example l~Q
(cis ) -3- I-I (2-P~nino-4-thi,azo-lyl ~ c~ r~y-l-
methylethoxy)imino]acetyl]amino]-4-~methoxy-
carbonyl)-2-oxo-1-azetidinesulfonic acid,
dipotassium salt
A slurry of (cis~-3-~12-Amino-4-thiazolyl)-
~ (diphenylmethoxycar~onyl~-l-methylethoxy)-
- imino~acetyl]amino~-4-~methoxycar~onyl~-2-oxo-1-
azetidinesulfonic acid, potassium salt in n . 4 ml
of anisole is stirred at -12C under argon, and
0.9 ml of cold (-10C) trifluoroacetic acid is
added. After 1.5 hours, 4 ml of ether and 2 ml
of hexane are added and the resulting slurry
is stirred for 15 minutes at -10C, then 15
minutes at ambient temperature. The solid is
isolated by centrifugation and washed with
ether. The pH of a suspension of this material
in O.S ml of cold water is adjusted to 6 with
lN potassium hydroxide and then applied to a
30 ml HP-20AG column. Elution with water yields
30 mg of the title compound after evaporation
(acetonitrile added and evaporated twice).
AnalysiS calc'd for C14H15K2N5O9S2 C~ 31-15;
H, 2.81; N, 12.98
Found: C, 29.08; H, 3.03; N, 12.19
f
-226-
1 338670
Example 191
(S)-(trans)-3-Amino-4-ethynyl-2-oxo-1-azetidine-
sulfonic acid
A) 2-(Trimethylsilyl)ethynylmagnesium bromide
To a flame-dried 50 ml flask maintained
under positive nitrogen pressure is added 20 ml
of dry tetrahydrofuran, 2.20 ml of trimethylsilyl
acetylene and 5.05 ml of 3.06 M solution of
methylmagnesium bromide in ether. The mixture
is stirred for 140 minutes yielding the title
compound.
B) (S)-(trans)-4-[2-(trimethylsilyl)ethynyl]-2-
oxo-3-[(triphenylmethyl)amino]azetidine
To a flame dried 250 ml 3-necked flask
is added 6.00 g of (S)-(cls)-4-(methylsulfonyl)-
2-oxo-3-[(triphenylmethyl)amino]azetidine. The
flask is flushed with nitrogen, and then
maintained under positive nitrogen pressure.
After the reaction mixture is cooled in a dry
ice/isopropanol bath, 4.65 ml of a 3.06 M
solution of methylmagnesium bromide in ether
is added dropwise via syringe with rapid
stirring. The solution of 2-(trimethylsilyl)-
ethynylmagnesium bromide prepared in part A
is added via a Teflon tube under positive
nitrogen pressure (the flask containing the
reactant is rinsed with 7 ml of tetrahydrofuran).
When the addition is complete the-cold bath is
removed. After 45 minutes, a solution of 3.5 g
-
- ~-C1~5f
-226a-
1 338670
.
of potassium bisulfate in 20 ml of water is
added. Most of the tetrahydrofuran is removed
on the rotary evaporator. The residue is
transferred to a separatory funnel with ether
and water. The water layer is separated and
extracted twice with ether. The combined
ether layers are washed once with saturated
aqueous sodium chloride, dried over sodium
sulfate, and filtered. Removal of the solvent
gives a foam which is chromatographed on a
silica column. Elution with 2 liters of
dichloromethane, 1 liter of 1% ether/dichloromethane,
2 liters of 2% ether/dichloromethane and 1.5
liters of 10% ether/dichloromethane (fraction
1=1000 ml; fraction 2,3=500 ml; fraction
4-end=250 ml) gives 1.30 g of the title compound
in fractions 2-8 and 1.80 g of the corresponding
trans-isomer in fractions 12-19. Fractions 9-11
contain 1.19 g of a mixture of cls and trans isomers.
C) (S)-(trans)-4-ethynyl-2-oxo-3-[(triphenyl-
meth~yl)amino]azetidine
(S)-(trans)-4-[2-(trimethylsilyl)ethynyl]-
2-oxo-3-[(triphenylmethyl)amino]azetidine (2.97 g)
is dissolved in 30 ml of dichloromethane and
330 mg of tetrabutylammonium fluoride (containing
20-25% water) is added. After 20 minutes, the
solvent is removed _ vacuo. The residue is
taken up in ethyl acetate and water. The organic
layer is separated, washed once with water and
once with saturated aqueous sodium chloride,
r l _ 5 f
-226b-
1 338670
dried over sodium sulfate, and filtered.
Removal of the solvent gives an oil which is
stirred for 15 minutes with 60 ml of pentane
to afford 2.35 g of the title compound as a
powder (after drying in vacuo).
D) (S)-(trans)-3-Amino-4-ethynyl-2-oxo-1-
azetidinesulfonic acid
(S)-(trans)-4-ethynyl-2-oxo-3-[(triphenyl-
methyl)amino]azetidine (404 mg) and 560 mg of
a complex of pyridine and sulfur trioxide
are added to a 25 ml flask. After the flask
is flushed with nitrogen, 4.0 ml of dry pyridine
is added and the mixture is heated at 80-85C
for 3 hours. The mixture is added to a rapidly
stirred mixture of 4.0 ml of concentrated
hydrochloric acid, 50 ml of water, and 50 ml
of ethyl acetate. The pH is adjusted to 3.15
with sodium carbonate. The water layer is
separated and extracted once with ethyl acetate.
The combined organic layer is washed once with
saturated aqueous sodium chloride, dried over
sodium sulfate and filtered. Solvent removal
ln vacuo gives a foam which is taken up in
10 ml of dichloromethane. Formic acid (98%, 8 ml)
is added, and after 15 minutes the mixture is
concentrated to 4 ml and 10 ml of dichloromethane
is added to give a solid suspended in solution.
Filtration gives 100 ml of the title compound
as a solid (obvious discoloration with melting
>180C).
GC155f
-226c-
1 338670
Example 192
I3S- [3~(Z),4B]]-3-~[(2-~mino-4-thiazolyl)(methoxy-
imino)acetyl]amino]-4-ethynyl-2-oxo-1-azetidine-
sulfonic acid, potassium salt
(Z)-2-Amino-a-tmethoxyimin~o)-4-thiazole-
acetic acid (100 mg), 85 mg of N-hydroxybenzo-
triazole monohydrate, and 113 mg of dicyclo-
hexylcarbodiimide are weighed into a 10 ml
flask. The flask is flushed with nitrogen and
cooled in an ice-water bath. Then 0.6 ml of
dimethylformamide is added and the mixture is
stirred for 10 minutes, at which point an
additional 0.6 ml of dimethylformamide is
added. (S)-(trans)-3-Amino-4-ethynyl-2-oxo-1-
azetidinesulfonic acid (95 mg; see example 191)
is added as a solid with 1.0 ml of dimethyl-
formamide and 56 ~1 of triethylamine. The cold
bath is removed and the mixture is stirred
for 22 hours. Acetone (3 ml) is added and
the solids present are removed by filtration
and washed with an additional 4 ml of acetone.
All solvents are removed in vacuo and the
residue is taken up in 5 ml of methanol and
162 mg of potassium perfluorobutanesulfonate
is added and dissolved. After standing/ a
solid is deposited and then isolated by
centrifugation to afford 68 mg of the title
compound, melting point >230C.
GC155 f -
- 26u
~ 338670
Exam le 193
(S)-3-[[[(2,5-Dichlorophenyl)thio]acetyl]amino]-
2-oxo-1-azetidinesulfonic acid, potassium salt
3-Amino-2-oxo-1-azetidinesulfonic acid
(100 mg) is dissolved in dry dimethylformamide
(2 ml) with triethylamine (0.083 ml). 2,5-
Dichlorophenylthioacetic acid (123 mg) 0.602 mmol,
N-hydroxybenzotriazole hydrate (81 mg) and
dicyclohexylcarbodiimide (124 mg) are added,
the mixture is stirred for 2 hours at room
temperature, and then 2 days at 5 C. Solvent
is removed in vacuo, the residue is taken up
in water, filtered through Celite, and the
filtrate is washed with ethyl acetate. The
aqueous layer is combined with dichloromethane,
tetrabutylammonium bisulfate (612 mg) is added,
the pH was raised to 3 with lN potassium
hydroxide solution, and after extracting a
total of three times with dichloromethane, the
combined extracts are dried (Na2SO4), and solvent
is removed _ vacuo yielding an oil. A solution
of the oil in acetone is added to a solution of
potassium perfluorobutanesulfonate (612 mg)
in acetone causing precipitation of the product.
After addition of a small amount of ether the
solid is collected by filtration, washed several
times with acetone, and dried to give a powder
(206 mg).
Anal. Calc'd for CllHgN2O5S2C12K:
C, 31.21; H, 2.14; N, 6.62; Cl. 16.75
Found: C, 27.90; H, 2.11; N, 5.84; Cl, 18.04
1 338670
_ ~ 5C155f
~ 226~-
Example 194
(3S-trans)-3-[[[(2,5-Dichlorophenyl)thio]acetyl]-
amino]-4-methyl-2-oxo-1-azetidinesulfonic acid,
potassium salt
(3S-trans~-3-Amino-4-methyl-2-oxo-1-
azetidinesulfonic acid (250 mg; see example 139)
is dissolved in dimethylformamide (2 ml) with
triethylamine (193 ~1). 2,5-Dichlorophenyl-
thioacetic acid (285 mg), N-hydroxybenzotriazole
hydrate (213 mg) and dicyclohexylcarbodiimide
(287 mg) are added. After stirring for about
16 hours at room temperature, the mixture is
filtered and solvent is removed in vacuo. The
residue is taken up in water and filtered.
The filtrate is washed with ethyl acetate,
layered with dichloromethane and tetrabutyl-
ammonium bisulfate (4.2 mmol) is added. After
a total of three extractions with dichloromethane,
the combined extracts are dried (Na25O4) and
solvent is removed ln vacuo yielding an oil
(920 mg). To a solution of the oil in acetone
is added potassium perfluorobutanesulfonate
(946 mg) dissolved in acetone. A solid slowly
precipitates, is collected, washed twice with
ether, and dried to give a powder (306 mg).
Chromatography on HP-20 resin (100 ml column),
eluting with 20% acetonitrile: 80% water, yields
the desired product, which crystallizes upon
evaporation of a ~Jater:methanol mixture.
Trituration of the residue with acétone gives
a powder (233 mg); melting point 212-213(dec.).
12 11 2 5 2 2
C, 32.95; H, 2.54; N, 6.41; Cl, 16.21; S, 14.66
Found: C, 32.91; H, 2.60; N, 6.42; Cl, 16.50;
S, 13.77
- rc~
-226f-
1 3386,~o
Examples 195-196
Following the procedure of example 138,
but substituting (3S-trans)-3-amino-4-methyl-
2-oxo-1-azetidinesulfonic acid for (3S-cls)-
3-amino-4-methyl-2-oxo-1-azetidinesulfonic
acid and the acid listed in column I for (Z)-2-
amino-~-(methoxyimino)-4-thiazoleacetic acid,
yields the compound listed in column II.
195. ~RJ-[(aminooxoacetyl)amino](4- [3S-[3~(R*),4~]]-3-~[[
hydroxyphenyl)acetic acid (aminooxoacetyl)amino]-
(4-hydroxyphenyl)acetyl]-
amino]-4-methyl-2-oxo-1-
azetidines~lfonic acid,
potassium salt
196. (R)-[(aminooxoacetyl)amino]- [3S-[3~(R*),4B]]-3-[[[-
phenylacetic acid (aminooxoacetyl)amino]-
phenylacetyl]amino]-4-
methyl-2-oxo-1-azetidine-
sulfonic acid, potassium
salt; melting point
187 C, dec.
f
-226g-
1 338670
Example 197
~3S(R*)~-3-[ L [ (Aminooxoacetyl)amino](4-hydroxy-
phenyl)acetyl]amino]-2-oxo-1-azetidinesulfonic
acid, potassium salt
Following the procedure described in
Example 28, but substituting (R)-L(aminooxoacetyl)-
amino](4-hydroxyphenyl)acetic acid for (Z)-2-amino-
-~[2-(diphenylmethoxy)-1,1-dimethyl-2-oxoethoxy]-
imino]-4-thiazoleacetic acid, yields the title
compound melting point 128C, dec.
Example 193
[3S(R*)]-3-~[(2-Amino-4-thiazolyl)[[[3-~(2-furanyl-
methylene)amino]-2-oxo-1-imidazolidinyl]carbonyl~-
amino]acetyl]amino]-2-oxo-1-azetidinesulfonic
acid, potassium salt
Following the procedure of example 6,
but substituting (R)-2-amino-~-~[[3-[(2-furanyl-
methylene)amino]-2-oxo-1-imidazolidinyl]carbonyl]-
amino]-4-thiazoleacetic acid for aminothiazoleacetic
acid yields the title compound, melting point
>250C.
GC155f
-
-227- 1 3 3 8 6 7 0
Example 195
Biological Production of EM5117
9 Liter Fermentation
Chromobacterium violaceum SC 11,378 A.T.C.C.
S No. 31532 is maintained on the following sterilized
agar medium (A):
Grams
Yeast Extract
Beef Extract
NZ Amine A 2
Glucose 10
Agar 15
Distilled H2O to 1 liter
The pH is adjusted to 7.3 before sterilization
lS at 121C for 30 minutes.
A loopful of surface growth of the micro-
organism is used to inoculate each of three
500 ml Erlenmeyer flasks, each contA;ning 100 ml
of the following sterilized medium (B):
Grams
Oatmeal 20
Tomato Paste 20
Tap H2O to 1 liter
Adjust pH to 7.0 before sterilization at 121 C
25 for 15 minutes.
The flasks are then incubated at 25 C on a rotary
shAker (300 rpm; 2 inch stroke~ for approximately
24 hours.
After the appropriate incubation as
30 described above, 1% (vol/vol) transfers are made
from the growth culture flasks to one hundred
500 ml Erlenmeyer flasks each containing 100 ml of
GC155f
-228- 1 338670
the following sterilized medium (C):
Grams
Oatmeal 20
Tomato Paste 20
S Glucose 30
Tap H2O to 1 liter
me pH is adjusted to 7.0 before sterilization
of 121C for 15 minutes.
After inoculation, the flasks are incubated at
25C on a rotary shaker (300 rpm; 2 inch stroke~
for approximately 18-24 hours. At this time the
contents of the flasks are pooled and the broth
is centrifuged yielding approximately 9 liters
of supernatant broth.
250 Liter Fermentation
A loopful of surface growth from an agar
slant (medium A) of Chromobacterium violaceum
SC 11,378 A.T.C.C. No. 31S32 is used to inoculate
each of five--500 ml Erlenmeyer flasks each
cont~; n; ng 100 ml of sterilized medium (B).
The flasks are then incubated at 25C on a
rotary sh~ker (300 rpm; 2 inch stroke) for
approximately 24 hours. After the appropriate
incubation, as described above, 1% (vol/vol)
transfers are made from the grown culture
flasks to five 4 liter Erlenmeyer flasks each
containing 1.5 liters of sterilized medium B.
After inoculation the flasks are then incubated
at 25 C on a rotary shaker (300 rpm; 2 inch
stroke) for approximately 24 hours. After the
GC155f
-229- 1 3 3 8 6 7~
appropriate incubation as described above, a
1% transfer (vol/vol) is made to an agitator
equipped fermentation tank containing 250
liters of sterilized medium (C). After inoculation
the fermentation is continued under the following
conditions: temperature - 25 C; pressure - 10 psig;
aeration - 10 cfm; agitation - 155 rpm. Ucon
is added as needed as antifoam agent. After
approximately 18-24 hours the fermentation is
completed. The fermentation broth is then
adjusted to pH 5.0 using HCl and the broth
contents of the tank is centrifuged yielding
approximately 230 liters of supernatant broth.
Isolation and Purification
The broth supernatant from the 250 liter
fermentation is adjusted to pH 5 using sulfuric
acid and filtered using 3-5% diatomaceous earth
(Celite). The broth filtrate is extracted
20 with two 30 liter portions of O.OOS M cetyl-
dimethylbenzylammonium chloride in methylene
chloride.
The combined lower phase is extracted with
6 liters of 0.05 M sodium iodide which has been
25 adjusted to pH 5 with acetic acid. The lower
phase is discarded and the upper phase is
concentrated ln vacuo to 500 ml.
The concentrated material is extracted with
400 ml of n-butanol. The upper phase is
GC155f
-230- 1 3 3 8 6 7 0
discarded and the lower phase is concentrated
to dryness in vacuo. The residue is dissolved
(to the extenl possible) in 150 ml of methanol.
The insoluble material is discarded and the methanol
solution is concentrated to dryness in vacuo,
yielding 38.6 g of crude antibiotic.
The crude product is dissolved in lQ ml of
methanol-water (1:1) and chromatographed on a 500 ml
~olumn of cross-linked dextran gel (Sephadex ~-lQ~ in
the same solvent mixture, eluting at 2 ml/minute and
collecting 20 ml fractions. Active fractions (19-26
are combined and concentrated in vacuo. The residue
(5.23 g) is mixed with 50 ml of methanol. Insoluble
material is filtered out and discarded.- The filtrate
is concentrated in vacuo.
The residue, 5.0 g of material,is dissolved
in 10 ml of pH 5 sodium 0.01 M phosphate buffer
and applied to a column of DEAE cellulose (Whatman
DE52 cellulose) packed and equilibrated in the
same buffer. The column is eluted at 5 ml/minute
with a linear gradient prepared from 4 liters of
pH 5 sodium 0.01 M phosphate buffer and 4 liters
of pH 5 sodium 0.1 M phosphate buffer, collecting
20 ml fractions. Active fractions (192-222) are
25 combined and concentrated in vacuo, and methanol-
insoluble material is removed, washing well with
methanol. Removal of solvent leaves 576 mg of
material.
GC155f
-231- 1 3386 7~
The 576 mg of residue is dissolved in 4 ml of
water and the pH adjusted to S with about 1 ml
of 0.1 N sodium hydroxide. The solution is
chromatographed on a column Df alkylated cross-
linke~ dextran gel (Sephadex LH-20) in water,
eluting at l ml/minute and collecting 10 ml fractions.
Active fractions (38-44) are combined and concen-
trated, giving 459 mg of residue.
Three hundred and forty eight ~348)mg of
the above residue is dissolved in water and the
solution placed on a column of macroreticular
styrene-divinylbenzene copolymer resin (Diaion HP20AG~;
the column has been first prepared by washing with -
methanolic potassium hydroxide, methanol, methanolic
15 hydrogen chloride, methanol and water, and then
packed in water. The column is eluted with water
at 1 ml/minute, collecting 10 ml fractions.
Active fractions (36-43) are combined and concen-
trated giving 186.4 mg of material. Chromatography
(in the same way) of another 100 mg of the 459 mg
of residue from the previous step yields 51.5 mg
of material. At this stage the 186.4 mg of material
and 51.5 mg of material are nearly pure EM5117
as shown by thin-layer chromatography and nuclear
25 magnetic resonance spectra.
The 186.4 mg portion of EM5117 from above
is dissol~ed in water and passed through a
column of ion-exchange resin ~Dowex 50W-X2,
100-200 mesh, potassium form), washing with two
30 bed volumes of water. Concentration of the effluent
.
-- GC155f
-232- 1 3 3 8 6 7 0
yields 189.0 mg of crystalline solid. Recrystal-
lization of this material is acco.nplished by
dissolving it in 0.38 ml of water and adding 3.42 ml
of methanol. The resulting mixture is cooled on
5 ice and filtered, yielding 145 mg of crystals.
Two further recrystallizations in this n~-nn~r
from water-methanol, 1:9, yields 95.9 mg of EM5117,
potassium salt, m.p. 194(dec.).
Optical rotation in water at 21C (c=l)
~ (mn) ~]
- - 589 +94.3
579 +98.6
546 +113.1
- 15 436 +203
365 +348
The following fermentation media are effective
for the production of EM5117, and may be substituted
for medium (B) and (C) in the above example.
- MEDIUM D
Grams
Nutrisoy Flour 30
Glucose 50
Yeastamine 2.5
CaCO3 7
Distilled H2O to 1 liter
MEDII~M E
Grams
Yeast Extract 4
Malt Extract 10
Glucose 30
Glycerol 2
Distilled H2O to 1 liter
Adjust pH to 7.3 before sterilization
GC155f
~~33~1 3 3 8 6 7 0
MEDI~ F
Grams
Gl~cerol 10
L-asparagine S
KH2 4
Na2HPO4 2
Glucose 50
gSO4 7H2O 0.2
Yeast Extract 2.5
Tap H2O to 1 liter
MEDIUM G
Grams
(NH4)2S04 2
L-asparagine 5
lS Glucose S0
Glycerol 10
KH2PO4 3
K2HPO4 7
MgSO4.7H2O 0.2
Yeast Extract 2.5
Distilled H2O to 1 liter
Ad~ust pH to 7.0
MEDIUM H
Grams
IC2HPO4 7
KH2PO4 . 3
Na Citrate 0.5
MgS04 0 . 1
(NH4)2SO4
Glucose 30
Yeast Extract 2.5
Distilled H2O to 1 liter
_ GC155
~234~
1 338670
MEDIUM I
- Grams
Nutrisoy Flour 10
~NH4)2S4 5
S Glucose 50
Yeast Extract 2.5
CaC03 5
Distilled H2O to 1 liter
MEDIUM J
- Grams
Gerber's Baby Oatmeal 20
Cont~i n~ Tomato Paste .S
Glucose 20
Tap H2O to 1 liter
lS Adjust pH 7.0
~EDIUM K
Grams
Gerber's Baby Oatmeal 5
Contadina Tomato Paste 20
Glucose 20
Tap H2O to 1 liter
Adj~st pH 7.0
~DIU~ L
Grams
Yeast Extract 4
Malt Extract 10
Glucose 34
MEDIU~ M
Grams
Amberex (1003) 5
Glucose 30
Tap H2O to 1 liter
GC155f
-235-1 338670
MEDIUM N
Grams
Amberex 5
Cerelose 33
Tap H20 to 1 liter
- l~S155f,
-~236- 1 3 3 8 6 7 0
Example 196
Biological Production of EM5210
Gluconobacter species SC11,435 A.T.C.C.
No. 31581 is maintained on the following
5 sterilized agar medium (A):
Grams
Yeast Extract
Beef Extract
NZ amine A 2
Glucose 10
Agar 15
Distilled H2O to 1 liter
~he pH is adjusted to 7.3 before sterilization at
121C for 30 minutes.
A loopful of surface growth from the agar
slant (medium A) of Gluconobacter species SC11,435
is used to inoculate each of three 500 ml Erlenmeyer
flasks each containing 100 ml of the following
sterilized medium (B):
Grams
Yeast Extract 4
Malt Extract 10
Dextrose 4
Distilled H2O1 liter
25 The pH is adjusted to 7.3 before sterilization
at 121C for 15 minutes.
After inoculation, the flasks are incubated
at 25C on a rotary shaker (300 rpm; 2 inch
stroke) for approximately 24 hours. After the
30 appropriate incubation, as described above,
1% (vol/vol) transfers are made from the grown
culture flasks to one hundred 500 ml Erlenmeyer
flasks each containing 100 ml of the following
. GC155f
-
-237-
1 338670
sterilized medium (C):
Grams
Yeast Extract 5
Glucose 10
Distilled H2O 1 liter
The.medium is sterilized at 121C for lS minutes.
After inoculation, the flasks are incubated
at 25C on a rotary shaker (300 rpm; 2 inch stroke)
for 18 hours. At this time the contents of the
fiasks are pooled and the broth is centrifuged
yielding approxLmately 9 liters of supernatant
broth.
Isolation and Purification (small scale)
Activity from the broth supernatant (1~0 liters)
is absorbed on a 500 g column of strongly basic
anion exchange resin with quaternary ammonium
groups attached to a styrene-divinylbenzene
copolymer lattice (Dowex AGl-X2(Cl-)),washed
with water and eluted with 5% sodium chloride
in aqueous 0.01 M NaH2PO4. The eluate is
concentrated in vacuo.
The residue is ~dsorbed on 250 g of charcoal
which is washed with water. EM5210 is eluted
with methanol-water (1:1). The active fractions
are combined and concentrated in vacuo yielding
crude EM5210.
The crude EM5210 is chromatographed on
. a 280 ml column of strong base anion exchange
resin (Bio.Rad AG l-X2 (Cl )) using a linear
gradient prepared from 1 liter of water and
*Trade Mark
., .~
~. ,~
GC155f
_ . .
-238- 1 3 3 8 6 7 0
1 liter of 2M pyridinium acetate (pH 4.5). The
active fractions are combined and concentrated
in vacuo.
The partially purified EM5210 is further
purified by gel filtration of the residue on
a 500 ml column of cross-linked dextran gel
(Sephadex G-10) eluting with water. The active
fractions are combined and concentrated yielding
26 mg of EM5210. The potassium salt of EM5210
is prepared by passing EM5210 through a column
of cation-exchange resin (Dowex 50-X2) in the
potassium form.
Isolation and Purification (large scale)
The broth filtrate of a 250 liter fermentation
(pH 3.7) of Gluconobacter species SC11,435 is
absorbed on 10.8 kg of strong base anion exchange
resin (Dowex l-X8(Cl-)). The resin is washed
with water and eluted with 5% sodium chloride
in 0.01 M sodium dihydrogen phosphate. The
active fractions are combined and concentrated
to a small volume. The precipitated salt is
removed and the filtrate is desalted by passing
it through a column of charcoal (1.1 kg of 20-40
mesh Darco) in water. The column is washed with
water and the EM5210 is elute~d with methanol-
water,l:l. Active fractions are combined and
concentrated. The residue (16 g) is dissolved
in water and chromatographed on a column (600 ml)
of strong base ion exchange resin (Bio.Rad AG 1-
X2(Cl-), 200-400 mesh), eluting with a linear
GC155f
-239- 1 3 3 8 6 7 0
gradient prepared from 1 liter of water and
1 liter of 10% sodium chloride in 0.01 M sodium
dihydrogen phosphate. Active fractions are combined !
concentrated in vacuo to a small volume, and
precipitated salts are removed by filtration.
The filtrate is applied to a column of macroreticular
styrene-divinylbenzene copolymer resin. The
column is eluted with water. The active fractions
are combined, concentrated to a small volume
and freeze-dried, yielding 120 mg of the sodium
salt of EM5210.
To transfonm the sodium salt to the lithium
-salt an ion-~YchA~ge resin (Dowex 50 W-X2, lithium
form) column is used. The sodium salt (100 mg)
is dissolved in 0.5 ml of water, applied to the
column and eluted with water. The active fracti~ns are com~
bined and directly freeze-dried, yi~l ~;ng 95 mg of the lithium
~lt of EMs~Io or as an ~I~L~ S solid.
The free acid (inner salt) of EM5210 is
prepared by passing a base salt of EM5210 through
a column of weak acid ion-exchange resin in
the H+ form. For example, about 2.5 mg of the
lithium salt can be applied to a column of
Bio.Rad Bio.Rex 70 (H+) and eluted with water
to give 1.45mg of the free acid (inner salt).
Chemical properties of EM5210
1) Ninhydrin positive.
2) Acid hydrolysis (6N-HCl at 115 C for 16 hours)
gives two major ninhydrin positive spots by
~ paper chromatography (.~atman No. 1, ~utanol-acetic
GC155f
-
- 240 -
1 33867~
acid-water (5:1:4), and one weak ninhydrin positive
spot that quenches W-excited fluorescence. The
two major ninhydrin pasitive spots are D-glutamic
acid and D-alanine.
Physical characteristics of EM5210
1) W spectrum of the sodium salt in water:
end absorption
2) IR - Major peaks of the lithium salt in KBr:
1770, 1640, 1530, 1384, 1242, and 1051 cm~l.
3) PMR - Chemical shifts of the lithium salt
in deuterated water, ppm down field from TSP:
1.40 (d,J=7Hz), ca. 2.14 (m), ca. 2.44 (m),
3.49 (5)
3.73 (t,J=6Hz), 3.94 (s), 4.28 (m)
Optical rotation of the free acid (inner
salt) in water at 24 C (C=0.15%) (pH 2.7):
~ (nm) [~]
- 589 +73
578 +79
54 6 - +91
4 36 +159
365 +263
- The following fermentation media have been
found effective for the production of EM5210 and
may be substituted for medium (B) and (C) in
the text.
MEDIUM D
Grams
Oatmeal 20
Tomato Paste 20
GC155f
- -241- 1 338670
MEDIUM D (continuçd)
Grams
Tap H2O to 1 liter
Adjust pH to 7.0 before sterilization of
121C for 15 minutes.
MEDIUM E ..
- ~rams
Yeastamine 5
Cerelose 11
Tap H2O to1 liter
- Sterilization at 121C for 15 minutes.
MEDIUM F
Grams
Glucose 5
Tartaric Acid 2
Yeast Extract0.5
(NH4)2PO4
(NH4)2SO4 2
K2HPO4 0.5
NaH2PO4 0.5
- g 4 2 0.2
CaC03
Distilled H2O1 liter
Adjust pH to 6.0 before sterilization of
121C for 15 minutes.
MEDIUM G
Grams
Nutrisoy flour~ 30
Glucose 50
Yeastamine 2.5
CaCO3 7
Distilled ~I2O to 1 liter
.
GC155f
-242- 1 3 3 8 6 7 0
MEDIUM G (continued)
Sterilization at 121C for 30 minutes.
MEDIUM H
Grams
NZ Amine A 10
Cerelose 33 .
YeastAr; n~ 2 . 5
Tap H2O to 1 liter
Sterilization at 121C for 15 minutes.
MEDIUM I
Grams
Nutrisoy flour 15
Soluble starch 15
Glucose 50
CoC12.6H2O 0.005
CaCo3 10
Distilled H2O to 1 liter
Sterilization at 121C for 30 minutes.
GC155f
-243- 1 3 3 8 6 7 0
Biological Activity
The following methDdology is used to
deterrine the m; n;mum inhibitory concentration
(hereinafter referred to as MIC) of the B-lactams
of this invention.
The test organisms are grown in approximately
15-20 ml of Antibiotic Assay broth (Difco) by
inoculating (in tubes) the broth with a loopful
of the organism from a BHI (Difco).agar slant.
The inoculated tubes are incubated at 37C for
18 to 20 hours. These cultures are assumed to
contain 109 colony forming units (hereinafter
CFU) per milliliter. The cultures are diluted
1:100 to give a final inoculum level of 104
CFU; dilutions are made with K-10 broth*.
The compounds are dissolved in the appropriate
diluent at a concentration of 1000 ~g/ml. Two-
fold dilutions are made in K-10 broth resulting
in a range from 1000 ~g/ml to 0.5 ~g/ml. 1.5 ml
of each dilution is placed into individual
square petri dishes to which 13.5 ml of K-10
agar** is added. The final drug concentration in
the agar ranges from 100 ~g/ml to 0.05 ~g/ml.
Organism growth control plates containing agar
only are prepared and inoculated before and
after the test plates. The organisms are applied
to the `agar surface of each plate with the Denley
Multipoint Inoculator (which delivers approximately
0.001 ml of each organism) resulting in a final
- inoculum level of 10 CFU on the agar surface.
.
GC155f
-244-
1 338670
The plates are incubated at 37 C for 18 hours
and the MIC's are deterrined. The MIC is the
lowest concentration of compound inhibiting growth
of the organism.
*K-10 broth is a yeast beef broth containing:
Beef extract 1.5 g
Yeast extract 3.0 g
Peptone 6.0 g
Dextrose 1.~ g
Distilled water q.s. 1 liter
**K-10 agar
Beef extract 1.5 g
Yeast extract3.0 g
Peptone 6.0 g
Dextrose 1.0 g
Agar 15.0 g
Distilled water q.s. 1 liter
The tables that follow are tabulated results
obtained when the ~-lactams of this invention are
tested against various organisms. The number
following each organism refers to the number of
the organism in the collection of E. R. Squibb &
Sons, Inc., Princeton, New Jersey. A dash (-) in
the tables means that the compound tested did not
show activity against the particular organism at
100 ~g~ml. The symbol "N.T." means not tested.
GC155f
-245- 1 3 3 8 6 7 0
o .o..,,,,,,,,o,,,,,,o,,
D ~ O ~ ~D O
U~o..,oooooooooo,,,,U~
~D ~ O ~ ~ O O ~ O O U~ U~ O U~ O
_, ~ _, , _, ._, .
.
. , , ooo,,, o,,,,,, oo,,
D ~ O O O u~ u~ O
_, ~ . - . - -, . ., o o o, , o o o o o, , , o U~
~-I~D~ _I~D OOO OOoOO O~
. .
o ~ o, , o o, , o ., o, , , , o, , o
U~ ~ ~ ~ ~ ~D U~ O u~ o ~ O U~ O
H
O - - - I - I U~ I I I I I I U~ I I I I I I I I I
I O _I ~D O ~1
a~
....IU~OIIIIIIIIIIIIIIIII
- - - I - o o Ul u~ o o I U~ I O O I I I O I
~ _I ~ ~ ~ ~ ~ In o ~ ~ o o ~ ~ u~ o o
- ~ I o o~ o~ ~ cc\ o o o o a~ o o~
u
~ C~
UJ'* r~ Ct ~ `
* t~ ~ ~ ~`
C u-~ t ~ ~~,
C~ t ~ I ~ ~ c
t r ~ ~ ~ ~ IUJ ~ ~ r~ ~ ~ C
C~ t~ C ~ ~* ~* ~ ~ '* ~* u~ ~ G ~ C
~i_ C ~ r_~ ~ ~ ~) . ~ 5 . ~ * r ~ n
O O O O ~ ~* ~ ~ ~ ~ C~
I~ ~~ C) C) ~ C~ ~ rC~ ~ CS~
to C~ G~ C~ G~ u~ ~
r~ C J C ~ rJ r '*~* ~ C~ C~* `~) ~ C~ Ul ~~ _; C~,
~ ~ J J O C C) rr rr rS rS ~ 1~ ~
Ul O O O O ~) ~ C) C~ C) C) O 1~ ~ ~ C~ C~ O r_
o ~ o ~ * ~* ~ ~ ~ r~
~15 r r rS` r ~ ~ C ~r~ r~ r~ u~ rJ~ ,rv V ~ r~
~ C~ C ~ ~ C) C~ C) C J ~ rv O ~ * ~~ J ~ ' *
4 ~ ,~ * cr, ~ tl tn ~ ~ S, -I ~ G rS' ~ ~ '* ;1~ ~ ~IJ C)
GC15 5;E
~246- 1 338670
o o o
CD O O L~ O I O O I I I I I I I I I I I I I I O Ll~
_I Ln 1~') ~ L~ In o
_I
Ln Ll~ In Ir~ ~ ~)
` . . . - I Ln I I O O I I I Ln I I I I I I O I
1 N N N ~D N O O N ~ 1~ ~o
- - - I Lr~ I I 00 1 1 oL
~ D N N~D N O OO N o o o N
_1 ~ U7 0000 1 o I I 1 1 1 1 1 1 1 1 1 1 1 '1 Lno I o
I o Ln 1-~ L~ N In N
X ~
3 Ln Ln
I N ~r N _I O _I O 0 ~ ~ I ~ U~
o ~ I o ~ o
O _l O O O ~ O O O O O O O O O O ~ ~ O ~ O
C.) ~ _I _I V V '
O
~5 Ln Lrl Ln L~ LO
O t~L~l L~ I I Ln Ll~ O O L~ L l Ln Ln I O I I O
h ~ ~ ~ ~ ~ ~ N Ln L~ i N ~ t~ _I N ~`1 ~ O Ln
n
Ln ~ ~ u~ D ~ er ~ ~ O ~r _I ~ er OD ~ Ln C~ Ln
... lo................. ~.......... -.-Ln
D O ~ ~ O O _I O O O O O O O O O O ~ ~ O
r~ v ~ ~
~D C~ O Ll~ _I 1~ Ln ~ ~ O 1~ Ln a~ D Ln a~ ~ Ln ~ ~1
I~ o~ o u~ Ln a~ o ~ t~l L~
~ 1 o t'~ Ln OD ~ ~ _~ ~r ~ o Ln 1--Ln ~ ~)
_I N N O ~n C~ N 0 O O O O 0~ ~1 co CJ~ _I CO ~ O C~ ~ cr, CO 00
,~ ~
-. C~
~ 5 J
r,o ~ r~ r' r
; \~ C n~ C ' ~
.) C' C~
r~
X ~ ~ C C ,~
C~ ;~ C C~ Q) ~ ~ ~ ~ r, r~ ~ r~ F, .. r~ C-
r ~ C' t 1 ~ r~ * ' ,~ r~
~n ~ O O o o
C '~ G ~ ~ * ~-~ C, C~ C~
J ~J ~ ~ rS~ ' S
U~O G O O ~ ~ O O C~ ~ ~) ~ ~ ~ Cl ~ Cl O
r ~r~ rJ~ rJ~ U
J , J~ rS r rC ~ cn
Cl r~ r) ~J
.. . . .
GC155f
-247- 1 3 3 8 6 7 ~ -
OU~---~Inoo
N U~ O ~ ~ 0 0 0 0 0 0 ~ It~ 0 ~ U~
~ o ~ o ~r o ~ ~ o o ~ e~
U~ ... ~O................................. In
OOOOOOOOOOOOOOOOOO~
V V V V V
- - - I - I 10011101111' 110011
~ ~D ~ 1' D ~ ~
a~ '
_I ~ o o o~o I o I I I I E~ I I I I I I I I I 1 I
Q~ n ~ ut O1- - o
_1 e _, z _,
X
000010100011111000100 01
~ 14 NIn O O O ~ O O O O O O O O D O
U J 11
r~Ln U~ t 0 0~ ~0 00 CO 00 0~ 0 ~0 X CO ~D ~ ~ ~
H ~ O O ~ . . . . . . . . . o 11~ L 1
I O O _ I ~D O O ~ O O O O O ~1 O O --1 0 ~ U~
o . . . I . In ~ O In o o o o I o o U) I o
N ~ ~ ~ ~ 1 O t~ ') U~ ~1 0 L~ U) ~`l Ul
Lr) ~ U~ U~
IOIIIIIIIIIIIIIIIl~ 00
_I ~ ~ o a~ ~ ~ x o o o o o~ o ~ a~
~ r~
~ C ,~ r r r ~
~ ~ C v~ r~ c.~ , * .,
;~~ C~ C3 r~ ~ CD ~ t` . . r`
~;~ C) V ~ r~ C
~ r~ CD r; r ~ CD ~ r~
- ~3 r~3 C3 r~-~ Cu~ r.~ C~ G ,, ` ``
Ct_ r ~ ~ ~ ~ ~ ~ O ~ D ~ n ~ ~ ~ C
C~ O O O O ~ 3
~3 r~ C) C) CD rq ~ r 3 ~ . 3 ~3
c c~ r~ C3 r~3 ~, r~3 ~3 L~ r~- I ~ d v, - ~
r~~C O S~ O C J '~ r~3 r~3 ~ -D ~ r~3 t~ r;3, '~3
V~O O O O J J ~ C) C) C) ~ ~ ~ r~3 ~ ~ ~ r~3 0
~rlt~ ~ ~ ~ G O ~ ~ r~D CD V~ ~ r~ r t~ 3
S ' ~ ~J ` C~' V~ V~ ~D ~D ~D ~ CD
Cù ~ c ~ rCD
h ~, ~ ~ ~ ~ r~ _3rr ~ r~
O t~3 r~ ~ r~ r~ r~ ~ h
.,
-
GC15 5f
-24~- 1 3 3 8 6 7 0
u~ . . . . I .. I . I o I I I I . o I I I I I In I
') ~ O_I N O ~ O ~
~r~..'.IIOOIIOIIIIIIIII
I ~ o o ~ u~ ~ ~ o
- - -o - I oo I ooo I I I I I I I
_I O _I ~ O O _I ~ O U~ O O O ~D ~
_I I I O O O I o O u~ o o o I I I I u~ I I
C~ ~ _I ~ `.0 ~D _I ~ 1~ ~ Irl O O ~ O 1~') 0 N
~ X
--, o .. ~oI ............... Io.oI
u~ er ~ co ~ er o o o ~ o ~ co co ~ o~
~ ~ ~ N O O O O O O O O O O O O O _1 0 0 ~ O
_I V V V V _1
o "~ ..... "~-~ - - .. - o ~ - o I
o ~ ~ ~ ~ ~1 ~ .--1 ~ O .--1 0 .') ~D u~ ~ ~1 ~) Lrl
~` U~ O I O U~ U~ O I O In ~ O I I O I I
~ ~ ~ ~ O ~ ~ O ~ ~ IS~ I ~ O O ~`1
~`J ~'1 ~r _1 0 N q`~ l CO 0 C~ ~ 11~ CO ~ ~ _I ~ ~ O U) I~ U~
_I ~ ~ o a~ o o o o a~ co oo o a~
~ ~ .
r~q ~ rl ~ ~ r ~ ~i
~q r~ r~ r~ rr
~;~ r~ C ~q rv ~ r ~ ~ ~ C
C) ~ U~ r , ,_
,Y ~ ~ r~ r~ ,, U~, U r~
rv ;~ r~ r~ ~ r, rn ~ ~ ~C
r~ r ~! r^ ~ O O O O ~ r.
~ ~ ~ r ~ O ~ ~ v ~ ~~ r~ ~ ~ J ~ r~
C r~ rJ C ~ * ~ ~r r~ r~ 'r~ ~) . r;~ C
U~ C~ r~ ~ ~ J rJ ~ rv rc) r~J r~ ~ ~ ~ r; ~ rc~ r~, r~
~_~ r~ ~ ~ ~ C v C~ r~ r~
rr r~ r~ ~ j r~ ;r;r~ r q
v~~ $r~ J r'~ r~ r ` r ~ r,~ ~ S
5'1 J~ ~ ~ ~ ~ ~~ ' r~ U U U U ~ t~ _1 ~1 .I r~ r ~ J U~ I rc)
- GC155f
-249- 1 33~6~O
~..... U~O......................... InU'~.OI
~ D ~ O ~ I ~ N ~D N N '1 0
u~ o I o u~ n o ~ IJ) O In 11~ o o o o I o
N N N O ~ N In N N N In N N ~ Ir) N N In Ir~ n N O
N 0 1 I 000 1 1 00 1 000 1 1 00 1 1
D N o - l ~D O ~ O It~ O O O O 1~
. -
_I _iIIOIOIIOOOOIIIOOI
~ d~ O U~ ~O Ul o o ~rl O
E~ "I
X
O-.o.................................... U~
4~ ~ ~ ~ ~ ~O O O ~ O _I O O O O O O O O O ~ I O ~O
. O ~1 V V V V
C~ ~
r)CO 00 CO _I ~ CO U~ In
H ~ ~ O I O I I I I O U~ I I I I I I I I
O O O ~1 u~ O O N o N N
~4
` O l ~
) ~ o o ~1 U~ o o
_i ~ ~
~..OII
~D ~ u') O ~D
I~ a~ o ~ ~ co a~ c~ o er N Ln r~ I 00 ~ N t
N ~ ~ _I O N ~r N C~ C~ ~r Lr~ OQ ~ ~r _I er N O U~
_I N N O C~ C~ N CO O O O O C~ t~ CO C~ _I 0 C~ O 5~ a~ Cl a~
~ c
u, * ,~
r~ Cr~ 4 ~ t~ r/ I U~ C
S S S t--~ C ~~~ C C ~ ~H C r
r~ r;~ r; ~ S. G
~1 ~ C~ t~ r ~ r C r~
* ~ .* ~ ~ ~* _~ U4 ~ ~ G
t~_~ C ~ ~ ~ ~ t--~ ~I * ~ ) t
ro U _ O O O O ~ * ~ ~1 C~
S U` ' ~~ C~ C) C) C) O ' r~ ~ r ~ ~ ~
J C) r~ r' ~ ~ rr ~ r~ ~ tr~ d - - ~
~ O r~ J _ * * .~ * ~) S ~
~ r~ rJ r~ ~ C , .S S ~ ~ ~ ~ ~ C~ ~
U~ r,~ o r~ " C) '~ C) Cl ~ ~ ~ ~ 3
* ~ r~ t4 t4 0 r~) t~
S~ J C ~ ~ ' ~ * ~ S
s r r ~ J J r ~ 0 ~ ~ ~
;' ~ r~ ~ r,~) r~ O O ~ t~~ * ~~
~ * ~ t, a ( ~ ~ I C)
O ~ '~ '~ rJ~ C~ ~ ~ L ~ ~ ~ P~
GC155f
-250-
1 338670
~o o ~ ~ o
Il~ I` ~D O O ~ i t~ N N ~ N ~D O I
~ . . . O . . - ~ - Il~ L o o In
U~ N N N O I ~ ~D N N N N N ~ N N N ~D N N U~ U~ N N I
U~
N 0: 0 0 0 U~ O O n ~ O O O O O O O O O O O
1~ II').ln O o I N N O O N N O U7 1 0 1 O O 11~ 0 ~ u~ I O
O o I I .01111110111111 I
p~ N N 11 1~ I O O O
X U~
-- O N N N N I I O I o ~ II o I I I I I I I o I
--~1 U _I N ~0 o O O U~
O
- U ~
H :~ co U~ I O O U) O o o o I o o I I O o I
~ J ~ ~ N N N ~ n N N O u~ o ~ U~ o o
~ .
~DOOOOIOOIIOIIIIIIIOIOOIO
O O O o 1~ L~ ) 0 Z O O O N O
In In
D CS) ~ co N ~ O ~ ~ o ~ C 00 ~ ~D _I ~ ~ u~
U)-U~I....................
I~oooo~ooooooo~_I~o~
V V _I
r~ ~ o ~ n a o ~ C~
--l N ~`1 O a~ ~ N C0 O o O O C~ t` oo cn ~I C~ 00 0 C~ C~ al CO
~ c~
U~ ' r~~ "
t)~ 4 -~ ~4 ~ Q~ '' r
S ~ ~S ~ C 1 l~ C~ r~ r
C~ C ~ r~ C~
r~ u ~ ~ O
S Q\ C, .~ UJ ~ rr~
Cl C~ r~ t ~ ~ r~ r;~ r~ UJ (~ r O
r~_ C ~ ~ r~ ~ r~ '~ t~ ~ t.~ .) ~ tn
O O O O ~r~ C) ~ r ~ r~
O OrO ~ C~
C~ --~ Cr ~ .~, ~ ~ r~!)
C r~ r ~ C~ C~ C~ ro ~~
~-*-~-r~ C~ C~ ; C~ tJ) ~ ~~ Q~ E
O ~ O ~ ~ ~ ~ r-~ C~ C~ o
rl ~ t~ r_~ r~ J ~ t~ Q r~ q rr~ ~ r~ r~
S- r ~ ~ S t~
ta r ~ r r~ r a ~ r r~ r ~ UJ !~ C r~ , r. ~.
p ~ J ~ ; .S .S r~ rQ ~ ~~
r~ r~, Q~ r~
-- GC155f -
-251-
1 338670
o o ~ o
In O _t ~D O ~ ~ 1 O O O _I ~ ~ N ~D ~ ~ O
O o o _I O O O _I O O _I O O O o ~ o _I o 0
V
U~
o ... oo............ ---------......... --.
OOOOOOOOO_I~OOO_I~O~10
_I V
D O ~ O _I O ~ O ~ D 0-
~ X U~ U~
'~ CO O----U~
OO~U~O_IOOOOOOOOOOOO~ I
O _t V V
~) ~ O
H ~ r~ Ll~ O O O ul I I I I I I I o I I I I I I I Q I o
~ ~ ~1 ~ 111 0 0 ~ I In o
In In In U'~ r~
~O IIIIOOIIOOIOOIIIOOI
1~1 ~ ~J ~ N ~D O O O Ul O O Ir~ Ll'~
u~ - o I o n o ~ o o u~ o o o I I I I
1~) D ~D ~ Ir) ') ~ 1~ N O ~ O Ll~ n O O
_I ~ ~ o a~ ~ ~ oo o o o o a~ I ~ ) o ~ a~
u
~ y
~ C .~ r~ C'
14 4 '* 1~ tl~ ~ ~. r.,
S ~ ~ S r~ C - rJ ~ Y .~ C ~
ù ,~ ù ù C~ C ~ ) C'~ , ~ r`
C~ t~ y * y ~* ;~ *~ t~ ,~ o ' ~
rr r r~ r O O O O ~ ~ " rU '~
O O U
rJ ; J ~ _ _ V ~ ) t" '~
~* -* ~* ~ ~ ~* ~ ~ ~4 ~ _~ O E
F r~ O ~ G C
O O O C ~ O ~) ~) ~ ~ ~ Cl ~ Cl ~
C ~J C ~ ~ '* t~ t~ 4 ~
~~ * ~ * S ~ ~ ~ o ~
t~ tr~ ~4 t~ t~
t~ ~ S_ U U C~ r . r r ; ~ ~ ~ ~ ~ r~
t~ C ~ , ~ tj) O o
O ~ C~ h tt t2. ~ ~ ;~ ) C~
GC15 5f
.
--2 5 2--
1 33867~
o,oo,",,o..................
t~ o o ~ o _, _, ~ o _, o , o o o o o _, , ~o ,
V _~
N O O ~ O ~ ~ I O 0 ~7 ~ I O
U~
a~ o o o I I u~ o ~ o I
o o _I o o o o o _I o
_I V
'.D ~ ~ ~ N _I O t`~ I O O O O ~ D O
X
0 U~ _I U _I ~ N
N t~l ~ ~ O ~ ~`J ~ O O O
O
O ~
r~ ~7
H ~ O I I I I O I I I I I I I I O I I I I I I O I
~ ~D O ' In o o ~D
- - - I - I O O O I I I O I O O I I I O I
D ~ O O O O O O O ~
---IOIIIIIIIIIIIII'IOOI
D 1~ 0 0
_I ~ ~ o ~ a~ o o o o o~ O ~ cn ~ co GO
~ ts .
~ C' ''1 ~S
` j t~ t~ , r~
C~ ` CO ~ '~ t`
c,~ ~ O ~J
S _ t~ t~ c~ o ( ~ t`
O O O O ~ Ci t~
~ C) C) C) C) ~ ~ ~ t` ~~ S
rJ J J C) ~r~ tS t~i tS ts S-~ ~ t~ J
G G ~tS t~ . tS
U~O G C O ~ ~ C) C) C) C) ~ ~ ~ ts ~ ~ ~- tS ~--
~ r~ O r . ~ ~ t~ t~ r~ tO CO ~ ` ~ tS ~,
. ~ ~ ') ~ ` CO tJ) ~ O ~ ~ ~ rl r~
* t~. ~ tC" U Ci) Q) ? '~ ~ ~
GC155f
-253- 1 338670
,~ o~_,_,o_,o~ooooo~_,~o~
--I --I V
r- I I I I I I I o o u~ ul o o ~n o o o ~ u~ o u~ o . o
1~ 1-') 0 ~ ~ O ~ In U7 C~l N O N U~
a~
--I E ~D ~ ~ o ~ ~ _l o ~ ~ o ~ ~ o ~ o
X
t~
~ ~ ~r u~ u~ o I ~ U u~ o u~ o o o u~ u~ o o o o o I
o r~~ t~l ~ U7 ~ ~ ~ ~1 ~ N In ~ O N ~1 0 0 U~ In U-)
H ~ ~ ~ 11~ U~ Ul
~, Or~ D N N ~1 D N N N N ~ N N ~1 N N N N ~ N N N O
h--1 H ~ 1 H
P~ .
, ..o o . .. . ~ O O
r~ ~N N Ir) O 0 ~D O~1 ~O _I O H 0--10 0 0 ~I N N ~ 0
_I O O ~ In O U^) O O O O Ir~ ~ ~ ~) Ir) Irl ~1 ~ ~ ~ ~ N Ir~ ~ N ~ ~ N ~ 1~ ) 0 IJ~ O N
O fn ~ o f~ O ~ .0 CO f~O f;~
O ~ ') f~ O ~ N Ln ~ ~ f;~ f-- _1 0 ~ N
N 0 q~ ~1 O N ~ N ~ CO C~ ~ L~ 00 ~ ~ --1 ~ N O u~0 ~
N ~1 O ~ C~ N 0 0 0 0 0 ~ 0 CO C~ _I CD 00 0 ~ a~ a~ CO 0
O~
,~ ?~
.~ ?~ C~
r~J rr~ q ~ f.~ ~
~? ~ . ~? ~ C ~ .? ~ r
.?;~ f~? .~ rr J
?~ ?~
?~ C -~Q r~ t~ G ,~ .
u " n , o c~ o o ~ ?, ~
^ q ~ ~ O C) ~ ?; ~ ? ? Sl
~ J ~ ~ r ~ S~ --~ ~ ~ Q\r ~
G O C O j f~j ~ * * * * C~? ?~ * "~
?_.Jr~ ,J ~ ~ rJ J rS. ~ S
U rJ o rj O J ' ~j ~ ~ ~ f;~ r;~
~ o r,~ C '* * ~ Q~ ~ ~J7 f~7 '' '~ ~ _ O
f~ S S ~1 ~ ~ r.~ ~ S " S ~
C) , `~ r ~ ~ J Q~ C~ ~
h~ ~ ~ ~ t~ ~ * ~ Cr- C7 r~ r.~ ? rS ~~ ~ ~ ~ ,7
O f~ f~ f~ ? 1~ ?~ ~ CrJ i~ ~ ~ C
GC155f
1 33 ~ ~ 7~
, _, _,
o o o ~ o U~ o o U~ o o o o
o o o, , ~ ~ o ~ ~ ~ o o ~ .,. U~ U~ ~, , , o,
, , _1 , ",
o o o o U~ U~ o U~
~ ,,,,,,o,
a o
.-1 E ~ o o o o
D~oo~Iu~IIIIIIoIIIIIIOI
X a~
o .......... -------o
J,J O ~ ~ N o I ~ ~ N O ~ D ~) O ~1 ~ ~D ~ ~ ~ ~ U~ I
H
. ~
~: O
S~ In ~r Lr)
U~ o o o o o o o o o n ..
CO ~ ~ U~ U~ I ~ O I ~ U~ U~ I O O ~ U~ U~ I I I I I ~ I I
~ O O O O O O O O U'l
OD t~l ~ ~1 U) I t~l O I ~1 In O I O O U~ O O I I I I I ~
_I ~ ~ o a~ o o o o o~ o a~ ~ a~ oo co
c
S S ~ S ' ~ O ~ u t .~
C C~ S r ~ S S
Cl C) C~ c) ~ & ~
~, c, c; c~ ~ s ~ . ~ O
O O O ~ r~ o ~
GC155f
-255- 1 338670
_, U~
o ~ U~ o o ~ o o o o o o o o o
_I ~ N U~ O I 11~ o U~ o It~ I o I o I o o I
o O o O o U~ U~ O O O U~ O O O O o O O o o O
_1 In In Ul O I ~I ~ O It'l 11~ ~ O O O It~ O 1~ I I O U~
~n ooo o o
a~ oo I I o o I I I I I I I I I I I I I I I I I
-
_ C~
_I ~ GO O O O O O
~ X ~~
-
_ ~ U'l
U J ~ ~ D o ~1 o ~ ~ D o ~ D O I
V ~1 ~1 ~-1
. ~
~: O
U ~
~, ~ O oo 00 00 0 0 0 0 0
a~ I I I I I ~ ~D I 0 1~ I I O O I I Ul Irl o ul 1~ Irl I Lrl
U~o o o o U~ o o o U~ o o o o o U ~C~ O
_I ~ ~ o a~ o~ ~ ~ o o o o ~ ~ a~ a) o ~ cn a
~
tD ~ ru
;~ t4 ~ ~ s~
* tr:
. ~ o tn ~ C
,t4 ~ *~1 0 ~ ~ c
t4 4 t4 t4 ~ O O O O S, ~ ~ ,~, ~ O ~ n,
i~ ~ ~ S~
t' O ~ O O O ~ ~
- p p " " ~ Qp q~
O tq ~ ~ ~01
GC155f
-- 6--
1 338670
. .
--I--I ~ a) c~ o ~ _I co o ~ o o ~
~ o . . . . . . . . . . . . . . . . . . . O
O _I t~ ~ ~ In O O O O O O O O O O O O O ~ O _I O N 11
_~ V V V _~
O ~ o~ o_la~oooooo ~ Do~
_I
D N ~ a a _1~ 0
O ~ ~ ~ ~ a o _~ o d o o o o o o o _I ~o ~ o
~ v v ,~ _~
--I m O O ~ ~ h
O ~ ~ ~ U~ O _I ~ o o o o o o o o O O o o O _I ~ O
W
er o O ~ O
C,) --I O ~ ~ I ~ ~ O ~ '1 ~1 0 0 0 ~ ~ C~ ~ ~ ~) Ir) I
H
. ~
u~ o~o~ooooo~r~x_I
4 r~ o . . . . . . . . . . . . . .. . . . o
O I I I I I U~ ~ o o o O o O o O O O O O O O O O ~ O
_I ~ V V V V V \~ V _I
~ I --I ~ _I--I o o o ~ o
O O O U~ I I . . . . . o ~
I ~,OOOOooOoOOOOOOO.O~--1.--1
V VV V V
,~ ~ ~ o a~ a~ ~ ~ o o o o o~ ~ o a~ ~ al ~ oo
.~ c
S ~ r
S ~V ~ X ~ o a~ tI S~ ' ~ ~
o-*-* *-* ~ E~ * ~O ~ C ;~ X C-
~` ~ ~ ~ ~ ~ I ;~ ~ ' p S~ * ~ ~ "' ~ S, C,
SOOOO ~*
;~ ~ X a~ ' ~ O O O O
, C,
E3 ~, ~ ~ o o ~ ~ ~
O Q ~ '- ~ O O U O O ~ ~ ~
b C` O ~ E E O
- ~;C155f
- 1 338670
e~257~
.
u - - -a - o a
_~ o o ~ a ~ ~ I ~ o ~ I N I
_I _I _I _I
t~ 0 N In d c~ _I o cl o ~ _I d o o o _I ~ ~ ~O ~0 ~ N
_I _I V ~
E ~ o o - - - - - - - - - . . In ~n
Ln o o O O o O O _I O O O O O O ~ ~ O N t~l
V V
~ I ~ CO ~ O ~D CO ~ ~D ~ ~ ~ _I ~ ~ u~ _~ ~ X U~
n O O ~D O ~1 0 ~ ') O ~ `1 ~ ~ O ~1 N
H ~ ~ V
,~
O
`1 0 0 ~ O ~ 1 0 0 ~D ~ ~ t~l t~ ~ O ~ I
U~
a~ - -o o - - .......... ..... ... O
O ~ ~ D O u~ _I O O O O O _I O O O O O O ~ ~ O ~ O
_~ ~ ~ o a~ o o o o a~ o a~
cS~ ~ ~c} ' C~ C ~ ~ ? c~
o o o o ~ C) ~5
S S S c~ o~ ~ O C~ C) C) ~ n CS ~ S~
C, C, c, ~, . C, ~ c5 C5 Cs C~ ~ 5C5 _~ ~ SO~ C~s
o C C C ~ ~ p p p p C~ C, p ~ ;S C5 ~o
O . r~ ~S- rS ~ ~ ~ E ~ C
0 C, C C O t C, C, O Cl C~ O ~ ~ ~ C5 ~5 C~ C~ C5 o O r~
_1 ~ ~ ~: ~ O O C P s~ r,~ 0 ~ ~ ~ ~ ~ C5 r.~ E E '
~ S S S ~ ~ ~ O ~ Q ~
rC~ ~ r~ c~ A- ~ r~ r~ r~ ~ r ~ ~ r ~ ~O
~ O ~ O O ~ O O O ~ ~ C C~p
GC15 5f
--258 -
1 338670
. .
O ~ D I O O ~O O _I O ~O O _I O O O _I ~ ~ ~ 0
V V _,
O . . . . . . . . . . a U~ ~ o
~ - -00 - ... - - - ............
~ ~oooo~o oo~Do~o ~ ~u~ oa~o
V V
I O CD ~ O t~ O ~ ~ ~ t~ co co ~r Q G 1
--I Ea~ - - o o - ~ - - - - - - - - - - - - - - o
D ~ o o o o o o o o o o o a o o o o o o o o ~
X" ~ v v v v
C~ t _~ ~1 ~ N O o ~ N O ~ O N ~ D O ~D ~D ~ N ~D N ~ Ul N
r
;
I ~ N ~C ~ O CO N ~ ~ CO ~9 0 ~D ~ ~ ~ ~D ~ ~
- I~ . 1-- . . . In O . . . . . ~ . . . . . . . . . ~ ~ . O O
N U') o ~ ~ O O O ~ --I O _I O ~ 0 O O
V V ~
- - - ~ o ~ o o o o o U ooo o - o
U O o o ~ c a o o I I ~ ~ I u~
o D r~ a~ o ~ ~ U7 1` r~ a~ ` r~ CO
_I ~ ~ o a~ a~ ~ co o o o o a~ ~ co a~ ,~ co co o a~
a
* ~l ~ ` * l ~` c
;~
* * ~* * ~ ~ * ~ o ~l ~ r
t4 Cl~ ;~ O O ~ - ~ O
S ~ r ~ ~ O O O O ~ S ~ t~
J ~ ~ ~ O D D
E3 ~ C ~ C o r~ e E ~
U~ C' O C, O t. ~ J O e~ O ~ O ~ o o ~
Q ~ O~ ~ ~ rO ~ ~ E E O
~ *
GC155f
-259- 1 3 3 8 ~ 7 ~
Co ~ O o ~ o
o.~ o U'~ . o o
aaoooooooooooa_~o~ I
_I _I V V _I
a a U ~ a ~ o
o~DU u a~o ~ ~d~u~N ~a
_I
F~ ~11 N 1~ ~ '1 ~ O O ~ t~ O U~ D ~ ~ O U~ ~0 ~ u~
X
-
O O ~ O ~ '
D U'l O O ~ D O ~) O ~ O' O ~--1 0 t~l ~ ~ L~l ~ ~ ~ ~ I
H
'5
. ~ U~ U~ ~ ` o O O U~ U~
U~ U~ U'l
,~, . . . ", . . . . .. . . . . . . . . . . . . . O
~ ~ ~ ~D ~ I _I _I _I O O O _1 0 0 0 0 0 0 0 ~ O _I O
_i V V V . .
_I ~ ~ o a~ o o o o a~ o a~ cn ~ ~ ~
r~O
S
., O
~*
.,., ., .~ rn ~* .~ ~0 r ~ ~j,
S S ;~ ~ C~ C~ Q S ~*
C~ C ~ r ~ C
S;~ S ~ C~O ~4~, ~. ro C _ S ~
Cl CS Cl CS C C~ '~ * * * * ~ E~ * ~ O ~ C ~ S C
S ~ ~ .* ~ .* ~ r~l~ tl~ ~ ~ C~
S O O O O ~ ~-* .~ 3
:S S ~ O O O O ~ CS ;:s~ S ~ C~ ~S
' O ~ . CS CS C3 CS ~ ~CS ~ C5~ o ~
e O ~ ~ O ~ E ~ O O ~ S
0 ~., " o ~ '- ~ '. o o o o ~ ~ ~ CS CS C~ O ~S o o ~
.a æ ~ s ~s.~
GC155f
-260- 1 3 3 8 6 7 0
.
_I ~ ~ ~ ~ _I ~ _I o o o o ~ ~ ~ ~ ~r ~
~D O O O - - - . . . o
oou~ odo o~o oclo oooo a~
v v v v v
u~u~ d ~o ~o d ~ c~ o o o a ~ ~ ~o
Ul
o a ~ . . . . . . . . . . . . O
I I I I u~c~a~ooooo-lo~D~u I
_~ _I v
a
_I
o o o o o
~ o 1 1 1 1 o u~ I I I I I I o
N - - O o ~ o
U ~ ~ ~ ~ ~ o ~ ~ o ~ o ~D ~ ~ o _~ o ~ ~ ~D N ~ ~ ~ O
H ~ ~ ~ V ,~
. ~
O
O ~ Ln O ~ o ~ U~ U~ ~ o
D Ll'l O O ~ N O ~ D O ~1 N N u~ N N N ~ ul
1~ ~D N ~D ~ ~ N.~ N N ~r co ~
~1 N N N O I N ~ _1 0 _I O _I O O O O O C ~ ~ ~ O N I
r~ ~ O ~ ~ O ~ N ~ ~ ~1 r ~ ~ ~ N
~S ~ ~ ~ o N ~ N ~ I ~ N O Ll~
_I N N O ~ o~ N C~ O O O O a~ O C~ ~ CSl . ) C~
~ C~
r~ t~ ~
S ;~ C O r~ r . C
5 ~ r~ C ~ ~ ~ rl _
;~S S ~ S ~ C ~ ~ o 6~ C O S ~
CS Cl ~ Cl ~ ~ * r~ ~ E r~ t ~ O ~; ~ r~ S C
~ O O O O ~ e. c3 $~ ~ )
~ S tO r ~ Cl C~
C~; O ~ ; ;S t3 C3 C3 C3 ~ D D ~ ~ L
C t-p ~ D E s ss ~.
U~ ~ O O ~ ~ " O O O U ~ 1-- ~ ~ C3 '3 ~ ~ O O ~:
O 0
d,3 ~ æ æ ~'
GC155f
-261- ~ 33867~
~r ~ ~ o ~ _I o o o o ~r co co co ~ a~ _~
U' o U~ - . - . . . o
, , , , , U~ ~ o o o o o o o o o o o o o o o o ~ .u~
V V V V V
o ~ ~ o ~ o U~ o
_, .
"~ o o o - - - - - - - - - ~ o o
~ o ~ o_~o~--Ioooo~ --~ ~
_1
I o ~ ~ _I o o o ~ ~ o o ~D I ~
X _, Z, I
O ~ ~ ~O O ~ ~ ~D ~ ~ el~ O ~ 00
o o . ... .. . ... ..
U ~ ~r ~ ~ ~ I I ~ ~D _I O ~ O ~ O O O O O O _I ~ ~ ~ O
u --l v v --l ~
H :1
. ~
o u~ In
I I O ~D ~ O O o O O O O o o O O _I O O _I ~ ~
_I V V ~--I
I O _~ O ~ O O O O O _I _I ~ --I
COO O O . . . . O U~
~ ou u~Daoooooooooooooooo~
_I _I V~ V V V V V V . . . . . . _~
o a~ a~ ~ ~ o o o o a~ ~ o a~
.~ r;s)
~, t ~, r~
rJ~ a~ r4 r" ~ ~ ~ ~ ` ~ '` ' r ``
S S S ~ C rJ~ t5 t~
S S ~ S t~ ~ O rJ~ r` ~ ' ~ ' r~ ~'`
C5 C5 ~ t5 r;~ ~ ~ ~ ~ '~ ~ ~ ~ ~ rn ~ -. G ~ S ~ S S t~
~- t' ~ ~ ~ ~ ~ ;~ ~ rO r~ C,
S O O O O S., ~ ~ ~ ~ ~ o 5
S S r~ ro ~ C) C) C)
G C; i C I t5 tt Ct tt ~ ~ ~ o 't5 G~
O ~ c ~ *-~ C~ S ~ rn ~ ~ r~ ~ C C~
~ ~ c, c, ~, O , ~ ~ ~ ~ ~ ~ E ~ ~ ~ C) ~ ~ S:
U~ I C C, O ~ O O ~ O ~ ~-- ~ C~ tt tt O C~ O O ~
o ~ D ~ A ~ C~ E E '
~ .; O O O c~ ~ ~D O0 0 ~
GC155f
-262- 1 3 3 8 6 7 0
~o ~ o ~ o
_I _I
It~ O O O
~1~ 1 1 1 1 1 1 1 1 It~ I I ' I I I O O I
_I _I _I
11 0 U~
~r o u~ u~ o ~ o o u~ o
U~ I I I I I I I 1~ ~ ~I t~l O ~ o ~ ~ ~ O O '1 0 1
p, u~ r o o o o o _I ~ co ~ ~ ~r
"
X ~
-
J Ir~ ~D ~ ~ O I U:~ ~ ~ O ~1 0 ~`I O O o O O ~1 o ~
r,) ~1 --1 ~ V ~ ~J
H
~ ~ D O ~ ~ ~ ~ 00 ~D O ~ 0 ~D ~ ~D ~ ~ n
-L~ O ' ' ' O ' ~ . . . . . . . . . . o
~D O I o ~ _I o o O ~ O o ~ O O O ~ ~ ~ ~ ~ ~ u~
~ ~l v v ~
a~. In . . . . . . . . . . . . . . . O O
D ~ I I ~ ~ ~ O o o ~ o _I--I O ~ ~ O ~ ~ ~ O
- ~ v - ~ v ~ - l
o u~ o 1` u~D 0 0 ~ U~
_I o ~ ~ ~ a:~ 0 ~ ~r In 0 ~ ~ ~ ~ o u~
o a~ al ~ co o o o o ~ ~ oo a~ _I ~ 0 o a~ 0 0
~
.~ ., ., ,, r~ , rJ~
;~ 5 S ;~ C~ r.~ *
r;~ r$~ r~ r; C ~ r~ * -. r;s r~ ~* rr
S S ~~ S
cs~ ~S CS ~ ~ ~ * * ~ * ~ r~ rr ;~ S n
"5 0 0 0 0
~ O C~ O O ~ S ~ r_~ CS
~, r ~ C~ C~ CJf C~ !a ~ rJ~ '
E~ c ~ ~ c ~ ~ * ~ C~ C- * -) s ~ n~ C~
tO C C ~ C ~ O O O O O ~ ~ ~ C' r ~ ~ ~ O O '
~ .3 0 0 0 ~ O O 0 ~ -p
1 338670 GC155f
--263--
~r _I co o N O O O O O _I _I 0 ~r ~ ~r ~
~O IIIII11Noooooooooooooooo_Io
u~I ~ u~ N ~ ~ N N ~ ~ ~ ~
U~ .--..oo
N N N I I ~O ~O ~ O N O _I O O O O O
- o U~ o o U7 o ~
~D OOoou~OOI~OIIInI~oIIIII~I
O er ~ ~ ~ 00 ~ ~ o ~ ~ ~ ~ ~ ~ ~ 0 u~
o ~ o
I O I O O O O O O O O O O O O O O O O ~ O
H
. ~
~4 a~ O o O
U~ II I I I I I I I O O I O I I I I I I I I I I I I
o o o U~ o O O o O o U~ o O o O o o
.
O ~ a~ O O O O a~ l c~ co O
S5 S `5 ~ C ~ '* ,,, r ;~
Cl C
SS S ~ ~ ~ ~ ~ ~
C~ ~ ~ '* '-' '~ '* ~ E ~ O ~, o S~ ~ C
O O O O 5~ o~5 ,~ , C
S ~ ~ o U C~ U ~ s~
o ~ ,, ~ ~ ~ ~ ~ ~ S~. C
~ IJ O ~'~ J * '* '-~ '~ ~ ~ '* ~
E~'. ~ ~ ''. o ~ .s~ ~ E ~ ~ ~ O ~ X ~
O rl r~ ; O O O O ~ O ~ C~ ~
~ O ~ * ~ m m m ~ ~
E 3 ~ ~ ~ .~ Q $ ~