Note: Descriptions are shown in the official language in which they were submitted.
- 1 ~ P' ~ ~ 3 ~
HOECHST AKTIENGESELLSCHAFT HOE 88/F 042 Dr.SW/St
Description
SpeciaL che-iluminescent acridine derivatives and the
use thereof in ~uminescence immunoassays
The present invention relates to chemiluminescent acri-
S dine derivatives, to processes for the preparation
thereof and to the use thereof in luminescence immuno-
assays.
Luminescent compounds already have a wide variety of
uses. They are employed as indicators in bioassays,
enzyme immunoassays and luminescence immunoassays (cf.
W.P. Collins "Alternative Immunoassays", published by
John Wiley ~ Sons Ltd., Chichester, 1985) but are also
used in nucleic acid hybridization assays (cf. J.A.
Matthews et al. "Analytical Biochemistry", 151, 205-209,
1985). In addition, chemiluminescent compounds are
employed in flow injection analysis, in post-column
detectors in liquid chromatography, in flow research and
in artificial light sources.
Chemiluminescent marker substances of two structural
types in particular have acquired relatively great sig-
nificance in chemiluminescence immunoassays. These are,
on the one hand, the derivatives of luminol and isolumi-
nol, which are described by H.R. Schroeder et al.,
"Methods in Enzymology", Academic Press Inc., Ne~ York,
Vol. LVII, 1978, 424 et seq., and in ~ritish Patents
2,008,247 and 2,041,920, German Patents 26 18 419 and
26 18 511, as well as European Patent Application 135,071.
A review of the use in practice of the isoluminol com-
pounds as luminescence indicators is to be found in
W.G. Wood, J. Clin.Chem. Clin. Biochem. 22, 1984 905-918.
On the other hand, acridinium ester compounds have also
2 ~ 3 ~ ~ Y ~ ~
been used as chemiluminescence marker substances. Such
acridinium esters are disclosed in US Patent 3,352,791,
British Patents 1,316,363 and 1,461,877 and European
Patent Application 82,636. The use of acridinium esters
as marker substances in immunoassays is described by
Weeks et al., Clin. Chem. 29/8 (1983), 1474-1479. The
use of phenanthridinium esters as marker substances in
luminescence immunoassays has also been disclosed, in
European Patent Application 170,415.
The chemiluminescence of acridinium esters can be initia-
ted by addition of alkaline H2~2 solution. A convincing
explanation of the mechanism of the chemiluminescence has
been given by F. McCapra, Acc.Chem. Res. 9, 201, 1976.
It is apparent from this that the nature of the leaving
group is crucial both for the quantum yield of light and
for the hydrolytic stability.
The acridinium esters which have hitherto been disclosed
have the advantage over the luminol and isoluminol com-
pounds that the quantum yield of light is higher and is
not adversely affected by proteins bound to the indicator
(cf. Weeks et al., Clin. Chem. 29/8 (1983), 1474-1479).
Although the acridinium phenyl esters disclosed in
European Patent Application 82,636 are distinguished by
a high detection sensitivity when the chemiluminescence
is excited by mild oxidizing agents, they have disadvan-
tages which interfere with practical use. In particular,
the phenyl ester linkage is very labile in aqueous
systems, even at room temperature. An additional factor
is that, under the oxidation conditions stated therein,
the acridinium phenyl esters show an emission of light
which has substantially, i.e. above 95%, disappeared only
after about 10 seconds. By comparison with this, other
non-isotopic assay methods have far shorter measurement
times and thus allow a higher sample throughput.
~ ~ 3 9 3 9 ~ z
-- 3
It has already been proposed to use chemiluminescent
acridinium derivatives which, together with a high quan-
tum yield of light, have more rapid reaction kinetics and
thus allow short measurement times for a luminescence
immunoassay.
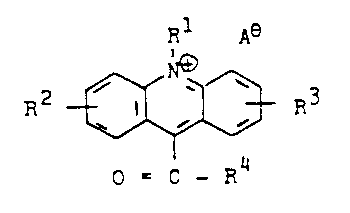
These take the form of acridinium derivatives of the
formula
R A~
RB --~3 RC
O = C-R
in which RA is hydrogen, an alkyl, alkenyl or alkynyl
radical having 1 to 10 carbon atoms, or a benzyl or aryl
group, RB and RC are hydrogen, an alkyl group having
1 to 4 carbon atoms, a substituted or unsubstituted amino
group, a carboxyl, alkoxy, cyano or nitro group or halo-
gen, RD represents a radical in which a sulfonamide
group is directly bonded via the nitrogen to the carbony~
group, or is a thioalkyl or thioaryl radical of the
formula
- S - Y - RE
where Y is a branched or unbranched aliphatic group or
an aromatic group which can also contain hetero atoms,
and RE is a reactive group which is able to undergo
bonding under mild conditions selectively with amino,
carboxyl, thiol or other functional groups in substances
of biological interest, and A~ is an anion which does
not adversely affect the chemiluminescence.
It has now emerged that special acridinium derivatives
are particularly suitable, by reason of their outstanding
stability and their unexpectedly high detection sensitiv-
ity, especially for use as chemiluminescent compounds.
.~
3 ~ 0
AccordingLy, the invention relates to chemiluminescent
acridinium derivatives of the formula I
R A~
R2 ~ R3 ( I )
O = C - R
in ~hich R1 is hydrogen, an alkyl, alkenyl or alkynyl
S radical having 1 to 10 carbon atoms, or a benzyl or aryl
group,
R2 and R3 are hydrogen, an alkyl group having 1 to 4
carbon atoms, a substituted or unsubstituted amino group,
a carboxyl, alkoxy, cyano or nitro group, or halogen,
R4 represents a radical of the formula II or III
R6
N/ (II)
SO -X-R
X-R
-N ~ ( III )
in ~hich R5 is a reactive group ~hich is able to undergo
bonding under mild conditions selectively with amino,
carboxyl, thiol or other functional groups in substances
of biological interest,
R6 is hydrogen, an alkyl, alkenyl or alkoxy radical
having 1 to 10 carbon atoms, a substituted amino group,
a benzyl group, an aryl group, a heteroalkyl group or a
heterocycle, each of ~hich can also be substituted by
hydroxyl, amino, alkylamino, alkyl, alkenyl or alkoxy
having 1 to 4 carbon atoms, polyalkoxy or aryloxy groups
or a heterocyclic group, it being possible for the last-
mentioned substituents in turn to be substituted by a
heterocyclic compound or an amine, or together to form a
~l3~9~
heterocycle having 0 and/or S and/or NH or N-alkyl, and
X denotesan arylene group ~hich is bonded to the nitrogen
or sulfur atom directly or via an alkylene or oxyalky~ne
group and is bonded to the radical RS via an alkylene or
S oxyalkylene group and vhich can also be substituted one
or more times by alkyl, alkenyl, hydroxyl, amino, alkoxy,
polyalkoxy or aryloxy groups and/or hetero atoms, or
denotes the radical of an aliphatic, araLiphatic or
aromatic, not necessarily natural, amino carboxylic acid,
or is a phenylene group when R6 is a phenyl group which
is substituted one or more times by C1-C6-alkyland the
quaternary ammonium compounds.
The substances of biological interest are to be under-
stood to include, in particular, antigens. This term
covers, for example, hormones, steroids, pharmaceuticals,
metabolites of pharmaceuticals, toxins, a~kaloids and
even antibodies.
The invention will now be described in relation to the
drawings, in which:
Figure 1 is a graph showing the result of a stability
test using the compounds of the invention in which the
intensity of the particular chemiluminescence signal was
measured after storage at elevated temperature (50~C);
Figure 2 is a graph showing the result of a stability
test using the compounds of the invention in which the
intensity of the particular chemiluminescence signal was
measured after storage at a elevated temperature of 4~C;
and
Figure 3 is a graph showing the typical shape of a
standard plot of an immunochemiluminometric assay (ICMA)
for human thyroid-stimulating hormone (h-TSH) using the
compounds of the invention.
_ 5 A -
Examples of suitable aminocarboxy(ic acids are glycine,
alanine, serine, phenylalanine, histidine, ~-aminobutyric
acid, methionine, valine, norvaline, leucine, iso-leucine,
norleucine, aspartic acid, glutamic acid, 4-aminobenzoic
S acid, 4-aminophenylacetic acid, 4-aminophenoxyacetic acid
and 3-t4-amino)phenylpropionic acid.
The anion ~hich does not adversely affect the chemi-
luminescence can be, for example, a tetrafluoroborate,
perchlorate, halide, alkylsulfate, halosulfonate, alkyl-
sulfonate or arylsulfonate anion. It is also possible
for any other anion to be employed as long as it does not
quench or diminish the chemiluminescence.
The heteroalkyl groups or heterocyclic groups preferably
contain hetero atoms ~hich can contribute to increasing
the solubility of the compounds according to the inven-
tion in uater, such as, for example, nitrogen, oxygen,
sulfur, phosphorus or combinations thereof. Examples of
particularly suitable heterocycles are morpholine,
3 ~ ~
,.
-- 6
piperazine, piperidine, tetrahydrofuran, dioxanes etc.
Particularly important acridinium derivatives are those
which are claimed in claim 1 and in which X is a group
of the formula IV
~,1 0 9
- R ~ 8 (IV)
R
R1 1
in which R7 is a substituent of the formula
-(CH2)n- or ((CH2)m-O)n- , with n = 0 to 4
and m = 1 to 6,
R8 is a substituent in the ortho, meta or para position
to R7 of the formula -(CH2)p-, a polyalkylene oxide
group of the formula -(0-(CH2)m-)p or -((CH2)m-0-)p,
preferably with p= 1-6 and m= 1-6, or a branched or un-
branched hydrocarbon radical having 1 - 4 carbon atoms,
and the substituents R9 - R11 are hydrogen or straight-
chain or branched hydrocarbon radicals having up to 30carbon atoms, it also being possible for one or more
-CH2- units to be replaced by 0, S, S0, S02, NH or N-
alkyl, and for two of these substituents to be linked to
- form a ring.
Particularly important for the possibilities of employing
the acridinium derivatives according to the invention is
the substituent R5. Suitable choice of this group
results in the acridinium derivative having a reactivity
which is so high that it is able to undergo bonding even
under mild conditions selectively with a functional group
of the biological substance which is to be detected.
Suitable reactive groups are shown in the list which
follows:
o ~
a ) - C - O - N,~
O SO ~~) Y(+)
~1 ~ ~ 3
b) -C - O - N
~ Y = H , alkali metal(
~ ~
c) -C - O - N
d )- S O - CH = CH
(+)
e ) 2 2 2 ~ halide
f) -N = C = S
(+)
h) C~ 2 halide(
\ O-R
o
o
Cl Cl ~ ~ 3~3~
k) -C - O - ~ - Cl
Cl Cl
1 ) e - O - ~ -NO
~
m) -C- N~
O N
Il ll2
n) -C - C - CF3
O ~
o) -C-N S\
S
In many cases, acridinium derivatives according to the
invention which have proven suitable are those in which
R5 is a group of the formula V
O
O ~ ( V )
- C - O - N
0~
Furthermore, acridinium compounds of the formula VI have
proven to be particularly suitable. Formula VI is
N~3 A
~ R1 3
~C ~ (VI)
\ SO - X- - C - O - N ~
in which X is a group o~ the formula VII
-~-(CH ) _ (VII)
with n= 2 or 4,
and R12 and R13 are, independently of one another,
hydrogen, an alkyl group, an alkoxy group having 1 - 4
carbon atoms, a (-0-CH2-CH2)n-OR group, where n has
the meaning 0 - 8 and R is a morpholinoethyl or an alkyl
group having 1 - 4 carbon atoms or an N,N-dimethylamino-
ethyl group,
or are together an ethylenedioxy group,
and A~ has the meanings mentioned in claim 1. These
compounds are products which are readily soluble in
water.
Among the last-mentioned compounds of the formula VII,
those which are in turn very particularly preferred are
those in which X is a p-ethylenephenyl group, R12= H and
R13 is a p-methoxy group, or R12 is an ortho-methoxy
and R13 is a para-methoxy group, or R12 and R13 are
together a 3,4-ethylenedioxy group, such as, for example,
the compounds of the formula
~3
/C ~ OCH3
So - ~ 2 2 ~ O N
Further particularly suitable acridinium derivatives have
the formula VIII
- 10 -
1 3
~ O
~Y o (VIII)
// ~ / ~ o N~
in which R6 is an alkyl group having 1-4 carbon atoms
or a phenyl group which can be substituted by up to three
alkyl or alkoxy groups, each having 1-4 carbon atoms, by
a -(-O-CHz-CH2)n-OR group, where n has the meaning
0-8 and R is a morpholinoethyl or an N,N-dimethylamino-
ethyl group or alkyl group having 1 - 4 carbon atoms, or
by an ethylenedioxy group, and X has the meanings men-
tioned in claim 1 or 2, or in which R6 is a phenyl group
which can be substituted by up to three alkyl groups each
having 1-4 carbon atoms, and X- is an ortho-, meta- or
para-phenylene group. These compounds are also products
which are readily soluble in water.
It is surprising that acridinium-9-carboxamides substitu-
ted by sulfonyl on the amide nitrogen exhibit excellent
chemiluminescence, because it is known that, in contrast
to the acridinium-9-carboxylic esters, acridinium-9-
carboxamides show no chemiluminescence whatever (cf.
F. McCapra in ~. Carruthers and J.K. Sutherland: Progress
in Organic Chem., Vol. 8, 231-277, 1973, Putterworth,
London).
A significant advantage of the acridinium compounds
according to the invention compared with the acridinium
phenyl esters disclosed in European Patent Application
82,636 lies in the considerably more rapid reaction
kinetics of light emission.
Another advantage is provided by the stability of the
tracers prepared with the aid of the compounds according
to the invention. Figure 1 shows the result of a stabil-
ity test in which the intensity of the particular chemi-
luminescence signal was measured after storage at ele-
vated temperature (50~C). Curve 1 relates to tracers
prepared from the compound a) N-(4-methoxyphenyl)-N-[4-
(2-succinimidyloxycarbonylethyl)benzenesulfonyl]-10-
methylacridinium-9-carboxamide fluorosulfonate (6),
curve b) relates to tracers prepared from the compound
N-(4-methoxyphenyl)-N-[4-(4-succinimidyloxycarbonylbutyl)-
benzenesulfonyl~-10-methylacridinium-9-carboxamide
fluorosulfonate (11) and curve c) relates to the tracers
prepared from the compound 4-(2-succinimidyloxycarbonyl-
ethyl)phenyl-10-methylacridinium-9-carboxylate metho-
sulfate (European Patent Application 82,636, page 10).
It is clearly evident that the tracers, according to the
invention, from the compounds a) and b) are more stable
than the corresponding compounds from c).
A similar result is obtained in a corresponding test at
4~C. Figure 2 shows the signal intensity after storage
at 4~C. Once again, the tracer prepared from compound
a) proves to be distinctly more stable than that prepared
from compound c).
The acridiniumsulfonamide derivatives according to the
invention can be prepared starting from acridine-9-car-
bonyl chloride (IX). To prepare the latter, for example
acridine is reacted with potassium cyanide in ethanol/
glacial acetic acid by the method indicated by Lehmstedt
and Hundertmark in Ber. 63, 1229 (1930) to give 9-cyano-
acridine. From this is obtained, preferably after re-
crystallization, by reaction with sulfuric acid and
sodium nitrite by the method described by Lehmstedt and
~irth in Ber. 61, 2044 (1928), acridine-9-carboxylic acid.
Reaction of acridine-9-carboxylic acid with, for example,
thionyl chloride results in the compound of the formula IX
- 12 -
[ ~ i (IX)
S O y
in which Y has the meaning of chlorine. It is also pos-
sible, in place of a halogen, to introduce for Y in the
compound IX a hydroxycarbonylalkyl, hydroxycarbonylaryl
or imidazolide group.
1 0
The acid chloride (IX) can then be reacted with a pro-
tected sulfonamide carboxylic acid of the formula X
6 H O
R - S02 - N - X - C - OZ (X)
or of the formula XI
2 0 R - l~t - SO - X - COOZ ( X I )
in which X and R6 have the abovementioned meanings, and
Z is a radical which protects the carboxyl group and is
subsequently eliminated. It is possible to employ for
25 this reaction, for example, N-(4-benzoxycarbonylphenyl)-
N-4-toluenesulfonamide. It is advantageous to use the
t-butyl esters, whose protective group can be introduced
and eliminated again under particularly mild conditions.
The acid produced after elimination of the protective
30 groups is then converted, using a suitable compound, for
example using N-hydroxysuccinimide, into the radical R5.
From this is obtained the chemiluminescent acridine
compound by alkylation on the nitrogen in the 10-position.
35 The resulting acridinium compounds can then be reacted
with a substance of biological interest, for example an
antigen, an antibody, a hormone, a pharmaceutical, a
metabolite of a pharmaceutical, a toxin or an alkaloid,
3 ~ ~
- 13 -
to give a luminescent compound. This entails the acrid-
inium derivative being bonded either directly or via a
bridging molecule, such as, for example, amino acids,
oligo- or polyamino acids, peptides or synthetic polymers,
to the biologically interesting substance, with the for-
mation of a stable immunologically active conjugate.
This conjugate is also called tracer and is employed in
the luminescence immunoassays described hereinafter.
Required for the luminescence immunoassay according to
the invention for the determination of an antigenic sub-
stance in a liquid sample by a competitive or a sandwich
method is at least one immunologically active component
which is immobilized on a solid phase, and, in addition,
the luminescent tracer.
After the immunological reaction and any washing steps
which are required are complete, the light emission is
initiated by successive or simultaneous addition of one
or more reagents, with at least one reagent containing
an oxidizing agent in bound or unbound form. It is now
possible to carry out the luminescence immunoassay in a
variety of ways.
One possibility comprises incubation of the immobilized
antibody, which reacts specifically with the antigen,
with a sample of the liquid which is to be investigated,
and with a conjugate composed of the antigen and of a
chemiluminescent acridinium derivative (antigen tracer),
separation of the sample and the unbound tracer, contact-
ing the bound tracer with the reagents necessary to bringabout light emission, and then determination of the
amount of antigen present from the measured intensity of
light emission.
Another possibility for carrying out the luminescence
immunoassay comprises incubation of an immobilized anti-
body, which reacts specifically with the antigen, with a
sample of the liquid which is to be investigated and with
~3~
- a conjugate composed of a second specifically reacting
antibody and of a chemiluminescence acridinium derivative,
separation of the sample and the unbound conjugate with
marker, contacting the bound conjugate with marker with
the reagents necessary to bring about light emission, and
determination of the amount of antigen present from the
measured intensity of light emission.
The abovementioned luminescence immunoassays can also be
carried out in such a way that the liquid which is to be
investigated is separated from the immobilized antibody
before the addition of the conjugate with marker.
In another luminescence immunoassay which can be carried
out according to the invention, it is not the antibody
but the antigen which is immobilized. Thus, it is pos-
sible for an immobilized antigen, which reacts specific-
ally with the antibody, to be incubated with a sample of
the liquid which is to be investigated and with a solu-
tion of a conjugate composed of the antibody and of achemiluminescent acridinium derivative, and for the
sample and the unbound conjugate with marker then to be
separated, and then the bound conjugate with marker then
to be contacted with the necessary reagents. Light
emission then occurs, and the amount of antigen present
can be determined from the intensity thereof.
Another variant comprises incubation of an immobilized
antigen, which reacts specifically with the antibody,
with a solution of a conjugate composed of the antibody
and of a chemiluminescent acridinium derivative, separat-
ing off the unreacted conjugate with marker, addition of
a sample of the liquid which is to be investigated, sub-
sequently separating off the sample again, contacting the
bound conjugate with marker with the reagents necessary
to bring about light emission, and then determination
from the latter of the amount of antigen present.
~ ~ 3 ~
- 15 -
FinaLly, the luminescence immunoassay can also be carried
out in such a way that an immobilized antigen, which
reacts specifically with the antibody, is incubated with
a solution of a conjugate composed of the antibody and
S of a chemiluminescent acridinium derivative, a sample of
the liquid which is to be investigated is added, the
sample and the unbound conjugate are separated, the bound
conjugate with marker is contacted with the necessary
reagents, and then the amount of antigen present is
determined from the measured light emission.
The preparation of the acridinium compounds according to
the invention is explained in detail in Examples 1 to 7.
Example 1:
N-(4-Methoxyphenyl)-N-[4-(2-benzyloxycarbonylethyl)-
benzenesulfonyl]acridine-9-carboxamide (3)
460 mg of 4-(N,N-dimethylamino)pyridine and 22.1 ml of
triethylamine are added to 17 g of benzyl 4'-tN-(4-meth-
oxyphenyl)sulfamido]-3-phenylpropionate (1) in 400 ml of
dichloromethane and, after 10 min, 11.12 g of acridine-
9-carbonyl chloride hydrochloride (2) are added, and the
mixture is refluxed for 6 hours. The cooled solution is
briefly stirred with 2 N NaOH, and the organic phase is
separated off, washed with H20, dried over magnesium
sulfate and concentrated. The residue is purified by
column chromatography.
Yield: 60% Melting point: 130 - 132~C
NMR (DMSO, 100 MHz):~ = 2.7-3.0 ppm (d,br,2H), ~= 3.0-
3.3 ppm (d,br,2H), ~=3.5 ppm (s,br,3H), ~=5.1 ppm (s,2H),
~= 6.5 ppm (d,br,2H),~ =7.1 ppm (d,br,2H),~ = 7.35 ppm
(s,5H),~= 7.5-8.3 ppm (m,12H).
N-(4-Methoxyphenyl)-N-~4-(2-carboxyethyl)benzenesulfonyl]-
acridine-9-carboxamide hydrobromide (4)
6.3 9 of (3) in 30 ml of 33% HBr/glacial acetic acid are
15a
~N ~ (3)
~ 'D ''
~\/ --CH3
O~N~/ ~\
O ~ 0~
~ O
~ ~ 3 ~
15b
,~N~ (41
~ O
~\/ CH3
O~N~/
o=~ OH
- 16 -
heated at 60~C for 2 hours and, after cooling, 60 ml
of diisopropyl ether are added, and the precipitate is
filtered off with suction and dried in vacuo:
Yield: 90% Melting point: decomposition 237~C
NMR (DMS0, 100 MHz):~ = Z.7 ppm (d,br,ZH),~ = 3.1 ppm
(d,br,2H),~ = 3.5 ppm (s,br,3H),~ = 6.5 ppm (d,br,2H),
~= 7.0-8.4 ppm (m,15H)
N-(4-Methoxyphenyl)-N-t4-(2-succinimidyloxycarbonylethyl)-
benzenesulfonyl]acridine-9-carboxamide (5)
1.41 ml of triethylamine are added to 3.1 g of (4) in
50 ml of tetrahydrofuran, the mixture is cooled to -20~C,
and 0.474 ml of ethyl chloroformate is added. After
stirring for 20 min, 575 mg of N-hydroxysuccinimide are
added, and the mixture is stirred at -20~C for 3 hours
and left to reach room temperature overnight ~hile stirr-
ing. The precipitate is filtered off with suction, the
filtrate is concentrated, the residue is taken up in
dichloromethane or ethyl acetate, and the resulting solu-
tion is ~ashed ~ith ~ater, NaHC03 solution and ~ater
and dried over MgS04. The organic phase is concentra-
ted, and the residue is recrystallized from toluene.
Yield: 50%
NMR (DMS0, 100 MHz):~ = 2.8 ppm (s,4H),~ = 3.2 ppm (s,br,
4H), ~ = 3.5 ppm (s,Br,3H),~ = 6.5 ppm (d,br,2H),~ = 7.2
ppm (d,br,2H),~ = 7.6-8.4 ppm (m,12H)
IR: 3400 cm 1 (br), 3060, 2930, 1815(w), 1780(~),
1740(s), 1690(m), 1600(~), 1510(m), 1370(m), 1250(m),
1203(m), 1175(m).
N-(4-Methoxyphenyl)-N-[4-(2-succinimidyloxycarbonylethyl)-
benzenesulfonyl]-10-methylacridinium-9-carboxamide
fluorosulfonate (6)
0.4 ml of methyl fluorosulfonate is added at -20~C to
1.27 g of (5) in 60 ml of dichloromethane. The mixture
is left to stir at -20~C for 2 hours and to reach room
16a
~,~N~ ~5
~,~~
.~/ --C H 3
O~N~ o
O ~;~.. ~ ~N~
O
O ~~_/
Q ~ ~ ~
16b
C H 3
,~/ --C H 3
~~-~(ag
- 17 -
- temperature overnight. Addition of toluene results in
precipitation of a yellow solid ~hich is filtered off
~ith suction and dried in vacuo.
Yield: 80%
NMR (DMS0, 100 MHz):~ = 2.9 ppm (s,4H),~ = 3.2 ppm (s,br,
4H),~ = 3.5 ppm (s,br,3H),~ = 4.8 ppm (s,br,3H),~ = 6.5
ppm (br,2H),~ = 7.2 ppm (br,2H),~ = 7.6-9.0 ppm (m,12H)
IR= 3400 cm 1 (br), 3160, 2970, 1810(w), 1785(~),
1740(s), 1695(m), 1600(~), 1555(~), 1510(m), 1370(m),
1290(m), 1250(m), 1210(m), 1170(m)
Mass spectrum: m/z: 652 M (cation)
Example 2:
The preparation of N-(4-methoxyphenyl)-N-~4-(4-succin-
imidyloxycarbonylbutyl)benzenesulfonyl]-10-methylacrid-
inium-9-carboxamide fluorosulfonate (11) starting from
benzyl 4'-tN-(4-methoxyphenyl)sulfamido]-5-phenylvalerate
(7) and acridine-9-carbonyl chloride hydrochloride (2) is
carried out in analogy to the synthesis of (6). The
yields in the individual steps in the synthesis, and the
spectroscopic characterization are indicated hereinafter.
N-(4-Methoxyphenyl)-N-[4-(4-benzyloxycarbonylbutyl)-
benzenesulfonyl]acridine-9-carboxamide (8)
Yield: 40% viscous oil, partially solidifies
NMR (CDCl3, 100 MHz): ~= 2.85 (m,4H), ~= 2.45 ppm (t,
br,2H), ~= 2.8 ppm (t,br,2H), ~= 3.5 ppm (s,3H),~ = 5.15
ppm (s,2H),~ = 6.3 ppm (d,2H),~ = 6.9 ppm (d,2H),~ = 7.3-
8.3 ppm (m,17H).
N-(4-Methoxyphenyl)-N-~4-(4-carboxybutyl)benzenesulfonyl]-
acridine-9-carboxamide hydrobromide (9)
Yield: 95X Melting point: decomposition 153-5~C
NMR (DMS0, 100 MHz):~ = 1.7 ppm (s,br,4H),~ = 2.3 ppm
(t,br,2H),~ = 2.8 ppm (s,br,2H),~ = 3.5 ppm (s,br,3H),
~= 6.5 ppm (br,2H),~ = 7.05 ppm (br,2H),
17a
~N~ ~)
,~\/ --CH3
O~N ~ ,~\
o=1~ ~~~
O O
17b
. .
~N ~ t~J
~~
~\/ --C H 3
O~N~/
o=~/ \> ~,~OH
- 18-
~= 7.5-8.5 ppm (m,12H).
N-(4-Methoxyphenyl)-N-[4-(4-succinimidyloxycarbonylbutyl)-
benzenesulfonyl]acridine-9-carboxamide (10)
Yield: 25% Melting point: decomposition 75 - 80~C
NMR (DMS0, 100 MHz): ~= 1.8 ppm (br,4H),~ = 2.3 ppm (s,
2H),~ = 2.85 ppm (s,br,6H),~ = 3.5 ppm (s,br,3H),~ = 6.5
ppm (d,br,2H),~ = 7.05 ppm (d,br,2H),~ = 7.5-8.3 ppm
(m,12H)
N-(4-Methoxyphenyl)-N-[4-(4-succinimidyloxycarbonylbutyl)-
benzenesulfonyl]-10-methylacridinium-9-carboxamide fluoro-
sulfonate (11)
Yield: 90%
NMR (DMS0, 100 MHz):~ = 1.8 ppm (br,4H),~ = 2.3 ppm (s,
br,2H),~ = 2.8 ppm (s,br,6H),~ = 3.5 ppm (s,br,3H),
~= 4.8 ppm (br,3H),~ = 6.5 ppm (br,2H),~ = 7.05 ppm (br,
2H),~ = 7.5-9.0 ppm (m,12H)
IR: 3340 cm 1 (br), 3060(w), 2930(m), 2870(w), 1810(w),
1785(w), 1740(s), 1695(m), 1600(w), 1550(w), 1510(m),
1460(w), 1370(m), 1290(s), 1250(s), 1205(s), 1170(s)
Mass spectrum: m/z = 680 M (cation)
Example 3:
The preparation of N-(2,4-dimethoxyphenyl)-N-[4-(2-
succinimidyloxycarbonylethyl)benzenesulfonyl]-10-methyl-
acridinium-9-carboxamide fluorosulfonate (16a) starting
from benzyl 4'-[N-(2,4-dimethoxyphenyl)sulfamido]-3-
phenylpropionate (12a) and acridine-9-carbonyl chloride
hydrochloride (2) is carried out in analogy to the syn-
thesis of (6). The yields in the individual steps in the
synthesis, and the spectroscopic characterization are
indicated hereinafter.
N-(2,4-Dimethoxyphenyl)-N-[4-(2-benzyloxycarbonylethyl)-
benzenesulfonyl]acridine-9-carboxamide (13a)
18a - -
(10)
~ O
~/ CH3
O~N~ O
0=~ 0 ~
O
~ 0~
Q
- 18b
H3C
,~N~ ~1 1)
,~\/ --C H 3
O=S~O
O
18c
,~N ~ (1 3aJ
~'
H3C~ ~~ ~CH3
O~N~/ D\
0=~ ~ ~
O
3 ~ ~
- 19 -
Yield: 50% Melting point: 74~C
NMR (DMS0, 100 MHz): ~= 2.9 ppm (d,br,2H), ~= 3.1 ppm
(d,br,2H), ~= 3.3 ppm (s,3H), ~= 3.4 ppm (s,3H),~ = 5.1
ppm (s,2H), ~= 5.9-6.2 ppm (m,2H), ~= 7.0 ppm (d,1H),
~= 7.35 ppm (s,5H), ~= 7.5-8.2 ppm (m,12H)
N-(2,4-Dimethoxyphenyl)-N-[4-(2-carboxyethyl)benzene-
sulfonyl]acridine-9-carboxamide hydrobromide (14a)
Yield: 95%
NMR (DMS0, 100 MHz): ~= 2.75 ppm (d,br,2H), ~= 3.05 ppm
(d,br,2H),~ = 3.3 ppm (s,3H),~ = 3.5 ppm (s,3H),~ = 5.95-
6.3 ppm (m,2H), ~= 7.05 ppm (d,1H), ~= 7.6-8.6 ppm
(m, 12H), ~= 9.2 ppm (s,br,2H).
N-(2,4-Dimethoxyphenyl)-N-C4-(2-succinimidyloxycarbonyl-
ethyl)benzenesulfonyl]acridine-9-carboxamide (15a)
Yield: 45% Melting point: -105~C decomposition
NMR (DMS0, 100 MHz): ~= 2.9 ppm (s,4H), ~= 3 ppm (br,2H),
~= 3.2 ppm (s,3H), ~= 3.4 ppm (s,3H), ~= 5.9-6.3 ppm
(m,2H), ~= 7.0 ppm (d,1H), ~= 7.5-8.4 ppm (m,12H)
IR (KBr disk): 3440 cm 1 (br), 3060 (w), 2930(w),
2850(w), 1815(w), 1785(w), 1740(s), 1695(m), 1600(w),
1510(m), 1460(w), 1440(w), 1365(m), 1320(w), 1290(w),
1240(m), 1210(s), 1165(m), 1085(m)
N-(2,4-Dimethoxyphenyl)-N-C4-(2-succinimidyloxycarbonyl
ethyl)benzenesulfonyl]-10-methylacridinium-9-carboxamide
fluorosulfonate (16a)
YieLd: 80% MeLting po;nt: 135~C decomposition
NMR (DMS0, 100 MHz): ~= 2.9 ppm (s, 4H),~ = 2.95-4.2 ppm
(m 10H), ~= 4.8-5.0 ppm (s,s,3H),~ = 6.05-6.25 ppm (m,1H),
~= 7.6-9.0 ppm (m,14H)
IR (KBr disk): 3430 cm 1(m), 2950(w), 2870(w), 2825(w),
1810(w), 1780(m), 1750(s), 1695(m), 1610(m), 1555(w),
1510(m), 1465(m), 1380(m), 1285(m), 1250(m), 1210(s),
l9a
,~N~ (1 4aJ 1~
~ '
H 3C~O~/ ~C H 3
~OH
O
Q
l9b
,~N~ (1 5a,
~S'
H3C~ ~ ~CH3
0~/~ 0
~=l~~-~
O
l9c
3 1 So3~ (16a)
H 3C'O~O~C H 3
C~N~~ O
- 20 -
1170(m)
Exa-ple 4:
The synthesis of N-(3,4-ethylenedioxyphenyl)-N-[4-(2-
succinimidyloxycarbonylethyl)benzenesulfonyl]-10-methyl-
acridinium-9-carboxamide fluorosulfonate (16b) starting
from benzyl 4'-~N-(3,4-ethylenedioxyphenyl)sulfamido]-3-
phenylpropionate (12b) and acridine-9-carbonyl chloride
hydrochloride (2) is carried out in analogy to Example 1.
The yields in the individual steps and the spectroscopic
characterization are indicated hereinafter.
N-(3,4-Ethylenedioxyphenyl)-N-[4-(2-benzyloxycarbonyl-
ethyl)benzenesulfonyl]acridine-9-carboxamide (13b)
Yield: 50% Melting point: 91.5~C
NMR (DMS0, 100 MHz):~ = 2.9 ppm (d,br,2H),~ = 3.1 ppm (d,
br,2H),~ = 4.0 ppm (s,Br,4H),~ = 5.1 ppm (s,2H), ~= 6.3-
6.8 ppm (m,3H),~ = 7.3 ppm (s,5H),~ = 7.6-8.3 ppm (m,12H).
N-(3,4-Ethylenedioxyphenyl)-N-~4-(2-carboxyethyl)-
benzenesulfonyl]acridine-9-carboxamide hydrobromide (14b)
Yield: 95% Melting point: > 200~C
NMR (DMS0, 100 MHz):~ = 2.7 ppm (m,2H),~ = 3.05 ppm (m,
2H),~ = 4.0 ppm (s,br,4H),~ = 6.3 - 6.8 ppm (m,3H),~ =
7.5-8.6 ppm (m,12H),~ = 9.6 ppm (s,br,2H).
N-(3,4-Ethylenedioxyphenyl)-N-[4-(2-succinimidyloxy-
carbonylethyl)benzenesulfonyl]acridine-9-carboxamide (15b)
Yield: 45% Melting point: 140~C decomposition
NMR (DMS0, 100 MHz): ~= 2.7-2.9 ppm (d,s, overlapping,
6H), ~= 3.0 ppm (d,2H), ~= 4.0 ppm (s,br,4H), ~= 6.3-6.8
ppm (m,3H), ~= 7.5-8.4 ppm (m,12H)
IR(KBr disk): 3420 cm 1 (br), 3060(m), 2980(m), 2930(m),
1810(w), 1790(w), 1740(m), 1695(s), 1590(m), 1460(w),
1430(w), 1410(w), 1370(m), 1300(m), 1225(s), 1175(s).
20a
~\/~ 113b~
~ O~
~0
O~\N~ /~
o=~ o
O
20b
~N~ (14b)
~ O~
~0
O/~N~/
O S~ ~OH
20c
,~N~ pSbJ
~ o~
~0
~ ~ 3 ~
N-(3,4-Ethylenedioxyphenyl)-N-[4-(2-succinlmidyloxy-
carbonylethyl)benzenesulfonyl]-10-methyl acridinium-9-
carboxamide fluorosulfonate (16b)
Yield: 80% Melting point: ~110~C, decomposition
NMR (DMS0, 100 MHz): ~= 2.85 ppm (s,4H),~ = 3.0-3.3 ppm
(s,s,br,4H),~ = 3.8-4.5 ppm (m,br,4H),~ = 4.75-5.1 ppm
(s, br with shoulder, 3H), ~= 6.3-9.0 ppm (m,15H).
Example 5:
N-(4-Carboxyphenyl)-4-toluenesulfonamide (5 - 1)
A mixture of 190.5 g (1 mole) of 4-toluenesulfonyl
chloride in 300 ml of i-propyl ether is added dropwise
at 20 - 30~C to one of 252 g (3 mole) of sodium bi-
carbonate and 139.1 g (1 mole) of 4-aminobenzoic acid in
2.5 l of water. The mixture is stirred vigorously for
2-4 hoùrs, until the sulfonyl chloride has been consumed.
The aqueous solution is separated off and then adjusted
to pH 1 with concentrated hydrochloric acid, and the
precipitate is taken up in propyl acetate. The extract
is washed 2x with 2N hydrochloric acid and 1x with water,
dried over sodium sulfate and evaporated. 240 g (82.5%
of theory) of N-(4-carboxyphenyl)-4-toluenesulfonamide
are obtained.
1H NMR (DMS0-d6): ~= 2.3 (s; CH3); 7.1 d, aromatic,
2H) 7.3 (d, aromatic, 2H); 7.6-7.9 (m, aromatic, 4H);
10.75 (broad); 12.7 (broad).
N-(4-Benzyloxycarbonylphenyl)-4-toluenesulfonamide (5 - 2)
A solution of 11.64 9 (40 mmol) of N-(4-carboxyphenyl)-
4-toluenesulfonamide, 5.06 g (40 mmol) of benzyl chloride
and 5.20 g (44 mmol) of di-i-propylethylamine in 100 ml
of dimethylformamide is heated at 140~C for 4 hours.
After the reaction is complete, the mixture is evaporated
in vacuo, the residue is taken up in propyl acetate, and
the solution is washed 2x with 2N hydrochloric acid and
21a
S ~3 ~ (1 6b)
,~ ~
~~
~0
O~N o
~=b~~-~7
o
21b
(5- 1 )
H3C~ COOH
O ~
21c
(5-2)
--S//
2x with saturated NaHC03 solution, dried over sodium
sulfate and evaporated. 11.8 9 (78% of theory) of N-(4-
benzyloxycarbonylphenyl)-4-toluenesulfonamide, which are
recrystallized from methanol, are obtained.
1H NMR (DMS0-d6):~ = 2.3 (s; CH3); 5.3 (s, CH2);
7.1-7.3 (dd, 4 aromatic H); 7.4 (s, C6Hs); 7.7-7.9
(dd, 4 aromatic H); 10.8 (broad, NH).
N-(4-Benzyloxycarbonylphenyl)-N-(4-toluenesulfonyl)-
acridine-9-carboxamide (5 - 3)
2.1 ml (15 mmol) of triethylamine in 10 ml of tetrahydro-
furan are added dropwise at 25~C to a solution of 1.5 9
(4 mmol) of N-(4-benzyloxycarbonylphenyl)-4-toluene-
sulfonamide, 1.23 9 (4.4 mmol) of acridinecarbonylchloride hydrochloride and 0.02 9 of dimethylaminopyri-
dine in 20 ml of anhydrous tetrahydrofuran. The tempera-
ture is raised to 60~C.
The product which has crystallized out is, after the
reaction is complete, stirred with methanol, filtered off
with suction and recrystallized from ethyl acetate.
Yield: 1.57 9 (67.0~ of theory)
1H NMR (CDCl3): ~= 2.5 (s, CH3); 5.2 (s, CH2); 7.3
(s, C6Hs), 7.0-8.2 (m, 16 aromatic H).
N-(4-Carboxyphenyl)-N-(4-toluenesulfonyl)acridine-9-
carboxamide hydrobromide (5 - 4)
1.17 9 (2 mmol) of N-(4-benzyloxycarbonylphenyl)-N-(4-
toluenesulfonyl)acridine-9-carboxamide are stirred with
10 ml of 33% strength solution of hydrogen bromide in
glacial acetic acid while heating at 60~C for 4 hours
After cooling, the precipitate is filtered off with
suction and dried in vacuo. Yield: 1.00 9 (87% of
theory)
H NMR (TFA):~=2.6 (s, CH3); 7.3-8.6 (m, 16 aromatic
H); 11.65 (s, NH); MS: 496 (M ).
22a
(5-3J
O=
~CH3
2 2 b
,~N~ (5-4)
~,
O -
//--
H O SS~ ~
o CH3
- 23 -
N-t4-Succinimidyloxycarbonylphenyl)-N-(4-toluenesulfonyl)-
acridine-9-carboxamide (5 - 5)
0.11 9 (1 mmol) of ethyl chloroformate is added, while
S stirring at -15~C, to a solution of 0.57 9 (1 mmol) of
N-(4-carboxyphenyl)-N-(4-toluenesulfonyl)acridine-9-
carboxamide hydrobromide and 0.21 9 (2 mmol) of triethyl-
amine in 25 ml of anhydrous tetrahydrofuran. The mixture
is stirred at the same temperature for 1 hour and then
0.12 9 (1 mmol) of N-hydroxysuccinimide is added. After
a further hour, the mixture is left to stand at room
temperature for 15 hours. It is evaporated in vacuo,
the residue is taken up in ethyl acetate, and the solu-
tion is washed with water, sodium bicarbonate solution
and water and dried over sodium sulfate. Evaporation
yields 0.42 9 (70.8% of theory) of the desired product.
1H NMR (TFA): ~= 2.6 (s, CH3), 3.1 (s, CH2-CH2),
7.0-8.6 (m, 16 aromatic H).
N-(4-Succinimidyloxycarbonylphenyl)-N-(4-toluenesulfonyl)-
10-methylacridinium-9-carboxamide fluorosulfonate (5 - 6)
0.85 9 (7.5 mmol) of methyl fluorosulfonate is added,
while stirring at 25~C, to a solution of 0.59 9 (1 mmol)
of N-(4-succinimidyloxycarbonylphenyl)-N-(4-toluene-
sulfonyl)acridine-9-carboxamide in 30 ml of 1,2-dichloro-
ethane. The reaction product precipitates out ~ithin 4
hours. Filtration with suction and drying result in
0.43 9 (60.8% of theory) of the desired product.
1H NMR (TFA): ~= 2.6 (s, Ar-CH3); 3.1 (s, CH2-CH2);
4.9 (s, N-CH3); 7.3-8.8 (m, 16 aromatic H).
Exa~ple 6:
N-(4-Succinimidyloxycarbonylmethylphenyl)-N-(4-toluene-
sulfonyl)-10-methylacridinium-9-carboxamide fluorosul-
fonate is obtained from N-(4-carboxymethylphenyl)-4-
toluenesulfonamide in the same way as in Example 5.
2 3 a . - ~ 'J
~5-5) ~N~
~CH3
23b
CH3
(5-61 ~ N~
O /~N~S~
~~ ~ ~// ~
~CH3
O
~t ~
- 24 _~-
N-(4-Carboxymethylphenyl)-4-toluenesulfonamide (6 - 1)
1H NMR (DMS0):~ = 2.3 (s, CH3); 3.5 (s, CH2); 7.0
(AB, C6H4); 7.3-7.6 (AB; C6H4); 10.2 (s, NH).
N-(4-Benzyloxycarbonylmethylphenyl)-4-toluenesulfonamide (6 - 2)
Yield: 35% of theory
1H NMR (DMS0):~ = 2.3 (s, CH3); 3.6 (s, COCH2); 5.1
(s, OCHz); 6.9-7.7 (m, 13 aromatic H); 10.2 (s, NH).
N-(4-Benzyloxycarbonylmethylphenyl)-N-(4-toluenesulfonyl)-
acridine-9-carboxamide (6 - 3)
Yield: 35% of theory
1H NMR (CDCl3):~ = 2.55 (s, CH3); 3.3 (s, COCH2);
5.0 (s, 0-CH2), 6.7-8.2 (m, 21 aromatic H).
N-(4-Carboxymethylphenyl)-N-(4-toluenesulfonyl)acridine-
9-carboxamide hydrobromide (6 - 4)
Yield: 95% of theory
1H NMR (TFA):~ = 2.65 (s, CH3); 3.55 (s,CH2); 6.8-8.6
(m, 16 aromatic H)
N-(4-Succinimidyloxycarbonylmethylphenyl)-N-(4-toluene-
sulfonyl)acridine-9-carboxamide (6 - 5)
Yield: 71% of theory
1H NMR (TFA):~ = 2.65 (s, toluene-CH3); 3.0 (s,
CH2-CH2); 3.6 (broad, COCH2); 6.9-8.5 (m, aromatic)
N-(4-Succinimidyloxycarbonylmethylphenyl)-N-(4-toluene-
sulfonyl)-10-methylacridinium-9-carboxamide fluoro-
sulfonate (6 - 6)
Yield: 80% of theory
1H NMR (TFA):~ = 2.6 (s, toluene-CH3); 2.9 (s, CH2-CH2);
3.6 (broad, COCH2); 4.9 (s, N-CH3); 6.8-8.9 (m,
aromatic)
Exa~ple 7
N-C4-(2-Succinimidyloxycarbonylethyl)phenyl]-N-(4-
to~uenesulfony~)-10-methylacridinium-9-carboxamide
24a
(6- ~)
H3C~ ~~OH
~S// ~
24b
(6-2)
o
H3C ~ ~o
N/~/
24c
(6-3) ~\~
'~
~ o
o// ~
~/\CH3
3 ~ ~
24d
(6-4~ ~N
O--
OH ,~/
--C H 3
7 ~ n
24e
(6-5) ,~\~N~
O ~ ~/
~C H 3
~ O
24f
(6-6) 1 + S03~ 1-
~ '~
O /~N~S~//
~0 ~ o//~
----6 CH3
fluorosulfonate is obtained from N-[4-(2-carboxyethyl)-
phenyl]-4-toluenesulfonamide in the same way as in
Example 5.
S N-[4-(2-Carboxyethyl)phenyl]-4-toluenesulfonamide(7 - 1)
Yield: 44% of theory
1H NMR (DMS0): ~= 2.3 (s, CH3); 2.4-2.8 (m, CH2-CH2);
7.0 (AB, 4H); 7.2-7.8 (AB, 4H); 10.1 (s, NH).
N-[4-(2-Benzyloxycarbonylethyl)phenyl]-4-toluenesulfon-
amide (7 - 2)
Yield: 78~ of theory
1H NMR (CDCl3): ~= 2.35 (s, CH3); 2.4-3.0 (m,
CH2-CH2); 5.1 (s, 0-CH2); 7.0-7.8 (13 aromatic H, NH)
N-[4-(2-Benzyloxycarbonylethyl)phenyl]-N-(4-toluene-
sulfonyl)acridine-9-carboxamide (7 - 3)
Yield: 77% of theory
1H NMR (CDCl3): ~= 2.2-2.8 (m, 7H); 5.0 (s, CH2);
6.65-7.0 (AB, 4 aromatic H); 7.3-8.3 (m, 17 aromatic H)
N-~4-(2-Carboxyethyl)phenyl]-N-(4-toluenesulfonyl)-
acridine-9-carboxamide hydrobromide (7 - 4)
Yield: 65% of theory
1H NMR (CD30D): ~= 2.1-2.8 (m, CH2-CH2); 2.5 (s,
CH3); 6.7-8.5 (16 aromatic H)
N-[4-(2-Succinimidyloxycarbonylethyl)phenyl]-N-(4-toluene-
sulfonyl)acridine-9-carboxamide (7 - 5)
Yield: 90% of theory
1H NMR (CDCl3): ~= 2.6 (s, CH3); 2.7 (broad, CHz-
CHz); 2.8 (s, CH2-CH2); 6.6-8.3 (16 aromatic H)
N-[4-Succinimidyloxycarbonylethyl)phenyl]-N-(4-toluene-
sulfonyl)-10-methylacridinium-9-carboxamide fluoro-
sulfonate.(7 - 6)
Yield: 84% of theory
1H NMR (TFA): ~= 2.6 (s, CH3); 2.3-3.3 ~broad background,
3 ~ ~
25a
(7-1)
~/\J~OH
25b
(7-2)
H3C
O ~--0
\~\S//
N~/ \/~
25c
(7-3J ~N~
O
/~/ ~S//
O
\~ ~/\C H 3
25d
(7-4) ~N
~~
o// ~
--C H 3
~ ~ 3 ~ 3 ~ ~
25e
(7-5~ ,~N~\/~
o /~N~S/
~C H 3
~ O
3 !~ ~
25f
(7-6) N~ 50'~
O=
O /~N~Y
~0 ~ ~//
~CH3
~O ~
26 -
11H; s,3.1); 4.9 (s, N-CH3); 6.7-8.8 (16 aromatic H)
Example 7a:
N-t4-(N-Methylmorpholino-N-2-ethoxy)phenyl]-N-C4-(2-
succinimidyloxycarbonylethyl)phenylsulfonyl]-10-methylacri-
dine-9-carboxamide diium difluorosulfonate is obtained
in a manner analogous to that in the previous exa~ples.
Differing steps in the process are described in the
1û following reaction stages.
3-t4-ChlorsulfonvlDhenyl)-propionic acid-tert~.-butylester (7 - al)
25 9 (0.1 mole) of 3-(4-chlorosulfonyl- phenyl)-Dropionic
acid, 12 ml of tert.-butanol, 6û ml of i-butene and 3 ml
of concentrated sulfuric acid are mixed at -15~C and
stirred vigorously in an autoclave at room temperature
for 24 hours. The reaction mixture is again cooled to
-15~C and stirred into excess sodium bicarbonate solu-
tion, which is then extracted with methylene chloride,and the extracts are finally evaporated in vacuo.
Yield: 2û.4 9 (67% of theory). The product is recrystal-
lized from hexane.
1H NMR (CDCl3): ~= 1.4 (s); 2.6 (m); 3.û (t); 7.4 (m);
7.95 (m),
MS; - - 305 (M H)
z
3-(4-Chlorsulfonylphenyl)-propionic acid was prepared
in a known manner from 2-phenylpropionic acid and chloro-
sulfonic acid.
4-(Morpholino-N-2-ethoxy)aniline (7 - a2)
25 9 (0.1 mole) of 4-(morpholino-N-2-ethoxy)nitrobenzene
are refluxed with 75 9 of granulated zinc in 400 ml of
50% concentrated hydrochloric acid for 4 hours. The mix-
ture is cooled and then poured into 400 ml of 33%
strength hydrochloric acid, the mixture is extracted with
26a
( 7-al)
Cl S ~ ~ \ O CH3
~ 6 CH3
26b - ~'
( 7-a2 )
O
N
NH2
t - 27 -
i-propyl ether, and the organic phase is dried and then
evaporated. 20 9 (90% of theory) of desired product are
obtained.
1H NMR (CDCl3):~ = 2.5 (t); 2.7 (t); 3.5 (broad);
3.7 (t); 4.0 (t); 6.6 (m)
MS: z = 222 (M )
The same product is obtained by hydrogenation of the
nitro compound on palladium/animal charcoal in methanol.
The nitro compound was prepared by the method of ~ull.
Soc. chim. France 1955, 1353-62.
N-t4-(MorphoLino-N-2-ethoxy)phenyl]-N-C4-(2-t-butoxy-
carbonylethyl)phenylsulfonamide] (7 - a3)
The solution of 9.3 9 (30 mmol) of t-butyl 4-chloro-
sulfonylphenylpropionate, 6.9 9 of 4-(morpholino-N-2-
ethoxy)aniline and 0.3 9 of dimethylaminopyridine in
150 ml of methylene chloride, ~hich is clear after stand-
ing at room temperature for 10 hours, is ~ashed withsaturated sodium bicarbonate solution, concentrated to
1/3 and chromatographed on a kieselguhr column ~ith a
mixture of 90% methylene chloride and 10% methanol. The
main fraction of the eluate is evaporated.
Yield: 9 9 (61.2% of theory)
1H NMR (CDCl3): ~= 1.35 (s); 2.5 (m); 2.7-3.0 (m);
3.7 (m); 4.0 (t); 6.7-7.0 (m); 7.2-7.7 (m)
MS: z = 491 (M H).
N-[4-(Morpholino-N-2-ethoxy)phenyl]-N-[4-(2-t-butoxy-
carbonylethyl)phenylsulfonyl]-9-acridinecarboxamide (7 - a4)
50ml of 33% strength sodium hydroxide solution, 30 mg of
dimethylaminopyridine, 1.2 9 of tetrabutylammonium chlor-
ide and 1.55 9 (5.6 mmol) of 9-acridinecarbonyl chloride
hydrochloride are successively added to a vigorously
stirred solution of 2.0 9 (4.1 mmol) of N-C4-morpholino-
N-2-ethoxy)phenyl]-N-[4-(2-t-butoxycarbonylethyl)-
27a
(7-a3)
\ O CH3
HN~ \ ~ CH3
~0~/
27b
~N~ (7-a4)
N~/ ~ H3
~0~/
- 28 -
phenylsulfonamide] in 50 ml of methylene chloride. After
6 hours, the organic phase is separated off, washed with
water, dried over sodium sulfate and evaporated.
Yield: 2.8 9 (98% of theory)
H NMR (CDCl3):~ = 1.4 (s); 2.3-2.5 (m); 2.5-2.8 (m);
3.0-3.3 (t); 3.6-3.9 (m); 6.3 (d); 6.9 (d); 7.4-8.3 (m).
MS: m = 695 (M )
N-[4-(Morpholino-N-2-ethoxy)phenyl]-N-[4-(2-carboxyethyl)-
phenylsulfonyl]-9-acridinecarboxamide (7 - aS)
0.7 9 (1 mmol) of N-[4-(morpholino-2-ethoxy)phenyl]-N-[4-
(2-t-butoxycarbonylethyl)phenylsulfonyl]-9-acridine-
carboxamide is dissolved in 5 ml of trifluoroacetic acid
and left to stand at room temperature overnight. The
mixture is evaporated under waterpump vacuum, the residue
is dissolved in water, and the filtered solution is
neutralized to pH 4 with sodium acetate. The resulting
product is extracted with methylene chloride, dried over
sodium sulfate and evaporated.
Yield: 0.6 9 (94% of theory)
1H NMR (DMS0):~ = 2.6-3.0 (m); 3.0-3.3 (m); 3.6-4.0 (m);
4.0-4.4 (m); 5.8-6.6 (m); 6.8-7.0 (m); 7.3-8.4 (m).
MS: mz 639 (M J
N-[4-(Morpholino-N-2-ethoxy)phenyl]-N-[4-(2-succinimidyl-
oxycarbonylethyl)phenylsulfonyl]-9-acridinecarboxamide (7 - a6)
Yield: 95% of theory
H NMR (CDCl3): ~= 2.3-2.7 (m); 2.8 (s); 2.9-3.4 (m);
3.5-3.9 (m); 3.9-4.3 (m); 6.2-7.0 (m); 7.3-8.3 (m).
MS: 736 (M )
N-C4-(N-MethylmorphoLino-N-2-ethoxy)phenyl]-N-C4-(2-
succinimidyloxycarbonylethyl)phenylsulfonyl]-1û-methylacri-
dine-9-carboxamide diium difluorosulfonate (7 - a7)
Yield: 84% of theory
The compound gives a clear yellow-colored solution in
water.
1H NMR (DMS0): ~= 2.85 (s); 3.1 (s); 3.2-4.4 (m); 4.75
28a
,~N~ (7-a5)
~/
/ ~ 0~ 0
\ / N~ )H
- ~0~/
~3~9~
28b
~- 7-a6
/~\ ~~~
~N ~/
~0~/
0
28c
H3 (7-a7)
~~~ 05 o
- ~0~
~1 39~
- 29 -
(s); 6.4-8.3 (m)
Exa-ple 8:
Preparation of tracer for the TSH chemiluminescence
immunoassay
91 ~l of antibodies (100 ~9), 20 ~l of the acridinium
derivative prepared as in Example 1 (compound (6)) (1 mg/
ml in acetonitrile) and 600 ~l of conjugation buffer
(0.01 M phosphate, pH 8.0) are incubated for 15 minutes.
Then 200 ~l of lysine (10 mg/ml in conjugation buffer)
are added, and the mixture is incubated for a further 15
minutes. This mixture is applied to a PD 10 column
(Sephadex(R) G 25 medium, (crosslinked dextran gel in
the form of beads, manufactured by Pharmacia, Sweden))
and eluted with 0.1 M phosphate, pH 6.3, as mobile phase.
10 drops/fraction are collected. The individual frac-
tions are diluted suitably and then tested for their
chemiluminescence activity (350 ~l of oxidizing agent:
0.1% H2~2 in 0.1 N NaOH). The tracer fractions
(1st activity peak) are pooled and stored at 4~C. The
tracer which is ready for use for the h-TSH chemilumines-
cence immunoassay is prepared by suitable dilution with
a phosphate buffer (0.1 M phosphate, pH 6.3, 1% Tween(R)
20 (polyethylene sorbitan monolaurate manufactured by,
for example, ICI American Inc., USA), 0.1% bovine serum
albumin, 0.1 M NaCl, 0.01% NaN3).
Exa-ple 9:
Procedure for the h-TSH chemiluminescence immunoassay
50 ~l of standard/sample and 200 ~l of tracer were shaken
at room temperature for 2 hours in tubes coated with mono-
clonal anti-TSH antibodies. ~ashing 3x with 1 ml of
buffer and distilled water is then carried out. The light
emission is effected by addition of, in each case, 300 ~l
of activating reagent (pH 1 buffer, 0.5% H202) and 300 ~l
of initiator reagent (0.2 N NaOH) via 2 dispensers in the
- ~1139~
,
- 30 -
luminometer into the tubes. The measuring time is 1 sec.
Figure 3 shows the typical shape of a standard plot of an
immunochemiluminometric assay (ICMA) for human thyroid-
stimulating hormone (h-TSH).