Note: Descriptions are shown in the official language in which they were submitted.
' 13~940~
FIELD OF THE INVENTION
The present invention relates to electrochemi-
luminescent (ECL) phenomena and more particularly relates to
a system and methods which improve the detection limit and
precision of an ECL measurement using an apparatus adapted
to measure electrochemiluminescent phenomena.
~AC~GROUND OF THF INVENTION
Electrochemiluminescent measurement techniques
derive from electrochemistry and chemiluminescent detection
techniques. Electrochemistry (EC) deals generally with the
relation of electricity to chemical changes and with the
interconversion of chemical and electrical energy.
Chemiluminescence (CL) base~ assay or detection techniques
include, for example, binding assay techniques which
generally comprise forming a mixture of a sample containing
an unknown amount o an analvte of interest to be determined
with a known amount of a reactant which is coniugated with a
chemiluminescent label. The mixture is incubated to allow
the labeled reactant to bind to the analyte. After
incubation, the mixture is separated into two fractions: a
bound and an unbound fraction. The bound fraction is
labeled reactant bound to analyte and the unbound fraction
is the remaining unbound reactant. The CL measurement is
1~940~ ' -
then taken. One or both fractions are chemically caused to
luminesce, for example by the addition of an oxidant to the
fractions. The bound and unbound fractions of the labeled
reactant will emit different amounts of light. The measured
level of chemiluminescence at the specific wavelength is
indicative of the amount o' the bound and/or unbound
fraction, respectively, and from such measurements one
skilled in the art can determine the amount of analvte in
the sample.
Electrochemiluminescent (ECL) detection techniques
provide a sensitive and controllable measurement of the
presence and amount of an analyte o' interest. In ECL
techniques, the incubated sample is e~posed to a
voltammetric working electrode, that is, an electrode to
which a voltage is applied and from which a current for a
redox reaction may be passed. The ECL mixture does not
react with the chemical environment alone, as does the CL
mixture, nor with an electric field alone as in EC, but
rather, electrochemiluminescence is triggered by a voltage
impressed on the working electrode at a particular time and
in a particular manner to controllably cause the LCL moiety
to emit light at the electrochemiluminescent wavelength of
interest. The measurement of interest is not the current at
the electrode, as in EC, but rather is the intensity of the
- 13~94~
light. The ECL operating conditions should be controlled to
enhance the accuracy and precision of this measurement.
The key to this control, however, is recognizing
which operating conditions have an effect on the ECL
measurements, how these effects appear and how the ECL
measurement process may be controlled so as to provide
measurement results which are reproducible w-thin very
strict limits. The chemistry involved in the ECL compounds,
the analytes of interest and/or the buffer solutions in
which they appear is highly complex. The ECL compounds must
react with a precursor component so as to emit light. While
the general nature of the chemical changes and reactions
which occur during the ECL measurement process is currently
believed to be known, the specific nature is not known with
sufficient accuracy to permit the theoretical prediction of
all factors which contribute to each measurement and to what
e~tent they contribute and/or combine.
Nevertheless, it would be highly advantageous to
provide an apparatus whose operation is controllable so that
at least the initial conditions for each measurement are
exactly and precisely obtained. One aspect of the control
of the initial conditions relates to the surface condition
of the workinq electrode which triggers the ECL reaction.
EC techniques may include cleaning and conditioning the
13394~
surface of the working electrode so as to improve its
measurement of current.
It is the discovery of the present inventors that
techniques which improve the measurement of current in
conventional EC techniques are not necessarily desirable or
useful for ECL techniques. The initial conditions for ECL
measurements must meet different criteria. The analysis of
results for cleaning by conventional EC techniques are based
on the current response, where ECL techniques use the
criteria of light intensity to evaluate the results.
It is also a discovery of the present inventors
that the precision and detection limit of the ECL
measurements of light are very sensitive to the condition
and the redox (reduction/oxidation) state of the working
electrode surface and in what manner that redox state is
achieved and maintained. Conventional procedures for
cleaning and/or conditioning solid voltammetric working
electrodes have involved, for example, flaming, polishing,
roughening the electrodes, usuallv followed by an
electrochemical pretreatment. These procedures,
disadvantageously, have frequentlv required the working
electrode to be removed from the cell and/or manually
cleaned.
~ 1339108
It is believed that non-reproducible results
during ECL measurements derive at least in part from the
non-reprodu~ible changing surface redox state of the working
electrode due to variable conditions during and following
the conventional cleaning/conditioning procedures.
Additionally, when ECL techniques are performed on
biological sample matrices, for example, serum or plasma
based samples, some of the biological molecules may react
with the working electrode during a measurement and cause
electrode fouling.
Another aspect of the control of the ECL
measurements is the relationship between the nature of the
analyte of interest, the manner in which the sample is
introduced into the ECL measurement cell and the use and
operation of the workin~ electrode therein. Previously ECL
measurements of samples with complex biological or
biochemical components, were performed onlv with the sample
at rest in a "beaker" or batch system. In order to provide
more rapid assay methods, continuous or flow systems which
need not be disassembled for cleaning are needed.
OBJECTS AND SUMMARY OF THE INVENTION
Accordingly, it is an object of the present
invention to provide a method and apparatus for improving
133~108
the control of electrochemiluminescent measurements which avoid
the above-described difficulties of the prior art.
It is another object of the present invention to
provide a method and apparatus for in-situ operation of a working
electrode of an ECL cell which provides improved precision in ECL
measurements.
It is yet another object of the present invention to
provide a method and apparatus for in-situ operation of a working
electrode of an ECL cell in which a redox state of the surface of
the working electrode formed during a conditioning procedure is
maintained until a measurement step begins.
It is yet a further object of the present invention
to provide a method in a flow-through ECL cell in which a working
electrode may be cleaned and conditioned between measurements.
According to one aspect, the present invention provides
a method of conducting an assay in a biological matrix, compris-
ing the steps of: (a) flowing a solution which includes a sample
of said biological matrix and an ECL moiety into a flow-through
' 1339408
cell including a source of electrochemical energy; (b) exposing
said source to said solution; (c) operating said source to expose
said solution to an amount of electrochemical energy so as to
induce said moiety to repeatedly emit electromagnetic radiation;
(d) detecting the intensity of emitted electromagnetic radiation;
and (e) flowing said solution out of said cell.
According to another aspect, the present invention
provides a method of conducting an assay in a biological matrix
comprising the steps of: (a) flowing a solution including a
sample of said biological matrix and an ECL moiety into a flow-
through cell: (b) exposing said sample to an amount of electro-
chemical energy so as to induce said moiety to repeatedly emit
electromagnetic radiation; (c) detecting the intensity of emitted
electromagnetic radiation; and (d) flowing said solution out of
said cell.
According to yet another aspect, the present invention
provides a method of conducting an assay in a biological matrix,
comprising the steps of: (a) flowing a solution into a flow-
through cell, said solution including an electrohemiluminescent
moiety, a tertiary alkyl amine component and a buffer component,
said moiety and said tertiary alkyl amine component being
chemically reactive in response to applied electrochemical energy
to make said moiety inducible for emitting electromagnetic
radiation; (b) inducing said moiety to repeatedly emit electro-
magnetic radiation; (c) detecting the intensity of emitted
electromagnetic radiation; and (d) flowing said solution out of
said cell.
1339408
According to still another aspect, the present
invention provides a method of conducting measurements of assay
samples including respective electrochemiluminescent moieties,
said method comprising the steps of: (a) initializing an assay
measurement cell; said step of initializing including cleaning
and conditioning a working electrode of said cell by flowing at
least one first solution into said cell, exposing said working
electrode to said first solution and flowing said first solution
out of said cell; and (b) measuring electrochemiluminescence of
a moiety; said step of measuring including flowing at least one
second solution including a first one of said samples into said
cell, exposing said working electrode to said second solution,
inducing the emission of electromagnetic radiation in said
sample by exposing the same to electrochemical energy from said
working electrode, detecting the intensity of emitted electro-
magnetic radiation and flowing said second solution out of said
cell; said steps of initializing and measuring being repeatable
in alternating fashion for conducting successive measurements of
successive ones of said samples.
These and other objects, aspects and features of the
present invention will become clear from the following detailed
description of preferred embodiments thereof taken in connection
with the accompanying drawings, throughout which like reference
numerals denote like elements and parts.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1 is a side cross-sectional view of an embodiment
of apparatus according to the present invention;
-8a-
~ 1339408
Fig. 2 is a block diagram of a voltage control connectable
to the apparatus of Fig. l;
Fig. 3 is a signal diagram illustrating exemplary voltage
signals applicable in accordance with the present invention;
Fig. 4 is a signal diagram of a voltage signal applied
in a first embodiment of a method according to the present
invention for a platinum/oxalate (Pt/oxalate) environment;
Fig. 5 is a signal diagram of a voltage applied in a
second embodiment of the method according to the present
-8b-
- 1339408
invention for a gold electrode/tripropylamine (Au/TPA)
environment.
Fig. 6 is a plot of data from a Au/TPA
environment; and
Fig. 7 is a plot of data from a Pt/TPA
environment.
DETAILED DESCRIPTIOM OF ~E PREFERRED EMBODIMEMTS
The present invention is directed towards the
operation of a working electrode in an ECL cell in such a
manner thzt the measurements are i~proved both in detection
limit and in precision.
The ECL technique developed by emplovees of the
assignee of the present application and under an obligation
of assignment thereto is a method of detecting in a volume
of a multicomponent, liquid sample an analyte of interest
present in the sample in relatively small concentrations and
which comprises: a) contacting a sample with a reagent (i)
capable of being induced to repeatedly emit electromagnetic
radiation upon exposure to an amount of electrochemical
energy from a suitable source effective to induce the
reagent to repeatedly emit radiation and (ii) capable of
combining with the analyte of interest, the contact being
effected under appropriate conditions such that the analyte
' 1339~08
and the reagent combine; b) exposing the resulting sample to
an amount of electrochemical energy from a suitable source
effective to induce the reagent to repeatedly emit
radiation, the exposure being effected under suitable
condition so as to induce the reagent to repeatedly emit
electromagnetic radiation; and c) detecting electromagnetic
radiation so emitted and therebv detecting the presence of
the analyte of interest in the sample.
The methods provided in this ECL technique may be
performed in heterogeneous assays, i.e., assays in which
unbound labeled reagent is separated from bound labeled
reagent prior to exposure of the bound or unbound label
reagent to electrochemical energy, and in homogeneous
assays, i.e., assays in which unbound labeled reagent and
bound labeled reagent are exposed to electrochemical energv
together. In homogeneous assays the intensitv of the
electromagnetic radiation (light) emitted by the bound
labeled reagent is either qreater than or less than the
intensity of electromagnetic radiation (light) emitted by
the unbound labeled reagent. The presence or absence of the
respective bound and unbound components can be determined bv
measuring the difference in intensity.
In one such ECL technique, anv reagent which is
not combined with the analyte of interest is separated from
the sample which had been contacted with the reagent prior
- 1 0 -
1339408
to exposure of the sample to electrochemical energy. In
another technique, prior to contacting the sample with the
reagent, the sample is treated so as to immobilize the
analyte of interest. Means for immobilizing analytes of
interest are well known in the art and include contacting
the sample with a part~cular surface.
The ECL techniques may be used in a variety of
detection and quantitative assav formats as are well known
in the art. In quantitative assays a known amount of ECL
reaaent is used and the amount of electrochemiluminescence
measured is correlated to known standards to calculate the
amount of analyte present. Forward, reverse, competitive
and sandwich assays can be performed by methods well known
to the skilled worker. In competitive assays, for example,
a method for quantitativelv determining the amount of an
analvte of interest in a volume of a multicomponent, liquld
sample is performed as follows. The sample is contacted
with a known amount of an electrochemiluminescent reagent
which is capable of competing with the analvte of interest
for bindin~ sites on a complementary materia' not normally
present in the sample and with a known amount of the
complementary material, the contact being effected under
appropriate conditions such that the analyte of interest and
the reagent competitively bind to the complementary
~ 1339408
material. The resulting sample is exposed to
electrochemical energv and the amount of radiation emitted
is quantitativelv determined, thereby quantitatively
determining the amount o' the analyte of interest present in
the sample.
The analyte of interest may be, for example, a
whole cell, subcellular particle, virus, prion, viroid,
nucleic acid, protein, lipoprotein, lipopolysaccharide,
glycoprotein, peptide, hormone, pharmacological agent,
non-biological polvmer, synthetic organic molecule,
organometallic mo~ecule or an inorqanic molecule present in
the sample. Moreouer the analyte of interest mav be a whole
cell, subcellular particle, virus, prion, viroid or nucleic
acid present in the sample.
The sample may be derived from, for example, a
solid, emulsion, suspension, liquid or gas. Furthermore,
the sample may be derived from, for example, water, food,
blood, serum, plasma, urine, feces, tissue, saliva, oils,
organic solvents or air. Moreover, the sample may comprise,
for example, acetonitrile, dimethyl sulfoxide, dimeth~Jl
formamide, N-methyl-pyrrolidine or tert-butyl alcohol.
The sample may comprise a reducing agent or an oxidizing
agent.
~ 1339408
The reagent which is contacted with the sample may
comprise, for example, an electrochemiluminescent chemical
moiety conjugated to a whole cell, subcellular particle,
virus, prion, viroid, lipid, fatty acid, nucleic acid,
polysaccharide, protein, lipoprotein, lipopolysaccharide,
glycoprotein, peptide, cellular metabolite, hormone,
pharmacological agent, tranquilizer, barbiturat~, alkaloid,
steroid, vitamin, amino acid, sugar, non-biological pol~rmer,
synthetic organic molecule, organometallic molecule,
inorganic molecule, biotin, avidin or streptavidin. In one
example, the agent is an electrochemiluminescent moiety
conjugated to an antibody, antigen, nucleic acid, hapten,
ligand, enzyme, biotin, avidin or streptavidin. The reagent
may be the analyte of interest conjugated to an
electrochemiluminescent chemical moiety or an analog of the
analyte of interest conjugated to an electrochemiluminescent
moietv.
The electrochemiluminescent chemical moiety mav
comprise, for example, a metal-containing organic compound
wherein the metal is selected from the group consisting of
ruthenium, osmium, rhenium, irridium, rhodium, platinum,
palladium, molybdenum, technetium and tungsten.
The above discussion illustrates the broad
applicability of ECL measurement techniques to many
- 1339408
different analytes of interest and the different methods and
assays for qualitatively and quantitatively detecting their
presence in the multicomponent, liquid sample. For a fuller
description of these ECL techniques, reference should be made to
published PCT Patent Application No. W087/06706, assigned in
common with the present application and published on November 5,
1987.
As noted above, these ECL techniques may include the
detection of a multiplicity, that is, two or more, analytes of
interest in the same sample. When the ECL measurements are being
quantitatively performed, the measurement should be precise and
accurate. The precision of the measurement refers to its
repeatability, that is, the extent to which a measurement with the
same initial conditions and the same sample will produce the same
result. The accuracy refers to the closeness of the measured
concentration to the actual concentration. In order to enhance
the precision of such ECL measurements, the present invention
conditions the electrode supplying the electrochemical energy in a
highly repeatable manner to produce a specific surface on the
electrode. This specific surface can be maintained, if
appropriate, until the measurement begins. This means that if a
particular sample is supplied for measurement, the result will be
repeatable.
'- 1339~0~
Each ECL measurement is taken by exposing the
sample to the electrode system of the apparatus, more
particularly to the voltammetric working electrode thereo r,
and impressing a known voltage waveform on the working
electrode so as to trigger electrochemiluminesence. This
known voltage waveform is frequently in the form of a
voltage sweep from a first voltage to a second voltage, back
through first ~oltage to a third voltage and then back again
to the first voltage. The nature of the ~CL reaction is
such that the samples will chemically change in a thin layer
adjacent the workinn electrode surface during the ECL
measurement process.
The qualitative nature Oc the chemical change in
the sample is believed to be as follows. The electrochemi-
luminescent moiety, termed a TAG in the above-referenced and
commonlv assigned application, may or may not be bound to an
analyte, but in either case it may be promoted to an excited
state as a result of chemical reactions triggered by the
electrical energy received from the working electrode. For
example, a TAG may be oxidized from a 2+ to a 3+ state in
the following reaction:
TAG --~ TA G + e (1)
This reaction is known to take place only in the thin layer
of sample fluid immediately touching the electrode surface.
-15-
~ 1339~08
The oxidized TAG (TAG 3+) will luminesce if it can
react with a strong reductant P~ which is able to reduce TAG
3+ back to TAG 2+, but in an electronically excited state.
This molecule is provided by mixing the TAG in a buffer
solution with a high concentration of a precursor P, which
may advantageously be oxalate or tripropylamine (TPA), as
discussed in detail below. The energy from the electrode
causes first the oxidation of the precursor P as follows:
P -> P + e (2)
Then the oxidized precursor P can rearrange unimolecularly
to give the strong reductant P~:
p+ ~ po (3).
The reactions o~ equations (1), (2) and (3) all
occur during a portion of the measurement sweep when the
voltage at the working electrode reaches a triggerinq value.
Now the TAG3 reacts chemically with the reductant P~ to
~ield TAG : in an electronicall~ exçited state denoted
with an asterisk (*) as follows:
TAG3+ + P~~ TAG2+ + PD (4 )
--16--
1339408
where PD is a modified precursor which is not reactive as in
equation (2). The excited TAG now luminesce, that is, it
emits light as follows: -
TAG2+ _~ TAG2 + hc/,\ (5)
where h is Planck's constant, c is the speed of light and
is the wavelength of the emitted light.
While the above qualitative description is
believed to be accurate, the quantitative nature and effect
of these reactions on the ECL measurement is unknown.
In order to provide an accurate description of the
present invention, the precise meaning of some terms as used
below will now be defined. While these terms have been used
loosely to mean various things in the prior art, they are to
be construed in the present application as now defined.
"CLEANING"
This term is used to define the physical,
electrochemical and/or che~ical removal of any undesirable
chemical species contained in a sample which had been
introduced into the apparatus and which now remains within
the sample probe, tubing leading to the ECL cell and the ECL
cell itself, including but not limited to the working
electrode thereof. Proteins are an example of such species
which may cause electrode fouling requiring electrode
cleaning. Cleaning may include the processing of the
~ 1339~08
surfaces in the presence of a cleaning solution.
Advantageous examples of such cleaning solutions may include
dilute acids, dilute bases or any buffer solution with added
constituents, for example, detergents and salts, present for
the cleaning purpose described above. Cleaning may also
include the controlled introduction of air mixed with the
cleaning solution, in which pulses of solution separAted by
pulses of air are forced throuqh the cell and across the
working electrode. Furthermore, as in the embodiments of
the present invention described below, part of the cleaning
process of at least the working electrode may include the
application to the working electrode and counter electrode
of a desirable electrochemical voltage waveform, combining a
selected voltage signal with a selected chemical
environment.
"CO~DITIONING"
This term is used to define a general contribution
of a desirable electrochemical voltage waveform during the
simultaneous exposure of at least the working electrode to a
conditioning solution. This conditioning solution may or
may not be different from the cleaning solution. Also, the
conditioning procedure serves as the method of preparing the
working electrode surface state for the sample measurement
and incorporates the means to provide reproducibility. When
-18-
- 1339408
the ECL measurement is a measurement of a TAG or TAGged
conjugate in accordance with the chemistry disclosed above,
the constituents of the conditioning solution are those
which provide the optimal subsequent measurement o' the TAG
or TAGged conjugate of interest and the blank, that is,
TAG-free, measurement solution. An advantageous
conditioning solution may be the blank measurement buffer.
"MEASVREMENT"
This term refers to the detection and subsequent
quantitative or qualitative measurement of
electrochemiluminescence from a desired chemical, which may
be the analyte of interest. In accordance with the
invention referred to above, this measurement may be of anv
TAG or TAGged conjugate or a blank, TAG-free measurement
solution. Again, the measurement solution may, or mav not,
be different from the cleaning and/or conditioning
solutions. Examples of the measurement solution are the
buffer constituents present in the particular examples of
ECL chemistries disclosed below. The analytes of interest
are aenerally presented in measurement solutions selected
for the purpose of enhancing the ECL TAG liqht output,
diminishing the TAG-free light output, aiding in measurement
reproducibility and providing general detergent wetting.
--19--
1339~08
"ELECTROCHEMICAL CONTRIBUTIONS"
The electrochemical contributions to the cleaning
and/or conditioning aspects of the total sampling cycle
include anv combination of applied voltage waveforms to the
working electrode which have been demonstrated to be
advantageous for the particular chemistry and measurement
involved while the working electrode is exposed to one or
more selected solutions. These waveforms include, without
limitation, pulses, sweeps, constant voltages or any
combination of the above. In these voltage waveforms, there
are at least three potentials of interest: an upper limit
voltage, a lower limit voltage and a preoperative or hold
potential voltage.
- Embodiments are disclosed below for two separate
mechanical/chemical environments. These environments are
defined as follows.
"Pt/Oxalate Environment"
This environment uses a platinum (Pt) working
electrode. The precursor is oxalate, which is a water
soluble salt of oxalic acid, advantaaeouslv a sodium or
potassium salt. The oxalate is combined in an aqueous
buf'ered solution with a conditioner/surfactant.
"Au/TPA Environment"
This environment uses a gold (Au) working
electrode. The precursor is tripropylamine (TPA) and is
-20-
~ 1339408
provided in an aqueous buffered solution with a
conditioner/surfactant.
The present invention is not limited to these two
environments. For example, the platinum electrode may be
used in the TPA chemistry or the gold electrode may be used
in the oxalate chemistry, or different electrode materials
mav be used. As another example, the TPA chemistry mav be
broadened to include any of the tertiary alkyl amines.
These and other environments are all within the scope of the
present invention.
Using the above definitions and in accordance with
an aspect of the present invention, a method for in-situ
operation of a working electrode of a cell adapted to
measure electrochemiluminescent phenomena comprises the
steps of (a) cleaning and conditioning the working electrode
by applying a variable voltage thereto in the presence of at
least one solution, (b) terminating the step of cleaning and
conditioning by varying the variable voltage in a
predetermined direction to reach a predetermined
preoperative potential, and (c) starting a measurement step,
in which the working electrode is exposed to a sample
solution, before varying the voltage applied to the working
electrode from the preoperative potential.
1339~8
In a development of this method, the measurement
step may start as soon as the variable voltage reaches the
preoperative potential to terminate the step of cleaning and
conditioning, or the step of terminating may be followed by
a step of continuously holding the working electrode at the
preoperative potential until the start of the measurement
step. The working electrode may also be continuously
exposed to solution while being continuouslv held at the
preoperative potential. Tf the working electrode is formed
of a selected metal, the step of cleaning and conditioning
may terminate with the working electrode in a redox state
selected for the metal, such as an oxidized state maintained
by the preoperative potential until the start of the
measurement step, for example where the working electrode is
formed of platinum, or a reduced state, for example where
the working electrode is formed of gold. The redox state
mav also be selected for the particular sample solution.
The step of cleaning and conditioning may include passing
pulses of a cleaning solution separated b~ pulses of air
across the working electrode, and one solution may be a
cleaning and conditioning solution.
The step of cleaning and conditioning may be
separated into a step of cleaning the working electrode by
applying a first variable voltage thereto in the presence of
1339408
a cleaning solution and a step of conditioning the working
electrode by applying a second variable voltage thereto in
the presence of a conditioning solution. The measurement
step may include at least one measurement sweep of a voltage
applied to the working electrode while the sam~ is exposed
to the sample solution, with each measurement sweep being
adapted to trigger luminescenc~ in the sample solution. The
measurement step may include two or more of such measurement
sweeps.
In accordance with this aspect of the present
invention, an apparatus for in-situ operation of a working
electrode of a cell adapted to measure
electrochemiluminescent phenomena comprises cell means for
conducting an electrochemiluminescent measurement and
adapted to receive a solution therein, working electrode
means associated with the cell mear.s and adapted to be
exposed to a solution within the cell means and fluid
transport means fnr providing a selected solution to the
cell means. The apparatus further comprises voltage control
means adapted to be connected to a voltage source for
supplying selected voltage signals to the working electrode
means during at least an electrochemical cleaning operation
during which the cell means has at least a cleaning solution
therein and a conditioning operation during which the cell
-23-
1339108
means has a least a conditioning solution therein, the
voltage control means including step control means for
terminating the conditioning operation by varying the
applied voltage signal in a predetermined direction to reach
a predetermined preoperative potential, the step control
means controlling the fluid transport means to provide a
sample solution to the cell means for a measurement step
before the voltage control means varies the voltage signal
applied to the working electrode means from the preoperative
potential.
In this apparatus the voltage control means may
continuously hold the working electrode means at the
preoperative potential from the termination of the
conditioning operation until the start of the measurement
step.
The above-described aspect of the present
invention wherein the working electrode can be cleaned and
conditioned in-situ to provide controllable initial
conditions is closely related to another aspect of the
present invention taking advantage of the flow-through
construction. This aspect includes a method of conducting
an assay in a biological matrix, which method compri~es the
steps of (a) flowing a solution which includes a sample of
biological matrix and an electrochemiluminescent moiety into
-24-
13394U8
a flow-through cell including a source of electrochemical
energy, (b) exposing the source to the solution, (c)
operating the source to expose the solution to an amount of
electrochemical energy so as to induce the moiety to emit
electromagnetic radiation, (d) detecting the intensity of
emitted electromagnetic radiation an~ (e) flowing the
solution out of the cell. Advantageously, the source
includes a working electrode. The assay may be a binding
assay or, more particularly, an immunoassay, and the ECL
moiety may be bound or free.
More broadly, this aspect of the present invention
is embodied in a method of conductina an assay in a
biological matrix comprising the steps of (a) flowing a
solution including a sample of the biological matrix and an
electrochemiluminescent moiety into a flow-through cell,
(b) exposing the solution to an amount of electrochemical
energy so as to induce the moiety to emit electromagnetic
radiation, (c) detecting the intensity of emitted
electromagnetic radiation and (d) flowing the solution out
of the cell.
An advantageous method of conducting an assay of a
biological matrix comprises the steps of (a) flowing a
solution into a flow-through cell, the solution including an
electrochemiluminescent moiety, a tertiary alkyl amine
-25-
1339408
component and a buffer component, the moiety and the
tertiary alkyl amine component being chemically reactive in
response to applied electrochemical energy to make the
moiety inducible for emitting electromagnetic radiation, (b)
inducing the moiety to emit electromagnetic radiation, (c)
detecting the intensit~ of emitted electromagnetic radiation
and (d) flowing the solution out of the cell. The tertiary
alkyl amine component advantageously may include
tripropylamine, the electrochemical energy may be applied by
a gold or platinum working electrode, and the solution may
be flowing during the step of inducing ECL.
In accordance with another aspect of the present
invention, a method of conducting measurements of assay
samples of biological matrices including respective
electrochemiluminescent moieties comprises the steps of
(a) initializing a measurement cell, where the step of
initializing includes cleaning and conditioning a working
electrode of the cell by flowing a first solution into the
cell, exposing the working electrode to the first solution
and flowing the first solution out of the cell, and (b)
measuring electrochemiluminescence of a moiety, where the
step of measuring includes flowing a second so,ution
including a first one of the samples into the cell, exposing
the working electrode to the second solution, inducing the
' 1339408
emission of electromagnetic radiation in the sample by
exposing the same to electrochemical energy from the working
electrode, detecting the intensity of emitted
electromagnetic radiation and flowing the second solution
out of this cell, the steps of initializing and measuring
being repeatable in alternating fashion for conducting
successive measurements of successive ones of the sample.
In this method, the first solution may be the same as the
second solution or it may be different from the second
solution.
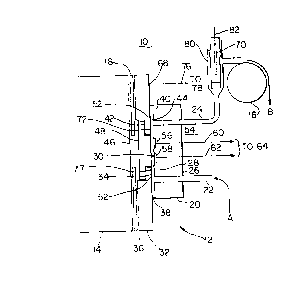
Turning now to the drawings and initially to Fig.
1 thereof, an advantageous ECL apparatus 10 is adapted to
perform the methods according to the present invention. It
will beco~e apparent from the discussion below that the
methods according to the present invention are not limited
to application in apparatus 10, but rather may
advantageously be employed in all tvpes of ECL apparatus
utilizing a working electrode or other triggering surface to
provide electrochemical energy to trigger the analvte of
interest into electrochemiluminescence. However, apparatus
10 is a flow-through cell, which provides distinct
advantages for all types of samples including binding assav
samples.
1339408
Apparatus 10 includes an electrochemical cell 12,
a light detection/measurement device 14, which may
advantageously be a photomultiplier tube (PMT), photodiode,
charge coupled device, photographic film or emulsion or the
like, and a pump 16, which is advantageously a peristaltic
pump, to provide for fluid transport to, through and from
cell 12. Alternatively, a positive displacement pump may be
used. A shutter mechanism 18 is provided between cell 12
and P~T 14 and is controllably operated to open only so far
as to expose PMT 14 to cell 12 during ECL measurement
periods. Shutter mechanism mav be closed, for example,
during maintenance. Also included in apparatus 10 but not
illustrated in Fia. 1 is a lightproof housing intended to
mou~t the various components therein and to shield PMT 14
from any external light during the ECL measurements.
Cell 12 itself includes a first mounting block 20
through which passes an inlet tube 22 and an outlet tube 24,
which mav be advantageousl~ constructed of stainless steel.
Mounting block 20 has a first, outer surface 26 and a
second, inner surface 28 defining one side of a
sample-holding volume 30 of cell 12 in which cell 12 holds
the cleaning and/or conditioning and/or measurement
solutions during corresponding operations of apparatus 10.
Inlet and outlet tubes 22, 24 pass through mounting block 20
-28-
1339408
from outer surface 26 to inner surface 28 and open into sample-
holding volume 30. A second mounting block 32, advantageously
constructed of stainless steel also has a first, outer surface
34 and a second, inner surface 36. Second mounting block 32
is separated from first mounting block 20 by an annular spacer
38, advantageously constructed of Teflon or other non-
contaminable material. Thus, outer surface 34 of mounting
block 30 defines part of the second side of the sample-holding
volume 30. Spacer 38 has an outer portion 40 and a central
aperture 42 whose inner edge 44 defines the side wall of sample-
holding volume 30. Outer portion 40 seals the inner surface 28
of first mounting block 20 to outer surface 34 of second mount-
ing block 32 to prevent any solution from passing out from
sample-holding volume 30 between the two surfaces 28, 34.
Mounting block 32 further has a central aperture 46 in which a
window 48 is seal-fitted to define the rest of the second side
of sample-holding volume 30 as a continuation of outer surface
34. Window 48 is formed of a material which is substantially
transparent at the wavelength of electrochemiluminescent light
emitted by the ECL moiety. Window 48 is therefore
advantageously formed of glass, plastic, quartz or the like.
*
Trade-mark
-29-
,~ ~
1339408
Inlet tube 22 intersects sample-holding volume 30
at a first end 50 thereof adjacent to spacer 38 and outlet
tube 24 intersects sample-holding volume 30 at a second end
52 thereof, adjacent spacer 38. The combination of inlet
tube 22, sample-holding volume 30 and outlet tube 24 thereby
provides a continuous flow path for the narrow,
substantially laminar flow of a solution to, through and
from cell 12.
Mounted on inner surface 28 of first mounting
block 20 is a working electrode system 54 which, in the
illustrated embodiment, includes first and second working
electrodes 56 and 58. In other embodiments, a sinale
working electrode may advantageousl~ be provided, or only
electrode 56 may be a working electrode. Working electrodes
56, 58 are where the electrochemical and ECL reactions of
interest take place. Working electrodes 56, 58 are solid
voltammetric electrodes and ma~ therefore be advantageously
constructed of platinum, gold, carbons or other materials
which are effective for this purpose. Wire connectors 60,
62 connected to working electrodes 56, 58, respectively,
pass out through first mounting block 20.
Connectors 60, 62 are both connected to a first,
"working electrode" terminal 64 of a t~oltage control 66,
illustrated in Fig. 2. Voltage control 66 advantageously
-30-
1339~
operates in the manner of a potentiostat to supply voltage
signals to working electrodes 56, 58 and optionally to
measure current flowing therefrom during an ECL measurement.
Alternatively, connectors 60, 62 may be connected to
separate terminals of voltage control 66 for individuzl
operation.
The potentiostat operation of voltage control 66
is further effected through a counter electrode 68 and,
optionally but advantageously, a reference electrode 70. In
the illustrated embodiment, mounting block 32 is made of
stainless steel and coun'er electrode 68 consists in exposed
.surfaces 7', 74 of mounting block 32. Counter electrode 72,
74 and workin~ electrodes 56, 58 provide the interface to
impress the potential on the solution within sample-holding
volume 30 which energizes the chemical reactions and
triggers electrochemiluminescence in the sample and/or
provides energy for cleaning and conditioning the surfaces
of cell 12. During the ECL measurement process, some
electrochemical reactions take place at counter electrode
72, 74 but they are not the type to stimulate the emission
of electrochemiluminesence and therefore need not be
considered. Counter electrode 72, 74 is connected by a wire
connector 76 to a second, "counter electrode" terminal 78 of
voltage control 66.
-31-
1339408
Reference electrode 70 provides a reference
voltage to which the voltage applied by the working
electrodes 56, 58 is referred, for example, +1.2 volts
versus the reference. Reference electrode 70 is
advantageously located in outlet tube 24 at a position 8
spaced from cell 12 and is connected through a wire
connector 8 to a third "reference electrode" terminal 84 of
voltage control 66. In the three electrode mode, current
does not flow through reference electrode 70.
Reference electrode 70 may be used in a three electrode mode
of operation to provide a poised, known and stable voltage
and is therefore advantageously constructed of silver/silver
chloride (Ag/AgCl) or is a saturated calomel electrode
(SCE). Voltage control 66 may be operable in a two
electrode mode of operation using onlv working electrode 56
and electrode 58 as a counter/reference electrode. In this
two electrode mode of operation, counter/reference electrode
58 is electrically connected to voltage control terminals 78
and 84 on voltage control 66. In this case, voltage control
66 operates essentially as a battery. Voltage control 66
supplies voltage signals to working and counter electrodes
56 and 58 and optionally measures the current flowing
through the respective electrodes. Reference electrode 70
may alternatively be a so-called "~uasi-reference" electrode
-32-
1339408
constructed of platinum, gold, stainless steel or other
material, which provides a less stable voltage, yet one that
is measurable with respect to the solution in contact. In
both the two and three electrode mode, the reference
electrode 70 or 58 serves the purpose of providing a
reference against which the voltage applied to working
electrodes 56 is measured. The poised voltage reference is
currently considered to be more advantageous. Voltage
control 66 in its potentiostat operation controls the
various electrodes by providing a known voltage at working
electrodes 56, 58 with respect to reference electrode 70
while measuring the current flow between working electrodes
56, 58 and counter electrode 72, 74. Potentiostats for this
purpose are well known, and the internal structure of
voltage control 66 may therefore correspond to any of the
conventional, commercially available potentiostats which
produce the above-reciting functions and so does not form a
part o~ the present invention per se. Indeed, apparatus 10
may alternatively be constructed without an internal voltage
control 66, and may be adapted to be connected to an
external potentiostat which is separately controlled for
providing the required voltage signals to electrodes 56, 58,
72, 74 and 70. These voltage signals, applied in a specific
manner as described below, provide repeatable initial
1339108
conditions for the surfaces of working electrodes 56, 58 and
advantageously for the surfaces of cell 12 as a whole, a
feature which contributes significantly to improved
precision in ECL measurements.
Pump 16 is advantageously positioned at outlet
tube 24 to "pull" solution from a sample volume in the
direction of arrow A into inlet tube 22. The solution will
flow through inlet tube 22, sample-holding volume 30 and
outlet tube 24 past reference electrode 70 and out in the
direction of arrow B. Alternatively, pump 16 mav be
positioned at inlet tube 22 to "push" the solution through
apparatus 10. Advantageously, this same flow path through
inlet tube 22, sample-holding volume 30 and outlet tube 24
is used for all solutions and fluids which pass throush cell
12, whereby each fluid performs a hydrodynamic cleaning
action in forcing the previous fluid out of cell 12. Pump
16 mav be controlled to suspend its operation to hold a
particular solution in cell 12 for anv period of time.
In accordance with an aspect of the present
invention, it has been found that the types of chemical/
mechanical environments defined above permit the measurement
of ECL phenomena in assay samples of biological matrices
using a flow-through cell, such as cell 12, without
destroying the advantageous results available using the
1~39~08
cleaning/conditioning routines described below. Such assays
include immunoassays in which the analyte of interest is an
antibody or antigen. Other assays include, for example,
binding assays such as avidin-biotin (protein binding),
lectin-carbohydrate, receptor-ligand and nucleic acid
hybridizations. The sample itself need not be fullv
reacted, i.e., it need not be at equilibrium. The complex
chemical/biological nature of these assay samples would
appear to be particularly prone to cause electrode fouling
to adsorption onto the electrode and other surfaces. A
flow-through cell, however, is not easily taken apart for
cleaning, and yet the present inventors have demonstrated
thzt ECL measurements with improved precision of such assay
samples are indeed possible in accordance with the present
invention, in that the present invention includes the
effective cleaning and conditioning operations which make
such assays of "dirty" samples viable in the flow-through
environment.
The flow-through construction permits working
electrodes to be impressed with a variable voltage or to be
continuously held at a preoperative potential while being
continuously exposed to one or more solutions, that is,
without exposing working electrodes 56, 58 (or counter and
reference electrodes 72, 74, 70) to air. Exposure to air,
39408
which opens the circuit to the reference electrode 70,
permits unknown, random voltage fluctuations which destroy
the reproducibilitv of surface conditions on working
electrodes 56, 58 which the advantageous
cleaning/conditioning methods described below are intended
to achieve.
Further, the flow-through construction permits the
rapid alternation between initializing steps, in which
electrode system 54 is cleaned and conditioned, and
measurement ~teps, in which one or more measurement
waveforms or sweeps trigger ECL.
In yet a further development, it has been found
that ECL measurements of binding assay and other samples in
the TPA solution with platinum working electrodes are
enhanced when the sample solution continues to flow past
working electrodes 56, 58 during the measurement step and
when an upper limit voltage, defined below, is sufficiently
high. The discovery that the light emission was enhanced in
a flowing Pt/TPA environment was both unexpected and
startling.
Turning now to Figs. 3-5, some of the types of
voltage signals which may be applied by voltage control 66
are illustrated in Fig. 3. These voltage signals may
include pulse signals, as illustrated in Section A of Fig.
-36-
- 1339408
3, triangular sweep signals as illustrated in Section B and
truncated triangular signals as illustrated in Section C.
Other waveforms, including constant voltages, sawtooth
signals and asymmetric signals may also be used. As shown
in Fig. 3, the various types of pulse signals varv between
an upper limit voltage Vu and a lower limit voltage Vl.
Limitations on the magnitude of the applied upper and lower
limit voltages are advantageous, for example, in preventing
the evolution of oxygen bubbles from the aqueous solution if
the voltage becomes too positive with respect to the
reference electrode. Although the three types of signals
illustrated in Fig. 3 all have the same upper and lower
limit voltages Vu, Vl, it will be understood that the
particular voltage signal mav include individual wave forms
with different upper and lower voltage limits. Furthermore,
while in Fig. 3 the upper voltage limit Vu is shown as a
positive voltage and the lower voltage limit Vl is shown as
a negative voltage, both voltages in a particular
application may be positive or négative. Fig. 3 is intended
merely to illustrate different types of wave forms which may
be applied to working electrodes 56, 58 from voltage control
66. It will be understood that the voltages illustrated in
Fig. 3, as well as Figs. 4 and 5, are the voltages appearing
at working electrodes 56, 58 with respect to reference
i339408
electrode 70. All voltages given below are referenced to
Ag/AgCl.
Fig. 4 illustrates an advantageous variable
voltage signal which may be applied to working electrodes
56, 58 in the Pt/oxalate environment. In this embodiment, a
single solution of oxalate is used for both the cleaning and
conditioning aspects.
As shown in Fig. 4, the voltage signal includes an
initial portion extending from time to to time tl during a
cleaning operation. This cleaning portion includes two
cycles of a pulse signal extending from -0.7 volts to +1.5
volts. Thereafter, during a conditioning step from time t
to t2, the working electrodes 56, 58, together with other
surfaces of cell 12, are conditioned by the application of a
constant conditioning potential of +l.S volts. This
conditioning step is terminated bv moving the potential in a
downw~rd direction at time t2 to reach a predetermined
preoperative or hold potential, which in this embodiment is
+1.1 volts. This preoperative potential, and hence the
redox state, may then be held by voltage control 66 for any
period of time until a measurement step begins. From time
t2 until time t3 when the measurement step then begins, the
conditioning fluid retained in cell 12 is replaced by
introducing the sample fluid. In an aspect of the present
-38-
- 1339408
invention, however, the applied voltage from voltage control
66 is not varied before the measurement step begins. It has
been found in accordance with the present invention that by
terminating the conditioning step by moving the applied
potential in a predetermined direction, here downwardly, to
reach a predetermined operative potential, here ~1.1 volts,
and by thereafter not varyinq the working electrodes 56, 58
from that pre-operative potential until the measurement step
begins, the ECL measurement is more precise and provides
more sensitive detection of ECL phenomena. The measurement
step may be considered to begin at any time intermediate
times t2 and t3, but may most easily be visualized as
heginning at time t3, with the actual recording of data
taking place during a measurement window indicated between
times t4 and t5 when the applied voltage has a magnitude
such as to trigger ECL.
A second embodiment of the method according to the
present invention is illustrated in Fi~. 5, when a
particular voltage signal applied during the
cleaning/conditioning/ measurement operations is adapted for
the Au/TPA environment. In this embodiment, the cleaning,
conditioning and measurement solutions are all different.
Specifically, at time t6 the cleaning step begins and a
cleaning solution of sodium hydroxide (NaOH) is introduced
-39-
133940~
into cell 12. The voltage is held at a constant value of O
volts, until time t7, at which time a single pulse of peak
magnitude +2.0 volts is applied, and then the voltage
returns to -0.2 volts at time t8 and is held there until
time tlo. At an intermediate time tg between time t8 and
time t1o, the cleaning solut.ion is removed by pump 16 and a
conditioning solution is provided to cell 12 to begin the
conditioning operation. This conditioning solution may
advantageously be the blank buffer solution in which the
analyte of interest is provided to cell 12. At time tlo,
approximately two periods of a triangular waveform are
applied for the conditioning step, beginning in a downwards
direction towards -1.0 volts and then upward to a peak of +2
volts. The conditionina step terminates by moving the
applied voltage downwardly to a preoperative potential of
+0.4 volts at time tll. Working electrodes 56, 58 may
thereafter be held at the preoperative potential for
measurement sample introduction until a time tl2. At that
time, the applied voltage is varied from the preoperative
potential in a downward direction to -1.0 volts and
thereafter upwardly during the measurement step, in which
the actual measurement is taken during the measurement
window from time t13 to time t14.
-40-
1~9~08
The above two embodiments of the method according
to the invention are specific to their particular chemical
environments, and it will be understood that the present
invention is not limited to these two specific examples.
These two embodiments, however, illustrate aspects of the
present invention by which it achieves its advantageous
result. Both embodiments include cleaning and conditionins
steps for achieving both cleaning and conditioning results.
Furthermore, both embodiments reach their respective
preoperatit~e potentials by varying the applied voltage in a
predetermined direction to reach the preoperative potential.
While the two embodiments both have this predetermined
direction as the downward direction, other embodiments of
the present invention may vary the applied voltage upwa-dlv
to reach the preoperative potential.
The significance of this termination by varying
the applied voltage in a predetermined direction to reach a
predetermined preoperative potential following the selected
cleaning and conditioning steps is that the surface of
working electrodes 56, 58 will thereby be placed in a
reproducibly obtainable controlled state, defining initial
conditions for the ECL measurement which are controllably
repeatable to improve the precision of the system.
Furthermore, having reached this controlled fiurface state of
i~ 1339408
working electrodes 56, 58, this surface state may thereafter
be maintained for any period of time until the measurement
step begins. By holding the applied voltage at the
preoperative potential, it is believed that the conditioned
surface state of working electro~es 56, 58 is thereby
maintained without change. It is believed that fluctuations
or variations in the voltages appearing at working
electrodes 56, 58 in anv prior art system in which a
preoperative position was not controllably reached and
maintained were a cause of the variation in results and the
conse~uent lack of precision found in the prior art.
Thus, as illustrated in dashed lines in Fig. 4,
the measurement step need not necessarily beqin at time t5,
but rather may begin at any earlier time t15 intermediate
time t2 and time t3. Indeed, as illustrated in dashed lines
in Fig. 5, the method according to the present invention
does not require any holding time at the preoperative
potential if the measurement step begins the instant the
conditioning step is over, that is, the instant when the
preoperative potential is reached at the end of the
conditioninq step by varying the voltage in the
predetermined direction. In the Pt/oxalate environment, the
surface condition of working electrodes 56, 58 at the
termination of the conditioning step is believed to be an
-42-
1339~08
oxidized condition. The predetermined potential of +1.1
volts is believed to hold the surface in this oxidized
condition until the measurement step.
Correspondingly, in the Au/TPA environment, the
condition of the surface of working electrodes 56, 58, at
the termination of the conditioning step is believed to be a
reduced state and the preoperative potential +0.4 volts is
believed to hold the surface at that reduced state.
Voltages which are impressed upon the electrodes can
broadlv range from -5 volts to +5 volts in each of the
cleaning, conditioning, and sample measurement steps.
Preferably the voltages vary from -1.5 volts to +3 volts in
each of those steps. It is well within the skill of the art
to determine the exact values employed.
The time which elapses from start to finish of each
of the cleaning, conditioning, and sample measurement steps
mav broadly vary from 10 microseconds to several minutes and
more typically is in the range of 1 millisecond to 40 seconds.
Again, one skilled in the art can determine in any given
system the optimum time period for each of the respective
steps.
Additional examples of the methods according to
the present invention will now be presented with a complete
description of the particular chemical/mechanical
environments.
1339408
Example I
PT/OXALATE ENVIRONME~T
Methods and Materials
l) Solutions
a) Sample Measurement ~uffer
Component Concentration Amount/Liter
NaH2PO4 2 0.10M 15.599g
Oxalic Acid 0.04M 3.602g
NaOH 50% (add to adjust to
pH=4.0)
Triton X-100(TM) 1.0~ 10.OmL (nonionic
surfactant)
b) Cleaning/Conditioning Solution: same as (a)
c) Calibration solutions of TAG Ru(bpy)3Cl2
(MW=749g/mole) in the sample measurement buffer
were prepared from stock solutions of lmM, luM,
and lnM. Ru (bpy) is Tris (2,2' - bipyridyl)
ruthenium (II).
2) Instrumentation
Three electrode cell operat.ion
a) Flow-Through Cell:
Working Electrode - one or both Pt disks
Counter Electrode - stainless steel faceplate
Reference Electrode - Ag/AgCl
1339~08
Teflon Gasket (.030" thick)
Stainless Steel/ Plexiglas Faceplate
Inlet Tubing = .042" id polypropylene
Aspiration Rates 2mL/min
b) Potentiostat:
Princeton Applied Research Model 273
c) Luminometer:
Berthold Biolumat LB9500 T (photon counting)
PMT = ~m~m~tsu R374 (low gain red sensitive tube)
PMT Voltage = +1375V
Current and photon output were recorded on a Kipp & Zonen
recorder.
Two electrode cell operation
The only change from the three electrode cell
operation was that only one of the Pt disks was used as a
working electrode while the other Pt disk was used as a
combination counter/reference electrode.
3) ECL Measurement Cycle
(three electrode cell operation)
a) Cleaning/Conditioning Procedure:
1.0 mL Cleaning/Conditioning solution was aspirated
into the flow-through ECL cell. With the solution stagnant,
pulses between +1.5V and -0.7V (vs. Ag/AgCl), were applied
from the potentiostat for 60 sec., with a pulse width of
Trade-mark
-45-
. ~
133~408
3 sec at each potential. The applied potential
was then stepped to +1.5V and held there for 10
sec, and then stepped to the hold or preoperative
potential of +l.lV. This preoperative potential
may be held until the measurement step begins, in
accordance with the present invention.
b) Sample Measurement Procedure:
With the applied potential held at +l.lV, l.OmL
sample solution was aspirated into the
flow-through ECL cell. The flow of sample
solution was stopped for stagnant measurement. A
measurement sweep was then per~ormed in which the
applied potential was swept from +l.lV to +l.9v at
50 mV/sec to measure ECL.
4) ECL Measurement Cycle (two electrode cell operation)
Here the potentials were varied, reflecting the change
in the reference potential.
a) Cleaning/Conditioning Procedure:
l.OmL Cleaning/Conditioning solution was aspirated
into the flow-through ECL cell. With the solution
stagnant, pulses between +2.2V and -2.2V (vs. Pt
quasi reference/counter electrode) were applied
from the potentiostat for 60 sec, with a pulse
width of 3 sec at each potential. The applied
-46-
1339408
potential was then stepped to +2.2V and held there
for 10 sec, and then stepped to the preoperative
potential of +1.5V. Again, this preoperative
potential mav be held until the measurement step
begins.
b) Sample Measurement Procedure:
With the applied potential held at +1.5V, l.OmL
sample solution was aspirated into the
flow-through ECL cell. The flow of sample
solution was stopped for stagnant measurement. A
measurement sweep was then performed in which the
applied potential was swept from +1.5V to +2.5V at
50 mv/sec to measure ECL.
DATA - EXA~PLE I FOR PT/OXALATE ENVIRONMENT
1) The following is a comparison of data taken using the
method in accordance with the present invention with
data taken using a different method to determine the
effect of turning the potentiostat off and exposing the
cleaned/conditioned electrode to air and water between
cleaning/conditioning and sample measurement steps.
The different method used was an operation of the
flow-through cell to simulate a "beaker" or batch ECL
method termed "OCC" for open circuit cleaning. In the
method in accordance with the present invention, the
1339~08
preoperative potential was maintained until the
measurement step began. This is termed ~CCC" for
closed circuit cleaning.
a) Two Electrode Mode: This experiment was first run
in the two electrode mode. The data are as
follows.
Average
Solution ECL CountsPrecision (% CV)
CCC lOOnM TAG75501.2% (n=6 data points)
OCC lOOnM TAG 42076.0~ (n=6 data points)
~CV = (standard deviation/mean) x 100%
This data clearlv shows that the precision of the
CCC data is vastly improved relative to the precision o~ the
OCC data. This effect is a hi~hly advantageous feature of
the present invention by which ECL measurements are made
truly repeatable.
-48-
133~408
h) Three Electrode Mode: In the three electrode mode,
the OCC method included turning the potentiostat
off, but the electrodes were not exposed to air or
water. Here the particular TAG solution had a 100
nM concentration. The data are as follows.
CCC TAG Counts OCC TAG Counts
12,200 5,750
12,300 3 050
12,00~
12,350 _____
1~,300 _____
_______________________________________
avg.=12,230 (1.1% CV) 4400
Once again, the CCC data displays excellent precision.
A % CV cannot be calculated for only two points, but
the two OCC points are wide apart.
-49-
13~9~08
2) The following is a comparison of data taken to
determine, for this particular environment, the effect
of having the sample measurement solution either
flowing or stagnant during the measurement step, that
is, while the ECL moiety is being induced to emit
light. The data are as follows.
Flow Rate (mL/min)ECL Counts
0.0 (stagnant) 15,380
2.1 13,100
3.7 12,560
5.0 12,420
Using lOOn~ TAG in buffer, the flowing ECL counts
decreased somewhat and leveled off at 8n% of the
stagnant ECL counts.
3) The following is a comparison of data taken to
determine the effect of performing a step of cleaning
and conditioning the electrode before each measurement
step vs. performing successive measurement steps
without cleaning or conditioning in between. Two
different measurement solutions were tested under these
two different conditions. The electrode was
cleaned/conditioned before the first measurement step
in all cases.
-50-
1339408
a) The measurement solution was the TAG in
buffer.
i) without cleaning/conditioning (x 1000 counts)
7.50, 6.00, 5.00, 4.25, 3.70
average = 5.29 (28.5% CV)
ii) with cleaning/conditioning (x lO00 counts):
7.45, 7.50, 7.60, 7.55, 7.50, 7.70
average = 7.55 (1.2% CV)
b) The measurement solution was the TAG in 2% serum
(GIBCO Labs)/98~ buffer
i) without cleaning/conditioning (x lO00 counts):
3.92, 1.72, 0.36, 0.20
average = 1.55 (111% CV)
il) with cleaning/conditionins (x 1000 counts):
3.92, 4.25
average = 4.09
In both cases, the measurements with cleanin~ and
conditioning display noticeably superior precision. It is
believed this is due to the controlled initial conditions
e.stablished by the step ~f cleaning and c~nditioning being
substantially identical before each measurement step.
1339408
Example II
Au/TPA ENVIRONMENT
Methods and Materials
1) Solutions
a) The TPA (tripropylamine) buffer composition was as
follows:
30.8g XH2 ~4 ~2~
g 2 ~4 7 2~
l9.lmL tripropyl amine
pH adjusted to 7.5 with NaOH (50%)
1.0 mL Triton X-100 (TM)
(nonionic surfactant)
1.0 mL Tween 20 tTM) (nonionic
surfactant)
water added to make 2.0 liter
b) Calibration solution:
TPA buffer (see above)
TAG (as in Example I)
TAG-Antibody (TAG-Ab)
The antibody was goat anti mouse antibody
obtained from Kirkegaard & Perry Laboratories,
Inc. (KPL), 2 Cessna Court, Gaithersburg,
MD 20879. The conjugation of antibody to TAG was
via an aldehyde functional group using normal
1339108
literature coupling procedures. The conjugate
TAG-Ab was purified.
c) Cleaning solution:
0.2M NaOH in H2O
0.5% Triton X-100
d) Sample measurement solution:
TPA buffer (100%)
5% hybridoma growth medium
TAG-Ab: Ru(bpy)3Cl2(MW=749g/mole) conjugated to an
antibody
5% hybridoma growth medium (HGM) is a diluted and
modified form of Iscove's Modified Dulbecco's Media (IMDM),
obtained from J.R. Scientific, Inc., One Harter Avenue,
Suite 8, Woodland, California 95695. See, Dulbecco, R. and
Freeman, G. (1959) Virology 8, 398, Smith, J.D., Freeman,
G., Vogt, M. and Dulbecco, R. (1960) Virology 12, 155.
Tissue Culture Standards Committee, In Vitro 5:2, 93, and
Iscove, N.N. and Melchers, F., J. Experimental Medicine
147,923. 100% HGM contains 200 ml IMDM (JR Scientific lot
C077201), 40 ml fetal bovine serum (batch 67, HI), 2 ml 5 x
10 3M 2-mercaptoethanol (batch 39), 2 ml kanamycin sulfate
(10,000 mg/ml, lot 13N2672, 4 ml HAT (10 M hypoxanthine, 4
x 10 5M aminopterin, 1.6 x 10 3M thymidine; stock GI~CO), 40
ml 1~MCM primary microphage conditioning media(ll/7/86,
1339408
harvest 4). It is diluted to 1 part in 20 with ~uffer
solution to prepare 5% HGM.
2) Instrumentation (three electrode cell operation only)
a) Flow-Through Cell:
Working Electrode - both Au disks
Counter Electrode - stainless steel faceplate
Reference Electrode - Ag/AgCl
Teflon Gasket (.030" thick)
Stainless S~eel/Plexiglas Faceplate
Inlet Tubing = .042" id polypropylene
Aspiration Rates 2ml/min
b) Potentiostat:
Oxford
c) Luminometer:
Berthold ~iolumat LB9500 T (photon counting)
PMT=Hamamatsu R374 (low gain red sensitive tube)
PMT Voltage=~1350V
The current and photon output were recorded on a Kipp &
Zonen recorder.
3) ECL Measurement Cycle (three electrode cell operation)
Cleaning/Conditioning/Sample Measurement Procedure:
The total cycle used in obtaining this data included 6
steps, each step using the same applied voltage
waveform. Each cycle had two conditioning steps (with
-54-
1339~08
the solution flowing), one sample measurement step
(with the measurement solution flowing or stagnant),
two cleaning steps followed by one conditioning step
(with the conditioning solution flowing). Each step
used the following applied voltage sweep at a constant
500mV/sec for the electrochemical cycle:
+0.3V to -0.7V to +2.2V and back to +0.3V.
Sample volume was 1.0 ml.
DATA - Au/TPA ENVIRONMENT
1) The following data corresponds to the data in Section 1
for Example I and was taken to determine the effect of
turning the potentiostat off between cleaning/
conditioning and sample measurement steps. Here the
electrode was not exposed to air or water.
The preoperative potential was +0.4v oxidized.
-55-
13394~8
a) CCC
Solution ECL Counts
i) TAG-Ab 1720
1640
1720
1600
1670
1720 average = 1678
% CV = 3.0%
ii) Blank Buffer 62
62
63
average = 61.4
% CV = 2.2%
-56-
1339408
b) O
Solution ECL Counts
i) TAG-Ab 1650
1850
1680
1660
1680 average = 1704
% CV = 4.8%
ii) Blank Buffer 55
51
67
63 average = 59.3
% CV = 9.6~
It will be seen that the % CV for the CCC method
measuring TAG-Ab is about 50~ less than that for the OCC
method, while the blank buffer ~ CV CCC/OCC ratio is even
less.
13~9408
To confirm these results, the tests were rerun.
The data were as follows.
a) CCC
Solution ECL Counts
i) TAG-Ab 1527
1527
1485
1485
1515
1557
1539
1527
lS27 average = 1521 (n = 9)
% CV = 1.5%
-58-
1333408
ii) Blank Buffer 57
S 5
5S
52
52 average = 55 (n = 10)
% CV = 3.3%
--59--
1339408
b) OCC
Solution ECL Counts
i) TAG-Ab 1801
2470
2541
2368
2505
2028
1945
1408
1491
1503
2446 average = 2046 (n = 11)
% CV = 21.7%
ii) Blank Buffer
62
52
48
57
57
52
52
average = 53 (n = 10)
% CV = 8.6%
-60-
1339408
The results here are even better than in the first
tests, demonstrating that the OCC method is not protected
against the random voltage fluctuations which destroy the
advantageous initial conditions.
2) The following data correspond to the data in
Section 2 of EYample I and were taken to determine the
effect of having the measurement solution flowing or
stagnant during the measurement step. Here the TAG was
Ru(phenanthroline), which is Tris(disulfonated
4,7-diphenyl-1,10-phenanthroline) ruthenium tII). The
applied voltage waveform was a pulse waveform with a
lower limit voltage constant at -0.5V vs the Ag/AgCl
reference. Separate measurements were taken for
different values of the upper limit voltage between
+1.35 to +2.55V.
The data are presented in Fig. 6. No enhancement is
seen for the Au/TPA environment when the solution is
flowing.
3) The following data were taken to determine the effect
of the choice of the preoperative potential and the
choice of direction of approach thereto on the
magnitude of the ECL data, and hence its detection
limit, i.e., the smallest concentration which can be
effectively measured. When the preoperative potential
-61-
1339408
is approached downwardly, that is, from a higher
voltage, the electrode surface is considered to be
relatively oxldized, and so the preoperative potential
obtained by a downward approach is labeled "oxidized".
Conversely, when the preoperative potential is
approached upwardly from a lower voltage, the electrode
surface is considered to be relatively reduced, and so
the preoperative potential obtained by an upward
approach is labeled "reduced."
For these tests, voltage waveforms of the type
illustrated in Fig. S were used. This exact waveform
was used for the oxidized preoperative potential, while
a waveform with an extra half cycle of the conditioning
sweep was used for the reduced preoperative potential.
The following data also demonstrate that different
preoperative potentials may be required by different
sample measurement solutions to obtain the best
results.
a) Here the measurement solution was the TAG
conjugated to an antibody (TAG-Ab) in 100% buffer
solution. The blank solution was 100~ buffer.
The data were as presented in Table 1.
-62-
1339408
Table 1
Preop.Pot. TAG-Ab(S) Blank (B) S/B S - B
+0.44V : 8107 : 64 : 127 :8043 :
oxidized : 2.5% : 4.7%
:
+0.44V : 2148 : 59 : 36 :2089 :
reduced : 3.1~ : 5.1%
:
O.OOV : 2215 : 50 : 44 :2165 :
reduced ; 1.3% ~ 12% ~ -
-0.70V : 2688 : 53 : 51 :2631 :
reduced : 1.9~ : 7.5%
In the (S), samples, and (B), blank, boxes the
upper data are the ECL counts and the lower data are the
%Cv~ Under these conditions with the preoperative potential
optimized at +0.44V oxidized, the TAG-Ab measurement (S) is
approximately four times greater than with any other tested
preoperative potential. Furthermore, the signal-to-blank
ratio S/B is also 2-5 times greater. Thus, lower
concentrations of the ECL moiety can be effectively detected
in accordance with the present invention.
b) The preoperative potential selected for the
solution of TAG-Ab in 100% buffer is not
necessarily the best preoperative potential for
another solution. To demonstrate this, the same
concentration of TAG-Ab was combined in 95% buffer
-63-
1~39408
solution and 5% HGM (hybridoma growth medium).
When the same tests were run with this one change,
the data were as presented in Table 2.
Table 2
Preop.Pot. TAG-Ab~s) B-lank (~) S/BS - B
+0.44V : 2250 : 141 : 16 :2109 :
oxidized 2.1% 5.0% ~
+0.44V : 1984 : 109 : 18 :1875 :
reduced 0.8% 2.8% ~ .
O.OOv : 1762 : lO0 : 18 :1662 :
reduced ~ 0.8~ ~ 4.0%
-0.70V : 2914 : 209 : 14 :2705 :
reduced : 1.2% : 5.3%
Since the concentration of TAG-Ab was unchanged,
it might have been expected that the detected TAG-Ab signal
S would be unchanged. However, now this signal is reduced
to approximately equal to the values taken for the other
preoperative potentials. Indeed, all the values of S are
approximately equal to the values in Table 1 for the
non-optimized preoperative potentials. It is estimated that
an optimized preoperative potential for this second
measurement solution would be about +0.60V oxidized.
4) The following is data taken to demonstrate that a
sample of a biological matrix, here an immunoassay, can
be effectively measured in an ECL flow-through cell.
-64-
1339408
Here the TAG was conjugated with anti-human IgG. Three
measurements were made, each preceded by a cleaning and
conditioning step in accordance with the present
invention, for each of 6 concentrations of the ECL
moiety. The data are presented in Table 3.
Example of Immunoassay/ECL
Detection of Anti Human Igb (ng/mL)
Table 3
[Anti-HuIgG]ng/ml
ECL COUNTS AVGE SD %CV
1 2 3
0 1,692 1,7041,7161,704 12 0.7%
1,572 1,6081,6081,596 21 1.3%
1,392 1,4161,4041,404 12 0.9%
100 1,140 1,1281,1341,134 6 0.5%
300 1,044 1,0501,0321,042 9 0.9~
1200 924 906 912914 9 1.0%
As may be seen, all the individual %CVs are low, -
even though the biological matrix includes proteins and
other molecules which would be expected to adsorb to the
working electrode. The measurements may therefore be ta~en
successively without disassembling the apparatus for manual
cleaning.
-65-
1339~0~
.
Example III
Pt/TPA ENVI~ONMENT
1) The following are data taken to compare the effect
of flowing the measurement solution during the measurement
step vs. having it stagnant.
For this test, the solutions and instrumentation
were identical to those in Data Section (2) in the Au/TPA
environment except that the working electrode disks were
made of platinum. The same applied voltage waveforms were
used.
The data are presented in Fig. 7. A dramatic
enhancement may be seen for measurements with an upper limit
voltage of +1.8V or more when the sample measurement
solution was flowing as compared with stagnant measurements.
1'339~08
Although embodiments of the present invention have
been described in detail herein with reference to the
accompanying drawings, it will be apparent that the
invention is not limited thereto, and that various changes
and modifications may be effected therein by one skilled in
the art without departing from the spirit and s~ope of the
invention as defined in the appended claims.
-67-