Note: Descriptions are shown in the official language in which they were submitted.
-
- 1~3989~
DIDEOXYDIDEHYDROCARBOCYCLIC NUCLEOSIDES
The present invention relates to dideoxycarbocyclic nucleoside
analogues. More specifically it is concerned with carbocyclic
2',3'-dideoxy-2',3'-didehydro purine nucleoside analogues and their
use in therapy, in particular as antiviral agents.
In view of the similarity between viral and host cellular
functions it is difficult to selectively attack a virus while leaving
the host cell intact. Thus, there are relatively few agents
effective against viruses per se and it is difficult to find antiviral
agents having an acceptable therapeutic index, i.e. sgents which have
a meaningful antiviral effect at a dose level at which the agent has
an acceptable toxicity, or side effect, profile.
One group of viruses which have recently assumed major
significance are the retroviruses responsible for the human acquired
immunodeficiency syndrome (AIDS). Such viruses have previously been
referred to by various terminologies but are now generally referred to
as human immunodeficiency viruses (HIV'~); two such viruses, HIY-I end
HIV-II, have been reproducibly isolated from patients suffering from
AIDS and related conditions such as AIDS related complex (ARC) and
persistent generalised lymphadenopathy.
Although a number of nucleosides have been taught as useful in
the treatment of conditions associated with HIV infections, only
zidovudine (AZT, Retrovir) has received regulatory approval for the
treatment of such conditions. However, it is known that zidovudine
has severe side effects, causing suppression of the bone marrow
leading to a drop in the white blood cell count with consequent
pronounced anaemia, and there is a need for effective agents which are
less cytotoxic.
We have now found a novel class of nucleoside analogues having
antiviral activity. There is accordingly provided in a first aspect a
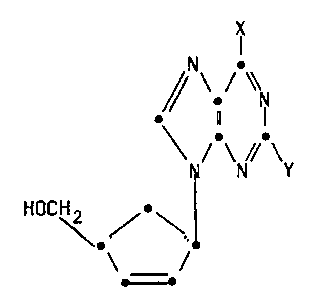
compound of formula (I)
1339896
- . N
~// \ / \\
Z N \N/ ( I )
HO-CH2 ~ ~ ~ ¦
\ _ /
wherein X is hydrogen, NRR , SR, OR or halogen;
Z is hydrogen, OR or NRR ;
R, R1 and R2 may be the same or different and are selected
from hydrogen, C1 4alkyl and aryl;
and pharmaceutically acceptable derivatives thereof.
Accordingly, the present invention provides the use of a
compound of formula (I) or pharmaceutically acceptable derivatives
thereof, as an anti-viral agent.
The present invention also provides a commercial package
containing as active ingredient a compound of formula (I), or
pharmaceutically acceptable derivatives thereof, together with
instructions for the use as an anti-viral agent.
It will be appreciated by those skilled in the art that
the compounds of formula ~I) are cis compounds and further that
the cyclopentene ring of the compounds of-formula (I) contains two
chiral centres (shown in formula (I) by *) and may thus exist in
the form of two optical isomers (i.e. enantiomers) and mixtures
thereof including racemic mixtures. All such isomers and mixtures
thereof including racemic mixtures are included within the scope
133989~
of the invention. Thus in the compounds of formula ~I) either the
chiral centre to which the base is attached is in the R
configuration and the chiral centre to which the CH20H moiety is
attached is in the S configuration ~hereinafter the D isomer~ or
the chiral centre to which the base is attached is in the S
configuration and that to which the CH20H moiety is attached is in
the R configuration (hereinafter the L isomer). Conveniently the
compounds will be in the form of either a racemic mixture or
substantially as the pure D isomer. The D isomers may be
represented by the formula ~Ia)
133~
. N
// \ / \\
1 11 \~ (I~)
Z N
H0-C~
\ _ /
where X and Z are as defined for formula (I). Reference
hereinafter to compounds of formula (I) includes compounds of
formula (Ia).
It will also be appreciated by those skilled in the art
that certain of the compounds of formula (I) may exist as a number
of tautomeric forms and all such tautomers are included within the
scope of the invention.
As used herein the term halogen refers to fluorine,
chlorine, bromine and iodine; when X is halogen it is preferably
chlorine.
As used herein Cl 4alkyl refers to a straight or
branched chain alkyl group for example methyl, ethyl, n-propyl,
lso-propyl, n-butyl, sec-butyl and t-butyl. Conveniently
Cl 4alkyl is methyl.
As used herein aryl refers to any mono- or polycyclic
aromatic moiety and includes unsubstituted and substituted aryl
(such as phenyl) and unsubstituted and substituted aralkyl
including aryl(Cl 4) alkyl such as phenyl(Cl 4)alkyl for example
benzyl or phenethyl. The substitutents are for example methyl and
methoxy. The substituted aryl includes tolyl, xylyl and anisyl.
D
1~39,~g~
3a
In the compounds of formula ~I)
is preferably amino.
In one preferred class of compounds of formula (I) X is
OR, in particular OH.
In a further preferred class of compounds of formula (I)
X is NRR in particular NH2, or hydrogen.
Particularly preferred compounds of formula (I) are
those wherein Z is NH2 and X is H, NH2 or, especially, OH. Such
compounds in particular have especially desirable therapeutic
indices as antiviral agents.
By "a pharmaceutically acceptable derivative" is meant
any pharmaceutically acceptable salt, ester, or salt of such
ester, of a
~D
~398~
- 4
compound of formula (I) or any other compound which, upon
administration to the recipient, is capable of providing (directly or
indirectly) a compound of formula (I) or an antivirally active
metabolite or residue thereof.
Preferred esters of the compounds of formula (I) include
carboxylic acid esters in which the non-carbonyl moiety of the ester
grouping is selected from hydrogen, straight or branched chain alkyl
(e.g. methyl, ethyl, n-propyl, t-butyl, n-butyl), alkoxyalkyl (e.g.
methoxymethyl), aralkyl (e.g. benzyl), aryloxyalkyl (e.g.
phenoxymethyl), aryl (e.g. phenyl optionally substituted by halogen,
Cl_4 alkyl or Cl_4 alkoxy); sulphonate esters such as alkyl- or
aralkylsulphonyl (e.g. methanesulphonyl); amino acid esters (e.g.
L-valyl or L-isoleucyl) and mono-, di- or tri-phosphate esters.
With regard to the above described esters, unless otherwise
specified, any slkyl moiety present advantageously contains 1 to 18
carbon atoms, particularly 1 to 4 carbon atoms. Any aryl moiety
present in such esters advantageously comprises a phenyl group.
Pharmaceutically acceptable salts of the compounds of formula (I)
include those derived from pharmaceutically acceptable inorganic and
organic acids and bases. Examples of suitable acids include
hydrochloric, hydrobromic, sulphuric, nitric, perchloric, fumaric,
maleic, phosphoric, glycollic, lactic, salicylic, succinic,
toluene-p-sulphonic, tartaric, acetic, citric, methanesulphonic,
formic, benzoic, malonic, naphthalene-2-sulphonic and benzenesulphonic
acids. Other acids such as oxalic, while not in themselves
pharmaceutically acceptable, may be useful in the preparation of salts
useful as intermediates in obtaining the compo~nds of the invention
and their pharmaceutically acceptable acid addition salts.
Salts derived from appropriate bases include alkali metal (e.g.
sodium), alkaline earth metal (e.g. magnesium), ammonium and NR 4+
(where R is Cl_4alkyl) salts.
~ References hereinafter to a compound according to the invention
includes both compounds of formula (I) and their pharmaceutically
acceptable derivatives.
Specific compounds of formula (I) include : -
(la,~ )-4-(6-Chloro-9H-purin-9-yl)-2-cyclopentenyl-carbinol;
133~896
-- 5
(la,4a)-4-(6-Hydroxy-9H-purin-9-yl)-2-cyclopentenyl-carbinol;
(1~,4a)-4-(6-Amino-9H-purin-9-yl)-2-cyclopentenyl-carbinol;
(1~,4a)-4-(6-Mercapto-9H-purin-9-yl)-2-cyclopentenyl-carbinol;
(la,4a)-4-~2-Amino-6-chloro-9H-purin-9-yl)-2-cyclopentenyl-
carbinol;
(1~,4a)-4-(2-Amino-6-hydroxy-9H-purin-9-yl)-2-cyclopentenyl-
carbinol;
(la,4a)-4-(2,6-Diamino-9H-purin-9-yl)-2-cyclopentenyl-
carbinol;
in the form of a racemic mixture or a single enantiomer.
The compounds of the invention either themselves possess
antiviral activity and/or are metabolizable to such compounds. In
particular these compounds are effective in inhibiting the replication
of retroviruses, including human retroviruses such as human
immunodeficiency viruses (HIV's), the causative agents of AIDS.
Certain compounds of the invention in particular those wherein Z
i8 H also posssess anticancer activity.
There is thus provided as a further aspect of the invention a
compound of formula (I) or a pharmaceutically acceptable derivative
thereof for use as an active therapeutic agent in particular as an
antiviral agent, for example in the treatment of retroviral
infections, or an anticancer agent.
In a further or alternative aspect there is provided a method for
the treatment of a viral infection, in particular an infection caused
by a retrovirus such as HIY, in a mammal including man comprising
administration of an effective amount of an antiviral compound of
formula (I) or a pharmaceutically acceptable derivative thereof.
There is also provided in a further or alternative aspect use of
a compound of formula (I) or a pharmaceutically acceptable derivative
thereof for the manufacture of a medicament for the treatment of a
viral infection or use as an anticancer agent.
The compounds of the invention having antiviral activity are also
useful in the treatment of AIDS related conditions such as
AIDS-related complex (ARC), progressive generalised lymphadenopathy
(~GL), AIDS-related neurological conditions tsuch as dementia or
tropical paraparesis), anti-HIV antibody positive and HIV- positive
conditions, Kaposi's sarcoma and thrombocytopenia purpura.
13~3~
The antivirsl compounds of the invention are also useful in the
prevention of progression to clinical illness of individuals who are
anti-HIV antibody or HIV-antigen positive and in prophylaxis following
exposure to HIV.
The antiviral compounds of formula (I3 or the pharmaceutically
acceptable derivatives thereof, may also be used for the prevention of
viral contamination of physiological fluids such as blood or semen in
vitro.
Certain of the compounds of formula (I) are also useful as
intermediates in the preparation of other compounds of the invention.
It will be appreciated by those skilled in the art that reference
herein to treatment extends to prophylsxis as well as the treatment of
established infections or symptoms.
It will be further appreciated that the amount of a compound of
the invention required for use in treatment will vary not only with
the particular compound selected but also with the route of
administration, the nature of the condition being treated and the age
and condition of the patient and will be ultimately at the discretion
of the attendant physician or veterinarian. In general however a
suitable dose will be in the range of from about 1 to about 750mg/kg
e.g. from about 10 to about 750mg/kg of bodyweight per day, such as 3
to about 120mg per kilogram body weight of the recipient per day,
preferably in the range of 6 to 90 mg/kg/day, most preferably in the
range of 15 to 60mg/kg/day.
The desired dose may conveniently be presented in a single dose
or as divided doses administered at appropriate intervals, for example
as two, three, four or more sub-doses per day.
The compound is conveniently administered in unit dosage form;
for example containing lû to 1500mg, conveniently 20 to lOOOmg, most
conveniently 50 to 700mg of Qctive ingredient per unit dosage form.
Ideally the active ingredient should be administered to achieve
peak piasma concentrations of the active compound of from about 1 to
about 75 ~M, preferably about 2 to 50 ~M, most preferably about 3 to
about 30~M. This may be achieved, for example, by the intravenous
injection of a 0.1 to 5~ solution of the active ingredient, optionally
~ 339~S
in saline, or orally administered as a bolus containing about 1 to
about lOOmg of the active ingredient. Desirable blood levels may be
maintained by a continuous infusion to provide about O.Ol to about 5.0
mg/kg/hour or by intermittent infusions containing about 0.4 to about
15 mg/kg of the active ingredient.
~ While it is possible that, for use in therapy, a compound of the
invention may be administered as the raw chemical it is preferable to
present the active ingredient as a pharmaceutical formulation.
The invention thus further provides a pharmaceutical formulation
comprising a compound of formula (I) or a pharmaceutically acceptable
derivative thereof together with one or more pharmaceutically
acceptable carriers thereof and, optionally, other therapeutic and/or
prophylactic ingredients. The carrier(s) must be 'acceptable' in the
sense of being compatible with the other ingredients of the
formulation and not deleterious to the recipient thereof.
Pharmaceutical formulations include those suitable for oral,
rectal, nasal, topical (including buccal and sub-lingual), vaginal or
parenteral (including intramuscular, sub-cutaneous and intravenous)
administration or in a form suitable for administration by inhalation
or insufflation. The formulations may, where appropriate, be
conveniently presented in discrete dosage units and may be prepared by
any of the methods well known in the art of pharmacy. All methods
include the step of bringing into association the active c~ ound with
liquid carriers or finely divided solid carriers or both and then, if
necess~ry, shaping the product into the desired formulation.
Pharmaceutical formulations suitable for oral administration may
conveniently be presented as discrete units such as capsules, cachets
or tablets each containing a predetermined amount of the active
ingredient; as a powder or granules; as a solution, a suspension or as
an emulsion. The active ingredient may also be presented as a bolus,
electuary or paste. Tablets and capsules for oral administration may
contain conventional excipients such as binding agents, fillers,
lubricants, disintegrants, or wetting agents. The tablets may be
coated accoraing to methods well known in the art. Oral liquid
preparations may be in the form of, for example, aqueous or oily
suspensions, solutions, emulsions, syrups or elixirs, or may be
13~98g~
-- 8
presented as a dry product for constitution with water or other
suitable vehicle before use. Such liquid preparations may contain
conventional additives such as suspending agents, emulsifying agents,
non-aqueous vehicles (which may include edible oils), or
preservatives.
The compounds according to the invention may also be formulated
for parenteral administration (e.g. by injection, for example bolus
injection or continuous infusion) and may be presented in unit dose
form in ampoules, pre-filled syringes, small volume infusion or in
multi-dose containers with an added preservative. The compositions
may take such forms as suspensions, solutions, or emulsions in oily or
squeous vehicles, and may contain formulatory agents such as
suspending, stabilising and/or dispersing agents. Alternatively, the
active ingredient may be in powder form, obtained by aseptic isolation
of sterile solid or by lyophilisation from solution, for constitution
with a suitable vehicle, e.g. sterile, pyrogen-free water, before
use.
For topical administration to the epidermis the compounds
according to the invention may be formulated as ointments, creams or
lotions, or as a transdermal patch. Ointments and creams may, for
example, be formulsted with an aqueous or oily base with the addition
of suitable thickening and/or gelling agents. Lotions may be
formulated with an aqueous or oily base and will in general also
contain one or more emulsifying agents, stabilising agents, dispersing
agents, suspending agents, thickening agents, or colouring agents.
Formulations suitable for topical administration in the mouth
include lozenges comprising active ingredient in a flavoured base,
usually sucrose and acacia or tragacanth; pastilles comprising the
active ingredient in an inert base such a~ gelatin and glycerin or
sucrose and acacia; and mouthwashes comprising the active ingredient
in a suitable liquid carrier.
Pharmaceutical formulations suitable for rectal administration
wherein the carrier is a solid are most preferably presented as unit
dose suppositories. Suitable carriers include cocoa butter and other
materials commonly used in the art, and the suppositories may be
convenientiy formed by admixture of the active compound with the
13398~
softened or melted carrier(s) followed by chilling and shaping in
moulds.
Formulation~ suitable for vaginal administration may be presented
as pessaries, tampons, creams, gels, psstes, foams or sprays
containing in addition to the active ingredient such carriers as are
known in the art to be appropriate.
For intra-nasal administration the compounds of the invention may
be used as a liquid spray or in the form of drops.
Drops may be formulated with an aqueous or non-aqueous base also
comprising one more more dispersing agents, solubilising agents cr
suspending agents. Liquid sprays are conveniently delivered fron
pressurised packs.
For administration by inhalation the compounds according to the
invention are conveniently delivered from an insufflator, nebuliser or
a pressurised pack or other convenient means of delivering an aerosol
spray. Pressurised packs may comprise a suitable propellant such as
dichlorodifluoromethane, trichlorofluoromethane,
dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In
the case of a pressurised aerosol the dosage unit may be determined by
providing a valve to deliver a metered amount.
Alternatively, for administration by inhalation or insufflation,
the compounds according to the invention may take the form of a dry
powder composition, for example a powder mix of the compound and a
suitable powder base such as lactose or starch. The powder
c IFosition may be presented in unit dosage form in, for example,
capsules or cartridges or e.g. gelatin or blister packs from which
the powder may be administered with the aid of an inhalator or
insufflator.
When desired the above described formulations adapted to give
sustained release of the active ingredient may be employed.
The pharmaceutical compositions according to the invention may
also contain other active ingredients such as antimicrobial agents, or
preservatives.
The compounds of the invention may also be used in combination
with other therapeutic agents for example other antiinfective agents.
1~39~
-- 10
In particular the compounds of the invention msy be employed together
with known antiviral agents.
The invention thus provides, in a further aspect, a combination
comprising 8 compound of formula (I) or a physiologically acceptable
derivative thereof together with another therapeutically active agent,
; in particular an antiviral agent.
The combinations referred to above may conveniently be presented
for use in the form of a phR qceutical formulation and thus
pharmaceutical formulations comprising a combination as defined above
together with a pharmaceutically acceptable carrier thereof comprise a
further aspect of the invention.
Suitable therapeutic agents for use in such combinations include
acyclic nucleosides such as aciclovir, interferons such as
a-interferon, renal excretion inhibitors such as probenicid,
nucleoside transport inhibitors such as dipyridamole,
2',3'-dideoxynucleosides such as 2',3'-dideoxycytidine,
2',3'-dideoxyadenosine, 2',3'-dideoxyinosine, 2',3'-dideoxythymidine
and 2',3'-dideoxy-2',3'-didehydrothymidine and immunomodulators such
as interleukin II (IL2) and granulocyte macrophage colony stimulating
factor (GM-CSF), erythropoetin and ampligen.
The individual components of such combinations may be
administered either sequentially or simultaneously in separate or
combined pharmaceutical formulations.
- When the compound of formula (I) or a pharmaceutically acceptable
derivative thereof is used in combination with a second therapeutic
agent active against the same virus the dose of each compound may
differ from that when the compound is used alone. Appropriate doses
will be readily appreciated by those skilled in the art.
The compounds of formula (I) and their pharmaceutically
acceptable derivatives may be prepared by any method known in the art
for the preparation of compounds of analogous structure.
- Suitable methods for preparing compounds of formula (I) and their
phermaceutically acceptable derivatives are described below; the
groups X and Z are as defined above except where otherwise indicated.
It will be appreciated that the following reactions may require the
use of, or conveniently may be applied to, starting materials hsving
133989~
11
protected functional groups, and deprotection might thus be required
as an intermediate or final step to yield the desired compound.
Protection and deprotection of functional groups may be effected using
conventional means. Thus, for example, amino groups may be protected
by a group selected from aralkyl (e.g. benzyl), acyl or aryl (e.g.
2,4-dinitrophenyl); subsequent removal of the protecting group being
effected when desired by hydrolysis or hydrogenolysis as appropriate
using standsrd conditions. Hydroxyl groups may be protected using any
conventional hydroxyl protecting group, for example, as described in
'Protective Groups in ûrganic Chemistry', Ed. J. F. W. McOmie (Plenum
Press, 1973) or 'Protective Groups in Organic Synthesis' by Theodora
W. Greene (John Wiley and Sons, 1981). Examples of suitable hydroxyl
protecting groups include groups selected from alkyl (e.g. methyl,
t-butyl or methoxymethyl), aralkyl (e.g. benzyl, diphenylmethyl or
triphenylmethyl), heterocyclic groups such as tetrahydropyranyl, acyl
(e.g. acetyl or benzoyl) and silyl groups such as trialkylsilyl (e.g.
t-butyldimethylsilyl). The hydroxyl protecting groups may be removed
by conventional techniques. Thus, for example, alkyl, silyl, acyl and
heterocyclic groups may be removed by solvolysis, e.g. by hydrolysis
under acidic or basic conditions. Aralkyl groups such as
triphenylmethyl may similarly be removed by solvolysis, e.g. by
hydrolysis under acidic conditions. Aralkyl groups such as benzyl may
be cleaved by hydrogenolysis in the presence of a noble metal catalyst
such as, palladium-on-charcoal. Silyl groups may also conveniently be
removed using a source of fluoride ions such as tetra-n-butylammonium
fluoride.
In a first process (A), compounds of formula (I) and
pharmaceutically acceptable derivatives thereof may be prepared by
reacting a compound of formula (II)
- 12 _ ~33989~)
~~ ~NH2
~ il
Z N NH ( I I )
Y-CH2\ ~-~
~
~_
(wherein X and Z sre substituents having the meaning in formula (I) or
are protected forms thereof snd Y is OH or a protected form thereof)or
8 pharmaceutically acceptable derivative thereof with a reagent
selected from formic acid snd reactive derivatives thereof, followed,
where necessary, by removal of unwanted groups introduced by said
reagent and/or by removal of any protecting groups present.
Examples of suitable derivatives of formic acid which may be used
in process (A) above include orthoformates (e.g. triethyl
orthoformate), dialkoxymethyl acetates (e.g. diethoxymethyl acetate),
dithioformic acid, formsmide, s-triazine or formamidine acetate.
Unwanted groups introduced by formic acid or a reactive
derivative thereof may conveniently be removed by mild hydrolysis, for
example using an inorganic acid such as aqueolJs hydrochloric acid.
When a trialkyl orthoformate such as triethyl orthoformate is
used this i8 conveniently also the solvent for the reaction. Other
solvents which may be used include amides (e.g. dimethylformamide or
dimethylacetamide), chlorinated hydrocarbons (e.g. dichloromethane),
ethers (e.g. tetrahydrofuran) or nitriles (e.g. acetonitrile).
In some cases (e.g. when a trialkyl orthoformste such as triethyl
orthoformate is used) the reaction may preferably be carried out in
the presence of 8 catalyst such as a strong acid (e.g. concentrated
hydrochloric, nitric or sulphuric acid). The reaction msy be effected
at a temperature in the range of -25~ to +150~C, e.g. 0~ to lOOqC,
and conveniently at ambient temperature.
~333~ô
- 13
In another process (B), compounds of formula (I) and their
pharmaceutically acceptable derivatives or a protected form thereof
are subjected to an interconversion reaction whereby the substituent X
initially present is replaced by a different substituent X and/or the
group Z initially present is replaced by a different group Z followed,
where necess~ry by removal of any protecting groups present.
In one embodiment of process (B), compounds of formula (I) in
which X represents a group RRl (where R and Rl are as defined
previously) may be prepared by amination of a corresponding compound
of formula (I) in which X represents a halogen atom (e.g. chlorine).
The amination may be effected by reaction with a reagent HNRRl (where
R and Rl are as defined previously) çonveniently in a solvent such as
an alcohol (e.g. methanol). The reaction may be carried out at any
suitable temperature and conveniently at an elevated temperature such
as under reflux or, when liquid ammonia is used, in a sealed tube at
about 5û to 80~C. Suitable conditions for the conversion of halides
to secondsry and tertiary amines have also been described by I. T.
Harrison et. al., Compendium of Organic Synthetic Methods,
Wiley-Interscience, New York (1971) at pages 2~0-252.
In another embodiment of process (B), compounds of formula (I) in
which X represents a group OR (where R is as defined previously) may
be prepared by displacement of the halogen (e.g. chlorine) atom with
an appropriate anion RO-. When R represents a hydrogen atom the
displacement reaction may be carried out by hydrolysis which may be
effected in water or in a mixture of water and a water-miscible
solvent such as an alcohol (e.g. methanol or ethanol), an ether (e.g.
dioxan or tetrahydrofuran), a ketone (e.g. acetone), an amide (e.g.
dimethylformamide) or a sulphoxide (eg. dimethylsulphoxide),
conveniently in the presence of an acid or base. Suitable acids
include organic acids such as p-toluenesulphonic acid and inorganic
acids such as hydrochloric, nitric or sulphuric acid. Suitable bases
include inorganic bases such as alkali metal hydroxides or carbonates
(e.g. sodium or potassium hydroxide or carbonate). Aqueous acid or
base may also be used as the reaction solvent. The hydrolysis may
conveniently be effected at a temperature in the range -10~ to +l50~C,
- 14 _ 1339~96
e.g. at reflux. When R represents a Cl_4alkyl or aryl group the snion
RO- is convèniently formed from a corresponding alcohol ROH using an
inorganic base such as an alkali metal (e.g. sodium metal) or an
alkali metal hydride (e.g. sodium hydride). The resction with the in
situ formed anion may conveniently be effected at ambient
temperature.
In a further embodiment of process (B), compounds of formula (I)
in which X represents a group SH may be prepared by reacting the halo
compound of formula (I) with thiourea in a suitable solvent such as an
alcohol (e.g. n-propanol) at an elevated temperature (e.g. reflux)
followed by alkaline hydrolysis. Suitable bases which may be used
include alkali metal hydroxides (e.g. sodium hydroxide). The reaction
may conveniently be carried out according to the method of G. G.
Urquart et. al. Org. Syn. Coll. Vol. 3, 363(1953) eg by refluxing the
intermediate product with aqueous NAOH for about 0.25 to about 5
hours.
In another embodiment of process (B), compounds of formula (I) in
which X represents a hydrogen atom may be prepared by reducing the
halo compound of formula (I) using a reducing system which will not
affect the rest of the molecule. Suitable reducing agents which may
be used to effect the desired dehalogenation reaction include zinc
metal/water using the method described by J. R. ~arshsll et. al., J.
Chem. Soc., 1004 (1951). Alternatively, the reaction may be effected
by photolysis in a suitable solvent such as tetrahydrofuran containing
10~ triethylamine and conveniently in a Rayonet photochemical reactor
(2537A) according to the method of V. Nair et. al., J. Org. Chem., 52,
1344 (1987).
In a yet further embodiment of process (B), compounds of formula
(I) in which X represents a halogen atom may be prepared from a
different halo c nF~nd of formula (I) by conventional methods of
halide-halide exchange. Alternatively, when X is chlorine this
substituent may be replaced by other halogen atoms by using various
p-(halo)benzene diazonium chlorides according to well-known
procedures.
Compounds of formula (I) in which X represents a group SR where R
is a Cl_4&lkYl or aryl group may be prepared from the corresponding
1~398g~
28476-2
thiols using standard methods of alkylation or arylation for
example as described in US Patent No. 4,383,114.
Compounds of formula (I) in which Z represents a
hydroxyl group may conveniently be prepared from a corresponding
compound of formula (I) in which Z represents NH2 by reaction with
nitrous acid, for example employing the procedure used by J.
Davoll in J. Amer. Chem. Soc., 73, 3174 (1951).
Many of the reactions described hereinabove have been
extensively reported in the context of purine nucleoside
synthesis, for example in Nucleoside Analogs - Chemistry, BioloqY
and Medical Applications, R. T. Walker et al., eds, Plenum Press,
New York (1979) at pages 193-223.
Pharmaceutically acceptable salts of the compounds of
the invention may be prepared as described in US Patent No.
4,383,114. Thus for example, when it is desired to prepare an
acid addition salt of a compound of formula (I) the product of any
of the above procedures may be converted into a salt by treatment
of the resulting free base with a suitable acid using conventional
methods. Pharmaceutically acceptable acid addition salts may be
prepared by reacting the free base with an appropriate acid
optionally in the presence of a suitable solvent such as an ester
(e.g. ethyl acetate) or an alcohol (e.g. methanol, ethanol or
isopropanol). Inorganic basic salts may be prepared by reacting
the free base with a suitable base such as an alkoxide (e.g.
sodium methoxide) optionally in the presence of a solvent such as
an alcohol (e.g. methanol). Pharmaceutically acceptable salts may
also be prepared from other salts, including other
pharmaceutically acceptable salts, of the compounds of formula (I)
133~b
15a
using conventional methods.
A compound of formula (I) may be converted into a
pharmaceutically acceptable phosphate or other ester by reaction
with a phosphorylating agent, such as POCL3, or a suitable
esterifying agent, such as an acid halide or anhydride, as
appropriate. An ester or salt of a compound of formula (I) may be
converted to the parent compound for example by hydrolysis.
- 16 _ 1 3 ~
The compounds of formula (II) and salts thereof are novel
compounds and form a further feature of the present invention.
The compounds of formula (II) in which Z represents hydrogen or
hydroxyl may be prepared directly from the compound 2a
H~ . ~IH2
/ \
~--
2a
by reaction with sn excess of a pyrimidine of formula (III)
/; \ / 2
N
U (III)
/;\ /-\
Z N Y
(wherein Y is a halogen atom, e.g. chlorine and Z is hydrogen or
hydroxyl) in the presence of sn amine base such as triethylamine and
in an alcoholic solvent (e.g. n-butanol), conveniently at reflux.
C~ ,ol~nds of formula (II) in which Z represents NH2 may be
prepared using the compound of formula 2a by reaction with an excess
of a pyrimidine of formula (IV)
X
!
// \
( I V )
/~/ \
H N N Y
(wherein Y is as defined in formula (III) above) under similar
conditions to those described just above for the preparation of
compounds of formula (II) in which Z represents hydrogen or hydroxyl
to give a compound of formula (V)
13~98~6
- 17
// \
11 i1
/;\ /-\
H2N N r H (V)
H~-CH2\ ~-~
.
which may be diazotized ~sing a diazonium salt ArN2+E~ (wherein Ar
represents an aromatic group, e.g. p-chlorophenyl, and E- represents
an anion, e.g. a halide such as chloride) in a solvent such as water,
an organic acid such as acetic acid or a mixture thereof, conveniently
at about ambient temperature to give a compound of formula (VI)
/-\ /N=N-Ar
H2N N ~H (VI)
HO-CH2~
.1
(wherein Ar is as defined just above) which may be converted to the
desired compound of formula (II) by reduction using for example a
reducing metsl such as zinc in the presence of an acid, e.g. acetic
acid. It will be appreciated that the choice of reducing agent will
depend on the nature of the group X.
The compound 2a may be prepared from the versatile precursor,
la-acetylamino-3a-acetoxy-methylcyclopent-2-ene (la) by hydrolysis in
the presence of a mild base, such as an alkaline earth metal
hydroxide.
A particularly convenient synthesis of compounds of formula (I)
via 6-chloro compounds of formula (II) is outlined below.
1~983~
Cl
Ni
Z N NH
NHAc ~ H 2
Acll . Hll ~ HO_ -
~ ~ ~ ~ ~ --
~_ ~ ~_ >
la - 2a ~a
Ci 1 Ci 1 x
H2 ~ N~
\0~ ~-Cl Ni ~ i i1 \C H
H2N N --? H2N N \~iH ~ Z N 1
NH HO~ HO~
_I
~_ -
6a (I)
5a ~
!
// \
N! u
/ ~ / \
H2N N
NH
HO~
133~89~
19
The compound 2a and compounds of formulae (V) and
(VI) are novel intermediates and form further features of the
present invention.
The compound Ia is a known compound described in US
Patent No. 4,138,562.
Where the compound of formula (I) is desired as a
single isomer it may be obtained either by resolution of the
final product or by stereospecific synthesis from isomerically
pure starting material or any convenient intermediate.
Resolution of the final product, or an intermediate
or starting material therefor may be effected by any suitable
method known in the art : see for example 'Stereochemistry of
Carbon Compounds' by E. L. Eliel (McGraw Hill, 1962) and
'Tables or Resolving Agents' by S. H. Wilen.
One convenient method for obtaining chirally pure
compounds of formula (I) is by enzymatic conversion of a
racemic mixture of the compound or a precursor thereof. sy
such a method both (+) and (-) compounds of formula (I) may be
obtained in optically pure form. Suitable enzymes include
deaminases such as adenosine deaminase.
The invention will be further described by reference
to the following detailed examples wherein elemental analyses
were performed by M-H-W Laboratories, Phoenix, AZ. Melting
points were determined on a Mel-Temp apparatus and are
corrected. Nuclear magnetic resonance spectra were obtained
1~398~
l9a
on Jeol FX 90QFT or Nicollet NT300 spectrometers and were
recorded in DMSO-D6. Chemical shifts are expressed in ppm
down-field from Me4SI. IR spectra were determined as KBr
pellets with a Nicollet 50XC FT-IR spectrometer, and W
spectra were determined on a Beckmann DU-8 spectrophotometer.
Mass spectra were obtained with an AEI Scientific Apparatus
Limited MS-30 mass spectrometer. Thin layer chromatography
(TLC) was performed on 0.25 mm layers of Merck silica gel
(230-400 mesh). All chemicals and solvents are reagent grade
unless otherwise specified. The term "active ingredient" as
used in the Example means a compound of formula (I) or a
pharmaceutically acceptable derivative thereof.
Trade-Mark
133989~
- 20
Example 1
(+)-(la,~ )-4-r(5-Amino-6-chloro-4-pyrimidinyl)-amino]-2-
cyclopentenylcarbinol (3a)
A mixture of la-acetylamino-3a-acetoxymethyl cyclopent-2-ene (la)
(3.09, 15 mmol) and aqueous barium hydroxide (0.5N, 300ml) was
refluxed overnight. After cooling, it was neutralized with dry ice.
The precipitate was filtered out, and the ~ueous solution was
concentrated to dryness. The residue was extracted with absolute
ethanol and concentrated again to yield 2a as a colourless syrup 1.69
(14mmol).
To this syrup, 5-amino-4,6-dichloropyrimidine (4.599 28 mmol),
triethylamine (4.29, 42 mmol), and n-butanol (50ml) were added and the
mixture was refluxed for 24 hr. The volatile solvents were removed,
the residue was absorbed on silica gel (79), packed in a flash column
(4.0 x 12cm) and eluted with CHC13-MeOH~(20:1) to yield 2.699 (74~) of
compound 3a; m.p. 130-132~C. An analytical sample was obtained by
recryst~llis~tion from ethyl acetate (EtOAc), m.p. 134-135~C, MS t30
ev, 200~C); m/e 240 and 242 (M+ and M~+2), 209 (Ml -31), 144 (B+?; IR:
3600-2600 (OH), 1620, 1580 (C=C, C=N); Anal. (ClUHl3ClN40) C, H, N.
Example 2
(+)-(1~,~ )-4-~(2-Amino-6-chloro-4-pyrimidinyl)-amino]-2-
cyclopentenylcarbinol (4a)
To 14 mmol of crude 2a (Example 1! 2-smino-4,6-dichloro-
pyrimidine (3.749, 22.8 mmol), triethylamine (15ml) and n-butsnol
(75ml) were added and the mixture was refluxed for 48 hr. The
volatile solvents were removed, residue was treated with methanol to
separate the undissolved by- product (the double pyrimidine
nucleoside). The methanol solution was absorbed on silica gel (89)
packed into a column (4.0 x 14cm) and eluted with CHC13-MeOH (40:1) to
yield 1.529 (42~) of crude 4a. The product was recrystallised from
ethyl acetate to yield 4a; m.p. 132-134~C, MS t30 ev, 200~C); m/e 24n
and 242 (M~ and M~+2), 209
(M+-31), 144 (B+); IR: 3600-3000 (NH2, OH), 1620,1580 (C=C, C=N);
Anal. (CloHl3ClN4) C,H, N-
1~3~,~9~
- 21
Example 3
(+)-(la,4a)-4-([(2-Amino-6-chloro-5-(4-chlorophenyl)-azo]-4-
pyrimidinyl-amino)-2-cyclopentenylcarbinol (5a)
A cold diazonium salt solution was prepared from p-chloroaniline
(1.479, 11.5 mmol) in 3N HCl (25ml) and sodium nitrite (870mg, 12.5
mmol) in water (lOml). This solution was added to a mixture of 4a
(2.409, 10 mmol), acetic acid ~50ml), water (50ml) and sodium acetate
trihydrate (209). The reaction mixture was stirred overnight at room
temperature. The yellow precipitate was filtered and washed with cold
water until neutral, then it was air-dried in the fumehood to yield
3.6ûg (94~), of 5a, m.p. 229~C (dec). The analytical sample was
obtained from acetone-methanol (1:2), m.p. 241-243~C (dec). MS (30ev,
260UC): m/e 378 and 380 (M+ and M+ + 2), 282 (B+); IR: 3600-3000 (NH~,
OH), 1620, 1580 (C=C, C=N); Anal. (Cl6H~6C12N60) C, H,'~N.
Example 4
(+)-(la,4 )-~ ~(2,5-Diamino-6-chloro-4-pyrimidinyl)-amino]-2-
cyclopentenylcarbinol ~6a)
A mixture of 5a (379mg, 1 mmol), zinc dust (0.659, 10 mmol),
acetic acid (0.32 ml), water (15ml) and ethanol (15ml) was refluxed
under nitrogen for 3 hr. The zinc was removed and the solvents were
evaporated. The residue was absorbed on silica gel (29), packed into
a column (2.0 x 18cm), and eluted with CHC13-MeOH (15:1). A pink
syrup was obtained. Further purification from methanol-ether yielded
6a as pink crystals, 170mg (66~), m.p. 168-170~C, MS (30 ev, 220UC);
m/e 255 and 257 (M+ and M+ + 2), 224 (M~ -31), 159 (B+); IR: 3600-3000
(NH2, OH) 162û,1580 (C=C, C=N); Anal. (CloHl4ClN~) C, H, N.
Example 5
(+)-(la,4a)-4-(6-Chloro-9H-purin-9-yl)-2-cyclopentenyl-carbinol (7a)
A mixture of 3a (1.309, 5.4 mmol), triethyl orthoformate (30ml)
and hydrochloric acid (12N, 0.50ml) was stirred overnight at room
temperature. The solvent was evaporated at 35~C in vacuo. To the
residue was added aqueous hydrochloric acid (0.5 N, 30ml) and the
mixture was stirred for lhr., the mixture was neutralised to pH 7-8
with lN sodium hydroxide and absorbed onto silica gel (89), packed in
1339~9~
- 22
a column (4.0 x 8cm), and eluted with CHCl3-MeOH (20:1) to yield-white
crystals of 7a, 1.129 (82X). The crude product was recrystallised
from ethyl acetate to yield 7a, m.p 108-110~C, MS (30 ev, 200~C); m/e
250 and 252 (M~ and M+ + 2), 219 (M+-~l), 154 (8~); IR; 3600-2800
(OH), 1600 (C=C, C=N); Anal- (cllHllclN40) C~ H~ N-
Example 6(+)-(la~4a)-4-(6-Hydroxy-9H-purin-9-yl)-2-cyclopentenyl-carbinol (8a)
A mixture of 7a (251mg, 1 mmol) and aqueous sodium hydroxide
(0.2N, lOml) was refluxed for 3hr. After cooling, the reaction
mixture was adjusted to pH 5-6 with acetic acid. The reaction mixture
was absorbed on silica gel (29) packed in a column (2.0 x llcm) and
eluted with CHC13-MeOH (10:1) to yield 105mg (45~) of 8a. The crude
white product was recrystallised from water-methanol (3:1) to yield
8a, m.p. 248-250~C (dec), Ms (30 ev, 300~C); m/e 232 (M+), 214
(M~ 18), 136 (B+), IR; 3600-2600 (OH), 1680,1600 (C=O, C=C, C=N);
Anal. (CllHl2N402) C, H, N-
.
Example 7(+)-(la,4a)-4-(6-Amino-9H-purin-9-yl)-2-cyclopentenyl-carbinol (9a)
Liquid ammonia was passed into a bomb containing a solution of 7a
(250mg, 1 mmol) in methanol (5ml) at -8d c. The bomb was sealed and
heated at 60~C for 24hr. Ammonia and methanol were evaporated and the
residue was recrystallised from water to yield off-white crystals of
9a, 187mg (81~), m.p. 198-200UC. MS (30 ev, 210~C): m/e 231 (M+), 213
(M+ -18), 135 (B+); IR: 3600-2600 (NH2, OH), 1700,1600 (C=C, C=N);
Anal. (CllHl3N~O) C, H, N.
Example 8
(+)-(la,~ (6-Mercapto-9H-purin-9-yl)-2-cyclopentenyl-carbinol
(lOa)
A mixture of 7a (125mg, 0.5 mmol), thiourea (40mg, 0.64 mmol) and
n-propanol (5ml3 was refluxed for 2hr. After cooling, the precipitate
was isolated by filtration, washed with n-propanol, and dissolved in
sodium hydroxide (lN, 5ml). The solution was adjusted to pH 5 with
acetic acid. The crude lOa (9Omg, 73~) was isolated again, m.p.
~3~9~96
- 23
260-262~C (dec) and was recryst~llised from N,N-dimethylformamide, to
yield 108, m.p. 263-265~C (dec). MS (30 ev, 290~C): m/e 248 (M+), 230
(M+ -18), 152 (B+); IR: 3600-3200 (OH), 3100,2400 (SH), 1600 (C=C,
C=N); Anal. (CllHl2N405) C, H, N.
Example 9
(+)-(la,4 )-4-(2-Amino-6-chloro-9H-purin-9-yl)-2-cyclopentenyl-
carbinol (13a)
A mixture of 6a (1.419, 5.5 mmol) triethyl orthoformate (30ml)
and hydrochloric acid (12N, 1.40ml) was stirred overnight. The
suspension wss dried in vacuo. Diluted hydrochloric acid (0.5N, 40ml)
was sdded and the mixture was reacted 8t room tempersture for lhr.
The mixture W8S neutralised to pH 8 with lN sodium hydroxide and
absorbed on silica gel (7.59) packed in a column (4.0 x lOcm) and
eluted by CHC13-MeOH (20:1) to yield off-white crystals of 13a, 1.189
(80~3. The crude product was recryst~llised from ethanol to yield
138, m.p. 145-147~C. MS (30 ev, 220~C): m/e 265 and 267 (M+ and
M++2-), 235 (Ml ~30), 169 (B+); IR: 3600-2600 (NH2, OH), 1620-1580
(C=C, C=N); Anal. (CllH12N~OCl.3/4 H20) C, H, N.
Example 10
(+)-(la,4x )-4-(2-Amino-6-hydroxy-9H-purin-9-yl)-2-
cyclopentenyl carbinol (14a)
A mixture of 13a (266mg, 1 mmol) and aqueous sodium hydroxide
(0.33N) was refluxed for 5hr, absorbed onto silica gel (29) packed in
a column (2.0 x 7.5cm) snd eluted with CHC13-MeOH (5:1). The crude
product was recrystallised from methanol-water (1:4) to yield white
crystals of 148, 152mg (61~), m.p. 254-256~C (dec). MS (30 ev,
200~C): m/e 247 (M+), 217 (M+ ~30), 151 (B~); IR: 3600-2600 (NH2, OH),
1700,1600 (C=O, C=C, C=N); Anal. (CllHl3N~02.3/4 H20) C, H, N-
Example 11
(+)-(1~ (2,6-Diamino-9H-purin-9-yl)-2-cyclopentenyl carbinol
(15a)
Liquid ammonis wss psssed into a solution of 138 (265mg, 1 mmol)
in methsnol (lOml) at -800C in a bomb- The bomb was sealed snd heated
133~896
- 24
at 75~C for 48hr. Ammonia and methanol were evaporated. The residue
was absorbed on silica gel (29), packed in a column t2.~ x lOcm) and
eluted with CHC13-MeOH (15:1). The crude product was recrystallised
from ethanol to yield 196mg t80~) of 15a, m.p. 152-155 ~. MS (30 ev,
200~C): m/e 246 (M+), 229 (M+ -17), 216 (M+ ~3~), 150 (B+); IR:
3600-3000 (NH2, OH), 1700,1650,1600 (C=O, C=C, C=N); Anal. (CllHl4N60)
C, H, N.
Example 12
(lS,4R)-4-(2,6-Diamino-9H-purin-9-yl)-2-cyclopentenyl carbinol
[(lS,4R)-4-(2,6-Diamino-9H-purin-9-yl)-2-cyclopentene methanol]
(a) Intermediate 1 : (lR,2S,3R,5R)-3-[6-Amino-9H-purin-9-yl]-5-
t((l,l-dimethylethyl)-dimethylsilyloxy)methyl~-1,2-cyclopentanediol
(-) Aristeromycin1 (12.5059), tert-butyldimethylsilyl chloride (7.89)
and imidazole (12.969) in dry dimethylformsmide (85ml) was stirred at
ambient temperatur~ for 2~ hours. The resulting solution was diluted
with ethyl acetate (500ml), then washed with water (3xlOOml) and brine
(50ml) before a white solid c~ystaIlised out. This was collected by
filtration, washed with ethyl acetate, then dried in vacuo to give the
title product (3.929); lH n.m.r. (DMSO-d6) 8.15 (lH), 8.09 (lH), 7.19
(2H), 5.00 (lH), 4.72 (lH), 4.69 (lH3, 4.36 (lH), 3.85 (lH), 3.67
(2H), 2.23 (lH), 2.09 (lH), 1.79 (lH), 0.89 (9H), 0.07 (6H).
1. Journal of the American Chemical Society 1983, vol. 105,
4049-4055.
(b) Intermediate 2 : (4R,3aS,6R,6aR)-4-[6-Amino-9H-purin-9-yl]-6-
[((l,l-dimethylethyl)-dimethylsilyloxy)methyl]-3a,5,6,6a-
tetrahydro-4H-cyclopenta-1,3- dioxole-2-thione
A stirred suspension of Intermediate 1 (3.459) in dry
dimethylformamide (56ml) waa treated with l,l'-thiocarbonyldiimid~zole
(3.39), giving a yellow solution. After 15~ hours at ambient
temperature the resulting solution was combined with that from a
previous experiment (6~ scale), and solvent was removed by
evaporation. The residual oil was diluted with ethyl acetate (lOOml),
then wsshed with water (2x20ml) and brine (2x20ml), dried (Mg504) and
~ 3 ~
- 25
evaporated to a yellow solid. This was washed with diethyl ether
(25ml), then collected by filtration, further washed with ether
(25ml), then dried in vacuo to give the title product as a pale cream
solid (3.619); ~max (ethanol) 240.0nm (ElCm 459); lH n.m.r. (DMSO-d6)
8.27 (lH), 8.13 (lH), 7.33 (2H), 5.81 (lH), 5.37 (lH), 5.28 (lH), 3.78
(2H), 2.60 (lH), 2.28 (2H), 0.90 (9H), 0.09 (6H).
(c) Intermediate 3 : (l'R,4'S)-9-t4-(((1,1-Dimethylethyl)
dimethylsilyloxy)methyl)-2-cyclopenten-1-yl]-9H-purin-6-amine
A solution of Intermediate 2 (3.57g) in dry tetrahydrofuran (25ml) was
treated with a solution of 1,3-dimethyl-2-phenyl-1,3,2-
~i~7~phospholidine (4.949) in dry tetrahydrofuran (lOml), then atirred
at ambient temperature for ~ hours. The solvent was removed by
evaporation. The residual oil was combined with that from a previous
experiment (40~ scale), then subjected to column chromatography on
silica (200g, Merck 7734), eluted with chloroform, then
chloroform-ethanol m~ixtures to give a white solid. This solid was
washed with diethyl ether (25ml), then collected by filtration. The
solid was further washed with ether (lOml), then dried in vacuo to
give the title product (1.479); ~max (ethanol) 261.4nm (ElCm 443); lH
n.m.r. (DMSO-d6) 8.14 (lH), 8.00 (lH), 7.20 (2H), 6.12 (lH), 5.95
(lH), 5.60 (lH), 3.66 (2H), 2.96 (lH), 2.69 (lH), 1.65 (lH), 0.74
(9H), 0.02 (6H).
(d) Intermediate 4 : (l'R,4'S)-9-[4-(((1,1-Dimethylethyl)
dimethylsilyloxy)methyl)-2-cyclopenten-1-yl]-9H-purin-6-amine,l-oxide
A solution of Intermediate 3 (1.37g) in chloroform (30ml) was treated
with 80-90~ m-chloroperoxybenzoic acid (1.299), then stirred at
ambient temperature for 3 hours. Solvent was removed by evaporation
and the residual gum was dissolved in ethyl acetate (lOml). A white
solid crystallised out. This solid and material recovered by
evaporation of the filtrate were dissolved in chloroform (lOOml), then
washed with saturated aqueous sodium bicarbonate solution (3xlOml) and
brine (2xlOml). The aqueous washings were back-extracted with
chloroform (50ml). The combined organic solutions were dried (MgS04),
,~
~33989~
- 26
then evaporated to e solid. This solid was washed with diethyl ether
(25ml), then collected by filtration. The white solid WQS further
wsshed with ether (lOml), then dried in vacuo to give the title
product (1.169); ~max (ethanol) 235.4nm (ElCml324), 263.2nm (ElCm
248), 300.2nm (Ell~Cm 75); lH n.m.r. (CDC13 ) 8.7Z (lH), 8.02 (lH), 7.16
(2H), 6.21 (lH), 5.87 (lH), 5.72 (lH), 3.68 (2H), 3.04 (lH), 2.82
(lH), 1.74 (lH), 0.89 (9H), 0.06 (6H).
(e) Intermediste 5 : (l'R,4'5)-7-~4-(((1,1-Dimethylethyl)
dimethylsilyloxy)methyl)-2-cyclopenten-1-yl]-2-imino-1,2-dihydro
[1,2,4]oxadiazolo[3,2-i]-9H-purine hydrobromide
A stirred, ice-chilled suspension of Intermediate 4 (1.089) in
methanol (20ml3 was treated with a solution of cyanogen bromide
(0.349) in methanol (2ûml) added over 5 minutes. After 15 minutes, the
suspension was allowed to warm to ambient temperature, giving a
solution. After 90 minutes, solvent was removed by evaporation. The
residue was washed with diethyl ether t25ml), then collected by
filtration. The solid was further washed with ether (25ml), then dried
in vacuo to give the title product (1.379); ~max (ethanol) 228.2nm
(ElCm530), 285.2nm (ElCm 445); 1 H n.m.r. (CDCl3) 10.20 (lH), 10.02
(lH), 8.37 (lH), 6.25 (lH), 6.01 (lH), 5.90 (lH), 3.69 (2H), 3.05
(lH), 2.86 (lH), 1.73 (lH), 0.86 (9H), 0.03 (6H).
(f) Intermediate 6 : (l'R,4'5)-9-[4-(((1,1-Dimethylethyl)
dimethylsilyloxy)methyl3-2-cyclopenten-1-yl]-6-cyanoimino-
1,6-dihydro-1-methoxy-9H-purine
A solution of Intermediate S (1.369) in dimethylformamide (lOml) was
stirred at ambient temperature, then treated with triethylamine
(1.2ml). After 40 minutes iodomethane (0.54ml) was added, giving a
yellow solution. After 3~4 hours solvent was removed by evaporation.
The reRidue wa~ partitioned between ethyl acetate (lOOml) and water
(20ml). The organic solution was further washed with water (2x20ml)
and brine (20ml), dried (MgS04) and evaporated to a solid. This solid
was washed with diethyl ether (25ml), then collected by filtration.
This white solid was further washed with ether (lOml), then dried in
- 27 - 1 ~ 3 ~ ~ g 6
vacuo to give the title product (0.8659); AmaX (ethanol) 227.2nm (ElCm
449), 287.0nm (ElCm 544); lH n.m.r. 8.23 (lH), 7.96 (lH), 6.24 (lH),
5.85 (lH), 5.65 (lH), 4.21 (3H), 3.66 (2H), 3.04 (lH), 2.77 (lH), 1.68
(lH), 0.88 (9H), 0.05 (6H).
(g) Intermediate 7 : (l'R,4'5)-9-t4-(((1,1-Dimethylethyl)
dimethylsilyloxy)methyl)-2-cyclopenten-1-yl]-6-methoxyamino-
9H-purin-2-amine
A solution of'Intermediate 6 (802mg) and 1,8-diazabicyclot5,4,0]undec-
7-ene (0.45ml) in ethanol (80ml) was stirred and heated at reflux.
Heating was stopped after 9 hours, and the solution was left at
ambient temperature overnight. Solvent was removed by evaporation.
The residual oil was combined with that from a previous experiment (4Z
scale), then subjected to column chromatography on silica (409, Merck
9385) eluted with chloroform, then chloroform-ethanol mixtures to give
a foam. This foam was triturated with diethyl ether (lOml) and the
resulting solid was collected by filtration. The solid was further
washed with ether (5ml), then dried in vacuo to give the title product
(594mg); ~ a (ethanol) 282.2nm (El% 409); lH-n.m.r. (DMSO-d6) 9.76
(lH), 7.32 (lH), 6.53 (2H), 6.08 (lH), 5.88 (lH), 5.26 (lH), -3.72
(3H), 3.61 (2H), 2.90 (lH), 2.50 (lH), 1.52 (lH), 0.83 (9H), 0.02
(6H).
(h) Intermediate 8 : (15,4R)-4-[2-Amino-6-methoxyamino-9H-purin-9-
yl]-2-cyclopentene-methanol
A solution of Intermediate 7 (356mg) in tetrahydrofuran (35ml) was
stirred at ambient temperature then treated with tetrabutylammonium
fluoride (l.OM solution in tetrahydrofuran, 1.4ml). After 90 minutes
the reaction was quenched with water (lml), then solvents were removed
by evaporation. The residual oil was subjected to column
chromatography on silica (209, Merck 77~4), eluted with chloroform,
then chloroform-ethanol mixtures to give the title product as a solid
(243mg); ~ (pH 6 buffer) 280.2nm (ElC 534); lH n.m.r. (DMSO-d6)
1';~,~9~6
28
9.75 (lH), 7.39 (lH), 6.52 (2H), 6.10 (lH), 5.84 (lH), 5.27
(lH), 4.73 (lH), 3.40 (2H), 2.83 (lH), 2.55 (lH), 1.52 (lH).
(lS,4R)-4-[2,6-Diamino-9H-Purin-9-Yl]-2-cYclopentenecarbinol
A stirred, ice-chilled solution of Intermediate 8
(210mg) in water (lOml) and tetrahydrofuran (50ml) was treated
with aluminium amalgam [from aluminium (237mg) and 0.5%
aqueous mercuric chloride solution], added in small pieces
over 15 minutes. After 40 minutes the stirred mixture was
allowed to warm to ambient temperature. After 15'hours the
resulting mixture was filtered through kieselguhr to remove
insolubles. These were washed with water:tetrahydrofuran
(1:5, 60ml). The combined filtrates were evaporated. The
residue was subjected to column chromatography on silica (lOg,
Merck 9385), eluted with chloroform-ethanol mixtures to give
the title product as a foam (159mg); [~]D -81 (c1.04,
methanol); ~max (pH 6 buffer) 255.0 nm (ElCm 302), 280.8nm
(ElCm 381), lH n.m.r. (DMSO-d6) 7.61 (lH), 6.66 (2H), 6.10
(lH), 5.87 (lH), 5.76 (2H), 5.38 (lH), 4.76 (lH), 3.45 (2H),
2.87 (lH), 2.60 (lH), 1.60 (lH).
Example 13
(lS,4R)-4-(2-Amino-6-hydroxy-9H-Purin-9yl)-2-cYcloPentenYl
carbinol
(l'R,4'S)-2-Amino-l,9-dihydro-9-[4-hydroxymethyl-2-
cyclopenten-l-yl]-6H-purin-6-one
A turbid solution of the title compound of Example
12 (144mg) in O.lM pH 6 buffer (lOml) (from 28.4g disodium
133~836
28a
orthophosphate in 2 litres of water, adjusted with ortho-
phosphoric acid) was treated with a solution of adenosine
deaminase (0.5ml, 778 units), in 50% glycerol - O.OlM
potassium phosphate, pH 6.0, then stirred and warmed to 37~.
After 18~ hours the resulting suspension was refrigerated.
The collected solid was recrystallised from water to give the
title product as a white solid (86mg); [~]D-49 (c 0.5,
dimethylsulphoxide); AmaX (pH 6 buffer) 252.6nm (E1Cm 531), 1H
n.m.r. (DMSO-d6) 10.60 (lH), 7.60 (lH), 6.47 (2H), 6.10 (lH),
5.86 (lH), 5.33 (lH), 4.72 (lH), 3.45 (2H), 2.59 (lH), 1.58
(lH).
133~9~
- 29
Example 14
Preparation of Enantiomers of (~ ,4a)-4-(2-Amino-6-hydroxy-9H-
purin-9-yl)-2-cyclopentenylcarbinol
(a) (lS, 4R)-~-(2-Amino-6-hydroxy-9H-purin-9-yl)-2- cyclopentenyl
carbinol
The diamino analog (lOOmg) (Example 11) was dissolved in 3ml of
0.05M K2PO4 buffer (pH 7.4) with heat (5d C). The solution was cooled
to room temperature and 4û units of adenosine deaminase (Sigma, Type
VI, cslf intestinal ~ ~osa) was added. After three days of incubation
at room temperature a precipitate formed and was removed by
filtration-, yield, 18.2mg. The filtrate was concentrated to 1.5ml snd
refrigerated for 2 days. Additional solid was obtained by filtration,
yield, 26.8mg. The two solid fractions were recrystallized from water
and gave t';he pure title product m.p. 269-2724C, ~ ]D4 - 62.1 (c 0.3
MeOH).
(b) (lR, 45)-4-(2-Amino-6-hydroxy-9H-purin-9yl)-2- cyclopentenyl
carbinol
The filtrates from the preparation of the lS, 4R isomer (Example
14a) were combined and evaporated to dryness. The unchsnged diamino
starting material was separated on a silica gel flash column using 10
methanol/chloroform. The diamino compound was dissolved in 0.05M
K2P04 buffer, pH 7.4 (15ml) and 800 units of adenosine deaminase were
added. The solution was incubated for 96 hours at 37~C. TLC
indicated some unreacted product remained. The solution was heated in
boiling wster for 3 minutes and filtered to remove denatured protein.
Another 800 units of adenosine deaminase were added and the process
was repeated. The deproteinated solution was evaporated to dryness
and
the product was crystallized from water. The title product as a white
solid was collected by filtration from water, m.p. 265-2704. ta]D +
61.1 (c0.3 MeOH).
13~989~
- 30
Example 15
(+) (la,~ )-4-(2-Amino-6-hydroxy-9H-purin-9-yl)-2-cyclopentenyl
acetoxycarbinol
To a suspension of the product of Exsmple 10 (130 mg, 0.50 mmol)
and 4-dimethylaminopyridine (5mg, 0.04mmol) in a mixture of
scetonitrile (6ml) and triethylamine (0.09ml, 0.66mmol) was sdded
acetic snhydride (0.06ml, 0.6 mmole). The mixture was stirred at room
temperature for 3 hrs. Methanol (lml) was added to quench the
reaction. The solution was concentrated and absorded on silica gel
(1.59), packed on a column (2.0 x 12cm), eluted with CHC13-Me~H
(20:1). The product fractions were collected and concentrated to get
white solid. The solid product was washed with MeOH-AcOEt: yield,
123mg (85~). Further purification from methanol gsve the title
product ss needle-like crystals, m.p. 237-239~C. Ansl.
(C13Hl~,N~,03)C,H,N.
Exsmple 16
(15,4R)-4-~2-smino-9H-purin-9-yl]-2-cyclopentenylcs-rbinol
A stirred, ice-chilled solution of (15,4R)-4-~2-smino-6-
methoxyamino-9H-purin-9-yl]-2-cyclopentene-methanol (intermediate 8,
Exsmple 12) (1.2029) in tetrahydrofursn (250ml~ and wster (50ml) was
treated with aluminium amslgsm (from sluminium (1.7619) snd 0.5~
aqueous mercuric chloride solution), added in small pieces over lhr
47min. After 35 min the stirred mixture wss allowed to wsrm to
ambient tempersture. After 16hr 50min more sluminium smalgsm (from
235mg aluminium) W8S sdded over 14min. After a further 4hr lOmin the
resulting mixture W8S filtered through kieselguhr to remove
insolubles. These were washed with tetrahydrofuran:water (5:1,
300ml). The combined filtrates were evaporated to leave a yellow
foam. The foam was subjected to column chromatography on silica
(33.89, Merck 7734) prepsred in chloroform snd eluted with
chloroform-ethanol mixtures to give seversl frsctions (578mg, 420mg
snd 40mg). The two larger fractions were separstely cryst~11ised from
iso-propsnol. The filtrstes were combined with the smsllest column
fraction and subjected to preparative thin layer chromstography (Merck
5717) developed three times in 10:1 chloroform:methanol. The plates
133989~
werè eluted with ethyl acetate and ethyl acetate-ethanol (1:1) to give
a brown solid (45mg). The solid was subjected to column
chromatography on silica (2.79, Merck 7734) prepared in chloroform and
eluted with chloroform-methanol-triethylamine mixtures to give a gum
(17mg). Following an unsuccessful cryst~lisation from iso-propanol
and charcoal treatment in methanol, an a~ueous solution of the
recovered material was freeze dried to give the title compound (15mg).
lHnmr (DMS0-d6) 1.62 (lH), 2.63 (lH), 2.89 (lH), 3.45 (2H), 4.73 (lH),
5.48 (lH), 5.91 (lH), 6.14 (lH), 6.50 (2H), 7.98 (lH), 8.57 (lH).
Mass spec, [MH]+ 232.
Example 17
Tablet Formulations
A. The following formulation is prepared by wet granulation of the
ingredients with a solution of povidone in water, drying and
screening, followed by addition of magnesium stearate and
compression.
mg/tablet
(a) Active ingredient 250
(b) Lactose B.P. 210
(c) Povidone B.P. 15
(d) Sodium Starch Glycollate 20
(e) Magnesium Stearate 5
500
B. The following formulation is prepared by direct compression; the
lactose is of the direct compression type.
13~9~9~
mq/tablet
Active ingredient 250
Lactose 145
Avicel 100
Magnesium Stearate 5
500
C. (Controlled Release Formulation) The formulation is
prepared by wet granulation of the ingredients (below) with a
solution of povidone in water, drying and screening followed
by the addition of magnesium stearate and compression.
mq/tablet
(a) Active ingredient 500
(b) Hydroxpropylmethylcellulose 112
(Methocel K4M Premium)
(c) Lactose B.P. 53
(d) Povidone B.P. 28
(e) Magnesium Stearate 7
700
Exam~le 18
Capsule Formulation
A capsule formulation is prepared by admixing the
ingredients below and filling into a two-part hard gelatin
capsule.
*Trade-Mark
~39~
3Za
mq/capsule
Active ingredient 125
Lactose 72.5
Avicel 50
Magnesium Stearate 2.5
250
X
13~890
Example 19
Injectable Formulation
Active ingredient 0.2009
Sodium hydroxide solution, O.lM q.s. to a pH of about 11.
Sterile water q.s. to lOml
The active ingredient is suspended in some of the water (which
may be warmed) and the pH adjusted to about 11 with a solution of
sodium hydroxide. The batch is then made up to volume and filtered
through a sterilising grade membrane filter into a sterile lOml glass
vial and sealed with sterile closures and overseals.
Example 20
Suppository
mg/suppository
Active ingredient (63~m) 250
Hard Fat, BP 1770
2020
One-fifth of the hard fat is melted in a steam-jacketed pan at
45~C maximum. The active ingredient is sifted through a 200~m sieve
and added to the molten base with mixing, using 8 high shear stirrer,
until a smooth dispersion is achieved. Maintaining the mixture at
45~C, the remsining hard fat is added to the suspension and stirred to
ensure a hl ~enous mix. The entire suspension is passed through a
250~m stainless steel screen ~nd, with continuous stirring, is allowed
to cool to 40~C. At a temperature of 38~C to 40~C, 2.029 of the
mixture is filled into suitable, 2ml pla~tic moulds. The
suppositories are allowed to cool to room temperature.
133989~
_ 34
Example 21 - ANTIVIRAL ACTIVITY
(A) Anti-HIV Assay
Compounds of formula (I) were screened for anti-HIV activity at
the National Cancer Institute, Frederick Cancer Research Facility,
Frederick, Msryland (FCRF). The following are the current screening
mode operational procedures utilized at FCRF. The protocol consists
of 3 areas, (I) preparation of infected cells and distribution to the
test plates, tII) preparation of drug dilution plates and distrubition
to the test plates, and (III) XTT assay procedure. See D. A. Scudiero
et al., "A New Simplified Tetrazolium Assay for Cell Growth and Drug
Sensitivity in Culture," Cancer Res., 48, 4827 (1988).
I. Infection and Distribution of ATH8 Cells to Microtiter Trays
Cells to be infected (a normal lymphoblastoid cell line which
expres~es CD4) are placed in 50ml conical centrifuge tubes and treated
for 1 hr with 1-2~g/ml of polybrene at 37~C. The cells a~e then
pelleted for 8min. at 1200 RPM. HIV virus, diluted 1:10 in media
(RMPl-1640, 10~ human serum or 15~ fetal calf serum (FCS), with IL-2
and antibiotics) is added to provide an MOI of .001. Medium alone is
added to virus-free control cells. Assuming an infectious virus titer
of 10-4, an MOI of .001 represents 8 infectious virus particles per
10,000 cells. About 500,000 cells/tube are exposèd to 400 ~ of the
virus dilution. The resultant mixture is incubated for lhr at 37~C in
Air-C02. The infected or uninfected cells are diluted to give 1 x
10-4 (with human serum or 2 x 10-4 (with fetal calf serum)
cel ls/100~
Infected or uninfected cells (100~1) are distributed to
appropriate wells of a 96 well, U-bottom microtiter plate. Each
compound dilution is tested in duplicate with infected cells.
Uninfected cells are examined for drug sensitivity in a single well
for each dilution of compound. Drug-free control cells, infected and
uninfected, are run in triplicate. Wells B2 through G2 served 8S
- 35 - 1 3 3 ~
reagent controls and received medium only. The plates are incubsted
at 37~C in Air-C02 until the drug is added.
!
II. Drug Dilution and Addition
Dilution plates (flat bottom 96 well, microtiter plates) are
treated overnight with phosphste buffered saline (PBS) or media
containing at least 1% FCS or 1% human serum (depending on the medium
used in the test), beginning the day before assay. This "blocking"
procedure is used to limit the adsorption of drug to the microtiter
tray during the dilution process. The wells are filled completely
with the blocking solution and allowed to stand at room temperature in
a humidified chamber in a hood.
The dilution process is begun by first diluting the test compound
1:20. Blocked, dilution plates are prepared by flicking out the
blocking solution ~nd blotting dry on sterile gauze. All wells of
each plate are then filled with 225~1 of the appropriate medium using
a Cetus liquid handling system. Twenty-five microliters (25~1) of
each 1:20 diluted compound is then manually added to row A of a
blocked and filled dilution plate. Four compounds, sufficient to
supply two test plates, are added per dilution plate. The four
compounds are then serially diluted 10 fold from row A through row H
using the Cetus liquid handling system. The starting dilution of each
compound in row A is, at this point, 1:200. The dilution plates are
kept on ice until needed.
Using a multi-channel pipettor with 6 microtips, 100~1 of each
drug dilution is transferred to the test plate which already contains
100~1 of medium plus cells. The final dilution, in the test plate,
starts at 1:400 (wells B4 through G4). This dilution (to .25% DMS0)
prevents the DMS0 vehicle from interfering with cell growth.
Drug-free, infected or uninfected cells (wells B3 through G3) and
reagent controls (B2 through G2) receive medium alone. The final 2
compounds are then transferred from wells H7 through H12 to a second
test plate suing the same procedure. Test plates are incubated at
370C in Air-C02 for 7-14 days or until virus controls are lysed as
determined macroscopically.
- 36
III. Quantitation of Viral Cytopathogenicity and Drug Activity
A. Materials
1. A solution of 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-5-[(
phenylamino)carbonyl]-2H-tetrazolium hydroxide. (XTT) - lmg/ml
solution in media without FCS. Store at 4~C. Prepare weekly.
2. Phenazine methosulfonate (PMS) stock solution - This can be
prepared and maintained frozen until needed at -20~C. It should be
made in PBS to a concentration of 15.3mg/ml.
B. Microculture Tetrazolium Assay (MTA)
1. Preparation of XTT-PMS Solution - The XTT-PMS is prepared
immediately prior to its addition to the wells of the culture dish.
The stock PMS solution is diluted 1:100 (0.153mg/ml). Diluted PMS is
added to every ml of XTT required to -give a final PMS concentration of
0.02mM. A 50~1 sliquot of the XTT-PMS mixture is added to each of the
appropriate wells, and the plate is incubated for four hours at 37 ~.
The plate lids are removed and replaced with adhesive plate sealers
(Dynatech cat 001-010-3501). The sealed plate is shaken on a
microculture plate mixer and the absorbance is determined at 450nm.
IV. Results
Figure 1 depicts a plot of the percentage of test cells over
uninfected cells (~) for both infected and uninfected cells as a
function of the increasing concentration of the compound of Example
10.
The dat~ plotted on Figure 1 permits the calculation of sn
effective concentration (EC~o) with respect to infected cells of about
0.15~g/ml, an inhibitory concentration (IC50) with respect to normal
cells of about lOO~g/ml, and a therapeutic index (TI~o)of about 667.
An earlier assay carried out at the Southern Research Institute
yielded at TI50 of about 200 when MT-2 cells were cultured with
H9/HTLV-IIIB.
1339~9~
_ 37
The inhibitory concentrations against HIY determined as described
above for the compounds of Examples 7, 9, 10, 11 and 14(b) are shown
in Table 1.
TABLE 1
Compound Example Cell Line ED50 ID~o TI~o
9a 7 MT-2 2.3 50 21.4
13a 9 MT-2 0.41 6.97 17.3
14a 10 MT-2 0.15 100 667
15a 11 MT-2 2.9 > 125 > 42.7
(-) 14a 14 (b) CEM 0.66 189 284
The compounds of Examples 5 and 8 also showed antiviral activity
in this screen.
(B) Activity against Feline Leukemia Virus
Antiviral screening for activity against FeLV-FAIDS was performed
in 96-well plates (Corning) using 81C indicator cells in Iscove's
Modified Dulbecco's medium supplemented with 10~ heat-inactivated
fetal bovine serum (FBS). Twenty hours prior to the assay, the plates
were seeded with the 81C cells at 5 x 103 cells/well. On the day of
the assay, the cells were pretreated for 30 minutes at 37~C with DEAE-
dextran (25~g/ml) in O.lml Hanks balanced salt solution. This was
removed and then O.lml of growth medium containing 32 TCID~o of
FeLV-FAIDS, or O.lml of growth medium alone, was added to each well.
The virus was allowed to adsorb for 1 hour, then 0.1 ml of test or
positive control compound (2',3'-dideoxycytidine; ddC), or growth
133g896
- 38
medium was added. Plstes were incubated at 37~C. Cells were fed
fresh growth medium containing compound on Day 4 post-infection.
Culture medium was completely changed and replaced with fresh medium
containing compound on Day 7 post-infectin. On Day 10 post-infection
the cells were fixed with formalin, stained with 0.1~ Coomsssie
Brilliant Blue R-250 and observed microscopically for CPE and drug
cytotoxicity.
The compound of Example 10 had an ED50 of 1.9~g/ml.
. .
(C) Activity against Murine AIDS
Falcon 6-well tissue culture plates were seeded with 1.75 x 105
cells per well in total volume of 2.5m1 EMEM containing 5~ heat-
inactivated FBS. Twenty hours after the cells were seeded, the medium
was decanted and 2.5ml DEAE-dextran (25~g/ml in phosphate-buffered
saline) was added to each well. The cultures were incubated at 37~C
for 1 hour, after which the DEAE-dextran solution was decanted and the
cell layers rinsbd once with 2.5ml PBS. Normal cell controls were
refed with 2.5ml medium alone (no virus or drug). Drug control
cultures-received 2.5ml of medium containing drug but no virus.
Virus-infected control cultures received 0.5ml of the appropriate
dilution of stock CAS-BR-M to produce countable plaques plus 2.Oml
medium. The test samples received 0.5ml of the appropriate virus
dilution'plus 2.0ml medium of the drug dilution. Six concentrations
of the test compound diluted in serial half-log10 dilutions were
tested. Three concentrations of the positive control drug, ddC, were
tested. Triplicate wells for eàch concentration of test compound and
6 virus and 6 cell control cultures were included in each assay. On
Day 3 post-virus inoculation toxicity of the drug for the SC-l cells
was determined by microscopic examination of stained duplicate cell
and drug control cultures. The remaining test and control cultures
were irradiated with an ultraviolet lamp for 20 seconds and XC cells
were added to each culture (5 x 105 cells/well in 2.5ml EMEM
containing 10~ heat-inactivated FBS). On Day 3 post-UV irradiation,
the cultures were fixed with formalin and stained with crystal violet.
The plaques were counted with the aid of a dissection microscope.
13398g6
- 39
Antiviral activity in the CAS-BR-M plaque reduction was expressed
in terms of the reduction in the mean number of plaques counted in the
drug-treated, virus-infected cultures compared with the mean number of
plaques counted in the untreated, virus-infected control cultures
(percent of control). The compound of Example 10 had an ED50 of
l.l~g/ml.
(D) Activity against Simian retrovirus SAIDS (SRV-2)
Antiviral screening against the SAIDS virus (D/Washington) was
performed by a syncytia-inhibition assay on Raji cells. The drug was
diluted in complete Iscove's medium and then 100~1 of each dilution
was added to the appropriate wells of a 96-well plate. Actively
growing Raji cells, 5 x 103 cells in 50~1 of complete Iscove's medium,
were then added to each well. This was followed by the addition of
50~1 of clarified supernate from an SRV-2/Raji cell co-culture. DDC
was included in this assay as the positive control drug. Plates were
incubated at 37~C in a humidified atmosphere containing 5% C02.
Syncytia were counted on Day 7 post-infection. Drug toxicity was
ascertained by comparing viable cell counts of the uninfected, drug-
treated sample to the viability of the uninfected, untreated control.
The compound of Example 10 had an EDso of 2.8~g/ml.
(E) Activity against Visna Maedi Virus
The antiviral activity agsinst Visna Maedi Virus (VMV) strain
WLC-l, was determined by measuring reduction of virus-specific
immunohistochemical staining. Monolayers of sheep choroid plexus
cells were infected with VMV and overlaid with serial dilutions of
test compounds. After incubation for five days, the monolayers were
further incubated with virus specific antisera conjugated to horse
rsdish peroxidase (HRP). Subsequent incubation of the monolayers with
a chromagenic substrate of HRP, strains areas of virus replication.
These discrete foci were counted and the concentration of test
compound required to reduce the number of foci to 50% of that of drug
untreated controls calculated.
The compound of Example 13 had an ED~o 0.2~g/ml.
1339~gS
- 40
Example 22
CYTOTOXIC ACTIVITY
The compounds of Examples 5, 7 and 8 showed cytotoxic activity
- when tested against P388 mouse leukemia cell culture assay as
described by R. G. Alonquist and R. Vince, J. Med. Chem, 16, 1396
(1973). The ED~ols (~g/ml) obtained were :-
Exsmple 5 12
Example 7 40
Example 8 3