Note: Descriptions are shown in the official language in which they were submitted.
1 340424
It is known that various representatives of 7-a-
aminoacyLcephalosporins with different substituents in
the 3-position of the molecule, thus, for example,
cephalexin [7-(D-~-phenylglycylamido)-3-methyl-3-cephem-
S 4-carboxylic acid, compare DE-OS (German Published
Specification) 2,432,485], cefaclor [7-(D-a-phenylglycyl-
amido)-3-chloro-3-cephem-4-carboxylic acid, compare DE-OS
(German Published Specification) 2,408,698 and 2,728,578]
and cefadroxil ~7-(D-a-p-hydroxyphenylglycylamido)-3-
methyl-3-cephem-4-carboxylic acid, compare DE-OS (German
Published Specification) 2,718,741~ have a good antibio-
tic activity.
3-Alkenyl-substituted cephalosporins are further-
more described as compounds with an oral action in DE-OS
(German Published Specification) 3,402,642 and U~S.
Patent 4,619,925.
3enzothiazolylglycylamido-substituted vinyl-
cephalosporins are known from DE-OS (German Published
Specification) 3,508,258. The present invention relates
to a selection of the substances described in DE-OS
(German Published Specification) 3,508,258.
The invention thus relates to B-lactam compounds
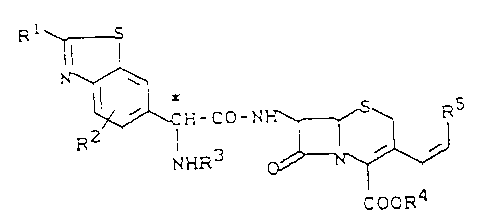
of the general formula (I)
R 1 ~5
~jH_CO_I~R5 ( I )
cooR4
in which
R1 represents hydrogen, or represents a straight-
chain, branched or cyclic alkyl radical which hasLe A 25 238
1340'12~
up to 8 carbon atoms and can be substituted by
halogen, hydroxyl, alkoxy with up to 4 carbon
atoms, carboxyl, phenyl or sulpho, or by a group
of the formula
~R6
~R7
~herein
R6 and R7 are identical or different and denote
hydrogen, alkyl ~ith up to 6 carbon atoms, aryl
with 6 to 12 carbon atoms, aralkyl ~ith 7 to 12
carbon atoms or acyl ~ith Z to 7 carbon atoms,
or
R1 represents phenyl,
halogen, alkoxy, alkylthio or alkylsulphonyl ~ith
in each case up to 6 carbon atoms, sulpho,
sulphamoyl, mercapto, hydroxyl, phenylthio,
phenyloxy, guanidino or amidino, or represents a
group of the formula
,~R6
~R7
wherein
2û R6 and R7 have the abovementioned meaning,
R2 represents hydrogen, alkyl, alkoxy or alkyl-
thio with in each case up to 6 carbon atoms,
trifluoromethyl, trifluoromethoxy, hydroxyl,
mercapto, nitro, cyano or halogen,
2S R3 represents hydrogen, or represents an amino-
protective group,
R4 represents hydrogen, or represents a carboxyl-
protective group, or represents an ester ~hich
can be split off in vivo and
R5 represents hydrogen, or represents straight-
chain or branched alkyl ~hich has up to 6 carbon
atoms and can be substituted by halogen, alkoxy
Le A 25 238
-- 2
134042~
or alkylthio with in each case up to 6 carbon
atoms, hydroxyl or amino, or by a radical of the
formula
N _ ~ ~ ~ CH3
CH3
S ~ ' ~ '
H2C--CH2
HCHO
' ~'CH3' 'CH3
CH3 ~CH3 ' 'CH3
CH3
~ CH3 ~CH3 -ClHC3H3
~0 and salts thereof.
An amino-protective group in the context of the
definition given above in general represents a protective
Le A 25 238
~ 340424
group customary in ~-lactam chemistry from the series comprising:
benzyl, tert.-butoxycarbonyl, benzyloxycarbonyl, 2-nitrobenzyloxy-
carbonyl, 4-nitrobenzyloxycarbonyl, 2.2.2-trichloro-ethoxycarbonyl,
fluorenyl-(9)-methoxycarbonyl, N-diphenyl-methoxycarbonyl, aceto-
acetyl, 2-nitrobenzoyl, 2-nitrophenylsulfenyl, phthaloyl, trityl,
vinyloxycarbonyl, formyl, benzoyl, allyloxycarbonyl, 2.4-dimethoxy-
benzyloxycarbonyl, 2-methylthio-ethoxycarbonyl, 1,3-dithian-2-yl-
methoxycarbonyl (Dmoc), trimethyl-, triethyl-, triphenylsilyl,
tert.-butyl-dimethylsilyl, tert.-butyldiphenylsilyl, l-methyl-2-
benzoyl-vinyl, 1-methyl-2-ethoxycarbonyl-vinyl, 1-methyl-2-methoxy-
carbonyl-vinyl, l-methyl-2-(2.6-dimethoxybenzoyl)-vinyl, 4-methoxy-
benzyloxycarbonyl (see E. Wunsch, Method of Organic Chemistry,
Hourben-Weyl, Vol. 15/I (1974)).
A carboxyl-protective group in the context of the defini-
tion given above represents the carboxyl-protective groups custo-
mary in ~-lactam chemistry. Groups which can easily be split off
may be mentioned as preferred, such as, for example: methyl, ethyl,
tert.-butyl, decyl, 2-chloroethyl, 2,2,2-trichloroethyl, cyanoethyl,
diphenylmethyl (benzhydryl), triphenylmethyl, acetoxymethyl, allyl,
benzyl, 4-methoxyphenyl, 4-nitrobenzyl, 2-nitrobenzyl, 4-methoxy-
benzyl, 2,4-dimethoxybenzyl, trimethylsilylethyl, trimethylsilyl,
tert.-butyl-dimethylsilyl, acetonyl, l-phenoxyethyl or 2-methyl-
2-propenyl (see a). E. Wunsch, Method of Organic Chemistry, Houben-
Weyl, Vol. 15/I (1974) and b) Th. W. Greene Protective Groups in
Organic Synthesis, J. Wiley & Sons (1981)).
If R4 represents an ester radial which can easily be
1~40424
split off in vivo, by these are meant pharmaceutically tolerated
ester radials which are easily hydrolyzed in vivo to give free
carboxylic groups (R = H).
Such ester radicals are well-known in the ~-lactam
field. In most cases, they improve the absorption
- 4a -
" ' 134042~
.
properties of the B-lactam compounds. rhe radical R4
should furthermore be such that it imparts pharmaceutic-
ally acceptable properties to a compound of the formula
(I) and liberates pharmaceuticaLly acceptable fragments
~hen split in vivo.
ExampLes of such groups are to be found in DE-OS
(German Published Specifications) 2 228 255 and
2 350 230. Preferred ester groups which can be split off in
vivo are those of the following formulae:
~ ~ ~ ~ ~
O O
RlO o
O -I;O-C-R and
R10 C
_C_o_c_o-Rl 2
Rl 1
~herein
R8 and R9 are identical or different and repre-
sent hydrogen or phenyl, or represent C1-C4-alkyl,
preferably methyl,
R10 and R11 are identical or different and
represent hydrogen, or represent C1-C4-alkyl,
preferably methyl, and
R1Z represents C1-C6-alkyl, preferably C1-C4-
alkyl.
~he compounds of the general formula (I) accord-
ing to the invention can be in the form of the free acids,
esters, inner salts
Le A 25 238
_ 5
.. . .
Rl~; 13 4 0 4 2 1
N~fH-CO-~ rJ ~:(amo~e of
COOe salt
or non-toxic physiologically tolerated salts with a
counter-cation
R ~
Example of
~2 ~ H-CO- ~ S ~ CH3 a salt with
NHR3 ~ a counter-
COOeN~ cation
S or, if R4 is a positively charged radical, non-toxic
physiologically tolerated salts with a counter-anion.
R ~
Example of
R2 ~ H-CO- ~ ~ ~ a salt with
Cle a counter-
COOH anion
( COOAlkyl )
Preferred counter-cations which may be mentioned
are alkali metal or alkaline earth metal cations, such
as, for example, sodium, potassium, magnesium or calcium
ions, or a(uminium or ammonium ions, and non-toxic sub-
stituted ammonium ions of amines such as di-lower alkyl-
amines, tri-lower alkylamines, procaine, dibenzylamine,
N,N'-dibenzylethylenediamine, N-benzyl-B-phenyl-ethyl
Le A 25 Z38
-- 6
...... .. . .. . .
134~42~
amine, N-methylmorpholine, 1-ephenamine, dihydroabietyl-
amine, N,N'-bis-dihydroabietylethylenediamine, N-lower
alkylpiperidines or other amines which can be used to form
salts of B-lactam compounds.
Preferred counter-anions which may be mentioned
are inorganic or organic acid radicals, such as, for
example, chloride, bromide, iodide, sulphate, hydrogen
sulphate, phosphate, hydrogen phosphate, carbonate or
bicarbonate, or sulphonates, such as methanesulphonate,
ethanesulphonate, toluenesulphonate, benzenesulphonate
or naphthalenedisulphonate, or carboxylates, such as
acetate, formate, oxalate, tartrate, citrate, maleate,
fumarate, benzoate, succinate and lactate.
The compounds of the general formula tI) exist
(in respect of the double bond) in the Z-(cis) and in the
E-(trans) configuration. The compounds with the Z-(cis)
configuration are preferred. Pecause of the presence of
the asymmetric carbon atom labelled with ~ (see formula
I), the B-lactam antibiotics of the general formula (I)
according to the invention include the D-, L- and D,L-
forms. Poth the diastereomer m;xtures and the D-form and
L-form of the compounds according to the invention can
be used for the treatment of bacterial infection dis-
eases. The D-forms of the compounds according to the
invention are particularly preferred.
Compounds of the general formula (I) which may
be mentioned as preferred are those
in which
R represents hydrogen, or represents straight-
chain, branched or cyclic alkyl with up to 6
carbon atoms, or represents phenyl, or represents
a group of the formula -NHR6,
wherein
R6 denotes hydrogen, phenyl, benzyl, acetyl,
benzoyl or alkyl with up to 6 carbon atoms,
R2 represents hydrogen, methyl, methoxy, tri-
Le A 25 238
-- 7
... . . .. . ... .
1 3 ~ 2 4
fluoromethyl, trifluoromethoxy, nitro, cyano,
fluorine, chlorine, bromine or hydroxyl,
R3 represents hydrogen, or represents an amino-
protective group from the series comPrising:
tert.-butoxycarbonyl, benzyloxycarbonyl, 2.2.2-
trichloroethoxycarbonyl, trityl, vinyloxycarbonyl,
allyloxycarbonyl, 2.4-dimethoxybenzyloxycarbonyl,
l-methyl-2-benzoyl-vinyl, 1-methyl-2-ethoxycarbo-
nyl-vinyl, l-methyl-2-methoxy-carbonyl-vinyl,
4-methoxy-benzyloxycarbonyl,
R4 represents hydrogen, or represents methyl,
ethyl, tert.-butyl, 2-chloroethyl, 2,2,2-tri-
chloroethyl, cyanoethyl, diphenylmethyl, tri-
phenylmethyl, acetoxymethyl, allyl, benzyl, 4-
methoxybenzyl, 2,4-dimethoxybenzyl, 1-phenoxy-
ethyl, 2-methyl-2-propenyl, 4-nitrobenzyl, 2-
nitrobenzyl, trimethylsilylethyl or tert.-butyl-
dimethylsilylethyl, or represents a radical of
the formula
o~3 ~,~ ~~
-H2C' I I 'CH3 -CH2-oco-c ( CH3 ) 3 -
or
O -CH ( CH3 ) -OCOOC2H5
-CHz-OCOCH3
Le A 25 238
-- 8 --
~ . ~, . . . .. . . ..
1 ~40424
, ~
and
R5 represents hydrogen, or represents straight-
chain or branched alkyl which has up to 4 carbon
atoms and can be substituted by fluorine, chlor-
ine, bromine, iodine, alkoxy with up to 3 carbon
atoms, hydroxyl or amino, or by a radical of the
formula
~ ~ y ~ ~3
CH3
C~ , ~ , ~,
~~ ' ~ ~CH '
or ~ ~
~ 'CH3
and salts thereof.
Compounds of the general formula (I) which may be
mentioned as particularly preferred are those
in which
R represents hydrogen, or represents straight-
chain or branched alkyl with up to 4 carbon atoms,
or represents a group of the formula -NHR6
wherein
R6 denotes hydrogen, methyl, ethyl, propyl or
isopropyl,
R2 represents hydrogen or hydroxyl,
Le A 25 238
. , ~ . .
13~042~
R3 represents hydrogen, or represents benzyloxycar-
bonyl, 1-methyl-2-benzoyl-vinyl-, 4-methoxybenzyl-
oxycarbonyl, 2-nitrophenylsulfenyl (NPS), trityl,
allyloxycarbonyl, tert.-butyl-dimethylsilyl, l-methyl-
2-methoxycarbonyl-vinyl (MMV) or tert -butoxycarbonyl (Boc),
R4 represents hydrogen, or represents methyl,
ethyl, tert.-butyl, diphenylmethyl, 2,2,2-tri-
chloroethyl, allyl, acetoxymethyl, 4-nitrobenzyl,
2-nitrobenzyl, 4-methoxybenzyl, benzyl or tri-
methylsilylethyl, or represents a radical of the
formula
-H2C'1 ~CH3
~0 , -CH(CH3)-OCOOC2H5
or -CH2-0C0-CtCH3)3, and
R5 represents hydrogen, oethyl, chloromethyl,
dihydroxyethyl or iodomethyl, or represents a
radical of the formula
~ Q H3 ~ H3C
-H2C CH3
CH
or ~
-H2CN~J
H
and salts thereof.
20~he compounds listed in the follo~ing table are
moreover especially preferred:
R ~--S
~ CH-CO- ~ R5.
Le A 25 238 COOR4
-- 10
..
1~40424
R 1 R2 R4 R5
H3C- H H -CH3
H2N - H H - C 2H5
H2N H H -CH2C 1
H2N- H H -CHz-OCH3
H2N- H H -CH2I
H- H H
.~
H3C- H H ~
p~H3
H2N- H H
H2C S H
H2N H CH20COCH3 CH3
H2N OH H CH3
H2N H -CH20COCH3 CH2-OCH3
H H2N H CH3
H2N C 1 H CH3
H2N H -CH20COC ( CH3 ) 3 I=~JN~
S~ CH3
H OH H CH3
,OH
H2N H H -CH
CH20H
D H H C~3
Le A 25 238
.. . . . ...
1~110424
R 1 R2 R4 RS
H3C--N
CH3 H2N H I ,~
S~
H2N OH - CH ( CH3 ) OCOOC 2HS CH3
A process has furthermore been found for the
preparation of the substituted vinylcephalosporin com-
pounds of the general formula (I) according to the
invention, which is characterized in that
~A] substituted cephalosporin compounds of the general
formula (II)
Rl~i 5
N~H-CO-Ni~ ( I I )
NHR3 o~~CH2-X
CoOR4
in which
R1 and R2 have the abovementioned meaning,
Le A 25 238
- 12 -
R3 represents an amino-protective group, 134042~1
R4 represents a carboxyl-protective group and
X represents a group of the formula
R13 oR13
-P(R13)3ze -P-RI4 or -P-oR14
0 11
o
wherein
R13 and R14 are identical or different and denote
alkyl, phenyl or tolyl and
Z denotes a halide anion, preferably chloride,
bromide or iodide,
are reacted with aldehydes of the general formula (III)
R5-CH0 (III)
in which
RS has the abovementioned meaning,
in inert solvents in the presence of bases, or in that
~B] phosphonium compounds of the general formula (IV)
R5-CHz-X (IV)
in which
R5 has the abovementioned meaning and
X represents a group of the formula
R13 IOR13
-P(RI3)3ze , _p_R14 _Il_oRl4
wherein
R13 and R14 are identical or different and denote
alkyl, phenyl or tolyl and
Z denotes a halide anion, preferably chloride,
Le A 25 238
- 13 -
1~4()42 1
bromide or iodide,
are reacted with cephalosporinaldehydes of the general
formula (V)
Rl~
~H-co- S
NHR3 ~HO
CoOR4
in which
R1 and R2 have the abovementioned meaning,
R3 represents an amino-protective group and
R4 represents a carboxyl-protective group,
in inert solvents in the presence of bases, or in that
~C] carboxylic acids of the general formula (VI)
R l~lr5
H-COOH ( V I )
NHR3 '
in ~hich
R1 and R2 have the abovementioned meaning and
R3 represents an amino-protective group,
after activation of the carboxyl group by conversion into
a mixed anhydride, for example with ethyl chloroformate,
isobutyl chloroformate or methanesulphonyl chloride, or
by conversion into the acid halide, or by conversion into
an activated ester, for example ~ith N-hydroxybenzotri-
azole and dicyclohexylcarbodiimide (DCC), are reactedwith the vinylcephalosporinamines of the general formula
(VII)
Le A 25 238
- 14 -
134042~
H2 ~ R(VII), (VII)
CoOR4
in which
R4 and R5 have the abovementioned meaning,
and, if appropriate, protective groups are then split off
and the desired salts are prepared or the free acids are
prepared from the salts.
The process according to the invention can be
illustrated by the following equation:
Process variant A:
H3C
CIH-CO- ~ ~
NH~oc ~ H2 P(C6Hs)3I
COOCH~C6H5)2
B~-
H3C~
C=O
~ H~
H3C ~
CIH-CO- ~ S~ CH3
NHBoc ~
COOCH(C6H5)2
* Boc = (H3C)3C-O-CO-
Le A 25 238
1 5
134042~
Process variant L:
H3C
H-CO- ~
NHBoc HO
COOCH(C6H5)2
B~ce
H3C H2C _ pe~ ~ C6H5 ) 3 I e
H3C ~
H-CO-N S CH3
NHBoc ~
COOCH(C6H5)2
* Boc = (H3C)3C-O-CO-
Process variant C:
H3C ~ ~ H-COOH ~ CH3
NHBoc CoocH(c6Hs)2
compling
H3C ~
H-CO- ~ CH3
NHBoc o ~
COOCH(C6H5)2
* Boc = (H3C)3C-O-CO-
Le A 25 238
Process variants A and B 134042~
Suitable inert solvents for process variants A
and B are the customary organic solvents which do not
change under the reaction conditions. These include,
S preferably, ethers, such as diethyl ether, butyl methyl
ether, dioxane or tetrahydrofuran, or hydrocarbons, such
as benzene, toluene, xylene or cyclohexane, or amides,
such as dimethylformamide or hexamethylphosphoric acid
triamide, or alcohols, such as methanol, ethanol, pro-
panol or isopropanol, or chlorohydrocarbons, such asmethylene chloride, chloroform or carbon tetrachloride,
or acetone, acetonitrile or ethyl acetate. It is also
possible to use mixtures of the solvents mentioned.
Suitable bases for process variants A and B are
the customary basic compounds. These include, prefer-
ably, alkali metal or alkaline earth metal hydroxides,
such as, for example, sodium hydroxide, potassium hydrox-
ide or barium hydroxide, or alkali metal carbonates, such
as sodium carbonate, sodium bicarbonate or potassium
carbonate, or alkali metal alcoholates, such as sodium
methanolate, sodium ethanolate, potassium methanolate,
potassium ethanolate or potassium tert.-butylate.
The choice of solvent and base depends on the
stability, hydrolysis-sensitivity and CH-acidity of the
corresponding phosphorus compound. Solvents ~hich are
particularly preferably used are chlorohydrocarbons, such
as, for example, methylene chloride, chloroform or carbon
tetrachloride, in the presence of dimethylformamide as a
co-solvent. Bases ~hich are particularly preferably used
are alkali metal carbonates, such as sodium carbonate,
sodium bicarbonate or potassium carbonate, or alkali
metal or alkaline earth metal hydroxides, such as, for
example, sodium hydroxide, potassium hydroxide or barium
hydroxide, particularly preferably in the form of their
aqueous solutions.
The reaction is in general carried out in a tem-
Le A 25 238
- 17 -
.
1340~2~
perature range from -30~C to +80~C, preferably from 0~C
to +30~C.
The reaction can be carried out under normal,
increased or reduced pressure (for example in a range
S from 0.5 to 5 bar). It is in general carried out under
normal pressure.
In carrying out process variants A and B, the
phosphorus compound (II) or (IV) is in general employed
in an amount of 1 to 3 mol, preferably in molar amounts,
per mol of the aldehyde (III) or (V). The bases are in
general employed in an amount of 1 to 5 mol, preferably
1 to 2 mol, per mol of the phosphorus compounds.
Process variants A and B are particularly prefer-
ably carried out as a Wittig reaction. In carrying out
the process according to the invention, it is also pos-
sible, instead of the phosphonium salts [X = -P(R13)3+Z ],
for the corresponding phosphoranes
Rl ~
R2 ~ H-CO-N ~ ~ ,P(R13)3
NHR3
CoOR4
which have first been prepared from the corresponding
phosphonium salts and base in a separate reaction, to be
employed directly. However, it has proved to be advan-
tageous to carry out the reaction with the triphenyl-
phosphonium salts (X = P (C6Hs)3Z ) in the presence
of bases as a one-pot process. As a particular variant
of a one-pot process, the reaction can also be carried
out in the form of a phase transfer-catalyzed reaction,
depending on the stability of the phosphorus compounds,
solvents which can be used being ethers, hydrocarbons and
Le A 25 238
- 18 -
. .
13~24
halogenohydrocarbons and bases which can be employed
being aqueous sodium hydroxide or potassium hydroxide
solutions.
Alternatively, if the reaction is carried out by
a procedure in which the corresponding phosphorane is
isolated as an intermediate compound and is reacted with
the aldehyde in a second step, it has moreover been
found that the yield and the ratio of Z/E isomer of the
end products of the generaL formuLa (I) are improved by
adding a suitabLe Lithium haLide, such as, for example,
lithium chloride, lithium bromide or lithium iodide. The
reaction here is preferably carried out with 10 to 15
equivalents of lithium haLide.
However, it is particuLarly preferable to carry
out process variants A and ~ as a one-pot reaction with-
out isolation of the intermediate product. ~he process
variants according to the invention can be carried out,
for example, by adding the base and then a corresponding
aldehyde, if appropriate in a suitable solvent, to the
phosphonium compounds, dissolved or suspended in a suit-
able solvent, and if appropriate warming the mixture.
~orking up is carried out in the customary manner by
extraction, chromatography and~or crystallization.
Other specific process variants for the ~ittig
reaction are described, inter alia, in the following
references: J. Fuhrhop and G. Penzlin: Organic Synthesis,
Ver~ag Chemie, 1983, pages 26 - 35; R.K. Mackie and D.M.
Smith: Guidebook to Organic Synthesis, Longman Group
Limited, 1982, pages 93 - 99; H.O. House: Stereochemistry
of the ~ittig Reaction vith stabilized ylides: J. Org.
Chem. 29, 3327 - 3333 (1964).
Process variant C
It has proved to be advantageous to activate
amino acids and then to coupLe them with B-Lactams, which
have been dissoLved as saLts with an amine.
Activation of carboxylic acids of the general
formula (VI) with (a) sulphonic acid derivatives of
Le A 25 238 - l9 -
13~04~
the general formula (YIII) or with (b) chloroformic acid
esters, preferably ethyl chloroformate, to give anhyd-
rides of the general formula (IXa,b), as illustrated in
the following equation, is particularly advantageous:
R
a) T-502-R15 (VIII )
Rl~ 7 R2~H-CO0502R1 5
NHR3
R2 H-COOH IXa
NHR 3 R 1
VI b ) ClCOOC2H5 1~
R2 H-COOCOOC2H5
NHR3
IXb
In this equation, in formula (VIII) and (IXa)
T represents the radical R15-So2-o- or halogen
and
R15 represents alkyl which has up to 10 carbon
atoms and is optionally substituted by fluorine,
chlorine, cyano/alkyl, alkoxycarbonyl, alkoxy
or alkyl with in each case up to 4 carbon atoms,
or represents phenyl, which is optionally substi-
tuted by fluorine, chlorine, bromine, cyano,
alkyl, alkoxy, alkylthio or alkoxycarbonyl with
in each case up to 4 carbon atoms, nitro, tri-
fluoromethyl or phenyl.
If R15 is substituted, 1 to 3 substituents are
preferably present, and those mentioned above are parti-~0 cularly preferably present.
R15 especially preferably represents a methyl
or p-tolyl radical.
The mixed anhydrides of the general formula
Le A 25 238
- 20 -
l3~o42~
(IXa,b) are prepared by dissolving the carboxylic acids
of the general formula (VI) and 1 to 1.4 e~uivalents of an
amine in a solvent and alloving the solution to react
vith 1 to 1.2 equivalents of a sulphonic acid derivative
S of the formula (VIII) or of a chloroformic acid ester.
Suitable solvents are all the solvents vhich do
not change under the reaction conditions. rhese include,
preferably, ethers, such as, for example, diethyl ether,
dioxane or tetrahydrofuran, or chlorohydrocarbons, such
as methylene chloride, chloroform or carbon tetrachloride,
or amides, such as dimethylformamide or hexamethylphos-
phoric acid triamide, or acetonitrile or acetone. It is
also possible to use mixtures of the solvents mentioned.
Suitable amines are tertiary amines, such as, for
example, triethylamine, ethyl-diisopropylamine or tri-
butylamine, but also sterically hindered secondary
amines, such as, for example, diisopropylamine. Mixtures
of the amines mentioned can also be used.
The reactions can be carried out at temperatures
betveen -80~C and room temperature. rhe activation is
advantageously carried out ~ith methanesulphonyl chloride
in dimethylformamide at -40~C to -60~C in the course of
0.2 to 24 hours, preferably O.S to S hours.
The solvents mentioned in the preparation of the
compounds of the formula (IX) or ~ater can be used to
dissoLve the vinylcephalosporinamines of the formula (VII) for
the coupling with the formula (IXa) or (IXb) to obtain the
formula (I), and the amines mentioned there can be used as the base.
Activation of the carboxylic acids of the general
formula (VI) by conversion into an activated ester vith,
for example, dicyclohexylcarbodiimide, if appropriate in
the presence of N-hydroxysuccinimide or 1-hydroxybenzo-
triazole, is also particularly advantageous.
Suitable solvents here are all the solvents vhich
are also suitable for the preparation of anhydrides of
the general formula (IXa,b) and have already been mentioned
Le A 25 238
- 21 -
1340~2 t
there.
The reactions can be carried out at temperatures
between -30~C and +100~C. Activation is advantageously
carried out with 1-hydroxybenzotriazole and dicyclohexyl-
S carbodiimide in dimethylformamide at room temperature for
2 to 6 hours, and the dicyclohexylurea which has precipi-
tated out is then fiLtered off with suction and reacted
with the vinylcephalosporinamines of the formula (VII)
in the form of a solution of their amine salt in the course
of 2 to 24 hours. The solvents mentioned for the pre-
paration of the compounds of the formula (IX) can be used
to dissolve the vinylcephalosporinamines of the formula
(VII), and the amines mentioned there can be used as the
base.
The aldehydes of the general formula (III) used
as starting substances are known or can be prepared by
known methods tHouben-Weyl's "Methoden der organischen
Chemie" ("Methods of Organic Chemistry") Volume VII/1;
E2].
The cephalosporin aldehydes of the general for-
mula (V) used as starting compounds are known or can be
prepared by known methods by oxidation of the correspond-
ing 3-hydroxymethyl-cephalosporin compounds with chromium
trioxide in acetone (Jones reagent), such as is described,
for example, by J.A. Webber, J.L. Ott and R.T. Vasileff
in J. Med. Chemistry 18, 986 (1987).
The phosphonium compounds of the general formula
(IV) used as starting substances are known or can be pre-
pared by known methods CHouben-~eyl's "Methoden der
organischen Chemie" ("Methods of Organic Chemistry")
Volume XII/1, 33, 167; Volume V/lb, 383, 872].
The substituted cephalosporin compounds of the
general formula (II) used as starting substances are new
in some cases and can be prepared by a process in which
halogenomethylcephalosporin compounds of the general
formula (X)
Le A 25 238
- 22 -
1340';2~
R ~
~H-CO-N~ (X)
N}IR3 0~ :
CoOR4
in which
R1, R2, R3 and R4 have the abovementioned
meaning and
S Z represents halogen, preferably chlorine, brom-
ine or iodine,
are reacted with phosphorus compounds of the general
formula (XI)
X¦ (XI)
~0 wherein
X¦ represents a phosphorus compound of the
formula XIa, XIb or XIC
R14 oR14
IP(R13)3 , ¦g-R13 or ¦P-OR13
o o
XIa XIb XIc
wherein
R13 and R14 are identical or different and
represent alkyl, phenyl or optionally substituted
phenyl,
without solvents or in inert solvents.
The process according to the invention can be~0 illustrated by the following equation:
Le A 25 238
- 23 -
CH3' ~ 13 4 0 4 2 4
N~"j~
H-CO-N ~
NHaoc ~ Cl
CoocH(c6Hs)2
H3C' S ~ N~l
H-CO-N ~ ~
NHBoc ~ I
CoocH(c6H5)2
~' IP~C6HS)3
H3C'I S
H-CO- ~
NHBoc ~ ~C6H5)3Ie
COOcH~c6Hs)2
Suitable inert solvents are the customary organic sol-
vents ~hich are not changed under the reaction condi-
tions. These include, preferably, ethers, such as
diethyl ether, buty~ methy~ ether, dioxane, tetrahydro-
furan or glycol dimethyl ether, or hydrocarbons, such asbenzene, toluene, xylene, hexane or cyclohexane, or
petroleum fractions, or halogenohydrocarbons, such as
methylene chloride, ch~oroform, carbon tetrachloride or
chlorobenzene, or ethyl acetate, acetone, dimethylform-
amide, hexamethylphosphoric acid triamide or dimethyl-
acetamide. It is also possible to use mixtures of the
solvents mentioned.
The reaction is in general carried out in a tem-
Le A 25 238
- 24 -
- 1~40~2~
perature range from 0~C to +150~C, preferably from +20~C
to +180~C.
The reaction can be carried out under normal,
increased or reduced pressure. The reaction is in
general carried out under normal pressure.
The reaction is in general carried out by a pro-
cedure in which the halogenomethylcephalosporin compound
and the phosphorus compound are mixed in an inert solvent
and the mixture is warmed, if appropriate. The phos-
phorus compound is ;n general employed here in an amountof 1 to S, preferably 1 to 2 mol/per mol of the chloro-
methylcephalosporin compound.
In carrying out the process according to the
invention, it has proved to be particularly advantageous
to use the corresponding iodine compound (X = I) as the
halogenomethylcephalosporin compound, this being obtained
from the corresponding chloromethyl or bromomethyl com-
pound by treatment with sodium iodide in dimethylform-
amide or acetone. It is moreover possible, if the
chloromethyl or bromomethyl compounds are used, to carry
out the conversion into the iodine compound and the
reaction with the phosphorus compound as a one-pot reac-
tion. For this, the corresponding bromomethyl- or
chloromethylcephalosporin compounds are reacted in a
suitable solvent, such as, for example, ethers, acetates,
hydrocarbons or chlorohydrocarbons, but preferably ace-
tone, with sodium iodide and the corresponding phosphorus
compounds.
The halogenomethylcephalosporin compounds of the
general formula (X) used as starting substances are new.
A process has been found for the preparation of
the halogenomethylcephalosporins of the general formula
(X), which is characterized in that carboxylic acids of
the general formula (VI)
Le A 25 238
- 25 -
13~042A
R ~
R2 ~ fH-COOH ~YI)
NHR~
in which
R1 and R2 have the meaning given and
R3 represents an amino-protective group,
after activation of the carboxyl group by conversion into
a mixed anhydride, for example ~ith ethyl chloroformate,
isobutyl chloroformate or methanesulphonyl chloride, or
by conversion into the acid halide, or by conversion into
an activated ester ~ith, for example, N-hydroxybenzotri-
a~ole and dicyclohexylcarbodiimide, are reacted ~ith aB-lactam compound of the general formula (XII)
H2N~ (XII)
CoOR4
in ~hich
R4 and Z have the abovementioned meaning,
if appropriate, protective groups are then split off and
the desired salts are prepared or the free acids are
prepared from the salts.
A large number of methods kno~n from cephalo-
sporin or penicillin chemistry can be used for coupling
carboxylic acids (VI) to the B-lactam compound (XII).
It has proved to be advantageous to activate the carboxy-
lic acids of the general formula (VI) ~ith an amine-
protective group (R3) and then to couple them uith the B-
lactam compounds of the formula (XII), ~hich have been
~ dissolved as salts ~ith an amine.
Activation of carboxylic acids of the general formula (VI)
with (a) sulphonic acid derivatives of the general formula
(VIII) or with (b) chloroformic acid esters, preferably
ethyl chloroformate, to geve anhydrides of the general
Le A 25 238 - 26 -
.. . .
13~042~
, ..
formula (IXa, b), as i11ustrated in the following equation,
is particularly advantageous
R 1 ~,_5
a) 1'-502-R15(VIII) 1~
Rl~ 5 R2 H-C00502R15
N~ NHR3
R2 H-COOH IXa
N~3 Rl S
VI b ) ClCOOC2H5 ~
R2~CH - COOCOOC 2H5
NHR3
IXb
In this equation, in formula (VIII) and tlXa)
T represents the radical R15-So2-o- or halogen
and
R15 represents alkyl ~hich has up to 10 carbon
atoms and is optionally substituted by fluorine,
chlorine, cyano/alkyl, alkoxycarbonyl, alkoxy
or alkyl ~ith in each case up to 4 carbon atoms,
or represents phenyl, ~hich is optionally substi-
tuted by fluorine, chlorine, bromine, cyano,
alkyl, alkoxy, alkylthio or alkoxycarbonyl ~ith
in each case up to 4 carbon atoms, nitro, tri-
fluoromethyl or phenyl.
If R15 is substituted, 1 to 3 substit~ents are
preferably present, and those mentioned above are parti-
cularly preferably present.
R15 especially preferably represents a methyl
or p-tolyl radical.
The mixed anhydrides of the general formula
(IXa,b) are prepared by dissolving the carboxylic acids
Le A 25 238
- 27 -
, ... . . . . . ... ..
13~0~2~
of the general formula (VI) and 1 to 1.4 equivalents of an
amine in a solvent and allouing the solution to react
~ith 1 to 1.2 e~uivalents of a sulphonic acid derivative
of the formula (VIII) or of a chloroformic acid ester.
Suitable solvents are all the solvents ~hich do
not change under the reaction conditions. These include,
preferably, ethers, such as, for example, diethyl ether,
dioxane or tetrahydrofuran, or chlorohydrocarbons, such
as methylene chloride, chloroform or carbon tetrachloride,
or amides, such as dimethylformamide or hexamethylphos-
phoric acid triamide, or acetonitrile or acetone. It is
also possible to use mixtures of the solvents mentioned.
Suitable amines are tertiary amines, such as, for
example, triethylamine, ethyl-diisopropylamine or tri-
tS butylamine, but also sterically hindered secondaryamines, such as, for example, diisopropylamine. Mixtures
of the amines mentioned can also be used.
The reactions can be carried out at temperatures
betveen -80~C and room temperature. The activation is
advantageously carried out ~ith methanesulphonyl chloride
in dimethylformamide at -40~C to -60~C in the course of
0.2 to 24 hours, preferably O.S to 5 hours.
The solvents mentioned in the preparation of the
compounds of the formula (IX) or ~ater can be used to
dissolve the B-lactams compound of the formula
(xII) for the coupling with the formula (IXa) or (IXb) to
obtain the formula (X), and the amines mentioned there can be used as the base.
Activation of the carboxylic acids of the general
formula (Vl) by conversion into an activated ester ~ith,
for example, dicyclohexylcarbodiimide, if appropriate in
the presence of N-hydroxysuccinimide or t-hydroxybenzo-
triazole, is also particularly advantageous.
Suitable solvents here are all the solvents which
are also suitable for the preparation of anhydrides of
the general formula (lXa,b) and have already been mentioned
there.
Le A 25 238
- 28 -
134042~
The reactions can be carried out at temperatures
between -30~C and ~100~C. Activation is advantageously
carried out with 1-hydroxybenzotriazole and dicyclohexyl-
carbodiimide in dimethylformamide at room temperature for
2 to 6 hours, and the dicyclohexylurea which has precipi-
tated out is then filtered off with suction and reacted
with the B-lactam compounds of the formula (XII) in
the form of a solution of their amine salt in the course
of 2 to 24 hours. The solvents mentioned for the pre-
paration of the compounds of the formula (IX) can be usedto dissolve the B-lactam compounds of the formula (XII),
and the amines mentioned there can be used as the base.
The carboxylic acids of the general formula (VI)
used as starting substances are known or can be prepared
by known methods tDE-OS (German Published Specification)
3,508,258].
The B-lactam compounds of the general formula
(XII) used as starting substances are known or can be
prepared by known methods ~U.S. Patent Specification
4,639,448 and DE-OS (German Published Specification)
3,402,642].
The amino-B-lactams of the general formula (VII)
used as starting substances are known or can be prepared
by known methods ~DE-OS (German Published Specification)
3,402,642; U.S. Patent Specification 4,639,448].
The compounds of the general formula I have a
broad antibacterial spectrum against Gram-positive and
Gram-negative germs and anaerobic bacteria, coupled with
a low toxicity. These properties enable them to be used
as chemotherapeutic active compounds in human and
veterinary medicine.
The compounds according to the invention are
active against a very broad spectrum of microorganisms.
Gram-negative and Gram-positive bacteria and bacteria-
like microorganisms and the diseases caused by thesepathogens can be prevented, alleviated andtor cured with
Le A 25 238
- 29 -
1~042~
the aid of these compounds.
The compounds according to the invention are
particularly active against bacteria and bacteria-like
microorganisms. They are therefore particularly suitable
S in human and veterinary medicine for the prophylaxis and
chemotherapy of local and systemic infections caused by
these pathogens.
For example, local and/or systemic diseases which
are caused by the following pathogens or by mixtures of
the following pathogens can be treated and/or prevented:
Gram-positive cocci, for example Staphylococci tStaph.
aureus and Staph. epidermidis) and Streptococci (Strept.
agalactiae, Strept. faecalis, Strept. pneumoniae and
Strept. pyogenes); Gram-negative cocci (Heisseria gonor-
rhoeae) and Gram-negative rod-shaped bacillae, such as
Enterobacteriaceae, for example Escherichia coli, Haemo-
philus influenzae, Citrobacter (Citrob. freundii, Citrob.
divernis), Salmonella and Shigella; and furthermore
Klebsiella (Klebs. pneumoniae and Klebs. oxytoca),
Enterobacter (Ent. aerogenes and Ent. agglomerans),
Hafnia, Serratia (Serr. marcescens), Proteus (Pr. mira-
bilis, Pr. rettgeri and Pr. vulgaris), Providencia,
Yersinia and the genus Acinetobacter. The antibacterial
spectrum moreover includes the genus Pseudomonas (Ps.
aeruginosa and Ps. maltophilia) and strictly anaerobic
bacteria, such as, for example, Bacteroides fragilis,
representatives of the genus Peptococcus, Peptostrepto-
coccus and the genus Clostridium; and furthermore Myco-
plasma (M. pneumoniae, M. hominis and M. urealyticum) and
mycobacteria, for example Mycobacterium tuberculosis.
The substances according to the invention have an action
in particular against Staphylococci, Streptococci,
Enterococci and Haemophilus influenzae. On parenteral
or, in particular, oral adm;nistration, the new compounds
have a very good action against microorganisms such as
Staphylococci, Streptococci, Enterobacteriaceae, Escheri-
Le A 25 238
- 30 -
1~40424
chia coli, Klebsiella, Salmonella, Shigella and Proteus.
The above list of pathogens is given merely by
way of example and is in no way to be interpreted as
limiting. Examples which may be mentioned of diseases
which can be caused by the pathogens or mixed infections
mentioned and can be prevented, alleviated or cured by
the compounds according to the invention are: infection
diseases in humans, such as, for example, otitis, pharyn-
gitis, pneumonia, peritonitis, pyelonephritis, cystitis,
endocarditis, systemic infections, bronchitis (acute and
chronic), septic infections, diseases of the upper res-
piratory tract, diffuse panbronchiolitis, pulmonary
emphysema, dysentery, enteritis, liver abscesses, ure-
thritis, prostatitis, epididymitis, gastrointestinal
infections, bone and joint infections, cystic fibrosis,
skin infections, postoperative wound infections, absces-
ses, phlegmons, wound infections, infected burns, burn
wounds, infections in the oral region, infections follow-
ing dental operations, osteomyelitis, septic arthrit;s,
cholecystitis, peritonitis with appendicitis, cholangitis,
intraabdominal abscesses, pancreatitis, sinusitis, mas-
toiditis, mastitis, tonsillitis, typhus, meningitis and
infections of the nervous system, salpingitis, endome-
tritis, genital infections, pelveoperitonitis and eye
infections.
As well as in humans, bacterial infections can
also be treated in other species. Examples which may be
mentioned are: pigs: coli-diarrhoea, enterotoxaemia,
sepsis, dysentery, salmonellosis, metritis-mastitis-
agalactiae syndrome and mastitis; ruminants (cattle,sheep, goats): diarrhoea, sepsis, bronchopneumonia,
salmonellosis, pasteurellosis, mycoplasmosis and genital
infections; horses: bronchopneumonia, joint ill, puer-
peral and postpuerperal infections and salmonellosis;
dogs and cats: bronchopneumonia, diarrhoea, dermatitis,
otitis, urinary tract infections and prostatitis;
Le A 25 238
- 31 -
.. , . . , . . ~ . ~,
. 1340A24
poultry (chickens, turkeys, quails, pigeons, ornamental
birds and others): mycoplasmosis, E. coli infections,
chronic respiratory tract infections, salmonellosis,
pasteurellosis and psittacosis.
~acterial diseases in the breeding and rearing
of stock and ornamental fish can also be treated, the
antibacterial spectrum being extended beyond the above-
mentioned pathogens to further pathogens, such as, for
example, Pasteurella, ~rucella, Campylobacter, Listeria,
Erysipelothrix, Corynebacteria, E~orellia, Treponema,
Nocardia, Rickettsia and Yersinia.
The minimum inhibitory concentrations (MIC values,
B ~g/ml) for Example 6 in comparison with Cefaclor ~
tM. Gorman et al., Cefaclor, Chronicles of Drug Discovery,
Volume 2, 49, J. Wiley ~ Sons (1983)] are given in the
following tables. The MIC values are determined by the
agar dilution test with the aid of a multipoint inocula-
tor, the reading being taken after incubation at 37~C
for 18 to 24 hours. Isosensitest agar is used as the
growth medium.
Ha~mophi lu~ inf lu-nzao Example 6 Cofaclor
No .
2 0,5 2
4 1 8
0,5 4
6 0,5 4
7 0~5 2
8 0,5
9 0,5
0,5 2
Le A 25 238
- 32 -
~r~e f~k
...... . . . .. ..
13~0~2~
ICB-No. Ex~ple 6 Cefaclor
S~aph. aureu~ 25 412 32 256
25 413 0,5 4
25 414 0,5 32
25 417 < 0,5 4
25 418 ~ 0,5 4
25 473 ~ 0,5 4
25 559 ~ 0,5 8
25 560 0,5 8
25 565 ~ 0,5 4
25 568 ~ 0,5 4
25 569 ~ 0,5 4
25 470 0,5 8
25 508 1 8
25 527 2 64
25 397 < 0,5 8
25 523 < 0,5 8
25 524 < 0,5 16
25 525 < 0,5 4
25 583 4 16
ICB-No. Ex~ple 6 C-faclor
S~rep. fsocalic 27 261 8 . > 256
27 249 16 ~ 256
27 250 16 > 256
27 251 32 ) 256
27 252 16 > 256
27 253 16 ~ 256
27 254 16 ) 256
27 255 16 > 256
27 256 4 ) 256
27 257 4 ) 256
Le A 25 238
- 33 -
. .
.. . . . . .
1~40 124
ICB- No. Example 6 Cefaclor
s
Klebs pneumoniae 6310 < 0,5
6318 4 2
6360 64 8
6362 < 0,5
6379 < 0,5
6380 < 0,5
ICB-No. Example 6 Cefaclor
E Coli 4895 2
4322 2
4800 2
4815 0,5 2
~naorobic bacterie Example 6Cefsclor
germ~
1 Bacteroid-c fragilir 1 256
4 Bac~-roid-s fragilis 0,5 128
6 Bacteroid-~ fragili~ 0,5 128
10 Bac~eroid-~ fr-gilir 0,5 128
13 Bacteroides ~hetaiotaomicron 4 ) 256
15 Bact-roide- di~aroni~ < 0,5 2
16 Bacteroid-~ o~atu~ 8 ) 256
17 Clo~tridium perfringen~ ~ 0,5 2
18 ~act-roid-~ ~ulga~u~ ~ 0,5 4
19 Clos~ridium p-rfring-n~~ 0,5 < 0,5
Le A 25 238
- 34 -
1340424
The present invention includes pharmaceutical
formulations which, in addition to non-toxic, inert
pharmaceutically suitable excipients, contain one or
more compounds according to the invention or consist of
one or more active compounds according to the invention,
and relates to processes for the preparation of these
formulations.
The present invention also includes pharmaceuti-
cal formulations in dosage units. This means that the
formulation is in the form of individual parts, for
example tablets, dragees, capsules, pills, suppositories
and ampoules, the active compound content of which
corresponds to a fraction or a multiple of an individual
dose. The dosage units can contain, for example, 1, 2,
3 or 4 individual doses or 1/2, 1/3 or 1/4 of an indivi-
dual dose. An individual dose preferably contains the
amount of active compound which is given in one adminis-
tration and which usually corresponds to a whole, one
half, one third or one quarter of a daily dose.
2û ~y non-toxic/inert pharmaceutically suitable
excipients there are to be understood solid, semi-solid
or liquid diluents, fillers and formulation auxiliaries
of all kinds.
Tablets, dragees, capsules, pills, granules,
suppositories, solutions, suspensions and emulsions,
pastes, ointments, gels, creams, lotions, dusting po~ders
and sprays may be mentioned as preferred pharmaceutical
formulations.
Tablets, dragees, capsules, pills and granules
can contain the active compound or compounds, in addition
to the customary excipients, such as (a) fillers and
extenders, for example starches, ~actose, sucrose, glu-
cose, mannitol and silicic acid, (b) binders, for example
carboxymethylcellulose, alginates, gelatine or polyvinyl-
pyrrolidone, (c) humectants, for example glycerol (d)disintegrating agents, for example agar-agar, calcium
Le A 25 238
- 35 -
1~0~2 1
carbonate and sodium carbonate, (e) solution retarders,
for example paraffin and (f) absorption accelerators,
for example quaternary ammonium compounds, (g) wetting
agents, for example cetyl alcohol and glycerol mono-
stearate, (h) adsorbents, for example kaolin and benton-
ite and (i) lubricants, for example talc, calcium stear-
ate and magnesium stearate and solid polyethylene glycols,
or mixtures of the substances mentioned under (a) to (i).
The tablets, dragees, capsules, pills and gran-
ules can be provided with the customary coatings andshells, optionally containing opacifying agents, and can
also be of such composition that they release the active
compound or compounds only or preferentially in a certain
part of the intestinal tract, if appropriate in a delayed
manner, examples of embedding compositions which can be
used being polymeric substances and waxes.
The active compound or compounds can also be in
microencapsulated form, if appropriate with one or more
of the abovementioned excipients.
Suppositories can contain, in addition to the
active compound or compounds, the customary water-soluble
or water-insoluble excipients, for example polyethylene
glycols, fats, for example cocoa fat, and higher esters
(for example C14-alcohol with C16-fatty acid), or
mixtures of these substances.
Ointments, pastes, creams and gels can contain,
in addition to the active compound or compounds, the
customary excipients, for example animal and vegetable
fats, waxes, paraffins, starch, tragacanth, cellulose
derivatives, polyethylene glycols, silicones, bentonites,
silicic acid, talc and zinc oxide, or mixtures of these
substances.
Dusting powders and sprays can contain, in addi-
tion to the active compound or compounds, the customary
excipients, for example lactose, talc, silicic acid,
aluminium hydroxide, calcium silicate and polyamide pow-
Le A 25 238
- 36 -
.
l~AOA2~
.
der, or mixtures of these substances. Sprays can addi-
tionally contain the customary propellants, for example
chlorofluorohydrocarbons.
Solutions and emulsions can contain, in addition
to the active compound or compounds, the customary
excipients, such as solvents, solubilizing agents and
emulsifiers, for example water, ethyl alcohol, isopropyl
alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol,
benzyl benzoate, propylene glycol, 1,3-butylene glycol,
dimethylformamide, oils, in particular cottonseed oil,
groundnut oil, maize germ oil, olive oil, castor oil and
sesame oil, glycerol, glycerolformal, tetrahydrofurfuryl
alcohol, polyethylene glycols and fatty acid esters of
sorbitan, or mixtures of these substances.
For parenteral administration, the solutions and
emulsions can also be in a sterile form which is isotonic
with blood.
Suspensions can contain, in addition to the
active compound or compounds, the customary excipients,
such as liquid diluents, for example water, ethyl alcohol
or propylene glycol, suspending agents, for example
ethoxylated isostearyl alcohols, polyoxyethylene sorbitol
and sorbitan esters, microcrystalline cellulose, alumin-
ium metahydroxide, bentonite, agar-agar and tragacanth,
or mixtures of these substances.
The formulation forms mentioned can also contain
colouring agents, preservatives and additives which
improve the smell and taste, for example peppermint oil
and eucalyPtus oil, and sweeteners, for example saccharin.
The therapeutically active compounds should
preferably be present in the abovementioned pharmaceuti-
cal formulations in a concentration of about 0.1 to 99.5,
preferably about 0.5 to 95% by weight of the total mix-
ture.
The abovementioned pharmaceutical formulations
can also contain other pharmaceutical active compounds
Le A 25 238
- 37 -
-" ' 13~0~2 1
in addition to the compounds according to the invention.
The abovementioned pharmaceutical formulations
are prepared in the customary manner by known methods,
for example by mixing the active compound or compounds
S with the excipient or excipients.
The formulations mentioned can be used on humans
and animals either orally, rectally, parenterally (intra-
venousLy, intramuscularly or subcutaneously), intracis-
ternally, intravaginally, intraperitoneally or locally
tdusting powders, ointments, drops) and for the therapy
of infections in hollow spaces and body cavities. Suit-
able formulations are injection solutions, solutions and
suspensions for oral therapy, gels, infusion formulations,
emulsions, ointments or drops. Ophthalmological and
dermatological formulations, silver salts and other
salts, eardrops, eye ointments, dusting powders or solu-
tions can be used for local therapy. In the case of
animals, intake can also be via the feed or drinking
water, in suitable formulations.
Gels, powders, dusting powders, tablets, sus-
tained release tablets, premixes, concentrates, granules,
pellets, boli, capsules, aerosols, sprays and inhalates
can furthermore be used on humans and animals. The com-
pounds according to the invention can furthermore be
incorporated into other carrier materials, such as, for
example, plastics (chains of plastic for local therapy),
collagen or bone cement.
In general, it has proved advantageous both in
human and in veterinary medicine to administer the active
compound or compounds according to the invention in total
amounts of about 0.5 to about 500, preferably 5 to 100
mg/kg of body weight every 24 hours, if appropriate in
the form of several individual doses, in order to achieve
the desired results. An individual dose preferably con-
tains the active compound or compounds according to theinvention in amounts of about 1 to about 80, in particu-
Le A 25 238
- 38 -
~ ... . . ., .. ~
1340'~24
lar 3 to 30 mg/kg of body weight. However, it may be
necessary to deviate from the dosages mentioned, and in
particular to do so as a function of the nature and body
weight of the subject to be treated, the nature and
severity of the disease, the nature of the formulation
and of the administration of the medicament and the
period or interval within which adm;nistration takes
place.
Thus, in some cases it may suffice to manage with
less than the abovementioned amount of active compound,
whilst in other cases the abovementioned amount of active
compound must be exceeded. The particular optimum dosage
required and mode of administration of the active com-
pounds can easily be specified by any expert on the basis
of his expert knowledge.
The new compounds can be administered in the
customary concentrations and formulations together with
the feed or with feed formulations or with the drinking
water. Infection by Gram-negative or Gram-positive bac-
teria can thereby be prevented, alleviated and/or cured,and a promotion in growth and an improvement in feed
conversion can thereby be achieved.
The compounds according to the invention can be
combined with other antimicrobial active compounds and
lactamase inhibitors, for example with penicillins which
are particularly penicillinase-resistant and clavulanic
acid, for the purpose of increasing the action spectrum
and in order to achieve an increase in action, especially
against ~-lactamase-forming bacteria. Such a combination
would be, for example, that with oxacillin or dicloxa-
cillin.
The compounds according to the invention can also
be combined with aminoglycoside antibiotics, such as, for
example, gentamicin, sisomicin, canamicin, amicacin or
tobramycin, for the purpose of broadening the action
spectrum and achieving an increase in action.
Le A 25 238
- 39 -
13404~
The present invention also includes a commercial package
containing as active ingredient a compound of the invention
together with instructions for the use thereof as an antibacterlal
agent.
39a
B~
134042 1
Preparation Examples
ExampLe 1
3enzhydryl 7-amino-3-chloromethyl-3-cephem-4-carboxylate
H2S~
Cl
COOcH(c6Hs)2
S 19.64 ml (0.242 mol) of pyridine are added to a
suspension of 50 9 (0.0972 mol) of benzhydryl 7-phenyl-
acetamido-3-hydroxymethyl-3-cephem-4-carboxylate in
S00 ml of methylene chloride at room temperature. After
cooling to -20~C, 40.48 9 (0.0972 mol) of phosphorus
pentachloride are added and the mixture is stirred at
-20~C for 5 minutes. The mixture is warmed to 0~C with
an icebath and stirred for 10 minutes, and is then warmed
to 15~C with a waterbath and stirred for 1 hour.
Thereafter, the mixture is cooled to -70~C and 720 ml
of cold methanol are quickly added. The mixture is then
stirred at -70~C for 5 minutes, at 0~C for 10 minutes
and at +15~C for 25 minutes. The solution is subse-
quently concentrated to a high degree in vacuo and
1,400 ml of saturated sodium bicarbonate solution are
added. The solution is extracted three times with
methylene chloride and the organic phase is dried with
sodium sulphate and concentrated in vacuo. The crude
product is chromatographed on 500 9 of silica gel 60
(0.04 - 0.063 mm) with methylene chloride.
Yield: 29.0 9 (72% of theory)
C21H~gClN203S ~414.9)
NMR (CDCl3): ~ = 2.06 (s, 2H); 3.45 (d, 1H); 3.62 (d,
1H); 4.25 - 4.41 (q, 2H); 4.75 (d, 1H); 4.93 (d, 1H);
6.97 (s, 1H); 7.25 - 7.46 (m, 10H) ppm.
Le A 25 238
- 40 -
.... . , . , .. . ., . ~ . , .
. -
13 1~2i.!
Example 2
~enzhydryl D-7-t2-(t-butoxycarbonylamino)-2-(Z-amino-
benzothiazol-6-yl)-glycylamido]-3-chloromethyl-3-cephem-
4-carboxylate
H2N~;
Il 1
~ H-CO- ~ ~
IH ~ H2Cl
COOC t CH3 ~ 3 coocH ( C6H5 ) 2
18.07 9 (0.0875 mol) of N,N'-dicyclohexylcarbo-
diimide (DCC), dissolved in 150 ml of tetrahydrofuran,
are added to a mixture of 28.3 9 (0.0875 mol) of D-~-t-
butoxycarbonylamino-~-(2-aminobenzothiazol-6-yl)acetic
acid and 24.1 9 (0.058 mol) of benzhydryl 7-amino-3-
chloromethyl-3-cephem-4-carboxylate (Example 1) in 136 ml
of tetrahydrofuran and 77 ml of dimethylformamide at
0~C and the mixture is then stirred at room temperature
for 2 hours and concentrated to dryness. The residue is
suspended in 1,200 ml of ethyl acetate, the suspension
is stirred for 10 minutes and undissolved constituents
are then removed by filtration with suction. After dis-
tilling off the ethyl acetate, the residue is chromato-
graphed on silica gel 60 (0.04 - 0.063 mm) with toluene/
ethyl acetate (1 : 1).
Yield: 17.6 9 (42% of theory)
C35H34ClN5S206 (720.3)
NMR (DMS0): ~ = 1.37 (s, 9H); 3.46 (d, 1H); 3.64 (d, 1H);
4.32 - 4.43 (q, 2H); 5.11 (d, 5 Hz, 1H); 5.31 (d, 1H);
5.8 - 5.86 (q, 1H); 6.98 ts, 1H); 7.15 - 7.5 (mm, 14H);
7.67 (s, 1H) ppm
Le A 25 238
- 41 -
, . . . . . . . .. .
Example 3 1 340 i24
Benzhydryl D-7-~2-(t-butoxycarbonylamino)-2-(2-amino-
benzothiazol-6-yl)gLycylamido~-3-iodomethyl-3-cephem-4-
carboxylate
H2
~CH - CO -Nl~
rH ~ H2I
COOC ( CH3 ) 3 COOCH ( C6H5 ) 2
A mixture of 20.3 9 (0.0282 mol) of benzhydryl
D-7-~2-(t-butoxycarbonylamino)-2-(2-aminobenzothiazol-6-
yl)-glycylamido]-3-chloromethyl-3-cephem-4-carboxylate
(Example 2) and 12.68 9 (0.0846 mol) of sodium iodide in
300 ml of acetone is stirred at room temperature for 2
hours and evaporated to dryness. The residue is taken
up in 500 ml of ethyl acetate and the mixtre is ~ashed
with aqueous sodium thiosulphate solution, ~ater and
sodium chloride solution. After drying over sodium
sulphate, the solvent is distilled off and the residue
is digested in ether.
Yield: 22 9
The compound is used directly in the next stage.
Example 4
Benzhydryl D-7-~2-(t-butoxycarbonylamino)-2-(2-amino-
benzothiazol-6-yl)glycylamido]-3-(triphenylphosphonio)-
methyl-3-cephem-4-carboxylate iodide
H2~S
~H-CO-Nl~s 13 e
I H ~H2P ( C6H5 ) 3I
COOC ~ CH3 ) 3 COOCH ( C6H5 ) 2
Le A 25 238
- 42 -
, . . . .. . .... . . . . . ....
~34~424
A mixture of 22 9 (O.OZ71 mol) of benzhydryl D-
7-t2-(t-butoxycarbonylamino)-2-t2-aminobenzothiazol-6-yl)-
glycylamido]-3-iodomethyl-3-cephem-4-carboxylate (Example
3) and 21.32 9 (0.0813 mol) of triphenylphosphine in
500 ml of ethyl acetate is stirred at room temperature
for 1 hour. After 30 minutes, the product precipitates
out. The mixture is concentrated to about 150 ml under
reduced pressure and 500 ml of ether are added to the
concentrate. rhe resulting precipitate is filtered off
with suction and rinsed with ether.
Yield: 19.6 9 t67~ of theory)
C53H49lN5~6PS2 (1074 1)
NMR (DMS0): ~ = 1.35 (S, 9H); 3.32 - 3.42 (dd, 2H); 4.81
- 4.93 (t, 2H); 5.2 (d, 1H); 5.33 (d, 1H); 5.72 - 5.79
(q, 1H); 6.24 (s, 1H); 7.Z - 7.49 (mm, 15H); 7.6 - 7.79
lm, 15H) ppm.
Example 5
Benzhydryl D-7-C2-(t-butoxycarbonylamino)-2-(2-amino-
benzothiazol-6-yl)glycylamido~-3-t(Z)-1-propen-1-yl~-3-
cephem-4-carboxylate
H2~
H-CO- ~ CH3
COOCtCH3)3 COOCH(C6H5)2
7.04 ml (0.126 mol) of acetaldehyde and 7.6 9
(0.007 mol) of benzhydryl D-7-t2-(t-butoxycarbonylamino)-
2-(2-aminobenzothiazol-6-yl)glycylamido~-3-(trtphenyl-
phosphonio)methyl-3-cephem-4-carboxylate iodide ~Example
4) are added to a cold solution of 6.08 9 (0.07 mol) of
lithium bromide in 50 ml of dimethylformamide and 150 ml
of methylene chloride at -5~C rhe mixture is stirred
at -5~C for 20 hours and then at room temperature for
Le A 25 Z38
- 43 -
. . .
13~04:~
5 hours. The solution is concentrated to about 50 ml in
vacuo and the concentrate is partitioned in a solvent
mixture of Z00 ml of ethyl acetate and 200 ml of water.
The upper layer is separated off and washed once with
S aqueous sodium chloride solution. After drying over
sodium sulphate and distilling off the solvent, the resi-
due is taken up in toluene and the mixture is introduced
onto a column packed with silica gel (0.04 - 0.063 mm).
The column is eluted first with toluene and then with the
solvent mixture toluene/ethyl acetate (5:1) and toluene/
ethyl acetate (1:1).
Yield: 2.9 9 (58~ of theory)
C37H37N506S2 (711-9~
NMR (CDCl3): ~ = 1.35 (dd, 3H); 1.43 (s, 9H); 3.15 (d,
1H); 3.31 (d, 1H); 4.97 (d, 1H); 5.3 (s, 1H); 5.46 - 5.55
(m, 1H); 5.71 (broad s, 2H); 5.78 - 5.85 (q, 1H); 6.03
(d, J = 11 Hz, 1H); 6.87 (s, 1H); 7.2 - 7.4 (mm, 12H);
7.5 (s, 1H) ppm.
Example 6
D-7-t(2-Aminobenzothiazol-6-yl)glycylamido]-3-t(Z)-1-
propen-1-yl]-3-cephem-4-carboxylic acid, cis-isomer
H2~
NH~ ~ CH3
COOH
2.9 9 (4.1 mmol) of benzhydryl D-7-t2-(t-butoxy-
carbonylamino)-2-(2-aminobenzothiazol-6-yl)glycylamido]-
3-t(Z)-1-propen-1-yl]-3-cephem-4-carboxylate (Example 5)
are dissolved in 20 ml of methylene chloride, 40 ml of
trifluoroacetic acid (TFA) are added and the mixture is
stirred with a magnetic stirrer at room temperature for
60 minutes. The methylene chloride and trifluoroacetic
Le A 25 238
- 44 -
. ~ . .. . . ..
-
1340424
acid are removed in vacuo, the semi-solid red oil which
remains is triturated in ether and the product is fil-
tered off with suction and washed with ether. The pale
yellow trifluoroacetate is dried in vacuo and then sus-
pended in 100 ml of water and the insoluble yellow flocksare filtered off with suction over kieselguhr and rinsed
with 30 ml of water. The still slightly cloudy solution
B is filtered again over a membrane filter (Millipore~
0.45 ~m). The filtrate is pumped onto an RP 18 column
(Hibar~250-25, Merck). The column is eluted first with
200 ml of water (fraction 1), then with 400 ml of 5%
strength methanol (fraction 2) and finally with 10%
strength methanol, in each case 300 ml fractions being
collected (fraction 3 to 12). The fractions are investi-
gated by means of analytical HPLC and fractions 6 to 10,which contain the desired peak, are combined, the methanol
is distilled off in vacuo and the residue is lyophilized.
Yield: 400 mg
ClgH19N504S2 (445 5)
NMR (DCOOD): ~ = 1.67 (dd, 3H); 3.41 (d, 1H); 3.55 (d,
1H); 5.3 (d, 1H); 5.78 (s, 1H); 5.81-5.91 (q and m, 2H);
6.25 (d, J = 11.6 Hz, 1H); 7~81 - 7.9 (q, 2H); 8.18 (s,
1H) ppm.
Analytical HPLC: Hibar 250-4, RP-8, 10 ~m, 254 nm
Mobile phase: 1,000 ml of CH3CN - 30 ml of acetic acid -
870 ml of water
Flow rate: 4 ml/minute, concentration: 1 mg/ml
Retention: 3.22 (content: 97.3%)
Le A 25 238
- 45 -
I rade-(nqrk
134042~
Example 7
D-7-[(2-Aminobenzothiazol-6-yl)glycylamido]-3-t(E)-1-
propen-1-yl]-3-cephem-4-carboxylic acid, trans-isomer
H2
~CH-CO-N~
NH2 ~H3
COOH
The E-isomer compound is obtained from the pre-
parative high pressure liquid chromatography of D-7-~(2-
aminobenzothiazol-6-yl)glycylamido]-3-~(Z)-1-propen-1-yl]-
3-cephem-4-carboxylic acid (cis-isomer; Example 6) by
further elution with 30X strength methanol.
Yield: 137 mg
C19H19NS04S2 (445 5)
NMR (DCOOD): ~ = 1.9 (d, 3H); 3.61 (s, 2H); 5.25 (d, 1H);
5.72 (s, 1H); 5.87 (d, 1H); 6.23-6.46 (m, 1H); 7.04 (d,
J = 15.8 Hz, 1H); 7.8 - 7.88 (q, 2H); 8.18 (s, 1H) ppm.
Analytical HPLC: Hibar 250-4, RP-8 10 ~m, 254 nm
Mobile phase: 100 ml of CH3CN - 30 ml of acetic acid -
870 ml of water
Flow rate: 4 ml/minute, concentration: 1 mg/ml
Retention: 5.20 (content: 67.2%)
Example 8
9enzhydryl 7-phenylacetamido-3-(triphenylphosphonio)-
methyl-3-cephem-4-carboxylate iodide
O~ H2-CO_NH~S~ e
o~CH2P ( C 6Hs ) 3 I
COOCH(C6H5)2
32.5 9 (0.0609 mol) of benzhydryl 7-phenylacet-
Le A 25 238
- 46 -
... .. .-- _ . . . .
134042~
amido-3-chloromethyl-3-cephem-4-carboxylate are dissolved
in 330 ml of acetone, and 10.1 9 (0.0674 mol) of NaI and
17.6 9 tO.0671 mol) of triphenylphosphine are added in
succession, with stirring. After the mixture has been
stirred at room temperature for 1.5 hours, the insoluble
material is removed by filtration with suction and the
clear mother liquor is stirred into 1,000 ml of ether.
The white flocculent material which precipitates out is
filtered off with suction, washed with 300 ml of ether
and dried in vacuo.
Yield: 51 9 (94~ of theory)
C47H40IN204PS (886.8)
NMR (DMSO): ~ = 3.51 - 3.61 (q, 4H); 4.93 - 5.05 (t, 1H);
5.Z2 - 5.33 (d and t, 2H); 5.7 - 5.76 (q, 1H); 6.26 (s,
1H); 7.21 - 7.46 (mm, 15H); 7.68 - 7.79 (m, 15H); 9.14
(d, 1H) ppm.
Example 9
I. Benzhydryl 7-phenylacetamido-3-~(Z)-propen-1-yl~-3-
cephem-4-carboxylate
H2-CO- ~ CH3
COOCH(C6H5)2
15.9 9 (17.9 mmol) of benzhydryl 7-phenylacet-
amido-3-(triphenylphosphonio)methyl-3-cephem-4-carboxyl-
ate iodide (Example 8) are taken in 100 ml of methylene
chloride and 12.8 ml (229.6 mmol) of acetaldehyde in a
250 ml three-necked flask. The mixture is cooled to~
0~C and 100 ml of water are added. 16.3 ml of 1N NaOH
are then added dropwise in the course of 4 hours, while
keeping the pH constant at 8.6. The reaction solution is
diluted with methylene chloride and the organic phase is
separated off, washed once with water and then dried over
sodium sulphate. After the drying agent has been
Le A 25 238
- 47 -
134042~
removed, a further 13 ml (233 mmol) of acetaldehyde are
added to the methylene chloride solution and the mixture
is stirred overnight. The reaction solution is then con-
centrated to dryness, the residue is dissolved again in
a little methylene chloride and the solution is intro-
duced onto a column filled with 500 ml of silica gel
(0.04 - 0.063 mm). 400 ml fractions are collected and
all the fractions are investigated for the cis-isomer
compound by means of analytical HPLC.
Analytical HPLC: Hibar 250-4, Lichrosorb Si 60, 5 ~m,
254 nm
Mobile phase: 100 ml of methylene chloride - 3 ml of
methanol
Flow rate: 2 ml/minute, concentration: 1 mg/ml
Retention: 5.80 (content 73.1%)
Yield: 4.75 9 (51% of theory)
C31H2gN2045 (524.6)
NMR (CDCl3): ~ = 1.4 (dd, 3H); 3.22 (d, 1H); 3.41 (d,
1H); 3.65 (q, 2H); 5.0 (d, 1H); 5.48 - 5.6 (m, 1H); 6.07
(d, 1H); 6.92 (s, 1H); 7.21 - 7.4 (m, 15H) ppm.
II. aenzhydryl 7-phenylacetamido-3-[(Z)-propen-1-yl]-3-
cephem-4-carboxylate (different process)
1?7.2 9 (0.2 mol) of benzhydryl 7-phenylacet-
amido-3-(triphenylphosphonio)methyl-3-cephem-4-carboxyl-
ate iodide are dissolved almost completely in 800 ml of
CH2Cl2 and 80 ml of CH30H, the solution is cooled
to ~5~C and 167.8 ml (3.0 mol) of acetaldehyde are
added; during this operation, the temperature should not
rise above 20~C.
26.4 9 (0.25 mol) of sodium carbonate, dissolved
in 200 ml of water, are then slowly added at 14~C in the
course of 15 minutes. The icebath is subsequently
removed and the solution is stirred at room temperature
for 2 1/2 hours.
The course of the reaction is checked by thin
layer chromatography in acetonitrile : water = 9:1 and
Le A 25 238
- 48 -
134042il
toluene : ethyl acetate = 8:2. When the ~ittig reaction
has ended, the organic phase is separated off and the
aqueous phase is washed again with CH2Cl2. The combined
CH2Clz phases are filtered over 150 9 of silica gel
(Merck, 0.04-0.063 mm) and the residue is rinsed with
CH2Cl2 (about 1,000 ml) until the filtrate is colour-
less.
The CH2Cl2 filtrate is dried over Na2S04 and
then concentrated to an oily residue and the residue is
stirred with 800 ml of ethanol. The ethanolic solution
is stirred on a rotary evaporator for 15 minutes, the
product gradually crystallizing out. Removal of ethanol
by distillation is continued and the crystal sludge
formed is stirred with about 90 ml of ether/100 ml of n-
pentane, filtered off with suction and rinsed with 60 mlof n-pentane. The product is dried over P4010 in vacuo
overnight.
Yield: 41.2 9 (39.2% of theory), analytical HPLC: Hibar
250-4; Lichrosorb Si 60, 5 ~m, 254 nm.
Mobile phase: 850 ml of toluene - 150 ml of ethyl acetate
Flow rate: 2 ml min 1
Retention: 4.73 (80.2%; Z-isomer)
4.00 (17.4%; E-isomer)
Example 10
I. Henzhydryl 7-amino-3-C(Z)-1-propen-1-yl]-3-cephem-4-
carboxylate
H2~CH3
COOCHtC6H5)2
6.4 (12.2 mmol) of benzhydryl 7-phenylacetamido-
3-C(Z)-propen-1-yl]-3-cephem-4-carboxylate (Example 9)
are dissolved in 64 ml of methylene chloride, the solu-
tion is cooled to -40~C with a dry ice bath and 2.47 ml
Le A 25 238
- 49 -
1~40424
(30.5 mmol) of pyridine and 2.54 9 (12.2 mmol) of phos-
phorus pentachloride are added in succession. After 5
minutes, the mixture is allowed to warm to -20~C, after
~hich the temperature should rise to -10~C in the course
of 20 minutes and then rise to +10~C. The solution is
now stirred at +10~C to +15~C for 1 hour. The mixture
is subsequently cooled to -40~C, 100 ml of methanol
(-30~C) are added and the mixture is stirred at +10~C
for a further 30 minutes. The reaction solution is con-
centrated gently, the oil obtained is dissolved in 600 mlof methylene chloride, the solution is stirred into
800 ml of sodium bicarbonate solution and the mixture is
stirred for 10 minutes. The methylene chloride phase is
separated off, ~ashed once with water and dried over
sodium sulphate. The methylene chloride filtrate is
chromatographed on 400 ml of silica gel (0.04 - 0.063 mm),
elution being carried out first with methylene chloride
and then with methylene chloride with the addition of
methanol (gradient up to 10%). The eluate is investiga-
ted by means of analytical HPLC and TLC (thin layerchromatography) (methylene chloride/methanol = 100:1).
rield: 4.2 9
C23H22N2~3S (406-5)
NMR (CDCl3): ~ = 1.4 (dd, J = 2 Hz and 7 Hz, 3H); 3.3
(d, J = 17 Hz, 1H); 3.48 (d, J = 17 Hz, 1H); 4.75 (d,
J = 4.5 Hz, 1H); 4.98 (d, J = 4.5 Hz, 1H); 5.45 - 5.55
(d and q, J = 10 Hz and 7 Hz, 1H); 6.07 (d, J = 11 Hz,
1H); 6.96 (s, 1H); 7.23 - 7.42 (m, 10H); 8.6 (d, 2H) ppm.
Analytical HPLC: Hibar 250-4, Merck, Lichrosorb Si 60,
5 ~m, 254 nm
Mobile phase: 1,000 ml of methylene chloride - 5 ml of
methanol
Flow rate: 2 ml/min, concentration 1 mg/ml
Retention: 12.75 (content: 70.4X)
Le A 25 238
- 50 -
_ . .
1~042~
II. 9enzhydryl 7-amino-3-C(Z)-1-propen-1-yl)]-3-cephem-
4-carboxylate hydrochloride (different process)
187.05 9 (0.8976 mol; 1.57 equivalents3 of phos-
phorus pentachloride are taken in 3,300 ml of CH2Clz in
a 4 l three-necked flask at room temperature and 66.42 ml
(0.821 mol; 1.44 equivalents) of pyridine, dissolved in
330 ml of CH2Clz, are added dropwise in the course of
5-10 minutes, whereupon the temperature rises to 24-27~C
and a clear colourless solution is formed. The solution
is cooled to -2~C and 300 9 (0.572 mol) of benzhydryl
7-phenylacetamido-3-C(Z)-propen-1-yl]-3-cephem-4-carboxyl-
ate are added, during which the temperature should not
rise above +2~C. Thereafter, the cooling bath is
removed and the mixture is stirred for 40 minutes, during
which the temperature of the reaction solution rises to
10-12~C (imino chloride solution).
255 ml (2.843 mol; 4.99 equivalents) of 1,3-
butanediol, dissolved in 1,650 ml of CH2Cl2, are cooled
to -20~C to -25~C (acetone/dry ice) in a 6 l three-necked
flask and the imino chloride solution is introduced into
this solution by means of a vacuum in the course of 5-10
minutes. The temperature should not thereby rise above
-20~C. The low temperature bath is then removed and
the reaction solution is stirred for 2 hours, during
which the temperature rises to 10~C.
The reaction solution is now washed with 1,200 ml
of ice-water and with 1,200 ml of 2N HCl and 1,200 ml of
saturated sodium chloride solution. The CH2Cl2 phase is
dried briefly over Na2S04, the drying agent is separated
off and the filtrate is concentrated to dryness. The
crystalline material which has precipitated out is
stirred with 1,200 ml to 2,000 ml of ethyl acetate and
then filtered off with suction and rinsed with ether.
The product is dried overnight under a high vacuum:
Yield: 173.9 9 (68.7% of theory)
C23H23clN2~35 (442.96)
Le A 25 238
- 51 -
13~042q
NMR (DMS0): ~ = 1.48 (dd,3H); 3.56 (d,1H); 3.79 (d,1H);
5.2 (d,1H); 5.31 (d,1H); 5.56-5.72 (m,1H); 6.21 (weak d,
1H); 6.28 (weak d,1H); 6.91 (s,1H); 7.25-7.47 (m,10H) ppm.
As well as the Z-isomer, the product contains
some E-isomer (Z/E = 91:9).
Example 11
Elenzhydryl D-7-[2-(t-butoxycarbonylamino)-2-(2-amino-
benzothiazol-6-yl)glycylamido]-3-[(Z)-1-propen-1-yl]-3-
cephem-4-carboxylate
H2N~S
H-CO-N~CH3
COOC ( CH3 ) 3 coocH ( C 6H5 ) 2
3.4 9 (10.5 mmol) of D-~-t-butoxycarbonylamino-
~-(2-aminobenzothiazol-6-yl)acetic acid and 4.0 9 (7 mmol)
of benzhydryl 7-amino-3-[(Z)-1-propen-1-yl]-3-cephem-4-
carboxylate (Example 10) are dissolved in 100 ml of
tetrahydrofuran. 2.2 9 (10.5 mmol) of DCC are added to
the clear yellow solution and the mixture is then stirred
at room temperature for 2 hours. After stirring for two
hours, the mixture is concentrated to dryness, the residue
is suspended in 150 ml of ethyl acetate, the suspension is
stirred with a magnetic stirrer for 10 minutes and the
insoLuble constituents are removed by filtration with
suction. The ethyl acetate filtrate is concentrated to
dryness, the residue is dissolved in S0 ml of methylene
chloride and the solution is chromatographed on 200 ml of
silica gel (0.04 - 0.063 mm). Elution is carried out
with methylene chloride and methylene chloride/methanol
mixtures in the following sequence:
1.) Methylene chloride (fraction 1 to 6): 200 mg,
discarded
Le A 25 238
- 52 -
13~042 1
2.) Methylene chloride - 2% methanol (fraction 7, 8):
200 mg, discarded
3.) Methylene chloride - 3% to 4~ methanol (fraction 9,
10)
4.) Methylene chloride - 4Z to 5% methanol (fraction 11
to 13)
Yield of fraction 9 to 13 : 3.1 9
5.) Methylene chloride - 5% to 10% methanol (fraction
14,15): 1.9 9
6.) Methylene chloride - methanol (1:1, fraction 16):
0.7 9, discarded
According to investigation by analytical HPLC,
fractions 9 to 13 contain the desired compound.
Yield: 3.1 9 (44% of theory)
C37H37NsO652 (711.9)
NMR (CDCl3): = 1.35 (dd, 3H); 1.43 (s, 9H); 3.15 (d,
1H); 3.31 (d, 1H); 4.97 (d, 1H); 5.3 (s, 1H); 5.46 - 5.55
(m, 1H); 5.71 (broad s, 2H); 5.78 - 5.85 (q, 1H); 6.03
(broad d, J = 11 Hz, 1H); 6.87 (s, 1H); 7.2 - 7.4 (mm,
12H); 7.5 (s, 1H) ppm.
Example 12
D-7-C(2-Aminobenzothiazol-6-yl)glycylamido]-3-C(Z)-1-
propen-1-yl]-3-cephem-4-carboxylic acid trifluoroacetate
H2~i~
H- CO-Nl~S CH3
NH2 ~1J
COOH x F'3C-COOH
3.1 9 (4.35 mmol) of diphenylmethyl D-7-C2-(t-
butoxycarbonylamino)-2-(2-aminobenzothiazol-6-yl)glycyl-
amido]-3-C~Z)-1-propen-1-yl]-3-cephem-4-carboxylate
(Example 11) are dissolved in 20 ml of methylene chloride,
50 ml of trifluoroacetic acid and 5 ml of anisole are
Le A 25 238
- 53 -
134042'1
added and the mixture is stirred at room temperature for
60 minutes. The mixture is concentrated in vacuo, 200 ml
of ether are added to the concentrate and the solid which
has precipitated out is filtered off and rinsed with
100 ml of ether. The pale yellow trifluoroacetate is
dried in vacuo.
Yield: 2.7 9
C21H21F3Ns06S2 (560.6)
Analytical HPLC: Hibar 250-4, Merck RP-8, 10 ~m, 254 nm
Mobile phase: 250 ml of CH3CN - 75 ml of glacial acetic
acid - 2,175 ml of water
Flow rate: 4 ml/min, concentration: 1 mg/ml
Retention 3.33 (content: 76.4%)
Example 13
D-7-t(2-Aminobenzothiazol-6-yl)glycylamido]-3-C(Z)-1-
propen-1-yl]-3-cephem-4-carboxylic acid
H2~S
Il 1
~,
~CH-CO -Nl~S~ CH3
NH2 ~J
COOH
2.6 9 (4.64 mmol) of D-7-t(2-aminobenzothiazol-
6-yl)glycylamido]-3-t(Z)-1-propen-1-yl]-3-cephem-4-
carboxylic acid trifluoroacetate (Example 12) are suspen-
ded in 100 ml of water and the small yellow insoluble
particles are removed over kieselguhr and rinsed with
30 ml of water. The still slightly cloudy solution is
filtered over a membrane filter (Millipore, 0.45 ~um) and
the 3-propenylcephalosporin is isolated from the filtrate
by means of preparative HPLC analogously to Example 6.
Yield: 500 mg
C1gH1gNs04S2 (445.5)
NMR (DCOOD): ~ = 1.67 (dd, 3H); 3.41 (d, 1H); 3.55 (d,
Le A 25 238
- 54 -
..... ~ ......... .. .. .
- ~ 134042~
1H); 5.3 (d, 1H); 5.78 (s, 1H); 5.81 - 5.91 (q and m,
2H); 6.25 (broad d, J = 11.6 Hz, 1H); 7.81 - 7.9 (q, 2H);
8.18 (s, 1H) ppm.
Example 14
S Sodium salt of D-~-[(1-methyl-2-methoxycarbonyl-vinyl)-
amino]-(2-aminobenzothiazol-6-yl)acetic acid
H2~5
~ H-COON~
I
H3C-C~CH-COOCH3
100 9 tcorresponding to 97.2 9 (0.435 mol)] of
D-2-aminobenzothiazol-6-yl)glycine (content = 97.2Z,
enantiomeric excess = 89.7%) are introduced into methanolic sodium hydrox-
ide solution prepared from 18.6 9 (0.465 mol, that is to
say 7X excess) of NaOH and 1,000 ml of methanol. ~hile
boiling under reflux and stirring, a clear solution
forms, and to this is added 64 ml (0.59 mol, about 40Z
excess) of methyl acetoacetate, dissolved in 100 ml of
methanol, in the course of 40 minutes (pH ~hen the drop-
wise addition has ended = 10.3; sample/water = 1:1). The
solution is then heated under reflux for 1 hour and
stirring is subsequently continued without heating for
several hours. The material uhich has crystallized out
is filtered off with suction and ~ashed ~ith toluene
(tuo portions of 200 ml). The residue on the filter is
heated to the boiling point in 1,000 ml of toluene for
30 - 40 minutes, 300 ml of toluene are then distilled
off and the mixture is vashed by drop~ise addition of
fresh toluene and is dried in a fresh air cabinet at
70~C overnight.
Yield: 81.9 9 (52.1Z of theory)
C14H14N304SNa ~ H20 (361.4)
Calculated C 46.53 H 4.46 N 11.62 S 8.87 Na 6.36
Le A 25 238
_ 5s _
13~0~2~
.
Found C 46.2 H 4.4 N 11.9 S 8.4 Na 5.6 Br 0.1
Cl 0.5
NMR (DMS0): ~ = 1.63 (s, 3H); 3.49 (s, 3H); 4.25 (s, 1H);
4.7 (d, J = 7.5 Hz, 1H); 7.12 (dd, 1H); 7.21 (d, 1H);
7.34 (s, 2H); 7.48 (s, 1H); 9.55 (d, 1H) ppm.
ee = 100X
Example 15
7-Amino-3-t(Z)-1-propen-1-yl]-3-cephem-4-carboxylic acid
(7-APCA)
H2 ~ ~ CH3
COOH
54.5 9 (0.123 mol of benzhydryl 7-amino-3-[(Z)-
1-propen-1-yl]-3-cephem-4-carboxylate (Example 10) are
added to a stirred solution of 500 ml of trifluoroacetic
acid (TFA) and 31 ml of anisole, which is cooled to 0~C.
The mixture is stirred at room temperature for 1 hour and
is then concentrated in vacuo at 30~C and the oily residue
is stirred with 600 ml of ether for 1 hour. The precipi-
tate is filtered off with suction and washed with 300 to
400 ml of ether and the residue on the filter is dried
in vacuo for 3 hours. The trifluoroacetate is suspended
in 300 ml of water, the suspension is cooled to +5~C and
the pH is brought to 0.2 - 0.4 with 12 N HCl. The result-
ing clear solution is cooled to +5~C and stirred with
4 9 of active charcoal for 10 minutes. The active char-
coal is filtered off with suction over kieselguhr andrinsed with about 50 ml of 0.1 N HCl. The filtrate is
brought to pH 2.1 at +5~C with 20% strength NaOH and the
product which has precipitated out is left to stand in a
refrigerator for 1 hour in order to bring the crystal-
lization to completion. The crystal sludge is filteredoff with suction, washed with 100 ml of water and 300 ml
Le A 25 238
- 56 -
.. . .. . ..
of acetone and dried in vacuo. 13~042'1
Yield: 16.4 9 (55.4% of theory)
C10H12N203S (240.3)
NMR (DCOOD): ~ = 1.8 (dd, 3H); 3.61 (d, 1H); 3.77 (d,
1H); 5.39 (d, lH); 5.52 (d, 1H); 5.91 - 6.06 (q, 1H);
6.5 (d, 1H) ppm.
Analytical HPLC: Hibar 250-4, Merck RP-8, 10 ~m, 254 nm
Mobile phase: 100 ml of CH3CN - 30 ml of glacial acetic
acid - 870 ml of ~ater
Flo~ rate: 2 ml/min, concentration: 0.25 mg/ml
Retention: 2.39 (85.7Z; Z-isomer)
3.16 (11.3%; E-isomer)
Example 16
I. D-7-C(2-Aminobenzothiazol-6-yl)glycyl-amido]-3-C(Z)-
1-propen-1-yl]-3-cephem-4-carboxylic acid
H2~
N~l H- CO -NH~CH3
COOH
a) Activation of the precursor acid:
12.09 9 Ccorresponds to 11.48 9 (33.4 mmol)] of
sodium D-~-C(1-methyl-2-methoxycarbonyl-vinyl)-amino]-(2-
aminobenzothiazol-6-yl) acetate (content = 95~, Example
14) are dissolved in 57 ml of dimethylformamide, and
23 ml of acetonitrile are then added. The solution is
cooled to -70~C, 115 ~l of 3-dimethylaminopropanol and
3.30 ml (34.4 mmol) of ethyl chloroformate are added in
succession and the mixture is stirred at -70~C for 20
minutes.
b) Preparation of the cephalosporin component (7-APCA)
9.61 9 (40 mmol) of 7-amino-3-C(Z)-1-propen-1-
yl]-3-cephem-4-carboxylic acid (Example 15) are suspended
Le A 25 238
- 57 -
134042 1
in 57 ml of dimethylfor0amide and 23 ml of acetonitrile
and the suspension is converted into a clear solution by
addition of 1 N sodium hydroxide solution (36.8 ml) to
pH 8.5 at room temperature. The solution is cooled to
-20~C to -30~C.
c) Coupling, deblocking and isolation of the crude
betaine
The cooled 7-APCA solution (-20~C to -30~C~
according to b) is slowly added dropwise to the solution
of the mixed anhydride of the precursor acid according
to a) at -70~C and the mixture is subsequently stirred
at -70~C for 10 minutes. The temperature of the solu-
tion is then allowed to come to 0~C (without cooling)
in the course of 45 minutes and the solution is stirred
with 1.2 9 of active charcoal and 1.2 9 of kieselguhr for
a further 10 minutes. The reaction mixture is filtered
over a Seitz filter, the residue on the filter is rinsed
with a little dimethylformamide and 6.9 ml of concentra-
ted hydrochloric acid are added to the filtrate. The
volume of the solution is concentrated to 115 ml, salts
which have precipitated out being separated off. The
filtrate is brought to pH 4.0 by stirring with 25%
strength NH3 solution with a magnetic stirrer and 800
ml of acetone are added, whereupon the crude betaine
precipitates out. The precipitate is stirred for 10
minutes, filtered off with suction and rinsed with ace-
tone and the material is dried in vacuo.
Yield: 12.65 9
Analytical HPLC: Hibar 250-4, Merck RP-8, 10 ~m, 254 nm
Mobile phase: 100 ml of CH3CN - 30 ml of glacial acetic
acid - 870 ml of water
Flow rate: 2 ml/min; concentration 1 mg/ml
Retention: 7.06 (73.8%; Z-isomer)
11.18 (11.5%; E-isomer)
The crude betaine is suspended in water and dis-
solved with half-concentrated hydrochloric acid at pH 1.2
Le A 25 238
- 58 -
134042l1
and the solution is stirred with 1.2 9 of active charcoal
for 15 minutes. The mixture is filtered with suction
over a kieselguhr bed, the residue on the filter is
rinsed with 20 ml of 0.1 N hydrochloric acid and the
filtrate is pumped onto an RP 18 column ~Hibar 250-25,
Merck). The column is eluted first with water and then
with 5% strength methanol. The fractions are investigated
by means of analytical HPLC and the fractions which con-
tain the Z-isomer derivative are combined, the methanol
is distilled off in vacuo and the aqueous solution is
lyophilized.
Yield: 5.2 9 (32.3X of theory)
C19H19N504S2 . 2H20 (481.56)
Calculated: C 47.39 H 4.81 N 14.54 S 13.22
Found : C 47.7 H 4.8 N 14.3 S 13.0
II. D-7-C(2-Aminobenzothiazol-6-yl)9lycyl-amido]-3-[tz)
1-propen-1-yl]-3-cephem-4-carboxylic acid (different
process)
a) Activation of the precursor acid:
100 9 [corresponding to 95 9 (0.277 mol) of pure
material; 1.2 equivalents] of sodium D-~-C(1-methyl-2
methoxycarbonylvinyl)-amino]-(2-aminobenzothiazol-6-yl
acetate (content = 95X, Example 14) are dissolved in
500 ml of dimethylformamide and Z00 ml of acetonitrile
to give a clear solution. The solution is cooled to
-60~C, 1 ml of 3-dimethylamino-1-propanol and 27.7 ml
(0.281 mol) of ethyl chloroformate are added in succes-
sion and the mixture is stirred at -60~C for 30 minutes.
b) Preparation of the cephalosporin component:
102 9 (0.2304 mol) of benzhydryl 7-amino-3-[(Z)-
1-propen-1-yl]-3-cephem-4-carboxylate hydrochloride
(Example 10tII) are dissolved in 500 ml of dimethylform-
amide and 100 ml of acetonitrile to give a clear solu-
tion, 31 ml of water and 32.2 ml (0.2289 mol) of tri-
ethylamine are added at room temperature and the mixture
is stirred for 5 minutes, while cooling with ice.
Le A 25 238
- 59 -
c) Coupling: 1340 124
The cephalosporin solution (b), cooled to 0~C,
is slowly added to the solution of the mixed anhydride
(a) at -60~C, whereupon the temperature rises from
-60~C to -30~C. The mixture is subsequently stirred
for a total of 30 minutes and the temperature of the
reaction solution is allowed to come to 0~C. 57 ml of
concentrated hydrochloric acid are then added and the
solution is stirred at 0~C for 15 minutes.
d) Isolation of the trifluoroacetate salt:
Acetonitrile is distilled off from the reaction
solution and the pH is brought to 7.5 with 25% strength
NH3 solution, while cooling with ice. The solution is
shaken in S l of ethyl acetate and 3 l of 10% strength
NaHC03 solution containing sodium chloride. The mixture
is stirred intensively for S minutes and then filtered
with suction over a Seitz filter. The ethyl acetate
phase is separated off and washed once with 4 l of
saturated NaHC03 solution and twice with 4 l of water.
Thereafter, the ethyl acetate phase is dried over Na2S04,
the drying agent is filtered off with suction and,
finally, the filtrate is concentrated to dryness in vacuo.
The rigid foam formed is dried under a high vacuum for
30 minutes. Yield: 170 9.
e) Deblocking:
The rigid foam is dissolved in 1,600 ml of
CHzCl2, the solution is cooled to 0~C and a mixture
of 750 ml of trifluoroacetic acid and 4 ml of anisole is
added. The solution is then stirred at room temperature
for 45 minutes and subsequently concentrated to an oil
and the oily residue is digested with 6 l of ether. The
material which has crystallized out is filtered off with
suction, washed with ether and dried overnight in vacuo.
Yield: 125 9 (97% of theory)
C19H19N504S2 . CF3 COOH (559.55)
Analytical HPLC: Hibar 250-4, Merck RP-8, 254 nm
Le A 25 238
- 60 -
.. . . ... .. . ....... . .
1~40~2~
Mobile phase: 100 ml of CH3CN - 30 ml of glacial acetic
acid - 870 ml of water
Flow rate: 4 mL/min.
Retention: 2.25 (90.1X; Z-isomer)
3.77 (8.3%; E-isomer).
f) Preparation in the pure form by means of adsorber
resin chromatography:
141 9 of D-7-C(2-aminobenzothiazol-6-yl)glycyl-
amido]-3-[(Z)-1-propen-1-yl]-3-cephem-4-carboxylic acid
trifluoroacetate [moist material = 128.8 9 (100% of
theory)] are suspended in 1,000 ml of water, the suspen-
sion is stirred intensively for 15 minutes and the in-
soluble material is then filtered off with suction and
rinsed with water. The filtrate (pH 1.3) is introduced
~nto a column filled with 8 l of adsorber resin LGP 4429
(Lewatit~C 1062, HAYER AG). The column is eluted first
with S l of water and then with in each case 2 l portions
of water to which acetone is added in an amount which
increases from 2% to 10%. A total of 15 fractions with
a volume of in each case 2,000 ml are collected:
Fraction Yield
(a = 2,000 ml) (9)
1 3.5
2 2.1
25 3/4 9.5
5/6 16.7
7/8 13.2
9/10 8.3
11 5.1
30 12 5.3
13 3.7
14 2.3
1.0
Fractions 3 to 10, which contain the desired pro-
duct in a highly pure form, are distilled in vacuo toremove the acetone and the residue is lyophilized.
Le A 25 238
- 61 -
q~
.. . ... ~ ~
Yield: 47.7 9 (43.0X of theory) 131~12~1
C19H19NS04S2 ~ 2H20 (481.56)
g) Formation of the methanol solvate:
168.3 9 of D-7-C2-aminobenzothiazol-6-yl)-glycyl
S amido]-3-cephem-4-carboxylic acid as the lyophilisate
(f) are stirred in 1,700 ml of methanol for 90 minutes,
filtered off with suction and rinsed with S00 ml of
methanol on the suction filter. The suction filter
residue is stirred again in 1,000 ml of methanol for 45
minutes, filtered off with suction and rinsed with S00 ml
of methanol on the suction filter. The product is dried
overnight under a high vacuum.
Yield: 110.2 9 (65.5% of theory); analytical HPLC: Hibar
250-4, Merck RP-8, 10 ~m, 254 nm
Mobile phase: 100 ml of CH3CN - 30 ml of glacial acetic
acid - 870 ml of water
Flow rate: 4 ml min 1; concentration: 1 mg ml 1
Retention: 3.58 (98.4%; Z-isomer)
6.54 (0.79%; E-isomer)
h) Formation of the hydrate:
109.6 9 of D-7-C2-aminobenzothiazol-6-yl)glycyl
amido~-3-C(Z)-1-propen-1-yl-3-cephem-4-carboxylic acid
methanol solvate (g) are introduced into 1,100 ml of
B water (obtained from Milli-Q Watersystem, Millipore GmbH),
with stirring and the mixture is stirred with a magnetic
stirrer in vacuo for 2 hours. The product is filtered
off with suction and washed three times with approxi-
mately equal portions of water (from the Milli-Q Water-
system). The substance is dried under a high vacuum
30 without a drying agent for 36 hours.
Yield: 94.7 9 (86.4%)
C19H19NS04SZ ~ 2H20 (463.541)
Calculated: C 49.23 H 4.57 N 15.11 S 13.83
Found : C 48.8 H 5.1 N 14.9 S 13.4
Analytical HPLC: Hibar 250-4, Merck RP-8, 10 ~m, 254 nm
Mobile phase: 100 ml of CH3CN - 30 ml of glacial acetic
Le A Z5 238
- 62 -
~a Je -~a~k
~3~0 12-1
acid - 870 ml of ~ater
Flow rate: 4 ml min 1; concentration: 1 mg ml
Retention: 3.56 (98.8X~.
Example 17
S Sodium D-7-t~2-aminobenzothiazal-6-yl)glycyl-amido~-3-
ttZ)-1-propen-1-yl]-3-cephem-4-carboxylate
H2N'II S
H- CO - NH~gC~t3
COON~
15.0 9 (0.0324 mol) of D-7-[(2-aminobenzothiazol-
6-yl)-glycyl-amido]-3-t(Z)-1-propen-1-yl]-3-cephem-4-
tO carboxylic acid (Example 16/II) are suspended in 300 ml
of water, with stirrin~, and the
pH is brought to 8.56 under pH-stat conditions ~ith 1N
sodium hydroxide solution (Memo-Titrator DL 40 RC). The
pale yello~ solution formed is filtered ~ith suction over
15 filterpaper and the filtrate is lyophilized.
Yield: 15.0 9 (92.1~ of theory)
C1gH18N5NaO4s2 ~ 2H2~ (503-54)
Calculated: C 45.32 H 4.40 N 13.91 S 12.74 Na 4.56
Found : C 46.0 H 4.9 N 14.0 S 12.9 Na 4.3
NMR (DCOOD): ~ = 1.64 (dd, 3H); 3.38 (d,1H); 3.51 (d,1H;
5.26 (d, 1H); 5.72 (s, 1H); 5.78-5.85 (m, 1H); 5.85 (d,
1H); 6.21 (d, 1H); 7.76-7.83 (q, 2H); 8.12 (s, 1H) ppm.
Analytical HPLC: Hibar 250-4, RP-8, 10 ~m, 254 nm
Mobile phase: 1ûO ml of CH3CN - 30 ml of glacial acetic
acid - 870 ml of uater
Flo~ rate: 2 ml/min
Concentration: 1 mg/ml
Retention: 4.36 (content: 98.7~)
Le A 25 238
-- 63 --
, .
134042il
Example 18
D-~-t-Butoxycarbonylamino-~-(benzothiazol-6-yl)acetic
acid
~1
N ~ ~
H-COOH
NH-COOc~cH3)3
12.2 ml (0.102 mol) of tert.-butyl nitrite, dis-
solved in 25 ml of tetrahydrofuran, are added dropwise
to a solution of 20 9 (0.0618 mol) of D-~-t-butoxycarbon-
ylamino-~-(2-aminobenzothiazol-6-yl)acetic acid in 160 ml
of tetrahydrofuran at +50~C to +60~C in the course of
30 minutes. The mixture is subsequently stirred at
+50~C to +55~C for 30 minutes, the solvent is distilled
off and the residue is partitioned in 300 ml of water and
300 ml of ethyl acetate. The mixture is acidified to pH
1.5 with 2 N HCl, while cooling with ice, and is stirred
for 5 minutes and the pH is then brought to 8.0 to 8.5
with 40% strength potassium carbonate solution. The
aqueous phase is separated off, washed again with ethyl
acetate and then acidified to pH 2.5 with 2 N HCl at
0~C. The acid solution is extracted twice with ethyl
acetate and the extract is washed with sodium chloride
solution and dried over sodium sulphate. The ethyl ace-
tate phase is concentrated to 50 ml and stirred into
petroleum ether.
Yield: 14.4 9 (76% of theory)
C14H16N2045 (308.4)
Calculated: C 54.5 H 5.2 N 9.1 S 10.4
Found : C 54.1 H 5.6 N 8.9 S 9.7
t~]20 = -130.3~C (c = 1, methanol)
589
NMR (DMS0): ~ = 1.43 (s, 9H); 5.31 (d, 1H); 7.6 (dd, 1H);
7.73 (d, 1H); 8.08 (d, 1H); 8.2 (weak d, 1H); 9.4 (s,
Le A 25 238
- 64 -
,, . . . ~
1H) ppm. 1~40 l2~
Example 19
p-Methoxybenzyl 7-phenyLacetamido-3-t(Z)-propen-1-yl]-3-
cephem-4-carboxylate
O~H2 -CO-N~CH3
COOCH2~oCH3
40.0 9 (82.1 mm~l) of p-methoxybenzyl 7-phenyl-
acetamido-3-chloromethyl-3-cephem-4-carboxylate and
22.66 9 (86.4 mmol) of triphenylphosphine are dissolved
in 300 ml of dimethylformamide, 12.95 9 (86.4 mmol) of
sodium iodide are added and the mixture is stirred at
room temperature for 2 hours. The reaction solution is
then concentrated to an oil under a high vacuum.
The oily residue of 4.2 9 is taken up in 130 ml
of methylene chloride (no complete clear solution),
69.0 ml (1,231.5 mmol) of acetaldehyde are added to the
mixture in a 500 ml three-necked flask and the mixture
is then treated with 1 N sodium hydroxide solution by
means of an autotitrator at pH 8.1 under pH-stat condi-
tions. After 1 hour, 8.7 ml of 1 N sodium hydroxide
solution have been consumed. A further 72.5 ml of 1 N
sodium hydroxide solution are then consumed at pH 8.3 in
the course of 20 hours. The aqueous phase is separated
off and the methylene chloride phase is washed twice with
water and dried over sodium sulphate. The methylene
Z5 chloride phase is then stirred again with 20 ml (358
mmol) of acetaldehyde at room temperature for 2 hours,
the methylene chloride is distilled off, and the oil which
remains is taken up in toluene and the mixture is intro-
duced onto a column containing 1 l of silica gel (0.04 -
0.063 mm).Le A 25 238
- 65 -
.. . . . . . . . .. ... ....
13~0 ~2l1
Elution is carried out first with toluene (frac-
tion 1 to S) and then with toluene/ethyl acetate (5:1,
fraction 6, 7~, 600 ml fractions being collected. Frac-
tions 3 to 6 are combined and concentrated to dryness
S and the resulting oil is triturated with 100 to 150 ml
of ether. The white material which has precipitated out
is filtered off with suction and washed with ether (S0
to 80 ml).
Yield: 12.1 9 ~31% of theory)
C26H26N2~SS (478-6)
NMR (CDCl3): ~ = 1.52 (d, 3H); 3.23 (d, 1H); 3.41 (d,
1H); 3.61 (q, 2H); 3.78 (s, 3H); 4.95 (d, 1H); 5.13 (s,
2H); 5.59 - 5.69 (dq, 1H); 5.75 - 5.81 (q, 1H); 6.08
(broad d, 1H); 6.45 (d, 1H); 6.87 (d, 2H); 7.25 - 7.36
(m, 7H) ppm.
Fractions 7 to 11 are concentrated to a slightly
reddish oil, which cannot be assigned to the desired
compound.
Example 20
p-Methoxybenzyl 7-amino-3-[(Z)-1-propen-1-yl]-3-cephem-
4-carboxylate
H2 ~ CH3
COOCH2 ~ oCH3
12.1 9 (25.28 mmol) of p-methoxybenzyl 7-phenyl-
acetamido-3-~(Z)-propen-1-yl]-3-cephem-4-carboxylate
(Example 15) are dissolved in 133 ml of methylene chlor-
ide, the solution is cooled to -50~C and 5.11 ml
(63.2 mmol) of pyridine and 5.26 9 (25.28 mmol) of phos-
phorus pentachloride are added in succession. The tem-
perature is then aLlowed to rise to -10~C in the course
of 25 minutes. After a further 20 minutes, the tempera-
Le A 25 238
- 66 -
, . , , . . ~
1~40 ~2~1
ture of the solution is 0~C; thereafter, the solution
is stirred to +15~C for a further 45 minutes. The mix-
ture is now cooled to -50~C, 200 ml of methanol (about
-30~C) are added all at once and the mixture is stirred
S for 30 minutes, without cooling. The reaction solution
is concentrated, the oily residue is dissolved in methyl-
ene chloride and the solution is introduced onto a column
packed with 400 ml of silica gel (0.04 - 0.063 mm). The
column is eluted first with methylene chloride and then
with the solvent mixtures methylene chloride - SX methanol
and methylene chloride - 10% methanol in succession. The
fractions which are eluted with the methylene chloride -
10% methanol mixture are concentrated to dryness.
rield: 10 9
The oil is taken up in 500 ml of ethanol, an
insoluble slime is separated off by decanting filtration,
the filtrate is concentrated to dryness and the oil is
dried in vacuo.
Yield: 6.8 9 (75% of theory)
C1gH20N204S (360.4)
NMR (CDCl3): ~ = 1.55 (dd, 3H); 1.95 (weak dd, 2H); 3.3
(d, 1H); 3.5 (d, 1H); 3.76 (s, 3H); 4.72 (d, 1H); 4.98
(d, 1H); 5.18 (s, 1H); 5.58 - 5.69 (dq, 1H); 6.1 (broad
d, 1H); 6.88 (d, ZH); 7.31 (d, 2H); 8.61 (d, 1H) ppm.
Example 21
p-Methoxybenzyl D-7-C2-(t-butoxycarbonylamino)-2-(ben
thiazol-6-yl)-glycylamido]-3-C(Z)-1-propen-1-yl]-3-
cephem-4-carboxylate
Le A 25 238
- 67 -
13~0~2~
H-CO-N ~ CH3
COOC(C~3)3 COO-CH2 ~ CH~
3.90 9 (18.9 mmol) of dicyclohexylcarbodiimide
are added to a solution of 5.83 9 (18.9 mmol) of D-~-t-
butoxycarbonylamino-~-(benzothiazol-6-yl)acetic acid and
6.8 9 (15.1 mmol) of p-methoxybenzyl 7-amino-3-[(Z)-1-
propen-1-yl]-3-cephem-4-carboxylate (Example 19) in
150 ml of tetrahydrofuran at room temperature, while
stirring with a magnetic stirrer, and the mixture is then
stirred for 2 hours. The dicyclohexylurea formed is
filtered off with suction and washed with tetrahydrofuran
and the mother liquor is concentrated to dryness. The
oil which remains is dissolved in 100 ml of methylene
chloride and the solution is chromatographed on 600 ml
of silica gel (0.04 - 0.063 mm), elution being carried
out first with methylene chloride (2 x 300 ml) and then
with 5% strength methanol in methylene chloride with
control by means of thin layer chromatography (methylene
chloride : methanol = 100 : 5). In each case 300 ml frac-
tions are collected. The desired fractions of the eluate
(11 and 12) with 5X strength methanol are combined and
concentrated to dryness.
Yield: 8.8 9 (89% of theory)
C32H34N407S2 (650.8)
NMR (CDCl3): ~ = 1.4 (s, 9H); 1.5 (dd, 3H); 3.12 (d,
1H); 3.32 (d, 1H); 3.75 (s, 3H); 4.91 (d, 1H); 5.13 (s
Le A 25 238
- 68 -
1340A2ll
and d, 3H); 5.58 - 5.68 (m, 1H); 5.73 - 5.79 (q, 1H);
6.03 (broad d, 1H); 6.86 (d, 2H); 7.28 (d, 2H); 7.52 (d,
1H); 8.01 (s, 1H); 8.1 (d, 1H); 9.4 (s, 1H) ppm.
Example 22
S D-7-C(Benzothiazol-6-yl)glycylamido]-3-t(Z)-1-propen-1-
yl]-3-cephem-4-carboxylic acid
NH2 ~ CH3
COOH
A mixture of 8.8 9 (13.5 mmol) of p-methoxybenzyl
D-7-~2-(t-butoxycarbonylamino)-2-(benzothiazol-6-yl)-
glycylamido]-3-[(Z)-1-propen-1-yl]-3-cephem-4-carboxylate
(Example 20), 3 ml of anisole and 100 ml of trifluoro-
acetic acid is stirred at room temperature for 1 hour.
The mixture is concentrated in vacuo, 40 ml of toluene
are added to the pale red, readily mobile oil and the
mixture is concentrated again in vacuo. The oil which
remains is triturated with 400 ml of ether and the solid
which has precipitated out is filtered off with suction,
washed with ether and dried in vacuo. 6.3 9 of tri-
fluoroacetate are obtained and are dissolved in 800 ml
of water, and the insoluble constituents are separated
off by filtration over kieselguhr. The solution is
B passed over an HP 20 column (600 ml, Diaion~adsorber
resin, Mitsubishi), which is eluted with water and then
with a mixture of water and methanol (gradient up to 50%).
The eluate containing the desired compound is lyophilized.
Yield: 1.9 9
The lyphilisate is dissolved almost completely
in 150 ml of water, while stirring with a magnetic
stirrer, 100 mg of active charcoal are added, the mixture
Le A 25 238
- 69 -
~ra~-ma~k
.
13~0~2'1
is stirred for 5 minutes and the solid is then filtered
off with suction over kieselguhr and rinsed with 50 ml of
water. The filtrate is filtered again with a syringe by
means of a Millipore membrane filter and is then pumped
onto a preparative column (Hibar 250-25, RP-18, Merck,
flow rate: 10 - 15 ml. minute 1). After application of
the sample, the column is eluted with the following
mobile phase systems in succession:
1.) 500 ml of water
2.) 500 ml of water with 10% of methanol
3.) 1,000 ml of water with 10% to 40Z of methanol: the
eluate is collected here in 50 - 100 ml fractions and
then investigated by means of analytical HPLC, it being
found that fractions 8 to 10 contain the cis-isomer
derivative.
Yield: 468 mg
C1gH1gN404S2 (430.5)
NMR (DCOOD): ~ = 1.61 (dd, 3H); 3.31 (d, 1H); 3.48 (d,
1H); 5.25 (d, 1H); 5.75 - 5.9 (m and q, 3H); 6.2 (broad
d, 1H); 8.13 (dd, 1H); 8.48 (d, 1H); 8.67 (s, 1H); 10.33
(s, 1H); ppm.
Analytical HPLC: Hibar Z50-4, RP-8, 10 ~m, 254 nm
Mobile phase system: 790 ml of water - 200 ml of
methanol - 10 ml of buffer, pH 7.0
Flow rate: 2 ml/min, concentration: 1 mg/ml
Retention: 7.53 (content: 92.4%)
Example 23
D-7-C(2-Aminobenzothiazol-6-yl)glycylamido]-3-vinyl-3-
cephem-4-carboxylic acid
H2N~
~ H-CO-
NH2
COOH
Le A 25 238
- 70 -
. . . .. ...
134042~
Z76.1 ~l (1.585 mmol) of ethyl diisopropylamine
and 122.7 ~l (1.585 mmol) of mesyl chloride are slowly
injected in succession into a solution, cooled to -50~C,
of 512.5 mg (1.585 mmol) of D-~-t-butoxycarbonylamino-~-
(2-aminobenzothiazol-6-yL)acetic acid in 5 ml of dimethyl-
formamide. The mixture is stirred at -50~C for 40
minutes and a solution (-20~C) of 622 mg (1.585 mmol)
of diphenylmethyl 7-amino-3-vinyl-3-cephem-4-carboxylate
and 276.1 ~l (1.585 mmol) of ethyldiisopropylamine in
5 ml of tetrahydrofuran and 3 ml of dimethylformamide is
added dropwise. The mixture is subsequently stirred at
-50~C for 5 minutes and then without cooling for a
further 50 minutes. Thereafter, the reaction solution
is stirred into 40 ml of water and 120 ml of ethyl ace-
tate, the ethyl acetate phase is separated off, theaqueous layer is extracted again with 60 ml of ethyl
acetate and the organic phases are combined and washed
with 0.1N hydrochloric acid, sodium bicarbonate solution
and sodium chloride solution. After drying and distill-
ing off the solvent, the residue is taken up in 20 ml ofmethylene chloride, 20 ml of trifluoroacetic acid with 1
drop of anisole are added and the mixture is stirred at
rog~ temperature for 45 minutes. The trifluoroacetic
acid/methylene chloride mixture is then distilled off in
vac~o, the residue is dissolved in 15 ml of 80Z strength
acetic acid, the solution is pumped onto an RP-18 column
(Hibar 250-25, Merck) and the column is eluted with 3X
strength acetic acid. The eluate, which contains the
desired substance, is freeze-dried.
Yield: 800 mg
The lyophilisate is dissolved again in 10 ml of
3% strength acetic acid and the solution is rechromato-
graphed on an RP-18 column (Hibar 250-25, Merck).
rield: 165 mg
C18H17N5~452 ~ 3H20 . 1/2CH3CooH (505.5)
NMR (DCOOD): ~ = 3.61 - 3.76 (dd, 2H); 5.28 (d, 1H);
Le A 25 238
- 71 -
.. . . . .. ..
1340 12~
5.52 (d, 1H); 5.69 (d, 1H); 5.75 (s, 1H); 5.91 (d, 1H);
7.18 - 7.28 (q, 1H); 7.84 (m, 2H); 8.18 (s, 1H) ppm.
Le A 25 238
- 72 -