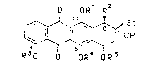Note: Descriptions are shown in the official language in which they were submitted.
BEHRINGWERKE ARTIENGESELLSCHAFT 88/B 031 - ~a 724
Dr. Ha/Li./Hch
Process for the preparation of 4-O-alkylrhodomycins
The present invention relates to a new process for the
preparation of 4-O-alkylrhodomycins, e~pecially 4-O-
alkyl-~-rhodomycinone~ and 4-O-alkyl epsilon-rhodomycin-
ones, and the use th~reof for the preparation of 7-O-
glycosylrhodomycins which, by virtue oP their cytostaticeffectiveness, ~re suitable for the treatmen~ of oncoses.
Anthracyclins are described in the ~pecialist literature.
It is known from the doxorubicin-daunorubicin group of
anthracyclin that 4-O-methyl ~ubstitution is necessary
in the anthracyclins for antitumoral action and tumor
~electivity. The corre~ponding 4-hydroxy derivativeæ,
~uc~ as carminomycins and ~-rhodomycins, are cytotoxic
and les~ tumor-selective.
The invention i~ based on the ob~ect of developing a
proce~s which gives 4-O-alkylrhodom~rcinone derivatives in
good yield~ and which means a s~nplifi~ation co~pared
with ~he known process, as well a~ the elaboration of a
protective group chemi~try on 4-O alkylrhodomycinones
whi~h make6 it pos~ible to u~e the aglycone for the
preparation of 7-O-glycosylrhodomycinones.
The present proce~s for the preparation of 4-O-alkyl-
rhodomycin~ i~ based on the surpxising findin~ that an
unprotected rhodcmycino~e aglycone can be reacted with
1,3-dichloro-1,1,3 9 3-tetrai~opropyldi6iloxane to ~ive a
6,7-O-(tetraisopropyldisiloxane-1,3 diyl)~rhodomycinone
and then with a trialkylsilyl halide to g~ve ~ 10-O-
trialkyl~ilyl compound. By thi6 route th~ ~ubseguent
alkylation can only t~ke place at the phenoli~ hydroxyl
group~ at the C-4 and C~ tom~ alkylation of the
phenolic group at the C-4 atom curpri~ingly is mainly
taking place.
- 2
Thi~ ob~ect is achieved in ~ccordance with the invention
by the proces~ for the preparation of a rhodomycin
derivative of the formula I
O ORl R
~ ~ E~ I
R30 D OR~ b R 3
in which
R~ denotes ~, C~-C4-alkyl or an acyl protecti~e group,
R2 denotes OH, COOCH3, O-Si(Cl-C4-alkyl)3 or an O~acyl
protective gxoup, acyl being acetyl, monohalogeno-
acetyl, dihalosenoac0tyl or trihalogenoacetyl with
fluorine or chlorine a6 the halogen, benzoyl or p-
nitroben~oyl,
R3 deno~e~ Cl-C4-alkyl,
R4 denotes H or
R4 and R5 together denote a tetrai~opropyldisiloxane-
1,3-diyl protective group, and
Rs denote H or a glycosyl radical of the formula II
R7 ~ ~
R~ ;
in which
R6 i8 OH, ~etoxy, ~HCOCF3, NHz, a mono-C~-C~-alkylamino
~roup or a di-Cl-C~-alkylEmino group or a 4-morpho-
linyl group and
R7 1~ H9 OH or an acetoxy, trifluoroaceto~y or p-
nitrobenzoylo~y group,
which compri~e6 reactin~ a rhodomycinone derivati~e of
the formula I in which
R2 is OH or COOCH3 ~nd
R3, R~ and R5 ~re H,
with 1,3Ydichloro-1,1,3,3-tetraisopropyldisiloxane a~d,
if appropriate, ~ubsequen~ly with a tri-C,-C~alkyl~ilyl
halide, pxeferably with a chloride deriv~tive, in the
,,
,~ , ,
8~7
-- 3 --
presence of a b~se, ~uch as pyricline or dimethylamino-
pyridine, and an organic ~olvent, 6uch as methylene
dichloride or chloroform, at a tempera ure between 15C
and 80C to give a 6,7 0-(tetrai~opropyldi~iloxans-1,3-
diyl)-rhodomycinone compound of the formula I in which
R1 and R3 are H,
R2 is COOCH3 or O-Si(Cl-C4-alkyl) 3 and
R4 and R5 together are a tetrai~opropyldi~iloxane-1/3-diyl
group,
then etherifying th~ resultin~ compound with an al~ylat-
ing agent, pxeferably a Cl-C4-alkyl bromide or iodide, in
the presence of an alkali metal carbonat~ and an organic
601vent, such a ace$onP or dioxa~e, at the phenolic
hydroxyl group on the C-4 a~om, if appropriate al~o at
the phenolic hydkoxyl group on the C-11 atom, then, if
appropriate, ~plitting off the trialkyl~ilyl protective
group st the C-10 atom by acid hydrolysis, preferably by
means of hyckochloric acid, and acylating the resulting
compound at the 10-hydroxyl group and, if appropriate, at
the ll hydroxyl group by means of an acylating agent,
preferably p-nitxobenzoyl chloride, tri~luoroacetic
anhydride or ~cetyl chloride, in the pre~ence of a ba~e,
~uch as pyridine, triethylamine or dimethylaminopyridine,
and an organic 501vent ~ ~uch as methylene dichloride or
chloroform, and ~plitting off the tetrai~opropyldi~
o~ne 1,3-diyl group by means of an ammonium fluoride,
preferably tetr~butylammonium fluoride, in a polar
organic solvent~ ~uch as tetrahydrofuran or dioxane, in
the course of which a compound of the formula III
O ORI R2
~ Jo~ III
R30 HO OH
in which
Rl denote~ ~, Cl C4-alkyl or an acyl protective group
~ndicated above,
R2 denotes COOCH3 or an O acyl protective group and
R3 denotes C,~C4~alkyl i8 formed. .
-- 4 --
The proce~s on which ~he inven~ion i~ based i~ illus-
trated in Scheme 1.
The use of a rhodomycinone compound of the formula III in
a proce6s for the praparation of a 7-0-sl~cos~ rhodomyci-
ncne compound of th~ forMula I compri6es reacting acompound of the formula III in a manner known per 6e with
a functionalized deoxy ~ugar of the formula IV or V
R 1
R6 R6
in which
R6 represents an acetoxy or trifluoroacetamido group,
R7 repre~ents a hydrogen atom or an acetoxy~ trifluoro-
ace~o~y or p-nitrobenzoyloxy group and
Y repre~ents an acetoxy or p-nitrobenzoyloxy group or
8 chloride, in the presence o:E a catalyst, such as
a tri-C~-C4-alkyl ilyl trifluoromethanesulfonate or
the ~ilver ~alt of trifluoromethane6ulfonic acid, to
give a 7-0-glycosyl-rhDdomycinone of the formula I in
which the radical~ Rl, R2, R3, R6 and R7 retain the
meaning defined in formulae III, IV and V, then
spli~ting off the acyl protect.ive groups by alkaline
hydroly~i~, in the course of which a compound of the
formula I in whieh
R1 denotes H or Cl-C4-~lkyl,
RZ denote6 OH or COOCH3,
~5 ~3 denotes Cl-C4-alkyl,
R~ denotes H and
R5 denotes a glyco~yl radical of the formula II in
which
R6 i~ ~H2 or OH and
R7 is H or OH i8 fonmed,
and, if appropriate, reacting a re~ulting compound of the
formula I containing an amino-~ugar under the conditions
of reductive alkylation with a C~-C~-aldehyde or diglycol
aldehyde in the presence of an alkali metal cyano-
borohydride to ~ive a further compound of the formulil I
iLn which
.
... , .,. ~ . ,
.,: ';!. ..
, ~ .: .
';, ' -' ~ , :
': .. : ~, .: ,
Scheme 1
~f Et -~~~ OH
6 , ~ r 1 d I~CH z Cl ;~ 6 7
-RMN ~ 5 ~ _ O_
/~
./ Me 3 SiCJ
~e .-~ pyrldine ~1~
rle-Si-~e ~le-Si-Me
OHO O O R10 O
Mel/ I~a2CO~ 0H
OH O 9 o ~e~D~C~ 2CI 2rqeO O O O
~si-o-sj <~ ~Si-O-Si~
CH 2Cl 2~,/"'/ [~ ~3
q ~J " O H O H O O - p N O z
neo o o o Butq~llF~THF ~ ~ = Et
~Si--o--si~ 6 ~7
~ 1eO O HO OH
pNBzCI I R = H ¦ :
yr/C~I 2Ct z C ¦ R2 = pNBZ ¦
pNBz = p 02N-PtlCO-
%~
-- 7 --
the radicals R1, R2, R3, R4, R5 and R7 retain the last-
mentioned meaning and RB represents a mono-Cl-C4-alkyl-
amino or di-Cl C4-alkylamino group or 8 4-morpholinyl
group.
S Example6
The present invention is described in greater detail in
the following e~ampl~s, without being limited thereto.
~he ~tructure o the compounds described in the following
examples was determined by means of NMR and M~ analytical
chemistry. ~he progre~ of the reactivns and the chemical
purity of the compounds were investigated by thin layer
chromatography or by EPLC.
~xample 1
The introduction of silyl protective groups into a
rhodomycinone in po~itions 6,7 ~nd, if appropriate, 10.
6,7-0-(1,1,3,3-Tetraisopropyldisiloxane-1,3-diyl)-~-
rhodomycinone
~Compound 1)
10 g (25.8 mmol) of ~-rhodomycinone were di~solved in
300 ml of 1:1 pyridine/methylene dichloride, and 1202 ml
(1.5 equivalents) of 1,3-dichloro~1,1,3,3-tetrai~opropyl-
d~iloxane, dis olved in 150 ml of methylene dichloride,
were added at 0C with the exclusion of moisture. ~he
re~ction mixture wa~ ~tirred for 24 hour~ Bt xoom tem-
perature and then for a further 72 hour~ at 60C. After
the further addition of 6.5 ml (O.8 equivalent~) of 1,3-
dichloro-1,1,3,3-tetraisopropyldi~iloxane, the reaction
mixture was ~tirred for a further 72 hours ~t 60~Co 50 ml
of methanol were added to the reaction ~i~ture ~nd the
product wa~ ev~poratPd in vacuo and ~ubject~d to two
further di~tillation~ with toluene. The re~idue which
remained was purified by column chromatography t~ilica
gel; mobile phase: 20sl chloroform/ethyl acetate)~
,
' '~
- 8 -
Yield: 10.8 g (67%); meltin~ point. 115-117C
~alpha~D = ~425 (C = 0.2 in chloroform)
~H-NMR (90 ~Hz, CDCl3, delta~: 13.91 and 13.07 (~,PhOH),
7.82 (br d, H-l), 7.65 (t, H-2), 7.30 (br d, H-3), 5.28
S (br 6, H-7), 4.9S (8, H-10), 4.75 (6, 9-OH)~ 3.66-3.54
(m, alkyl-Si3
6,7-0-(1 t 1,3,3~Tetrai~opropyldi6iloxane-1,3-diyl)-ep~i-
lon-rhodomycinone
(Compound 2)
5 g (11.67 mmol) of epsilon-rhodomycinone a~d 9.2 g ~2~5
equivalents) of 1,3-dichloro-1,1,3,3-tetrai~opropyl
disiloxane were dissol~ed in 150 ml of 1:1 methylene
dichloride/pyTidine. After a reaction time of 8 day~ the
title compound wa~ i~olated a de~cribed in the in~truc-
tions for ~he preparation of compound 1.
Yield: 6.1 g (78~)
6 t 7-0-~1,1,3,3-Tetraisopropyldi6iloxane~1,3-diyl)-10-O-
trimethylsilyl-~-rhodomycinone
(Compound 3)
19.0 g (30.2 mmol) of compound 1 were di~solved in 250 ml
of methylene dichloride a~d 250 ml of pyridine, and 4.2 g
~1.5 equivalents) o~ chlorotrimethylsilane t di~olved in
50 ml of methylene dichloride, we:re added at 0C. The
reaction mixture was ~tirred for 1 hour at room tempera~
ture. The r~sction mixture wa~ then evaporated in vacuo
and sub~ected to two further distillation6 ~ith toluene.
The residue wa~ puri~ied by column chromatography (silica
gel; mobile phase: 7:3 methylene dichloride/petrol~um
ether),
Yield: 1~.3 g (77~); ~elting points 194-196C
(alpha)D - +530~ (c = 0.2 in chloroform)
H-NMR (300 ~Hz, CDCl3, delt~): 13.87 and 13.12 t~, phOH),
7.79 ~dd, ~-1), 7.61 (t, H-~), 7.25 (dd, H-3), 5.23 ~dd,
~-7), 2.13 (dd, H-8~ .06 (dd, H-8b), 4.86 (br ~, H-
10), 1.64 (m, H-13a and H-13b), 1002 (t, H-14), 0.2 and
1.2-1.3 (m, alkyl-Si)
-- 9 -
Example 2
Alkylation of a rhodomycinone in po~ition 4 or, if
appropriate, 11.
4-0-Methyl-6,7-0-(1,1,3,3-~etraisopropyldisiloxane 1,3-
diyl)-10-0-trimethylsilyl-~rhodomycinone
(Compound 4) and
4,11-Di-0 methyl-6,7~0-(1,1,3,3-tetraisopropyldi~iloxane-
1,3-diyl)-10-O-trimethyl6ilyl-~-rhodomycinone
(Compound 5)
10 Process a):
0.75 g (1.07 mmol) of compound 3 were di~solved in 30 ml
of acetone and 7 ml of methylene dichloride~ and 4.4 g
(30 equivalent~) of pota~sium carbonste and 10 ml ~150
equivalent~ Qf methyl io~ide were added. The reaction
15 mixture wa~ ~tirred for 3 day6 at room temperature, the
progres~ of the xeaction being followed by thin layer
chromatography. Methylene dichloride wa~ added to the
reaction mixture and the product was extracted by w~shing
succes~ively with water, 1 N hydrochlori~ acid and water.
20 The organic phase was dried over sodium sula~e and
evaporated in ~acuo. The ~ompounds 6 and 7, ~hich were
present in the residue, were ~eparated by column chroma-
tography over 35 g of ~ilica gel u~i.n~ methylene dichlor-
ide as the mobile phase.
25 Compound 4:
Yield: 0.46 y (60~); melting point: 213-214C
(alpha~D = ~472 ~c = 0.05 in chloro~orm)
lH-NMR (30G MHz, CDCl~, delta): 13.22 (8, PhOH), 7.89 (dd,
H-l), 7.65 ($, H-2), 7.29 (dd, H~3), 5.35 (ddt H-7), 2.17
30 (dd, H-8a), 2.07 (dt, H-8h), 4~89 (d, H 10), 1.39
(m, ~-13a~, 1.15 (m, H-13b), 1.05 (t, H-14)~ 3.96 (~,
OMe), 0.1 and 1.18-1.33 (m, alkyl-si)~ 4.45 ts~ 9-OH)
Compound 5s
Yield: 0.27 g (35%); melting point: 103-105C
35 (alpha)D = +124 (c = 0.05 in chloroform)
, :
2~ 37
, -- 10 ~
lH-NMR ~400 MHz, CDCl3, delta): 7.72 (dd, H-l~, 7.59 (t/
H-2), 7.21 (ddl H 3), 5.34 (dd, ~ 7), 2.17 (dd, H-8a),
2.05 (dt, H-8b), 4.70 (d, H-10), 1.40 (m, H-13a), 1.31
(m, H-13b), 1.04 ~t, H-14), 3.93 and 3.91 (~, OMe), 0.1
S and 0.9-1.3 (alkyl-Si), 4.50 (æ, 9-OH)
Pxocess b):
1 g (1.42 mmol) of compound 3 were dis601ved in 4.5 ml of
acetone and 18 ml of methylene dichloride, and 4.2 g
(12.9 mmol) of cesium carbonate and 5 ml (71.5 mmol) of
methyl iodide were added with ~tirring at 4C. After a
reaction time of 3 days the reaction mixture was worked
up as described above, compound 4 (yialds 0.76 ~ (75~))
and compound 5 ~yield: 0.1 g (10~)) being isolated.
4 O-Met~yl 6,7-0-(1,1,3,3-tetrai~opropyl-di~iloxsne-1,3-
diyl)-epsilon-rhodomycinone
(Compound 6)
The title compound was prepared by the instructions for
the preparation of compound 4, ~tarting ~rom 6.03 g
(9 mmol) of compound 2, 37.49 g (30 equi~alent.) of
potassium carbonate and 56.5 ml (~00 mmol) of methyl
iodide.
Yield: 3.2 g ~52~)
Example 3
Selective deblocking of the txialkyl~ilyl protective
g~oup
4-0 ~ethyl-6,7-0-(1 t 1,3,3-tetrai~opropyldi6iloxane-1,3-
diyl)-~-rhodomycinone
(Compound 7)
2 ~ (2.8 ~mol) of compound 4 were di~601ved ~n 100 ml of
~athylane dichloride and 100 ml of msthanol, &nd 20 ml of
0.1 N hydrochloric acid were added. After stirring ~or 30
minutes ~t room temperature, the reaction mixture was
diluted with methylene dichloride and extracted by
shaking with water. The organic pha e wa~ dried by maans
. : .
o~ ~
of sodium ~ulfate and evaporated in vacuo. The r~sidue
was also filtered ~hrough 50 g of silica g~l (mobile
phase: 20.1 methylene dichloride/acetone).
Yield: 1.7 ~ (96%); melting point: 166-168C
~alpha)D = +386~ (o - 0.05 in chloroform)
~xample 4 ;~
Introduction of acyl protective groups
4-0-~ethyl-10-0-p-nitrobenzoyl-6,7-0~(1,1,3,3-tetraiso-
propyldi~iloxane-1,3-diyl)-~-rhvdomycinone
(Compound 8)
0.68g (1.05 mmol~ of compound 7 wa~ di~olved in 40 ml of
2:1 chloroform/pyridine, and 0.3 g ~1.5 e~uivalents) of
p-nitrobenzoyl chloride was added. After stirring at 0C
for 15 houxs, 10 ml of methanol were added to the reac
tion mixture. The reaction mixture was evapora~ed in
vacuo and di~tilled again with toluene. The residue was
dissolved in chloroform and washed twice with water, and
the organic phase was dried over sodium sulfate and
evaporated in vacuo~ The crude product which remained was
purified fuxther by column chromatography over 120 g of ::.
silica gel (mobile pha~e: 17:10:1 petroleum ether/methyl-
ene dichloride/acetone).
Yield: 340 m~ ~81.1g); melting point~ 114-116C
(alpha)~ = ~384D (c = 0.05 in ~hloroform)
1H-NMR (300 ~Hz, CDCl3, delta): 13.02 1~, PhOH), 7.84 (dd,
H-l~, 7.~5 (t, H-2~, 7.32 (dd, H~3), 5.51 (dd, H-7~, 2.38
(dt, H-8a), 2.07 (dd, H-8b), 6056 ~d, H-10), 1.50
(m, H-13a), 1.83 (m~ H-13b)~ 1.07 (t, H-14~, 3.98 (8, 4-
OMe), 8.08 and 8.22 (m, aryl), 0.64-1.2 (m, alkyl-Si)
10,11-Bis-~-(p-nitrobenzoyl)-4-0-methyl-6,7-0-tl,1,3,3-
tetrai~opropyldisiloxane-1,3-diyl~ ~-rhodom~cinone
(Compound 9)
0.34 g (0052g mmol) of compound 7 wa~ reacted with 0.3 g
(3 eguivalent~ of p nitrobenzoyl chloride as described
~ .
,
, ~ , , "
8~
12 -
above to give the title compound.
Yi~ld: 0.35 ~72~)
Example 5
Deblocking the tetrai~opropyldisil~xane-1,3-diyl protec-
tive group
4-O-Methyl-10-O-p-nitrobenzoyl-~-rhodomycinone
(Compound 10~
0.6 g (0.76 mmol) of ~ompound 8 was di~solved in 30 ml of
THF, and 1 ml of a 1-molar solution in THE of tetrabutyl-
ammonium fluoride was add~d at 0C. After the reactionmixture had been ~tirred for 30 minutes, 30 ml of 0.1 N
hydrochloric acid were added and the mixture was extrac-
ted twice with chloroform. The or~anic pha~e wa~ dried
over ~odi~m sulfate and ~vapora~ed in vacuo. The crude
lS product ohtained was purified by chromatography over 65 q
of silica gel (mobile pha~e: 15:1 methylene dichloride~
acetone).
Yield: 0.3~8 g (91%); meltins point: 153-155C
(alpha)D = ~275 (c = 0.02 in chlorofonm)
10,11-gis_o_(p-nitxobenzoyl)~4-O-methyl-~-rhodomycinone
~Compound 11)
0.15 g (0.15~ mmol) of compound 9 W~15 reacted with 0.2 ml
of a l-~olar ~olution in ~HF of tetrabutylamonium fluor-
ide a~ de~cribed above to give the title compound.
Yields 95.8 mg (88%)
4-O-~ethyl epsilon-rhodomycinone
(Compound 12)
The title compound wa~ prepared by the in~truction~ for
the prep~ration of compound 10, starting from 3.2 g (4.6
mmol) of compound 6.
Yield. 1.6 g (83%~; ~AB m/e = 443 ~M~Ht)
,
:~ .
- ~3 -
Example 6
Formation of glycosides of the rhodomycins with function-
alized amino-~ugar~
4-0-Methyl-10-0-p-nitrobenzoyl-7-0-(4'-0-p-nitro~enzoyl~
3'-N-trifluoroacetyl-alpha-L-daunosaminyl)-~ rhodomycin-
one
(Compound 13)
1.84 ~ ~3.36 mmol) of compound 10, 3.64 g (6~72 mmol) of
1,4-di~O-p-nitrobenzoy' 3-N-~rifluoroacetyl-alpha,~
daunosamine and 5 g of mol~cular ~ieve 4 A were ~u~p~n-
ded, with the exclu6ion of moisture, in 300 ml of a
~olvent mixture composed of lOtl chloroform/acetone. The
reaction mixture wa~ cooled to -50C, 3 ml of trimethyl-
fiilyl trifluoromethane~ulfonate were added and ~tirring
was continued ~or 4 hours at -45C. The progre~ of the
reaction wa~ monitored by thin layer chromatography
(mobile phase; 20:1, methylene dichloride/acetone). The
reac~ion mi~ture wa~ neutraliz~d (pH 7) with triethyl-
amine and was filtered. ~fter the 801vent had been
evaporated the residue wa~ purified by column chroma-
tography ~6ilica ~el; mobile phase: 20:1 methylene
dichloride~acetone).
Yield: 2~3 g (74%)
1H-NMR, HjH-COSY t300 MHz, C6H6~CDCl3 1:1, delta): 7.60 ~d~
H-l), 7.05 (tl H-2), S.58 (d, H-3)~ 5.12 (dd~ H~7)~ 2.29
(d, ~-8a), 1.90 (dd, H-8b) ~ 6.57 (6, H-10), 1.81
(m, H 13a), 1.38 (m, H-13b), 0.99 tt, H-14), 13.93 and
13.17 (8, PhOH~, 3.41 (~, OMe), 3.SO (8, 9~0H), 7.28-7.72
(~, p-N02Bz~, 5.49 (d, H-l'), 1~69 (ddd, H-2'a), 1~85
~dd, H-2'b), ~.13 (m, H 3~, 5.06 (br ~, H~4'), 4~21 (q,
H-5'), 1.00 (d, H 6'), 6.12 (d, NH)
10,11-Bis-0 (p-nitrobenzoyl)-4-0-methyl-7-0-(4'-0-p-
~itrokenzoyl~3'-N-trifluoroacetyl-alpha-L-daunosaminyl)-
~-rhodomycinone
(Compound 14)
- 14 -
The ~itle compound was prepared by ~he glyoo6idation
process for the preparation of compound 13, starting from
the aglycone compound 11 and 1,4-di~O-p-nitroben~oyl-3,N-
trifluoroacetyl-alpha,~-L-dauno~amine.
4~0-Methyl-7-0-l4'-0-p~-nitrobenzoyl-3'-N-~rifluroacetyl-
alpha-L-dauhosaminyl)-epsilon-rhodomycinone
(Compound 153
The title compound was prepared by the glyco~idation
proces~ for the preparation of the compvund 13, starting
from 4-0-methyl-epsilon-rhodomycinone (compound 12) and
1,4-di-0-p-nitrobenzoyl-3,N~trifluoroacetyl-alpha,~-L-
dauno~amine.
~xampl~ 7
Deblocking r~actions
4~0-Methyl-10-0-p-nitrobenzoyl-7-0-(3'-N-trifluoroacetyl
alpha-L-daunosami~yl)-~-rhodomycinone
(Compound 16)
0.12 g (0.13 ~mol) of compound 13 wa6 di&solved in 2 ml
of lol me~hylene dichloride/methano:l, and 0.5 ml of 0.1 N
N~OH was added. The reaction mixture was stirred for 30
minutes at room temperature. The reaction wa~ Etopped by
adding O.5 ml of 0.1 N HCl. Chloro:Eorm wa~ added to the
mi~ture and it was then extracted by washing with water.
The organic pha~e wa~ dried over ~odium ~ulfate and
evaporated in vacuo and the r86 ? d~e which re~ul ed ~as
purified by column chrom~tography (6ilica gel; 6:1
methylene dichloride/acetone).
Yield$ 0.08 g (80~), melting point: 186-188C
(alpha)D = ~700 (c = 0.~ in chloroform)
7-0-(~lpha~L~Dauno~aminy~)-4-0-methyl-~-rhodo~ycinone
(Compound 17)
O.3 g (0.32 mmol) of compound 13 wa~ dis~olved in 10 ml
of 1:1 chloroform/methanol, and 3 ml of 1 N NaOH wer~
added with ~tirring. After 2 hour~ the reaction mixture
. - ~5 -
was neutralized with 3 ml of 1 N HCl, dilu~ed with
chloroform and butanol and extracted by washing with
water. The organic phase wa~ dried over sodium ~ulfate
and evaporated in vacuo. The residue wa~ purified over
RP-18 silica gel.
Yield, 0.14 g (83%); MSs F~B m/e = 530 (~H~)
7-O-(alpha-L-Daunosaminyl)-4-O-methyl-ep~ilon-rhodomycin-
one
(Compound 18)
The title compound was prepared by the process for the
preparation of compound 16, starting from compound 15.
Example 8
~odification of the rhodomycins at the 3'-amino group
7-0-(3'-N-DLmethyl-alpha-L-daunosaminyl)-4 O-methyl~-
lS rhodomycinon~
(Compound 19)
200 mg [0.37 mmol) of compound 17 were dissolved in 20 ml
of methanol, and 0.6 ml of 37~ ~;trength formaldehyde
solution wafi added with stirring. After 142 mg of ~odium
~yanoborohydride had ~san added, the reaction mixture was
stirred for a further 3 hours. The re~ulting ~rude
product ~as purified by column chromatography (silica
gel, 3s1 chloroform/methanol).
~ield: 161 mg (78~); MS: FAB m/e = 558 (M+~+)
4 O-~ethyl-7-0-(3'~(morpholin-4-yl)-2~3,6-tri-deoxy-
~lpha-~-ly~ohexopyrano~yl~ rhodomycinone
(Compound 20)
200 mg (0.37 mmol) of compound 17 were di~olved in 20 ml
of 10:1 methanol/chloroformO and 77.2 m~ of 2,2'-hydroxy-
bis-acetaldehyde which had been freshly pr~pared from
1~4-anhydroerythritol by periodate oxidation ~ere added.
After 25 mg of ~odiuM syanoborohydride h~d been ~ddedl
the rea~tion mixture lwas ~tirred for 2 hours at room
temperature and then evaporated in vacuo. The re3idue wa~
8~3
- 16 -
purified by column chro~atography over ~P-18 silica gel.
Yields 165 mg (74%); MS: PAB m/e = 600 (M+H'~
4-O-~ethyl-(3'-morpholin-4-yl)-2,3,6~trideoxy alpha-L-
lyxohexopyrano~yl)-ep~ilon-rhodomycinone
(Compound 21)
The title compound wa~ prepared by the proees~ for the ~:
preparation of compound 20, starting from compound 18 and
2,2'-hydroxybis-acetaldehyde.