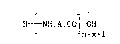Note: Descriptions are shown in the official language in which they were submitted.
2~ 3.'~08
-- 1 --
TRIALKYLSILYL ESTERS OF AMINO ACIDS AND THEIR USE
IN THE SYNTHESIS OF PEPTIDES
This invention relates to novel procedures for synthesising
peptides and to novel reagents for use in these procedures. The invention
also provides kits of reagents for use in carrying out the procedures of
the invention.
In recent years there has been an escalating need for synthetic
peptides in a wide variety of applications, including the development of
synthetic peptide vaccines, the detailed study of antigen-antibody
interactions, the preparation of analogues of biologically active
peptides, the optimisation of peptide antigens of clinical diagnostic
utility, the mapping of protein products of specific genes and the study
of conformational parameters. In a ma~ority of these studies, the
limiting factor has been the availability and cost of desired peptides.
Clearly, these studies would be greatly facilitated if synthetic methods
were available that would permit the synthesis of peptides rapidly and in
a cost efficient manner. Similarly for commercial exploitation, a
protocol suitable for large scale operation is required.
Peptides are linear polymers derived from amino acids and
generally have the formula
H2N.Al.Co.NH.A2.Co.NH.A3.Co.NH.A4.Co.An l.CO.NH.A COOH (I)
Where Al, A2, A3 ... An are the residues of the amino
acids making up the peptide.
Such amino acids may be represented by the formulae
H2N.Al.COOH, H2N.A2.COOH ... etc (II)
Peptides may additionally include sub-units derived from imino
acids (also termed "heterocyclic amino acids") such as, for example,
proline. These sub-units may be represented by the formula
.N<A.CO. (III)
wherein N<A represents a heterocyclic group.
ZC~3308
-- 2 --
The corresponding amino acids have the formula
HN<A.COOH (IV)
An important class of amino acids are the -amino acids that form
the sub-units of proteins. These amino acids, which in nature are of the
L-configuration, have been described as "naturally occurring amino
acids".
Essentially all proteins occurring in nature, whether from
unicellular prokaryotic microorganisms or from higher forms of life i.e.
eukaryotes, are constructed from the same set of twenty alpha-amino
acids. Nineteen of these amino acids can be depicted as having the
structure
H2NCHRCOOH (V)
where CHR may be referred to as the amino acid residue and R as the side
chain. In glycine, the only amino acid of the series which is not
optically active, R is hydrogen. Proline cannot be represented by
formula (V) as it is an imino acid of formula (IV) and its residue forms
part of a pyrrolidine ring.
Generally, the side chain R may be a hydrocarbyl group, for
example an an alkyl or aryl group, as in alanine, valine, leucine,
isoleucine, methionine and phenylalanine. The residue R may contain
polar, but non-ionizable groups, as in asparagine, glutamine, threonine
and serine, or ionizable groups, such as in aspartic acid, glutamic acid,
lysine, arginine, histidine, tyrosine, tryptophan and cysteine.
Peptides may be synthesised by two distinct routes, biological
and chemical. This invention is concerned with the second route.
Many naturally occuring pharmacologically active peptides or
protein hormones, including insulin, gastrin, oxytocin, vasopresin and
bradykinin have been synthesized by chemical means.
However a fundamental problem in the chemical synthesis of
peptides is that the groups which react to form the desired peptide bonds
are likely to enter into unwanted side reactions with other functional
groups. These other functional groups may be groups in the amino acid
reactants other than those forming the desired peptide bond or they may
be functional groups of reagents.
2C~)3308
- 3 --
Consequently, most known procedures for synthesising peptides
require the use of protective or blocking groups to prevent such
sensitive functional groups from reacting with the peptide-forming
reagents. Thus, the addition of each amino acid in the synthesis of a
peptide chain requires several steps to attach and then remove protective
or blocking groups, in addition to thè steps actually involved in forming
the peptide bond.
The expedient of utilizing blocking groups was first conceived by
Emil Fisher, and the classical approach for the synthesis of peptides of
defined structure was developed by Bergmann and coworkers. In the
Bergmann procedure, the amino group of the N-terminal amino acid
precursor is blocked or protected, typically by reaction with
benzyloxycarbonyl chloride in the presence of alkali (Schotten-Baumann
reaction):
(VI) (V)
6H5CH2c(NHcHRcooH ( 1 )
The next amino acid precursor is added and couples with the N-terminal
benzyloxycarbonyl-proteced amino acid precursor:
VII + H2NCHR'COOH C6H5CH20CONHCHRCONHCHR'COOH (2)
This sequence of steps is repeated to prepare tri- or higher
peptides. Since the procedure is effected in solution, it is necesary to
separate intermediate peptide precursors from by-products, unreacted
reagents, etc. In the last step, the terminal C6H5CH20CO blocking
group is cleaved by catalytic hydrogenation (toluene and carbon dioxide
are formed as by-products) or by use of HBr/acetic acid to yield the
desired peptide.
Such chemical methods of peptide synthesis need to take account
of three important factors. First in order to synthesise a desired
polypeptide, the synthesis must be conducted in a step-wise fashion with
desired amino acid residues attached to an incipient peptide chain
sequentially in the desired order.
Z(~)3308
-- 4 --
Secondly in order to form a peptide bond
-Al.co.NH.A2_ (IX)
by linking
-AlCOOH to NH2A ~ (X)
the carboxyl group .COOH needs to be converted to an active species .CO~
where ~ is a leaving group. In this active species, the carbonyl group is
activated and is susceptible to nucleophilic attack by the free amino
group of the entity NH2A2-.
Thirdly, any reactive groups (other than the -NH2 group and
-CO~ group) which might enter into undesired side reactions need to be
protected from attack by other active species.
Techniques have been established which take account of these
factors, the most important being the solid state methods devised by
Merrifield and Sheppard. In this regard solid-phase peptide synthesis has
been thoroughly reviewed by MerriPield (1969, 1973), Meienhofen (11973)
and Erickson and Merrifield (1976). Procedures described since then, with
varying points of emphasis, include those of Atherton (1979), Sheppard
(1977) and Marnett et aZ, (1976).
The key ideas and features of the solid phase principle are four-
fold:
1) The peptide is synthesised while it is covalently attached to
a polymeric support. This allows ready separation of the
product from by-products.
2) Reactions of the polymer-supported peptide chains can be
driven to completion through the use o~ excess reagents.
3) Mechanical losses are avoided by retaining the peptide polymer
beads in a single reaction vessel throughout the synthesis.
4) Physical manipulations are amenable to automation.
The Merrifield strategy utilises an N-protected amino acid
HOOC.A.NHProt (XI)
which is converted to an activated form
~.CO.A.NHProt (XII)
Z~3308
Traditionally tertiary butoxycarbonyl (tBoc) has been the
protective group of choice. In the Merrifield procedure, the initial
(C-terminal) amino acid NH2.An.COOH is covalently bonded to a resin
via its carboxyl group. This may be achieved by reacting a resin having
free -CH2Cl groups with a protected amino acid caesium salt
CsO.CO.An.NHProt (XII)
to form resin particles having the protected nth amino acid attached,
i.e.
Resin.CH20CO.AnNHProt (XIII)
In the next step (or ~cyclen) the protecting group Prot is
removed, leaving a free amino group
Resin.CH20.CO.An.NH2 (XIV)
and the resin particles, with the nth amino acid attached, are reacted
with the next (n-l)th activated protected amino acid
~.CO.An l.NHprot (XII')
which is to form the (n-l)th amino acid of the desired peptide, i.e.
Resin.CH OCO.An.NH I ~.COAn lNHProt -----------------------------9
2 (XIV) 2 (XII') n n-l
Resin.CH OCO.A .NHCOA NHProt.
2 (XV)
The sequence of steps is repeated until the desired resin-bound
peptide is formed. The peptide is then cleaved from the resin and
de-protected.
As indicated, in solid phase synthetic peptide chemistry,
tertiary butoxy oxycarbonyl (tBoc) has been the protective group of
choice. However, this protective group requires the use of strong acids
in both deprotection and final cleavage. Furthermore, such use of acidic
conditions has necessitated the use of acid-stable protecting moieties
(e.g. benzyl groups) for protecting the side chains of amino acids such
as the basic amino acids (Lys, His and Arg).
In more detail, in the solid phase procedure for the synthesis of
peptides developed by Merrifield, the peptide chain is built amino
acid-by-amino acid starting with the one intended to be the carboxyl or
C-terminal residue of the peptide chain.
2(~\330~3
- 6 -
~ This C-terminal amino acid is covalently bonded via its -COOH
group to an insoluble resin support. A chloromethylated polystyrene
resin is most often used because the benzyl ester group formed with the
C-teminal amino acid may be readily cleaved.
t-Boc-NHCHRCOOH ~ ClCH2-Resin
(XVI) (XVII)
- ~ t-Boc-NH ~ -Resin
(XVIII) ~
The next amino acid to be used in the peptide chain being formed,
after having its amino group blocked with a t-butyloxycarbonyl (t-Boc)
group, is activated with a coupling activator, such as dicyclohexyl-
dicarbondiimide (DCC), and coupled to the deprotected amino group of the
amino scid attached to the resin - (H2NCHRCOO-CH2-Resin XVIII')
(t-Bu-OCO)20 ~ H2NCHR'COOH
(XIX) (XX)
;~ t-Boc-NHCHR'COOH (IV)
(XXI)
DC~
XVIII' I XXI ;~ t-Boc-NHCHR'CONHCHRCOO-CH2-resin
(XXIII)
The t-Boc group is then removed by treatment with trifluoracetic
acid, and after neutralisation the growing peptide chain attached to the
resin is ready for addition of the next amino acid precursor. This
sequence is repeated until a peptide chain having the required structure
is synthesized. Having the growing peptide chain attached to resin
particles large enough to be separated from a liquid phase by filtration,
simplifies removal of excess reagents and washing of the resin particles
in the many repetitious steps involved, and makes the procedure more
canvenient forthe synthesis of larger peptides and proteins. When the
peptide chain is complete, it is cleaved from its resin support by a
reaction that does not affect the peptide linkages. Typically, hydrogen
fluoride is utilized for cleaving the peptide from the resin.
Recent years have seen the introduction of fluorenylmethoxy
carbonyl (Fmoc) as protective group for the activated protected amino
acid ~.COAnProt. The most significant difference in the Fmoc approach
is that the protecting group may be removed by base, typically using
piperidine.
933~)8
-- 7 --
However, in principle, there is a great similarity between the
Fmoc and tBoc approaches. Each cycle of the synthesis involvec
deprotection of the last attached amino acid residue followed by the
coupling of the next activated amino acid. In both Fmoc and tBoc
approaches, protected amino acid symmetrical anhydrides have generally
been favoured as the active species. N-hydroxybenzotriazole derivatives
and pentafluorophenyl esters of amino acids may also be used. The reason
for favouring symmetrical anhydrides is mainly due to their greater
reactivity compared to the corresponding amino acid active esters.
However the necessity to form Fmoc symmetrical anhydrides immediately
prior to use has prevented this activation protocol from being
implemented in automated instruments. This is also true of automated
peptide sythesis based on tBoc amino acids.
Available methods of peptide synthesis have a number of important
disadvantages which severely limit their applicability, particularly
in automated systems.
First, the tBoc and the Fmoc systems require the use of
respectively stringent acidic conditions and basic conditions in order to
remove the tBoc and Fmoc protective groups. Thus in the tBoc system,
strong acids such as trifluoroacetic acid have to be used and bases such
as, for example, piperidine in DMF, are required in the Fmoc system. The
use of trifluoroacetic acid in order to remove the tBoc protective group
is particularly troublesome as the repeated contacting of the resin
particles at each cycle with the trifluoroacetic acid reagent causes
degradation of the resin.
Secondly, in both the tBoc and Fmoc systems repeated washing
cycles are required to remove acidic and basic reagents used.
Thirdly the use of certain activated protected amino acids
(symmetrical anhydrides) prevents the ready application of the tBoc and
Fmoc procedures to automated systems. Such systems require the use of
reagents which are relatively stable because supplies of all the reagents
necessary to synthesise a desired peptide need to be stored for
relatively long periods. However, activated protected amino acids are
generally unstable and susceptible to decomposition in the presence of
even trace amounts of water and many commercially available activated
protected amino acids are prohibitively expensive.
Z~3308
-- 8 --
Fourthly the tBoc and Fmoc procedures require the use of an
excess of the activated protected amino acid rea8ent in each cycle in
order to ensure that the N-terminal amino acid residues of the
resin-bound peptide chain react to completion with said reagent. Thus,
for example, when a symmetrical anhydride is used as the activated
protected amino acid reagent, the reagent needs to be present in a mole
ratio of at least 4 moles of reagent per mole of N-terminal amino
residues. As each mole of reagent is formed of two moles of amino acid,
this represents a requirement for an 8x excess of amino acid starting
material.
The present invention overcomes the problems associated with
prior art solid state peptide synthesis procedures by binding the first
amino acid of the desired peptide to a resin via its amino group,
activating the carboxyl group of the thus-coupled amino acid and
utilising a carboxy-protected amino acid reagent to produce a peptide
bond by nucleophilic attack on the activated carboxy group of the bound
amino acid. Subsequent amino acids are coupled in a similar manner.
In the above procedure, reactive side chains of the carboxy-
protected amino acid may be protected (if necessary) by known
procedures.
The carboxy-protective groups utilised in accordance with the
invention are O-silyl ester groups. Apart from incidental exceptions
referred to below, amino acids having O-silyl ester protective groups are
novel and form a further aspect of the invention.
As indicated, certain silylated amino acids have been described
in recent years, particularly for the derivatisation of carboxy moieties
of N-acetyl amino acids for gas chromatography (Early et aZ, 1987).
Also in the 1960's Birkofer et aZ, Angew, Chem. 77, 414 (1965);
Kricheldorf et aZ, Liebigs Ann. Chem., 763, 17-38 (1972); R~hmann et
aZ., Liebigs Ann. Chem. 683, 211 (1965) describe trimethylchlorosilane
derivatives of amino acids and peptides. In a typical experiment
(Birkofer 1960, for example), an amino acid was heated at 130-140 c in
excess of hexamethyldisilane containing a few drops of conc. H2S04
until a solution was obtained. It was then cooled and benzene added
followed by triethylamine and trimethylsilyl chloride and the mixture
kept at room temperature for 12-16 hours.
XC~3308
g
The solid obtained was filtered off and the filtrate evaporated
and distilled under reduced pressure to give the desired N-trimethyl
silyl amino acid trimethyl silyl ester.
Furthermore, Barlos e~ at, J. Org. Chem. 47, 1324 (1982)
postulated formation of trimethyl-silyl esters of amino acids as
intermediates in the synthesis of N-trityl amino acids. However, no
definitive evidence of the existence, preparation or isolation of such
structural intermediates was presented.
As indicated, the present invention, in its process aspects, i5
based on the realisation that distinct advantages ensue from the
synthesis of peptides by a procedure in which an initial amino acid
forming the N-terminus of the desired peptide forms the starting point
for the step-wise synthetic route, the carboxyl group of this amino acid
is activated and the activated carboxy group is reacted with the free
amino group of a carboxy-protected amino acid.
20~3~08
- 10 -
According to one aspect thereof, the present invention provides
a process for producing a peptide of the formula
H ~ NH.A.C ~ H (XXIV)
n+xll
which comprises the steps of
(A) reacting a solid phase reactant comprising a solid support-
bound amino acid or a solid support-bound peptide, said
solid phase reactant having the formula
~ NH.A.CO ~ OH (XXV)
with a carboxyl group activating agent to form an activated
solid phase reactant of the formula
~ NH.A.C ~ ~ (XXVI)
(B) reacting the activated solid phase reactant from step (A)
,`r with a carboxy-protected amino acid of formula
H.NH.A.CO.Prot (XXVII)
to form a chain-extended product of formula
~ NH.A.CO~ OiProt (XXVIII)
(C) removing said protective group Prot,
(D) optionally repeating said steps (A), (B) and (C) x times,
~, .
2C~3308
and (E) cleaving the resulting peptide of formula
H ~ NH.A.CO~ OH (XXIX)
nlx~l
from the support
wherein n is a positive integer,
x is O or a positive integer,
~ is a leaving group,
Prot is a silyl group,
each A, which may be the same or different, either represents the
residue of an amino acid or the structure NH.A is the residue
N<A of an imino acid HN<ACOOH
and wherein any reactive side chains on residues A are protected and are
subsequently deprotected and said steps (A) and (B) are carried out
successively or in a single operation.
When carried out in a single operation, the process of the
invention comprises the following steps:
(a) reacting a solid phase reactant comprising a solid support-
bound amino acid or a solid support-bound peptide, said
solid phase reactant having the formula
~ NH.A.CO~ OH (XXV)
simultaneously with a carboxyl group activating agent
and with a carboxy-protected amino acid of formula
/
H.NH.A.CO.Prot (XXVII)
to form a chain-extended product of formula
~ NH.A.C ~ iProt (XXVIII)
(b) removing said protective group Prot,
(c) optionally repeating said steps (A), (B) and (C) x times,
)8
- 12 -
and (d) cleaving from the support the resulting peptide of formula
H ~ NH.A.CO~ OH (XXIX)
n~x l 1
n, x, ~, Prot and A being as previously defined and wherein any resctive
side chains on residue A are protected and are subsequently deprotected.
Preferably said solid phase reactant
~ NH.A.C ~ OH (XXX)
wherein n is 1, is produced by reacting a carboxy protected amino acid of
formula
H.NH.A.CO.OProt
with a support material having groups capable of forming a covalent bond
with the amino or imino group of the carboxyprotected amino acid, and
removing said protective group Prot.
In the above reaction schemes, the amino acid residues A may be
derived from a wide range of amino acids, which is not restricted to the
amino acids commonly occuring in natural proteins.
Such amino acids may be represented by the general formula
H.N ~ COOH
wherein N ~ represents either the group .NHB wherein B represents a
saturated or unsaturated Cl 10 hydrocarbyl group, optionally
substituted by one or more substituents selected from hydroxy, oxo, thio,
Cl 4alkylthio, Cl 4alkoxy, carboxy, acetamido or -NR4R5 (wherein
R4 and R5, which may be the same or different represent C1 4 alkyl
groups), or N~ represent~ a heterocyclic ring containing 4-7 ring
atoms selected from N, C and O.
2C~3308
- 13 -
Thus the invention includes the production of peptides derivedfrom amino acids in one or more of the following classes
(i) the twenty L-~-amino acids glycine, alanine, valine, leucine,
isoleucine, serine, threonine, aspartic acid, asparagine,
glutamic acid, glutamine,llysine, histidine, arginine,
phenylalanine, tyrosine, tryptophan, cysteine, methionine and
proline,
(ii) D-analogues of amino acids in class (i),
(iii) dehydro derivatives of amino acids in class (i),
(iv) amino acids selected from the following:
hydroxylysine, thyroxine, hydroxyproline, ~-alanine,
y -aminobutyric acid, homoserine and statine.
In general, where the amino acid is an ~-amino acid of formula
H2N.CHR.COOH, R may be defined as being selected from Cl 10
hydrocarbyl groups optionally substituted by one or more substituents
selected from hydroxy, thio, Cl 4 alkylthio, Cl 4 alkoxy, carboxy,
acetamido, guandidyl, 3-indolyl and 2-imidazolyl.
The aforementioned hydrocarbyl groups include branched and
straight chain alkyl groups (which preferably contain from 1 to 6 carbon
atoms) and aryl groups, particularly phenyl groups, which may be
unsubstituted or substituted by one or more moieties selected from Cl 4
alkoxy, nitro and halogen.
The groups of the support which are capable of forming covalent
bonds with the amino or imino groups of the carboxy protected amino acid
are preferably activated carboxy groups of formula -C0~ or activated
oxycarbonyl groups of formula -OC0~ (~ is as defined above).
Examples of suitable silyl groups are those having the formula
Si(Rl,R2,R3) (XXXI)
wherein Rl, R2 and R3, which may be the same or different represent
saturated or unsaturated hydrocarbyl groups containing 1 to 20 carbon
atoms, which may be unsubstituted or substituted by one or more groups
selected from C4-alkoxy, nitro, tri(Cl 4alkyl)silyl and halogen.
The aforementioned hydrocarbyl groups include branched and
straight chain alkyl and alkenyl groups (which preferably contain from 1
to 6 carbon atoms) and aryl groups, particularly phenyl groups.
20~)3~08
- 14 -
Preferably said silyl groups have the formula
Si(Rl,R2,R3) (XXXI)
wherein Rl, R2 and R3, which may be the same or different represent
Cl 20 alkyl groups. The alkyl groups may be straight and branched (and
a combination of both may be present).; Examples include methyl, ethyl,
n-propyl, n-butyl, sec-butyl, t-butyl and dodecyl
The carboxy protected amino acids XXVII may be formed from the
corresponding amino acid or protected amino acid by converting a free
carboxyl group to a silyl ester group using known silylating agents. Such
agents may generally have the structure X-Si(RlR2R3) where Rl,
R2 and R3 are as defined above and X is a leaving group.
Examples of suitable leaving groups X include
(i) Cl, Br and I (as in tri-ethyIsilyl chloride)
(ii) alkoxy, e.g. ethoxy and methoxy (as in trimethylethoxy
silane)
(iii) secondary amino, for example di-Cl 4-alkylamino. A typical
secondary amino group is dimethylamino (as in
N-trimethylsilyldimethyl amine)
(iv) disilazano (as in hexamethyl disilazane)
The leaving group ~ should be selected according to known
criteria such that the carbon atom to which it is attached is activated
sufficiently that it can undergo nucleophilic attack by a lone electron
pair on the free amino or imino group of the carboxy protected amino or
imino acid. Preferably group ~ should be what is termed ~good leaving
group".
Particularly effective leaving groups ~ are strongly electron
withdrawing.
Also it is desirable that the reaction(s) which introduce leaving
group ~ minimise racemisation and that also the activated intermediate
containing ~ should not be susceptible to racemisation.
Examples of leaving groups ~ are groups having the structure
Q~ 6 (a)
where R5 and R which may be the same or different represent
Cl 10 hydrocarbyl groups. In a well studied example of such groups R5
and R are both cyclohexyl.
ZC~3308
- 15 -
Other leaving OEoups ~ include pentafluorophenoxy, i.e.
F F
~ (b)
and the group F F
--N~l (C)
Compounds having leaving groups (a) may be produced by reacting acompound having a carboxyl-function with a corresponding carbodiimide.
Compounds having leaving groups (b) and (c) may be made from
compounds containing leaving groups (a) by reaction with
pentafluorophenol and l-hydroxybenzotriazole.
In other words, the groups -CO~ represent carboxyl groups
activated by known procedures, for example by reaction with a diimide
such as for example, dicyclohexylcarbodiimide, by formation of a
hydroxybenzotriazole ester or pentafluorophenyl ester~or by using
benzotriazole-l-yl-oxy-tris-(dimethyl-amino)phosphonium
hexafluorophosphate.
As indicated carboxyl activation and addition of the
carboxy-protected amino acid may be carried out successively or
simultaneously. Thus:
(i) the carboxyl OEoup can be activated prior to addition of
protected amino acid, e.g. carboxyl activated by formation of the
hydroxybenzotriazole ester or pentafluorophenyl ester. The
"activated" resin is then washed and the protected amino acid
then added or
(ii) the "activation" step (A) and the "addition of amino acid
step" - Step (B) can be achieved in one step using e.g.
benzotriazole-l-yl-oxy-tris(dimethyl-amino)phosphonium
hexafluorophosphate (known as BOP or Castro's reagent), base and
protected amino acid (stoichimetry 1:2:1).
2~3308
16 -
The invention may also be used to synthesise peptides in the
liquid phase, thus according to a further aspect of the invention there
is provided a process for producing a peptide of the formula
H ~ NH.A.C ~ OH (XXIX)
n+x+1
which comprises the steps of
(A) reacting a carboxy-activated reactant comprising an NH2-
protected amino acid or N terminal NH2-protected peptide,
said carboxy-activated reacting having the formula
Prot' ~ NH.A.CO~ ~ (XXXII)
wherein Prot' represents an -NH2 protecting group with a
carboxy protected amino acid of formula
H.NH.A.CO.OProt
wherein Prot is a silyl group to form a chain-extended
product of formula
Prot' ~ NH.A.CO~ OiProt (XXXIII)
(B) removing said protecting group Prot
(C) optionally repeating steps (A) and (B) x times and
i~
X(~13308
- 17 -
(D) removing the -NH2 protecting groups Prot'
wherein n is a positive integer,
x is O or a positive integer,
~ is an leaving group,
each A, which may be the same or different, either represents the
residue of an amino acid or the structure NH.A is the residue
N<A of an imino acid HN<ACOOH,
and wherein any reactive side chains on residues A are protected and are
subsequently deprotected. The protective groups Prot' are preferably
t-butoxycarbonyl (tBoc), fluorenylmethoxycarbonyl (Fmoc) or
triphenylmethyl (trityl or Trt) groups.
In the above procedures, the silyl groups are preferably of the
formulae specified above, i.e. of the formula
Si(Rl,R2,R3) (XXXIII)
wherein R1, R2 and R3, which may be the same or different represent
hydrocarbyl groups containing 1 to 10 carbon atoms, preferably of the
formula
Si(R1,R2,R3) (XXXIII)
wherein R1, R2 and R3, which may be the same or different represent
Cl_4 alkyl groups.
Certain of the silyl esters suitable for use in accordance with
the process aspects of the invention are novel. These include the
following:
(A) silyl esters of an L-amino acids selected from
L-valine, L-isoleucine, L-serine, L-threonine, L-aspartic acid,
r L-asparagine, L-lysine and L-methionine, said ester being substantially
free of the corresponding D-isomer;
X(~3308
- 18 -
(B) silyl esters of a protected amino acid being:
(a) an 0-protected amino acid selected from 0-protected
serine, 0-protected threonine and 0-protected tyrosine.
(b) a ~- and r-carboxy-protected amino acid selected from
~-carboxy protected glutamic acid and ~-carboxy protected
aspartic acid;
(c) ~-amino protected lysine and guanino-protected arginine;
(d) (indo)-protected tryptophan (n(indo)-protected" indicates
protection of the indole nitrogen)
(e) thio-protected cysteine; and
(f) imidazole-protected histidine;
Examples of classes (d), (e) and (f) include
N-formyl tryptophan, 4-methoxy benzyl cysteine, and dinitrophenyl
histidine.
(C) silyl esters of an a-NH2-protected amino acids selected
from a-amino protected glycine, a-amino protected alanine, a-amino
protected valine, a-amino protected leucine, a-amino protected
isoleucine, a-amino protected serine, c-amino protected threcnine,
a-amino protected aspartic acid, a-amino protected asparagine, a-amino
protected glutamic acid, a-amino protected glutamine, a-amlno protected
lysine, a-amino protected histidine, a-amino protected arginine, a-amino
protected phenylalanine, a-amino protected tyrosine, a-amino protected
tryptophan, a-amino protected cysteine, a-amino protected methionine and
a-amino protec~ed proline, characterised in that said a-NH2-protected
group is other than a silyl group; and
(D) trialkyl silyl esters of amino acids of the formula
H.NH.A.C0ØSi(RlR2R3) (XXXIV)
wherein Rl, R2 and R3, which may be the same or different represent
alkyl groups having 1-20 carbon atoms, and H.NH.A.C0.0 is the residue of
an a-amino acid selected from glycine, alanine, valine, leucine,
isoleucine, serine, threonlne, aspartic acid, asparagine, glutamic acid,
glutamine, lysine, histidine, arginine, phenylalanine, tyrosine,
tryptophan,cysteine, methionine and proline, with the proviso that where
the amino acid is other than lysine, aspartic acid or glutamic acid, at
least one of Rl, R2 and R3 is other than methyl.
Z~330~3
- 19 -
Where the novel silyl esters of the invention posess protective
groups of the amino function such groups are preferably t-butoxycarbonyl
(tBoc) groups, fluorenylmethoxycarbonyl (Fmoc) groups or triphenylmethyl
(Trt) groups.
In classes (B) and (C) above,' the amino-protecting groups are
preferably t-butoxycarbonyl, fluorenylmethoxycarbonyl or triphenylmethyl
groups.
Further aspects of the invention include (a) the use of silyl
esters of amino acids in peptide synthetic procedures and (b) kits
comprising such esters, particularly kits of reagents for use in the
synthesis of peptides comprising supplies of silyl esters of amino acids,
and a supply of a reagent for activating carboxyl groups to convert them
to activated carboxyl groups capable of reacting with amino groups to
form peptide bonds.
The solid phase procedure for the synthesis of a peptide of
predetermined structure by the process of the present invention typically
involves the following steps:
(a) covalently bonding a trialkylsilyl ester of an alpha-amino acid
to an insoluble resin via the amino group of the amino acid;
(b) cleaving the trialkylsilyl ester;
(c) activating the carboxyllc acid group formed by cleavage of the
trialkylsilyl ester;
(d) coupling a trialkylsilyl ester of an alpha-amino acid via its
amino group to the activated carboxylic acid group;
A (e) repeating steps (b), (c), and (d) in sequence until a peptide chain
of the predetermined structure has been formed; and
(f) cleaving the peptide chain from the resin, and cleaving the
trialkylsilyl ester.
Although the process aspect of the invention described above is
directed to solid state synthesis of peptides utilising a solid phase
reactant comprising a support, use of the preferred carboxyl protective
groups (silyl groups) in liquid phase systems is possible.
2(~3308
- 20 -
The liquid phase procedure for the synthesis of a peptide of
predetermined structure by the process of the present invention typically
involves the following steps:
(a) activating the carboxylic acid'group of an N-protected
alpha-amino acid;
(b) coupling the amino OEoup of a trialkylsilyl ester of an
alpha-amino acid to the activated carboxylic acid group of the
N-protected amino acid;
(c) cleaving the trialkylsilyl ester;
(d) activating the carboxylic acid group resulting from cleavage of
the trialkylsilyl ester;
(e) coupling the amino group of a trialkylsilyl ester of an
alpha-amino acid to the activated carboxylic acid group;
(f) repeating steps (c), (d), and (e) in sequence until a peptide
chain of the predetermined structure has been formed and
(g) cleaving the trialkylsilyl ester group and the N-protecting
OEoup.
Both the liquid phase and the solid phase procedures for the
synthesis of peptides of the present invention are more convenient than
the prior art procedures. The overall synthesis requires less time,
requires less steps and uses lesser amounts of milder reactants under
milder reaction conditions.
/
- 21 -
General Methods of Preparation
Trialkylsilyl esters of amino acids are most conveniently
prepared according to the invention by the reaction of a trialkylsilyl
chloride with an N-blocked amino acid.l Many N-blocked amino acids may be
obtained commercially, however in general they may be prepared in the
well known manner from the corresponding amino acid. Thus, for example a
t-Boc amino acid may be produced by reacting an amino acid with
di-t-butyldicarbonate.
The blocking group may be any conventional amine blocking group
such as t-butoxycarbonyl, fluorenylmethoxycarbonyl, phenylacetyl,
acetoacetyl, N-benzylidine, benzoyl, benzyl, t-amyloxycarbonyl,
benzyloxycarbonyl, p-toluenesulfonyl, choroacetyl, carbamyl
triphenylmethyl and the like, which can be readily cleaved, at the
appropriate time, by acid or base, by hydrogenation or by enzymatic
action.
As indicated, in preparing the silyl esters of amino acids in
accordance with the invention, it i~ necessary first to block the amino
group or groups. Preferably this is achieved by introducing a
t-butoxycarbonyl (t-Boc) group by reaction with di-t-butyl dicarbonate.
An appropriate procedure is to dissolve the amino acid in aqueous
dimethyl formamide or dioxan (2:1 vol. ratio of solvent:water) and add an
equivalent amount of triethylamine (one mole/carboxyl group).
Then more than one equivalent of di-t-butyl dicarbonate is added
with stirring for 1/2 - 2 hours at room temperature. The same volume of
water is added and the mixture extracted with diethyl ether to remove
unreacted reagent.
The reaction mixture is then acidified to pH 3 with citric acid
or lN HCl and extracted 2 x with ethyl acetate. The organic phase is then
evaporated to isolate the t-Boc-protected amino acid.
For amino acids with more than one amino groups the individual
amino groups generally require different blocking groups so that only the
amino group destined to form the peptide bond is subsequently
de-blocked.
2C~3308
- 22 -
Thus for lysine, the ~ -amino group is preferably blocked with a
benzyloxycarbonyl group prior to blocking the a-amino OE oup with t-Boc.
This may be achieved by forming a copper complex with the a-NH2 and
COOH groups, reacting with benzyloxycarbonyl chloride (Z-chloride) and
then decomplexing and reacting with t-butyl dicarbonate as described
above.
A similar procedure may be used for arginine, except that the
guanidino group is blocked with a p-toluene sulphonyl group.
Amino acids having additional carboxyl groups are preferably in
the form of their benzyl esters.
The hydroxyl groups of serine and threonine can be blocked, if
necessary, by formation of benzyl esters and the hydroxyl group of
tyrosine by formation of the 2-bromobenzyloxycarbonyl derivatives.
Similarly, the indoly] nitrogen of tryptophan may be blocked by
formylation and the thio group of cysteine by 4-methylbenzylation.
Histidine may be protected as a dinitrophenyl derivative.
These methods of blocking the a-amino groups and reactive
functional groups are standard and most of the protected amino acids are
available commercially.
The reaction between the N-protected amino acid and the
trialkylsilyl chloride, preferably trimethylsilyl chloride or
t-butyldimethyl silyl chloride, is effected in an inert solvent,
preferably an aprotic solvent such an ether, for example diethyl ether or
tetrahydrofuran or in dimethyl formamide. The solvent should be
carefully dried prior to use.
Most conveniently, the trialkylsilyl chloride, neat or dissolved
in the solvent, is added dropwise over a period of time to a solution of
the N-t-Boc blocked amino acid in a reaction vessel chilled in a cold
water or water-ice bath. The reaction medium preferably contains a
tertiary amine, e.g. triethyl amine, pyridine or imidazole, to serve as a
scavenger for the hydrogen chloride formed during the course of the
reaction.
After the trialkylsilyl chloride has been added, the reaction
mixture is generally allowed to stand for about an hour to permit the
reaction to reach completion. If the reaction mixture had been cooled,
it may at this stage be permitted to warm to room temperature.
ZC~3308
- 23 -
The solid tertiary amine hydrochloride, formed during the course
of the reaction, may then be separated, most conveniently by filtration.
Removal of the solvent from the filtrate under reduced pressure gives an
almost theoretical yield of highly pure trialkylsilyl ester of the
N-protected amino acid.
The silylation reaction is preferably carried out in an aprotic
solvent in the presence of one equivalent of base (triethylamine,
pyridine or imidazole). For the preparation of the trimethylsilyl esters,
diethyl ether is the preferred solvent. Dimethylformamide is preferred
for production of the t-butyldimethylsilyl esters. The reaction is
generally complete in one hour at room temperature for the trimethylsilyl
esters and in 2-8 hours for the t-butyldimethyl silyl esters.
In the case where the N-blocking group is t-butoxycarbonyl, to
prepare the N-unblocked trialkylsilyl ester, the intermediate N-t-Boc
blocked ester may be dissolved in dry ether and dry hydrogen chloride
passed through the solution, preferably chilled to about 0-5 C, for about
30 minutes. The product can be isolated either by filtration or by
evaporation of the solvent.
~3~0~
- 24 -
Using the procedure decribed above, the following trialkylsilyl
esters of L-a-amino acids were prepared;
Tyr (2-Br-Z) - trimethylsilyl ester
Ala - trimethylsilyl ester
Phe - trimethylsilyl ester
Leu - trimethylsilyl ester
Pro - trimethylsilyl ester
Lys (2-Cl-Z) - trimethylsilyl ester
Gly - trimethylsilyl ester
Met - trimethylsilyl ester
Arg(Tos) - trimethylsilyl ester
Glu(OBz) - trimethylsilyl ester
Ile - trimethylsilyl ester
Ser(Bzl) - trimethylsilyl ester
Thr(Bzl) - trimethylsilyl ester
Trp(Formyl) - trimethylsilyl ester
Gln - trimethylsilvl ester
Asn - trimethylsilyl ester
Cys(4-Methyl Bzl) - trimethylsilyl ester
Asp(OBzl) - trimethylsilyl ester
Val - trimethylsilyl ester
His(DNP) - trimethylsilyl ester
from
Z~93308
- 25 -
t-Boc - Tyr(2-Br-Z) - trimethylsilyl ester
t-Boc - Ala - trimethylsilyl ester
t-Boc - Phe - trimethylsilyl ester
t-Boc - Leu r trimethylsilyl ester
t-Boc - Pro - trimethylsilyl ester
t-Boc - Lys (2-Cl-Z) - trimethylsilyl ester
t-Boc - Gly - trimethylsilyl ester
t-Boc - Met - trimethylsilyl ester
t-Boc - Arg(Tos) - trimethylsilyl ester
t-Boc - Glu(OBz) - trimethylsilyl ester
t-Boc - Ile - trimethylsilyl ester
t -Boc - Ser(Bzl) - trimethylsilyl ester
t-Boc - Thr(Bzl) - trimethylsilyl ester
t-Boc- Trp(Formyl) - trimethylsilyl ester
t-Boc - Gln - trimethylsilyl ester
t-Boc - Asn - trimethylsilyl ester
t-Boc - Cys(4-Methyl Bzl) - trimethylsilyl ester
t-Boc - Asp(OBzl) - trimethylsilyl ester
t-Boc - Val - trimethylsilyl ester
t-Boc - His(DNP) - trimethylsilyl ester
Trp(Formyl), His(DNP) Arg(Tos) and Lys(2-Cl-Z) trimethyl silyl
esters were found to be unstable on prolonged storage at room temperature
due to their hygroscopic nature.
This problem was however circumvented by substituting a more
stable t-butyldimethylsilyl moiety in place of trimethylsilyl group.
N-t-Boc and Trt protected t-butyldimethyl silyl esters of alpha amino
acids were prepared in N,N-dimethylformamide using imidazole as a base.
Using this methodology the esters specified in Table 3 (below)
were prepared.
To prepare a peptide by the liquid phase procedure of the present
invention (Scheme 1), the carboxylic acid group of an N-blocked amino
acid is activated for coupling by treatment with an activator such as
dicyclohexylcarbodiimide.
XC~3308
- 26 -
Then a trialkylsilyl ester oP the next amino acid in the peptide
chain being formed is added. The amino group of the amino acid moiety of
the trialkylsilyl ester then couples with the activated carboxylic acid
group of the N-blocked amino acid in solution.
The trialkylsilyl ester of the~intermediate peptide precursor is
readily cleaved with methanol or methanol/acetic acid and the liberated
carboxylic acid group activated for coupling with a conventional
activator such as DCC/HOBT. This sequence is repeated until a peptide
chain of the required structure is formed. Then, the trialkylsilyl ester
at the C-terminal end of the chain is cleaved and the N-blocking group
removed with hydrochloric acid/diethyl ether to liberate the peptide. In
order to prepare a pure peptide product, it is generally necessary to
isolate and/or separate unreacted reagents from one or more of the
intermediate peptide precursors produced during the course of the
synthesis.
Tyr-Ala-Ala-Phe-Leu-OH and Ala-Ala-OH were prepared using the
liquid phase procedure described above.
In the solid phase process of the present invention unlike that
of the Merrifield procedure wherein an N-tBoc blocked amino acid is
coupled via its carboxylic acid group to the resin, the trialkysilyl
ester of the initial amino acid in the peptide chain to be formed is
bound via its amino group to the insoluble resin. Most conveniently, the
resin is a polystyrene resin bearing a -CH20COCl substituent, e.g.
(Alk)3SiOOCCHRNH2 + ClOCOCH2-Resin
(Alk)3SiOOCCHRNHOCOCH2-Resin
The carboxyl group is then deprotected by cleaving the trialkylsilyl
ester with methanol or methanol/acetic acid, and the thus liberated
carboxy group activated with DCC/HOBT. A trialkylsilyl ester of a second
amino acid is added, snd the amino group of the second amino acid couples
with the activated carboxyi group of the first amino acid, which remains
bound to the resin.
2(~)3308
- 27 -
This sequence of reactions is repeated until a peptide chain of
the desired structure is formed. Finally, the last trialkylsilyl ester
is cleaved with methanol or methanol/acetic acid, and the peptide is
liberated from the resin under acidic conditions e.g. B r/acetic acid,
trifluoromethanesulphonic acid (TFMSA) in trifluoracetic acid (TFA) and
HF.
When preparing peptides using the process of the present
invention, lesser amounts of reagents are required in each of the cycles
in which an amino acid residue is added to the peptide chain, and the
cost of reagents and the overall time required to complete the synthesis
are reduced.
The reagents and reaction condition utilized are milder than
those customarily utilized (in the present process, methanol may be used
to cleave the protecting group; (c.f. the Merrifield procedure:
trifluoroacetic acid is used to remove the N-t-Boc blocking group and
Sheppard's Fmoc system: the base piperidine is used to remove the
N-terminal blocking group).
Accordingly, there is less damage to the resin matrix in solid
phase procedures or risk of adverse reactions with the constituent amino
acid in the peptide being formed. The reaction of trialkylsilyl chloride
with amino acid provides stable C-blocked amino acids, convenient to use
as intermediates in the synthesis of peptides, and the blocking group is
readily removable when the desired peptide chain has been prepared.
A further advantage of the method of the invention is that it is
possible to test for completion of the individual reaction stages by
measuring the conductivity of the reaction medium.
. .
Z(~308
- 28 -
The following examples illustrate the invention
EXAMPLE 1
Preparation of t-Butyloxycarbonyl (tBOC) Amino Acid
Trimethylsilyl Esters
li
1.1 t-~oc-L-Alanine-Trimethylsilyl Ester
The trimethylsilyl ester of L-alanine was prepared by the
following procedure.
N-t-Boc-L-alanine (10 m.moles) was dissolved in anhydrous diethyl
ether (40 ml) and pyridine (10 m.moles) was added. The solution was
cooled to O C on an ice bath and stirred.
Trimethylsilyl chloride (11 m.moles) was then added dropwise.
Immediately a white precipitate began to form (pyridine-HCl). The mixture
was stirred for 1 hour and then allowed to warm to room temperature. The
white solid was removed by filtration and the organic layer was
evaporated under reduced pressure to give N-t-Boc-L-alaninetrimethylsilyl
ester as a liquid in a yield of 94%, which was stored under nitrogen.
All operations were carried out under anhydrous conditions.
The product was characterised by its infra-red (IR) spectrum,
H Nuclear Magnetic Resonance (NMR) spectrum and by thin layer
chromatography (tlc) analysis. The H NMR spectrum were recorded on a
R-1500 Hitachi (60 MHz) instrument using tetremethylsilane (TMS) as the
internal standard).
j The Infra-red spectrum showed bands at max (thin film):
r 760(st.), 840(st.), 1695(st.), 1740(st.) and 3400 cm 1.
(st. refers to a strong band).
The lH NMR spectrum (CDC13) showed the following ô values:
ô 0.25 (s., 9H), 1.45 (s, 9H), 4.0-4.25 (t., lH)
and 5.2 (br., lH).
Thin layer chromatography on a silica plate using 70% diethyl
ether-light petroleum (60-80 C.) as solvent gave a Rf value of 0.31
2~3308
- 29 -
1.2-20 Trimethylsilyl Ester~ of (2~ t-Boc Glycine,
(3) t-Boc L-Valine, (4) t-Boc L-Phenylalanine,
(5) t-Boc L-I~oleucine, (6) T-Boc L-Leucine,
(7) t-Boc (2-C1-Z)-L-Lysine, (8) t~Boc L-Methionine,
(9) t-Boc L-Asparagine, (10) t-Boc L-Proline,
(11) t-Boc (4-MBzl)-L-Cysteine,
(12) t-Boc (2-Br-Z)-L-Tyrosine,
(13) t-Boc (Bzl)-L-Threonine,
(14) t-Boc (0-Bzl)-L-Glutamic acid,
(15) t-Boc (0-Bzl)-L-Aspartic acid,
(16) t-Boc L-Glutamine, (17) t-Boc (Tos)-L-Arginine,
(18) t-Boc (Formyl)-L-Tryptophan,
(19) t-Boc (DNP)-L-~istidine and
(20) t-Boc (Bzl)-L-Serine
Trimethylsilyl esters of the title amino acids and protected
amino acids were prepared by the procedure described in Example 1.1
above, but substituting the following amino acids and protected amino
acids for N-t-Boc-L-alanine: t-Boc-glycine, L-valine, L-phenylanine,
L-isoleucine, L-leucine, N-2-Cl-Z-L-lysine, L-methionine, L-asparagine,
L-proline, S-4-MBzl-L-cysteine, 0-2-Br-Z-L-tyrosine, 0-Bzl-L-threonine,
0-Bzl-L-glutamic acid, 0-Bzl-L-aspartic acid, L-glutamine,
N-Tos~L-arginine, N-formyl-L-tryptophan, N-DNP-L-histidine and
0-Bzl-L-serine.
~ iethylether was used as solvent in all cases except for Met,
Asp, Glu, Arg and His for which dichloromethane was used.
The trimethylsilyl esters were obtained in the yields reported in
Table 1.
.~ .
20~1330~3
- 30 -
TABLE 1
t-BOC-AMINO ACID-TRIMETHYLSILYL ESTER
t-Boc-amino Side-chain Solvent Rf Nature ofYield
Acid Protection Product %
1 ~ Alanine _ DEE`. 0.31 Liquid 94
2 Glycine _ DEE 0.30 Liquid 95
3 L-Valine _ DEE 0.28 Liquid 90
4 L-Phenylamine _ DEE 0.29 Liquid 90
5 L-Isoleucine _ DEE o.3 Liquid 90
6 L-Leucine _ DEE 0.32 Liquid 92
7 L-Lysine 2-Cl-Z DEE 0.29 Liquid 9~
8 L-Methionine _ DCM _ Liquid 94
9 L-Asparagine _ DCM 0.28 Liquid 95
10 L-Proline _ DEE 0.31 Liquid 9o
11 L-Cysteine 4-MBzl DEE 0.28 Liquid 92
12 L-Tyrosine 2-BrZ DEE 0.26 Liquid 94
13 L-Threonine Bzl DEE 0.4 Liquid 9o
14 L-Glutamic acid O-Bzl DEE 0.27 Liquid 91
15 L-Aspartic acid O-Bzl DEE 0.3 Liquid 9o
16 L-Glutamine _ DCM 0.27 Liquid 91
17 L-Arginine Tos DCM 0.25 liquid)95
18 L-Tryptophan Formyl DEE _ (hygroscopic 90
(CHO) liquid)
19 L-Histidine DNP . DCM _ (hygroscopic 91
liquid)
20 L-Serine Bzl DEE o.39 Liquid 95
2-Cl-Z - 2-chlorobenzyloxycarbonyl Tos - p-Toluenesulphonyl
4-M-Bzl - 4-Methylbenzyl CHO - formyl
2-Br-Z - 2-Bromobenzyloxycarbonyl DNP - 2,4-dintrophenyl
Bzl - Benzyl DEE - Diethylether
O-Bzl - Benzyl ester DCM - Dichloromethane
2~!~3308
- 31 -
EXAMPLE 2
Cleavage of t-Butyloxycarbonyl (tBOC) Amino Acid Trimethylsilyl
Esters to Yield Amino Acid Trimethylsilyl Esters
2.1 L-Alanine-Tri ethylsilyl Ester Hydrochloride.
N-t-Boc-L-alanine trimethyl silyl ester (10 m.moles) prepared as
described in Example 1.1 was dissolved in anhydrous diethyl ether
(50 ml). Dry hydrogen chloride gas was then bubbled into the solution
until saturation. A white precipitate formed which was filtered, washed
with diethyl-ether to remove traces of HCl and was dried under reduced
pressure. The solid was then stored under inert atmosphere (N2).
The resulting L-alanine-trimethyl silyl ester hydrochloride was
characterised by its Infra-red spectrum, and by melting point
determination.
Infra-red spectrum (Nujol mull) showed bands at max 760 (st.),
845-860 (st.), 1175 (st.), 1200 (st.), 1755 (st.), 2985 and 3400 cml.
2.2-20 Tri ethylsilyl Ester Hydrochlorides of (2) Glycine, (3) L-Valine,
(4) L-Phenylalanine, (5) L-Isoleucine, (6) L-Leucine,
(7) 2-Cl-z-L-Lysine, (8) L-Methionine, (9) L-Asparagine,
(10) L-Proline, (11) (4-MBzl)-L-Cysteine,
(12) (2-Br-Z)-L-Tyro~ine, (13) (Bzl)-L-Threanine,
(14) (O-Bzl)-L-Glutad c acid,
15) (O-Bzl)-L-Aspartic acid,
(16) L-Guta ine, (17) (Tos)-L-Arginine,
(18) (For yl)-L-Tryptophan, (19) (DNP)-L-Histidine and
(20) (Bzl)-L-Serine
Trimethylsilyl esters of the title amino acids and protected
amino acids were prepared by the procedure described in Example 2.1
above, but substituting the corresponding t-Boc amino acid and t-Boc
protected amino acid trimethylsilyl esters (2) to (20) referred to in
Table 1 for t-Boc L-alanine trimethylsilyl ester.
The trimethylsilyl ester hydrochlorides were obtained in the
yields reported in Table 2.
Z~3308
- 32 -
TABLE 2
AMINO ACID TRIMETHYLSILYL ESTER HYDROCHLORIDE
Side-chain Melting Point Yield
Amino Acid Protection (Uncorrected) C %
1 L-Alanine _ 1 225-228 85
2 Glycine _ 165-167 75
3 L-Valine _ 230-233 80
4 L-Phenylalanine _ 227~229 87
L-Isoleucine _ 176-177 78
6 L-Leucine _ 232-234 81
7 L-Lysine 2-Cl-Z 205-208 81
8 L-Methionine _ 222 223 89
9 L-Asparagine _ 162-164 77
10 L-Proline _ 125 75
11 L-Cysteine 4-M-Bzl 210-212 75
12 L-Tyrosine 2-Br-Z 221-222 88
13 L-Arginine Tos hygroscopic 72
14 L-Threonine Bzl . 80
15 L-Glutamic acid O-Bzl 145-148 75
16 L-Aspartic acid O-Bzl 186-188 78
17 L-Glutamine _ _ 80
18 L-Tryptophan CHO _ 72
19 L-Histidine DNP hygroscopic 70
20 L-Serine Bzl - 81
(Abbreviations as in Table 1)
Z0~3308
- 33 -
EXAMPLE 3
Preparation of t-Butyloxycarbonyl (tBOC) Amino Acid
t-Butyl-Dimethylsilyl Esters
3.1 t-Boc-L-Alanine t-Butyldimethylsilyl Ester
t-Boc-L-alanine-t-butyldimethylsilyl ester was prepared by the
following procedure.
N-t-Boc-L-alanine (10 m.moles) was dissolved in
N,N-dimethylformamide (20 ml) and imidazole (20 m.moles) added to act as
base and catalyst.
The solution was stirred at room temperature and then
t-butyldimethylsilyl chloride (11 m.moles, 1.6 g) was added slowly.
The reaction mixture was stirred for a further period of 2 hours at room
temperature and a precipitate of imidazole hydrochloride formed.
The mixture was diluted with diethyl ether (60 ml) and washed
with 10X sodium bicarbonate (1 x 20 ml), water (1 x 20 ml), 0.1 M
hydrochloric acid (1 x 20 ml), and water (2 x 20 ml). The organic layer
was then dried over anhydrous sodium sulphate and was evaporated under
reduced pressure to give N-t-Boc-L-alanine-t-butyldimethyl silyl ester as
an oil.
The N-t-Boc-L-alanine-t-butyldimethyl silyl ester was
characterised by its Infra-red spectrum, lH nuclear magnetic resonance
spectrum and by thin layer chromatography (tlc).
Infra-red spectra showed bands at max. (thin film) 760-780 (st.), 845( t.), 1180 (st.), 1260 (st.), 1695 (st.), 1720 (st.),
2960, and 3400 cm 1.
H n.m.r. (CDC13) ô 0.21 (s., 6H),
0.9 (s., 9H),
1.45 (s., 9H)
Z~308
- 34 -
3.2-11 t-Butyl-Dimethyl~ilyl Esters of (2) t-Boc Phenylalanine
(3) t-Boc (0-Bzl)-L-Aspartic acid,
(4) t-Boc L-Leucine,
(5) t-Boc (2-Br-Z)-L-Tyro~ine, (6) t-80c L-proline
(7) t-Boc L-Valine, `(8)t-Boc (DNP)-L-Histidine and
(9) t-Boc L-Isoleucine, (10) t-Boc L-Glycine,
(11) t-Boc (Tos)-L-Arginine,
t-Butyl-dimethylsilyl esters of the title t-Boc amino acids and
protected amino acids were prepared by the procedure described in Example
3.1 above, but substituting the following t-Boc amino acids and protected
amino acids for N-t-Boc-alanine:
t-Boc Phenylalanine t-Boc 0-Bzl-L-Aspartic acid, t-Boc
L-Leucine, t-Boc 2-Br-z-L-Tyrosine, t-Boc L-proline t-Boc L-Valine,
t-Boc DNP-L-Histidine and t-Boc L-Isoleucine, t-Boc L-Glycine,
t-Boc Tos-L-Arginine.
Dimethylformamide was used as solvent in all cases.
The t-butyldimethylsilyl esters were obtained in the yields
reported in Table 3.
Cys(4-methylBzl)-t-butyldimethylsilyl ester and Lys(2-Cl-Z)-
t-butyldimethylsilyl ester have also been prepared by the above
procedure.
"r
,,
2(~)3~08
~ 35 -
TABLE 3
t-BOC-AMINO ACID t-BUTYLDIMETHYLSILYL ESTERS
Side-chain Solvent Nature of Yield
t-Boc-Amino Acid Protection (reaction) Product
1 L-Alanine _ DMF Liquid 60
2 L-Phenylalamine _ DMF Liquid 55
3 L-Aspartic acid O-Bzl DMF Liquid 45
4 L-Leucine _ DMF Liquid 60
5 L-Tyrocine 2-Br-Z DMF Liquid 5o
6 L-Proline _ DMF Liquid 45
7 L-Valine _ DMF Liquid 5o
8 L-Histidine DNP DMF Liquid 5o
9 L-Isoleucine _ DMF Liquid 55
10 Glycine _ DMF Liquid 45
11 L-Arginine Tos DMF Liquid 51
(Abbeviations as in Table 1)
3~08
- 36 -
EXAMPLE 4
Cleavage of t-Boc-group - Preparation of Amino
Acid t-Butyldimethylsilyl Esters
4.1 L-Alanine t-Butyldimethylsilyl Ester
t-Boc-L-alanine t-butyldimethylsilyl ester (10 m.moles) was treated with
25% trifluoroacetic acid in dichloromethane (25 ml) for 30 minutes at
room temperature. The solvents (i.e. dichloromethane, trifluoroacetic
acid) were then removed under reduced pressure at room temperature and an
oil was obtained in high yield.
The resulting L-alanine t-butyldimethylsilyl ester was characterised by
its Infra-red spectrum and lH n.m.r. spectrum.
Infra-red spectrum (thin film) showed bands at max. 700 (st.),
800-860 (st.), 1200 (st.), 1740-1790 (st.), 2960, 3050
and 3240 cm 1,
H n.m.r. (DMS0-d6) ô 0.19 (s., 6H), C.9 (s., 9H).
4.2 Cleavage of t-Boc-group - Preparation of
t-Butyl~imethylsilyl Esters of (2) Phenylalanine
(3) (0-Bzl)-L-Aspartic acid, (4) L-Leucine,
(5) (2-Br-Z)-L-Tyrosine, (6) L-proline
(7) L-Valine, (8) (DNP)-L-Histidine,
. (9) L-Isoleuclne, (10) L-Glycine,and
(11) (Tos)-L-Arginine,
The t-Boc t-butyldimethylsilyl esters of the title t-Boc amino
acids and protected amino acids may be cleaved by the procedure described
in Example 4.1 above.
. .
2C~Q~
~ 37 -
EXAMPLE 5
Production of Resin
A resin containing -CH20.CO.Cl groups was prepared from a
standard, commercially available "Merrifield" resin (bearing -CH2Cl
groups) by the following procedure (see Scheme 2).
Merrifield resin (25g was reacted with sodium acetate in
dimethoxyethane for 48 hours at 80 C to give the ester in high yield.
The identity of the ester was confirmed by its infra-red spectrum (1730
cm 1 and an undetectable amount of chlorine. Reduction of the ester
with LiAlH4 in diethylether gave after the usual work-up procedure the
methylol resin. The methylol resin was then treated with phosgene in
toluene at room temperature and afforded the desired chloroformate resin
in high yield. Chlorine analysis indicated that there was 0.9 mmoles/g
of Cl on the resin
EXAMPLE 6
Production of Peptides
Solid Phase Synthesis of Tetrapeptide
The tetrapeptide H-Leu-Ala-Gly-Val-OH was synthesised manually by
the following procedure. All amino acids were of L-configuration. The
reaction sequence is outlined in Schemes 3 and 4.
The resin as prepared in Example 5 was placed in a reaction
vessel and washed with dimethylacetamide (DMA) (3 x 10 ml).
Leu t-butyl dimethylsilyl ester was added in DMA (10 ml) followed
by triethylamine and the reaction vessel was shaken at room temperature
for 2 hours. The liquid phase was drained out and the resin washed with
DMA (2 x 10 ml). Estimation of unrescted Leu derivative indicated that
0.61 mmol/g had been bound to the resin. The remaining free chloroformate
on the resin was capped with diethylamine in DMA.
The t-butyldimethylsilyl Leu resin was treated with warm (40 C.)
methanol (10 ml) for 30 minutes to remove the t-butyl dimethysilyl group.
The resin was washed with DMA (3 x 10 ml) and was shaken with
H-Ala-OSi-(Me)2t-Bu in DMA in the presence of DCC/HOBT for 45 minutes.
.3308
- 38 -
The solvents were drained and the resin washed with DMA
(3 x 10 ml). 100 mg resin was withdrawn from the reaction vessel. The
resin was then coupled as described above to H-Gly-OSi(Me)2t-Bu and
100 mg of
Resin-(C6H5)-CH20-CO-NH-Leu-Ala-Gly-OSi(Me)2tBu
was withdrawn as usual. It was then coupled to H-Val-OSi(Me)2tBu in a
similar manner and the desired resin peptide was obtained. The peptides
Leu-Ala, Leu-Ala-Gly, and Leu-Ala-Gly-Val were released from the resin by
the standard HF cleavage procedure.
These peptides were also synthesised by Merrifield solid phase
procedure. Comparative TLC and HPLC have shown that di-, tri-, and tetra-
peptides synthesised by both methods are identical. There was no
indication of any racemisation during solid phase silyl methodology in
the qynthesis of these peptides.
,r
/