Note: Descriptions are shown in the official language in which they were submitted.
115
730-0062
CAPSAICrN DBPIVATXV~S
The present invention relates to novel compounds having pharma-
ceutical properties, to processes for their production, pharmaceutical
compositions comprising them and their use as pharmaceuticals.
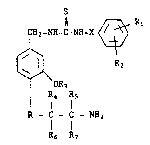
The present invention provides in one aspect compounds of formula I
CH2-NH-C-NH-X ~ R1
R2 (I)
OR3
R4 Rs
R - C - C - NH2
R6 R7
wherein:
Rl i9 halogen, Cl_4alkyl, phenyl, benzyl, substituted or unsub-
stituted benzyloxy, nitro, cyano, trifluoromethyl, formyl-
amino or C~_ 16 alkoxy,
R2 is hydrogen or has any one of the meanings given for Rl,
R3 is hydrogen or Cl_4alkyl,
R4 and Rs~ independently, are hydrogen, halogen, Cl_salkyl,
substituted C1_~alkyl, substituted or unsubstituted aryl,
COOH, COOR8 or CONR9RIo wherein R8 is Cl_salkyl, and each
illS
700-OQ~2
of Rg and Rlo independently is hydrogen or C1_salkyl,
R6 and R" independently have the meanings given for R4 and Rs
or, together with the carbon atoms to which they are
attached form a substituted or unsubstituted C3_7cyclo-
alkyl group,
R is 0, S or NH and
X is ~(CH2)n- or -(CH2)m-CH=CH-(CH2)r- wherein n is 1, 2 or 3
and each of m and r is independently zero or an integer of
from 1 to 3,
and physiologically hydrolysable and acceptable esters or amide and
pharmaceutlcally acceptable salts thereof.
The compounds of formula I wherein X is -(CH2 3m-CH=CH~(CH2 )r~ may
exist in both cis or trans isomeric forms, i.e. as Z and E isomers. The
present in~ention is to be understood as embracing both the individual
cis and trans isomers as well as mixtures thereof.
Substituted benzyloxy groups as R1 include be~zyloxy substituted by
halogen or phenyl. Substltutions may be, e.g. at the 2- and/or the
4-position of the benzyloxy group. Suitably the benzyloxy ~roup is
mono-substituted. Preferably the benzyloxy group is mono-substituted at
the 4-position.
Alkyl groups as Rl through R1o as well as the alkyl moiety of alkoxy
groups as Rl or R2 may be branched or straight chain. Preferably such
alkyl groups and moieties are straight chain.
By halogen is meant fluorine, chlorine, bromine or iodi~e.
Preferred aryl grouys as R4 to R7 are phenyl or naphthyl. Such
groups may be unsubstituted or substituted~ e.g. mono-, di- or tri-
substituted phenyl or naphthyl. Preferred substituents are selected from
3L15
700-0062
halogen, hydroxy, amino and carboxy. Especially preferred aryl groups
as R4 to R~ are phenyl or substituted phenyl.
A group of compounds of formula I comprises those compounds in which
R1 is as defined above but excluding substituted benzyloxy and R, R2 to
R1o ar.d X are as defined above.
In the compounds of formula I, the following significances are
preferred either individually or in any combination or sub-combination:
1. R1 is halogen, C1_4alkyl, C~_~6alkoxy or substituted or unsubstitu-
ted benzyloxy, especially halogen, C~_~6alkoxy or substituted or
unsubstituted benzyloxy. Uhen R1 is C1_16alkoxy this is suitably
n-octyloxy. When Rl is substituted benzyloxy, this is suitably
halo- or phenyl-benzyloxy, especially 4-halo- or 4-phenyl-benzyloxy.
R1 is preferably in the 4-position.
2. R2 is hydrogen, halogen or C1_4alkyl, especially hydrogen or halo-
gen. Uhen R2 i9 other than hydrogen it is preferably in the 2-
position.
3. R3 is hydrogen or methyl, suitably methyl.
4. R4 to R7, independently are hydrogen, methyl, ethyl, C~_5alkyl
substituted by Off or NH2, phenyl or C1_5alkyl-phenyl. More prefer-
ably at least one of R4 to R6 is hydrogen. Yet more preferably at
least three of R4 to R7, e.g. R4, Rs and R6 are hydrogen. Most
preferably all of R4 to R7 are hydrogen.
S. R is -0-.
6. X is -(Cff2)n-, wherein n has the meanings given above. Preferably n
is 1 or 2, especially 2.
115
700-0062
7. X is -(CH2 )~-CH~CH-(CH2 )r~ in which one of m and r is zero and the
other is 1. More preferably both m and r are zero. Most preferably
such groups X are in Z isomeric form.
An especially preferred group of compounds of formula I are those
whereln X has the meanings given under 6 above and especially such com-
pounds wherein Rl and R2 are as defined under 1 and 2 above. A further
preferred group of compounds of formula I are those wherein X has the
meanings given under 6 above and each of R4 to R, is hydrogen, especia-
lly such compounds wherein R1 and R2 are as defined under 1 and 2 above.
By the term "physiologically-hydrolysable and -acceptable esters or
amides" as used herein is meant esters (e.g. compounds of formula I
wherein one or more of R4 to R7 is -COOH and/or R3 is hydrogen) or
amides which are hydrolysable under physiological conditions to produce
acids or alcohols which are themselves non-toxic, i.e. have no signifi-
cant undesirable side effects at desired dosage levels. Such esters
include esters with mono- and di-carboxylic acids, e.g. esters of the
compounds of formula I in which the 3-hydroxy group is acetylated and
esters with lower, e.g. C1-4 alkanols. Such amides include those deri-
ved from organic carboxylic acids e.g. acetic acld amldes including
amino acids e.g. glycine amides.
The present invention also provides a process for the production of
the compounds, amides, eseers and salts of the invention which process
comprises:
a) for the production of a compound of formula I reacting a compound of
formula II
115
700-0062
ll fi~ R
CH2 -NH-C-NH-X~
~ r (II)
RZ
OR3 0
R4 Rs
I I ~
R - C - C - N
R6 R7 ~
wherein R, R~ to R, and X are as defined above,
with hydrazine
b) for the production of a compound of formula I, removing at least one
amino protecting group which is present in an amino protected com-
pound of formula I,
c) for the production of a physiologically-hydrolysable and -acceptable
ester or amide of a compound of formula I, eseerifying or amldating
a compound of formula I, for example in which R3 is hydrogen, by
reaction with an appropriate acylating or amidating agent, respec-
tively,
d) for the production of a compound of formula I wherein X is
-(CH2)m-C~-CH-(C~2)r- in Z-isomeric form isomerising a compount of
formula I wherein X is -(CH2)m-CH.CH-(CH2)r~ in ~-isomeric form
and recovering a compound of formula I thus obtained in free or salt
form.
Process step a) may be carried in analogy with known methods, for
example by reaction of the compound of formula II with hydrazine hydrate
in an inert solvent or diluent, e.g. alkane/alkanol, at ambient or ele-
vated temperatures, e.g. at a temperature of from 0C to reflux. Pro-
61~5
700-006~
cess step b) may be carried out in accordance with standard techniques
for the removal of amino protecting groups as known in the art. Sui-
table amino protecting groups include p-toluene sulphonyl, benzyl,
formyl and trifluoroacetyl.
Process step c) may also be performed employing conventional acy-
latlon or ~midstlon method~ e.g. by reaction wlth an approprlate e.g.
Cl _ 4 acyl halide or anhydride.
The trans isomers of compounds of formula I wherein X is
-(CH2 )m-CH.CH-(CH2 )r~ may be converted into the cis (Z) isomers in con-
ventional manner, e.g. by irradiation. Individual cis and trans isomers
may be obtained in accordance with techniques known in the art, e.g.
separation of cis/trans isomer mixtures, for example by chromatography.
Compounds of formula II used as starting materials in process step
(a) may be produced by either of the following Reaction schemes, A or B
34~5
700-0062
R~ACTION SCHENe A
CH2-NH-C-NH-X- ~ 1 (VIII)
R2
~ \
I OR3 \ R4 Rs
R'H \ Y - C - C ~ Y (IX)
\ R6 R7
\ tetrabutylammonium hydroxide
S R
CH2-NH-C-NH-X ~
Rz
OR3 (X)
/ R ,R4 R~s
/ R6 R7
~ a) DM~
/ b) potassium phthalimide
L~
S Rl
C~2 -NH-C-N~-X ~_R2
(II)
OR3 0
] ~ - C - C - N
R6 R
o
wherein R, Rl to R7 and X are as defined above, Y is halogen, e.g
bromine and R' is O or S.
Compounds of formula VIII, in which R is 0, are described, along
700-0062
with processes for their production in, for example, published GB patent
application 2,206,347A. Other compounds of formula VIII may be prod~lced
in similar or analogous fashion to the known compounds.
In Reaction Scheme B, on the following page, R, R1 to R7 and X are
as defined above and Y is halogen, e.g. bromine.
The intermediates in Reaction Schemes A and B of formulae X, VIII,
VI, V, II and III are new and also form part of the present invention
per se.
f~O~6~5
700-0062
NH3+Cl- H
N ~ o-C(CH3)3
OR3 Et3N ~ ~ (VI)
RH di-t-butyl- ~
dicarbonate I OR3
(VII) RH \ R4 Rs
\ R6 R~
\ tet~abutylammonium-
~ hydroxide
R~ACTION SCEeHE B H
N~O-C(~H3 )3
O
a) potassium ~ OR3 (V)
phthalimide, ~R4 ,Rs
DMF ~ R - C - C - Y
NH3'TFA- / R6 R7
b) TFA, CH2C12
~ O
OR3
R,4 R,s /
R6 R7 \
(III) g
b) N-X ~ \ a) Et3N
ll R2 \ S
¦¦ (IV) ~ CH2-NH-C-NH-X ~ lR2
~\ O
OR3
R4 R, 5 /~
R6 R
_ g _
70Q-0062
Insofar as the production of the starting materials is not particu-
larly described, the compounds may be prepared by analogy with known
methods.
The compounds of formula I as defined above may exist in free or
salt form. Suitable pharmaceutically acceptable salts of compounds of
formula I, e.g. wherein one or more of R4 to R7 is -COOH for use in
accordance with the present invention include for example the sodium,
potassium and calcium salts as well as quaternary ammonium, e.g. tri-
ethylammonium salts. Such salts also include pharmaceutically accepta-
ble acid addition salts, e.g. salts with inorganic or organic acids such
as hydrochloric acid, acetic acid, fumaric acid, citric acid and benzoic
acid.
The following Examples are illustrative of the process of the pre-
sent invention.
The following abbreviations are used:
DMF = dimethylformamide
TLC s thin layer chromatography
XAHPLl~ 1: N-14-(2-Aminoethoxy)-3-l~ethoxy-Benzyl]-N'-[2-~4-Chloro-
phenyl)-RthyllThiourea
2.0 g N-14-(2-Phthalimidoethoxy)-3-methoxybenzyl]-N'-l2-(4-chloro-
phenyl)ethyl]thiourea (0.0038 mole) is suspended in 10 ml ethanol in a
round bottomed flask and heated to 60~ C until the solution becomes
homogeneous. 0.10 ml 1-Hexene and 0.95 ml of a 64% aqueous solution of
hydrazine hydrate is added and the mixture heated for 90 ~inutes. After
15 minutes a white precipitate begins to form and a small amount of
ethanol is added to keep the mixture mobile. The reaction is cooled,
transferred to a separating funnel and 10 ml methyl-butyl ether, 5 ml
water, S ml lN NaOH and 0.5 ml 50~ NaOH are added. The resulting mixture
_ 10 --
700-0062
is shaken thoroughly and the organic layer extracted twice as above,
then washed with brine. The organic layer is dried over Na2SO4, filtered
and the solvent removed by evaporation. The residue is purified via
flash column chromatography, eluting first with CH2Cl2 : MeOH (10:1) and
then with MeOH. The product fractions are evaporated and dried in vacuo
(30 C, 0.1 mm Hg) to give the title compound as a solid colourless
glass.
The following compounds of formula I may be obtained analogously:
Example R _1 _2 _3 _4 to R7 X
2 0 4-F H CH3 H (CH2)2
3 0 4-C1 2-Cl CH3 H (CH2)2
and also the following compounds of formula I
S
CH2-NH-C-NH-(CH2)~ ~ O-R1l
(I)
¦ OCH3
O-CH2 -CE~2 -NH2
EXAMPLE Rl1 n
4 benzyl 2
4-bromobenzyl 2
6 N-octyl 2
7 N-octyl
8 4-chlorobenzyl 2
9 4-phenylbenzyl 2
115
700-0062
CHARACTERISING DATA
1. 1H NMR data [400 MHz]
Example 1: [D6DMSOl
~2.80(2H,t,J.7.2Hz), 2.99(2H,t,J~5.5HZ), 3.61(2H,S,broad),
3.75(3H,s), 4.00(2H,t,J~5.5Hz), 5.57(2H,s,broad),
6.79(1H,d,J,1.8Hz), 6.94(1H,d,J-8.1Hz), 6.96(1H,d,J-1.6Hz),
7.26(2H,d,J=B.2Hz), 7.34(2H,d,J_8.1Hz), 7.75~1H,s,broad),
7.99~1H;s,broad)
Example 2: ICDCl3~
~1.55(3H,s,broad), 2.84(2H,t,J,6.9Hz), 3.10(2H,t,J-5.3Hz),
3.74(2H,s,broad), 3.83(3H,s), 4.02(2H,t,J_5.3Hz), 4.41(2H,s,broad),
5.72(1H,s,broad), 6.18(1H,s,broad), 6.70-6.81(2H,m),
6.93-6.98(2H,m), 7.06-7.10(2H,m)
Example 3: lCDCl3l
~1.52(3H,s,broad), 2.99(2H,t,H.7.0Hz), 3.11(2H,t,J.5.3Hz),
3.75(2H,s,broad), 3.84(3H,s), 4.03(2H,t,J-5.3Hz), 4.4S(2H,s,broad),
5.82(1H,s,broad), 6.17(1H,s,broad), 6.79-6.84(4H,m),
7.10-7.17(3H,m), 7.35(2H,d,J.1.8Hz)
Example 4: [CD3ODl
~2.80(2H,t,J.7.1Hz), 3.09(2H,t,J-5.1Hz), 3.68(2H,s,broad),
3.83(3H,s), 4.06(2H,t,J~5.1Hz), 4.60(2H,s,broad), 5.04(2H,s),
6.80-7.13(5H,m), 7.29-7.43(4H,m)
Example 5: [CD3OD]
~2.78(2H,t,J,7.15Hz), .13(2H,t,J~5.1Hz), 3.68(2H,s,broad),
3.84(3H,s), 4.08(2H~t,Jz5.1Hz), 4.60(2H,s,broad), 5.01~2H,s),
6.81-7.12(7H,m), 7.43(4H,dd,J=8.3Hz,J=64.5Hz)
700-0062
Example 6: [CD3OD]
~0.92(3H,m), 1.28-1.38(8H,m), 1.46(2H,m), 1.75(2H,m),
3.27(2H,t,J=5.0Hz), 3.83(3H,s), 3.94(2H,t,J=6.4Hz),
4.16(2H,t,J,5.0Hz), 4.63(4H,s,broad), 6.82-7.90(5H,m),
7.19-7.21(2H,m)
Example 7: ICD 3 0D]
~0.90(3H,m), 1.28-1.35(8H,m), 1.46(2H,m), 1.74(2H,m),
2.79(2H,t,J=7.25Hz), 3.16(2H,t,J=5.15Hz), 3.68(2H,s,broad),
3.85(3H,s), 3.92(2H,t,J=6.45Hz), 4.11(2H,t,J=5.15Hz),
4.63(2H,s,broad), 6.70-7.11(7H,m)
Example 8: ICD 3 OD]
~2.8(2H,t,J~7.15Hz), 3.02(2H,t,J.5.25Hz), 3.68(2H,s,broad),
3.83(3H,s), 4.03(2H,t,J~5.25Hz), 4.59(2H,s,broad), 5.03(2H,s),
6.80-6.97(5H,m), 7.11(2H,d,J=8.45Hz), 7.38(4N,dd,J=8.5Hz,J.13.4Hz)
Example 9: ~d6DMSOl
~2.74(2H,t,J~7.3Hz), 2.94(2H,t,J,5.65Hz), 3.56(2H,s,broad),
3.74(3H,s), 3.95(2H,t,J.5.65Hz), 4.56(2H,s,broad), 5.12(2H,s),
6.78(1H,m), 6.80-6.97(4H,m), 7.15(2H,d,J_8.5Hz), 7~37(1H,m),
7.45-7.54(5H,m), 7.66-7.69(4H,m)
The compound of Example 9 has a melting point of 90-92C.
HPLC Retention time
_
The retention times of the compounds of Examples 1 to 9 were
measured on a C-18 MicrobondapakR reverse phase column usi~g the follow-
ing gradient and conditions:
~V~6~5
700-0062
Solution A: 0.1% trifluoroacetic acid
Solution B: acetonitrile
TIME FLOW RATE XA %B
(min) (ml/min)
0 2.25 90 10
2 2.25 9~ 10
6 2.25 45 55
22 2.25 40 60
29 2.25 0 100
31 2.25 90 10
The following results were obtained:
EXAMPLERETENTION TIME (min)
1 9.404
2 9.415
3 9.740
4 9.752
1~.297
6 10.827
7 11.317
8 10.080
9 10.482
~ ollowing the steps of Reaction Scheme B the compounds used as
starting materials may be prepared as follows:
- 14 -
611S
700-0062
a) t-Boc-vanillylamine
18.0g Vanillylamine hydrochloride (0.095 mole) and 10.6g triethyl-
amine (0.11 mole) are dissolved in 250ml water and placed in a lL round
bottomed flask. 20~5g di-tert-butyldicarbonate (0.095 mole) in 200ml
dioxan is added with stirring, over a period of 15 minutes. The resul-
ting mixture is stirred overnight at room temperature.
The dioxan is removed in vacuo and the aqueous residue extracted
with CHCl3 (5xlOml). The combined extracts are drled over MgSO4, fil-
tered and the solvent removed in vacuo to leave a broun oil which is
purified via flash column chromatography (cyclohexane : EtOAc / 5:2) to
give a colourless oil which crystallises on standing. It.l.c. cyclohex-
ane : EtOAc / 1:1; r.f. = 0.5]
b) t-Boc-(4-(2-bromoethoxy)-3-methoxybenzylamine
21.0g t-Boc-vanillylamine (0.083 mole), 250ml 1,2.dibromoethane,
66ml 40% KOH and 6.6ml 40% tetrabutylammonium hydroxide are combined in
a 500ml round bottomed flask and the resulting mixture is heated at 50C
for 3 hours with rapid stirring.
The mixture is cooled, diluted with 200ml CH2Cl~, washed with water
(3x200ml) and the combined aqueous washings extracted once with 600ml
CH2Cl2. The combined organic layers are washed with brine, dried over
MgSO4, filtered and the solvent removed in vacuo to leave a uhite solid
which is used uithout further purification. It.l.c. cyclohexane : EtOAc
/ 1:1; r.f. = 0.6]
c) t-Boc-(4-(2-phthalimidoethoxy)-3-methoxybenzylamine
23.0g t-Boc-(4-(2-bromoethoxy)-3-methoxybenzylamine (0.064 mole) and
11.8g potassium phthalimide (0.064 mole) are suspended in 500ml dry DMF
_ 15 -
G~15
700-0062
in a round bottomed flask. The resulting suspension is heated at 50C
for 2 hours with rapid stirring. After 30 mins of this heating/-
stirring, the mixture becomes homogeneous. The mixture is then cooled
and the DMF removed under high vacuum. The resulting solid residue is
purified via flash column chromatography (cyclohexane : EtOAc / 1:1) to
give a white solid. It.l.c. (cyclohexane : EtOAc / 1:1) rf . 0.45.
d) 4-(2-phthalimidoethoxy~3-methoxybenzylamine.T~A
26.5g t-Boc-(4-(2-phthalimidoethoxy)-3-methoxybenzylamine (0.062
mole) is dissolved in 200ml CH2Cl2 in a 500ml round bottomed flask.
15ml trifluoroacetic acid is added dropwise with stirring. On comple-
tion of the addition, the mixture is stirred for a further 2 hours at
room temperature (the reaction is shown to be completed by the loss of
staring material (cyclohexane : EtOAc / 1:1)). The solvent is removed
in vacuo, initially on the water pump and then high vacuum. The resul-
ting colourless oil solidifies on standing and is used wlthout further
purification.
e) N-[4-(2-phthalimidoethoxy)-3-methoxybenzyl)-N-12-(4-chlorophenyl)-
ethyl]thiourea
2.0g 4-(2-phthalimidoethoxy)-3-methoxybenzylamine.TPA (0.005 mole)
and 0.5g Et3N (0.005 mole) are suspended in 30 ml dry EtOAc under an
atmosphere of dry nitrogen. O.9g 2-(4-chlorophenyl)ethylisothiocyanate
(0.0045 mole) in dry lOml EtOAc is added dropwise and the mixture
stirred at room temperature for 3 hours.
The EtOAc is removed in vacuo, the residue suspended in 50ml water
and then extracted with 3x50ml CH2C12. The combined organic layers are
dried over MgSO4, filtered and the solvent removed in vacuo to leave a
brown oil which is purified via column chromatography Itlc (cyclohexane
: EtOAc / 1:1) rf = 0.2]. Melting point = 59-61C.
_ 16 -
115
700-006~
The compounds and amides and esters of the invention have pharma-
cological, in particular analgesic and anti-inflammatory, activity and
are thereiore indicated to be useful as pharmaceuticals, e.g. for
therapy.
In particular the compounds exhibit pharmacological activity as
indicated in standard test models, for example as follows:
1. TAIL-~LICR TBST IN T~B ~OUSB
The method is based on that of D'Amour et al. J. Pharmacol. Exp.
Ther. 72, 74-79 (1941), but employing unstarved mice (o + o, 16-25 g).
Animals are divided into control and test groups, control animals
receiving a vehicle injection only. Each test animal is placed in an
individual perspex cylinder to prevent movement with its tail protruding
along a narrow groove. The tail of each animal is exposed to a beam of
radiant heat at ca. 35 mm from the tail root, from a lamp of known out-
put and temperature, place directly under the tail. Test substance is
administered p.o. or s.c. 30 mins. post introduction into the cylinder.
The time in seconds taken by the mouse to flick its tail out of the
ligh~ beam is recorded 30 to 15 mins prior to administration of test
substance. Animals whose reaction times differ by more than 25 % are
discarded. Reaction time is re-determined 15 and 30 mins post admini-
stration. Extension of reaction time by > 75 ~ over mean pre-treatment
values in the same animal are taken as indicative of analgesic response.
Three doses are employed per test substance and 10 animals per dose.
EDso values (95 ~ confidence limits) are estimated in accordance with
the method of Litchfield and Wilcoxon and represent the dose prolonging
treatment reaction time by > 75 ~ in 50 ~ of test animals.
Compounds and amides and esters of the invention are active in the
above test model at dosages of the order of from about 1.0 to about
120.0 ~m/kg, s.c.
_ 17 -
llS
700-0062
2. Y~AST INDUCED IN~LAHHATION T~ST IN TRB HOUSB
~1, 20 % fresh yeast suspension is injected into the plantar
region of one hind paw and saline is injected into the other. Degree of
inflammation is estimated by the relative increase in paw weight (yeast
in~ected v.s.sallne) 2 hrs. post-in~ection. Test substance is admini-
stered s.c. at varying dosage at the same time as yeast/saline treat-
ment. 5 animals are used~dose and testing at each dose is repeated 2 - 3
times, control and test values being compared statistically as above.
EDso values are taken as the do~age required to effect 50 % inhibition
of inflammation as compared with control animals not receiving test
substance, and are established from dose response curves plotting
inflammation vs. dose. Compounds and esters of the invention are active
in the above test model at dosages of the order of from about 2.5 to
100 ~M/kg, s.c.
The compounds, esters, amides and pharmaceutically acceptable salts
of the invention are accordingly useful as pharmaceuticals, e.g. as
analgesics for the treatment of pain of various genesis or aetiology,
for example dental pain and headache, particularly vascular headache,
such as migraine, cluster, and mixed vascular syndromes as well as non-
vascular, tension headache, and as anti-inflammatory agents for the
treatment of inflammatory diseases or conditions, for example the treat-
ment of arthritis and rheumatic diseases, Raynaud's disease, inflamma-
tory bowel disorders, trigeminal or herpetic neuralgia, inflammatory eye
disorders e.g. uveitis, psoriasis, cystitis as well as other chronic
inflammatory conditions.
Having regard to their analgesic/anti-inflammatory profile they are,
in particular, useful for the treatment of inflammatory pain, for the
treatment of hyperalgesia and, in particular, the treatment of severe
chronic pain, e.g. for the treatment of deafferentation pain as an al-
ternative to surgical procedures.
2Q~ S
700-0062
According to a further embodiment, the compounds of formula I, their
esters, amides and pharmaceutically acceptable salts are also useful for
the prophylactic or curative treatment of epithelial tissue damages or
dysfunction, e.g. spontaneous lesions, and for the control of disturban-
ces of visceral motility at respiratory, genitourinary, gastrointestinal
and vascular level, e.g. for treating wounds, burns, skin allergic reac-
tions, pruritus and vitiligo, for the prophylactic or curative treatment
of gastrointestinal disorders such as gastric ulceration, duodenal
ulcers and diarrhoea, for the prophylactic or curative treatment of
gastric lesions induced by necrotising agents, for example ethanol, for
the treatment of vasomotor or allergic rhinitis and for the treatment of
bronchial disorders or bladder disorders. The utility in treating as
epithelial tissue damages or dysfunction may be shown in standard test
models, for example as follows:
BT~ANOL-INDUC~D GASTRIC LeSIONS
The tests are carried out employing male rats (200-250g) fasted
overnight but with free access to water. The test substance ~s admini-
stered s.c. or orally by a metal stomach tube. Absolute ethanol is given
orally 30 min after the administration of the test substance and the
animals are killed 1 hour later. The stomach is cut open along the
greater curvature and pinned flat. Haemorrhagic erosions are quantified
in two ways: area and length of the erosions.
On administration of a compound of formula I as test compound at a
dosage of from ca. 0.1 to 20 mg/kg, substantial inhibition of the gas-
tric lesions induced by ethanol is observed compared with results for
control groups receiving placebo in lieu of the test compound.
For the above uses the required dosage will of course vary depending
on the mode of administration, the particular condition to be treated
and the effect desired. An indicated daily dosage in the range of from
_ 19 --
G115
700-0062
about 2 to about 1,000 or 2,000 mg p.o., e.g. from about 75 to 750 or
1,500 mg p.o. for analgesic use, and of the order of from about 10 to
about 2,000 mg p.o. e.g. from about 75 to 1,500 mg p.o., for anti-
inflammatory use, conveniently administered once, in divided dosages 2
to 4 times/day, or in sustained release or retard form. Dosage forms
suitable for oral administration accordingly comprise from about 0.5 to
about 500 or 1,000 mg, e.g. from about 20 to about 375 or 750 mg (anal-
gesic use) or from about 2.5 to about 1,000 mg, e.g. from about 20 to
about 750 mg, (anti-inflammatory use) active ingredient (i.e. compound,
ester, amide or pharmaceutically acceptable salt of the invention) ad-
mixed with an appropriate solid or liquid, pharmaceutically acceptable,
diluent or carrier therefor.
In accordance with the foregoing the present invention also pro-
vides a method:
A. i) for the treatment of pain of various genesis or aetiology, for
example for the treatment of any such disease or conditions as here-
inbefore set forth;
ii) for the treatment of inflammatory diseases andJor lnflammatory
pain, for example for the treatment of any such disea~e or condlti-
ons as hereinbefore set forth;
iii) for the prophylactic or curative treatment of epitheleal tissue
damage or dysfunction, gastrointestinal disorders or gastric lesions
induced by necrotising agents;
iv) for the control of disturbances of visceral motility at respira-
tory, genitourinary, gastrointestinal and vascular level; or
v) for the treatment of rhinitis and bronchial or bladder disor-
ders,
_ 20 -
700-0062
in a subject in need thereof, which method comprises administering
to said subject an effective amount of a compound of formula I or
physiologically hydrolysable and acceptable ester or amide thereof
as hereinbefore defined, or a pharmaceutically acceptable salt
thereof;
B. A compound of formula I or physiologically-hydrolysable and -accep-
table ester or amide thereof as hereinbefore defined, or a pharma-
ceutically acceptable salt thereof, for use as a pharmaceutical, for
example for use in a method as defined under any one of A(i) to A(v)
above or for use in the preparation of a pharmaceutical composition
for use in a method as defined under any one of A(i) to A(v) above;
as well as
C. A pharmaceutical composition comprising a compound of formula I or
physiologically-hydrolysable and -acceptable ester or amide thereof
as hereinbefore defined, or a pharmaceutically acceptable salt
thereof, together with a pharmaceutically acceptable diluent or
carrier therefor.
Suitable pharmaceutically acceptable salts of the compounds and
esters of the invention include for example the sodium and pota~sium
salts.