Note: Descriptions are shown in the official language in which they were submitted.
2007085
4-17407/+/CGC 1391
CERTAIN PYRROLYLPHENYL-SUBSTlTUTED HYDROXAMIC ACID
DERIVATIVES
Summary of the Invention
The invention relates to the pyrrolylphenyl-substituted hydroxamic acid derivatives as
defined herein which are particularly useful as selective lipoxygenase inhibitors, methods
for preparation thereof, pharmaceutical compositions comprising said compounds, and a
me$hod of inhibiting lipoxygenase and of treating diseases in mammals which are respon-
sive to lipoxygenase inhibition using said compounds or pharmaceutical compositions
comprising said compounds of the invention.
The compounds of the invention are particularly useful for the treatment of various
inflammatory and allergic conditions, e.g. bronchial allergies and inflammatory disorders
such as asthma, ocular allergies and inflammation, and dermatological allergies and
infl~nmation such as psoriasis; also for the treatment of rheumatic disorders such as
rheumatoid arthritis; and also for the treatment of ischemic conditions such as myocardial
infarction and cerebral ischemia.
Detailed Description of the Invention
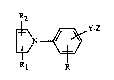
More particularly the invention relates to the compounds of formula I -
R2
wherein R represents hydrogen, lower aL~yl, halogen or lower alkoxy; Rl and R2 indepen-
dendy represent hydrogen, lower aL~yl or aryl; Y represents a direct bond, lower aL~cylene,
lower aLkenylene, lower aL~cadienylene, (thio, sulfinyl or sulfonyl)-lower alkylene or oxy-
lower aLtcylene; Z represents
200708S
R4 (a)
wherein R3 represents hydrogen or acyl; R4 represents lower aLkyl, C3-C7-cycloalkyl, aryl
or aryl-lower alkyl; or Z represents
~6 f ~s
-(~ N-OR3 (b)
R7
wherein R3 represents hydrogen or acyl; Rs represents lower aLlcyl, C3-C7-cycloaLkyl, aryl,
aryl-lower alkyl, amino or N-(mono- or di-lower alkyl)-arnino; R6 and R7 represent
hydrogen or lower aLkyl; and pharmaceutically acceptable salts thereof provided that R3
represents hydrogen.
A particular embodiment of the invention relates to the compounds of formula I wherein Z
represents (a), i.e. of formula II
O R4
R2 ll l
~N ~y--C--N--O--R3
Rl R
. j . .
wherein R represents hydrogen, lower aLkyl, halogen or lower alkoxy; Rl and R2 indepen-
dently represent hydrogen, lower aLkyl or aryl; R3 represents hydrogen or acyl; R4 repre-
sents lower alkyl, C3-C7-cycloalkyl, aryl or aryl-lower alkyl; Y represents a direct bond,
lower aLkylene, lower aLkenylene, lower alkadienylene, (thio, sulfinyl or sulfonyl)-lower
alkylene or oxy-lower alkylene; and pharmaceutically acceptable salts of said compounds
provided that 1~3 represents hydrogen.
.
r, ~
-
2007085
- 3 -
Preferred are the compounds of formula II wherein Y represents lower alkenylene, lower
alkadienylene, lower alkylene, thio-lower alkylene or oxy-lower aLkylene; R represents
hydrogen or halogen; Rl and R2 independently represent hydrogen or lower aLkyl; R3
represents hydrogen or acyl; R4 represents lower aLkyl; and pharmaceutically acceptable
salts of said compounds provided that R3 represents hydrogen.
Further preferred are said compounds of formula II wherein Y represents lower
alkenylene, lower aLkylene or oxy-lower alkylene; R represents hydrogen; Rl and R2
represent hydrogen or lower alkyl; R3 represents hydrogen, lower alkanoyl or aroyl; R4
represents lower aLkyl; and pharmaceutically acceptable salts thereof provided that R3
represents hydrogen.
Particularly preferred are said compounds of formula II wherein Y represents lower
alkenylene; and pharmaceutically acceptable salts thereof provided that R3 represents
hydrogen.
Also particularly preferred are said compounds of formula II wherein Y represents lower
aIkylene; and pharmaceutically acceptable salts thereof provided that R3 represents
hydrogen.
A particular preferred embodiment of the invention represented by compounds of
formula II relates to the compounds of formula III
.
O R4
~N ~ CR~= CRg - C - N - OR3 (~
Rl
wherein Rl, R2 and R8 and Rg independently represent hydrogen, methyl or ethyl; R4
represents cl-c3-aLkyl;R3 represents hydrogen; and pharmaceutically acceptable salts
thereof.
,:i ,;j,,~ " " ~ ~ ~ " ~ , ~ " . ', " ', "
200708S
A further embodiment thereof relates to the compounds of formula IV
R9
R2 1 o R4
~N ~C - N - O - R3 (rV)
R~ R8
wherein Rl-R4, R8 and Rg have meaning as de~med above for compounds of formula III;
and pharmaceutically acceptable salts thereo
Particularly prefeIred are said compounds wherein Rl, R2 and R4 represent methyl or
ethyl; R3, R8 and Rg represent hydrogen; and pharmaceutically acceptable salts thereof.
The invention also relates to the compounds of formula I wherein Z represents (b), i.e. of
formula V
R2 R6 1--R5
~N~Y - C - N - OR3 (V)
Rl R
wherein R represents hydrogen, lower alkyl, halogen or lower aLIcoxy; Rl and R2 indepen-
dently represent hydrogen, lower alkyl or aryl; Y represents a direct bond, lower alkylene,
lower alkenylene, lower alkadienylene, (thio, sulfinyl or sulfonyl)-lower alkylene or
oxy-lower alkylene; R3 represents hydrogen or acyl; Rs represents lower alkyl, C3-C7-
cycloalkyl, aryl, aryl-lower alkyl, amino or N-(mono or di-lower alkyl)-amino; R6 and R7
represent hydrogen or lower alkyl; and pharrnaceutically acceptable salts thereof provided
that R3 represents hydrogen.
Preferred are said compounds of formula V wherein Y represents a direct bond, thio-lower
alkylene, oxy-lower alkylene, lower alkenylene or lower alkylene; R represents hydrogen;
Rl and R2 independently represent hydrogen or lower alkyl; R3 represents hydrogen,
lower alkanoyl or aroyl; R5 represents lower aLkyl, aryl-lower alkyl, N-(mono- or di-lower
,~
,''' ::
,.
~j~,, ' .
:,.
.,
2007085
alkyl)-amino or amino; R6 represents hydrogen; R7 represents hydrogen or lower alkyl;
and pharmaceutically acceptable salts thereof provided that R3 represents hydrogen.
Further preferred are said compounds wherein Y represents a direct bond, lower alkylene
or lower aL~cenylene.
Particularly preferred are the compounds of the formula VI
H
R2
I~\N ~Y--f N--OR3 (VI)
4~ \J R7 c--R5
Rl O
wherein R~ and R2 represents hydrogen or lower alkyl; Y represents a direct bond,
methylene or ethylene; R3 represents hydrogen; Rs represents Cl-C3-aLkyl, N-(mono- or
di-lower aLtcyl)-amino or amino; R7 represents hydrogen or Cl-C3-aLtcyl; and pharma-
ceutically acceptable salts thereof.
Also preferred are the compounds of formula VII
R2 - H
~N ~ CR~= CRg--C--NI--OR3 (VII)
Rl R7 1--R5
o
wherein Rl, R2, R7, R8 and R9 represent hydrogen, methyl or ethyl; R3 representshydrogen; Rs represents Cl-C3-aLkyl, N-(mono- or di-lower aLkyl)-amino or amino; and
pharmaceutically acceptable salts thereof.
Generally preferred are the compounds of the invention, e.g. of formula I, II, III, V and VI,
wherein the point of attachment of the grouping Y is either meta or para to the pyrrole
ring, and also said compounds wherein Rl and R2 are attached at the 2 and 5 positions of
the pyrrole ring.
2007085
- 6-
The general definitions used herein have the following meaning within the scope of the
present invention.
The term "lower" referred to above and hereinafter in connection with organic radicals or
compounds respectively defines such as branched or unbranched with up to and includ-
ing 7, preferably up to and including 4 and advantageously one or two carbon atoms.
A lower alkyl group preferably contains 1-4 carbon atoms, advantageously 1-3 carbon
atoms, and represents for example ethyl, propyl, butyl or most advantageously methyl.
A lower alkoxy group preferably contains 1-4 carbon atoms and represents for example
ethoxy, propoxy, isopropoxy or advantageously methoxy.
Halogen preferably represents chloro or fluoro but may also be bromo or iodo.
Aryl represents preferably carbocyclic aryl.
Carbocyclic aryl represents for example phenyl or phenyl mono- or di-substituted by one
or two radicals selected from lower alkyl, lower alkoxy, halogen, cyano and trifluoro-
methyl; or 1- or 2-naphthyl.
Aryl-lower aLI~yl represents for example benzyl or phenylethyl.
Acyl is preferably lower alkanoyl or aroyl.
Lower alkanoyl represents preferably C2-C4-alkanoyl such as acetyl or propionyl.
Aroyl represents preferably benzoyl or benzoyl mono- or di-substituted by one or two
radicals selected from lower aLkyl, lower alkoxy, halogen, cyano and trifluoromethyl; or 1-
or 2-naphthoyl.
C3-C7-cycloaLkyl represents cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cyclo-
heptyl, preferably cyclohexyl or cyclopentyl.
Lower alkylene represents either straight chain or branched Cl-C7-alkylene and represents
preferably a methylene, ethylene, propylene or butylene chain, or said methylene,
2007085
ethylene, propylene or butylene chain monosubstituted on the same or different carbon
atoms by Cl C3-alkyl ~advantageously methyl), the total number of carbon atoms being up
to and including 7.
Lower alkenylene represents C2-C7-aLkenylene, may be straight chain or branched and
represents preferably straight chain Cl-C4-alkenylene or said straight chain Cl-C4-
alkenylene substituted on either saturated or unsaturated carbon atoms in the chain by one
or two of Cl-C3-aL~cyl, advantageously methyl, the total number of carbon atoms being up
to and including 7.
Lower alkadienylene represents C4-C7-aLlcadienylene and represents preferably 1,3-buta-
dienylene (C4-alkadienylene), unsubstituted or substituted by Cl-C3-alkyl (advantageously
methyl), the total number of carbon atoms being up to and including 7.
Pharmaceutically acceptable salts of the acidic compounds of the invention (provided that
R3 represents hydroxy) are salts formed with bases, namely cationic salts such as alkali
and alkaline earth metal salts, such as sodium, lithium, potassium, calcium, magnesium, as
well as ammonium salts, such as ammonium, trimethyl-ammonium, diethylammonium,
tris-(hydroxymethyl)-methylammonium salts.
The compounds of the invention exhibit valuable pharmacological properties in mammals,
and are particularly useful as selective 5-lipoxygenase inhibitors for the treatment of e.g.
inflammatory, allergic and ischemic conditions.
The above-cited properties are demonstrable in in vitro and in vivo tests, usingadvantageously mammals, e.g. rats, guinea pigs, dogs, rabbits or isolated organs, tissues,
and enzyme preparations thereof, as well as cells and fluids isolated from mammalian,
including human, blood. Said compounds can be applied in vitro in the forr,n of solutions,
e.g. preferably aqueous solutions, and in vivo either enterally or parenterally,advantageously orally, e.g. as a suspension or in aqueous solution. The dosage in vitro
may range between about 10-5 molar and 10-8 molar concentrations. The dosage in vivo
may range, depending on the route of administration, between about 0.01 and 30 mg/kg.
5-HETE and various leukotriene products are formed from arachidonic acid by means of
the enzyme 5-lipoxygenase. Leukotrienes (LTs) B4, C4, D4 and E4 are a group of media-
tors with potent leukocyte-chemoattractant, smooth muscle-constricting and vascular
," ., - . . . . . . , . . ~ . ,
.,,, . ~ . . . ..
2007085
permeability-enhancing properties. LTB4 is among the most potent leukocyte chemotactic
agents known. LTC4, LTD4 and LTE4 are the components of the "slow-reacting substance
of anaphylaxis" (SRS-A) and are potent inducers of broncho-constriction that are released
during an antigen challenge in lungs. Leukotrienes have been implicated in the patho-
genesis of a variety of vascular and pul~onary disorders involving leukocyte and smooth
muscle activation. Since these products are derived from the biotTansforrnation of arachi-
donic acid (AA) through the 5-lipoxygenase pathway, selective inhibition of 5-lipoxy-
genase will suppress biosynthesis of leukotrienes in leukocytes and various organ systems.
5-Lipoxygenase inhibition is determined e.g. by measuring the percent inhibition of the
synthesis of 5-HETE [(5S)-5-hydroxy-6,8,11,14-eicosa-tetraenoic acid] and leukotriene B4
(LTB4,5,12-dihydroxy-6,8,10,14-eicosa-tetraenoic acid) in A-23187-stimulated guinea
pig polymorphonuclear leukocytes, essentially according to radiometric thin-layer
chromatogTaphic assays described by Walker and Dawson (J. Pharm. Pharmacol. 31: 778,
1979) and Jakschik and Lee (Nature 287: 51, 1980) used to measure the formation of
5-HETE and LTB4-like products from l4C-arachidonic acid. `IC50 values are determined
graphically as the COnCentTation of test compound at which the synthesis of 5-HETE and
LTB4-like products is reduced to 50 % of their respective control values.
The inhibition of LTB4 formation can also be determined in vitro in whole blood from
dogs. One ml samples of blood aTe preincubated at 37C for 5 minutes with the desired
concentration of test compound added as a solution in 10 microliters of dimethylsulfoxide.
LTB4 synthesis is then stimuIated by the addition of A-23187 and N-formyl-met-leu-phe
(f-MLP). The amount of LTB4 is measured in the separated plasma fraction by Tadio-
immunoassay. ICso values are determined graphically as the concentration of testcompound causing 50 % inhibition of LTB4 formation seen in control whole blood.
Furthermore, the inhibition of 5-lipoxygenase is determined after oral or i.v.
administTation to rats or dogs by measuring ex vivo in whole blood the decrease of
A-23187-stimulated LTB4 formation as compared to non-treated control animals.
Antiinflammatory activity is demonstrated by measuring the inhibition of the edema and
inhibition of the influx of polymorphonuclear (PMN's) and mononuclear leucocytes(monocytes) after oral administration in the rat model in which pleurisy is first induced by
injecting CaTTageenin into the pleural cavity, e.g. according to A.P. Almeida et al, J.
Pharmacol. Exp. Therap. 214,74 (1980).
,~
.',~
' ~
'": ",,
.
2007085
Illustrative of the invention, the compound of example 1, (E)-3-[4-(2,5-dimethyl-lH-
pyrrol-l-yl)-phenyl]-N-hydroxy-N-methyl-2-propenamide, inhibits the formation ofS-HETE [(5S)-5-hydroxy-6,8,11,14-eicosa-tetraenoic acid] and leukotriene B4 (LTB4,
5,12-dihydroxy-6,8,10,14,eicosa-tetraenoic acid) in A-23187-stimulated guinea pig
polymorphonuclear leukocytes, e.g. at an ICso of about 0.7 micromolar (7x10-7M). Said
compound also inhibits by 50 % LTB4 formation as determined ex vivo when administer-
ed at a dose of about 1 mg/kg p.o. to the dog.
Furthermore, the compound of example 1, at 10 mg/lcg p.o. administered for two days at
-1, 6, 24 and 45 hours relative to the carrageenin injection, causes inhibition of exudate
volume and lowers the cell count of leukocytes 48 hours after injection of carrgeenin in
the rat pleurisy model of inflammation. ~ ;
The compounds of the invention are thus useful, particularly for the treatment and
amelioration of diseases and conditions in mammals, including man, in which lipoxy-
genase activity or the accumulation of leukocytes (e.g. neutrophils) is involved,
particularly allergic and inflammatory disorders, e.g. pulmonary allergies and
inflammatory disorders such as asthma), dermatological allergies and inflammatory dis-
orders (such as psoriasis), also arthritic inflammatory disorders (such as rheumatoid
arthritis), ocular allergies and inflammatory disorders, as well as ischemic conditions
(such as in myocardial infarction).
The compounds of the invention can be prepared by the following synthetic processes~
(a) for compounds of forrnula I condensing a compound of the formula VIII
H2N ~ (VIII)
R
wherein R, Y and Z have the meanings as defined hereinbefore (preferably whereinhydroxy groups are in protected form) with a compound of the formula IX
Rl-cocHRl-cHR2-co-R2 (IX)
f'
2007085
- 10-
or a compound of the forrnula IXa
Rl~ R2
Rl~l J~R2 (IXa)
RbO--~o~ ~ORa
wherein Rl and R2 have the meanings as defined hereinbefore with the proviso that one of
the two substituents Rl and one of the tWO substituents R2 has to be hydrogen and Ra and
Rb represent lower aL~yl, to form the pyrrolyl substituted compounds of formula I or
(b) for compounds of forrnula II condensing a compound of folmula X
L~ ~,Y-COOR (X)
Rl R
or a reactive ffinctional derivative thereof, wherein R, Rl, R2 and Y have the meanings as
defined hereinbefore, with a compound of the formula XI
R4-NH-OR3 (XI)
wherein R3 and R4 have the meanings as defined herein, optionally in protected form
whon R3 represents hydrogen; and
(c) for compounds of formula V condensing a compound of the formula XII
~, Y--C--NH--OR3 (Xll)
Rl R
wherein R, Rl, R2, R3, R6 and R7 have the meanings as defined hereinbefore with a
compound of the formula XIII
Rs-COOH (xm)
,. ~
2007085
or a reactive functional derivative thereof, wherein Rs represents lower alkyl, C3-C7-
cycloalkyl, aryl, aryl-lower alkyl or di-lower alkylarnino; or
(d) condensing a compound of the formula XII above with phosgene followed by
ammonia, a mono lower alkylamine or a di-lower alkylamine; or
(e) condçnsing a compound of the formula XII above with a lower alkyl isocyanate or
tri(lower alkyl)silyl isocyanate and subsequent removal of the silyl protecting group.
In the above cited processes, the said process is carried out while, if necessary, -
temporarily protecting any interfering reactive group(s), and then liberating the resulting
compound of the invention; and, if required or desired, a resulting compound of the
invention is converted into another compound of the invention, and/or, if desired, a
resulting free compound is converted into a salt or a resulting salt is converted into a free
compound or into another salt; and/or a mixture of isomers or racemates obtained is
separated into the single isomers or racemates; and/or, if desired, a racemate is resolved
into the optical antipodes. -
In starting compounds and intermediates which are converted to the compounds of the
invention in a manner described herein, functional groups present, such as amino and
hydroxy groups, are optionally protected by conventional protecting groups that are
cornmon in preparadve organic chemistry. Protected amino and hydroxy groups are those
that can be converted under mild conditions into free amino and hydroxy groups without
the molecular framework being destroyed or other undesired side reactions taking place.
The purpose of introducing protecting groups is to protect the functional groups from
undesired reactions with reaction components and under the conditions used for carrying
out a desired chemical transformation. The need and choice of protecting groups for a
particular reaction is known to those skilled in the art and depends on the nature of the
functional group to be protected (hydroxy group, amino group, etc.), the structure and
stability of the molecule of which the substituent is a part and the reaction conditions.
Well-known protecting groups that meet these conditions and their introduction and
removal are described, for example, in JP.W. McOmie, "Protective Groups in Organic
2007085
Chemistry", Plenum Press, London, New York, 1973, T.W. Greene, "Protective Groups in
Organic Synthesis", Wiley, New York, 1984.
In the processes cited herein, reactive functional derivatives of carboxylic acids represent,
for example, anhydrides especially mixed anhydrides, acid halides, acid azides, lower
alkyl esters and activated esters thereof. Mixed anhydrides are preferably such from
pivalic acid, or a lower alkyl (ethyl, isobutyl) hemiester of carbonic acid; acid halides are
for example chlorides or bromides; activated esters are for exarnple succinimido, phthalo-
imido or 4-nitrophenylesters; lower alkyl esters are for example the methyl or ethyl esters.
Also, a reactive esterified derivative of an alcohol in any of the processes cited herein
represents said alcohol esterified by a strong acid, especially a strong inorganic acid, such
as a hydrohalic acid, especially hydrochloric, hydroboromic or hydriodic acid, or sulphuric
acid, or by a strong organic acid, especially a strong organic sulfonic acid, such as an
aliphatic or aromatic sulfonic acid, for example methanesulfonic acid, 4-methylphenyl-
sulfonic acid or 4-bromophenylsulfonic acid. A said reactive esterified derivative is espe-
cially halo, for example chloro, bromo or iodo, or aliphatically or aromatically substituted
sulfonyloxy, for example methylsulfonyloxy or 4-methylphenylsuifonyloxy (tosyloxy).
The above processes for the synthesis of compounds of the invention can be carried out
according to methodology generally known in the art for the preparation of hydroxamic
acids and derivatives thereof.
The synthesis according to process (a) involving the condensation of a compound of
formula vm with a compound of formula IX or ~a is carried out in a inert solvent such
as toluene, optionally in the presence of an anhydrous acid so as to form the pyrrole
substituted compounds wherein E~l and R2 are located at the 2 and 3 or 4 and 5 positions of
the pyrrole ring.
The compounds of formula VIII, diketones or dialdehydes of formula IX and tetrahydro-
furan derivatives of formula IXa are either known in the art or can be prepared according
to the method known in the art.
The synthesis according to process (b) involving the condensation of a free carboxylic
acid of formula X with an optionally hydroxy protected hydroxylamine derivative of
formula XI can be carried out in the presence of a condensing agent, e.g. diethyl
~, : .. . . .
2007085
- 13-
phosphorocyanidate, 1,1 '-carbonylcliimidazole or dicyclohexylcarbodiimide, in an inert
polar solvent, such as dimethylformamide or methylene chloride.
The synthesis according to process (b) involving the condensation of a reactive functional
derivative of an acid of formula X as defined above, e.g. an acid chloride or mixed
anhydride with an optionally hydroxy protected hydroxylamine derivative of formula XI,
or a salt thereof in presence of a base such as triethylamine can be carried out, at a
temperature ranging preferably from about -78 to +75, in an inert organic solvent such
as dichloromethane or toluene.
The synthesis according to process (c) involving the condensation of a carboxylic acid of
formula XIII or a reactive functional derivative thereof with a hydroxylamine derivative of
formula XII optionally hydroxy protected when R3 represents hydrogen) is essentially
carried out as generally described for process (b).
In the case of acylation of the compounds of formula XII wherein R3 represents hydrogen,
e.g. with 2 mole equivalents or excess of a functional derivative of a compound of
formula XIII the N,O-bis-acylated compounds of formula V, narnely those wherein R3
represents COR5, are obtained. The N,O-diacylated compounds of formula V, e.g. wherein
Rs represents lower aLIcyl and R3 represents the corresponding COR5 group, can be
selectively O-deacylated under basic conditions, e.g. with potassium carbonate in a
hydroxylic solvent such as methanol to yield the corresponding compounds of formula V
wherein R3 represents hydrogen.
Processes (d) and (e) are directed to the preparation of urea derivatives, the compounds of
formula I wherein Z represents group (c), i.e. of formula V wherein Rs represents amino
or substituted amino, from hydroxylamines of formula XII.
The preparation according to process (d) can be carried out by reacting the hydroxylamine
derivative of formula XII preferably in hydroxy-protected forrn, with phosgene in an inert
solvent such as toluene in the presence of e.g. triethylamine, followed by condensation
with the appropriate amine at a temperature of about -25C to +50C.
The preparation according to process (e) involves the condensation of a hydroxylamine of
formula XII preferably in hydroxy-protected form, with a lower aL~cyl isocyanate in an
.,~ ~
. ., ~ ,
200708S
inert solvent such as toluene at a temperature Tanging from room temperature to reflux
temperature.
Protected forms of hydroxylamines of formula XI and XII (wherein R3 represents
hydrogen) in the above processes are those wherein the hydroxy group is protected for
exarnple as a benzyl ether or tetrahydropyranyl ether. Removal of said protecting groups is
carried out according to methods well known in the art, e.g. hydrogenolysis or acid
hydrolysis, respectively.
The starting materials of formula X can be prepared analogously as under process (a) e.g.
by condensing an ester of a compound of the formula
,~,Y-COOH
H2N ~ (XIV)
wherein R and Y have meaning as defined hereinabove with a compound of the forrnula
Rl-cocH2cH2co-R2 (XV)
or a compound of the formula
-
Rb~2Ra (XVa)
wherein Rl and R2 have meaning as previously defined, and Ra and Rb represent lower
aLkyl, in an inert solvent such as toluene, optionally in the presence of an anhydrous acid
so as to form the pyrrolyl substituted compounds wherein Rl and R2 are located at the
2 and 5 positions of the pyIrole ring. Starting materials wherein Rl and R2 represent lower
alkyl at other positions can be prepared from compounds wherein, in formula XV or XVa,
Rl and R2 are located at appropriate positions as indicated for example in process (a).
The starting materials of formula X for the preparation of e.g. of compounds of forrnula III
can also be prepared by condensing a compound of formula XVI
.... . ..
2007085
- 15-
R2
~ ~ (XVI) ~
Rl R
wherein Rl and R2 have meaning as defined hereinabove and X represents halogen, e.g.
bromo, with an optionally substituted acrylic acid or a functional derivative thereof under
conditions of the Heck reaction, e.g. in the presence of palladium chloride and tri-O-tolyl~
phosphine.
The acids of formula XIV, diketones or dialdehydes of formula XV and tetrahydrofuran
derivatives of formula XVa are either known in the art or can be prepared according to
methods known in the ar~
The hydroxylamine derivatives of formula XI are known or are prepared according to
methods well-known in the art for the preparation of hydroxylamines e.g. by condensing
corresponding halides with e.g. benzyl or tetrahydropyranyl O-pro~ected hydroxylamine,
by reduction of oximes orreduction of nitro compounds (particularly if R4represents aryl).
The starting hydroxylamines of forrnula X~ may be prepared from a corresponding
reactive derivative of an alcohol of formula XVII
Rl 6
~ R7 (XVII)
Rl R
wherein R, Rl, R2, Y, R6 and R7 have meaning as defined herein, such as the
corresponding bromide, tosylate or mesylate derivative, by condensing such with e.g.
` ~ O-benzylhydroxylamine or O-tetrahydropyranylhydroxylamine.
.
,: .
~ Alternately hydroxylamines of forrnula XII wherein at least one of R6 and R7 represents
;~; hydrogen can be prepared from the coIresponding aldehyde or ketone by conversion to the
oxime with e.g. hydroxylamine hydrochloride according to known methods, followed by
; ' .
~,~,,,,. i . . .
~; :,: - : : . : i
s,,
2~07085
- 16-
reduction to the hydroxylamine with e.g. diborane or sodium cyanoborohydride in acidic
medium.
The alcohols of forrnula XVII or corresponding aldehydes or ketones may be prepared e.g.
from the corresponding acids of formula X or ester derivatives thereof according to
methods well-known in the art. For example, such can be reduced to the alcohol wherein
R6 and R7 represent hydrogen using an appropriate reducing agent such as lithiumaluminum hydride, aluminum hydride and the like. Alternately the intermediates of
formula XVII may be prepared from the corresponding anilines by condensation thereof
with a compound of formula XV or XVa as described hereinabove for the preparation of
intermediates of formula X.
The carboxylic acids of formula XIII and reactive derivatives thereof are known in the art
or can be prepared according to methods well-known in the art.
Starting materials of formula vm for process (a) can be prepared according to process (b),
(c), (d) or (e) starting with appropriate protected aniline derivatives in reaction sequences
described above.
The above-mentioned reactions are carried out according to standard methods, in the
presence or absence of diluent, preferably such as are inert to the reagents and are solvents
thereof, of catalysts, condensing or said other agents respectively and/or inert atmos-
pheres, at low temperatures, room temperature or elevated temperatures (preferably at or
near the boiling point of the solvents used), and at atmospheric or super-atmospheric
pressure. The preferred solvents, catalysts and reaction conditions are set forth in the
appended illustrative examples.
The invention further includes any variant of the present processes, in which anintermediate product obtainable at any stage thereof is used as starting material and the
remaining steps are carried out, or the process is discontinued at any stage thereof, or in
which the starting materials are formed under the reaction conditions, or in which the
reaction components are used in the form of their salts or optically pure antipodes.
Advantageously those starting materials are used in said reactions that lead to the
forrnation of those compounds indicated above as being preferred.
~' ,':' ' ' - ~ .
2007085
Compounds of the invention can also be converted into each other according to methods
generally known per se, è.g. by hydrogenation of one or more double bonds.
Furthermore, compounds of forrnula I wherein R3 represents hydrogen can be converted to
compounds of formula I wherein R3 represents acyl using conventional O-acylationmethods. Conversely, compounds of formula I wherein R3 represents acyl can be
converted to compounds of formula I wherein R3 represents hydrogen using conventional
methods of ester hydrolysis.
The invention also relates to any novel starting materials and processes for their
manufacture.
Depending on the choice of starting materials and methods, the new compounds may be in
the form of one of the possible isomers or mixtures thereof, for example, as substantially
pure geometric (Z or E, cis or trans) isomers, optical isomers (antipodes), racemates, or
mixtures thereof. The aforesaid possible isomers or mixtures thereof are within the
purview of this invention.
Any resulting mixtures of isomers can be separated on the basis of the physico-chemical
differences of the constituents, in known manner, into the pure geometric or optical
isomers, diastereoisomers, racemates, for example by chromatography and/or fractional
crystallization.
Any resulting racemates of final products or intermediates can be resolved into the optical
andpodes by known methods, e.g. by separation of the diastereoisomeric salts thereof,
obtained with an optically active acid or base, and liberating the optically active acidic or
basic compound. The hydroxamic acids (wherein R3 represents hydrogen) can thus be
resolved into their optical antipodes e.g. by fractional crystallization of d- or
l-alpha-methylbenzylamine, cinchonidine, cinchonine, quinine, quinidine, ephedrine,
dehydroabietylamine, brucine or strychnine)-salts.
Finally, acidic compounds of the invention are either obtained in the free form, or as a salt
thereof.
Acidic compounds of the invention may be converted into salts with pharmaceutically
acceptable bases, e.g. an aqueous aLIcali metal hydroxide, advantageously in the presence
2007085
of an etheral or alcoholic solvent, such as a lower aL~anol. From the solutions of Ihe latter,
the salts may be precipitated with ethers, e.g. diethyl ether. Resulting salts may be
converted into the free compounds by treatment with acids. These or other salts can also
be used for purification of the compounds obtained.
In view of the close relationship between the free compounds and the compounds in the
form of their salts, whenever a compound is referred to in this context, a corresponding
salt is also intended, provided such is possible or appropriate under the circumstances.
The compounds, including their salts, can also be obtained in the form of their hydrates, or
include other solvents used for their crystallization.
The pharmaceutical compositions according to the invention are those suitable for enteral,
such as oral or rectal, transdermal and parenteral amdinistration to mammal, including
man, to inhibit 5-lipoxygenase and for the treatment of disorders responsive thereto,
comprising an effective amount of a pharmacologically activè compound of the invention,
alone or in combination, with one or more pharmaceutically acceptable carriers.
The pharmacologically active compounds of the invention are useful in the manufacture of
pharmaceutical compositions comprising an effective amount thereof in conjunction or
admixture with excipients or carriers suitable for either enteral or parenteral application.
Preferred are tablets and gelatin capsules comprising the active ingredient together with a)
diluents, e.g. lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine; b)
lubricants, e.g. silica, talcum, stearic acid, its magnesium or calcium salt andlor poly-
ethyleneglycol; for tablets also c) binders e.g. magnesium aluminum silicate, starch paste,
gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and or polyvinyl-
pyrrolidone; if desired d) disintegrants, e.g. starches, agar, alginic acid or its sodium salt,
or effervescent rnixtures; and/or e) absorbents, colorants, flavors and sweeteners. Inject^
able compositions are preferably aqueous isotonic solutions or suspensions, and supposi-
tories are advantageously prepared from fatty emulsions or suspensions. Said composi-
tions may be steriliæd and/or contain adjuvants, such as preserving, stabilizing, wetting or
emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or
buffers. In addition, they may also contain other therapeutically valuable substances. Said
compositions are prepared according to conventional mixing, granulating or coating
methods, respectively, and contain about 0.1 to 75 %, preferably about 1 to 50 %, of the
active ingredient.
;,
-~
2007085
- 19-
Suitable formulations for transdermal application include an effective amount of a com-
pound of the invention with carrier. Advantageous carriers include absorbable pharma-
cologically acceptable solvents to assist passage through the skin of the host. Characteri-
stically, transdermal devices are in the form of a bandage comprising a backing member, a
reservoir containing the compound optionally with carriers, optionally a rate controlling
barrier to deliver the compound of the skin of the host at a controlled and predetermined
rate over a prolonged period of time, and means to secure the device to the skin.
The invention also relates to a method of inhibiting 5-lipoxygenase activity in mammals
and treating diseases and conditions responsive thereto, particularly inflammatory and
allergic disorders, which comprises administering to a mammal in need thereof aneffective amount of a compound of the invention or of a pharmaceutical composition com-
prising a said compound in combination wi~h one or more pharmaceutically acceptable
carriers.
The dosage of active compound adrninistered is dependent on the species of warm-blooded animal (mammal), the body weight, age and individual condition, and on the form
of administration.
.
A unit dosage for a mammal of about 50 to 70 kg may contain between about
10 and 100 mg of the active ingredient.
The following examples are intended to illustrate the invention and are not to be construed
as being limitations thereon. Temperatures are given in degrees centrigrade. If not
mentioned otherwise, all evaporations are performed under reduced pressure, preferably
between about 15 and 100 mm Hg. The structure of final products, intermediates and
starting materials is confrmed by standard analytical methods, e.g. microanalysis and
spectroscopic characteristics (e.g. MS, IR, NMR).
Example 1
(a) A solution of 12.05 g of (E)-3-[4-(2,5-dimethyl-lH-pyITol-l-yl)-phenyl]-2-propenoic
acid in 350 ml of dichloromethane and 9.7 ml triethylamine, is cooled in an ice bath. To
this stirred solution is added 7.0 ml of ethyl chloroforrnate dropwise. After four hours at
ice-bath temperature, 20.7 ml of triethylamine is added, followed by 12.6 g of N-methyl-
hydroxylamine hydrochloride and the mixture is kept at room temperature for 16 hours.
,~
2007085
- 20 -
This mixture is washed with cold 0.3 N aqueous hydrochloric acid, ice cold sodium
bicarbonate and brine. The organic phase is dried over MgS04, treated with charcoal and
filtered. The solvent is evaporated at 50 under reduced pressure and the product is
crystallized from ether to give (E)-3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-N-
hydroxy-N-methyl-2-propenamide, m.p. 143-145, the compound of formula IV wherein
Rl and R2 represent methyl at the 2 and 5 positions, R4 represents methyl, and R3, R8 and
Rg represent hydrogen; the compound may also be named (E)-3-[4-(2,5-dimethyl-lH-pyrrol- l-yl)-phenyl]-N-methyl-2-propenohydroxamic acid. The product can also berecrystallized from ethyl acetate (with charcoal and silica gel).
(b) The sodium salt is prepared as follows:
A solution of 7.4 ml of lN sodium hydroxide in methanol is added to a solution of 2.0 g of
the hydroxamic acid in 50 ml of dichloromethane. This solution is evaporated at 60 under
reduced pressure to give sodium (E)-3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-N-
methyl-2-propenohydroxamate, m.p. 235.
(c) Similarly prepared is potassium (E)-3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-N-
methyl-2-propenohydroxamate, m.p. 200-202.
(d) The tromethamine salt is prepared as follows:
(E)-3-[4-(2,5-dimethyl-lH-pysrol-l-yl)phenyl]-N-methyl-2-propenohydroxamic acid
(1.85 g) is added to a solution of 0.85 g of tromethamine in 150 ml of methanol. The
resulting solution is treated with charcoal, filtered and evaporated to dryness. The residue
is crystallized from ether to obtain (E)-3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)phenyl]-N-
methyl-2-propenohydroxamic acid trometharnine salt, m.p. 134-136.
The starting material is prepared as follows:
A mixture of 21.4 g of methyl 4-aminocinnamate and 50 g of acetonylacetone in 500 ml of
toluene is heated under reflux using a Dean Stark water separator for 16 hours. The solu-
tion is washed with ice cold dilute hydrochloric acid, brine, and then dried over
magnesium sulfate. The organic phase is filtered through silica gel, evaporated at 60
under reduced pressure and the residue is crystallized from hexane to yield methyl 4-(2,5-
dimethyl-lH-pyrrol-l-yl)-cinnamate, m.p. 73-75. A solution of the ester in 150 ml of
methanol and 100 ml lN aqueous sodium hydroxide is stirred at room temperature for
,~ .~.: , . .
. ~
.~".-, `;..,:
Z007085
- 21 -
16 hours. Methanol is evaporated at 60 under reduced pressure. The basic solution is
washed with ethyl acetate, then made acidic with ice cold SN aqueous hydrochloric acid,
and extracted with ethyl acetate. the extract is washed with brine, dried over magnesium
sulfate, treated with charcoal, and evaporated to dryness at 60 under reduced pressure.
The residue is crystallized from etherlhexane (2:1) to yield (E)-4-(2,5-dimethyl-lH-
pyrrol-l-yl)-cinnamic acid, m.p. 189-191, also named (E)-3-[4-(2,5-dimethyl-lH-pyrrol-
l-yl)-phenyl]-2-propenoic acid.
3-[4-(2,5-Dimethyl-lH-pyrrol-l-yl)phenyl]-2-propenoic acid can also be prepared as
follows:
To a stirred solution of 50.0 g 1-(4-bromophenyl)-2,5-dimethylpyrrole (Meakins et al, J.
Chem. Soc., Perkin Trans. I, 1984, 2801-2807) and tri-O-tolylphosphine (2.5 g) in triethyl-
amine (100.0 ml), under a nitrogen atmosphere, is added decolorizing carbon (5.0 g) and
palladium chloride (0.35 g). To this stirred slurry is added acrylic acid (20.0 g) followed
by triethylamine (39.0 ml). The reaction mixture is refluxed for 4.5 hours, cooled to room
temperature and diluted with water (700 ml). The pH of the reaction mixture is adjusted to
ca. 2 by the slow addition of conenctrated hydrochloric acid (63 ml). The solids are
collected by filtration and the ~llter cake is washed with water until the pH is ca. 7 and
then dried. The dried filter cake is suspended in boiling ethanol (350 ml), clarified, cooled
to room temperature and diluted with water (450 ml) to induce crystallization. The slurry
is cooled to -10C and the product collected by filtration. The filter cake is dried in vacuo
at 45. The dried solid is slurried in cold toluene, filtered and to dried to afford 3-[4-(2,5-
dimethyl-lH-pyrrol-l-yl)phenyl]-2-propenoic acid, m.p. 184-185.
Example 2
A solution of 1.0 g of 2-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenylthio]-acetic acid in 75 ml
of dichloromethane and 8.0 ml triethylamine, is cooled in an ice bath. To this solution is
added 0.5 ml of trimethylacetyl chloride and the solution is stiIred at room temperature
overnight. Triethylamine (2 ml) is added followed by 1 g of N-methylhydroxylamine
hydrochloride and the- mixture is kept at room temperature for 20 hours. The mixture is
washed with 0.3 N aqueous hydrochloric acid, ice cold dilute sodium bicarbonate and
brine. The organic phase is dried over MgSO4 and filtered. The solvent is evaporated at
50 at reduced pressure, the residue is dissolved in diethyl ether and the solutions filtered
through 10 g of silica gel to yield 2-[4-(2,5-dimethyl-lH-pyrrol-l-yl)phenylthio]-N-
hydroxy-N-methylacetamide; NMR (DMSO-d6): ~ 7.1, 5.85, 3.25, 1.95.
-,. ~ - .. .. ~, -
2007085
- 22 -
The StaTting material is prepared by treatment of ethyl 4-aminophenylthioacetate with
acetonylacetone according to previously described procedure to yield ethyl 2-[4-(2,5-di-
methyl-lH-pyrrol-l-yl)-phenylthio]-acetate which is then hydrolyzed to the acid.
Example 3
Prepared in a similar manner as described in the previous examples are:
(a) 2-[4-(2,5-dimethyl-lH-pyrrol-l-yl~-phenoxy]-N-hydroxy-N,2-dimethylpropanamide,
m.p. 122-124.
The starting material is prepared as follows:
A mixture of 4.0 g of p-aminophenol and 4.0 g of acetonylacetone is heated under reflux
in toluene for 3 hours using a Dean Stark water separator to yield 4-(2,5-dimethyl-lH-
pyrrol-l-yl)-phenol. Condensation with ethyl alpha-bromoisobutyrate in ethyl alcohol in
the presence of potassium carbonate gives ethyl 2-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-
phenoxy]-2-methylpropanoate. Hydrolysis with 1.5 N sodium hydroxide solution yields
2-[4-(2,5-dimethyl- lH-pyrrol- 1 -yl)-phenoxy]-2-methylpropanoic acid, m.p. 178- 180.
(b) 2-[4-(2,5-dimethyl-lH-pyIrol-l-yl)-phenoxy]-N-hydroxy-N-methylacetamide, m.p.
148-150.
The starting material is prepared as described under (a) using ethyl bromoacetate instead
of ethyi alpha-bromoisobutyrate.
(c) 3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-N-hydroxy-N-methylpropanamide, oil;
NMR (DMSO-d6): ~ 7.1,7.3, 5.85, 3.25, 2Ø
~ j .
The starting material is prepared as follows:
A solution of (13)-3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-2-propenoic acid in ethyl
acetate is hydrogenated at 3 atmospheres pressure using 10 % palladium on charcoal to
yield 3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-propanoic acid, as an oil.
7 ~" . f '`~
Z007085
(d) 2-[4-(2~-dimethyl-lH-pyrrol-l-yl)pheny~]-N-hydroxy-N-methylacetamide~ oil; NMR
(DMSO-d6): ~ 7.15, 7.35, 5.85, 3.4, 2Ø
The starting material is prepared from ethyl 4-aminophenylacetate and acetonylacetone
according to previously described procedures for a si;nilar condensation.
(e) 5-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-N-hydroxy-N-methylvaleramide; oil,
NMR (DMSO-d6): ~ 7.1, 7.25, 5.88, 3.3, 2Ø
The starting material is prepared as follows:
Triethyl 4-phosphonocrotonate is condensed with p-nitrobenzaldehyde in the presence of
sodium hydride in toluene/tetrahydrofuran to yield 5-(p-nitrophenyl)-penta-2,4-dienoic
acid ethyl ester, m.p. 150-152. Hydrogenation in ethyl acetate at one atmosphere pressure
and room temperature using Adams catalyst yields ethyl 5-(4-aminophenyl)valerate.
Condensation with acetonylacetone yields ethyl 5-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-
phenyl] -valerate.
(f) (E)-3-[4-(lH-pyrrol-l-yl)-phenyl]-N-hydroxy-N-methyl-2-propenamide.
The starting material is prepared by condensing methyl 4-aminocinnamate with 2,5-di-
ethoxytetrahydrofuran in glacial acetic acid under reflux for 1 hour. See e.g. J. Med.
Chem. 31, 802 (1988).
(g) 3-[4-(lH-pyrrol-l-yl)-phenyl]-N-hydroxy-N-methylpropanamide by hydrogenation of
the compound under (f).
(h) 2-[4-(lH-pyrrol-l-yl)-phenyl]-N-hydroxy-N-methyl acetamide.
,
The star~ng material is prepared by condensing ethyl p-aminophenylacetate with 2,5-di-
ethoxytetrahydrofuran.
(i) 2-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-3-chloro-phenyl-N-hydroxy-N-methylpropanamide.
The starting material is prepared from ethyl alpha-(3-chloro-4-aminophenyl)-propionate
(U.S. patent 3,868,391) and acetonylacetone.
~, ,: ~ - ,.... .
,~
2007085
- 24 -
(j) 2-[3-(lH-pyrrol-l-yl)-phenyl]-N-hydroxy-N-methyl acetamide. The starting material is
prepared from methyl 3-(lH-pyrrol-l-yl)-phenylacetate (J. Med. Chem. 31, 802 (1988)
and acetonylacetone.
Example 4
a) Acetyl chloride (1 ml) is added while stirring to a solution of 1.4 g of N-hydroxy-1-[4-
(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-ethylamine and 2 ml of triethylamine in 100 ml of
methylene chloride at 0. The reaction mixture is stirred for 2 hours, washed with ice cold
dilute hydrochloric acid, water, dried over magnesium sulfate, treated with charcoal,
filtered and evaporated to dryness to yield N-acetyl-N-acetyloxy-1-[4-(2,5-dimethyl-lH-
pyrrol-l-yl)-phenyl]-ethylamine as an oil.
The starting material is prepared as follows:
A mixture of 4.0 g of p-aminoacetophenone and 4.0 g of acetonylacetone is heated under
reflux for 4 hours, and filtered to yield 4-(2,5-dimethyl-lH-pyrrol-l-yl)-acetophenone.
A mixture of 4-(2,5-dimethyl-lH-pyrrol-l-yl)-acetophenone (22.7 g), hydroxylamine
hydrochloride (25.0 g), pyridine (130 ml) in 100 ml of methanol is heated under reflux for
20 hours. Workup in the usual manner yields 4-(2,5-dimethyl-lH-pyrrol-l-yl)-aceto-
phenone oxime.
Sodium cyanoborohydride (6.9 g) is added to a rnixture of 4-(2,5-dimethyl-lH-pyrrol-l-
yl)-acetophenone oxime in 400 ml of methanol and ethanolic hydrochloric acid is added to
adjust to pH 3. The reaction mixture is stilred at room temperature overnight, poured over
ice, rendered basic and extracted with ether. The ether extract is washed with brine, dried
and evaporated to dryness. The residue is chromatographed over silica gel first with
methylene chloride and then with 15 % methanol, 85 % methylene chloride to yield in
fractions obtained with second eluent N-hydroxy-1-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-
phenyl]-ethylarnine, m.p. 109-111.
b) Similarly prepared is N-acetyl-N-acetyloxy-1-[3-(2,5-dimethyl-lH-pyrrol-l-yl)-
phenyl]-ethylamine as an oil.
rrr~
2007085
- 25 -
c) Similarly prepared is N-benzoyl-N-benzoyloxy-1-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-
phenyl] -ethylamine.
d) Similarly prepared is N-(N',N'-dimethylcarbamoyl)-N-(N,N'-dimethylcarbamoyloxy-
1-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-ethylamine using dimethylcarbamyl chloride
as the acylating agent.
e) Similarly prepared is N-acetyl-N-acetyloxy-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-cinnamyl-
amine. The starting material is prepared by first condensing p-aminobenzaldehyde with
acetonylacetone to obtain 4-(2,5-dimethyl-lH-pyrrol-l-yl)-benzaldehyde which is in turn
condensed with diethyl phosphonoacetaldehyde diethylacetal to obtain 4-(2,5-dimethyl-
lH-pyrrol-l-yl)-cinnamaldehyde. The aldehyde is then converted to the oxime and
reduced to the hydroxylamine according to previously described procedures.
Example 5
a) A mixture of 1.4 g of N-acetyl-N-acetyloxy-1-[4-(2,5-dimethyl-lH-pyIrol-l-yl)-
phenyl]-ethylamine and 7.0 g of potassium carbonate in 70 ml of methanol is stirred at
room temperature overnight and then evaporated to dryness. The residue is suspended in
water and the product is extracted with ethyl aceta~e. The ethyl acetate extract is washed
with brine, dried and evaporated to dryness. The residue is purified by chromatography on
silica gel to yield N-acetyl-N-hydroxy-1-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-ethyl-
amine; NMR (DMSO-d6): ~ 7.15, 7.45, 5.85, 2.0, 2.15.
b) Similarly prepared is
N-acetyl-N-hydroxy-1-[3-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-ethylamine; NMR
~DMSO-d6): ~ 7.1-7.5, 5.85, 2.0, 2.15.
c) Similarly prepared is N-benzoyl-N-hydroxy-1-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyi] -ethylamine.
d) Similarly prepared is N-acetyl-N-hydroxy-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-cinnamyl-
amine.
Example 6
a) A solution of 1.4 g of N-hydroxy-1-[4-(2,5-dimethyl-lH-pyIrol-l-yl)-phenyl]-ethyl-
amine in 60 ml of toluene is stiTred and treated first with one mole equivalent of 4.0 M
200708S
ethanolic HCI, followed by 15 ml of 2.0 M phosgene in toluene. The solution is stirred for
7 hours, 50 ml of concentrated ammonium hydroxide is added and the mixture is stirred
overnight. Excess water is then added and the mixture is again stirred for 2 hours. The
organic layer is separated, washed with brine, dried over magnesium sulfate, treated with
charcoal and evaporated to dryness. The residue is crystallized from ether to yield
N-carbamoyl-N-hydroxy-1-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-ethylamine.
b) Similarly prepared is N-carbamoyl-N-hydroxy-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-cinnamylamine.
N-Carbamoyl-N-hydroxy-4-(2,5-dimethyl-lH-pyrrol-l-yl)-cinnamylamine can also be
prepared as follows:
A mixture of 1.1 g of N-hydroxy-4-(2,5-dimethyl-lH-pyrrol-l-yl)-cinnamylamine and
5 ml of trimethylsilyl isocyanate in 100 ml ether is stirred at room temperature overnight.
Water is added, the mixture is stirred for an additional 4 hours, the ether layer is separated,
washed, dried and evaporated to dryness. The residue is purified by chromatography to
yield the product as a thick oil; IR 1685, 1662, 970 cm~l.
c) Similarly prepared is
N-carbamoyl-N-hydroxy-4-(2,5-dimethyl- lH-pyrrol- 1 -yl)-alpha-methylcinnamylarnine,
m.p. 94-
The starting material is prepared as follows:
A mixture of 4-(2,5-dimethyl-lH-pylrol-l-yl)-benzaldehyde (2.8 g), 5.0 g of l-triphenyl-
phosphoranylidene-2-propanone and 120 ml of tetrahydrofuran is treated under reflux for
3 days. The mixture is evaporated to dryness, the residue is stirred in hexane and the
suspension filtered. The hexane solution is evaporated to a small volume leading to
crystallization of 1-[4-(2,5-dimethyl-lH-pyrrol-l-yl)]-buten-3-one, m.p. 108-110, which
is in turn converted to the corresponding oxime and reduced to the hydroxylarnine.
Exarnple 7
a) A solution of 0.7 g of N-(N'-methylcarbamoyl)-N-benzyloxy-1-[4-(2,5-dimethyl-lH-
pyrrol-l-yl)-phenyl]-ethylamine in 50 ml of ethyl acetate is hydrogenated at room
~.,.; ,i - . . s .
... .
.:~
2007085
- 27 -
temperature and 3 atmospheres pressure for 3 hours to yield N-(N'-methylcarbamoyl)-N-
hydroxy-l-[4-(2~s-dimethyl-lH-pyrrol-l-yl)-phenyl]-ethylamine; m.p. 131-133.
The starting material is prepared as follows:
O-Benzylhydroxylamine hydrochloride (7.7 g) is added to a solution of 9.0 g of 4-(2,5-di-
methyl-lH-pyrrol-l-yl)-acetophenone in 200 ml of methanol, and the mixture is stirred for
3 hours. The solvent is removed under reduced pressure, the residue is dissolved in 300 ml
of hexane and the solution is washed first with 2N aqueous hydrochloric acid and then
brine. The organic layer is dried over magnesium sulfate, treated with charcoal and
evaporated to dryness to yield O-benzyl 4-(2,5-dimethyl-lH-pyrrol-l-yl)-acetophenone
oxime.
A solution of 6.6 g of O-benzyl 4-(2,5-dimethyl-lH-pyrrol-l-yl)-acetophenone oxime in
150 ml ethyl acetate is treated with 8 g of borane-pyridine complex followed by 45 ml of
6N aqueous hydrochloric acid. The mixture is stirred for 16 hours, poured over ice, made
aLl~aline to pH 9 and extracted with ether. The ether extract is washed with brine, dried
over magnesium sulfate and filtered to yield N-benzoyloxy-1-[4-(2,5-dirnethyl-lH-
pyrrol- 1 -yl)-phenyl]-ethylamine.
Methyl isocyanate (5 ml) is added to a solution of 2.1 g of N-benzyloxy-1-[4-(2,5-di-
methyl-lH-pyrrol-l-yl)-phenyl]-ethylamine in 200 ml of ether and the solution is stirred at
room temperature for 24 hours. Excess water is added and the mixture is stirred for
3 hours. The organic layer is separated, washed with brine, dried over magnesium sulfate
and evaporated to dryness. The residue is crystallized from ether-hexane (3:1) to yield
N-(N'-methylcarbamoyl)-N-benzyloxy-1-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-ethyl-
amine, m.p. 140-142.
b) Similarly prepared is N-(N'-methylcarbamoyl)-N-hydroxy-4-(2,5-dimethyl-lH-pyrrol-
l-yl)-cinnamyl-amine, m.p. 73-75.
Example 8
Triethylamine (2.06 g) is added to a mixture cf 5.0 g of (E)-3-[4-(2,5-dimethyl-lH-
pyrrol-l-yl)-phenyl]-N-hydroxy-N-methyl-2-propenamide in 100 ml of methylene
chloride and the mixture is stirred for 15 minutes. Acetic anhydride (2.07 g) is added and
the mixture is stirred at room temperatu~e overnight. The reaction mixture is washed with
2007085
- 28 -
water and sodium bicarbonate solution, the organic layer is separated, treated with
charcoal, dried and evaporated to dryness. The residue is crystallized from ethanol, water
(1:1) to yield (E)-3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl~-N-acetoxy-N-methyl-2-
propenamide, m.p. 120-121.
Example 9
A mixture of 2.5 g (E)-3-(4-aminophenyl)-N-acetoxy-N-methyl-2-propenamide and 2.5 g
acetonylacetone is heated at reflux in toluene using a Dean Stark water separator for
4 hours. The mixture is cooled, washed with water, brine and dried over magnesium
sulphate. Concentration yields after purification (E)-3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-
phenyl]-N-acetoxy-N-methyl-2-propenamide, the compound of Example 8.
The starting material is prepared as follows:
To a mixture of 34.4 g of p-bromoaniline in 400 ml of chloroform is added 200 ml of
saturated sodium bicarbonate, followed by 51 g di-t-butyl di~arbonate in 50 ml of chloro-
form. The mixture is first stirred at room temperature for 4 hours and then heated under
reflux overnight ~o yield 4-(t-butoxycarbonylamino)-bromobenzene, m.p. 101-103.
To a mixture of 27 g of 4-(t-butoxycarbonylamino)-bromobenzene, 0.18 g of palladium
chloride, 1.25 g of tri-O-tolylphosphine and 50 ml of triethylamine is added 9.5 ml of
acrylic acid followed by 25 ml of triethylamine. The reaction mixture is heated under
reflux for 3 hours. Methylene chloride (250 ml) is added, the reaction mixture is filtered
and evaporated to dryness. The residue is suspended in water, the suspension is washed
with toluene, concentrated HCl i3 added while cooling to pH 5. The resulting 4-(t-butoxy-
carbonylamino)-cinnamic acid is collected, washed with water and dried; m.p. 175-177.
Under conditions similar to those described in Example 1, 4-(t-butoxycarbonylamino)-
cinnamic acid is converted to (E)-3-(4-t-butoxycarbonylaminophenyl)-N-hydroxy-N- ~ -
methyl-2-propenamide, which is recrystallized from isopropanol-water; m.p. 220-221
dec.
Acetyl chloride (0.7 ml) is added dropwise with stirring to an ice cold solution of 2 g
(E)-3-(4-t-butoxycarbonylamino-phenyl)-N-hydroxy-N-methyl-2-propenamide and tri-ethylarnine (1 ml) in dichloromethane. The mixture is stirred for 2 hours, then washed
with ice cold lN HCl, water, brine and dried over magnesium sulphate. Concentration to
.,~
.- - . ~,
?' ~
2007085
- 29 -
dryness yields (E)-3-(4-t-butoxycarbonylaminophenyl)-N-acetoxy-N-methyl-2-propen-
amide.
Dry hydrogen chloride gas is bubbled for 45 minutes through a stirred solution of 1.5 g
(E)-3-(4-t-butoxycarbonylaminophenyl)-N-acetoxy-N-methyl-2-propenamide in an-
hydrous ethyl acetate which is cooled to -15. Nitrogen gas is then bubbled through the
solution to remove excess HCl and the solution is concentrated to yield the amine
hydrochloride.
The salt is partitioned between ethyl acetate and saturated aqueous sodium bicarbonate
solution. The organic phase is dried over magnesium sulphate and concentrated to yield
the free base, (E)-3-(4-aminophenyl)-N-acetoxy-N-methyl-2-propenamide which is used
immediately for the condensation with acetonylacetone.
Example 10
To a solution of 3.12 g of (E)-3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)phenyl]-N-acetoxy-N-
methyl-2-propenamide in 2-propanol (50 ml) cooled to 0, lN aqueous lithium hydroxide
(13 ml) is added with stirring. The mixture is stirred at room temperature for 30 minutes,
then partitioned between ethyl acetate and 2N HCl. The organic phase is washed with
brine, dried over magnesium sulphate and concentrated to dryness. Recrystallization from
ethyl acetate yields (E)-3-[4-(2,5-dimethyl-lH-pyrrol-l-yl)-phenyl]-N-hydroxy-N-methyl-2-propenamide of Example 1.
Example 11
a? Preparadon of 10,000 tablets each containing 20 mg of the active ingredient, having the
formula as follows:
(E)-3-[4-(2,5-dimethyl-lH-pyrrol-1-yl)-phenyl]-
N-hydroxy-N-methyl-2-propenamide 200.00 g
Lactose 2,535.00 g
Corn starch 125.00 g
Polyethylene glycol 6t ~ 150.00 g
Magnesium stearate 40.00 g
Purified water q.s.
~ '''~ '
.,
2007085
- 30-
Procedure: All the powders are passed through a screen with openings of 0.6 mm. The
drug substance, lactose, magnesium stearate and half of the starch are mixed in a suitable
mixer. The other half of the starch is suspended in 65 ml of water and the suspension
added to the boiling solution of the polyethylene glycol in 250 ml of water. The paste
forrned is added to the powders, which are granulated, if necessary, with an additional
amount of water. The granulate is dfied overnight at 35 broken on a screen with 1.2 mm
opening,s and compressed into tablets, using concave punches uppers bisected.
Analogously tablets are prepared, containing about 1-100 mg of one of the other com-
pounds disclosed and exemplified herein.
b) Preparation of 1,000 capsules each containing 10 mg of the active ingredient, having
the formula as follows:
(E)-3-[4-(2,5-dimethyl-lH-pyrrol-1-yl)-phenyl]-
N-hydroxy-N-methyl-2-propenamide ` 10.00 g
Lactose 207.00 g
Modified starch 80.00 g
Magnesium stearate 3.00 g
~E All the powders are passed through a screen with openings of 0.6 mm. Then
the drug substance is placed in a suitable mixer and mixed first with the magnesium
stearate, then with the lactose and starch until homogeneous. No. 2 hard gelatin capsuled -
are filled with 300 mg of said mixture each, using a capsule filling machine.
Analogously capsules are prepared, containing about 1-100 mg of the other compounds
disclosed and exemplified herein.