Note: Descriptions are shown in the official language in which they were submitted.
7 ~ X
~ BACRGROUND OF THE lNv~ ON
The present invention relates to pradimicin amides
which are active as antifungal agents, to pharmaceutical
compositions containing them, and to their use for treating
fungal infections.
Few examples of benzota]naphthacene quinones derived
from microbial sources have been reported and these include
compounds designated G-2N and G-2A, and KS-619-1. While no
biological activity was reported for G-2N and G-2A, KS-619-1
was disclosed as inhibitor of calcium ion and calmodulin-
dependent cyclic nucleotide phosphodiesterase. Recently,
published European Patent Application 277,621 discloses
antifungal antibiotics BU-3608 (Ia), BU-3608 B (Ib), and
BU-3608 C (Ic). Antibiotics benanomicins A and B were
reported in J. Antibiotics, 1988, 41:807-811; benanomicin B
appears to be the same as BU-3608 C whereas benanomicin A
has a hydroxyl group in place of the sugar amino group.
CONHCHCC2H
O ~ ~
CH~J~,
R-
R=CH3 R=H
Ia: R'=CH3; R"=B-D-xylosyl Id: R'=CH3; R"=~-D-xylosyl
Ib: R'=CH3; R"=H Ie: R'=H; R"=~-D-xylosyl
Ic: R'=H; R"=B-D-xylosyl
R=CHzOH
If: R'=CH3; R"=~-D-xylosyl
Ig: R'=H; R"=B-D-xylosyl
~ ~ ~ o ~
I Our co-pending application S.N. 602,076, filed 7 June,
1989 discloses BU-3608 D (Id) and BU-3608 E (Ie).
BU-3608 FA-l (If) and FA-2 (Ig) and derivatives thereof are
disclosed in our co-pending application S.N. 2,001,714,
filed 27 October, 1989. N-alkylated derivatives of the
BU-3608 group of compounds are disclosed in our co-pending
application S.N. 605,139, filed 7 July, 1989.
SUMMARY OF THE lNV~l~ lON
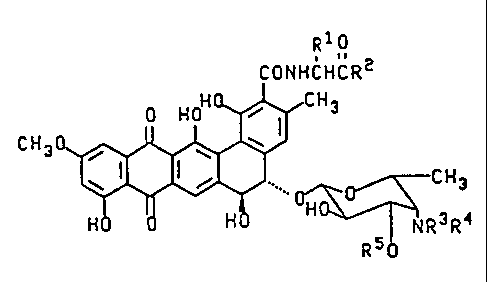
The present invention provides compounds of formula II
R 1 o
CONHCHCR2
C H 3 0 ~ N R 3 R
II
wherein R1 is selected from the group consisting of H, methyl
and hydroxymethyl, and when R1 is methyl or hydroxymethyl the
resulting amino acid residue has the D-configuration; R2 is
selected from the group consisting of -NR6R7, -NHNR6R7,
-NHCH2CO2H and (D)-NHCH(CH3)CO2H; R6 is H and R7 is selected
from the group consisting of H, C16alkyl, C36cycloalkyl,
C610aryl and C715aralkyl; or R6 and R7 are independently
C16alkyl; or R6, R7 and the nitrogen to which they are
attached form a 3- to 6-membered ring; R3 and R4 are each
independently selected from the group consisting of H and
C16alkyl; and R5 is H or B-D-xylosyl; or a pharmaceutically
acceptable salt thereof.
--' 2011117
There is provided by a further aspect of the present
invention pharmaceutical compositions for treatment of
fungal infections comprising an antifungal effective amount
of a compound of Formula II and a pharmaceutically
acceptable carrier.
A further aspect of the present invention provides a
method for treatment of fungal infections in a mammalian
host comprising administering to said infected host an
antifungal effective amount of a compound of ~ormula II.
DETAILED DESCRIPTION OF THE INVENTION
Pradimicin A refers to the antifungal agent formerly
known as BU-3608; BU-3608 B, C, D, and E is now pradimicin
B, C, D, and E, respectively. As used herein, the term
"alkyl" includes both straight and branched alkyl chains;
"pharmaceutically acceptable salt" includes acid addition
salts formed with, for example, hydrochloric acid,
hydrobromic acid, sulfuric acid, phosphoric acid, nitric
acid, acetic acid, tartaric acid, citric acid,
methansulfonic acid, succinic acid and the like; base salts
with an alkali metal base such as sodium or potassium
hydroxide, carbonate, and bicarbonate; and when possible,
internal salt.
In a preferred embodiment of compounds of Formula II,
R is selected from the group consisting of amino,
C1 6~1kylamino, di(C1 6)alkylamino, hydrazino, glycyl and
D-alanyl. Alkyl is more preferably of from one to four
carbon atoms.
--- 2 ~ 1 1 7
In another preferred embodiment of compounds of Formula
II, Rl is methyl.
In yet another preferred embodiment of compounds of
Formula II, R3 is H and R is H or methyl, or R3 and R4 are
both methyl; more preferably R is H and R4 is methyl.
Compounds of the present invention are prepared by
reacting a pradimicin of formula
Rl
CONHCHCOzH
CH30~CH3
~N R R
R50
wherein Rl is H, methyl or hydroxymethyl; R3 and R4 are
independently selected from the group consisting of H and
C1 6alkyl; and R is H or ~-D-xylosyl; or an acylating
equivalent thereof with an appropriate amine.
The amine reactant may be ammonia, primary amine,
secondary amine, hydrazine, mono- or disubstituted
hydrazine, or an amino acid. Examples of suitable amines
include, bu~ are not limited to, ammonia, methylamine,
ethylamir,e, propylamine, isopropylamine, butylamine,
pentylamine, hexylamine, dimethylamine, ethylmethylamine,
phenylamine, benzylamine, phenethylamine, cyclobutylamine,
cyclohexylamine, piperiaine, pyrrolidine, hydrazine,
methylhydrazine, l,l-dimethylhydrazine, glycine, and
D-alanine. Where 'he amine is an amino acid, the
5 ~ t- ~ ~
non-reacting functional groups, e.g. the carboxyl group, are
preferably protected. Suitable carboxyl protecting groups
are for example lower alkyl esters, benzyl and benzhydryl
esters and t-butyl esters.
The starting material pradimicins A, B and C are
produced by fermentation of Actinomadura hibisca strains
P157-2 (ATCC 53557) and Q278-4 (ATCC 53646) as disclosed in
applicant's Canadian Patent No. 1,309,045; the production of
pradimicins D and E by the mutant strain A2660 (ATCC 53762)
derived from the parent strain P157-2 is disclosed in S.N.
602,076 and the production of pradimicins FA-1 and FA-2 by
mutant strains A2493 (ATCC 53815) and B0012 (ATCC 53816)
capable of incorporating supplemented D-serine and derived
from the parent strain P157-2 is disclosed in S.N.
2,001,714. The general procedure for alkylating the sugar
amino group via reductive alkylation is provided in S.N.
605,139. The preparation of desxylosyl pradimicins by acid
hydrolysis is provided in the specification of the above-
mentioned applications.
The acylating species may be the carboxylic acid of the
pradimicin component or it may be a reactive derivative
thereof, for example an acid halide, an active ester or a
mixed anhydride. The acid halide may be generated by
reacting the carboxylic acid with a halogenating agent such
- 5 -
G'~
2011117
as thionyl chloride, phosphorous trichloride, phosphorous
pentachloride and oxalyl chloride in the presence of a
tertiary amine base to neutralize the acid produced. Active
esters may be prepared by reacting the carboxylic acid with
an al¢ohol such as a lower alkanol, an aromatic alcohol,
benzyl alcohol or an N-hydroxy compound such as
N-hydroxybenzotriazole and N-hydroxysuccinimide, in the
presence of a catalytic amount of an acid such as sulfuric
acid or toluenesulfonic acid or in the presence of a
coupling agent dicyclohexylcarbodiimide (DCC). Mixed
anhydrides may be obtained by treating the pradimicin with
an acid chloride derived from for example alkanoic acids or
aromatic carboxylic acids. When the pradimicin reactant is
used in the free acid form, the coupling reaction is
advantageously carried out in the presence of a condensing
agent such as DCC.
The sugar amino group of the pradimicin reactant is
optionally protected. Protection of the amino group is
preferred when a coupling agent is used in conjuction with
the free acid in the acylation step or when the acid
chloride is employed. The protecting groups for the amino
group are not particularly limited but may be any that can
be put on and removed easily without adversely affecting the
rest of the molecules. Suitable amino protecting groups
include benzyloxycarbonyl, t-butoxycarbonyl,
toluenesulfonyl, trifluoroacetyl and chloroacetyl; in our
experience, the benzyloxycarbonyl (CBZ) group has served as
a converient amino protecting group. The selection of amino
and/or car~oxyl protecting groups, and methods of blocking
and deblocking the non-reacting amino and/or carboxyl groups
are discussed in monographs such as "Protective Groups in
Organic Chemistry" J.F.W. McOmie, Plenum Press, 197~ and
-- 6 --
- 2011117
"Peptide Synthesis" M. Bodansky et al, Wiley, 1976, and are
generally within the skills of one of ordinary skill in
organic synthesis.
The condensation reaction is carried out in an organic
solvent such as tetrahydrofuran, dimethylformamide, acetone,
a lower alkanol, methylene chloride and acetonitrile, or an
aqueous mixture thereof. The temperature at which the
reaction is conducted is not particularly restricted and may
range from 0~C to 100~C although room temperature is
preferred. The reaction time needed depends on the
particular reactants used and the temperature at which the
reaction is carried out and may range from several minutes
to several days.
After the completion of the reaction am_no and/or
carboxyl protecting groups are removed using conventional
methods and the choice of deblocking method depends on the
protecting groups used. Far example, the CBZ group can be
readily removed by catalytic hydrogenation, and ester of the
D-amino acid moiety can be saponified to give the free
carboxyl group. The desired product of the present
invention may then be recovered and purified using standard
techniques such as solvent partition, recrystallization, and
column chromatography.
BIOLOGICAL PROPERTIES
Antifungal activities of representative compounds of
the present invention were evaluated both in vitro and ~n -
vivo. The minimum inhibitory concentrations (~iICs) against
various fungi were determined by serial agar dilution method
2011117
using Sabouraud dextrose agar. Thus, approximately 0.003 ml
of fungal suspension containing 106 cells/ml was applied to
the surface of agar plates containing the test antibiotics.
The MIC value~ recorded after the cultures had been
incubated for 44 hours at 28~C are set forth below in Table
I.
Table I. In vitro Antifungal Activity
Compound of MIC (~/ml)
Ex~mple C.albicsns C.neoformans A.fum~tu~ T.ment~Rrophytes
1 25 6.3 >100 >lOO
2 12.5 1.6 6.3 12.5
3 50 3.1 25 25
4 6.3 1.6 6.3 12.5
6.3 1.6 6.3 6.3
6 100 3.1 SO lOO
7 >50 I2.5 >50 >50
In vivo activity of compounds of the present invention
was tested against Candida albicans A9540 infection in mice.
Test organisms were cultured for 18 hours at 28~C in YGP
medium (yeast extract, glucose, peptone, K2HP04, MgS04) and
then suspended in saline. Male ICR mice weighing 20 to 24 g
were infected intravenously with about 10 times the me~ian
lethal dose of the test fungus. The antibiotic at various
dose levels was administered to groups of 5 mice each
intravenously just after the fungal infection. The dose
that protects 50% of the animals from infection (PD50,
mg/kg) was calculated from survival rates recorded on the
20th dcy after the fungal challenge. All control animals
-- 8 --
2011117
died within 7 to 15 days after infection. The PD50 for
compounds of Examples 2, 4, and 5 are 18 mg/kg, 11 mg/kg and
8.8 mg/kg, respectively.
Accordingly, another aspect of the present invention
provides a method for treating fungal infections which
comprises administering to a host infected with a
susceptible fungus an antifungal effective amount of a
compound of the present invention. For treatment of fungal
infections in animals and human beings, the antibiotics of
the present invention may be given in an antifungally
effective amount by any accepted routes of administration;
these include, but are not limited to, intravenous,
intramuscular, oral, intranasal, and for superficial
infections, topical administration. Preparations fo-
parenteral administration include sterile aqueous or
non-aqueous solu_ions, suspensions or emulsions. They may
also be manufactured in the form of sterile solid
compositions which can be dissolved in sterile water,
physiological saline, or some other sterile inject~ble
medium immediately before use. Oral formulation may be in
the form of tablets, gelatin capsules, powders, lozenges,
syrups, and the like. For topical administration, the
compound may be incorporated ,nto lotions, ointments, g~ls,
creams, salves, tinctures, and the like. Unit dosage forms
may be prepared using methods generally known to those
skilled in the art of pharmaceutical formulations.
It will be appreciated that when treating a host
infected with a fungus susceptible to the antibiotics of
this invention, the actual preferred route of administration
and dosage used will be at the discretion of the a~tending
clinician skilled in the treatment of fungal or viral
g _
2011117
infections, and will vary according to the causative
organism, its sensitivity to the antibiotic, severity and
site of the infection, and patient characteristics such as
age, body weight, rate of excretion, concurrent medications,
and general physical condition.
The following examples serve to illustrate the present
invention without limiting its scope which is solely defined
by the claims appended to the end of the specification.
Preparation Of N-benzyloxycarbonyl Pradimicin A (N-Cbz
Pradimicin A)
Benzyl chloroformate (5 ml) was added dropwise to a
solution of pradimicin A ~ HCl (2.5 g) and sodium carbonate
(7.5 g) in 50% agueous acetone (800 ml) at O~C. The mixture
was stirred for 1 hr at 0~C and 3 hrs at 10~C and then
concentrated to 200 ml. The resulting solution was mixed
with methanol (200 ml) and 6N sodium hydroxide (70 ml), and
kept for 15 hrs at room temperatu:-e to deblock the
0-benzyloxycarbonyl groups. After which time methanol was
removed and the residue was diluted with water. The
solution was adjusted to pH 9.0 to deposit
N-benzyloxycarbonyl pradimicin A (~.2 g) as a red solid.
Example 1
Preparation Of D-alanyl Pradimicin A (II, R =R =CH3;
R2=NHC~(CH3~C02H; R-=H; R =~-D-xylosyl).
A suspension of D-alanine benzyl este. tosylate (116
mg) and triethylamine (46 ~1) in tetrahydrofuran (2 ml) was
mixed with a so'ution of N-C~.z pradimicin A (300 mg), N-
- 10 -
~ O ~ 7.
hydroxybenzotriazole (49 mg) and dicyclohexylcarbodiimide
(68 mg) in tetrahydrofuran (20 ml). After the mixture had
been stirred for 2 hrs at O'C and then 13 hrs at room
temperature, it was diluted with water (100 ml) and
extracted with ethyl acetate (100 ml). The organic solvent
was evaporated to give a solid residue which was dissolved
in a mixture of methanol (30 ml), ethanol (10 ml) and water
(20 ml), and hydrogenated in the presence of 5% palladium on
carbon for 15 hours. The catalyst was filtered off and the
filtrate was concentrated and applied on a reversed-phase
silica gel column (~ 2.2 x 45 cm). Elution was carried out
with acetonitrile - 0.15% KH2P04 (21:79, pH 3.5) and
fractions containing the homogenous product were pooled,
concentrated and desalted by HP-20 chromatography (acetone-
lN HC1, pH 3) to yield D-alanyl pradimicin A ~HC1
(27 mg). The hydrochloride salt was dissolved in water and
the solution adjusted to pH 5.5 to provide D-alanyl
.
pradlmlcln A (20 mg).
MP 213 - 221~C (Dec.)
IR (KBr) 3400, 1620, 1445, 1260 cm~1
W ~x (50% methanol) nm (~) 221 (26,200) 276 (22,400), 502
(10,400).
SI-MS m/z 912 (M + H)~.
~.
Example 2
PreParation of Pradimicin A Dimethylamide (II, R1=R3=CH3;
R2=N(CH3)2; R4=H; R5=~-D-xylosyl)
A mixture of N-CBZ pradimicin A (224 mg, 0.23 mmol),
N-hydroxysuccinimide (32 mg, 0.28 mmol) and
dicyclohexylcarbodiimide (58 mg, 0.28 mmol) in
tetrahydrofuran (5 ml) was stirred for 1 hr at room
temperature and the resulting precipitate was filtered off.
The filtrate was added to a 50% aqueous solution of
dimethylamine (0.04 ml, 0.4 mmol) and the mixture was
stirred at room temperature overnight and then concentrated
in vacuo. The residue was chromatographed on a silica gel
column (5 g, chloroform/methanol = 10/1) to yield 215 mg
(93%) of N-benzyloxycarbonyl pradimicin A dimethylamide.
MP: 130 - 140~C
IR ~x (KBr) cm~1: 1712, 1623.
W ~x (MeOH) nm (E1% ): 286 (203), 482 (79)
lcm
1H NMR (400MHz, DMSO-d6 + D2O) ~: 1.01 (3H, d, J=5.6 Hz,
5'-CH3), 1.25 (3H, d, J=6.9 Hz, CHCH3), 2.59 (3H, s,
4'-N-CH3), 2.87 & 3.10 (3H, each, s, N(CH3)2), 4.89
(lH, q, J=6.9 Hz, CHCH3).
A mixture of N-benzyloxycarbonyl pradimicin A
dimethylamide (100 mg, 0.1 mmol), methanol (6 ml), water
(1.5 ml) and acetic acid (1.5 ml) was stirred with 10% Pd-C
(30 mg) under hydrogen atmosphere overnight. The solid was
filtered off and the filtrate was concentrated in vacuo.
- 12 -
C
- 2011117
The residue was dissolved in a small volume of 0.lN HCl and
charged on a reversed phase column (25mm x 150mm), eluted
with water, and then successively with 20%, 30% and 40%
aqueous acetonitrile. The desired fractions eluted with 40%
aqueous acetonitrile were combined, concentrated to a small
volume and lyophilized to yield 53 mg (61%) of a dark violet
powder.
MP: >230~C (grad. dec.).
IR vmax (KBr) cm 1 1628.
UV ~max (0.OlN NaOH) nm (E1%m): 319 (167), 496 (157)
lH NMR (400MHz, DMSO-d6 + D20) ~: 1.25 (2x3H, d, J=6.9 Hz,
5'-CH3 and CHCH3), 2.25 (3H, s, 3-CH3), 2.86 & 3.11 (3H
each, s, N(CH3)2), 3-91 (3H, s, ll-OCH3), 4.91 (lH, q, J=6.9
Hz, CHCH3), 6.71 (lH, d, J=2.6 Hz, 10-H), 6.84 (lH, br-s,
4-H), 7.12 (lH, d, J=2.6 Hz, 12-H), 7.62 (lH, br-s, 7-H).
Anal Calcd for C42H49N3~17 3H20
Found: C 54.54, H 5.63, N 4.72.
Example 3
Preparati~n Of Pradimicin A Hydrazide (II, Rl=R =CH3;
R =NHNH2i R =H; R =B-D-xylosyl)
A mixture of pradimicin A (210 mg, 0.25 mmol),
dicyclohexylcarbodiimide (60 mg, 0.3 mmol) and
N-hydroxysuccinimide (30 mg, 0.3 mmol) in dimethylformar,ide
(4 ml) was stirred at room tem~erature for 1.5 hr and then
filtered. The filtrate was stirred with hydrazine hydrate
- 13 -
. .
? ~
- 2011117
(13 mg, 0.3 mmol) at room temperature overnight and then
subjected to reversed phase column chromatography. The
column was washed with water and then eluted successively
with 20,'o, 30% and 40% aqueous acetonitrile. The desired
fractions eluted with 30% and 40% aqueous acetonitrile were
combined and f~rther purified by preparative HPLC (25%
acetonitrile) to yield 9 mg (4%) of the title compound.
MP: >220~C (grad. dec.)
IR vmax (KBr) cm : 1626, 1557.
UV ~max (O.OlN NaOH) nm (E1%m): 319 (142), 496 (129).
lH NMR (400 MHz, DMSO-d6) ~: 1.15 (3H, d, J=6.8 Hz, 6'-CH3),
1.30 (3H, d, J=7.3 Hz, CHCH3), 2.20 (3H, s, 3-CH3), 2.43
(3H, s, N-CH3), 3.91 (3H, s, OCH3), 4.35 - 4.42 (2H, m),
4.48 (lH, d, J=9.4 Hz, 1"-H), 4.60 (lH, d, J=6.8 Hz, l'-H),
6.71 (lH, d, J=2.1 Hz, 10-H), 6.86 (lH, s, 4-H), 7.12 (lH,
d, J=2.6 Hz, 12-H), 7.70 (lH, s, 7-H).
Example 4
Prepara'ion Of Pradimicin A Amide (II, R =R =CH3; R =NH2;
R =H; R =~-D-xylosyl).
A methanol solution (2 ml) of pradimicin A methyl
ester-HCl (95 m~) was added drop~ise to a stirred solution
of 28% ammonia (25 ml) and stirring was continued for 4
hours at room temperature. The solution was evaporated and
the solid residue was treated with 0.25N NaOH in aqueous
methanol (30 ml) at room temperature for 4 hours. The
solution was then acidified to pH 3.5, concentrated and
- 14 -
2011117
desalted by HP-20 column chromatography (lOO ml) to yield
semi-pure amide HCl (105 mg). The solid (100 mg) was
applied on a column of reversed phase silica gel (~20 x
450mm). Elution was carried out with a mixture of
CH3CN-0.15% KH2P04, pH 3.5 (21:79). The fractions
containing homogenous BU-3608 amide were pooled,
concentrated and desalted by HP-20 chromatography (100 ml,
eluent:acetone-lN HCl, pH 3) to yield pure amide HCl (59
mg). An aqueous solution of the hydrochloride was adjusted
to pH 6.0 to deposit the free form of pradimicin A amide (50
mg).
MP: 205 - 208~C (dec.)
IR (KBr)cm 1 3400, 1675, 1440, 1250.
W ~max (O.OlN NaOH -50% MeOH) nm (~) 245 (30,100), 320
(13,000), 496 (11,800)
SI-MS m/z 840 (M+H)
Example 5
Preparation Of Pradimicin A Methylamide (II, Rl=R3=CH3;
R =NHCH3; R =H; R =~-D-xylosyl)
The title compound (117 mg) was prepared according to
the general procedure described in Example 4 using as
starting materials pradimicin A methyl ester HCl (160 mg)
and 40~ aqueous methylamine (15 ml).
MP: 202 - 205~C (dec)
2 ~ 7
~ IR (KBr) cm~1 3400, 1620, 1440, 1290
W ~x (0.OlN NaOH - 50~ MeOH) nm (~) 245 (30,600), 320
(13,500), 496 (12,300)
SI-MS; m/z 854 (M+H)'
Example 6
Preparation Of Pradimicin A Butylamide (II, R1=R3=CH3;
R2=NH(CH2)3CH3; R4=H; R5=~-D-xylosyl)
The title compound (62 mg) was prepared according to
the general procedure described in Example 4 using as
starting materials pradimicin A methyl ester HCl (135 mg)
and butylamine (10 ml).
MP: 200 - 205~C (dec.)
IR (KBr) cm~1 3400, 1625, 1600, 1440, 1255
W ~x (0.OlN NaOH - 50% MeOH) nm (~) 245 (31,600), 320
(13,300), 496 (12,400)
SI-MS m/z 898 (M+3H)~.
Example 7
Pre~aration of Glycyl Pradimicin A (II, R1=R3=CH3;
R2=NHCH2C02H; R4=H; R5=~-D-xylosyl)
A mixture of N-Cbz pradimicin A (107 mg, 0.11 mmol),
N-hydroxysuccinimide (17 mg, 0.15 mmol) and DCC (31 mg, 0.15
~t - 16 -
~'
- 2011117
mmol) in THF (2 ml) was stirred at room temperature for 1.5
hr. The reaction mixture w~s filtered and to the filtrate
was added a mixture of glycine ethyl ester hydrochloride (21
mg, 0.15 mmol) and triethylamine (21 ~1, 0.15 mmol) in THF
(O.S ml). The mixture was stirred at room temperature
overnight and then diluted with ethyl acetate, washed
succesively with dil. HCl and water, dried over MgS04 and
concentrated in vacuo. The residue was chromatographed on a
silica gel column (5 g) and eluted with 10% CH30H-CHC13 to
yield 118 mg of the protected dipeptide of pradimicin A,
which was dissolved in a mixture of EtOH (10 ml), MeOH (20
ml), water (5 ml) and acetic acid (1 ml) and hydrogenated in
the presence of 10% Pd-C (30 mg) at room temperature
overnight. The mixture was filtered and the filtrate was
concentrated in vacuo. The residue was chromatographed on a
C-18 column, which was washed with water and eluted with 50%
CH3CN. Fractions containing the desired product were
combined, concentrated and lyophil~zed to yield 18 mg of
-glycyl pradimicin A ethyl ester. The ethyl ester was
hydrolysed in a mixture of MeOH (3 ml) and lN NaOH (3 ml) at
room temperature for 2 hr. The mixture was concentrated to
remove MeOH and subjected to C-18 column chromatography
(elution, water and 30% CH3CN) to give 9.5 mg (Y. 11%) of
the title compound.
MP: >195~C (grad. dec.).
IR vmax(KBr) cm 3366 (broad), 1733, 1607, 1558.
- 17 -
-- 2011117
lH NMR (DMSO-d6) ~ 1.28 (3H, d, J=6.4 Hz, 5'-CH3), 1.31 (3H,
d, J=7.3 Hz, 17-CH3), 2.29 (3H, s, 3-CH3), 3.83 (2H, d-ABq,
J=5.6 & 17 Hz, NHCH2-COOH), 3.97 (3H, s, OCH3), 6.97 (lH, d,
J=2.6 Hz, 10-H), 7.16 (lH, s, 4-H), 7.32 (lH, d, J=2.6 Hz,
12-H), 8.05 (lH, S, 7-H), 8.20 (lH, t, J=5.6 Hz, N_-CH2C02H,
disappeared in D20).