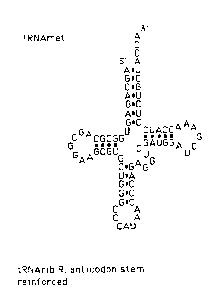Note: Descriptions are shown in the official language in which they were submitted.
,w 2 ~ ~ 2 31 2
55 141.304
Genetic Units for inhibiting the function of RNA
This invention relates to the specific inhibition of RNA
by interaction with RNA.
The specific inhibition of genes by oligonucleotides,
e.g. in order to achieve a therapeutic blocking of
deregulated oncogenes or viral genes, is based on the
ability of such complementary RNA or DNA so-called
antisense-oligonucleotides to hybridise with mRNAs,
processing signals or pre-mRNAs and in this way
interrupt the transfer of information from genes to
proteins.
The use of antisense DNA results in the breakdown of
complementary target RNA by RNAse H-cleaving of the
hybrid formed, resulting in the irreversible destruction
of the complementary RNA.
When antisense RNA is used, a so-called hybrid arrest of
translation or processing occurs, the RNA/RNA hybrids
constituting the structural obstacle. It is assumed
that such hybrids accumulate in the cells; their
subsequent fate has not hitherto been investigated. As
far as is known at present, this mechanism is largely
thought to be a reversible event. The use of antisense-
RNA molecules has the advantage that these molecules may
either be synthesized in vitro and introduced into the
cells or that the genes coding for them can be
introduced into the cells so that the inhibiting RNA can
be produced within the cell. However, nobody has
hitherto succeeded in bringing such genes into a form
which makes it possible to produce an effective quantity
of antisense RNA in the cell.
2012312
2
Very recently, a third principle of RNA inhibition has
been discovered and made available for use in vitro.
This principle is based on the ability of RNA molecules
the so-called ribozymes, to recognise certain RNA
sequences, bind to them and cleave them. It was derived
from the autocatalytic cleavage reactions of RNA
molecules in plant viroids and satellite RNA observed in
vivo.
On the basis of certain structural requirements for the
ribozyme catalysed RNA cleavage, it is now possible to
construct de novo ribozymes which have an endonuclease
activity directed in tr_ ans to a certain target sequence.
Since these ribozymes, of which the ones which have been
most carefully researched are known as hammer-head
ribozymes on account of their structure, can act on
numerous different sequences, the corresponding ribozyme
can be "made to measure" for virtually any RNA
substrate. This makes ribozymes interesting and
extremely flexible tools for inhibiting specific genes,
with the result that they are a promising alternative to
antisense constructs, which have already demonstrated
potential therapeutic use.
One ribozyme model currently known which has so far been
researched most thoroughly has three structural domains;
on the basis of this model, ribozymes against CAT-mRNA
have already been successfully constructed (Haseloff et
al., 1988: Uhlenbeck et al., 1987):
The three domains comprise:
a) a highly conserved region of nucleotides flanking
the cleavage site in the 5' direction. This
usually means the sequence GUC, although
modification in the GUA or GUU also showed a
substantially undiminished cleaving activity.
201 2312
3
Cleaving was also found after the sequences CUC,
and to a lesser extent for AUC and UUC as well (the
requirements for efficient cleaving have not yet
been fully explained).
b) the highly conserved sequences contained in
naturally occurring cleavage domains of ribozymes,
forming a sort of base-paired stem;
c) the regions which flank the cleavage site on both
sides and ensure the exact arrangement of the
ribozyme in relation to the cleavage site and the
cohesion of the substrate and enzyme (in the
experiments carried out hitherto, 8 bases were
selected on each side).
RNA enzymes can be constructed according to this model
and have already proved suitable ~n vitro for the
efficient and specific cleaving of RNA sequences
(Haseloff et al., 1988).
Very recently, further types of autocatalytic RNA
cleavage activity were discovered which may be used for
the targeted RNA inhibition. One of these models is the
so-called hairpin ribozyme, the active site of which is
derived from the minus strand of the satellite RNA of
tobacco ring spot virus (Hampel and Tritz, 1989). Other
self-cleaving RNA activities are associated with
hepatitis delta virus (Kuo et al., 1988: Sharmeen et
al., 1988; Wu et al., 1989) and with RNAseP (Altmann et
al., 1988).
The experiments which preceded the studies for the
present invention served to compare the activities of
antisense RNA, antisense DNA and ribozymes. These
experiments were carried out using the snRNP U7-
dependant histone-preRNA-processing reaction, in that an
in vitro system which processes U7-dependent histone-
preRNA (Mowry et al., 1987, Soldati et al., 1988). It
201 2312
4
was found that antisense RNA is the most potent inhibitor, and
the inhibition is reversible. The inhibitory effects of
antisense DNA and of ribozymes of the hammer-head type, both
of which are irreversible, were within the same order of
magnitude, each requiring an approximately one thousand-fold
excess over the substrate RNA to achieve total inhibition.
Whereas the previous tests had been carried out with
ribozymes with naked RNA in protein-free systems, the
preliminary tests for the present invention were the first
experiments to demonstrate that synthetically produced
ribozymes directed to a specific sequence show a cleaving
activity even in a medium which contains protein. This fact
provided a first indication of a potential use in vivo.
One of the limiting factors in the use of ribozymes
to inhibit the expression of specific genes would be in the
build-up of a ribozyme concentration which is sufficient to
efficiently rule out a specific biological reaction; the
reasons for this would be, among other things, the inadequate
stability of the RNA, as in the use of antisense RNA.
An object of the present invention is to provide a
system for using mRNA as an in vivo inhibitor of mRNA, which
overcomes the previous restrictions in the use of RNA, by
making an effective concentration of inhibition RNA available
in the cell.
One aspect of the invention provides DNA molecule
optionally occurring in multiple copies, containing sections
of a gene transcribed by polymerase III, and a DNA sequence
v
27855-24
241'2312
4a
coding for an inhibiting RNA molecule characterized in that it
contains the transcription units of a tRNA gene necessary for
transcription by polymerase III, including sequences which
determine the secondary structure of the tRNA, and the DNA
sequence coding for an inhibiting RNA molecule is arranged
inside the DNA molecule in such a way that the inhibiting RNA
molecule is part of the transcript.
27855-24
2Q~23~2
The idea which served as a basis, for this invention was
the realisation that, by introducing a gene which
produces the inhibiting RNA, as against importing the
RNA as such, considerable amplification of the RNA would
be ensured and consequently there would be a supply of
RNA sufficient to inhibit the biological reaction.
The inhibiting RNA may be any desired ribozyme or
another mRNA-inhibiting RNA, e.g. an antisense RNA.
Theoretically, it would be possible to carry out
efficient transporting of RNA or the DNA sequence coding
for it by means of viruses or viral vectors such as
retroviruses.
However, this system may have some serious
disadvantages, such as the mobilization of endogenous
viruses, recombination with endogenous retroviruses,
activation of endogenous genes by integration,
restriction with regard to host organism and type of
tissue.
By contrast, therefore, carrier genes are preferably
prepared according to the invention which do not have
these disadvantages.
The genes proposed as carrier genes for RNA genes within
the scope of the present invention have the following
advantages; they have a compact structure, they are
easier to transport in the cell, being smaller in size,
they have a high transcription rate and are not
restricted to specific tissues in their expression but
are expressed ubiquitously, i.e. in virtually all types
of cell.
A further advantage of the polymerase III genes is the
presence of a very powerful transcription termination
201 2312
6
signal. This reduces the probability of a nearby
cellular gene being undesirably activated.
The genes transcribed by the polymerase III have the
following features: they are genes wherein the promoter
is not located upstream in front of the gene but is
inside the gene (Geiduschek et al., 1988). These
internal control regions which are essential for the
binding of the polymerase III have a discontinuous
structure; they consist of two so-called boxes (the A
box and B box) which are essential for recognition by
the transcription factors, and an intermediate gene
section the length of which is critical (Hofstetter et
al., 1981). The length of this sequence is 31 to 74
base pairs in tRNA genes (Clarkson).
Examples of these genes which are transcribed by
polymerase III are the tRNA genes, 5S-RNA and a number
of other small nuclear and cytoplasm RNA genes: 7SK,
7SL, M, U6 and 4,5S RNA, as well as the adenovirus genes
VA1 and VA2 (Geiduschek et al., 1988). Something common
to these genes is their reduced size, compact structure,
high transcription rate and ubiquitous transcription.
It has surprisingly been found that using the genetic
units according to the invention it is possible to
achieve increased stability of the inhibiting RNA
without having to accept a reduction in their
effectiveness in terms of activity.
It has already been shown by Jennings and Molloy, 1987,
that a promoter recognised specifically by
polymerase III is suitable for guiding the synthesis of
antisense RNA. For this purpose, the XbaI/BamHl
fragment of the VA1 gene, containing the promoter, was
cloned in the 5' direction in front of the antisense
DNA, corresponding to the conventional principle when
201 2312
using polymerase II promoters. Endeavours were made to
obtain a transcript which was short in comparison with
polymerase II transcripts. The transcript has only one
slight modification which makes base-pairing of the ends
possible in order to protect the ends from digestion by
single strand exonucleases.
In contrast to this suggestion, in which only the
promoter sequence of a specific polymerase III gene is
used (the region between positions 30 and 73, whereas
the wild-type VA1 gene extends as far as the terminator
sequence at positions +160 to +200), according to the
present invention those sequences of the polymerase III
gene which determine the secondary structure of the
transcript are additionally used in controlled manner in
order to exploit them for stabilising the inhibiting RNA
sequences. In contrast to the proposal described above,
the genetic sequence coding for the inhibiting RNA is
arranged inside the gene transcribed by polymerase III
so as to obtain, according to the invention a "gene
cassette". Furthermore, the carrier genes used in the
present invention are non-toxic, unlike the viral VA1
gene.
The genes transcribed by polymerase III can be used
flexibly within the scope of the invention. With
respect to their function as carrier genes for RNA-
inhibiting sequences, the following criteria should be
considered when selecting or modifying natural genes or
artificial genes during construction:
1) the A and B boxes are highly conserved.
2) a section of 5 to 7 T residues downstream of the B
box is responsible for terminating the transcription.
3) the distance between the A and B boxes cannot be
enlarged at will, the maximum spacing currently being
assumed to be about 90 bp.
201 2312
8
4) in some systems, the 5' flanking sequence has an
influence on transcription.
5) there are indications that an intact anticodon stem
region is responsible for the stability of the
transcript.
tRNA genes are particularly suitable as carrier genes
for the production of the genetic unit according to the
invention. The RNAs coded by these genes are also
regarded as particularly suitable for stabilising the
inhibiting RNA, owing to their clover leaf structure.
Using these genes, it is possible to produce compact
RNA-producing genetic units, by inserting the sequence
which codes for the inhibiting RNA between the A and B
blocks (Fig. 1 shows a diagrammatic representation of a
tRNA ribozyme gene).
Experiments within the scope of the present invention
were carried out using the start methionine-tRNA, which
has an ApaI restriction site in the anticodon stem and
loop region located between the A and B blocks (the
start methionine-tRNAs of all higher eukaryotes have
this restriction site in the anticodon loop).
In addition to the region located between the A and B
blocks other insertion sites are also possible within
the carrier DNA sequences (tRNA genes or other
polymerise III genes), provided that care is taken to
ensure that the transcription activity of the
polymerise III and the stability of the transcript are
maintained.
However, theoretically, the genetic units according to
the invention may be produced with all tRNAs; if
necessary, a suitable restriction site may be
constructed, into which the sequence coding for the
inhibiting RNA is then inserted. The only condition is
2p12312
9
that during insertion no base exchange must be allowed
in the anticodon base region which will affect the
transcription rate of the gene or the stability of the
resulting tRNA (Folk et al., 1983). Furthermore, it
must be born in mind that inserts larger than 60 by may
effect the transcription (Ciliberto et al., 1982).
With regard to the choice of suitable carrier genes it
is fundamentally desirable to use tRNA genes of the same
species or other genes transcribed by polymerase III
which are not toxic.
The experiments within the scope of the present
invention were carried out by introducing ribozyme
sequences into normal tRNA genes. If the tRNA ribozyme
transcribed by this gene is located primarily in the
cytoplasm but it is desirable for specific applications,
e.g. in order to inhibit nucleus-specific RNAs, e.g.
those from snRNP particles, to locate the tRNA ribozyme
or the tRNA ribozyme or tRNa antisense RNA in the
nucleus, it is possible to use mutants which are located
predominantly in the nucleus, e.g. the met i tRNA gene
described by Zasloff et al., 1982 and by Zasloff, 1983,
which has a single base exchange.
The genetic units according to the invention may be
prepared as follows, for example:
A copy of the gene transcribed by polymerase III, e.g. a
tRNA gene, contained on a bacterial plasmid, is cleaved
with a suitable restriction enzyme at the site provided
for insertion, e.g. between the A and B blocks, and the
double stranded DNA produced by conventional methods,
which codes for the inhibiting RNA, is ligated therein.
Suitable host organisms are transformed therewith,
selected, replicated and the amplified plasmid-DNA is
obtained. The plasmids are checked for the presence of
w 2012312
to
the genetic unit according to the invention; this can be
done by restriction digestion, sequence analysis or by
detecting a functional in vitro transcript of the
plasmid DNA.
The genetic units thus obtained may be used both in the
form of the circular plasmid and also in the form of the
genetic unit cut out of the plasmid, which contains all
the information required for the transcription by
polymerase III.
The particular form chosen would generally depend on the
field of use and on the transport system selected for
introducing the genetic unit into the cell.
The genetic units according to the invention may occur
as multiple copies (tandem structures in which the
genetic units are arranged one behind another, with the
inhibiting inserts being identical or different).
Transcription of a tandem of this kind yields separate
RNA units, owing to the promoter and termination signals
contained in each individual unit. The orientation of
the individual units with one another is also irrelevant
owing to this property of the fragments to occur as
complete transcription units. When producing such
tandems, plasmids are used which contain multimeric
copies of the region coding for the inhibiting units.
Vectors which contain multimeric tRNA ribozymes genes
may be produced, for example, starting from the erbB-cut
series described in example 1, by ligating the
individual fragments using T4 DNA ligase to produce
polymers and recloning the polymeric genes into a
corresponding restriction site of a suitable plasmid
vector. The production of such tandems is made possible
by the small size of the units according to the
invention, which preferably amount to 200 to 350 bp.
2012312
Using a tandem inhibitor of this kind, after the
preparation of a heteromeric complex of antisense RNAs
or ribozymes, the effectiveness of the set of inhibitors
can be tested in a single experiment. For example, a
mixture of 5 to 10 ribozymes can be introduced into a
cell system using this method. After inhibition has
occurred as a result of the mixture, the individual
ribozymes can be tested for their activity. This is
particularly advantageous owing to the fact that the
criteria for selecting target RNA sequences which are to
be inhibited have not yet been thoroughly investigated.
Good target sequences include, for example, those
regions which have no secondary structure, regions near
cleavage signals, regions following the initiation
codon, regions without binding sites for specific
proteins (such as for example the Sm binding site in
snRNA molecules). Using the present invention,
particularly in tandem form, it is possible to come to
some conclusions regarding these target sequences and
consequently to inhibit them efficiently.
The use of tandem units is also advantageous when it is
necessary to cleave the RNA, e.g. viral RNA, at several
sites in order to inactivate biological processes.
Multiple cleaving can be achieved for example by
constructing a fairly large number of different
ribozymes and introducing a vector containing the
genetic units with the DNA sequences coding for them
into cells. When these sequences are transcribed, the
ribozyme units are added to the corresponding target
sequences, as a result of which the mRNA is split into
fragments.
Multimeric copies can be used when larger quantities of
a low molecular ribozyme are required for achieving
satisfactory results in specific applications, for
2412312
12
example when using the transferrin-polycation transport
system. Plasmids which carry multimeric copies of the
genetic unit and which contain the DNA sequence coding
for this ribozyme improves the production of the
fragment by increasing the yield to a multiple of what
it was.
Tandem units can also advantageously be used when it is
simultaneously desired to produce inhibiting RNAs
directed against various types of RNA.
When the invention is applied to antisense RNA care must
be taken to limit the size of the insert, in the case of
tRNA genes. For efficient transcription the order of
magnitude is about 60 bp; therefore, when larger
antisense-RNA constructs are used, other genes
transcribed by polymerase III which have a larger
capacity, with a view to transcribing longer sequences,
should be considered.
The carrier genes which can be used within the scope of
the invention may also be produced synthetically,
provided that they meet the condition of having the
transcription units necessary for transcription by
polymerase III. The use of such synthetic genes may
bring the following advantages:
a) such genes are not recognised by aminoacyl-
synthetases and by ribosomes and consequently
interference with the translation machinery of the
circle is avoided.
b) there is the possibility, by creating synthetic
constructs, of producing inhibiting RNAs with a
greater stability and higher transcription rate
than are obtained with a natural gene.
c) the cloning process can be made more flexible by
the creation of synthetic sequences.
2012312
13
It has been established, within the scope of the present
invention, that the stability of the ribozyme-tRNA
molecules can be increased by lengthening the anticodon
base region of the carrier tDNA molecule. It has been
demonstrated that, by lengthening the anticodon base
region of the wild-type tRNA gene, which has 5 base
pairs, to a total of 9 base pairs, it is possible to
obtain six times as much transcript compared with the
shortened wild-type tRNA gene, and this can be put down
to an increase in stability and the processability which
can be obtained as a result.
It has also been established that an increase in the
stability of the genetic units according to the
invention in the form of tRNA-ribozyme genes or tRNA-
antisense genes can also be achieved if the DNA
sequences coding for the inhibiting RNA function are
present as parts of introns (known as "ribintrons" in
the case of ribozymes). The starting premise was that
naturally occurring introns do not change the secondary
structure of tRNA precursor molecules and the tRNA
introns show no sequence conservation, and consequently,
by claiming ribozyme or antisense sequences as part of
introns, the structural changes in the resulting tRNA
precursor are minimised and the stability is
consequently maximised. By means of the tyrtRNA gene
used according to the present invention, wherein the
splicing of the tRNA precursor molecules is effected
only slowly, it is possible to demonstrate that by using
tDNA molecules which contain "ribintron" sequences the
activity of ribtRNA can be effectively increased. It is
then possible to launch a more powerful attack on RNAs
located in the cytoplasm. This system provides a simple
method of incorporating suitable structural elements for
increasing the stability of the "ribintrons" against
degradation by exonucleases. This may be achieved for
2012312
14
example by additional base-pairings or by larger hairpin
regions adjoining the ribozyme sequences. In
modifications of this kind it must be ensured that the
structures which are crucial to the splicing of the
intron are maintained.
The natural intron sequences can be modified, e.g. by
the insertion of suitable restriction cutting sites, in
order to permit the cloning of oligonucleotides or, if
they are not already contained in the naturally
occurring gene, by the insertion of nucleotides which
permit base pairing with the anticodon triplet, in order
to make available an additional stabilising structural
feature.
It has been shown, within the scope of the present
invention, that the expression of ribozymes as a
constituent of introns does not essentially involve any
deterioration in the tRNA secondary structure, and
consequently the transcript produced can be accumulated
in high concentrations and processed correctly. If the
intron released during processing proves to be
insufficiently stable, suitable structural features can
be provided which will increase the stability to
exonuclease degradation.
A number of methods may be used to introduce the genetic
units according to the invention into the cells:
The standard method of inserting DNA into tissue culture
cells makes use of the formation of a co-precipitate
between the DNA and calcium phosphate (Graham et al.,
1973). The precipitate is added to cells which take up
a certain amount, possibly by a pinocytosis process. A
similar method uses a positively charged material, DEAE-
dextrane, which makes it easier for DNA to be absorbed
by the cell. Methods have also been developed in order
212312
to introduce DNA into the cell by electroporation (in
which pores are temporarily produced by a pulsating
electrical field) (Langridge et al., 1987, Fromm et al.,
1987). Microinjection techniques for introduction into
large cells (Kressmann et al., 1980) and tissue culture
cells (Perpperkok et al., 1988) can also be used.
However, these methods are only suitable for laboratory
use or for in vitro applications. Recently, a synthetic
cationic peptide (DOTMA) was developed which
spontaneously forms liposomes with DNA and thus makes it
easier for the DNA to be carried into the cells (Felgner
et al., 1987). Theoretically, as already mentioned,
retroviral vectors are also suitable for the transfer of
genetic material into the cell (Stewart et al., 1986,
1987): however, these systems have the disadvantages
already mentioned.
Another transporting mechanism is based on the use of
"disarmed" toxins as transporting vehicles.
The methods of transport used hitherto all suffer from
the defect of being unable to convey sufficient
inhibiting nucleic acid into the cell. With the aid of
the present invention it is now possible, owing to the
smallness and compact nature of the molecules, to
increase the number of active inhibitor units in
relation to the transporting capacity. Because of the
small size and compact structure of the genetic units
according to the invention, after slight modifications,
e.g. conjugation with cholesterol, lipophilic counter-
ions or nucleus locating peptides, there is no need for
a transporting system at all.
Preferably, within the scope of the present invention, a
soluble system is used for transportion, using receptor-
mediated endocytosis. It is particularly preferable for
a transferrin-polycation conjugate to be complexed with
2012312
16
the genetic unit according to the invention; the complex
is taken up by the transferrin receptor, which is
present on virtually all growing cells.
The fields of application for the present invention are
numerous: e.g. transgenic animals can be produced which,
because of the presence of the genetic units according
to the invention in their genetic material, will have an
intracellular immunity to viruses, e.g. foot and mouth
virus, Newcastle Disease virus, bovine papilloma virus,
pseudorabies or infectious gastroenteritis.
Accordingly, intracellular immunity, e.g. against the
potato virus PVX, can also be produced in transgenic
plants. Furthermore, the genetic units according to the
invention may also be introduced into somatic cells in
order to use ribozymes or antisense-RNAs directed
against pathogenic viruses such as HIV or related
retroviruses in order to fight these viral pathogens.
Hence the invention provides, as a further aspect
thereof, a method of inhibiting RNA in vivo in a subject
comprising administering to the subject an effective
amount of a genetic unit according to the first aspect
of the invention.
Another field of use is in gene therapy by the use of
RNA constructs with complementarity to oncogenes or
other key genes which control the growth and/or
differentiation of cells. In such applications, the
high specificity of RNA inhibition which can be
effectively achieved with the aid of the present
invention and by means of which it is possible to
distinguish, for example, between protooncogene and
oncogene transcripts, acquires some importance.
Moreover, the genetic units according to the invention
may be used in this way to prevent the expression of
specific genes in plants or animals, in order to bring
. 27855-24
17 2012312
out desirable characteristics. Accordingly, the
invention includes within its scope methods of producing
transgenic animals and plants using the genetic units of
the invention.
The inhibitory effect of RNA can also be used to combat
diseases so that the production of undesirable genetic
products is suppressed, e.g. the production of the major
plaque protein which occurs in Alzheimer's Disease or
the proteins which cause autoimmune diseases.
The present invention may also be applied in those cases
in which the regulatory protein which interacts with RNA
is supposed to be eliminated by the addition of RNA.
The invention also includes pharmaceutical compositions
which contain the genetic units according to the
invention as their active component, possibly in the
form of lyophilised materials. The use thereof covers
the ranges of indications specified above. The compositions
often also contain pharmaceutically acceptahle carriers.
Using the experiments carried out within the scope of
the present invention it was possible to demonstrate the
transcription activity of the tDNA ribozyme gene. To do
this, a tDNA ribozyme gene construct was prepared by
inserting a DNA sequence coding for a 53 by long
ribozyme directed against the snRNA U7 sequence into the
ApaI restriction site between the A box and B box of the
start methionine tDNA (the A box and B box are the two
recognition sequences for the polymerase; transcription
begins 15 by upstream of the A box and ends at an oligo
T sequence downstream of the B box). After
microinjection of this gene, transcription was detected;
the concentration of the tRNA/ribozyme hybrid was 10 to
20% of the concentration of tRNA, which was produced by
a co-injected wild-type tRNA gene.
C
-- 2012312
18
RNA molecules, synthesized in vitro from the tRNA
ribozyme gene, cleave the target RNA at the site
envisaged. The addition of the tRNA structure to the
ribozyme sequence does not block the ribozyme activity.
tRNA ribozyme molecules synthesized from genes which
have been injected into oocytes also cleave the target
RNA at the site envisaged and with the same
effectiveness as ribozymes synthesized in vitro without
the additional tRNA structure. This proves that in vivo
synthesis and processing of a tRNA ribozyme are not
accompanied by modifications which interfere with the
activity of the ribozyme.
Within the scope of the present invention, the activity
of a ribozyme has been detected in vivo for the first
time. To do this, tDNA/ribozyme genes together with
radioactively labelled GTP were injected into the
nucleus of oocytes. After 8 hours incubation which was
provided for the synthesis of ribozyme, the
radioactively labelled substrate RNA (U7 RNA) was
injected into the cytoplasm of the oocytes. After
another 2 hours, the nucleic acid was removed from the
oocytes: in the oocytes in which ribozyme synthesis had
taken place, no remaining substrate RNA could be
detected. By contrast, in those oocytes which had not
been injected with the tDNA/ribozyme gene or in which
the gene had missed the nucleus, the substrate RNA was
stable.
Within the scope of the present invention it was also
possible to demonstrate that the tDNA ribozymes
according to the invention are capable of inhibiting the
transforming effect of an oncogene. Using erythroid
chicken cells, transformed with the erbB oncogene, the
activity of a tRNA ribozyme was detected by means of the
differentiation of the cells in erythrocytes which
occurred as a result of inhibition of the erbB
212312
19
expression.
The efficacy of the genetic units according to the
invention can also be tested by observing the resistance
of mouse cells to infections, e.g. polyoma infections,
after the use of genetic units according to the
invention which are directed against the virus (possibly
against several regions), e.g. against the papilloma
virus.
The invention will now be described by way of non-
limiting examples with reference to the drawings in
which:-
Figure 1 illustrates a typical tRNA molecule and a tRNA-
ribozyme-model;
Figure 2 illustrates plasmids containing sequences
coding for tRNA-ribozyme;
Figure 3 illustrates three mRNAs;
Figures 4 to 9 illustrate autoradiograms of
electrophoretic gels;
Figure 10 shows photomicrographs of fluorescing cells;
Figures 11 and 12 illustrate truncated and reinforced
tRNA-ribozymes;
Figure 13 illustrates the insertion of ribozyme
sequences into a plasmid;
Figures 14, 16 and 18 illustrate autoradiograms of
electrophoretic gels;
Figure 15 illustrates the primary structure of two tRNA
2012312
molecules; and
Figure 17 illustrates the insertion of ribozyme-
sequences into a plasmid.
Example 1
Construction of tRNA ribozyme genes.
a) Construction of pSPTl8metl
The methionine initiator 1-tRNA gene of xenopus, present
on a 284 by EcoRI fragment which was cloned in pBR322
(the HinfI H-G fragment (Hofstetter et al., 1981,
Tellford et al. 1979), was isolated by EcoRI digestion
of the pBR322 vector, purified by gel electrophoresis
(2% agarose/TBE) and ligated into the EcoRi site of the
bacterial plasmid pSPTl8 (Boehringer Mannheim) in such a
way that when the plasmid was transcribed with SP6
polymerise a sense-tRNA transcript was obtained.
Standard cloning method as described in (Maniatis) were
used for this purpose. The main advantage of the
recloning of the tRNA gene in pSPTl8 consists in the
presence of opposing SP6 and T7-RNA polymerise promoters
in the plasmid. Therefore, by in vitro transcription it
is possible to obtain specific RNA transcripts which
contain either the tRNA ribozyme sequence or the
complementary sequence (Melton et al., 1984). These
transcripts are useful for testing the cleaving activity
of the RNA molecule or for detecting the presence of
tRNA ribozymes in cell extracts which express the tRNA
ribozyme, by "RNase Protection Mapping".
b) Construction of tRNA ribozyme genes
The tRNA gene on pSPTi8metl was cleaved at the single
ApaI site in the anticodon stem and loop region (see
Fig. 2). This figure shows plasmids which contain the
-- 2012312
21
sequences coding for tRNA ribozyme. pSPTl8metl contains
the 284 by EcoRl fragment which carries the Xenopus
Laevis initiator tRNA gene. This is the G-H fragment
(Hofstetter et al., 1981), cloned into the EcoRl site of
the polylinker of pSPTl8. The complementary
oligonucleotides coding for ribozymes which are directed
against U7snRNA (CD33 and the two sequences of the erbB-
mRNA (ES13, ES53), are shown. The cloning strategy used
here reverted in the removal of the projecting ends of
the ApaI site in the tRNA gene. The part of the insert
coding for the ribozyme and the regions complementary to
the target RNA (anti-U7, anti-erbB) is marked, as are
the A and B boxes, the section of the 5 T residues
(termination signal) and the transcription initiation
sites. The plasmid contains the ColEI replication
origin, and ampicillin resistance marker and the
promoters for T7 and SP6-RNA polymerase.
In order to produce double-stranded synthetic DNA
oligonucleotides coding for the viroid cleavage sequence
(Haseloff et al., 1988) flanked by the sequences which
are complementary to the target mRNA, first of all
single-stranded oligonucleotides were produced according
to standard methods (Applied Biosystems DNA
synthesizer). Complementary oligonucleotides were
phosphorylated, annealed and ligated into the ApaI-
cleaved pSPTl8metl plasmid using standard methods
(Maniatis). The ligation mixture was used to transform
E.coli HP101, bacterial clones containing the new
plasmid were isolated and the presence of active
ribozyme sequences on the bacterial plasmid was
confirmed by two methods:
1) RNA molecules originating from the in vitro SP6
transcription of cloned DNA plasmids were incubated
with a radioactively labelled RNA containing the
target sequence for the ribozyme and tested for
2012312
22
specific cleaving of the target RNA (see Fig. 4).
2) the presence of correctly inserted DNA sequences
was confirmed by dideoxy-DNA sequencing by means of
the insertion site.
Fig. 4 shows the in vitro ribozyme activity of the tRNA
ribozymes. Plasmid DNA molecules which carried the erB-
cut tRNA ribozymes ES13 and ES53 were digested with
PvuII and transcribed with SP6-RNA polymerase. This
transcription yielded RNA molecules 230 nucleotides
long, containing the tRNA ribozyme sequence and in
addition the 5' and 3' flanking sequences which
originate from the flanking xenopus sequences and the
flanking bacterial plasmid sequences (see Fig. 2). The
ribozyme transcripts were incubated with 20,000 cpm (20
fM) of an RNA molecule with the region of the erbB-mRNA
including the initiation codon. The RNA molecule has
the target sequences both for ES13 and ES53 (see
Fig. 3). After 2 hours incubation of the ribozyme plus
target RNA at 37°C in the presence of 10 mM MgCl2, 20 mM
Tris-HC1, pH 7.5 and 150 mM NaCl, EDTA was added to a
concentration of 15 mM, the sample was dried, dissolved
in 80% formamide/TBE, heated to 95°C for 30 seconds and
separated on a 9.5% acrylamide/8.3 M urea/TBE gel.
After electrophoresis, the labelled RNA molecules were
detected by autoradiography.
Trace M: molecular weight marker: pBR322 DNA, cleaved
with HpaII and radioactively labelled using the Klenow
fragment of DNA polymerase with alpha 32P CTP (Maniatis).
The molecular weight markers were dissolved in 80%
formamide/TBE immediately before being applied to the
gel and heated to 95°C for 3 minutes. The molecular
weights of some of the fragments (in nucleotides) are
shown on the left.
Trace 1: erbB-target mRNA (20,000 cpm, 20 fMj without
incubation.
Trace 3: erbB-target mRNA (20,000 cpm, 20 fM) incubated
2012312
23
with MgCl2 at 37°C without ribozymes.
Trace 4: erbB-target mRNA (20,000 cpm, 20 fM) incubated
with ES13-RNA (1 fM).
Trace 5: erbB-target mRNA (20,000 cpm, 20 fM) incubated
with ES53-RNA.
On the right of the figure the molecular weights (in
nucleotides) of the erbB target mRNA and the 5' and 3'
cleavage products of both ribozymes splitting reactions
are given (see also Fig. 3).
Fig. 3 shows the complementarity between ribozymes and
target RNAs: CD33 (A) against U7snRNA Cotten et al.,
1988, ES13 (C) and ES53 (B) against sequences of
erbBmRNA (Venustrom et al., 1980).
The tRNA part of the tRNA ribozymes is not shown, for
the sake of clarity. The cleavage sites for the
ribozyme are marked, as are the initiation codons of the
erbB-mRNA.
Example 2
tRNA ribozyme transcription in xenopus oocytes.
The transcription of the tRNA ribozyme genes,
microinjected into xenopus oocytes, was carried out
using the method described for tDNA in Kressmann et al.,
1978, Hofstetter et al., 1981, Kressmann et al., 1980.
The following procedure was used: Stage VI oocytes were
obtained from HCG (Human Chorionic Gonadotropin)-
stimulated adult Xenopus Laevis females. The oocytes
were briefly centrifuged in order to bring the nucleus
to the edge of the oocytes. Each nucleus was injected
with about 50 nl of a solution containing 0.3 ~,g/~1 of
supercoiled plasmid DNA (containing the tRNA ribozyme
gene according to Example 1) and 2 ~,Ci/~,1 of 32P-GTP.
After 5 to 8 hours incubation at 20°C the individual
2012312
24
injected oocytes were digested for 45 minutes at 37°C in
1% SDS, 1 mg/ml of Proteinase K, 300 mM NaCl. 20 mM
Tris, pH 8, 20 mM EDTA (400 ~C1/oocyte), extracted once
with phenol and once with phenol/chloroform and
precipitated with ethanol. The collected ethanol
precipitates were dissolved in 80~ formamide/TBE,
briefly heated to 95°C in order to denature them and
separated by electrophoresis on a 10% acrylamide/8.3 M
urea/TBE gel and made visible by autoradiography. In
all the experiments with tRNA ribozyme genes the
injection solutions contained the wild-type metRNA genes
with a concentration of 1/6 of the concentration of the
tRNA ribozyme gene. TBE buffer (Tris, borate, EDTA) was
prepared according to the instructions described in
(Maniatis).
The results of these experiments are shown in Fig. 5:
Trace m: molecular weight marker: as in Fig. 4. The
molecular weights of some of the fragments (in
nucleotides) are shown on the left of the drawing.
Traces 1,2,3: the nucleic acid of individual oocytes,
injected with the met-tRNA gene and the met-tRNA
ribozyme gene metribo 33. On the right of the drawing
the positions of the met tRNA (met, 77 nucleotides long)
and of the tRNA ribozyme (metribo), 128 nucleotides
long) are given.
Example 3
Determining the ribozyme activity of tRNA ribozyme
synthesized from oocytes by comparison with ribozyme
synthesized in vitro which contains no tRNA sequences.
A tRNA ribozyme directed against U7 and synthesized in
microinjected oocytes was obtained by separation using
electrophoresis, made visible by autoradiography, cut
201232
out of the polyacrylamide gel and eluted by incubation
overnight in an Eppendorf Vibratot~in HEP (Heidelberg
Extraction buffer: 0.75 M ammonium acetate, 10 mM
magnesium acetate, 1% (vol./vol.) phenol, 0.1%
(weight/vol.) SDS, 0.1 mM EDTA). The eluted RNA was
extracted once with phenol/chloroform and once with
chloroform and precipitated with ethanol in the presence
of 10 ~,g E.coli tRNA as carrier. The precipitate was
taken up and quantitatively determined by Cerenkov
10 counting of the 3zP-labelling using the values for the
specific activity (Kressmann et al., 1982). Samples of
the tRNA ribozyme were incubated with 32P-labelled RNA
containing the U7 sequence (10,000 cpm/sample, 10 fM
plus the stated quantities of unlabelled U7-RNA) for 2
hours at 37°C in the presence of 150 mM NaCl, 10 mM
MgCl2 and 20 mM Tris HC1, pH 7.5. The reaction was
stopped by the addition of EDTA to 15 mM, the samples
were dried, dissolved in 80% formamide/TBE, heated to
95°C for 30 seconds and separated on a pre-heated 9.5%
20 acrylamide/8.3 M urea/TBE gel. The radioactively
labelled types of RNA were made visible by
autoradiography at -80°C with a Dr. Goose"special"
intensifying film.
The ribozyme CD32 was obtained by T7 polymerase
transcription of a plasmid containing the insert of CD33
(see Fig. 2) and cloned into the Hind III/sal I sites of
pSPTl9 (Boehringer Mannheim). This transcript contains
only the ribozyme sequence plus the sequences
complementary to U7, flanked by short sections of the
vector sequence. The cleaving activity of this ribozyme
was used as a comparison with the cleaving activity of
the oocyte-synthesized tRNA ribozyme CD 33, in order to
assess the influence of the secondary structure of the
tRNA and of the in vivo synthesis and modification on
the ribozyme activity. It was found that both ribozymes
(CD 32 and CD 33) cleaved the RNA containing the U7
*Trade-mark
I%vi~_.
27855-24
Y':
°~... ~,,".
,.-
201 231 2
26
sequence (94 nucleotides long) into a 5' cleavage
product with 25 nucleotides and a 3' cleavage product
with 69 nucleotides. The results of these experiments
are shown in Fig. 6:
Traces m: molecular weight markers analogously to
Fig. 5.
Trace 1: U7-RNA (10,000 cpm, 10 fM), incubated without
ribozyme.
Trace 2: U7-RNA (10,000 cpm, 10 fM), incubated with
fM of the oocyte-synthesized tRNA ribozyme CD33.
Trace 3: U7-RNA (10,000 cpm, 1 pM), incubated with 10 fM
of oocyte-synthesized tRNA ribozyme CD33.
Trace 4: U7-RNA (10,000 cpm, 100 fM), incubated with
10 fM of ribozyme CD32 synthesized in vitro with T7
polymerase.
Trace 5: U7-RNA (10,000 cpm, 100 fM), incubated with
1 fM of ribozyme CD32 synthesized in vitro with T7
polymerase.
Example 4
Determining the cleaving of ribozyme substrate in
oocytes.
A mixture of 32P-GTP, anti-U7-tRNA ribozyme gene and the
met-tRNA gene was injected into oocyte nuclei. The
injected oocytes were incubated at 20°C for 8 hours as
described in Example 2 in order to allow transcription
to take place. Then radioactively labelled U7-RNA (50
nl, 100,000 cpm/~1, 100 fM/~,l) was injected into the
cytoplasm of the oocytes. The oocytes were then
incubated for 2 hours. The preparation of the nucleic
acids of individual oocytes and the separation thereof
by gel electrophoresis were carried out as described
above.
2412312
27
The results of these experiments are shown in Fig. 7:
Traces m: molecular weight markers as in Fig. 5.
Trace 1: nucleic acids from an oocyte injected with the
met and metribo genes.
Traces 2 and 3: oocytes injected with met and metribo,
followed by U7-RNA injection.
Traces 4 and 5: oocytes injected only with U7-RNA.
Trace 6: 1 aliquot of the U7-RNA used for the injection.
Trace 7: U7-RNA (10 fM) incubated with the ribozyme CD32
(10 fM) for 2 hours at 37°C in the presence of 150 mM
NaCl, 10 mM MgCl2 and 20 mM Tris-HC1, pH 7.5. Owing to
the gel conditions only the 3' cleavage product (69
nucleotides) is shown.
Example 5
Transcription activity of tRNA ribozymes in chicken
cells.
Plasmid DNA molecules containing the tRNA ribozyme genes
were introduced into chicken cells in order to determine
the transcription activity of the genes. It was shown
that the tRNA ribozyme gene (derived from a xenopus-tRNA
gene) is efficiently transcribed in chicken cells. The
following procedure was used: 106 primary chicken embryo
fibroblast cells (Zenke et al., 1988) were seeded out in
each 10 cm dish using the standard method and left to
grow overnight. In the morning, each dish was
transfected with 10 ~cg of plasmid DNA (containing either
erbB-cut 13 or ebrB-cut 53) using the calcium phosphate
co-precipitation method (Graham et al., 1973). The
cells were exposed to the precipitate overnight, the
next morning washed twice with fresh medium and
incubated for a further 48 hours in fresh medium. The
medium was then removed, the cells were taken up in
PK/SDS buffer (see above) and the nucleic acid was
201232
28
recovered. The nucleic acid was then subjected to RNAse
protection mapping using 32P-labelled antisense erbB-cut
13 or erbB-cut 53 RNA probes in order to detect the
presence of tRNA ribozyme transcripts. Labelled RNA
(10,000 cpm/10 fM antisense-RNA per sample) were added
to the dried ethanol precipitate, the sample was dried
again and dissolved in 10 ~,1 of 80% deionized formamide,
400 mM NaCl, 20 mM PIPES, pH 6,5,10 mM EDTA. The
samples were coated with sterile paraffin oil, heated to
95°C for 3 minutes and rapidly transferred into a 45°C
water bath in which they were incubated overnight. In
the morning 0.3 ml of ice cold NaCl (300 mM), 30 mM of
Tris, pH 7.5, 1 mM EDTA, 0.05 mg/ml of RNAs A and 80
units/ml of RNase T1, were added, with rapid careful
agitation. The samples were incubated at ambient
temperature for 45 minutes. Proteinase K and SDS were
added to 1 mg/ml and 0.5% and incubation was continued
at ambient temperature and then at 56°C, for 20 minutes
in each case. The sample was precipitated with ethanol
after the addition of 10 ,ug of tRNA. The precipitate
obtained was taken up in 80% formamide/TBE and separated
on a pre-heated 9.5% acrylamide/8.3 M urea/TBE gel. The
results of this experiment are shown in Fig. 8:
Traces m: molecular weight marker as in the previous
examples.
Trace l: antisense ES13 probe, hybridized with E.coli
tRNA ( 10 /~,g ) .
Trace 2: antisense ES53 probe, hybridized with E.coli
tRNA.
Traces 3 and 4: mapping of the nucleic acids of 10,000
and 100,000 cells which had not been transfected with
plasmid DNA hybridized with the ES13 probe.
Traces 5 and 6: mapping of the nucleic acids of 10,000
and 100,000 cells, transfected with ES13, hybridized
with the ES13 probe.
Traces 7 and 8: mapping of the nucleic acids of 10,000
2012312
29
and 100,000 cells, transfected with ES53, which had been
hybridized with the ES53 probe.
Example 6
Weakening of the activity of the V-erbB oncogenes by
tDNA ribozyme genes which have been introduced into v-
erbB transformed erythroblasts using polylysine-
transferrin conjugates.
Using this example it was possible to show that tDNA
ribozymes directed against the erbB oncogene are
introduced into erbB-transformed chicken erythroblast by
means of polylysine-transferrin-conjugates and are able
to weaken the transforming activity of the oncogene.
Preliminary test 1
Preparation of transferrin-polylysine conjugates
The coupling was carried out according to methods known
from the literature (cf. G. Jung, W. Kohnlein and G.
Liiders, Biochem. Biophys.Res. Commun. 101 (1981), 599)
by the introduction of disulphide bridges after
modification with succinimidylpyridyl dithiopropionate.
Pyridyldithiopropionate-modified transferrin 1:
6 ml of a solution of 120 mg (1.5 ~,mol) of transferrin
(from chicken albumin, Sigma, conalbumin-type I, iron-
free), which had been gel-filtered over sephadex G-25,
in 0.1 M sodium phosphate buffer (pH 7.8) were mixed
with 200 ~l of a 15 mM ethanol solution of
succinimidylpyridyl dithiopropionate (SPDP, pharmacia)
with thorough shaking and the mixture was left to react
for 1 hour at ambient temperature with occasional
shaking. Lower molecular reaction products and traces
_.. ~ 2 Q 1 2 3 1 2
of reagent were removed by means of gel column (sephadex
G-25, 14 x 180 mM, 0.1 M sodium phosphate buffer pH 7.8)
and 7 ml of the product fraction were obtained; the
content of pyridyldithiopropionate residues bound to
transferrin was determined by means of one aliquot after
reduction with dithiothreitol by photometric measurement
of the quantity of pyridine-2-thione released and
amounted to about 2.6 ~mol.
Mercaptopripionate-modified polylysine 2:
10 A solution of 18 mg (about 1.0 ~mol) of poly(L)lysine
hydrobromide (Sigma, fluoresceinisothiocyanate (= FITC)-
labelled, molecular weight about 18,000 - corresponding
to an average degree of polymerisation of about 90) in
3 ml of 0.1 M sodium phosphate (pH 7.8) was filtered
over sephadex~'G-25. The polylysine solution was diluted
with water to 7 ml, 270 ~1 of a 15 mM ethanolic solution
of SPDP were added with thorough shaking and the mixture
was left to react for one hour in the dark at ambient
temperature with occasional shaking. After the addition
20 of 0.5 ml of 1 M sodium acetate buffer (pH 5.0, the
mixture was filtered over sephadex G-25 to separate off
low molecular substances (eluent: 20 mM sodium acetate
buffer pH 5.0). The product fraction (stained with
ninhydrin, fluorescent) was concentrated by evaporation
in vacuo, adjusted to a pH of about 7 with buffer, a
solution of 23 mg (150 ~mol) of dithiothreitol in 200 ~cl
of water was added and the mixture was left to stand for
one hour at ambient temperature under an argon
atmosphere in the dark. Excess reducing agent was
30 removed by further gel filtration (sephadex G-25,
14 x 130 mM column, 10 mM sodium acetate buffer pH 5.0)
and 3.5 ml of product solution of fluorescent-labelled
polylysine were obtained containing 3.8 mol of mercapto
groups (photometric determination) using Ellman's
reagent, 5,5'-dithiobis(2-nitrobenzoic acid).
*Trade-mark
27855-24
2012312
31
Transferrin-polylysine conjugate 3:
The solution of modified transferrin 1 obtained as
described above (7 ml in 0.1 M sodium phosphate buffer
pH 7.8, about 1.5 ~mol of transferrin with about 2.5
~,mol of pyridyldithiopropionate residues) was rinsed
with argon; 2.0 ml of the above described solution of
mecapto-modified polylysine 2 (in 10 mM of sodium
acetate buffer pH 5.0, corresponding to about 0.6 ~,mol
of polylysine with about 2.2 ~,mol of mecapto groups)
were added, the mixture was rinsed with argon, shaken
and left to react for 18 hours at ambient temperature in
the dark and under argon. The reaction mixture was
diluted to 14 ml with water and separated by ion
exchange chromatography (pharmacia mono S column HR
10/10, gradient elution, buffer A: 50 mM HEPES pH 7.9,
buffer B: A + 3 M sodium chloride, 0.5 ml/min). Non-
conjugated transferrin was eluted first, product
fractions at about 0.66-1.5 M sodium chloride. The
conjugated products (ninhydrin staining, protein
absorption in UV at 280 nm, and fluorescence) were
collected in 6 fractions each containing about 10 mg of
transferrin. First, the fractions were dialysed against
a 100 mM iron (III) citrate solution (adjusted to pH 7.8
with sodium hydrogen carbonate) and then twice more
against 1mM HEPES buffer (pH 7.5). Sodium dodecyl
sulphate gelelectrophoresis (10% SDS, 8% polyacrylamide)
showed roughly the same content of transferrin in all
six fractions when pre-treated with 2-mecaptoethanol,
whereas in non-reduced samples no bands were visible for
free transferrin but only for less wide ranging
conjugates.
Preliminary test 2
Transport of transferrin-polylysine conjugates in living
2012312 .__
32
cells
In order to show that the transferrin-polylysine
conjugates described in preliminary test 1 are
efficiently taken up in living erythroblasts, these
conjugates were labelled with FITC. It is known
(Schmidt et al., 1986) that FITC-labelled transferrin
was detectable in vesicles within the cell after some
hours incubation with erythroblasts from which
transferrin had previously been removed (examination
under a fluorescence microscope).
In the present example, erythroblasts (transformed by an
EGF receptor retrovirus, Kahazaie et al., 1988) were
incubated for 18 hours in transferrin-free
differentiating medium (composition in Zenke et al.,
1988) at 37°C (cell concentration 1.5 x 106/ml). After
the addition of the various transferrin-polylysine
conjugates (or as a control, the corresponding amount of
sterile twice-distilled water) the cells were incubated
at 37°C in the presence of 10 ng/ml of EGF (in order to
maintain the transformed state). After 24 and 48 hours,
about 5 x 105 cells were taken, washed once in phosphate-
buffered physiological saline solution (PBS; pH 7.2),
fixed with fifty times the volume of a mixture of 3.7~
formaldehyde and 0.02% glutaraldehyde in PBS (10 min,
40°C), washed once in PBS, embedded in Elvanol and
investigated under a fluorescence microscope (Zeiss
Axiophot, narrow band FITC and TRITC excitation). At
the same time, the growth rate of the cells was
detenained in other aliquots of the various mixtures.
100 ~C1 of cell suspension were taken and the
incorporation of 3H-thymidine (8 ~Ci/ml, 2 hours) was
determined, as described in Leutz et al., 1984. Fig. 10
shows that the erythroblasts incubated with transferrin-
polylysine have 2 to l0 strongly fluorescing vesicles
after 24 hours, which cannot be detected in the
x
2p12312
33
controls. Table A (at the end of the description) shows
that with the exception of fraction 6 all the conjugates
have been absorbed by virtually all the cells.
The fact that the cells grow equally fast in all the
samples (as measured by the incorporation of tritiated
thymidine (3H TdR); Table A) shows that the cells are not
damaged by the polylysine constructs and consequently
non-specific uptake (e. g. through cell membranes which
have become permeable) can be ruled out.
Preliminary experiment 3
Polylysine-transferrin constructs can functionally
replace the native transferrin-ion complex in the in
vitro induced maturation of chicken erythroblasts to
form erythrocytes.
The objective of this experiment was to show that the
transferrin-polylysine conjugates used here can be used
by the cell-like native transferrin, i.e. they can pass
through the normal transferrin cycle with similar
efficiency. Erythroblasts which can be induced to
mature into normal erythrocytes by "switching off" the
transforming oncogene are particularly suitable as a
test system for this purpose (Beug et al., 1982). The
literature shows that such cells require high
concentrations of transferrin-iron complex for normal
maturation (100-200 ~g/ml, three times lower
concentrations prevent the maturation of the cells and
will result in the death of the cells after a few days
(Kowenz et al., 1986). It has also been shown (Schmidt
et al., 1986) that recycling, i.e. the reuse of
transferrin receptors and hence a transferrin cycle
proceeded at optimum speed is indispensable for normal
in vitro differentiation.
201 2312
34
Erythroblasts (transformed by the EGF receptor
retrovirus) are induced to differentiate by the removal
of EGF and addition of an optimum amount of partially
purified chicken erythropoietin (Kowenz et al., 1986,
free from transferrin). Incubation was carried out at a
cell concentration of 1 x 106/ml in transferrin-free
differentiating medium at 42°C and 5% COz. At the start
of incubation, either native transferrin-iron complex
(Sigma, 100 ~g/ml) or the iron-saturated transferrin-
Polylysine conjugates (concentration again 100 ~,g/ml)
were added. The growth and state of maturity of the
cells were analyzed after 24 and 48 hours in the
following way:
1. by determining the number of cells (in the Coulter
Counter, Model ZM, Beug et al., 1984)
2. by recording cell size distributions (in a Coulter
Channelyzer~Mod. 256) and
3. by photometric measurement of the haemoglobin
content of the cells (Kowenz et al., 1986)
In addition, aliquots of the mixtures were centrifuged
after 72 hours in a cytocentrifuge (Shandon) on an
object carrier and subject to histochemical
investigation to detect the haemoglobin (staining with
neutral benzidine plus Diff-Quik~rapid staining for
blood cells, Beug et al., 1982).
The results in Table B (at the end of the description)
clearly show that cells induced to differentiate in the
presence of the polylysine transferrin conjugates
fractions 1 to 5 mature just as efficiently and at the
same speed as those which were incubated with native
transferrin-iron. The cells in the transferrin-free
controls, on the hand, showed a much slower cell growth
and accumulated only small quantities of haemoglobin.
Investigation of the cell phenotype on stained cytospin
*Trade-mark
27855-24
2012312
preparations showed that the cells incubated with
polylysine-transferrin conjugates were matured to
produce late reticulocytes (late reticulocytes, Beug et
al., 1982) in just the same way as the cells treated
with native transferrin whilst the cells incubated
without transferrin constituted a mixture of
disintegrated and immature cells resembling
erythroblasts (Schmidt et al., 1986). Only the cells
treated with transferrin-polylysine fractions 6 showed a
lower haemoglobin content and a higher percentage of
immature cells (Table B). This shows that fractions 6,
conjugated with a particularly large quantity of
polylysine, functions less well in the transferrin
cycle. At the same time, this result indicates the
sensitivity of the test method.
Preliminary test 4
Polylysine-transferrin conjugates permit the uptake of
DNA in chicken erythroblasts.
The present experiment was intended to investigate
whether DNA can be efficiently transported into the
interior of the cell by transferrin-polylysine
conjugates in a size corresponding to that of tDNA
ribozymes (see example 1). In the present example, tDNA
was used with an insert of the sequence
CGTTAACAAGCTAACGTTGAGGGGCATGATATCGGGCC
CCGGGCAATTGTTCGATTGCAACTCCCCGTACTATAGC,
molecular weight about 300,000, terminally labelled with
gamma 32P ATP (Maniatis). Approximately 0.3 ~.g of this
DNA, dissolved in 20 ~,1 of TE buffer, were mixed either
with 10 ~,g of native transferrin, with 10 ~.g of
transferrin-polylysine conjugate fraction 3, each of
them dissolved in 50 ~1 of twice-distilled water, plus
400 ug/ml of bovine serum albumin (Beug et al., 1982) or
2012312
36
with 50 ~.1 of this solvent without transferrin. The
DNA-protein mixtures were added to 2 ml of transferrin-
free differentiating medium, 4 x 106 chicken
erythroblasts (which had been transformed with an EGF
receptor retrovirus and pre-incubated for 18 hours in
transferrin-free medium in the presence of EGF (Kahazaie
et al., 1988)) were added and the mixtures were
incubated for 8 hours at 37°C and 5% COZ. Then the cells
were removed by centrifuging, the supernatant was
removed and the cells were washed three times in
transferrin-free medium. The cell sediment and culture
medium were taken up in 1% SDS, lmg/ml Proteinase K, 300
mM NaCl, 20 mM Tris pH 8.0, 10 mM EDTA (PK/SDS buffer),
incubated for 30 minutes at 37°C, extracted with
phenol/chloroform, and the DNA was isolated by ethanol
precipitation. Isolated DNA with a radioactivity of
2000 cpm in total was separated on a non-denaturing 3.5%
acrylamide gel (TBE, Maniatis) and the DNA was detected
by autoradiography. The figure shows fluorescence
images of chicken erythroblasts which had been incubated
for 24 hours without (A) or with FITC-labelled
transferrin-polylysine conjugates (B, C). When activated
with blue light (B, in order to detect FITC),
significantly more fluorescent vesicles can be seen in
each cell. The specificity of this fluorescence is
shown by the fact that the vesicle fluorescence does not
occur on activation with green light (in which a non-
specific fluorescence of the cells can be seen similar
to that in A) (C).
This figure shows that in the cell sample treated with
transferrin-polylysine, approximately five to ten times
more DNA has been absorbed by the cells than in the
control samples with native transferrin or with no
transferrin.
Two tRNA ribozyme genes, directed against the
2Q12312
37
translation initiation region of erbB were constructed
(cf. Figs. 2 and 3, Example 1). About 100 dug of each
plasmid containing the gene was digested with EcoRI in
order to free the tRNA ribozyme gene on a 225 by
fragment. The digestion products were terminally
labelled with klenow fragment and purified by gel
electrophoresis using a 2~ agarose/TBE gel. The vector
fragment and the tRNA ribozyme gene fragments were
located by ethidiumbromide staining, cut out and
obtained by electroelution, phenol/chloroform and
chloroform extraction and ethanol precipitation. The
purified, radioactively labelled DNA fragments were then
used, making use of the transferrin-polylysine transport
system, to determine the uptake and inhibition of the
erbB-RNA. The vector pSPTl8 was used as control-DNA.
The test cell system chosen was a chicken erythroblast
cell line transformed by a temperature sensitive mutant
(ts 34, Graf et al., 1978) of the avine erythroblastosis
virus AEV (Beug et al, 1982 b). (The erbA oncogene
which is also expressed in these cells can be inhibited
by a specific protein kinase inhibitor (H7)). It has
been established that the v-erbA oncogene is
phosphorylated in vivo and in vitro (i.e. as a
bacterially expressed protein) at two sites, namely Ser
28 and Ser 29, by protein kinase C or by cAMP-dependent
protein kinase. Mutation of these serines into alanines
prevents phosphorylation and destroys the v-erbA-
oncogene activity. H7 is a specific inhibitor of these
two kinases and is capable of selectively stopping the
changes caused by v-erbA (e.g. blocking of
differentiation) in erythroblasts which contain v-erbA-
v-erbB.
It is known that erythroblasts in which the erbB
oncogene is inactivated (e. g. by a temperature increase
in the case of a temperature-sensitive erb8 mutant) can
201231 2
38
be induced to cause erythrocytes to mature. One of the
first indications of this process is an induction of
haemoglobin synthesis which can be detected by sensitive
investigation (acidic benzidine staining, Orkin et al.,
1975, Graf et al., 1978) at the level of the single
cell. Thus, a specific increase in the number of
benzidine-positive cells could be expected as a
phenotypical effect of a ribozyme directed against erbB
in this test system.
The test series on which this example is based was
carried out as follows: the various DNA preparations
(see above and Table C which Table is to be found at the
end of the description), dissolved in 30 ~1 of TE
buffer, were mixed with 10 ~g of native transferrin-iron
complex or transferrin-polylysine conjugate (dissolved
in 50 ~C1 of twice-distilled water) and incubated for 30
minutes at 37°C.
In the case of the vector DNA (10 ~,g) correspondingly
more (100 ~cg) of transferrin preparations was used. The
DNA-transferrin-DNA mixtures were each added to 1 ml of
transferrin-free differentiating medium (Zenke et al.,
1988). The test cells (per batch 3 x 106) were incubated
for 60 minutes at 42°C before the test, in transferrin-
free differentiating medium (to increase the uptake of
transferrin) and added to the mixtures which contain
DNA-transferrin. After 6 hours, 18 hours and 68 hours
(for the treatment of the cells see below) samples were
taken as described, separated into supernatant and cell
sediment, taken up in PK/SDS buffer and the DNA was
analyzed (Figure 9).
After the end of incubation (6h) the cells were
centrifuged off and incubated for a further 72 hours in
a transferrin-containing differentiating medium with
erythropoietin and insulin (Kowenz et al., 1986, Zenke
2012312
39
et al., 1988), 2 ml per batch, at 37°C, i.e. in the
presence of an active v-erbB protein).
The following results were obtained:
1. as in preliminary test 4, an increase uptake of DNA
was observed in the size of the erb-cut DNAs in the
cell samples treated with transferrin-polylysine
(approximately 5-fold).
2. by transfection of chicken fibroblasts with erb-cut
DNA it was demonstrated that the erb-cut ribozyme
tDNA is expressed in chicken cells (see example 5).
3. Table C shows that in every case where erb-cut
ribozyme tDNA was introduced, with the aid of
polylysine-transferrin constructs, into erbB
transformed erythroblasts, the percentage of
benzidine-positive cells was significantly
increased (approximately doubled) the reference
used was the samples treated with vector DNA, in
which the use of polylysine-transferrin conjugates,
as expected, did not result in an increase in the
number of benzidine-positive cells).
Example 7
Extending the anticodon stem region increases the yield
of ribtRNA.
The human met tRNA gene (cloned as a bamHI/RsaI
fragment, complemented by EcoRI linker in the EcoRI
cutting site of the Bluescript vector) was cleaved at
its single ApaI cutting site in the anticodon stem
region. The single-strand overhangs were removed by
treatment with T4-DNA polymerase and oligonucleotides
containing the ribozyme sequences were inserted. The
ribozyme insertion used in the preceding examples
resulted in a ribtRNA molecule containing a three-base
212312
stem (Fig. 11 shows the wild-type tRNA met and the tRNA
rib with the shortened anticodon stem). In the second
construction an oligonucleotide was used which differs
from the first by the sequence GGTTAT, complimentary to
the wild-type sequence, at the 5' end. The wild-type
strain was reestablished and extended by four additional
base pairs (Fig. 12 shows the construction of the tRNA
rib with the strengthened anticodon stem. The sequence
of the ribozymes is shown in Figs. 13; at the left hand
(5') end are shown the differences in the nucleotide
sequences between the region coding for the shortened
stem and the region coding for the strengthened stem.
Fig. 13 also shows the sequence of the met tRNA gene).
The two ligation products were transformed in E.coli HB
101 using the standard method described in Maniatis, the
plasmid DNA was isolated and sequenced in order to
determine the correct structure. The two resulting
ribtRNA genes were investigated by microinjection of the
cloned DNA into xenopus oocytes, carried out as
described in Example 2 or 3, in the presence of 32P-GTP,
for any evidence of transcription activity and
accumulation of the ribtRNA molecules. The wild-type
tRNA gene was co-injected at a ten times lower
concentration. The injected oocytes were incubated for
7 hours at 20'C and the resulting RNA was harvested (cf.
Example 2) and separated by electrophoresis (10%
acrylamide/8.3 M urea/TBE gel) and the RNA molecules
were made visible by autoradiography (two days exposure
at -70'C) (Fig. 14). The shortened ribtRNA gene yields
about one tenth of the quantity of RNA transcribed by
the wild-type gene, whilst in contrast the gene which
codes for the ribtRNA molecule with the longer stem
yields six times as much RNA.
Example 8
Expression of ribozyme genes as a constituent of
2012312
41
introns.
The starting gene used was a Xenopus Laevis tRNAt''rC gene
(oocyte-type) containing an intron comprising 13
nucleotides. The natural intron sequence was modified
as follows: first of all, a suitable restriction cutting
site was inserted (ApaI; GGGCCC), to permit subsequent
cloning of oligonucleotides, and secondly complimentary
nucleotides to the anticodon triplet were inserted in
order to have an additionally stabilising structural
feature by extending the intron sequence. The size of
the intron in the modified gene is increased from 13 to
15 nucleotides (Fig. 15).
The modification of the intron sequence was carried out
using the polymerase chain reaction (PCR; Ho et al.,
1989). Four primers were synthesized, two of which
contain the altered intron sequence (complimentary to
one another) whilst two were directed against the 5' or
3' end, respectively, of the gene, in order to introduce
an EcoRI or SalI restriction cutting site. The wild-
type gene was present as an Hha1 fragment(258 bp),
cloned in pBR327 (Stutz et al., 1989). The resulting
PCR product was purified over an agarose gel, cut with
the abovementioned restriction enzymes and ligated with
the vector pAALM (= pSP64 + T7 promoter; Vieira and
Messing, 1982). The construct was transformed in E.coli
HB 101 and clones containing the desired insert were
identified by sequence analysis.
The activity of the modified gene (tRNA°y=M), was
compared with that of the wild-type gene by
microinjection into xenopus oocytes, whilst an as an
internal standard a 5S-RNA gene (fifty times lower
concentration), which was present on the plasmid pUC-10-
5S (Caroll and Brown, 1976), was co-injected in the
presence of 32PGTP. The injected oocytes were incubated
20~ 23~ 2
42
for 20 hours at 20°C, the RNA was separated on an 8%
acrylaraide/8.3 M urea/TBE gel and autoradiographed (Fig.
16). In addition to the 5S-RNA at 120 nt the primary
tRNA°yr transcript was visible at 100 (102) nt, the 5'
and 3' processed precursor form at 90 (92) nt and the
finished processed tyrosine tRNA at 76 nt. The splicing
of the 90 nt precursor form appears to be~the limiting
factor, with the result that the majority of the
transcript formed (about 80%) is present in this form.
As expected, the biological activity was not reduced by
a modification from the wild-type gene.
In another experiment the capacity of the system was
tested. Two oligodeoxyribonucleotides which contained
ribozyme sequences were synthesized, which already
contain ApaI ends and were thus able to be cloned
directly into the intron sequence of the modified tRNAt''r
gene. In an oligonucleotide, 12 nt were inserted at
both ends in order to form stable "hairpins" in
ribintrons and thus counteract the degradation of
exonuclease. The overall size of the resulting introns
was 80 nt (ribozyme HP) as compared to 65 nt in the case
of the unprotected ribozyme sequence (ribozyme C). the
sequences of the ribozymes and the cloning plan are
shown in Fig. 17.
For the microinjections into xenopus oocytes, analogous
with those described above, in addition to the two
constructs already described a third was used which
contains the ribozyme HP in dimeric form and thus
increases the intron size to 163 nt (ribozyme D). The
concentration of the co-injected 5S standard was 1:20
for constructs HP and D and 1:1 for construct C
(Fig. 18). The experiment shows, that, in spite of
substantially enlarged introns, the constructs HP and C
are very actively transcribed and processed with the
same efficiency as the wild-type tRNAt''r gene. In the
.. 2012312
43
case of construct D, only a minimal quantity of
transcript is detectable since obviously the secondary
structure required for the formation of a PoI III
transcription complex was destroyed by the long intron
sequence.
It was possible to demonstrate that the expression of
ribozymes as introns of tRNAs does not result in any
major deterioration in the tRNA secondary structure, as
a result of which the transcript produced can be
accumulated in a high concentration and processed
correctly.
- 2p12312
0
0
0
U U U U U U U U U
~1
'.' .O O O O O O O O O
~1'
''~ E' O ~ c~ ~ M .-t M In
U1 ~ 'd~ N ('] t0 tn tl7 l W
W M D
.-1 .-~ .-, .-1 ~ .-~ .-1
.-,
t~
O
+ + + + +
.C + + + + + t O
.1..1 ~C d~ do + + + + + + U
d d~ d~ op oh do
O U CO .-i .-1 N
Ov ~ pv pv p~ ~ U
d~ v v n n n n n n
v
v
+ + + + + 0
+
'''~ dp dP + + + + +
O ' dp dP dF v
+ + +
~ w . .-i .-1 GOp tOp d~ U
~' oW ,
d~ v v pt ~ c0 ctf c~ UI
Q1 n n U U U v
N n
O
.~ 1
.~ >~
to ..~
Ei m
O
'- .-! N M d~ u7 V .L~
1 O Gr 4r GL L.a Ia, Lrr tll
w
~ ~, cw a a a a a a
o ., -~,
to 1~ zi x w ~..~1w' w w w
H H H H H H
w ~., " ~ .-1 .~ .-1 .-a .-~ .-1 v
o ~ ~. x
b ~ ~r v ~r ~r
t~ o ~3 ~r ~r ~r v
H i?, .-i .-a .-t .~ .-.~.-.1
H O
+~
v
E
U
E E E E E E E E
N N N N N N N N W
+
+
+
i
U
i b
ca
p4 .-j f'1 M d~ U1 v0 ~ cp +
... - 45 - 20 1 2 3 1 2
_ ; I
i
w o
'-'
O w .-~
O a. ~
>~ ~-' U O
O O w-! dP ('~
-~-I
W N l-v
+~
oa cb rJ c? y t7
S-a ~
N
r.
b~ ~ S-a '~ ~
O O ~ O -C~ f~ ~
G1 E da b .--t.-t CJ . p - O O
N .
l~ n v v n ~ n ~ ~ ~ N
x
v
0
.c ~ c~ M ~ .J rr m t~ c~ M
!~ U1 -~7 L~ N . W tn tn tn d' Ov
O
b
~
O
N
C N O O N N N N N .-i
O O
Sa .~
i.~ O N N
v s~ x a~ ~C m In o .o .n oo c~ .-t ~ o
w .C ~ M c'~ ~ c~ .-i tn u7 ~ .-~ p
c'~
N
C ~, W N .--tO O .-a ~ .~ rl ri .-1
~ f.~ -
S-~ v
H
w !~
O O
crs
E !~ +~
S-a O U
a7 vo N d~ ~ d W vD
O t
'
,~ ~ C tn C' d~ N In In N
~
~ .--i
Cp
CC U v ~' ~" N N d~ ~f' Q~ C' d' d'
~
tts ~ U
~ C1
fly ~ w O
Er
v O
X
N v O
-_ ~p N C O ~ N b ~ p
?, U z v
N ~ ~ t~ ~'7 t~ ~ C~ tf7
N s~
('~ N N C'~ ('~ C~ M ,c'~(~ M
>~ -..~
O I
O I
Yi
U 1-~
~ -r-i .(~ C
W ~ f~
w O i-r
-.-I tr O
~ -.-i +~ 'U 0
f0 ~i w N
U N 'D Vi ~i cue:c ~ tn
:
~
+~ W O L~. (:. L~. 1J Lr ~.- ~
G
, U
~
H a a a a a a a
I o s.~,a cz, u. c.~.a,
~
a
U V' w w
V- C-- I x E -i E-~ H H E. v
~ . ,
~
O
I N
w: i i
H
H I
O
U
-,-1 .--i'--ir-i .-I r-i '-i .-i .-( r-l ~
1:..
z7 E E E E E E E E E
N N N N N N N N N t0
p
a
.
O
Gr
O
N c'' '~l"In ~O t~ a0 Cn
..
- 46 - 20 1 2 3 1 2 _ __ .
__ _ ~ ~ _;
_
>~
v v
O
I U
, t~
'p +~ N
. CT
v s~ .c a
: ~ s
.rs v .u _ v
- -~
_ _ _ _
S~ ~ ~ c~ .-. ~ ._
p ~ w r N N N N N N N
- -rl
tn O O ,~ ~. v ~. ~ .-. .r ..
co
,O U a..t r3
' '
to N '01 N ( d' N r-1 f N N N
~ ~
p i 1 1 1 1 1 1 i
'p
v rl rI + + + + + + +
~ v
.O +.t tf~ L~ In N M N .~ Cfl
f~
t>j O w1 r! f~ N ~ N N N t7
- -r1
r-Wl~
~
fs O
I
U
.~, v ~ .>~
v
x -- .-t .-t .~ ,-~ ~, ~ .~ ~, o
.~
'd' v v v v v v v v
m
v
''' Ot t7t t7t to ~ ~ C7t t~
O o
O
0 0 0 0 0 0 0 0
v w v
z ~ m nn W n
w w w w '~
~
v H ,
v
~' o a~ v a a a a
.~ 4- ~' Q, ~. a. n,
>,
it cn ~ w w w w w w a-c w
N
N >r o E' E, E. N E H H
U
>",
U
+s ~
~
1 O
~ 4-~
pt C3: O O
G~ ~ O
O O - '
U
.-1 .-i .-4 ~-t O O
U
V7
O
I ~ O
:~
U
N
v
tt7 tn ~ N
O O O Sr
U1
_ .-i .-I .-i O v
c0
~ 3
U z N N N G7 E
a cn
M ch
j ' U
~,. 'v
+~ +~
p c'~ c'~ M t'~ ~ ~ M ~ o
N
~-i e-1 if7 lf7 ~ ~ '"i ''~
N
N N
'
1~ ,~,~ a
C
G
.,-1 .,-1 lCS
ri
~ .c
o Q, ~ ~ ~ ~ s~ ~ fa ~ ~ +~
~ w
a., U U U U O N O N U U O
H I I 1 I .1-t +t 1 I U
O O
.
.t7 ..CI~ .s7 U .Q U ~ ~ .L7 Q
O
v
v r~
z
~s ,
..
'
.-t N M ~ N ~O 1~ C'~ ~O
ll